

Abstract
The rostral ventrolateral medulla (RVLM) is a key region in cardiovascular regulation. It has been demonstrated that cholinergic synaptic transmission in the RVLM is enhanced in hypertensive rats. Angiotensin-converting enzyme 2 (ACE2) in the brain plays beneficial roles in cardiovascular function in hypertension. The purpose of this study was to determine the effect of ACE2 overexpression in the RVLM on cholinergic synaptic transmission in spontaneously hypertensive rats (SHRs). Four weeks after injecting lentiviral particles containing enhanced green fluorescent protein and ACE2 bilaterally into the RVLM, the blood pressure and heart rate were notably decreased. ACE2 overexpression significantly reduced the concentration of acetylcholine in microdialysis fluid from the RVLM and blunted the decrease in blood pressure evoked by bilateral injection of atropine into the RVLM in SHRs. In conclusion, we suggest that ACE2 overexpression in the RVLM attenuates the enhanced cholinergic synaptic transmission in SHRs.
Keywords: Hypertension, Renin-angiotensin system, Gene transfer, Acetylcholine, Sympathetic nerve activity
Introduction
Hypertension is the most important risk factor for cardiovascular and cerebrovascular diseases. However, the mechanism underlying hypertension, especially essential hypertension, is still unknown [1]. Although there are several ways to return blood pressure (BP) to normal, such as antihypertensive drugs and life-style improvements, the main complications of hypertension, such as stroke and heart failure, are harmful to health [2]. It is of vital clinical importance to understand the mechanism of essential hypertension. Sympathetic nerve overactivation, the main cause of essential hypertension, has a close relationship with the death rates from cardiovascular accidents [3]. So clarifying the mechanism of sympathetic nerve overactivation is important for the prevention of hypertension.
The rostral ventrolateral medulla (RVLM) is a critical region where sympathetic premotor neurons reside. The RVLM is involved in the regulation of cardiovascular functions and plays an important role in maintaining resting BP and sympathetic tone [3–5]. Considering the key role of the RVLM and its pre-sympathetic neurons in the integration of peripheral cardiovascular activity, its dysfunction plays a vital role in the development and progression of essential hypertension. Studies have shown that several factors, such as the inflammatory response and oxidative stress, increase the excitability of neurons in the RVLM and lead to overactivation of sympathetic outflow [6, 7].
The enhancement of excitatory synaptic transmission (mainly glutamate) in the RVLM is one of the important mechanisms for the enhanced sympathetic outflow and BP elevation [5]. Acetylcholine (ACh), an important excitatory neurotransmitter, is also involved in the mechanism of sympathetic outflow overactivation [8]. Previous studies have shown that a cholinergic mechanism is involved in the regulation of the cardiovascular system by the RVLM [9–11]. It has been reported that the pressor response evoked by activation of M2 muscarinic receptors in the RVLM is greater in spontaneously hypertensive rats (SHRs) than in normotensive Wistar Kyoto (WKY) rats [12]. Furthermore, ACh release and choline acetyl transferase (ChAT) activity in the RVLM are both enhanced in SHRs [13]. These findings suggest that cholinergic transmission in the RVLM is enhanced under hypertension, and this may contribute to the maintenance of a high BP and high levels of sympathetic tone.
Increasing evidence has indicated that the brain renin–angiotensin system (RAS), especially angiotensin II (Ang II), is enhanced under hypertension and critical in its development and maintenance [3, 6]. As an important component of the RAS, angiotensin-converting enzyme 2 (ACE2) catalyzes the formation of Ang-(1–7) and other related peptides in the peripheral system [14]. Further discoveries of ACE2 in both the peripheral and central cardiovascular systems have shown that it provides a long-term beneficial outcome to the cardiovascular system and is considered to be a novel therapeutic target for cardiovascular diseases such as hypertension [14, 15]. It has been demonstrated that ACE2 is widely expressed in cardiovascular regulatory regions [16, 17]. Furthermore, it has been reported that ACE2 expression and activity in the RVLM are decreased in SHRs, while ACE2 overexpression in the RVLM improves the cardiovascular dysfunction under hypertension [18]. However, the exact mechanism by which central ACE2 acts on hypertension remains unclear and requires investigation.
Interestingly, previous evidence has indicated a possible interaction between the RAS and cholinergic transmission in the central nervous and peripheral systems [19]. However, the specific mechanism involved in the effect of the RAS on enhanced cholinergic transmission in the RVLM is still unclear. Although ACE2 overexpression in the RVLM results in improvement of the cardiovascular dysfunction in SHRs [20, 21], there is no direct evidence for a relationship between ACE2 and cholinergic transmission in the RVLM. Therefore, the major purpose of the present study was to determine whether the enhanced cholinergic transmission in the RVLM is depressed by ACE2 overexpression in hypertension.
Materials and Methods
Animals
Sixteen-week-old normotensive male WKY rats and SHRs were purchased from Sino-British SIPPR/BK Laboratory Animal Co., Ltd (Shanghai, China). The rats were kept in a temperature-controlled (25 °C) room under 12 h of light and darkness, with free access to food and water. All procedures were approved by the Institutional Animal Care and Use Committee of the Second Military Medical University.
ACE2 Transfection and General Procedures
The processes of cloning human hACE2 into lentiviral vector and the construction of Lenti-ACE2 were as follows. The Lenti-ACE2 virus particles were packaged by Shanghai Innovation Biotechnology Ltd. (Shanghai, China), following the description in our previous study [22]. hACE2 cDNA (Accession No. NM_021804) was used as a template for polymerase chain reaction amplification using the well-designed primers: forward 5’ ATGCTGCGCGCCGCACTCAGCAC-3’ and reverse 5’ TTACGAGTTCTTCTGTGGCACTT-3’. The lentivirus cloning vector pLenO-DCE (Invabio Biotechnology Ltd. Shanghai, China) carrying a green fluorescent protein (GFP) reporter gene was used to clone the amplification fragment of hACE2. The constructed control lentivirus particles (Lenti-GFP) contained all the other sequence elements except for hACE2. In brief, to facilitate transfection of the transgene plasmid (pLenO-DCE-ACE2) into 293T cells, the lentivirus packaging helper plasmids pRSV-Rev, pMDLg/pRRE, and pMD2.G were selected. The media of 293T cells were collected at 48 h post-transfection and the lentivirus particles were enriched as follows: the collected virus-containing medium was concentrated by ultrafiltration and then ultracentrifugation, aliquoted, and stored at − 80 °C. The number of GFP-positive cells in 293T cell suspensions by FACS analysis was used to determine the number of transducing units, each defined as an infectious particle, and the final infectious titer used for the subsequent animal experiments was 5 × 109 transducing units/mL.
Lenti-ACE2 Delivery into the RVLM
After rapid induction of anesthesia with 5% isoflurane, rats were maintained in an anesthetic state with 3% isoflurane and the surgical procedures were as in our previous studies [23, 24]. After drilling two symmetrical holes in the skull, a 32-gauge Hamilton springe (5 μL) was used to deliver lentiviral particles (250 nL) slowly (20 min) into the RVLM (10 mm relative to the skull surface) in a stereotaxic frame. The speed of injection was regulated by an ultramicropump (World Precision Instruments, Sarasota, FL). After viral injection, the rats were kept individually in clean conditions, and were divided into two groups (SHR-lenti-GFP and SHR-lenti-ACE2).
Measurement of BP in Conscious Rats with a Tail-Cuff System
BP and heart rate (HR) in the conscious state were monitored weekly via a computer-assisted noninvasive tail-cuff system (Shanghai Alcott Biotech Co., Ltd, Shanghai, China), as previously described [25]. Before measuring BP and HR, rats were placed in a recording chamber to adapt to the restricted environment; and before treatment, the values of BP kept stable between two measurements and the fluctuations did not exceed 5 mmHg. Two parameters were simultaneously measured: systolic BP and HR. Rats were adapted to the recording chamber for 10 min–15 min before BP was measured, so they were resting comfortably in the recording chamber during the monitoring procedure. The cuff was placed around the tail. The cuff was connected to a sensor, and the BP signal was amplified and recorded by a data-collection system. All values were expressed as an average of at least 10 consecutive measurement cycles. BP and HR were monitored during injection of lentivirus particles to the RVLM and every subsequent week.
Measurement of ACh Concentration
RVLM microdialysis was done as previously described [26]. Three dialysates were collected via the microdialysis system for 10 min and stored at − 80 °C. The concentration of ACh was measured using a Choline/Acetylcholine Quantification Kit (MAK056, Sigma, St Louis, MO). After euthanasia by overdose with pentobarbital sodium (200 mg/kg), each rat’s brain was removed rapidly and frozen in liquid nitrogen. Extracted RVLM tissues were lysed with cell lysate, ultrasonicated, and centrifuged in a cryogenic centrifuge. Supernatants were collected and used to measure the concentration of ACh following the manufacturer’s instructions. The concentration of choline was assessed via a coupled enzymatic reaction, which produced a colorimetric (570 nm)/fluorometric product (lex = 535/lem = 587 nm), proportional to the choline present. ACh levels were determined by acetylcholinesterase, which hydrolyzes ACh to choline and acetate.
Measurement of Norepinephrine (NE) in Urine by High-Performance Liquid Chromatography (HPLC)
As previously described [27], the excretion of NE in 24-h urine was assessed by HPLC (model 582 pump, ESA, USA) with electrochemical ion detection (model 5300, ESA). In brief, metabolism cages were used to collect 24-h urine samples. The samples were acidified with glacial acetic acid in 15-mL centrifuge tubes, which were kept at low temperature in crushed ice. After centrifugation of samples, the supernatant (40 µL) was delivered into the HPLC column [reverse phase, ESA, 150 × 3.2 mm, 3 µm C18 (P/N 70–0636)], and the mobile phase (80 mmol/L citric acid monohydrate, 73.4 mmol/L citric acid trisodium salt, 0.12 mmol/L 1-octanesulfonicacid sodium salt, and 0.1 mmol/L EDTA, adjusted to pH 4.3 with phosphoric acid) was used to elute NE. Dihydroxybenzylamine (Sigma) served as the internal standard. The flow rate was set at 0.5 mL/min.
Western Blot
As in our previous study [28], Western blot was used to assess the protein expression of vesicular ACh transporter (VAChT) and muscarinic acetylcholine receptor 2 (M2AChR) in the RVLM. Five rats in each group were used for protein expression analysis by Western blot. These rats were euthanized by overdose with pentobarbital sodium (200 mg/kg), and then the brain was rapidly removed. The RVLM tissues were extracted by punching coronal sections according to the standard rat atlas [29]. The RVLM tissue was added to a tissue lysate solution (protease inhibitor, phosphatase inhibitor, and 1% Triton X-100 diluted in PBS at 1:100), homogenized, and centrifuged to extract the supernatant. The protein concentration in the samples was measured with a BCA kit, and protein samples (30 μg) were run on a 10% SDS-PAGE gel, then the protein bands were transferred to a polyvinylidene fluoride (PVDF) membrane (Millipore, USA). The loaded PVDF membrane was blocked with 5% milk for 1 h, and then subjected to immunoblot analyses with VAChT antibody (C-20) sc-7716, Santa Cruz Biotechnology, Inc., Santa Cruz, CA) and M2AChR antibody (ab2805, Abcam, UK), then incubated with secondary antibodies [for VAChT (C-20), peroxidase-conjugated donkey anti-goat IgG; for M2AChR, HRP-conjugated Affinipure Goat Anti-Mouse IgG(H+L); both from ProteinTech Group, Chicago, IL]. After 3 washes, the PVDF membrane was infiltrated with chemiluminescent agent. The Syngene Bio Imaging system (55000, Gene Co., USA) was used to visually detect the protein bands, which were analyzed by GeneTools software (Gene Co.). β-tubulin (T6074, Sigma) served as control to normalize the levels of VAChT and M2AChR proteins.
Fluorescence Microphotography
The procedure for GFP expression in the RVLM with immunohistochemistry was performed as in our previous study [22]. Rats were anesthetized by intraperitoneal injection of an overdose of pentobarbital sodium (200 mg/kg), and then the anesthetized rats were perfused successively with normal saline and 4% polyoxymethylene. After fixing and dehydration, the brain was rapidly frozen before sectioning. The brainstem was cut at 20 μm on a cryostat, and the sections were floated on 0.01 mol/L PBS (pH 7.4). After floating, the sections were mounted on slides with antifade medium. The GFP fluorescence (excitation at 488 nm and emission at 530 nm) was measured on a laser confocal microscope (TCS-SP5, Leica, Germany).
Brain Microdialysis In Vivo
In vivo microdialysis was performed on anesthetized rats to collect the extracellular fluid in the RVLM for measurement of ACh, as previously described [26]. Using the rat atlas [29], after drilling two symmetrical holes in the skull, an intact microdialysis probe (MAB6.14.2, Sweden) was slowly inserted into the RVLM (2.0 mm lateral to the midline, 3.0 mm posterior to lambda, and 10 mm deep to the skull surface). RVLM microdialysis was conducted through a probe perfused with artificial cerebrospinal fluid driven by a microdialysis pump (Bioanalytical Systems, West Lafayette, IN) at 2 μL/min. The total volume of each microdialysis sample (10 min) was 20 μL, and samples were collected at least 60 min after insertion of the microdialysis probe. Three samples were collected and stored at − 80 °C for ACh assessment.
Measurement of BP and HR
BP and HR were measured as described in our previous study [27]. In brief, rats were anesthetized with urethane (800 mg/kg i.p.) and α-chloralose (40 mg/kg, i.p.), and then the trachea was cannulated and connected to a ventilator (SAR-830, CWE, USA) to assist respiration. A CO2 analyzer (Capstar 100, CWE) was used to monitor end-tidal CO2, which was kept at 4% by regulating the ventilator parameters. The right femoral artery was catheterized and connected to a PowerLab (8SP, ADInstruments, Australia) for BP and HR measurements. The mean arterial pressure and HR were calculated from the BP wave. Body temperature was kept at 37 °C by a temperature controller.
Recording of Renal Sympathetic Nerve Activity (RSNA)
RSNA was recorded as in our previous study [27]. The left renal sympathetic nerves were separated using glass needles, and placed gently on a pair of platinum-iridium recording electrodes. The discharge of the renal nerve was amplified and recorded by the PowerLab system. The pre-injection level (baseline) of RSNA was expressed as 100% from the absolute value after subtracting the noise level.
Microinjection of Atropine into the RVLM
Acute microinjection was performed as in our previous study [27]. The rats were fixed horizontally in a stereotaxic frame (Shanghai Alcott Biotech Co., Ltd, Shanghai, China). The cerebellum was removed and the dorsal surface of the medulla exposed. A three-barrel micropipette delivered study drugs into the RVLM at 2.0 mm–2.5 mm rostral to the caudal point of the area postrema, 2.0 mm–2.2 mm lateral to the midline, and 3.0 mm–3.2 mm deep to the dorsal surface of the brainstem. A pressor response (at least 20 mmHg) induced by microinjection of L-glutamate (2 nmol, 100 nL) into the brainstem was considered as a marker of the RVLM. A 2 nmol (100 nL) dose of atropine was selected to block M2AChRs. The changes in BP, HR, and RSNA were continuously monitored for at least 60 min after microinjection of atropine. The peak changes in BP, HR, and RSNA were calculated before and after the acute microinjection. After each experiment, the rat was euthanized (pentobarbital sodium, 200 mg/kg, i.v.), and the noise level of RSNA recorded for 15 min–20 min. Finally, the injection sites were marked by microinjection of pontamine sky blue, and the rat was perfused intracardially with 4% polyoxymethylene (100 mL). The brainstem was rapidly removed and fixed in 4% polyoxymethylene. Frozen 20-μm coronal sections were cut on a freezing microtome and mounted on slides. Injection sites in the area of the RVLM were confirmed by reference to the standard rat atlas of Paxinos and Watson [29].
Data Analysis
Data are expressed as mean ± SEM. Student’s t-test was used to analyze the differences between experimental groups; however, the data collected by the tail-cuff system were analyzed by repeated-measures analysis of variance; one-way analysis of variance was used to analyze the differences in protein expression of M2AChR in the RVLM of WKY rats, SHRs, and SHRs after injection of Lenti-GFP or Lenti-ACE2. Differences were considered to be significant at P < 0.05.
Results
Cholinergic Transmission in Untreated WKY Rats and SHRs
The concentration of ACh in the RVLM of SHRs was significantly higher than that in WKY rats (12.97 ± 1.67 vs 5.69 ± 0.52 μg/mL, P < 0.05; Fig. 1A). We also found that the level of VAChT was markedly higher (increased by 81.9%; P < 0.05) in SHRs than in WKY rats (Fig. 1B). The function of cholinergic transmission in the RVLM was determined by blocking the muscarinic receptors with atropine (2 nmol) microinjection. Before microinjection, the baseline BP and HR in anaesthetized rats were higher in SHRs than in WKY rats (Table 1). Original traces of the changes in BP, HR, and RSNA induced by microinjecting atropine into the RVLM are shown in Fig. 2A. The peak reduction in BP (− 33% ± 2% vs − 13% ± 1%) and RSNA (− 21% ± 4% vs 7% ± 3%) evoked by atropine was notably enhanced in SHRs relative to WKY rats (P < 0.05, Fig. 2). However, there was no difference in the HR changes in SHRs and WKY rats (Fig. 2C).
Fig. 1.

Concentrations of ACh (A) and protein expression of VAChT (B) in the RVLM of untreated WKY rats and SHRs. Values are mean ± SEM, n = 5/group, *P < 0.05 vs WKY.
Table 1.
Baseline values of BP and HR before microinjection of atropine into the RVLM in the anesthetized rats.
| BP (mmHg) | HR (bpm) | |
|---|---|---|
| WKY | 113 ± 3 | 334 ± 12 |
| SHR | 165 ± 5a | 395 ± 11a |
| SHR-Lenti-GFP | 170 ± 4 | 399 ± 16 |
| SHR-Lenti-ACE2 | 149 ± 4b | 344 ± 12b |
aP < 0.05 vs WKY
bP < 0.05 vs SHR-Lenti-GFP
Fig. 2.

Effects of microinjection of atropine into the RVLM on cardiovascular activity in WKY rats and SHRs. Original traces (A) and maximum changes of BP (B), HR (C), and RSNA (D) in response to microinjection of atropine into the RVLM. Values are mean ± SEM, n = 5/group, *P < 0.05 vs WKY.
Transfection Efficiency and Cardiovascular Effect of Lenti-ACE2 Gene Transfer into the RVLM
The region of the RVLM transfected with the Lenti-GFP gene is shown in Fig. 3A. We confirmed that the protein level of ACE2 expression in SHRs transfected with the hACE2 gene was notably higher (an increase of 50.7%) than in the SHR-Lenti-GFP group after 4 weeks (P < 0.05) (Fig. 3B). Three weeks after Lenti-ACE2 injection, the levels of BP and HR began to decline in the SHR-Lenti-ACE2 group compared with SHR-Lenti-GFP group (Fig. 4A). This reduction in BP and HR lasted until the termination of the experiment (5 weeks). Compared with the SHR-Lenti-GFP group, ACE2 overexpression in SHRs markedly decreased the NE in 24-h urine by ~ 72% (P < 0.05) (Fig. 4B).
Fig. 3.
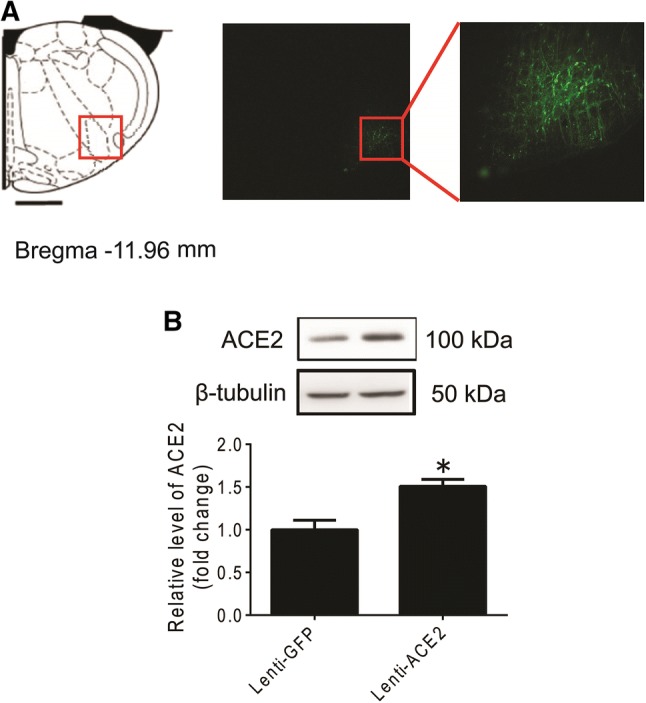
Efficiency of ACE2 overexpression in the RVLM. (A) According to the rat atlas (left), green fluorescent protein (GFP) was expressed in the RVLM (right). (B) Bar graphs show the protein levels of ACE2 in the RVLM in SHRs after injection of Lenti-ACE2 into the RVLM. Values are mean ± SEM, n = 5/group, *P < 0.05 vs Lenti-GFP.
Fig. 4.

Effect of ACE2 overexpression in the RVLM on cardiovascular activity of SHRs.
(A) Time courses of systolic blood pressure (SBP) (left) and HR (right) in SHRs after injection of Lenti-GFP or Lenti-ACE2 into the RVLM (arrows). Values are mean ± SEM, n = 5/group, *P < 0.05 vs day 0, #P < 0.05 vs Lenti-GFP. (B) NE in 24-h urine in SHRs injected with Lenti-GFP or Lenti-ACE2. Values are mean ± SEM, n = 5/group, *P < 0.05 vs Lenti-GFP.
Effects of ACE2 Overexpression on the Release of ACh and Expression of VAChT in the RVLM
The concentration of ACh in the microdialysis fluid from the RVLM was significantly lower (7.02 ± 0.93 vs 13.12 ± 1.67 μg/mL, n = 5, P < 0.05) in SHRs 4 weeks after RVLM injection of Lenti-ACE2 than with Lenti-GFP injection (Fig. 5A). Furthermore, ACE2 overexpression produced a notable reduction of 42% in the protein level of VAChT in the RVLM of SHRs (Fig. 5B).
Fig. 5.

Concentration of ACh (A) and the protein expression of VAChT (B) in the RVLM of SHRs after injection of Lenti-GFP or Lenti-ACE2. Values are mean ± SEM, n = 5/group, *P < 0.05 vs Lenti-GFP.
Effect of ACE2 Overexpression on the Depressor Response in BP, HR, and RSNA to Blocking Muscarinic ACh Receptors in the RVLM
The baseline BP and HR in anaesthetized rats were decreased in SHRs after ACE2 overexpression in the RVLM for 4 weeks (Table 1). Original traces of BP, HR, and RSNA in response to microinjection of the M2AChR antagonist atropine (2 nmol) into the RVLM are shown in Fig. 6A. The peak decreases in BP (− 15% ± 4% vs − 32% ± 2%) (Fig. 6B) and RSNA (− 12% ± 4% vs − 23% ± 4%) (Fig. 6D) induced by atropine in the RVLM were significantly lower (n = 5, P < 0.05) in SHRs transfected with Lenti-ACE2 than in those transfected with Lenti-GFP. However, there was no difference in the degree of decrease of HR in the Lenti-ACE2 and Lenti-GFP groups (Fig. 6C).
Fig. 6.

Effects of ACE2 overexpression in the RVLM on changes in cardiovascular activity (BP, HR, and RSNA) induced by microinjection of atropine into the RVLM in SHRs. Original tracings (A) and maximum changes in BP (B), HR (C), and RSNA (D) in response to microinjections of atropine into the RVLM of SHRs after injection with Lenti-GFP or Lenti-ACE2. Values are mean ± SEM, n = 5/group, *P < 0.05 vs Lenti-GFP.
Effects of ACE2 Overexpression on the Expression of M2AChRs in the RVLM
There were no differences in the protein expression of M2AChRs in the RVLM between WKY rats and SHRs; moreover, treatment with Lenti-ACE2 in the RVLM of SHRs had no influence on protein expression of M2AChRs (Fig. 7).
Fig. 7.

Protein expression of M2AChRs in the RVLM of WKY rats, SHRs, and SHRs after injection of Lenti-GFP or Lenti-ACE2. Values are mean ± SEM, n = 5/group.
Discussion
The major findings of the present study were as follows: (1) overexpression of ACE2 in the RVLM improved the cardiovascular function of SHRs; (2) ACE2 overexpression notably decreased the release of ACh in the RVLM of SHRs; and (3) ACE2 overexpression markedly inhibited the BP and RSNA responses to blockade of the M2AChR in the RVLM of SHRs. On the basis of these results, we conclude that ACE2 overexpression effectively attenuates the enhanced cholinergic transmission in the RVLM, and this may be an important underlying mechanism involved in the effects of central ACE2 on hypertension.
Evidence suggests that the RVLM is a primary premotor center that controls the sympathetic outflow and maintains resting BP [30, 31]. It has also been proposed that the neurons in the RVLM called C1 neurons, which contain phenylethanolamine-N-methyltransferase and can synthesize adrenaline, show greater sensitivity to electrical and chemical stimulation [32]. Excitation of these C1 neurons by local microinjection of the excitatory amino-acid L-glutamate can induce a pressor response, while inhibition of C1 neurons via microinjection of the inhibitory amino-acid γ-aminobutyric acid (GABA) can lead to a fall of arterial pressure [33]. It is known that C1 neurons in the RVLM are the key factor in generating sympathetic outflow and maintaining BP [34]. It has been reported that neurons which contain ChAT, the key enzyme for ACh synthesis, are distributed throughout the C1 area and make direct synaptic contacts with C1 neurons [35]. It has also been shown that blocking cholinergic transmission in the RVLM can reduce the arterial pressure and this suggests that cholinergic transmission contributes to the regulation of cardiovascular activity.
Considering the importance of ACh in the RVLM in maintaining the resting BP, we first determined its concentration and found that it was significantly higher under hypertension. Previous studies have shown that cholinergic transmission in the RVLM is involved in the pathogenesis of hypertension [36]. We assessed the ACh content using dialysate, which showed that the release of ACh was enhanced in SHRs. As to the source of the ACh release in the RVLM, a previous study showed that there are two sources [37]. One is the neurons in the RVLM that contain ChAT, and the other is the terminals of cholinergic neurons that project to the RVLM from other nuclei. We also determined the protein expression of VAChT and found it was significantly higher in SHRs than in WKY rats. Enhanced expression of VAChT may be responsible for the increased ACh release in the RVLM. The cholinergic neurons in other important nuclei with axon terminals projecting into the RVLM may also affect the enhanced ACh release in the RVLM. The hypothalamic paraventricular nucleus (PVN), another critical nucleus responsible for the regulation of water intake, urinary volume, and cardiovascular activity, controls sympathetic outflow via projections to the RVLM [38]. Furthermore, studies have shown that the neurons in the PVN that project to the RVLM include cholinergic neurons [39]. However, it was a limitation of our study that we did not examine the synthesis state of ACh in the PVN and this needs to be clarified in future.
In order to confirm the effect of endogenous ACh in the RVLM, we also microinjected atropine into the RVLM to block cholinergic transmission. It has been reported that the pressor response to microinjection of the acetylcholinesterase inhibitor into the RVLM is greater in SHRs than in WKY rats [19, 40]. We also found that the depressor response to blocking muscarinic receptors was significantly higher in SHRs than in WKY rats, which also confirmed that basal cholinergic transmission is enhanced under hypertension. As to the receptor that mediates the effect of ACh, there are two types, ionotropic (nicotinic, nAChRs) and metabotropic (muscarinic, mAChRs) [41]. Studies have suggested that cholinergic neurons in the RVLM contain both muscarinic and nicotinic receptors [42]. In previous studies, it was reported that microinjection of an mAChR antagonist into the RVLM area elicits a decrease in basal BP, while an antagonist of nAChRs has no effect on BP [43]. This suggests that the cholinergic transmission mediating basal BP works through acting on mAChRs. However, studies have also shown that the pathological change of cholinergic transmission is mainly in the presynaptic mechanisms (the synthesis, transport, and release of ACh), while the protein expression of mAChRs is unchanged under hypertension [44]. So we focused on the presynaptic mechanisms involved in cholinergic transmission in the RVLM. Whether improvement of the presynaptic transmission of ACh can contribute to a reduction of sympathetic outflow stimulated our interest.
ACE2, which converts the octapeptide Ang II into the heptapeptide Ang 1–7, is a key enzyme in the angiotensin system [45]. Previous studies have shown that the ACE2-Ang 1–7-Mas receptor axis has a potent cardiovascular protective effect. In peripheral studies, Ang 1–7, an endogenous counter-regulator of Ang II, produces effects opposite to Ang II, such as vasodilation, natriuresis, and cardioprotection [46]. In the central nervous system, Ang 1–7 also has critical neuronal protective effects and plays an important role in the improvement of stroke and neuro-inflammation [47]. Interestingly, the benefits of ACE2 overexpression lie not only in rectifying the concentrations of Ang II and Ang 1–7, but also in regulating the expression of Mas and Ang II receptor type 1 receptors [48].
As to the anti-hypertensive effect of ACE2 overexpression, peripheral studies have shown that ACE2 improves the myocardial hypertrophy, fibrosis, and diastolic dysfunction induced by hypertension [49]. Considering the central effects of ACE2, it has been demonstrated that ACE2 overexpression in the RVLM significantly reduces BP and sympathetic activity in SHRs [18]. However, the mechanism by which ACE2 overexpression decreases the release of ACh in the RVLM under hypertension is still unclear. Our previous study showed that ACE2 overexpression in the RVLM significantly decreases the concentration of glutamate and downregulates the protein expression of the glutamate receptor, which plays a key role in maintaining basal BP [23]. Our data showed that a reduction of the release of ACh after ACE2 overexpression in the RVLM improved the sympathetic overactivation under hypertension. However, the mechanism by which ACE2 overexpression decreases the release of ACh in the RVLM under hypertension is still unclear.
Previous studies have shown that the central nitric oxide (NO) system in the RVLM may modulate cardiovascular activity by activating adenosine A receptors and decreasing Ca2+ influx. Since ACh is released in a Ca2+-dependent manner, the mechanism involved in decreasing the release of ACh in the RVLM may be mediated by NO via reducing Ca2+ influx [50]. Our previous work has demonstrated that exogenous injection of Ang 1–7 into the NTS produces a significant increase in NO release, which is mediated by the PI3K-Akt-NOS signaling pathway [51]. Other studies have also indicated that ACE2 overexpression enhances the activity and increases the concentration of NO in the brain [52, 53]. So the mechanism involved in ACE2 overexpression depressing the enhanced cholinergic transmission may be associated with the NOS-NO-adenosine pathway. This is a limitation of the present study and needs to be defined in future. On the other hand, ACh also has a complex relationship with another important neurotransmitter, GABA, which plays a key role in inhibitory projections to the RVLM. Previous studies have shown that the release of GABA and the expression of GABA receptors are down-regulated under hypertension [54]. ACE2 overexpression facilitates GABA release and enhances inhibitory transmission in the RVLM [55]. However, GABA can cause a decrease in ACh release [56, 57]. So the GABA mechanism may also be involved in the improvement of cholinergic transmission mediated by ACE2 overexpression.
We also found that the protein expression of VAChT was significantly down-regulated by ACE2 overexpression. As the key transporter for assembling ACh vesicles, the decreased expression of VAChT made some contribution to the reduced ACh release. However, we did not examine the expression of ChAT, the key enzyme for ACh synthesis. It is interesting that a previous study showed that the gene encoding VAChT is localized within the first intron of the gene encoding ChAT and is in the same transcriptional orientation. This means that these two proteins might be under the same regulation. Studies have also shown that several classes of factors, such as leukemia inhibitory factor and retinoic acid, can induce ChAT activity and increase the levels of multiple ChAT mRNA species in these neurons [58]. So whether ACE2 has any effect on the transcriptional regulation of VAChT and ChAT is still unclear and needs further investigation.
Although previous studies have shown that the expression of muscarinic receptors in the RVLM does not change under hypertension [42], we also determined whether ACE2 overexpression has any effect on their expression. The data showed no difference in protein expression of mAChRs after ACE2 overexpression. However, we did not overlook the other type of cholinergic receptor, nAChRs, which are neurotransmitter-gated ion channels present on skeletal muscle cells, neurons, and a number of non-neuronal cells throughout the body, such as lymphocytes and epithelial cells [59]. Studies have referred to the protective effect of nAChRs in neurons by showing that they mediate an anti-inflammatory effect which leads to a decrease in the level of oxidative stress [60–62]. It is known that the state of oxidation is higher in the RVLM under hypertension. Our previous work also determined that ACE2 overexpression decreases the level of neuroinflammation in the RVLM [22]. So whether the nAChRs involved in the neuroprotective effect of ACE2 overexpression is still unknown and needs further study.
In summary, the present results have shown that ACE2 overexpression in the RVLM attenuates the enhanced cholinergic inputs to the RVLM in SHRs. We suggest that improvement in the increased cholinergic input to the RVLM neurons is a critical mechanism responsible for the beneficial effects of ACE2 on cardiovascular dysfunction under hypertension (Fig. 8). The present work provides a potential role of cholinergic synaptic transmission in the effect of the ACE2-Ang 1–7-MasR axis on central autonomic networks for cardiovascular activity.
Fig. 8.

Schematic of the relationship between ACE2 and cholinergic signaling involved in the regulation of sympathoexcitation in the RVLM.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81470534, 81770419, 81630012, and 81570385) and the Key Laboratory of Medical Electrophysiology (Southwest Medical University), Ministry of Education of China - No201709 (KeyME-2017-09).
Compliance with Ethical Standards
Conflict of interest
All authors claim that there are no conflict of interest.
References
- 1.Wang JG, Liu L. Global impact of 2017 American College of Cardiology/American Heart Association Hypertension Guidelines: a perspective from China. Circulation. 2018;137:546–548. doi: 10.1161/CIRCULATIONAHA.117.032890. [DOI] [PubMed] [Google Scholar]
- 2.Mankin LA. Update in hypertension therapy. Med Clin North Am. 2016;100:665–693. doi: 10.1016/j.mcna.2016.03.005. [DOI] [PubMed] [Google Scholar]
- 3.Malpas SC. Sympathetic nervous system overactivity and its role in the development of cardiovascular disease. Physiol Rev. 2010;90:513–557. doi: 10.1152/physrev.00007.2009. [DOI] [PubMed] [Google Scholar]
- 4.Guyenet PG. The sympathetic control of blood pressure. Nat Rev Neurosci. 2006;7:335–346. doi: 10.1038/nrn1902. [DOI] [PubMed] [Google Scholar]
- 5.Sved AF, Ito S, Sved JC. Brainstem mechanisms of hypertension: role of the rostral ventrolateral medulla. Curr Hypertens Rep. 2003;5:262–268. doi: 10.1007/s11906-003-0030-0. [DOI] [PubMed] [Google Scholar]
- 6.Diz DI, Arnold AC, Nautiyal M, Isa K, Shaltout HA, Tallant EA. Angiotensin peptides and central autonomic regulation. Curr Opin Pharmacol. 2011;11:131–137. doi: 10.1016/j.coph.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chan SH, Chan JY. Brain stem NOS and ROS in neural mechanisms of hypertension. Antioxid Redox Signal. 2014;20:146–163. doi: 10.1089/ars.2013.5230. [DOI] [PubMed] [Google Scholar]
- 8.de Wildt DJ, Porsius AJ. Central cardiovascular effects of physostigmine in the cat; possible cholinergic aspects of blood pressure regulation. Arch Int Pharmacodyn Ther. 1981;253:22–39. [PubMed] [Google Scholar]
- 9.Giuliano R, Ruggiero DA, Morrison S, Ernsberger P, Reis DJ. Cholinergic regulation of arterial pressure by the C1 area of the rostral ventrolateral medulla. J Neurosci. 1989;9:923–942. doi: 10.1523/JNEUROSCI.09-03-00923.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brezenoff HE, Giuliano R. Cardiovascular control by cholinergic mechanisms in the central nervous system. Annu Rev Pharmacol Toxicol. 1982;22:341–381. doi: 10.1146/annurev.pa.22.040182.002013. [DOI] [PubMed] [Google Scholar]
- 11.Kubo T, Fukumori R, Kobayashi M, Yamaguchi H. Altered cholinergic mechanisms and blood pressure regulation in the rostral ventrolateral medulla of DOCA-salt hypertensive rats. Brain Res Bull. 1998;45:327–332. doi: 10.1016/s0361-9230(97)00380-8. [DOI] [PubMed] [Google Scholar]
- 12.Kubo T, Ishizuka T, Fukumori R, Asari T, Hagiwara Y. Enhanced release of acetylcholine in the rostral ventrolateral medulla of spontaneously hypertensive rats. Brain Res. 1995;686:1–9. doi: 10.1016/0006-8993(95)00433-q. [DOI] [PubMed] [Google Scholar]
- 13.Lin Q, Li P. Rostral medullary cholinergic mechanisms and chronic stress-induced hypertension. J Auton Nerv Syst. 1990;31:211–217. doi: 10.1016/0165-1838(90)90187-n. [DOI] [PubMed] [Google Scholar]
- 14.Zisman LS, Keller RS, Weaver B, Lin Q, Speth R, Bristow MR, et al. Increased angiotensin-(1-7)-forming activity in failing human heart ventricles: evidence for upregulation of the angiotensin-converting enzyme Homologue ACE2. Circulation. 2003;108:1707–1712. doi: 10.1161/01.CIR.0000094734.67990.99. [DOI] [PubMed] [Google Scholar]
- 15.Der Sarkissian S, Huentelman MJ, Stewart J, Katovich MJ, Raizada MK. ACE2: A novel therapeutic target for cardiovascular diseases. Prog Biophys Mol Biol. 2006;91:163–198. doi: 10.1016/j.pbiomolbio.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 16.Feng Y, Xia H, Santos RA, Speth R, Lazartigues E. Angiotensin-converting enzyme 2: a new target for neurogenic hypertension. Exp Physiol. 2010;95:601–606. doi: 10.1113/expphysiol.2009.047407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Morais SDB, Shanks J, Zucker IH. Integrative physiological aspects of brain RAS in hypertension. Curr Hypertens Rep. 2018;20:10. doi: 10.1007/s11906-018-0810-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yamazato M, Yamazato Y, Sun C, Diez-Freire C, Raizada MK. Overexpression of angiotensin-converting enzyme 2 in the rostral ventrolateral medulla causes long-term decrease in blood pressure in the spontaneously hypertensive rats. Hypertension. 2007;49:926–931. doi: 10.1161/01.HYP.0000259942.38108.20. [DOI] [PubMed] [Google Scholar]
- 19.Kubo T, Hagiwara Y, Endo S, Fukumori R. Activation of hypothalamic angiotensin receptors produces pressor responses via cholinergic inputs to the rostral ventrolateral medulla in normotensive and hypertensive rats. Brain Res. 2002;953:232–245. doi: 10.1016/s0006-8993(02)03297-3. [DOI] [PubMed] [Google Scholar]
- 20.Agarwal D, Welsch MA, Keller JN, Francis J. Chronic exercise modulates RAS components and improves balance between pro- and anti-inflammatory cytokines in the brain of SHR. Basic Res Cardiol. 2011;106:1069–1085. doi: 10.1007/s00395-011-0231-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xia H, Suda S, Bindom S, Feng Y, Gurley SB, Seth D, et al. ACE2-mediated reduction of oxidative stress in the central nervous system is associated with improvement of autonomic function. PLoS One. 2011;6:e22682. doi: 10.1371/journal.pone.0022682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tan X, Jiao PL, Wang YK, Wu ZT, Zeng XR, Li ML, et al. The phosphoinositide-3 kinase signaling is involved in neuroinflammation in hypertensive rats. CNS Neurosci Ther. 2017;23:350–359. doi: 10.1111/cns.12679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang YK, Shen D, Hao Q, Yu Q, Wu ZT, Deng Y, et al. Overexpression of angiotensin-converting enzyme 2 attenuates tonically active glutamatergic input to the rostral ventrolateral medulla in hypertensive rats. Am J Physiol Heart Circ Physiol. 2014;307:H182–H190. doi: 10.1152/ajpheart.00518.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peng J, Wang YK, Wang LG, Yuan WJ, Su DF, Ni X, et al. Sympathoinhibitory mechanism of moxonidine: role of the inducible nitric oxide synthase in the rostral ventrolateral medulla. Cardiovasc Res. 2009;84:283–291. doi: 10.1093/cvr/cvp202. [DOI] [PubMed] [Google Scholar]
- 25.Zha YP, Wang YK, Deng Y, Zhang RW, Tan X, Yuan WJ, et al. Exercise training lowers the enhanced tonically active glutamatergic input to the rostral ventrolateral medulla in hypertensive rats. CNS Neurosci Ther. 2013;19:244–251. doi: 10.1111/cns.12065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang W, Zou Z, Tan X, Zhang RW, Ren CZ, Yao XY, et al. Enhancement in tonically active glutamatergic inputs to the rostral ventrolateral medulla contributes to neuropathic pain-induced high blood pressure. Neural Plast. 2017;2017:4174010. doi: 10.1155/2017/4174010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hao F, Gu Y, Tan X, Deng Y, Wu ZT, Xu MJ, et al. Estrogen replacement reduces oxidative stress in the rostral ventrolateral medulla of ovariectomized rats. Oxid Med Cell Longev. 2016;2016:2158971. doi: 10.1155/2016/2158971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peng JF, Wu ZT, Wang YK, Yuan WJ, Sun T, Ni X, et al. GABAergic mechanism in the rostral ventrolateral medulla contributes to the hypotension of moxonidine. Cardiovasc Res. 2011;89:473–481. doi: 10.1093/cvr/cvq289. [DOI] [PubMed] [Google Scholar]
- 29.Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. 3. New York: Academic; 1998. [DOI] [PubMed] [Google Scholar]
- 30.Gaede AH, Pilowsky PM. Catestatin in rat RVLM is sympathoexcitatory, increases barosensitivity, and attenuates chemosensitivity and the somatosympathetic reflex. Am J Physiol Regul Integr Comp Physiol. 2010;299:R1538–R1545. doi: 10.1152/ajpregu.00335.2010. [DOI] [PubMed] [Google Scholar]
- 31.Madden CJ, Sved AF. Cardiovascular regulation after destruction of the C1 cell group of the rostral ventrolateral medulla in rats. Am J Physiol Heart Circ Physiol. 2003;285:H2734–H2748. doi: 10.1152/ajpheart.00155.2003. [DOI] [PubMed] [Google Scholar]
- 32.Milner TA, Hernandez FJ, Herrick SP, Pierce JP, Iadecola C, Drake CT. Cellular and subcellular localization of androgen receptor immunoreactivity relative to C1 adrenergic neurons in the rostral ventrolateral medulla of male and female rats. Synapse. 2007;61:268–278. doi: 10.1002/syn.20370. [DOI] [PubMed] [Google Scholar]
- 33.Schreihofer AM, Stornetta RL, Guyenet PG. Regulation of sympathetic tone and arterial pressure by rostral ventrolateral medulla after depletion of C1 cells in rat. J Physiol. 2000;529(Pt 1):221–236. doi: 10.1111/j.1469-7793.2000.00221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Drolet G, Aslanian V, Minson J, Morris M, Chalmers J. Differences in the central hypotensive actions of alpha-methyldopa and clonidine in the spontaneously hypertensive rat: contribution of neurons arising from the B3 and the C1 areas of the rostral ventrolateral medulla. J Cardiovasc Pharmacol. 1990;15:118–123. doi: 10.1097/00005344-199001000-00019. [DOI] [PubMed] [Google Scholar]
- 35.Falquetto B, Tuppy M, Potje SR, Moreira TS, Antoniali C, Takakura AC. Cardiovascular dysfunction associated with neurodegeneration in an experimental model of Parkinson’s disease. Brain Res. 2017;1657:156–166. doi: 10.1016/j.brainres.2016.12.008. [DOI] [PubMed] [Google Scholar]
- 36.Kubo T, Taguchi K, Sawai N, Ozaki S, Hagiwara Y. Cholinergic mechanisms responsible for blood pressure regulation on sympathoexcitatory neurons in the rostral ventrolateral medulla of the rat. Brain Res Bull. 1997;42:199–204. doi: 10.1016/s0361-9230(96)00256-0. [DOI] [PubMed] [Google Scholar]
- 37.Samano C, Zetina ME, Cifuentes F, Morales MA. Segregation of met-enkephalin from vesicular acetylcholine transporter and choline acetyltransferase in sympathetic preganglionic varicosities mostly lacking synaptophysin and synaptotagmin. Neuroscience. 2009;163:180–189. doi: 10.1016/j.neuroscience.2009.06.010. [DOI] [PubMed] [Google Scholar]
- 38.Cham JL, Badoer E. Exposure to a hot environment can activate rostral ventrolateral medulla-projecting neurones in the hypothalamic paraventricular nucleus in conscious rats. Exp Physiol. 2008;93:64–74. doi: 10.1113/expphysiol.2007.039560. [DOI] [PubMed] [Google Scholar]
- 39.Kubo T, Hagiwara Y, Sekiya D, Chiba S, Fukumori R. Cholinergic inputs to rostral ventrolateral medulla pressor neurons from hypothalamus. Brain Res Bull. 2000;53:275–282. doi: 10.1016/s0361-9230(00)00343-9. [DOI] [PubMed] [Google Scholar]
- 40.Stornetta RL, Macon CJ, Nguyen TM, Coates MB, Guyenet PG. Cholinergic neurons in the mouse rostral ventrolateral medulla target sensory afferent areas. Brain Struct Funct. 2013;218:455–475. doi: 10.1007/s00429-012-0408-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Beker F, Weber M, Fink RH, Adams DJ. Muscarinic and nicotinic ACh receptor activation differentially mobilize Ca2+ in rat intracardiac ganglion neurons. J Neurophysiol. 2003;90:1956–1964. doi: 10.1152/jn.01079.2002. [DOI] [PubMed] [Google Scholar]
- 42.Kumar NN, Ferguson J, Padley JR, Pilowsky PM, Goodchild AK. Differential muscarinic receptor gene expression levels in the ventral medulla of spontaneously hypertensive and Wistar-Kyoto rats: role in sympathetic baroreflex function. J Hypertens. 2009;27:1001–1008. doi: 10.1097/hjh.0b013e3283282e5c. [DOI] [PubMed] [Google Scholar]
- 43.Padley JR, Kumar NN, Li Q, Nguyen TB, Pilowsky PM, Goodchild AK. Central command regulation of circulatory function mediated by descending pontine cholinergic inputs to sympathoexcitatory rostral ventrolateral medulla neurons. Circ Res. 2007;100:284–291. doi: 10.1161/01.RES.0000257370.63694.73. [DOI] [PubMed] [Google Scholar]
- 44.Zhou P, Zhu Q, Liu M, Li J, Wang Y, Zhang C, et al. Muscarinic acetylcholine receptor in cerebellar cortex participates in acetylcholine-mediated blood depressor response in rats. Neurosci Lett. 2015;593:129–133. doi: 10.1016/j.neulet.2015.03.036. [DOI] [PubMed] [Google Scholar]
- 45.Ohishi M, Yamamoto K, Rakugi H. Angiotensin (1-7) and other angiotensin peptides. Curr Pharm Des. 2013;19:3060–3064. doi: 10.2174/1381612811319170013. [DOI] [PubMed] [Google Scholar]
- 46.Frantz EDC, Giori IG, Machado MV, Magliano DC, Freitas FM, Andrade MSB, et al. High, but not low, exercise volume shifts the balance of renin-angiotensin system toward ACE2/Mas receptor axis in skeletal muscle in obese rats. Am J Physiol Endocrinol Metab. 2017;313:E473–E482. doi: 10.1152/ajpendo.00078.2017. [DOI] [PubMed] [Google Scholar]
- 47.Chang AY, Li FC, Huang CW, Wu JC, Dai KY, Chen CH, et al. Interplay between brain stem angiotensins and monocyte chemoattractant protein-1 as a novel mechanism for pressor response after ischemic stroke. Neurobiol Dis. 2014;71:292–304. doi: 10.1016/j.nbd.2014.08.005. [DOI] [PubMed] [Google Scholar]
- 48.Pandey A, Goru SK, Kadakol A, Malek V, Gaikwad AB. Differential regulation of angiotensin converting enzyme 2 and nuclear factor-kappaB by angiotensin II receptor subtypes in type 2 diabetic kidney. Biochimie. 2015;118:71–81. doi: 10.1016/j.biochi.2015.08.005. [DOI] [PubMed] [Google Scholar]
- 49.Wang X, Ye Y, Gong H, Wu J, Yuan J, Wang S, et al. The effects of different angiotensin II type 1 receptor blockers on the regulation of the ACE-AngII-AT1 and ACE2-Ang(1–7)-Mas axes in pressure overload-induced cardiac remodeling in male mice. J Mol Cell Cardiol. 2016;97:180–190. doi: 10.1016/j.yjmcc.2016.05.012. [DOI] [PubMed] [Google Scholar]
- 50.Jiang MY, Chen J, Wang J, Xiao F, Zhang HH, Zhang CR, et al. Nitric oxide modulates cardiovascular function in the rat by activating adenosine A2A receptors and inhibiting acetylcholine release in the rostral ventrolateral medulla. Clin Exp Pharmacol Physiol. 2011;38:380–386. doi: 10.1111/j.1440-1681.2011.05521.x. [DOI] [PubMed] [Google Scholar]
- 51.Wu ZT, Ren CZ, Yang YH, Zhang RW, Sun JC, Wang YK, et al. The PI3K signaling-mediated nitric oxide contributes to cardiovascular effects of angiotensin-(1–7) in the nucleus tractus solitarii of rats. Nitric Oxide. 2016;52:56–65. doi: 10.1016/j.niox.2015.12.002. [DOI] [PubMed] [Google Scholar]
- 52.Feng Y, Xia H, Cai Y, Halabi CM, Becker LK, Santos RA, et al. Brain-selective overexpression of human angiotensin-converting enzyme type 2 attenuates neurogenic hypertension. Circ Res. 2010;106:373–382. doi: 10.1161/CIRCRESAHA.109.208645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zheng H, Liu X, Patel KP. Angiotensin-converting enzyme 2 overexpression improves central nitric oxide-mediated sympathetic outflow in chronic heart failure. Am J Physiol Heart Circ Physiol. 2011;301:H2402–H2412. doi: 10.1152/ajpheart.00330.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.de Brito Alves JL, de Oliveira JM, Ferreira DJ, Barros MA, Nogueira VO, Alves DS, et al. Maternal protein restriction induced-hypertension is associated to oxidative disruption at transcriptional and functional levels in the medulla oblongata. Clin Exp Pharmacol Physiol. 2016;43:1177–1184. doi: 10.1111/1440-1681.12667. [DOI] [PubMed] [Google Scholar]
- 55.Wang L, de Kloet AD, Pati D, Hiller H, Smith JA, Pioquinto DJ, et al. Increasing brain angiotensin converting enzyme 2 activity decreases anxiety-like behavior in male mice by activating central Mas receptors. Neuropharmacology. 2016;105:114–123. doi: 10.1016/j.neuropharm.2015.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shi J, Li Q, Wen T. Dendritic cell factor 1-knockout results in visual deficit through the GABA system in mouse primary visual cortex. Neurosci Bull. 2018;34:465–475. doi: 10.1007/s12264-018-0211-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li Z, You Z, Li M, Pang L, Cheng J, Wang L. Protective effect of resveratrol on the brain in a rat model of epilepsy. Neurosci Bull. 2017;33:273–280. doi: 10.1007/s12264-017-0097-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cervini R, Berrard S, Bejanin S, Mallet J. Regulation by CDF/LIF and retinoic acid of multiple ChAT mRNAs produced from distinct promoters. Neuroreport. 1994;5:1346–1348. [PubMed] [Google Scholar]
- 59.Kawashima K, Fujii T. Expression of non-neuronal acetylcholine in lymphocytes and its contribution to the regulation of immune function. Front Biosci. 2004;9:2063–2085. doi: 10.2741/1390. [DOI] [PubMed] [Google Scholar]
- 60.Roh HT, So WY. The effects of aerobic exercise training on oxidant-antioxidant balance, neurotrophic factor levels, and blood-brain barrier function in obese and non-obese men. J Sport Health Sci. 2017;6:447–453. doi: 10.1016/j.jshs.2016.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Roh HT, Cho SY, So WY. Obesity promotes oxidative stress and exacerbates blood-brain barrier disruption after high-intensity exercise. J Sport Health Sci. 2017;6:225–230. doi: 10.1016/j.jshs.2016.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Han Z, Shen F, He Y, Degos V, Camus M, Maze M, et al. Activation of alpha-7 nicotinic acetylcholine receptor reduces ischemic stroke injury through reduction of pro-inflammatory macrophages and oxidative stress. PLoS One. 2014;9:e105711. doi: 10.1371/journal.pone.0105711. [DOI] [PMC free article] [PubMed] [Google Scholar]