

Abstract
Federation is a popular concept in building distributed cyberinfrastructures, whereby computational resources are provided by multiple organizations through a unified portal, decreasing the complexity of moving data back and forth among multiple organizations. Federation has been used in bioinformatics only to a limited extent, namely, federation of datastores, e.g. SBGrid Consortium for structural biology and Gene Expression Omnibus (GEO) for functional genomics. Here, we posit that it is important to federate both computational resources (CPU, GPU, FPGA, etc.) and datastores to support popular bioinformatics portals, with fast-increasing data volumes and increasing processing requirements. A prime example, and one that we discuss here, is in genomics and metagenomics. It is critical that the processing of the data be done without having to transport the data across large network distances. We exemplify our design and development through our experience with metagenomics-RAST (MG-RAST), the most popular metagenomics analysis pipeline. Currently, it is hosted completely at Argonne National Laboratory. However, through a recently started collaborative National Institutes of Health project, we are taking steps toward federating this infrastructure. Being a widely used resource, we have to move toward federation without disrupting 50 K annual users. In this article, we describe the computational tools that will be useful for federating a bioinformatics infrastructure and the open research challenges that we see in federating such infrastructures. It is hoped that our manuscript can serve to spur greater federation of bioinformatics infrastructures by showing the steps involved, and thus, allow them to scale to support larger user bases.
Keywords: computational genomics, cyberinfrastructure, federation, identity management, MG-RAST, genomic privacy
Need for federated systems in computational genomics
Federation is a popular data-sharing concept in instantiating distributed cyberinfrastructures, with computational resources provided by multiple institutions through a common, unified portal. Clients access these resources without being made aware of which of the partnering organizations is providing the resource. Thus, the clients are relieved of much complexity, such as, negotiating access rights with individual organizations, moving data back and forth among multiple sites and portability of programs to process the data that are resident at multiple organizations. This concept has found practical instantiation in several widely used cyberinfrastructures, such as XSEDE, supported by the National Science Foundation (NSF) for providing leading edge compute resources to scientists across the country, and the Open Science Grid, supported jointly by the NSF and the Department of Energy, geared toward high energy physicists. Federation has been used in bioinformatics only to a limited extent, namely, federation of datastores, e.g. Gene Expression Omnibus (GEO) for microarray, next-generation sequencing and other forms of high-throughput functional genomics data. In this article, we argue for the need to federate a greater number of bioinformatics cyberinfrastructures. We show what are the available computational tools that have been used successfully for federation in other domains and then discuss the open research challenges to adapt them to bioinformatics in general and genomics in particular.
The field of computational genomics has grown in terms of the sizes of data that it needs to process, raising the bar significantly in terms of the computational and storage infrastructure that it needs. This has resulted in some popular portals for providing such resources to the community at large, such as cBioPortal for Cancer Genomics, originally developed at Memorial Sloan Kettering Cancer Center [1], the JGI Genome Portal from the US Department of Energy [2] and the metagenomics-RAST (MG-RAST) portal at Argonne National Laboratory for metagenomics data storage and analysis [3]. It is a metagenomics sequence data analysis platform, which accepts raw sequence data submission from freely registered users and has an automated workglow with a series of bioinformatics tools to process, analyze and interpret the data before returning analysis results to users. MG-RAST is popular among a number of scientific communities, such as microbiology, medical science, pharmacology, environmental science, ecology, archeology and anthropology. It currently hosts roughly 280 K data sets and has been highlighted in almost 2000 citations [4]. MG-RAST currently supports roughly 50 K annual users, with roughly 500 of them actively using the infrastructure on a daily basis. In terms of data, MG-RAST handles >4 terabase pairs (Tb) of data per month and typically >300 user submissions per day. One can readily imagine the significant amount of computational resources—processing, storage and networking that have been brought to bear to support a public computational infrastructure such as MG-RAST.
The model followed in all such portals, so far, has primarily been for centralized compute and storage resources, provided by the hosting organization. There are significant efficiencies that have resulted from such centralization. Notably, there exists a core well-trained staff, capable of managing the infrastructure and lowering the costs for procuring and managing the infrastructure, resulting from the economies of scale. However, increasingly, this model is being put under strain because of the exponentially increasing sizes of the data sets and consequent increase in the processing demands. This in turn is because of the democratization of the process of generating genomics data, sparked by the advances in next-generation sequencing technologies. Now, more than ever, there are large numbers of domain scientists who can generate the primary data and wish to perform analysis of the data using a validated pipeline of mature bioinformatics software packages, on a remote computation-rich environment.
In addition to this need for remote resources, there is an impetus to leverage resources available at the local organization. First, the volume of data is large such that it is not unusual to have petabytes of sequencing data being processed regularly at medium-sized laboratories. Therefore, it may be more efficient to have a local instantiation of the pipeline, so that data does not have to leave the premises. Another common use case is the need to share some specialized software packages that are locally installed, either with the community at large or with a select group of remote collaborators. Finally, there are privacy concerns about genomics data, and because of policies, either organizational or even national, it may be advisable to keep the sensitive portion of the data local. These use cases point directly to the need for federation of genomics pipelines.
Federation has been defined in the context of clouds [5] and that definition is useful in our context:
‘Federation is the ability of multiple independent resources to act like a single resource. Cloud computing itself is a federation of resources, so the many assets, identities, configurations and other details of a cloud computing solution must be federated to make cloud computing practical’.
Going further back [6], federation in the context of enterprise architectures was defined as:
‘Federated architecture describes an approach to enterprise architecture that allows interoperability and information sharing between semi-autonomous business units’.
A federation is thus seemingly a simple idea: it allows the end users to transparently access a set of resources and services, distributed among several independent service providers. However, there are several subtleties and technical design options in this space, which can be summarized under four orthogonal factors:
Which resources are to be federated?
What degree of access to allow to which end users?
What guarantees about reliability, security and privacy are to be provided to end users?
What level of integration is to be achieved among resources at various organizations contributing resources to the federation?
Achieving federation involves multiple technical and policy steps, including simple policy definitions for granting privilege and runtime access and federated identity management (FIM) for the authentication and authorization of principals in the federated infrastructure. It also involves definition of simple privacy policies, specialized to the genomics world, and seamless integration of privacy-preserving transforms into the genomics workflow. We describe our vision of federating genomic workflows, using the existing, widely used metagenomics portal, MG-RAST, as our exemplar. We describe the initial steps we are taking toward that vision, the broad challenges that we see moving forward to federating any computational genomics infrastructure and the technical solution approaches that can be adapted to solve these challenges.
Genomic data deluge and federation of genomics pipelines
Genomics has recently been described as a Big Data domain that presents itself as a ‘four-headed beast’ [7], given its need for huge resources for acquisition, storage, distribution and analysis. Briefly, data acquisition in genomics is both distributed, given the democratization of next-generation sequencing instruments, and highly heterogeneous, stemming from the idiosyncratic genomic data formats. As per the current data generation estimates, which records a doubling every 7 months, it is estimated that we will reach one zettabase (roughly 1000 exabases) of sequence per year by the year 2025. This has resulted in similar data-wrangling strategies that particle physicists at Conseil Européen pour la Recherche Nucléaire (CERN), and astronomers have dealt with in the past decades. Further, although in this article we will focus on genomic and metagenomic data, biological data are intrinsically even more diverse, with proteomic, chemogenomic and clinical data, such as from medical records [e.g. electronic health record (EHR) data sets], well within its ambit. Now, narrowing down to just genomic data, even a single human genome with 3.2 billion DNA base pairs, is roughly 140 gigabytes in size, which is on the lower end of the spectrum, considering a lower-than-desired 20× coverage. This would also include the information that is stored about the bases that are sequenced. While there are ways to compress these data [8] and the storage density continues to increase [9], this is still a pain point. Beyond storage of the data, consider the computational cost of analyzing the genomic data. A simple and ubiquitous form of analysis is aligning two genomic sequences to determine similarities and differences. Using the de facto standard software, BLAST, for local alignment of two 100 Mbp sequences takes >1 h [10] on a 4 node, 64 core cluster, and the computational time grows exponentially >2 Mbp sequences. For reference, a single human chromosome, chromosome 1, the largest of the lot, is ∼250 Mbp. Suffice it to say, the computational burden of analyzing genomic sequences has become prohibitive for most small- to mid-sized organizations. Recently, we have come up with a creative way of recognizing common kernels across a large set of computational genomics algorithms and combining these kernels using an engineered compiler—Sarvavid [11], which performs genomic domain-specific optimizations. This is in stark contrast to existing genomic applications, which are primarily written in a monolithic manner, and therefore, not easily amenable to automatic optimizations. Along similar lines, use of federated cyberinfrastructures both for generated data—confronting the small-N problem, often plaguing genomics data sets—and for computational resources, given the ready expansion and evolution of genomics data, is an attractive solution.
General architecture of federated systems
In Figure 1, we show the four aspects of federating a genomics infrastructure, which has computation and storage assets. We outline the basic motivation for why, we and others in the community, are considering federation, the foundational technologies that are crucial to federation, a possible step-wise deployment strategy to move a current production nonfederated infrastructure to a federated one and, finally, the technical challenges, as we see them.
Figure 1.

The quad chart shows the motivation—why we want to explore federation for computational infrastructures for genomics workflows. It then shows the supporting technologies that federation of genomics pipelines can build on. Next, we show a step-wise deployment strategy to take a current production, nonfederated infrastructure, and make it federated. Finally, we show the challenges in federating such an infrastructure.
Federation of heterogeneous computing resources and storage systems requires us to address two broad issues in the area of security and distribution, which are (i) access privilege controls and (ii) use of a distributed set of resources. The access privilege control becomes necessary because a user is intended to simultaneously use multiple resources where access privilege to the user may vary and the user can follow the bring-your-own-identity (BYOI) paradigm. A federation system must provide an autonomic way by which a user can access each resource at the right level of privilege. The distribution issue is fundamental to federation because the computational resources are distributed and potentially may belong to different participating organizations. A sub-issue here is the data exchange with potentially different data representations across resources. A federation system is responsible for interconnecting heterogeneous resources and providing an abstraction layer that transforms data from one format to another. This issue is particularly relevant in our context because of the presence of multiple data formats in the area of genomics, all belonging to different standards [12].
To deal with these issues, a federation system is typically structured as shown in Figure 2. An end user is first authenticated by a single sign-on (SSO) server using login credentials (e.g. user id and password). Then, the SSO server maintains a session that authenticates the user for all federated resources. The federation management middleware is an abstraction that hides integration complexities, and provides users with unified interfaces to heterogeneous resources. When a user tries to use a resource, the middleware layer retrieves the specific authentication credentials from the SSO server and accesses the resource with the predefined privileges for that level of user. Resource-specific communication protocols and data representations are also converted by the middleware, which communicates with the underlying resources and transforms the query and response between users and resources and between heterogeneous resources.
Figure 2.

Overview of resource federation. The SSO server authenticates end users, and eliminates further prompts when the users switch between resources. The federation management middleware provides users with unified interfaces to heterogeneous resources, potentially located at different organizations.
Foundational technologies to establish a federated infrastructure
Federated identity management
A starting point to accessing the federated system will be the use of an FIM system, such as InCommon [13], for the local resource providers and consumers alike. The workflow pertaining to such a system would be as follows: first, a user clicks on a Service Provider’s resource. Using federation SSO software, the user is authenticated by her Identity Provider, which releases only enough identity data to allow the Service Provider to make a decision about whether to allow access to the user. Second, the Service Provider uses the minimum identity information necessary to control access to the resource. Challenges would involve negotiation between the service and identity provider and some understanding of an OASIS Security Assertion Markup Language (SAML, pronounced SAMeL), such as, Shibboleth [14].
FIM versus SSO
FIM means all the policies, protocols and technologies in place that enable users of one security domain to seamlessly access resources in another domain. The SSO is one particular form of FIM pertaining only to authentication. With SSO, a user is uniquely recognized by each of the domains enrolled in the federation system. When the user signs into the gateway system (i.e. SSO server), the SSO solution replays log-in credentials for every domain, eliminating further log-in prompts when the user switches domains. Thus, this approach would be useful when we federate security domains, each of which has already has a unique log-in system.
FIM and decentralized identity management
Another form of FIM is what is often referred to as identity federation. Although identity federation looks like SSO to end users, it is different in how authentication is made. In the identity federation system, log-in credentials of each user are only known to a front-end system, and the federation server passes on the initial token using one of the standard identity protocols, such as SAML, OpenID, WS-Trust, WS-Federation or OAuth. Each security domain joining the federation is not aware of the user identity. Instead, it trusts and accepts the credential passed on from the front-end server. Such a solution has an advantage over SSO in that a user is required to sign up only once at the front-end server. Thus, it is suitable when we add a newly established resource into an existing federation, or when many organizations agree to join a new federation system. Figure 3 summarizes the difference between the SSO and the identity federation. Use of FIM standards can reduce development cost and increase security and privacy compliance. FIM platforms that use formal Internet standards include OAuth [15], OpenID [16] and InCommon [13].
Figure 3.
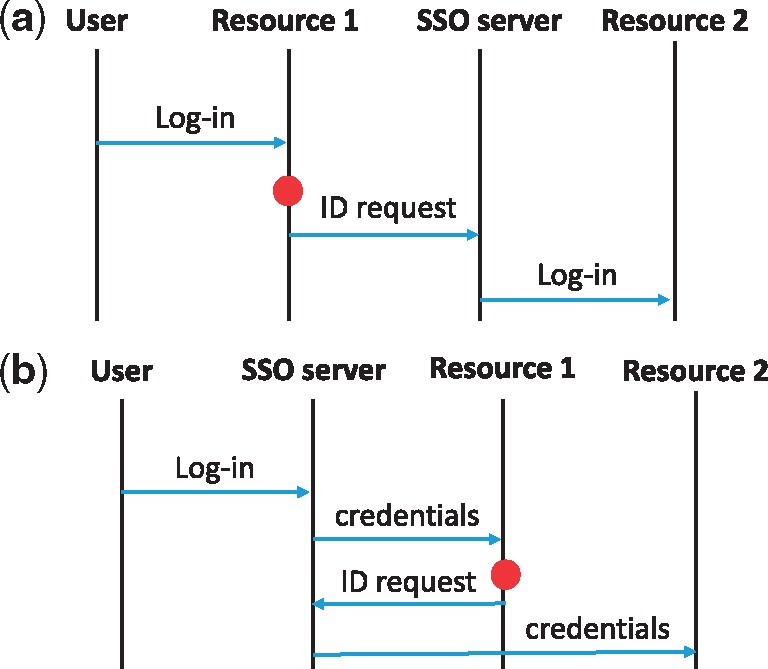
High-level comparison between SSO and identity federation. Here, the (red) dot denotes when a user needs to switch from resource 1 to resource 2.
Software-defined network
Federation systems often require to apply a different policy to route each individual flow between two end points. This is because Quality of Service requirements may vary depending on user privileges. For example, one flow may need a lot higher bandwidth than others because of its higher priority or larger volumes of streaming data. However, with the static architecture of traditional networks, it is hard to change the routing policy dynamically. This issue can be addressed with an emerging architecture of networking technology, called software-defined networking (SDN) [17]. This architecture decouples the network control layer and the data layer (the traditional routers that forward packets efficiently). By separating the control logic from forwarding devices, the idea is that the control of networks can be centralized and simplified.
Figure 4 shows a high-level overview of an SDN framework. In the figure, the control layer offers a centralized view of the overall network and enables network administrators to directly program the underlying forwarding devices (routers and switches) deciding how to forward each and every network traffic. Southbound application program interfaces (APIs) mean the interface by which the control layer relays information to forwarding devices. There exist several software solutions for this purpose, which include OpenFlow [18] and Cisco Open Network Environment [19]. Northbound APIs enable the control layer to communicate with the application. These help network administrators to shape traffic and deploy services according to application requirement.
Figure 4.
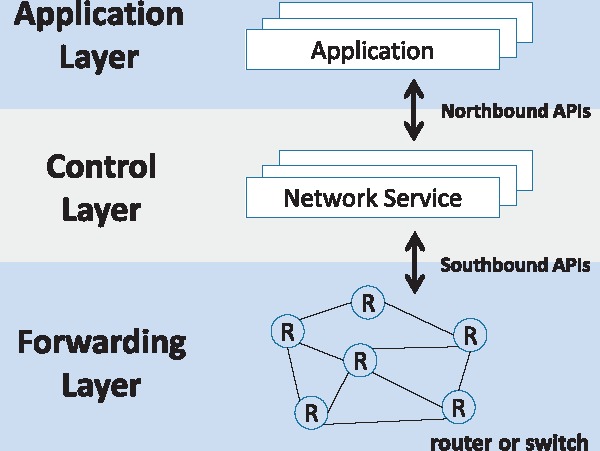
SDN framework.
With programmable networks that can dynamically provision resources to address the changing needs of applications, SDN can resolve some issues that arise with federation systems. First of all, SDN can enable efficient routing among resources. Depending on the characteristic of a data flow, we can set up the fastest route between two resources, or a certain number of intermediate nodes to provide in-network processing, e.g. to filter the data, thus reducing the pressure on the network. The centralized control enabled by SDN will also be able to improve the security of a federation system. For example, the central controller can redirect specific suspect flows to an intrusion detection system or a distributed denial-of-service detection system, allowing us to monitor events in a dynamic environment. In federation systems, we need to specify which user can access what resource. This kind of access privilege control is written by access control list (ACL) rules that define permissions attached to objects, which could be programs, processes, files, port numbers or IP addresses. SDN can provide a convenient way to execute the ACL rules by making the control layer decide whether a certain resource request from an application should be forwarded.
Data federation platforms
Data federation platforms (DFPs) include communication protocol wrapper and data representation wrapper that provide a uniform user interface, enabling exchange of data in different formats across heterogeneous resources. Another important functionality of DFPs is to create a virtual database and then map a user query into multiple suboperations, each of which is redirected to a corresponding federated database. This is akin to shards of data being kept in different datastores, but here, the volume is higher for each database, and they may be geographically distributed (and often will be). Thus, DFPs should be able to identify which databases are involved in the response to a user query, and figure out how to transform the heterogeneous representations of the sources. To this end, DFPs collect metadata that describes the structure of the original data and places it into the virtual database as well. Metadata is critical in genomics and metagenomics contexts and represents the in-depth, controlled description of the sample that the submitted sequence was taken from. Essentially, metadata captures the ‘what, where, how, and when’ of the user’s study from collection to sequence generation, plus contextual data such as environmental conditions (latitude, longitude, temperature) or clinical observations.
Resource scheduler
To manage the federated resources efficiently, one will need to assign the optimal resources required to deliver the job requested by a user. The software package responsible for this is called a resource scheduler. Formally, resource scheduling can be said to find the optimal mapping by which user-requested jobs are assigned to multiple resources available in such a way that a vector of objectives is maximized in a Pareto-optimal sense. The objectives considered in the resource scheduler typically include time to complete a job, energy consumption, cost-effectiveness and fair use of resources from various institutions. There are many resource schedulers available for distributed infrastructures, such as Portable Batch System and SLURM. The domain-specific challenge here will be that many bioinformatics infrastructures are not built to a standard specification that they can interface with these existing schedulers that were built for the High Performance Computing domain. Further, because of privacy concerns (detailed later in ‘Considerations for privacy with ubiquitous data storage and federation’ section), the schedulers may have an additional constraint, such as data cannot leave the computational assets of a particular organization.
Experiences from MG-RAST
The field of metagenomics encompasses the sequencing and analysis of the total microbial DNA, sampled directly from the environment. This culture-independent analysis of the metagenome, i.e. of the genetic material from this microbial potpourri, has transformed the study of microbial communities, affording the ability to study >99% of unculturable prokaryotes, present in various environments. Since 2008, the metagenomics RAST (MG-RAST) pipeline has served as a de facto repository and analysis provider for the exponentially increasing amount of metagenomic data sets [4]. Today, it is the most widely used metagenomic analysis resource and data portal. The system supports computing, informatics and Big Data Science for microbial communities. On a daily basis, >300 registered users use the system for complex data analysis. The rapid growth in the total number of users of the MG-RAST system can be seen from Figure 5, showing a 230% growth in the past 5 years (April 2012 to April 2017). This historical trend, coupled with the expectation of future growth, is an important driver toward the design of a federated system for MG-RAST.
Figure 5.

The growth in the total number of users in the MG-RAST metagenomics system. The continued growth and the resultant pressure on the computational resources are important motivators for moving toward federating the system.
Motivation for federation
The success of MG-RAST motivated us to explore the possibility of federating this resource. In Figure 6, we show the MG-RAST annotation pipeline, which supports amplicon (‘who is there’), shotgun data analysis (‘what is the genomic functional potential of the microbes’) and metatranscriptomics (‘who is doing what’). The MG-RAST pipeline is composed of three conceptual steps: quality control, data reduction and analysis. Some examples of computational tools being used are Bowtie, a sequence alignment tool [20]; gene prediction uses FragGeneScan, a hidden Markov model-based gene prediction tool for short and error-prone sequences [21]; and clustering uses UCLUST [22] or CD-HIT [23], a search-based clustering tool. The protein and RNA identification uses another sequence alignment tool named BLAT [24], and more recently, being replaced by DIAMOND [25]. One common characteristic of these tools is that they are computationally demanding. An approximate measure of our computational cost for the entire pipeline is 430 000 core hours per terabase pair of input sequences. The approximate computation cost, assuming Amazon’s EC2 pricing, would be $1500 per gigabase pair (Amazon EC2 standard prices for general-purpose workload m4.16xlarge as of May 2017). As the volume of user requests for sequencing increases, there is a need for a greater number of compute cores, and this stresses the current infrastructure. Currently, the computational resources are provided by the DOE Magellan cloud computing environment at Argonne National Laboratory. It is a large high-performance computing platform used to run data-intensive computing workloads. In raw form, it is made up of 504 general-purpose Intel Nehalem nodes, 200 ‘active’ compute/storage nodes and 50 GPU nodes. It has over 42 TB of RAM and almost 2 PB of storage. However, it is used in shared mode by MG-RAST, and therefore, there is pressure to use more federated computing resources.
Figure 6.

The MG-RAST pipeline showing the various software stages involved with quality control, data reduction and analysis. Each stage of the pipeline may run on computational resources at participant sites and access data at participant sites, under the full federation model.
The popularity of MG-RAST is also straining the storage resources. The repository today has grown to 40 696 public and 279 663 total metagenomes, containing >1 trillion sequences and 131.28 Tb, which translates to roughly 600 Tb of storage required. Further, the size of the derived data relative to the primary sequence data uploaded by the user is often an order of magnitude (or more) larger in size. This happens because of the additional annotations and metadata generation that happens, such as the phylogenetic and functional assignments of the metagenome being analyzed. This has the multiplier effect on the storage requirements, as more data sets are uploaded by users for analysis and storage.
To accommodate the increasing demands on compute and storage resources, we plan to federate the MG-RAST infrastructure in three stages. These three stages form a template for federating any Web-based bioinformatics resource, where the following three conditions need to be satisfied.
There is pressure on multiple categories of resources.
The resource is a production resource and a downtime because of federation, either at deployment or during operation, is not desirable.
There is some notion of authoritative results or data set at a ‘master’ site for the purposes of public download. Thus, not all contributing sites in the federation are peers; the master site enjoys an elevated status.
Stages in federating a production infrastructure
Federation is best rolled out in stages, to understand the challenges, in a point-by-point manner, from the stance of the production engineers as well as to educate its wide and diverse userbase, facilitating the usage and value proposition of the newly minted federated entity. Thus, we propose to move a centralized infrastructure to a federated one using a progressive, three-stage approach. These stages are in increasing order of complexity and equivalently, increasing degree of decentralization. The goal is to use resources (compute and storage in our case) at the participating institutions to support the end-user functionality (metagenomic uploads, queries and downloads in our case). We refer to the sites as ‘central site’ and ‘participant sites’. We summarize the three stages across various dimensions in Table 1.
Table 1.
Characterization of the three stages of federation, as would be applicable to a production bioinformatics computation and data infrastructure
| Stage | Degree of federation | Scalability | Complexity deployment | Complexity operation | Technology maturity |
|---|---|---|---|---|---|
| Stage 1: Central compute and data, local data access | Low | Medium | Low | Medium | High |
| Stage 2: Central compute and data, peer data access | Medium | Medium | Medium | Medium | High |
| Stage 3: Central compute and data, peer compute and data access | High | High | Medium | Hi | Medium |
Note: Deg. of fed.: degree of federation (i.e. the complement of the degree of centralization). scalability: Ability to scale with increasing number of users. Complexity-d: Complexity of the deployment. Complexity-o: Complexity of the operation. Technology maturity: Maturity level currently of the technologies needed to support this stage of federation.
Stage 1: Central compute and data, local data access
In this, the compute resources and the data are at the central site, with some local data staying at the participant sites. These are data owned by users at the participant site. Some stages of the computational pipeline (executed at the central site in its entirety) can access local data and store the output at the participant site. The data at the central site are thus not all inclusive. This best represents the current stage of the MG-RAST federation process with Argonne National Lab serving as the central site and Purdue University serving as the participant site.
Stage 2: Central compute and data, peer data access
In this stage, the compute resources and the data are at the central site, with some local data staying at the participant sites. The local data can be accessed by other participant sites, in a peering relationship. The central site has a directory-like structure for the data at the local structures that facilitates discovery of the data by the participant sites.
Stage 3: Central compute and data, peer data and compute access
In this stage, in addition to Stage 2, some parts of the pipeline may execute on computational resources at participant sites. With a more decentralized approach and with less sensitive data, the compute resources at participant site may process data belonging to a different participant site.
Common to all the three stages is that the central site keeps some of the authoritative data. This includes the golden data sets for benchmarking purposes (such as for execution time or the accuracy of any stage of the pipeline or of the overall pipeline), the results of the benchmarking, and reference data sets that are used for similarity computation.
Technologies specific to MG-RAST federation
In addition to the foundational technologies mentioned earlier (‘Foundational technologies to establish a federated infrastructure’ section) that can facilitate federation in any genomics pipeline, we comment on some specific aspects of MG-RAST that eases the path to federation. For further details of these MG-RAST elements, the reader is referred to [4].
Sharded object store: MG-RAST uses SHOCK, a specialized object storage for metagenomic data. Each object has metadata associated with the data, stored alongside the data and retrievable through a single query. The fine-grained object store helps in distribution, relative to a monolithic object store. Different parts of the object store can be stored in different organizations’ storage infrastructures.
Workflow engine: MG-RAST uses a workflow management system called argonne workflow engine (AWE) that has been specialized to its processing pipeline. It manages and executes workflows submitted by MG-RAST users. AWE models application-related concepts into three hierarchical elements: job, task, and work unit. A job, characterized by its input data, and workflow description (e.g. data dependencies), are parsed into task. A task represents a certain data analysis operation. A task can be split into multiple work units running the same command on different parts of the input data. AWE manages and executes these elements in a coordinated and automated fashion. With a federated infrastructure, the identical pipeline in its entirety can be executed at different organizations’ compute infrastructure on local data sets. Under a more nuanced use case, different parts of the same pipeline may be executed at different organizations.
Use of containerization solutions along with orchestration: A container solution, like Docker or LXC (Linux Containers), helps in packaging up the software, along with its dependencies, and instantiating it at different organizations’ infrastructures. The necessary orchestration for the containers—which container goes where, how to start one, how to terminate one, how to change access levels to one, how to update one, etc.—can be done by another software package such as Kubernetes. In its current instantiation, MG-RAST uses Skyport [26], which is built on top of Docker containers to solve software deployment problems and resource utilization inefficiencies inherent to all existing scientific workflow platforms. Working in concert with AWE/Shock, Skyport reduces the complexity associated with providing the environment necessary to execute complex workflows.
Use of APIs: The entire end-to-end functionality is implemented as a Web service so as to make it easy to integrate with other genomics analysis pipelines, thus enabling automation. API calls can now be made from multiple organizations. The idea of using API is important for two reasons: use of existing code and, importantly and somewhat atypically, use of precomputed data products, e.g. if someone has precomputed high fidelity gene similarity scores, and my data have a subset of those genes, I do not need to perform on my infrastructure, the expensive, and perhaps, lower-quality, gene similarity computation.
Considerations for privacy with ubiquitous data storage and federation
In the health-care field, we can envision the personalization pyramid, akin to specializations in consumer services such as in online marketing over the past decade or so. This trend started off with the conventional universal services, with individualized services next, and finally topping off with personalized services. The difference between individualized and personalized services is that while individualized services are primarily context-based, in the latter, one has to combine the context-based data with the EHRs for the patient. While the diagnosis that such specific data sets will result in may be precise, it also means that the access to and update of such EHR should be seamless, and preferably, amenable to health-care providers in the network. Imagine a person from Purdue (West Lafayette, Indiana, USA) travels to Rio (Brazil) and falls sick there. The greater the flexibility and accessibility we desire in this scenario, the greater would the risk of adversarial attack be, making the data sets widely available, of course, in an authenticated manner. Thus, although the economic and reparative potential of personalization seems appealing to general human health and welfare, the possible leakage of personal health-care data has been a continuous worry to consumers or patients, and rightly so, because breaches have occurred and with some regularity [27, 28]. Thus, with the exponential increase of personal digital health-care data, propelled by the slashing costs, and the need for better data security, without marginalizing accessibility and control, comes the need for sophisticated privacy-preserving transforms in the area. Interestingly, in the specific metagenomics case, for example, it has been found that ‘gut print’ or collective microbiomes colonizing a human body can uniquely identify individuals [29]. Thus, with the microbiome’s influence on human health and behavior coming to the fore, given its health-predictive abilities and publicly available data from projects, such as National Institutes of Health (NIH) human microbiome project, privacy concerns are inevitable yet again. Thus, while an overreaction could slow microbiome research, putting in place privacy-enabling platforms from the get-go is prudent, rather than removing information after loopholes are identified, what some call the whack-a-mole reaction. To this end, we highlight below some of the privacy guarantees to propagate our vision of adaptive and seamless privacy, which may be imperative in the genomics context, albeit, sacrificing some functionality, access or accuracy.
Configurable data desensitization
While transparent data encryption (TDE) is an important technology used to solve the problems of data security, TDE can protect data but only if the data stay within the approved science workflow. The goal here is that data can be accessed when an authorized user executes a certified program on an approved science workflow or program. However, sometimes, it is necessary to share the data outside the context of the workflow, e.g. with the scientific public, or with a broad class of recipients who want to use programs and computing platforms, but whose credentials have not been verified. In such cases, configurable data desensitization support aims at data protection. For different kinds of data, different requirements for desensitization exist. For some data relating to individuals, de-identification in the form of removing explicit identifiers and generalization of the data to satisfy k-anonymization might suffice. For other kinds, it may be desirable to process the data in a way that satisfies differential privacy.
Possible data desensitization approaches
Many data privacy techniques have been developed in the research community. They were designed to achieve different privacy objectives. While it has been recognized that syntactic privacy properties, such as k-anonymity [30] and l-diversity [31], have some fundamental limitations, specific anonymization algorithms can nonetheless provide privacy protection [32]. No single privacy notion or technique is universally applicable. For example, if one’s goal is to prevent the adversary from learning some attribute value in a record, then satisfying differential privacy may be insufficient because of correlation among the different attribute values or records. On the other hand, if one’s goal is to approximate the effect of opting out, then applying differential privacy, with appropriate parameters, achieves the effect. Below, we discuss some techniques in the research biosphere.
Anonymization approaches
Several microdata anonymization techniques have been proposed. The most popular ones are generalization [33, 30] for k-anonymity [30] and bucketization [34, 35, 36] for l-diversity. Generalization transforms the quasi-identifiers (QI) values in each bucket into ‘less specific but semantically consistent’ values, so that tuples in the same bucket cannot be distinguished by their QI values. In bucketization, one separates the sensitive attributes (SAs) from the QIs by randomly permuting the SA values in each bucket. In our prior work [37], we introduced a technique called slicing, which combines generalization and bucketing and partitions the data set both vertically and horizontally. Vertical partitioning is done by grouping attributes into columns based on the correlations among the attributes. Each column contains a subset of attributes that are highly correlated. Horizontal partitioning is done by grouping tuples into buckets. Finally, within each bucket, values in each column are randomly permuted (or sorted) to break the linking among different columns. Integration of such data anonymization techniques would work well in science workflows.
Approaches for satisfying differential privacy
Differential privacy is appropriate when one’s goal is to approximate the effect of individuals ‘opting out’. For a low-dimensional data set, a standard approach is to publish a histogram of the data set; this problem has been extensively studied [38–46].
When going beyond data sets with one or two dimensions, publishing data sets becomes challenging. In [37], we have studied data sets with binary attributes and introduced the PriView approach, which selects a number of ‘views’, then projects the data set onto these different views and publishes noisy versions of these views. From these noisy views, one can reconstruct an arbitrary marginal table of the input data. This provides a useful summary of the data and enables many kinds of statistical analysis on the data. For genomic data privacy, one would need to extend this technique to deal with nonbinary attributes, by developing techniques to deal with numerical attributes. We propose to also exploit the exponential mechanism [47] in the view selection mechanism to select marginal tables that are not well approximated by existing views.
Future research challenges to federation in bioinformatics infrastructures
We can discuss the future challenges for federation in bioinformatics infrastructures broadly under four categories.
Policy issues: This refers to challenges in ironing out policies that guide access to the resources, and specifically the data sets. The particular importance of data-sharing policy is because of the fact that in many cases, the data may be sensitive, e.g. because of having private genetic information. The policies will have to spell out the access rights to different classes of users as well as secondary uses of the data, including policies for sharing of derived data products.
Diversity of computational codes and data formats: There is a wide variety of computational codes that are meant to achieve almost equivalent functionality. These differ because of various factors, such as compatibility with some hardware (e.g. sequencing platform), dealing with different error rates, dealing with different data formats or simply because competitive tools arose in multiple different organizations. Alongside this, there is a wide variety of data formats for representing similar data. This raises the challenge that in a federated infrastructure, different software would have to coexist, exchanging data in one or more from the varied set of formats.
Predictable resource utilization: Till date, it has been a challenge to predict how much computational resource will be required to satisfy a set of user requests. This arises because the amount of processing is often dependent on the content of the query [48]. Further, there has not been much work in characterizing the resource utilization of bioinformatics software under various operational environments, such as with parallel software running on a cluster of machines, or with software running on accelerator-equipped nodes (such as a CPU equipped with a GPU card). This means that it is currently a challenge to schedule workflows on available federated resources, while guaranteeing (even probabilistically) properties like minimum job completion time or fair use of resources.
Personnel issues: There is the need for specialized in-house computer administration expertise, geared toward handling federation issues. Such administrators will have to learn the foundational technologies mentioned above that they may be unaware of. It has often been observed that the bioinformatics organizations are chronically understaffed with respect to computer administrators. The new requirements put in by federation may exacerbate the situation.
Key Points
Federation of cyberinfrastructures allows multiple organizations to contribute resources (computation, storage, data sets, etc.) to a shared pool. Users can use the cyberinfrastructure without concern for which organization has contributed which resource.
Federation in the bioinformatics area has only been used to a limited context, primarily, to store large biological data sets.
For genomics, because of the large volumes of sequencing information being made available through sequencers, it is important to federate computational resources, software and datastores, to keep the throughput of the analysis pipeline high.
We lay out the design choices in designing a federated infrastructure for genomics, using as an example, MG-RAST, the most popular metagenomics portal and analysis pipeline.
We lay out the technological building blocks that are available today, and the further research challenges needed to enable a true federated bioinformatics infrastructure.
Acknowledgements
Any opinions, findings and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the NIH.
Funding
The National Institutes of Health (NIH) (grant number 1R01AI123037, in part, 5R01AI114814) (2016–21). NSF Project: CNS-1718637, CNS-1527262.
Biographies
Somali Chaterji is a biomedical engineer and computational biologist by training. She is currently a visiting assistant professor in Computer Science at Purdue University. Here, she works at the intersection of computational genomics and machine learning.
Jinkyu Koo is a research scientist at Purdue University. His research interest is applying machine learning techniques to various application domains including gene regulation, cyber-physical system security and software engineering.
Ninghui Li is a professor of Computer Science at Purdue University. He received a PhD degree in Computer Science in 2000 from New York University. His research interests are in security and privacy.
Folker Meyer is a computational biologist at Argonne National Laboratory, where he serves as the deputy division director of the Biology Division. He is also a professor at the Department of Medicine and a senior fellow at the Computation Institute at the University of Chicago. His interest lies in building software systems to answer complex biological questions, and he is the driving force behind the MG-RAST project.
Ananth Grama is the associate director of the Center for Science of Information, a Science and Technology Center of the National Science Foundation, and a professor of Computer Science at Purdue. His research interests are in parallel and distributed computing algorithms.
Saurabh Bagchi is a professor in the School of Electrical and Computer Engineering at Purdue University. He is the founding director of a university-wide resiliency center at Purdue called CRISP. His interests are in dependable distributed system design and implementation.
References
- 1. Gao J, Aksoy BA, Dogrusoz U. Integrative analysis of complex cancer genomics and clinical profiles using the cbioportal. Sci Signal 2013;6(269):pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Joint Genomics Institute. JGI genome portal. 2017. http://genome.jgi.doe.gov/ (20 May 2017, date last accessed).
- 3. Meyer F, Paarmann D, D’Souza M. The metagenomics rast server–a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics 2008;9(1):386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wilke A, Bischof J, Gerlach W. The mg-rast metagenomics database and portal in 2015. Nucleic Acids Res 2016;44(D1):D590–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rak M, Ficco M, Luna J. Security issues in cloud federation In: Achieving Federated and Self-Manageable Cloud Infrastructures: Theory and Practice. Hershey, PA: IGI Global, 2012, 176–94. [Google Scholar]
- 6. Burke B. US Federal CIO faces a daunting challenge. 2009. http://blogs.gartner.com/brian_burke/2009/03/11/us-federal-cio-faces-a-daunting-challenge/
- 7. Stephens ZD, Lee SY, Faghri F. Big data: astronomical or genomical? PLoS Biol 2015;13(7):e1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Loh PR, Baym M, Berger B.. Compressive genomics. Nat Biotechnol 2012;30(7):627–30. [DOI] [PubMed] [Google Scholar]
- 9. Church GM, Gao Y, Kosuri S.. Next-generation digital information storage in DNA. Science 2012;337(6102):1628. [DOI] [PubMed] [Google Scholar]
- 10. Mahadik K, Chaterji S, Zhou B Orion: scaling genomic sequence matching with fine-grained parallelization. In: SC14: Proceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis. New York, NY: Institute of Electrical and Electronics Engineers (IEEE), 2014, 449–60.
- 11. Mahadik K, Wright C, Zhang J Sarvavid: a domain specific language for developing scalable computational genomics applications. In: Proceedings of the 2016 International Conference on Supercomputing. New York, NY: Association for Computing Machinery (ACM), 2016, 34.
- 12. Hung JH, Weng Z.. Data formats in bioinformatics. Cold Spring Harb Protoc 2016;2016(8):pdb.top093211. [DOI] [PubMed] [Google Scholar]
- 13. InCommon. Security, privacy and trust for the research and education community. 2017. https://www.incommon.org/ (20 May 2017, date last accessed).
- 14. Internet2. Shibboleth federated identity solutions. 2017. https://shibboleth.net/ (20 May 2017, date last accessed).
- 15. OAuth. Oauth 2.0. 2017. https://oauth.net/ (20 May 2017, date last accessed).
- 16. OpenID. Openid Foundation. 2017. http://openid.net/foundation/ (20 May 2017, date last accessed).
- 17. Nadeau TD, Gray K.. SDN: Software Defined Networks: An Authoritative Review of Network Programmability Technologies. Sebastopol, CA: O’Reilly Media, Inc., 2013. [Google Scholar]
- 18. Open Networking Foundation. Openflow. 2017. http://archive.openflow.org/ (20 May 2017, date last accessed).
- 19. Cisco. Cisco open network environment. 2017. http://www.cisco.com/c/en/us/products/collateral/switches/nexus-1000v-switch-vmware-vsphere/white\_paper\_c11-728045.html (20 May 2017, date last accessed).
- 20. Langmead B, Salzberg SL.. Fast gapped-read alignment with bowtie 2. Nat Methods 2012;9(4):357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rho M, Tang H, Ye Y.. Fraggenescan: predicting genes in short and error-prone reads. Nucleic Acids Res 2010;38(20):e191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Edgar RC. Search and clustering orders of magnitude faster than blast. Bioinformatics 2010;26(19):2460–1. [DOI] [PubMed] [Google Scholar]
- 23. Huang Y, Niu B, Gao Y. Cd-hit suite: a web server for clustering and comparing biological sequences. Bioinformatics 2010;26(5):680–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kent WJ. Blatthe blast-like alignment tool. Genome Res 2002;12(4):656–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Buchfink B, Xie C, Huson DH.. Fast and sensitive protein alignment using diamond. Nat Methods 2015;12(1):59–60. [DOI] [PubMed] [Google Scholar]
- 26. Gerlach W, Tang W, Keegan K Skyport: container-based execution environment management for multi-cloud scientific workflows. In: Proceedings of the 5th International Workshop on Data-Intensive Computing in the Clouds. New York, NY: Institute of Electrical and Electronics Engineers (IEEE), 2014, 25–32.
- 27. Perakslis ED. Cybersecurity in health care. N Eng J Med 2014;371(5):395. [DOI] [PubMed] [Google Scholar]
- 28. Munro D. Data breaches in healthcare totaled over 112 million records in 2015. New York, NY: Forbes, 2015, 31 December 2015.
- 29. Franzosa EA, Huang K, Meadow JF. Identifying personal microbiomes using metagenomic codes. Proc Natl Acad Sci USA 2015;112(22):E2930–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sweeney L. k-anonymity: a model for protecting privacy. Int J Uncertainty Fuzziness Knowl Based Syst 2002;10(05):557–70. [Google Scholar]
- 31. Machanavajjhala A, Kifer D, Gehrke J, Venkitasubramaniam M.. l-diversity: privacy beyond k-anonymity. ACM Trans Knowl Discov Data 2007;1(1):3. [Google Scholar]
- 32. Li N, Qardaji W, Su D. On sampling, anonymization, and differential privacy or, k-anonymization meets differential privacy. In: Proceedings of the 7th ACM Symposium on Information, Computer and Communications Security. New York, NY: Association for Computing Machinery (ACM), 2012, 32–3.
- 33. Samarati P. Protecting respondents identities in microdata release. IEEE Trans Knowl Data Eng 2001;13(6):1010–27. [Google Scholar]
- 34. Martin DJ, Kifer D, Machanavajjhala A Worst-case background knowledge for privacy-preserving data publishing. In: Proceedings of the IEEE 23rd International Conference on Data Engineering, 2007 (ICDE 2007). New York, NY: Institute of Electrical and Electronics Engineers (IEEE), 2007, 126–35.
- 35. Xiao X, Tao Y. Anatomy: simple and effective privacy preservation. In: Proceedings of the 32nd International Conference on Very Large Data Bases, Seoul, South Korea. VLDB Endowment, 2006, 139–50.
- 36. Zhang Q, Koudas N, Srivastava D, Yu T. Aggregate query answering on anonymized tables. In: Proceedings of the IEEE 23rd International Conference on Data Engineering, Istanbul, Turkey, 2007 (ICDE 2007). IEEE, 2007, 116–25.
- 37. Qardaji W, Yang W, Li N. Preview: practical differentially private release of marginal contingency tables. In: Proceedings of the 2014 ACM SIGMOD International Conference on Management of Data, Snowbird, Utah, USA. ACM, 2014, 1435–46.
- 38. Acs G, Castelluccia C, Chen R. Differentially private histogram publishing through lossy compression. In: Proceedings of the IEEE 12th International Conference on Data Mining (ICDM), Brussels, Belgium, 2012. IEEE, 2012, 1–10.
- 39. Cormode G, Procopiuc C, Srivastava D, Tran TTL. Differentially private summaries for sparse data. In: Proceedings of the 15th International Conference on Database Theory, Berlin, Germany. ACM, 2012, 299–311.
- 40. Hardt M, Ligett K, McSherry F. A simple and practical algorithm for differentially private data release. In: Proceedings of the Advances in Neural Information Processing Systems. 2012, 2339–47.
- 41. Hay M, Rastogi V, Miklau G, Suciu D.. Boosting the accuracy of differentially private histograms through consistency. Proceedings VLDB Endowment 2010;3(1–2):1021–32. [Google Scholar]
- 42. Li C, Hay M, Miklau G, Wang Y.. A data-and workload-aware algorithm for range queries under differential privacy. Proceedings VLDB Endowment 2014;7(5):341–52. [Google Scholar]
- 43. Qardaji W, Yang W, Li N.. Understanding hierarchical methods for differentially private histograms. Proceedings VLDB Endowment 2013;6(14):1954–65. [Google Scholar]
- 44. Xiao X, Wang G, Gehrke J.. Differential privacy via wavelet transforms. IEEE Trans Knowl Data Eng 2011;23(8):1200–14. [Google Scholar]
- 45. Xu J, Zhang Z, Xiao X. Differentially private histogram publication. VLDB J 2013;22(6):797–822. [Google Scholar]
- 46. Zhang J, Cormode G, Procopiuc CM Privbayes: private data release via Bayesian networks. In: Proceedings of the 2014 ACM SIGMOD International Conference on Management of Data. New York, NY: Association for Computing Machinery (ACM), 2014, 1423–34.
- 47. McSherry F, Talwar K. Mechanism design via differential privacy. In: Proceedings of the FOCS’07 48th Annual IEEE Symposium on Foundations of Computer Science, 2007. New York, NY: Institute of Electrical and Electronics Engineers (IEEE), 2007, 94–103.
- 48. Gardner MK, Feng WC, Archuleta J Parallel genomic sequence-searching on an ad-hoc grid: experiences, lessons learned, and implications. In: Proceedings of the International Conference for High Performance Computing, Networking, Storage, and (Supercomputing). New York, NY: Institute of Electrical and Electronics Engineers (IEEE), 2006, 1–14.