

Abstract
Inflammation and oxidative stress are considered major factors in the pathogenesis of ischemic stroke. Increasing evidence has demonstrated that Schizandrin A (Sch A), a lignin compound isolated from Schisandra chinesnesis, exhibits prominent anti-inflammatory and antioxidant activities. In this study, we investigated the anti-inflammatory and antioxidant effects of Sch A against cerebral ischemia/reperfusion (I/R) injury as well as the underlying molecular mechanisms. Sch A treatment significantly improved the neurological score and reduced infarct volume 24 h after reperfusion. It dose-dependently inhibited the expression of cyclooxygenase-2 and inducible nitric oxide synthase, reduced the release of pro-inflammatory cytokines (tumor necrosis factor-α interleukin [IL]-1β and IL-6), and increased anti-inflammatory cytokines (transforming growth factor-β and interleukin-10). Furthermore, it increased the activity of superoxide dismutase and catalase, decreased reactive oxygen species production and 4-hydroxynonenal and 8-hydroxy-2’-deoxyguanosine levels. Transcription of nuclear factor erythroid 2-related factor 2 (Nrf2) and downstream genes (heme oxygenase-1 and NAD[P]H: quinone oxidoreductase 1) increased. Knockdown of Nrf2 by siRNA inhibited the neuroprotective effects of Sch A. In addition, Sch A increased phosphorylation of adenosine monophosphate-activated protein kinase (AMPK) both in vivo and in vitro. Activation of the Nrf2 pathway as well as the protective effects of Sch A in an oxygen and glucose deprivation-induced injury model was abolished by AMPK knockdown. Our study indicates that Sch A protects against cerebral I/R injury by suppressing inflammation and oxidative stress, and that this effect is regulated by the AMPK/Nrf2 pathway.
Keywords: Schizandrin A, inflammation, oxidative stress, AMPK/Nrf2 pathway
Introduction
Ischemic cerebral stroke is associated with a high incidence of morbidity and mortality [1]. The most effective treatment for stroke is thrombolytic treatment, but treatment is limited to a small proportion of patients because of the narrow therapeutic time window and the strict exclusion criteria due to the complex sequence of pathophysiological events involved in stroke [2]. There is still a lack of efficient curative treatments. Inflammation has recently been shown to play an essential role in secondary brain injury following cerebral ischemia [3]. Oxidative stress is another important factor closely related to the pathogenesis of ischemic stroke [4]. Post-reperfusion pronounced oxidation of lesions occurs because of large amounts of reactive oxygen species (ROS), which lead to neural dysfunction, cell death, and the inflammatory response [5]. Thus, anti-inflammation and antioxidants have been considered for preventing and treating ischemic stroke.
Adenosine monophosphate-activated protein kinase (AMPK) is a major regulator of cellular and organismal energy homeostasis and its activation decreases oxidative stress and inhibits inflammation [6]. AMPK is a highly effective therapeutic target for protecting against cerebral ischemia [7], as phosphorylated AMPK increases glucose uptake and cellular energy for metabolism to induce apoptotic cell death in a neuronal context [8]. Nuclear factor erythroid 2-related factor 2 (Nrf2), which is a basic leucine zipper transcription factor, regulates the expression of numerous ROS detoxifying and antioxidant genes [9]. Overproduction of ROS or dysfunction of antioxidant enzymes results in oxidative stress and cellular damage [10]. Nrf2 prevents cells from oxidative stress via antioxidant enzymes. The potential for crosstalk between the Nrf2 and AMPK pathways has been noted because there are reports about natural anti-inflammatory agents and antioxidants providing neuroprotection by activating Nrf2 and upregulating AMPK expression in experimental stroke models [11].
Schizandrin A (Sch A) is a bioactive lignin compound isolated from Schisandra chinesnesis [12]. It shows several cytoprotective activities, including anti-inflammatory [13], antioxidant [14], and anti-liver injury activities [15]. It has potential neuroprotective activity by inhibiting the TRAF6-NF-κB and Jak2-Stat3 signaling pathways [16]. It prevents oxygen and glucose deprivation followed by reperfusion (OGD/R)-induced cell death in primary cultures of rat cortical neurons [13]. We hypothesized that it may have neuroprotective effects against cerebral ischemia-reperfusion-induced inflammation and oxidative injury, and that the AMPK/Nrf-2 pathway might play an important role in this effect.
Materials and methods
Drugs and reagents
Sch A (purity > 99%) was supplied by the National Institutes for Food and Drug Control (Beijing, China). Fetal bovine serum (FBS), Dulbecco’s modified Eagle’s medium (DMEM), Ham’s F12, and trypsin were purchased from Gibco (Carlsbad, CA, USA). 3-[4,5-Dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) and 2,3,5-triphenyltetrazolium chloride (TTC) were obtained from Sigma-Aldrich (St. Louis, MO, USA). The enzyme-linked immunoassay (ELISA) kits for superoxide dismutase (SOD), catalase (CAT), 4-hydroxynonenal (4-HNE) and 8-hydroxy-2’-deoxyguanosine (8-OHdG) were purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). The anti-Nrf2, anti-p-AMPK, and anti-AMPK antibodies were purchased from Abcam (Cambridge, UK). Anti-β-actin and anti-histone H3 were obtained from Beyotime Biotechnology (Shanghai, China).
Animals and focal cerebral ischemia model
Adult male Sprague-Dawley rats (weight, 220-240 g) were obtained from the Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). Experimental protocols were approved by the Ethics Committee for Animal Experimentation of the Shaanxi University of Chinese Medicine (Xianyang, China).
Focal cerebral ischemia was induced by intraluminal occlusion of the right middle cerebral artery (MCAO) for 2 h followed by reperfusion by dislodging the nylon suture for 24 h. Sham-operated rats were subjected to the same surgical procedure, but the MCA was not occluded. Sch A was injected immediately before reperfusion, i.e., 2 h after MCAO, via the vena caudalis. The animals were divided randomly into five groups (10 rats/group): sham operated (Sham), I/R, and the I/R + Sch A groups (low, medium, high dosages). Sch A was dissolved in 2 mL PBS for injection, and the volume of administration was 2 mL/kg. The Sham group was administered vehicle (PBS).
Neurological deficit score
Neurological deficit was evaluated 2 h after the surgery using a 5-point scoring system as described previously [17]. Scoring was as follows: No deficit, 0; failure to stretch the contralateral torso and forelimb fully, 1; turning to the ipsilateral side when lifted by tail, 2; falling to the affected side, 3; no spontaneous walking with depressed consciousness, 4.
Infarct volume evaluation
All rats were euthanized 24 h after ischemia, and their brains were harvested for further examination. Six slices of 2 mm coronal brain sections obtained from the entire brain were incubated in a 1% 2,3,5-triphenyltetrazolium chloride (TTC; Sigma-Aldrich) solution at 37°C for 30 min. Infarct volume was quantified by a method described previously.
Measurement of mitochondrial-generated ROS
The 2’,7’,-dichlorofluorescein diacetate (DCFH-DA) assay was used to measure total ROS. The DCFH-DA itself has no fluorescence and is free to cross the cell membrane. After entering the cell, the cells’ esterases hydrolyze 2’,7’,-dichlorofluorescein (DCFH). DCFH does not penetrate the cell membrane, so the probe accumulates in the cell. Intracellular ROS oxidize non-fluorescent DCFH to produce fluorescent DCF. Fluorescence intensity was measured at an excitation wavelength of 485 nm and an emission wavelength of 525 nm. ROS production is expressed as a percentage of fluorescence relative to the Sham group.
Measurement of SOD, CAT, 4-HNE and 8-OHdG
SOD and CAT are important antioxidant enzymes and the mitochondrial targets of ROS; thus, their activities might reduce ROS exposure. 4-HNE is a reaction product of lipid hydroperoxide break down that occurs in response to oxidative stress. 4-HNE rapidly modifies proteins on several amino acid residues, leading to loss of protein function. 8-OHdG, one of the most abundant oxidative modified lesions in DNA, is produced when nucleic acids are exposed to oxidative stress. Ischemic cerebral cortex 4-HNE, 8-OHdG and SOD activities were determined using specific ELISA kits according to the manufacturers’ instructions.
Measurement of IL-1β, IL-6, TGF-β, TNF-α and IL-10
The concentrations of interleukin (IL)-1β, IL-6, transforming growth factor (TGF)-β, tumor necrosis factor (TNF)-α, and IL-10 in the cerebral cortex were determined using ELISA kits, according to the manufacturer’s instructions (Nanjing KeyGEN Biotech. Co., Ltd., Nanjing, China).
Cell culture
SH-SY5Y (a human neuroblastoma cell line) was obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). SH-SY5Y cells are normally induced to differentiate into neuron-like cells by stimulation with ATRA, which has been used to imitate the responses of neurons. In the present study, SH-SY5Y cells were cultured in a humidified atmosphere of 5% CO2 in a mixture of 1:1 Ham’s F12 and DMEM supplemented with 10% (v/v) FBS and 100 U/mL penicillin and streptomycin. When the cells reached 80-90% confluence, 10 μM ATRA was added to the medium to stimulate cell differentiation.
Sch A treatment and oxygen glucose deprivation
After the Sch A treatment at different concentrations from 5 to 100 μM for 6 h before OGD, the cells were exposed to model ischemia-like conditions in vitro. Briefly, the culture medium was replaced with glucose-free Earle’s balanced salt solution, and the cells were placed in an oxygen deprived (95% N2/5% CO2) incubator at 37°C for 2 h. Control cells were incubated in Earle’s balanced salt solution with 10 mM glucose under normoxic conditions (95% O2/5% CO2) for the same duration. The cells were returned to normoxic conditions in regular medium to terminate OGD and start the 24 h of reperfusion.
Cell viability assay
Cell viability was determined using the MTT (Sigma) assay. In brief, the cell line was plated in 96-well plates (3,000 cells/plate), the MTT solution was added (0.5 mg/mL), and the plates were incubated for an additional 4 h at 37°C. Then the medium was removed and dye crystals were dissolved in 150 µL DMSO. Absorbance was measured at 490 nm.
Transient transfection of siRNA
The AMPK and Nrf2-specific small interfering RNAs (siRNAs), as well as the corresponding non-targeting scrambled control siRNA were designed and chemically synthesized by GenePharma (Shanghai, China). Differentiated SH-SY5Y cells were transfected with target-specific siRNA or control siRNA using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. After 48 h of transfection, the cells were treated with Sch A and subjected to a 2 h OGD challenge followed by reoxygenation for 24 h. The cell samples were collected for the MTT assay.
Western blotting
After 24 h of MCAO or OGD, cytoplasmic and nuclear proteins were extracted from the ischemic cerebral cortex and differentiated cells, using nuclear and cytoplasmic extraction kits, according to the manufacturer’s instructions. A bicinchoninic acid kit was used to detect protein concentrations. Nuclear Nrf2, p-AMPK, and AMPK expression was tested briefly as follows: after quantifying the protein concentration, equal amounts of nuclear or cytoplasmic protein samples (each well, 30 µg per sample) were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis and electrotransferred to polyvinylidene fluoride membranes using 100 V transfer-molded voltage lasting for 45-70 min. Then the samples were incubated at room temperature for 1 h with 5% bovine serum albumin and then with primary antibodies (1:1,000 dilution) at 4°C overnight. The samples were washed three times (5 min/time) with Tris-buffered saline Tween 20. The corresponding secondary antibody was added and incubated at room temperature for 1 h. Then the membranes were washed three times (5 min/time). The samples were developed with chemiluminescence reagents, and the bands were scanned and quantified by densitometric analyses.
Quantitative real-time polymerase chain reaction (qPCR)
Total RNA was extracted from ischemic penumbras of rat brains and reversed based on the protocol. qPCR was carried out according to the manufacturer’s instructions. All data were quantified using the threshold cycle normalized to GAPDH. The primers used in the study were listed: COX-2 forward: 5’-GAT GAC GAG CGA CTG TTC CA-3’; reverse: 5’-TGG TAA CCG CTC AGG TGT TG-3’; iNOS forward: 5’-TCA GGC TTG GGT CTT GTT AGC-3’; reverse: 5’-TGT TGG GCT GGG AAT AGC AC-3; HO-1 forward: 5’-GAA CTG TGG TCG GTA GAG GC-3’; reverse: 5’-ATC AAA GTG GCC ATG ACG CT-3’; NQO-1 forward: 5’-ATT GTA TTG GCC CAC GCA GA-3’; reverse: 5’-CGA CCA CCT CCC ATC CTT TC-3’; Keap-1 forward: 5’-GGA AAC AGA CGT GGA CTT TCG TA-3’; reverse: 5’-TCC AGG AAC GTG TGA CCA TCA TA-3’; SOD1 forward: 5’-AGG GCG TCA TTC ACT TCG AG-3’; reverse: 5’-CCT CTC TTC ATC CGC TGG AC-3’; GAPDH forward: 5’-AGG AGT CCC CAT CCC AAC TC-3’; reverse: 5’-CCC ACA ACA CTG CAT TCA CAC-3’.
Statistical analysis
All the values are presented as means ± SD and were analyzed by SPSS 21.0 software. Mean difference among multiple groups was compared with one-way ANOVA. A value of P < 0.05 was considered as statistically significant.
Results
Sch A pretreatment protects the rat brain against ischemia-reperfusion injury
Infarct volume and neurological score are the specific markers for evaluating brain injury. As shown in Figure 1A and 1B, the infarct volume in the different groups was examined by postmortem TTC staining 24 h after reperfusion. No infarction was found in the Sham group, and the infarct volumes in the MCAO group were the largest. The Sch A treatment, particularly the high-dose group, markedly reduced infarct volume compared to the MCAO group. No neurological deficit was found in the Sham group, while the other groups had different degrees of neurological findings. All Sch A dosages improved neurological function and decreased the neurological scores, particularly in the high-dose groups (Figure 1C). These results indicate that Sch A ameliorates cerebral ischemia.
Figure 1.
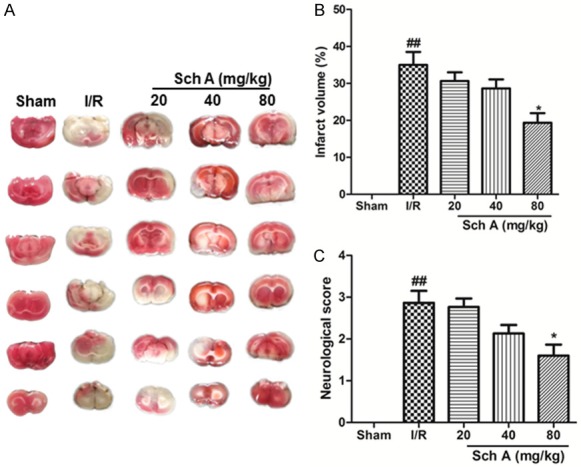
Sch A pretreatment protects the brain against cerebral I/R injury in rats. A and B. Effects of Sch A on cerebral infarct volume. C. Effects of Sch A on neurological scores. All data are mean ± SD. (n = 6, ##P < 0.01 vs. Sham group; *P < 0.05 vs. I/R group).
Sch A treatment reduces the inflammatory reaction in rat tissues after cerebral I/R injury
To access the anti-inflammatory effects of Sch A, we evaluated transcription of proinflammatory genes (inducible nitric oxide synthase [iNOS], cyclooxygenase-2 [COX-2]) and the release of proinflammatory and anti-inflammatory cytokines. As shown in Figure 2A and 2B, iNOS and COX-2 mRNA levels increased significantly in response to the I/R injury. However, Sch A dose-dependently downregulated the transcription of iNOS and COX-2. In addition, increased production of inflammatory cytokines, such as IL-1β, IL-6 and TNF-α, induced by I/R injury decreased significantly with Sch A treatment, particularly at the high dosage. The release of anti-inflammatory cytokines was also tested. Sch A effectively increased the levels of both TGF-β and IL-10. Taken together, our results demonstrate that Sch A inhibits the inflammatory reaction in the cerebral I/R injury model.
Figure 2.
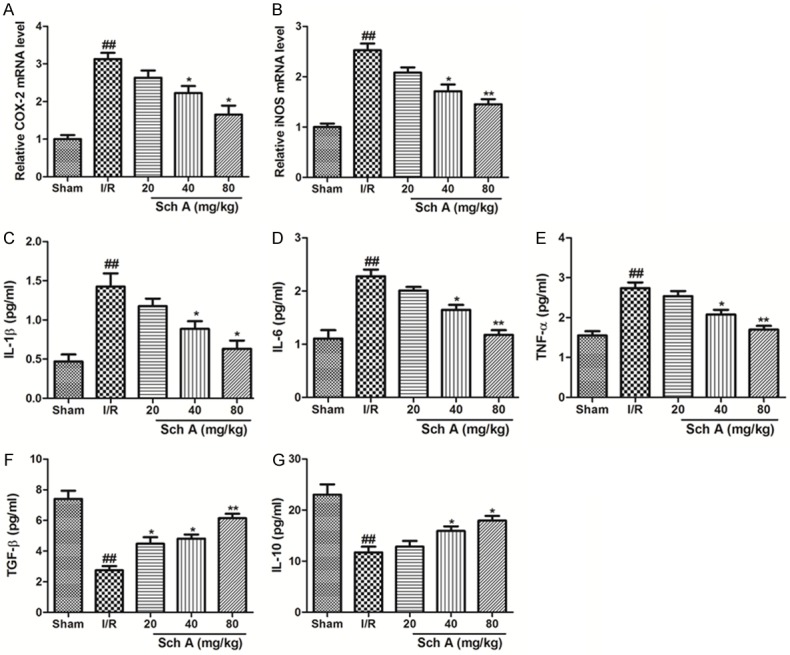
Sch A reduces the inflammatory reaction in rat tissues after I/R injury. (A and B) The relative mRNA levels of COX-2 and iNOS. (C) IL-1β (D) IL-6 (E) TNF-α (F) TGF-β, and (G) IL-10. All data are mean ± SD. (n = 6, ##P < 0.01 vs. Sham group; *P < 0.05 vs. I/R group).
Sch A activates antioxidant reactions in rat tissues after cerebral I/R injury
ROS, SOD, CAT, 4-HNE and 8-OHdG levels were assessed to identify the antioxidant effects of Sch A (Figure 3). A significant increase in ROS production was observed in the I/R group compared to the Sham group. ROS production decreased markedly after treatment compared to that in the I/R group (P < 0.05). SOD and CAT activities decreased markedly in the I/R group compared to the Sham group (P < 0.01), and Sch A reversed this downregulation. The levels of 4-HNE and 8-OHdG in the cortex of I/R group rats were significantly higher than those in the Sham group. Treatment with Sch A significantly reduced 4-HNE and 8-OHdG levels, particularly in the high-dose group (P < 0.05 vs. I/R group). Thus, Sch A activates the antioxidant reactions to protect tissues against I/R injury.
Figure 3.
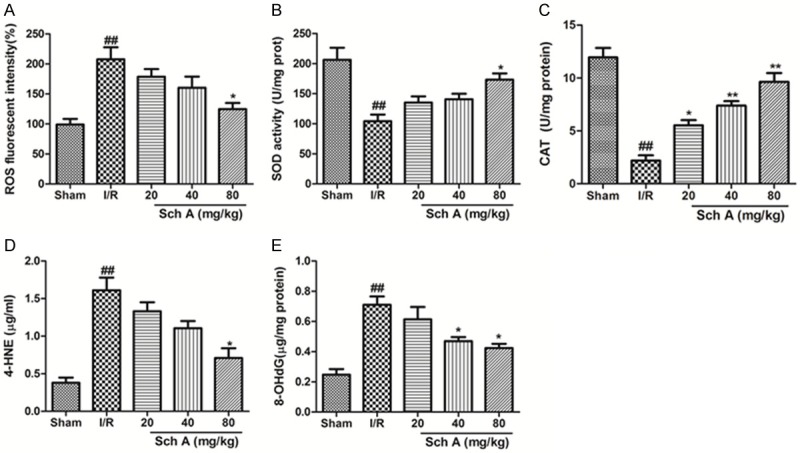
Sch A reduces oxidative damage. A. Mitochondria-generated ROS levels. B. SOD activity. C. CAT activity. D. 4-HNE content. E. 8-OHdG content. All data are mean ± SD. (n = 6, ##P < 0.01 vs. Sham group; *P < 0.05 vs. I/R group).
Sch A induces activation of the Nrf2 signaling pathway in rat tissues
Western blotting was used to analyze the expression of Nrf2 in the ischemic cerebral cortex (Figure 4). Sch A clearly increased nuclear Nrf2 protein expression 24 h after MCAO. We further tested HO-1 and NQO-1 expression to study the activation of Nrf2 signaling. Sch A significantly increased HO-1 and NQO-1 protein levels dose-dependently. Taken together, these data demonstrate that Sch A activates the Nrf2 signaling pathway.
Figure 4.
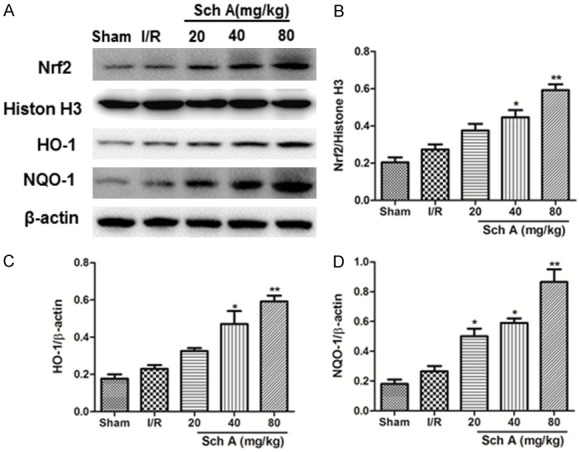
Sch A induces Nrf2 activation in tissues. Protein expression of Nrf2, HO-1, and NQO-1 (A) was evaluated by Western blotting analyses. (B-D) Quantification of Nrf2, HO-1, and NQO-1 levels normalized to that of β-actin. All data are mean ± SD. (##P < 0.01 vs. Sham group; *P < 0.05, **P < 0.01 vs. I/R group).
Sch A protects cells against OGD injury
An OGD model was utilized with differentiated SH-SY5Y cells to further investigate the neuroprotective effects of Sch A. The MTT assay (Figure 5) showed that OGD induced a significant decrease in cell viability to 49.32% compared to control cells (P < 0.01), whereas the Sch A treatment significant increased cell survival to 50.3%, 55.6%, 72.3% and 80.4%, respectively. Sch A (100 μM) significantly reversed cell death caused by OGD injury.
Figure 5.
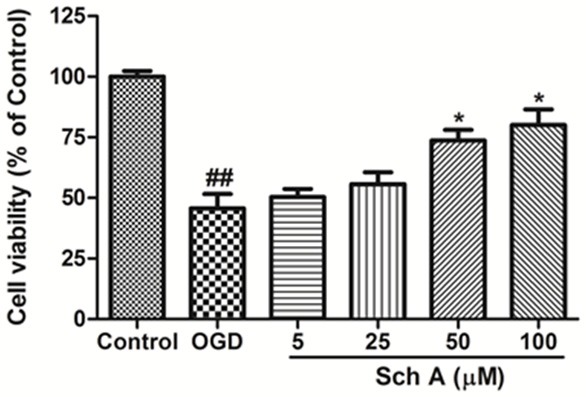
Sch A protects cells against OGD injury. Cell viability was determined using the MTT assay. All data are mean ± SD. (##P < 0.01 vs. Control group; *P < 0.05 vs. OGD group).
Neuroprotective effects of Sch A are Nrf2-dependent in the OGD model
Nrf2 expression was investigated after OGD injury in differentiated SH-SY5Y cells by Western blotting (Figure 6A). Nuclear Nrf2 protein expression increased slightly (P > 0.05) compared to the control group. The Sch A treatment markedly increased the Nrf2 expression levels dose-dependently compared to those in the OGD group.
Figure 6.
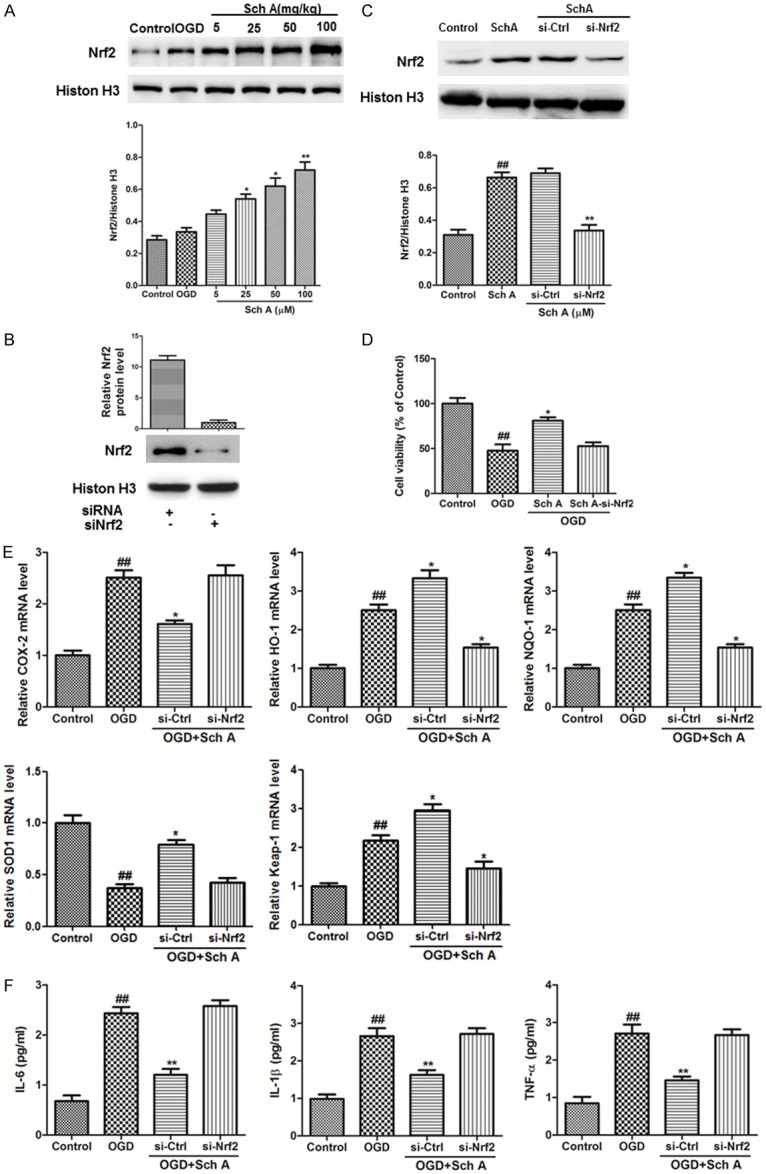
SchA exhibits neuroprotective effects in an Nrf2-dependent manner in OGD model. A. Effect of Sch A on Nrf2 expression in differentiated SH-SY5Y cells. The cells treated with 5-100 μM Sch A for 6 h before OGD. All data are mean ± SD. B, C. The cells were transfected with control or Nrf2 siRNA for 48 h, followed by treatment with 100 μM Sch A for 6 h. Nrf2 expression levels were analyzed by Western blotting. D. The viability of cells in different groups was evaluated by the MTT assay. E. Relative COX-2, HO-1, NQO-1, SOD1 and Keap-1 mRNA levels were determined by real-time RCR. F. The release of IL-6, IL-1β, and TNF-α was detected by ELISA. (##P < 0.01 vs. Control group; *P < 0.05, **P < 0.01 vs. OGD group).
Moreover, siRNA was used to knockdown Nrf2 expression and verify the role of Nrf2 in Sch A-induced neuroprotection. First, we determined knockdown efficiency in transfected differentiated SH-SY5Y cells after 48 h by Western blotting. Nuclear translocation of Nrf2 was significantly reduced after Nrf2 siRNA interference, but nonsense siRNA did not have this effect (Figure 6B). Nrf2 knockdown inhibited the increased cell viability in advance of the 6 h Sch A treatment under OGD (Figure 6D).
Furthermore, HO-1, NQO-1 and Keap-1 mRNA levels increased in response to the OGD and Sch A treatments but were downregulated by Nrf2 siRNA interference, but SOD1 mRNA level decreased in the OGD group and Sch A treatments were downregulated by Nrf2 siRNA interference. The expressions of the COX-2 gene as well as the pro-inflammatory cytokines IL-6, IL-1β, and TNF-α increased markedly after OGD injury and were downregulated by Sch A treatment. However, the effects of Sch A were partially blocked by knockdown of Nrf2 (Figure 6E and 6F). Taken together, these data indicate that the neuroprotective effects of Sch A are Nrf2 dependent.
Neuroprotective effects of Sch A involve the AMPK pathway
Western blotting analyses of ischemic cerebral cortex 24 h after MCAO showed that Sch A clearly increased p-AMPK protein expression. Similar results were observed in the OGD injury model. The MTT assay results showed that cell viability downregulated by OGD injury was reversed by Sch A treatment (Figure 7E). However, the effects were blocked by AMPK siRNA (Figure 7C-E). Thus, the protective effects of Sch A may involve activation of the AMPK pathway.
Figure 7.
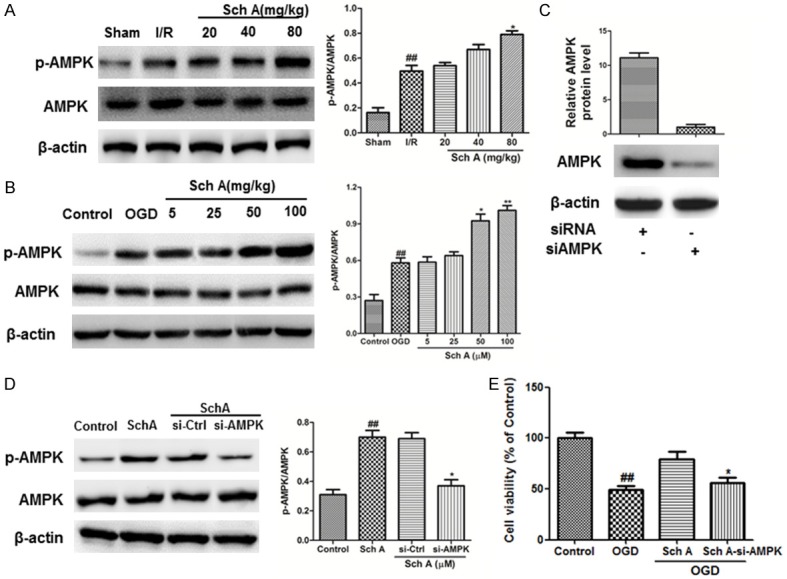
Neuroprotection of Sch A involves the AMPK pathway. A, B. The effects of Sch A on p-AMPK and AMPK expression were assessed by Western blotting. C, D. Cells were transfected with control or AMPK siRNA for 48 h, followed by treatment with 100 μM Sch A for 6 h; AMPK expression levels were analyzed by Western blotting. E. The viability of cells in different groups was evaluated using the MTT assay. Data are mean ± SD. (##P < 0.01 compared to Control cells; *P < 0.05, **P < 0.01 compared to si-Control (si-Ctrl) cells).
Nrf2-dependent neuroprotective effects of Sch A are regulated by the AMPK pathway
We further determined if Nrf2 nuclear translocation is regulated by AMPK. As shown in Figure 8, the levels of nuclear Nrf2 protein were downregulated by OGD injury and restored by Sch A treatment. However, upregulation was effectively blocked by AMPK siRNA. These results demonstrate that the Nrf2-dependent neuroprotective effects of Sch A are regulated by the AMPK pathway.
Figure 8.
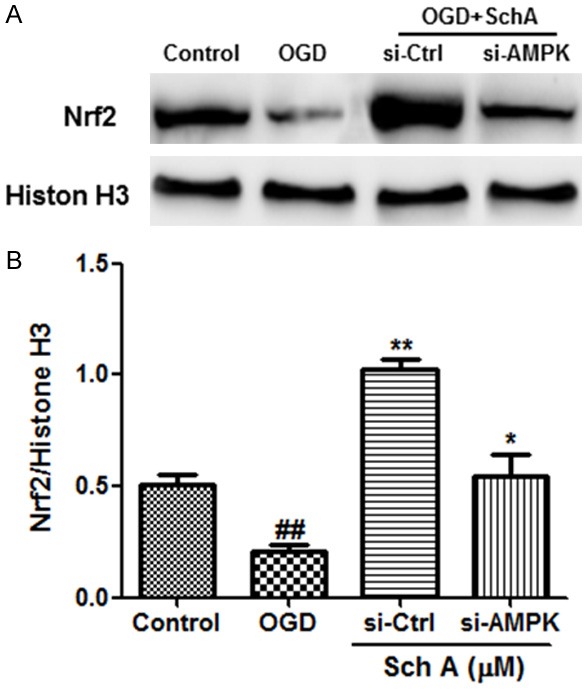
Nrf2-dependent neuroprotective effects of Sch A are regulated by the AMPK pathway. A, B. The effects of si-AMPK on nuclear Nrf2 protein levels were tested by Western blotting. Data are mean ± SD. (##P < 0.01 compared to Control cells; *P < 0.05, **P < 0.01 compared to si-Control (si-Ctrl) cells).
Discussion
Although the mechanism of ischemic stroke is complex, overproduction of free radicals is one of the most important initiating factors that cause severe damage to biological macromolecules, leading to cell and tissue damage [18]. Therefore, anti-inflammatory agents and antioxidants have been considered as treatments for preventing and treating stroke, and certain anti-inflammatory agents with antioxidant effects have demonstrated neuroprotective effects [19]. Here, our in vitro and in vivo studies show for the first time that Sch A has protective effects against cerebral I/R injury.
Sch A improved neurological scores, reduced infarct volume ratios, reduced the number of necrotic neurons, increased SOD and CAT activities, decreased ROS production, and decreased 4-HNE and 8-OHdG contents by inhibiting oxidative stress. It also inhibited release of inflammatory cytokines, including IL-1β, IL-6 and TNF-α, and increased the concentrations of anti-inflammatory cytokines such as TGF-β and IL-10. These results demonstrate that Sch A protects rats against I/R injury by activating antioxidant and anti-inflammatory reactions.
Studies have not thoroughly investigated the anti-inflammatory signaling pathways, particularly the network around the Nrf2-mediated antioxidant pathway. AMPK has gained much attention, as it acts as an attractive target to control inflammation [20]. Activation of AMPK inhibits inflammation by inhibiting inflammatory signaling, such as the NF-κB pathway, and serves as a potential target for treating inflammation disorders, whereas downregulation of AMPK activity increases inflammation [21]. Accumulating evidence suggests that oxidative stress is a major mechanism involved in brain diseases [22]. Many studies have established that the underlying mechanism of acute organ IR injury, including cerebral I/R injury, mainly involves a burst of ROS/oxidative stress from ischemic organs/tissues during reperfusion of ischemic tissues that can trigger the opening of the mitochondrial permeability transition pores, mitochondrial depolarization, decreased ATP synthesis, and increased generation of ROS/oxidative stress [23]. As a crucial regulator in response to oxidative stress and activation of endogenous antioxidant enzymes, a growing body of evidence from both in vitro and in vivo studies has established that transcriptional activation of the Nrf2 signaling pathway protects cells against oxidative/electrophilic stress, which might lead to inflammation, apoptosis, premature aging, and cellular transformation [24].
In the current study, we found that Sch A markedly increased the expression of nuclear Nrf2 and phosphorylation of AMPK in the ischemic cerebral cortex 24 h after MCAO. Similar results were found in the OGD model in vitro.
To further examine if AMPK/Nrf2 is involved in Sch A-induced neuroprotection, we used RNA interference directed against Nrf2 and AMPK. Knockdown of Nrf2 abolished the protective effects of Sch A on cell viability and reversed the downregulation of inflammatory reactions and Nrf2 downstream gene levels induced by Sch A. In addition, the neuroprotective effects of Sch A were blocked when AMPK was knocked down, and nuclear translocation of Nrf2 was also inhibited. Taken together, these data indicate that the neuroprotective effects of Sch A are regulated by the AMPK/Nrf2 signaling pathway.
In conclusion, our study elucidates a novel pathway by which Sch A protects against I/R-induced inflammation and oxidative injury in vivo and in vitro. Our results demonstrate that Sch A has a neuroprotective effect on neonatal hypoxic ischemic brain injury and that the effect is related to the anti-inflammation and antioxidant effects of the AMPK/Nrf2 pathway. These results provide a better understanding of the molecular mechanisms associated with the neuroprotective effects of Sch A and may provide new insight into a better design of neuroprotective agents against ischemic stroke. More work is needed before Sch A therapy for stroke can be advanced to the clinic.
Acknowledgements
This study was sponsored by National Science Foundation of China (81873387).
Disclosure of conflict of interest
None.
References
- 1.Zhang Q, Zhang L, Yang X, Wan Y, Jia J. The effects of exercise preconditioning on cerebral blood flow change and endothelin-1 expression after cerebral ischemia in rats. J Stroke Cerebrovasc Dis. 2014;23:1696–702. doi: 10.1016/j.jstrokecerebrovasdis.2014.01.016. [DOI] [PubMed] [Google Scholar]
- 2.Guo C, Wang S, Duan J, Jia N, Zhu Y, Ding Y, Guan Y, Wei G, Yin Y, Xi M, Wen A. Protocatechualdehyde protects against cerebral ischemia-reperfusion-induced oxidative injury via protein kinase Cε/Nrf2/HO-1 pathway. Mol Neurobiol. 2017;54:833–845. doi: 10.1007/s12035-016-9690-z. [DOI] [PubMed] [Google Scholar]
- 3.Wicha P, Tocharus J, Janyou A, Jittiwat J, Changtam C, Suksamrarn A, Tocharus C. Hexahydrocurcumin protects against cerebral ischemia/reperfusion injury, attenuates inflammation, and improves antioxidant defenses in a rat stroke model. PLoS One. 2017;12:e0189211. doi: 10.1371/journal.pone.0189211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li P, Stetler RA, Leak RK, Shi Y, Li Y, Yu W, Bennett MVL, Chen J. Oxidative stress and DNA damage after cerebral ischemia: Potential therapeutic targets to repair the genome and improve stroke recovery. Neuropharmacology. 2018;134:208–217. doi: 10.1016/j.neuropharm.2017.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ledoux SP, Druzhyna NM, Hollensworth SB, Harrison JF, Wilson GL. Mitochondrial DNA repair: a critical player in the response of cells of the CNS to genotoxic insults. Neuroscience. 2007;145:1249–1259. doi: 10.1016/j.neuroscience.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin XH, Pan JB, Zhang XJ. Anti-inflammatory and anti-oxidant effects of apigetrin on LPS-induced acute lung injury by regulating Nrf2 and AMPK pathways. Biochem Biophys Res Commun. 2017 doi: 10.1016/j.bbrc.2017.07.071. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 7.Rao Z, Pan X, Zhang H, Sun J, Li J, Lu T, Gao M, Liu S, Yu D, Ding Z. Isoflurane preconditioning alleviated murine liver ischemia and reperfusion injury by restoring AMPK/mTOR-mediated autophagy. Anesth Analg. 2017;125:1355–1363. doi: 10.1213/ANE.0000000000002385. [DOI] [PubMed] [Google Scholar]
- 8.Ashabi G, Khodagholi F, Khalaj L, Goudarzvand M, Nasiri M. Activation of AMP-activated protein kinase by metformin protects against global cerebral ischemia in male rats: interference of AMPK/PGC-1α pathway. Metab Brain Dis. 2014;29:47–58. doi: 10.1007/s11011-013-9475-2. [DOI] [PubMed] [Google Scholar]
- 9.Xu W, Li F, Xu Z, Sun B, Cao J, Liu Y. Tert-butylhydroquinone protects PC12 cells against ferrous sulfate-induced oxidative and inflammatory injury via the Nrf2/ARE pathway. Chem Biol Interact. 2017;273:28–36. doi: 10.1016/j.cbi.2017.05.021. [DOI] [PubMed] [Google Scholar]
- 10.Wang CH, Wu SB, Wu YT, Wei YH. Oxidative stress response elicited by mitochondrial dysfunction: implication in the pathophysiology of aging. Exp Biol Med (Maywood) 2013;238:450–60. doi: 10.1177/1535370213493069. [DOI] [PubMed] [Google Scholar]
- 11.Zhou J, Ma X, Cui Y, Song Y, Yao L, Liu Y, Li S. Methyleugenol protects against t-BHP-triggered oxidative injury by induction of Nrf2 dependent on AMPK/GSK3β and ERK activation. J Pharmacol Sci. 2017;135:55–63. doi: 10.1016/j.jphs.2017.09.003. [DOI] [PubMed] [Google Scholar]
- 12.Su X, Gao C, Shi F, Feng X, Liu L, Qu D, Wang C. A microemulsion co-loaded with Schizandrin A-docetaxel enhances esophageal carcinoma treatment through overcoming multidrug resistance. Drug Deliv. 2017;24:10. doi: 10.1080/10717544.2016.1225854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang CP, Li GC, Shi YW, Zhang XC, Li JL, Wang ZW, Ding F, Liang XM. Neuroprotective effect of schizandrin A on oxygen and glucose deprivation/reperfusion-induced cell injury in primary culture of rat cortical neurons. J Physiol Biochem. 2014;70:735. doi: 10.1007/s13105-014-0342-3. [DOI] [PubMed] [Google Scholar]
- 14.Liu GT. Pharmacological actions and clinical use of fructus schizandrae. Chin Med J. 1989;102:740–749. [PubMed] [Google Scholar]
- 15.Wang O, Cheng Q, Liu J, Wang Y, Zhao L, Zhou F, Ji B. Hepatoprotective effect of Schisandra chinensis (Turcz.) Baill. lignans and its formula with Rubus idaeus on chronic alcohol-induced liver injury in mice. Food Funct. 2014;5:3018–25. doi: 10.1039/c4fo00550c. [DOI] [PubMed] [Google Scholar]
- 16.Song F, Zeng K, Liao L, Yu Q, Tu P, Wang X. Schizandrin a inhibits microglia-mediated neuroninflammation through inhibiting TRAF6-NF-κB and Jak2-Stat3 signaling pathways. PLoS One. 2016;11:e0149991. doi: 10.1371/journal.pone.0149991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- 18.Zhao Y, Fang Y, Li J, Duan Y, Zhao H, Li G, Luo Y. Neuroprotective effects of Chrysophanol against inflammation in middle cerebral artery occlusion mice. Neurosci Lett. 2016;630:16–22. doi: 10.1016/j.neulet.2016.07.036. [DOI] [PubMed] [Google Scholar]
- 19.Wang C, Pei A, Chen J, Yu H, Sun ML, Liu CF, Xu X. A natural coumarin derivative esculetin offers neuroprotection on cerebral ischemia/reperfusion injury in mice. J Neurochem. 2012;121:1007–13. doi: 10.1111/j.1471-4159.2012.07744.x. [DOI] [PubMed] [Google Scholar]
- 20.Park EJ, Kim YM, Chang KC. Hemin reduces HMGB1 release by UVB in an AMPK/HO-1-dependent pathway in human keratinocytes HaCaT cells. Arch Med Res. 2017;48:423–431. doi: 10.1016/j.arcmed.2017.10.007. [DOI] [PubMed] [Google Scholar]
- 21.Chen Y, Zheng Y, Liu L, Lin C, Liao C, Xin L, Zhong S, Cheng Q, Zhang L. Adiponectin inhibits TNF-α-activated PAI-1 expression via the cAMP-PKA-AMPK-NF-κB axis in human umbilical vein endothelial cells. Cell Physiol Biochem. 2017;42:2342–2352. doi: 10.1159/000480006. [DOI] [PubMed] [Google Scholar]
- 22.Du F, Zhang Z, Li H, Yu Q, Douglas JT, Bratasz A, Kuppusamy P, Yan SD. Increased electron paramagnetic resonance signal correlates with mitochondrial dysfunction and oxidative stress in an Alzheimer’s disease mouse brain. J Alzheimers Dis. 2016;51:571–80. doi: 10.3233/JAD-150917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xia F, Xia Y, Chen S, Chen L, Zhu W, Chen Y, Papadimos TJ, Xu X, Liu L. Lipid emulsion mitigates impaired pulmonary function induced by limb ischemia/reperfusion in rats through attenuation of local cellular injury and the subsequent systemic inflammatory. BMC Anesthesiol. 2017;17:83. doi: 10.1186/s12871-017-0375-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Speciale A, Chirafisi J, Saija A, Cimino F. Nutritional antioxidants and adaptive cell responses: an update. Curr Mol Med. 2011;11:770–89. doi: 10.2174/156652411798062395. [DOI] [PubMed] [Google Scholar]