

We discovered a new CK2 inhibitor and revealed its mechanism of action, connecting the circadian clock and cancer regulation.
Abstract
Compounds targeting the circadian clock have been identified as potential treatments for clock-related diseases, including cancer. Our cell-based phenotypic screen revealed uncharacterized clock-modulating compounds. Through affinity-based target deconvolution, we identified GO289, which strongly lengthened circadian period, as a potent and selective inhibitor of CK2. Phosphoproteomics identified multiple phosphorylation sites inhibited by GO289 on clock proteins, including PER2 S693. Furthermore, GO289 exhibited cell type–dependent inhibition of cancer cell growth that correlated with cellular clock function. The x-ray crystal structure of the CK2α-GO289 complex revealed critical interactions between GO289 and CK2-specific residues and no direct interaction of GO289 with the hinge region that is highly conserved among kinases. The discovery of GO289 provides a direct link between the circadian clock and cancer regulation and reveals unique design principles underlying kinase selectivity.
INTRODUCTION
The circadian clock is an intrinsic timekeeping mechanism that controls daily rhythms of many physiological processes, including sleep/wake behavior, body temperature, hormone secretion, energy metabolism, and the cell cycle. Circadian rhythms are generated in a cell-autonomous manner, and within each cell, clock genes form transcriptional regulatory networks. The transcription factors CLOCK and BMAL1 activate expression of Period (Per1 and Per2) and Cryptochrome (Cry1 and Cry2) genes. After translation, complex formation, and nuclear localization, PER and CRY proteins inhibit CLOCK-BMAL1 function, resulting in rhythmic gene expression. In parallel, the Bmal1 gene is regulated by nuclear hormone receptors REV-ERB and ROR, whose gene expression is controlled by the CLOCK-BMAL1 complex to form an interconnected feedback loop (1–3).
In addition to transcriptional regulation, posttranslational modification of clock proteins forms an indispensable layer necessary for circadian rhythmicity. Most clock proteins undergo rhythmic phosphorylation (4), and the functional importance of clock protein phosphorylation has been proven by spontaneous gene mutations, resulting in alterations of circadian period. The tau mutant hamster with short-period behavioral rhythms has a missense mutation in the casein kinase I (CKI) ε gene (5), and human familial advanced sleep phase (FASP) with early sleep times has been attributed to missense mutations in the PER2 and CKIδ genes (6, 7). CKIε and CKIδ phosphorylate PER to trigger proteasomal degradation, and the tau and FASP mutations lead to faster degradation of PER, consistent with the short-period phenotype (8, 9). Phosphorylation of CRY1 also regulates circadian period through degradation-dependent and degradation-independent pathways (10). To fully understand and characterize the phosphorylation network controlling the circadian clock, it is necessary to identify the phosphorylation sites and determine the responsible kinases for each clock protein.
Perturbations of clock function by genetic mutations or environmental factors, such as shift work, have been implicated in sleep disorders, cancer, and cardiovascular and metabolic diseases in humans and animal models (1–3). Therefore, identification of small molecules modulating circadian clock function will provide new opportunities for treatments of clock-related disorders. Cell-based high-throughput chemical screening approaches have led to the discovery of a number of compounds that affect circadian rhythms (11, 12). Consistent with the functional importance of CKIδ/ε, multiple compounds that inhibit CKI activity have been identified to lengthen circadian period (13–17). Among these, well-known inhibitors for a number of different kinases [cyclin-dependent kinase, Cdc2-like kinase (CLK), p38 mitogen-activated protein kinase, c-Jun N-terminal kinase, casein kinase 2 (CK2), and vascular endothelial growth factor receptor] have been shown to be potent inhibitors of CKI (14), highlighting a key but complex issue of kinase inhibitor selectivity (18). The development of new CKI inhibitors with higher specificity through target-based approaches (19, 20) or unbiased approaches (15, 16) enabled the conditional manipulation of CKI and modulation of circadian period in CKIεtau mutant mice (21). We further discovered a unique chemical modulator of CRY by screening for period-changing compounds followed by affinity-based target deconvolution. This compound, KL001, inhibits proteasomal degradation of CRY, lengthens circadian period in a variety of cells and tissues, and blocks glucagon-dependent induction of gluconeogenesis in cultured hepatocytes (22). Consistent with these observations, a bioavailable derivative of KL001 exhibits antihyperglycemic activity in db/db diabetic mice (23). Moreover, small-molecule agonists of REV-ERB and ROR improve energy homeostasis (24, 25), and REV-ERB agonists impair cancer growth to improve survival in a mouse model of glioblastoma (26). The discovery of new compounds that target the circadian system offers a previously unidentified strategy to control clock-related processes, and there are still numerous clock-modulating compounds whose target proteins have not yet been identified.
We aimed to expand the chemical tools enabling dissection and control of the circadian clock system for a better understanding of the molecular mechanism. Here, we report the discovery of a new period-lengthening compound, GO289, whose target protein was identified as CK2 through an affinity-based proteomics approach. CK2 is one of the most well-studied protein kinases because of its ubiquitous expression, pleiotropy, and constitutive activity. On the basis of its major function in cell growth and apoptosis, CK2 is known as one of the key players in the pathogenesis of cancer (27, 28). Small-molecule inhibitors of CK2, such as TBB, DMAT, and CX-4945, have played important roles in understanding the functions of this kinase, but their selectivity is not optimal as either tool molecules or potential therapeutic agents (29–31). In mammals, the role of CK2 in circadian period regulation is less well characterized compared to CKI, partly because of the lack of genetic models due to lethality and the lack of selective chemical tools. The discovery of GO289 overcomes some of these issues. Through its high potency and selectivity, GO289 enabled manipulation of clock protein phosphorylation and cancer cell growth. Furthermore, x-ray crystallographic studies revealed the molecular basis underlying the potency and selectivity of GO289 through a binding modality relatively distinct from most known protein kinase inhibitors.
RESULTS
Identification of a new compound that lengthens circadian period
To identify small molecules that modulate the cell-autonomous function of the circadian clock, we conducted a cell-based chemical screen using human U2OS osteosarcoma cells harboring a Bmal1 promoter-luciferase (Bmal1-dLuc) reporter. Confluent cells were treated with an in-house collection of drug-like compounds, and circadian luminescence rhythms were measured. We found a number of compounds with different chemical scaffolds that strongly changed the period of circadian rhythms (15, 16, 22). In this study, we focused on the guaiacol derivative, GO289 (Fig. 1A), with no known biological activity. This compound caused dose-dependent lengthening of circadian period not only in Bmal1-dLuc reporter cells but also in Per2-dLuc reporter cells with a phase opposite to that of Bmal1-dLuc (Fig. 1B). GO289 also lengthened periods in cells differentiated from embryonic stem (ES) cells of Per2::LucSV knock-in mice harboring a PER2-LUC fusion protein reporter (Fig. 1C) and in lung explants from Per2::Luc mice (fig. S1A). These results indicate that GO289 reproducibly causes strong period lengthening regardless of the reporter or cell type in human and mouse.
Fig. 1. GO289 lengthens circadian period.
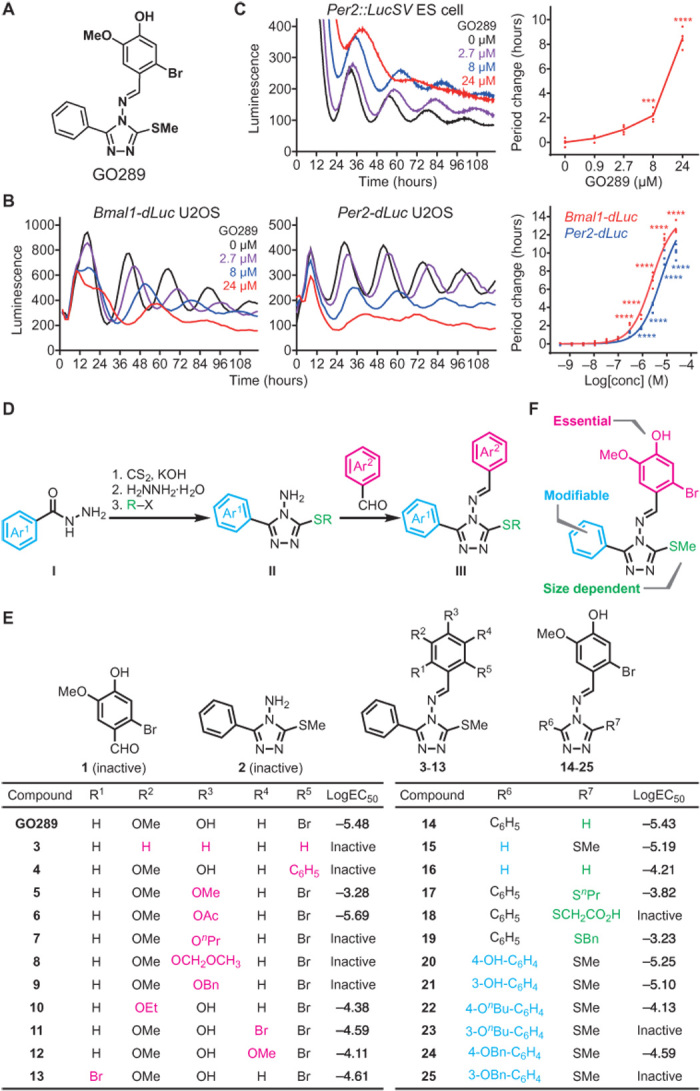
(A) Chemical structure of GO289. (B and C) Effect of GO289 on circadian rhythms in Bmal1-dLuc and Per2-dLuc U2OS cells (B) and cells differentiated from Per2::LucSV knock-in ES cells (C). Luminescence rhythms were monitored in the presence of various concentrations of GO289 and shown in the left (Bmal1-dLuc) and middle (Per2-dLuc) panels of (B) and the left panel of (C) (mean of n = 4). Period changes compared to a dimethyl sulfoxide (DMSO) control are plotted in the right panel of (B) and (C) (n = 4). ****P < 0.0001 and ***P < 0.001 against the DMSO control. (D) General synthetic scheme for GO289 derivatives. (E) Period-lengthening activity of GO289 derivatives. Luminescence rhythms of Bmal1-dLuc cells were monitored in the presence of various concentrations (threefold, 12-point dilution series) of GO289 derivatives (n ≥ 2), and the concentration required for half-maximal period lengthening is shown as logEC50. Modified part of the compound is shown in color. C4 and C3 positions of the benzene ring at R6 correspond to the para and meta positions, respectively. (F) Summary of the SAR study.
We previously demonstrated that the period-lengthening compounds longdaysin and KL001 inhibited CKI activity and CRY degradation, respectively (15, 22). In contrast, GO289 showed minor effects on CKIδ and CKIα activity in vitro (fig. S1B) and no stabilization of CRY1 in a cell-based degradation assay (fig. S1C), suggesting an alternative mechanism of action from previously known clock modulators.
Structure-activity relationship of GO289
GO289 consists of a triazole structure linked to bromoguaiacol, methyl thioether, and phenyl groups (Fig. 1A). To investigate functional sites of GO289 responsible for its effect on circadian period, we performed a structure-activity relationship (SAR) study. The general synthetic scheme for GO289 derivatives is shown in Fig. 1D. Arylhydrazides (I) were converted to potassium aroyl dithiocarbazates and then to amino triazole (II) by a ring-closure reaction with hydrazine and subsequent alkylation (32). Imine formation of II with aryl aldehyde furnished the core structure of GO289 (III). Each group was modified by substituting I, alkyl reagent, and aryl aldehyde.
We analyzed the effect of GO289 derivatives on circadian period in Bmal1-dLuc U2OS cells (Fig. 1E). Both triazole and bromoguaiacol groups were required for the activity, as either group alone did not show any effect on period (1, 2). Removal of all three substituents in the bromoguaiacol (Br, hydroxy, and methoxy) caused a complete loss of activity (3). Addition of bulkier substituents also resulted in a severe reduction in period-lengthening activity (4, 5, 7, 8, 9, 10), with the exception of acetylation of the hydroxy group that slightly increased activity (6). Addition of groups at an unsubstituted meta or ortho position also led to decreased activity (11, 12, 13), indicating that the bromoguaiacol cannot be modified. In contrast, removal of either the methyl thioether group or the phenyl group was tolerated (14, 15), although removal of both groups caused a severe reduction in activity (16). Addition of bulkier groups to the methyl thioether group strongly reduced activity (17, 18, 19). Similarly, modification of the phenyl group at the meta position decreased activity, while addition at the para position had little effect (20, 21, 22, 23, 24, 25). Together, the SAR analysis indicated that the bromoguaiacol is essential for activity, and the para position of the phenyl group is amenable for modification (Fig. 1F).
Target identification of GO289
To identify molecular targets of GO289, we used an affinity-based proteomics approach. On the basis of the SAR data (Fig. 1E), we attached a tetraethylene glycol linker to the para position of the phenyl group on GO289 (Fig. 2A). The compound GO457 retained significant period-lengthening activity (Fig. 2B) and therefore was used as an affinity probe. Proteins interacting with an agarose conjugate of GO457 were affinity-purified and analyzed by liquid chromatography–tandem mass spectrometry (LC-MS/MS). From two independent experiments, CK2α (CSNK2A1), CK2α′ (CSNK2A2), CK2β (CSNK2B), and GSK-3β (GSK3B) were highly enriched with the affinity probe. These interactions were blocked by free GO289 (Fig. 2C), indicating that they are candidates for GO289-target proteins. All of these proteins are known to be involved in regulating circadian period, as knockdown of CK2α, CK2α′, and CK2β lengthens period (33, 34) and GSK-3β knockdown causes period shortening (13) in U2OS cells. The spectral numbers for CK2α and CK2α′ under competition (−) conditions were much higher than those for CK2β and GSK-3β, suggesting that CK2α and CK2α′ are the primary targets of GO289 (Fig. 2C). CK2 is a Ser/Thr kinase, whose catalytic subunits (CK2α and CK2α′) and regulatory subunit (CK2β) exist as monomers, as well as α2β2 tetramers. By immunoblotting with specific antibodies against CK2α and CK2β, we confirmed their enrichment with the affinity probe and competition by free GO289 (Fig. 2D). Furthermore, recombinant CK2 (α2β2 tetramer) also interacted with the affinity probe, and this interaction was blocked by increasing concentrations of free GO289 (Fig. 2E), indicating a direct interaction of GO289 with CK2.
Fig. 2. GO289 interacts with CK2.
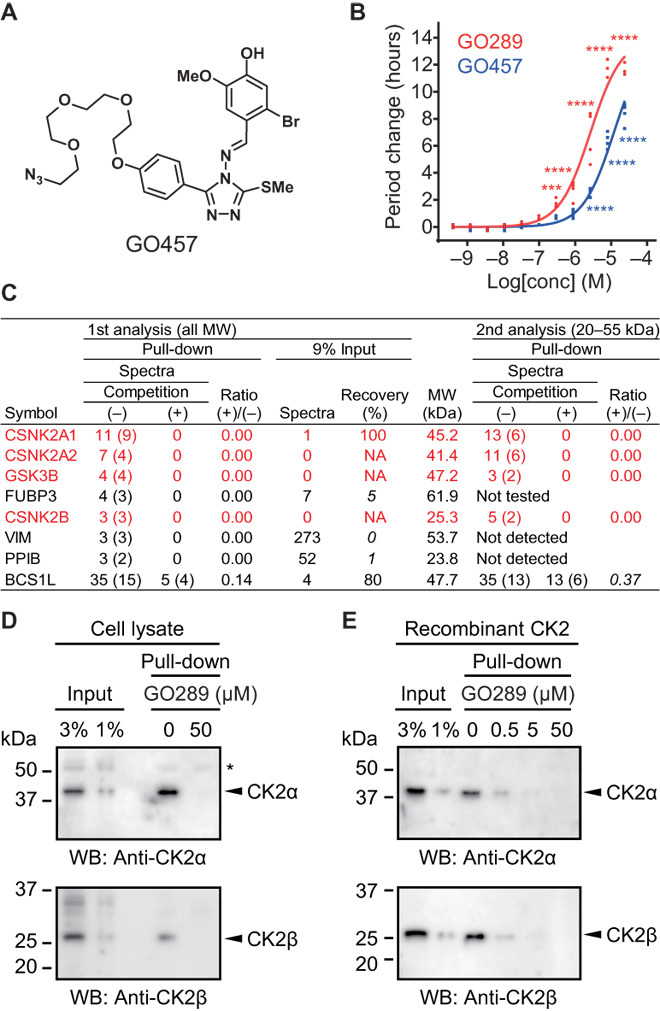
(A) Chemical structure of GO457. (B) Effect of GO457 on circadian period in Bmal1-dLuc U2OS cells. ****P < 0.0001 and ***P < 0.001 against the DMSO control (n = 4). (C) GO289-interacting proteins. Agarose-conjugated GO457 was incubated with a U2OS cell lysate in the presence of 0 and 50 μM GO289 [competition (−) and (+), respectively]. Affinity-purified proteins were analyzed by LC-MS/MS. Proteins identified by ≥3 tandem MS spectra from ≥2 unique peptides (numbers in parentheses) and showed ≥5-fold signal reduction upon competition with GO289 [ratio (+)/(−) ≤ 0.2] in the first analysis are shown (columns 2 to 4). Enrichment with the affinity probe was estimated by comparing with the MS spectra number in the input sample (columns 5 and 6). Low enrichment efficiency (recovery ≤ 10%) is indicated in italics. The result of the second analysis focusing on 20- to 55-kDa proteins is shown in columns 8 to 10. Proteins fulfilling the criteria in both analyses are indicated in red. MW, molecular weight. (D and E) Interaction of GO289 with CK2. Affinity-purified proteins from a U2OS cell lysate (D) or recombinant CK2 (E) were analyzed by immunoblotting with specific antibodies. The asterisk indicates a nonspecific protein that interacted with the probe, independent of GO289.
Potent and highly selective inhibition of CK2 by GO289
The effect of GO289 on CK2 activity was analyzed using an in vitro kinase assay. GO289 potently inhibited CK2 at a half-maximal inhibitory concentration (IC50) of 7 nM (Fig. 3A). In contrast, 5 μM GO289 showed only a moderate or minor effect on the activity of 59 kinases from a variety of classes (Fig. 3B). The second most affected kinase was PIM2, with an IC50 of 13 μM (Fig. 3C), which was >1000 times higher than that for CK2. The minor effect of GO289 on GSK-3β activity (Fig. 3B) supported the idea that GSK-3β is most likely not a direct target. Together, the protein interaction and kinase inhibition profiles indicate high potency and selectivity of GO289 toward CK2.
Fig. 3. GO289 potently and selectively inhibits CK2.

(A to D) Effect of GO289 on kinase activity in vitro. Activity of CK2 (A) and PIM2 (C) was analyzed in the presence of GO289 at various concentrations (n = 2). A panel of 60 kinases (B) and DYRK, HIPK, and PIM family kinases (D) were screened with 5 μM GO289 (n = 2). In (D), the effect of GO289 on multiple kinases is compared to published values for CK2 inhibitors TBB, DMAT, and CX-4945. ND, not determined. (E) Effects of CK2 inhibitors on circadian period and reporter signal intensity in Bmal1-dLuc U2OS cells. Changes in period (left) and luminescence intensity (right) compared to the DMSO control are plotted (n = 4). P values are summarized in table S3. (F and G) Effect of GO289 on cellular CK2 activity. HEK293T cells (F) or U2OS cells (G) were treated with GO289 at various concentrations for 24 hours and subjected to immunoblotting with anti-phosphorylated (anti-phospho) CK2 substrate antibody. The membrane was reprobed with anti-phospho PKA (protein kinase A) substrate and anti–β-actin antibodies (F) or stained with CBB (Coomassie Brilliant Blue) (G).
Well-known CK2 inhibitors, TBB and DMAT, strongly inhibit DYRK, HIPK, and PIM family kinases, in addition to CK2 (Fig. 3D) (29). CX-4945 is also a potent CK2 inhibitor with high selectivity, yet still inhibits DYRK, HIPK, and PIM family kinases (Fig. 3D) (30, 35). In contrast, GO289 showed little effect on these other kinases, even at a concentration of 5 μM, which is 700 times higher than its IC50 for CK2 (Fig. 3D). The IC50 ratios of TBB, DMAT, and CX-4945 against CK2 over PIM family kinases are 5.7 (PIM3), 0.7 (PIM3), and 46 (PIM1), respectively, while that of GO289 was >1000 (PIM2) (fig. S2A). Furthermore, CX-4945 is reported to inhibit CLK family proteins (30, 31). In our assays, the IC50 values of CX-4945 against CK2 and CLK2 were 6 and 23 nM, respectively (fig. S2, B and C), while GO289 showed no effect on CLK2 (IC50 of >50 μM). Consistent with the lower selectivity of DMAT and CX-4945, they both caused a severe reduction in luminescence in a dose-dependent manner in our cell-based circadian assay (Fig. 3E), possibly because of cytotoxicity arising from multitarget effects. In contrast, GO289 lengthened circadian period by more than 12 hours with little effect on luminescence compared to DMAT and CX-4945. These results together highlight the potency and selectivity of GO289.
We further analyzed the effect of GO289 on cellular CK2 activity by measuring phosphorylation of CK2 substrates by immunoblotting. Human embryonic kidney (HEK) 293T cells were treated with GO289, and cell lysates were analyzed using an anti-phospho CK2 substrate antibody. This antibody recognizes the CK2-consensus sequence pS/pT-D-X-E, in which pS/pT represents Ser/Thr residues phosphorylated by CK2. The antibody detected many protein substrates, and the phosphorylation signal was reduced by GO289 in a dose-dependent manner (Fig. 3F, left). In contrast, neither the CKIδ inhibitor LH846 nor the GSK-3 inhibitor CHIR-99021 altered the CK2-specific phosphorylation pattern (fig. S3). Furthermore, the signal detected by the anti-phospho PKA (protein kinase A) substrate antibody was not affected by GO289 (Fig. 3F, right), demonstrating the specificity of both GO289 and the anti-phospho CK2 substrate antibody. Inhibition of cellular CK2 activity by GO289 was also observed in U2OS cells at concentrations similar to those that affected circadian period (Fig. 3G), linking inhibition of CK2-dependent phosphorylation to period lengthening.
Regulation of clock protein phosphorylation by GO289
CK2 has been reported to regulate circadian clock oscillations across species, including Neurospora, Arabidopsis, Drosophila, and mammals. In mammals, CK2 phosphorylates PER2 and leads to nuclear accumulation (33) or degradation (36) and also phosphorylates BMAL1, which results in nuclear accumulation (37). Previous in vitro phosphorylation studies identified the CK2-dependent sites T12/S13 and S53 of PER2 and S90 of BMAL1. However, mutation of each of these sites has little effect on circadian period (33, 36, 37) compared to the prominent period-lengthening effects of GO289. We therefore conducted an extensive search for cellular CK2 phosphorylation sites in core clock proteins by inhibiting CK2 activity with GO289. HEK293T cells expressing Flag-tagged PER2, CRY1, CLOCK, and BMAL1, together with CK2α, were treated with GO289. After immunoprecipitation with an anti-Flag antibody, the clock proteins were subjected to phosphoproteomic analysis by LC-MS/MS, in which combinations of four proteases were used to obtain high-sequence coverage. In PER2, CRY1, CLOCK, and BMAL1, we identified 15, 3, 3, and 9 residues, respectively, whose phosphorylation was reduced by GO289 (Fig. 4A). Among these, S693 of PER2 exhibited the strongest signal. Because a previous study suggested that S693 or S697 of full-length PER2 was phosphorylated by both CKIδ and CK2 in vitro (33), we synthesized a peptide PER2(680-705) encompassing both of these amino acids and performed an in vitro kinase assay. This peptide was phosphorylated by CK2, but not by CKIδ (Fig. 4B). Mutation of T687, S693, and/or S697 to Ala identified S693 as the site phosphorylated by CK2 in the PER2(680-705) peptide (Fig. 4C). These observations indicate that CK2, but not CKIδ, is responsible for phosphorylation of PER2 S693 that is inhibited by GO289 in cells. In contrast, previously reported CK2 phosphorylation sites T12/S13 and S53 of PER2 were either unchanged or up-regulated by GO289 in our cell-based assay using full-length PER2 (table S1).
Fig. 4. GO289 affects phosphorylation of clock proteins.
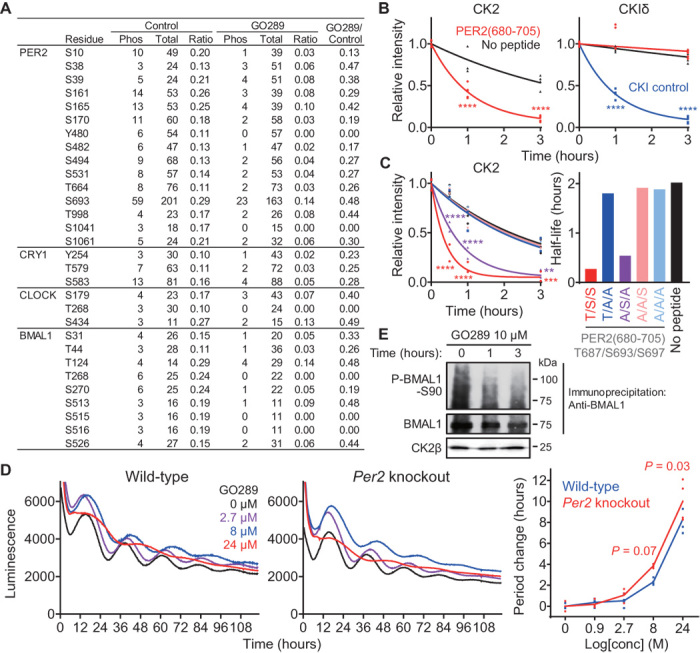
(A) Phosphorylation sites inhibited by GO289. HEK293T cells expressing Flag-tagged mouse clock proteins and CK2α were treated with GO289. Immunoprecipitated clock proteins were subjected to LC-MS/MS analysis. Phosphorylation level was calculated by dividing spectra number of phosphorylated peptides with total peptides (ratio). Effect of GO289 was defined as the ratio of the GO289-treated sample to the control sample (GO289/control). Sites abundantly phosphorylated (ratio of control sample ≥ 0.1) and inhibited by GO289 (GO289/control ≤ 0.5) are shown. (B and C) Phosphorylation of PER2(680-705) peptide by CK2. (B) Activity of CK2 and CKIδ was analyzed by measuring ATP levels (n = 3 to 6). (C) Ser/Thr residues were mutated to Ala (n = 2 to 4), and ATP half-life is plotted in the right panel. ****P < 0.0001, ***P < 0.001, **P < 0.01 against no peptide control. (D) Effect of GO289 on circadian rhythms of Bmal1-Luc reporter in wild-type and Per2 knockout cells. Period changes compared to the DMSO control are plotted in the right panel (n = 3 to 4). P values are against wild-type cells. (E) Effect of GO289 on BMAL1 S90 phosphorylation. Mouse NIH-3T3 cells were treated with 10 μM GO289 for different times and subjected to immunoprecipitation/immunoblotting with specific antibodies.
To evaluate the role of PER2 phosphorylation by CK2 in period regulation, we applied GO289 to Per2 knockout cells differentiated from ES cells. Period lengthening of Bmal1-Luc rhythms by GO289 was slightly enhanced in Per2 knockout cells compared to wild-type cells (Fig. 4D), supporting a functional interaction between CK2 and PER2. However, since the period-changing effects of GO289 were not abolished in Per2 knockout cells, CK2 substrates other than PER2 might also be involved in GO289-dependent changes in circadian period. Our phosphoproteomic analysis identified candidate phosphorylation sites in CRY1, CLOCK, and BMAL1, in addition to those in PER2 (Fig. 4A). As for the previously reported CK2-site BMAL1 S90, only one spectrum of the phosphorylated peptide was observed in both control and GO289-treated conditions, but immunoblot analysis using a specific antibody against S90-phosphorylated BMAL1 revealed its down-regulation by GO289 as represented by reduction of immunoreactivity (Fig. 4E). The band of S90-phosphorylated BMAL1 became broad possibly because of phosphorylation-dependent acetylation and SUMOylation of BMAL1 (38). Thus, it is likely that CK2 targets multiple protein substrates for its strong effect on circadian period, and GO289 provides an important tool to further investigate CK2-dependent phosphorylation of clock proteins and how this might regulate circadian period.
Regulation of cancer cell growth by GO289
In addition to regulating the circadian clock, CK2 plays important roles in cell growth and apoptosis, and elevated CK2 levels are associated with tumorigenesis (27, 28). Because perturbation of circadian clock function is linked to cancer (39), CK2 may be a key molecule connecting them. We therefore analyzed the effect of GO289 on cell growth in human renal cell carcinoma (RCC) cell lines, whose circadian properties have been characterized previously (40). Growth of Caki-2, A498, and 769-P cells was strongly inhibited by GO289, while that of 786-O, RCC4+vec, RCC4+VHL, A704, and ACHN cells was less affected (Fig. 5A). Similarly, CX-4945 inhibited growth of Caki-2 cells but had less of an effect in A704 and ACHN cells (fig. S4A), demonstrating cell type–dependent regulation of growth by CK2. We observed that Bmal1 reporter induction (40) correlated well with GO289 sensitivity (Fig. 5B), suggesting an interaction between clock function and CK2-dependent growth inhibition.
Fig. 5. GO289 inhibits cancer cell growth.
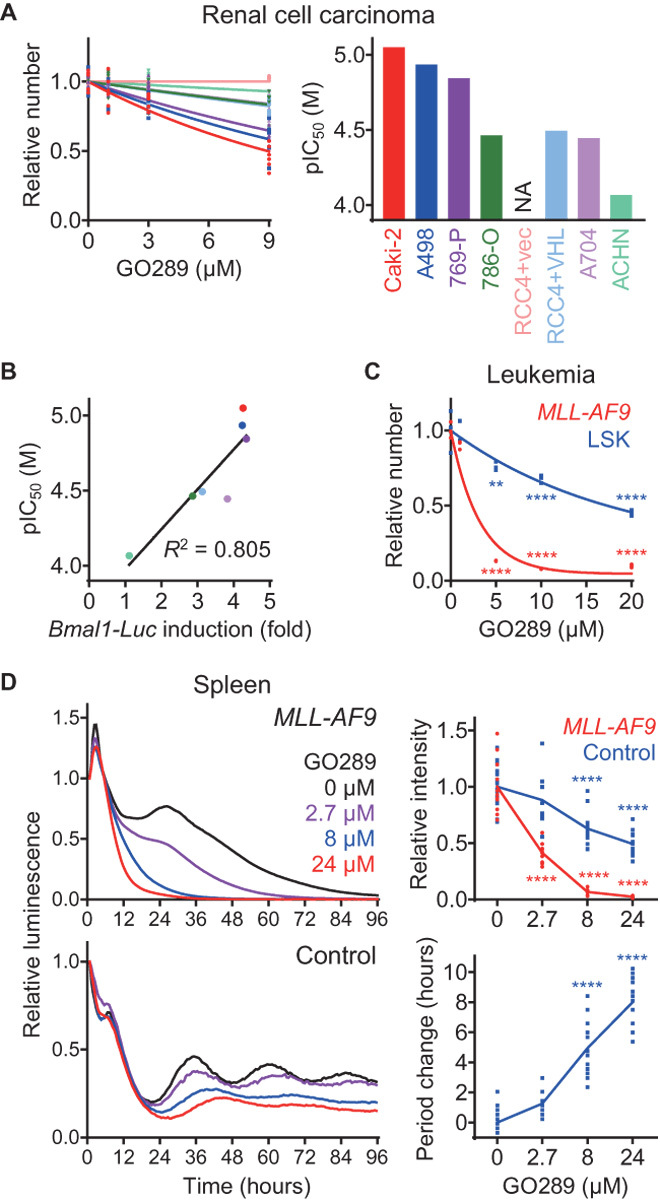
(A and C) Effect of GO289 on growth of human RCC lines (A) and mouse AML MLL-AF9 cells or normal LSK cells (C). Cell numbers are plotted in the left panel by setting the DMSO control to 1 [n = 6 in (A) and n = 3 in (C)]. For (A), pIC50 values are plotted on the right, and P values are summarized in table S3. (B) Correlation of growth inhibition by GO289 with Bmal1 reporter induction. Degree of Bmal1 reporter induction (40) was calculated by dividing intensity of the peak with time 0 and plotted against pIC50 values from (A). (D) Effect of GO289 on circadian period and reporter signal intensity in spleen explants of MLL-AF9 mice. Luminescence rhythms of the Per2::Luc knock-in reporter were monitored in the presence of GO289 and indicated in the top left (MLL-AF9 mice, mean of n = 11) and the bottom left (control mice, mean of n = 10 to 20). Changes in intensity (top right) and period (bottom right) compared to the DMSO control are plotted. ****P < 0.0001, **P < 0.01 against the DMSO control.
We further analyzed the effect of GO289 in a mouse model of acute myeloid leukemia (AML). A screen of genes whose knockdown inhibited growth of mouse MLL-AF9 leukemia cells has been reported, and CK2α (Csnk2a1) and CK2β (Csnk2b) were identified as one of the hit genes (41). Consistent with this, we found that pharmacological inhibition of CK2 with GO289 significantly reduced growth of mouse MLL-AF9 leukemia cells, while Lin−Sca1+c-Kit+ (LSK) hematopoietic progenitor cells as a normal counterpart were much less affected (Fig. 5C). Furthermore, the human AML cell line OCI-AML3 was also sensitive to GO289 (fig. S4B). We then investigated circadian rhythms in MLL-AF9 leukemia cells that were prepared from bone marrow cells of Per2::Luc reporter mice and transplanted into nonreporter mice. We found that circadian rhythms in spleen and bone marrow explants were severely disrupted in MLL-AF9 leukemia mice compared to control mice that were transplanted with wild-type bone marrow cells from Per2::Luc reporter mice (Fig. 5D and fig. S4C). GO289 caused a severe reduction in luminescence in MLL-AF9 tissues relative to the control, consistent with its anticancer effect. Furthermore, the compound lengthened circadian period in control spleen and bone marrow explants. Together, these results demonstrated that GO289 inhibits growth of cancer cells in a cell type–dependent manner and directly links the circadian clock with cancer (see Discussion).
Molecular basis of GO289 potency and selectivity against CK2
To understand the molecular basis of the potency and selectivity of GO289 for CK2, we determined the x-ray crystal structure of human CK2α in complex with GO289 at a resolution of 1.68 Å (table S2). The overall structure of the CK2α-GO289 complex showed a typical CK2α architecture with the N-terminal domain containing five β strands and an α helix (Fig. 6A). The C-terminal domain consisted of an α helix–rich domain with two long helices, multiple small helices, and a couple of two-strand β sheets that were present at the domain interface. The N-terminal domain was connected to the C-terminal domain via a hinge region, followed by a short helix. GO289 was located at the adenosine 5′-triphosphate (ATP)–binding site at the interface of the N- and C-terminal domains, indicating that the compound is an ATP-competitive inhibitor of CK2α. GO289 stacked flat into the binding site with a small rotation of the phenyl group that interacted with the imidazole of H160 in the up conformation (Fig. 6B). The oxygen atoms in the hydroxy and methoxy groups of bromoguaiacol formed hydrogen bonds with K68 (Fig. 6C). The hydroxy oxygen of the bromoguaiacol also interacted with one of the crystal waters (W1). The bromine atom of the bromoguaiacol interacted with another crystal water (W2) that formed hydrogen bonds with the backbone CO of E114 and NH of V116 at the hinge region. The methyl of the methyl thioether group made hydrophobic interactions with V66 and M163 from the top and bottom of the cavity, respectively. These extensive polar and nonpolar interactions between GO289 and CK2α support high potency of the molecule without any direct interaction with the hinge region.
Fig. 6. X-ray crystal structure of the CK2-GO289 complex.

(A) Overall structure of CK2α in complex with GO289. (B) Surface of the GO289 binding pocket. GO289 stacked flat in the binding site with a slight rotation of the phenyl ring forming a stacking interaction with H160. (C) Interactions of GO289 with CK2α. Hydrogen bond, halogen bond, and stacking interactions are shown as dotted lines in blue, yellow, and green, respectively. (D) Period-lengthening activity of GO289 derivatives. Luminescence rhythms of Bmal1-dLuc cells were monitored in the presence of various concentrations (threefold, 12-point dilution series) of GO289 derivatives (n ≥ 2), and the concentration for half-maximal period lengthening is shown as logEC50. The modified part of the compound is shown in red. (E) Structure-based sequence alignment of the binding pocket. Data for CK2α, PIM1, PIM2, CLK2, and DYRK2 are from published x-ray crystal structures, while those for HIPK1 are based on its primary sequence. Conserved residues are shown in blue. (F) Comparison of the binding mode of GO289, TBB, DMAT, and CX-4945. DMAT and CX-4945 interact directly with V116 of the hinge region.
The structure of the CK2α-GO289 complex correlates remarkably well with our SAR results (Fig. 1E). All groups of the bromoguaiacol made crucial interactions with CK2α (Fig. 6C), and their removal led to a complete loss of activity (3). Addition of bulky groups might cause steric interference, resulting in a substantial decrease in activity (4, 5, 7, 8, 9, 10, 11, 12, 13). The only exception to this was acetylation of the hydroxy group, which slightly increased cellular activity (6). By performing in vitro CK2 kinase assays, we found that derivative 6 had severely reduced activity (fig. S5A), suggesting that cleavage of the acetyl group in cells might lead to the release of active GO289. The methyl thioether group made close hydrophobic interactions with V66 and M163, and its removal (14, 16) or the introduction of larger groups (17, 18, 19) caused a reduction in activity. The phenyl group made a stacking interaction with H160, and its loss decreased activity (15, 16). This phenyl group was exposed to a solvent with minimal steric hindrance, consistent with a high tolerance for substitutions (20, 21, 22, 24) and suitability for linker attachment at the para position (GO457).
To further characterize the CK2α-GO289 interactions, we synthesized GO289 derivatives and analyzed their activity in a cell-based circadian assay (Fig. 6D). Modification of the C== N bond connecting bromoguaiacol and the triazole ring caused complete loss of activity (26, 27, 28), suggesting a crucial role for planarity of the GO289 molecule (Fig. 6B). Replacement of the phenyl ring with thiophene (29) or pyridine (30) that can participate in stacking interactions maintained the activity, consistent with its interaction with H160 (Fig. 6B). Elimination or changing the position of bromine atom at the bromoguaiacol group led to inactivity (31, 32), indicating an interaction with crystal water W2 (Fig. 6C). Introduction of polar groups at the methyl thioether position decreased activity (33, 34, 35), supporting its hydrophobic interactions with V66 and M163 (Fig. 6C). Replacement of the methyl thioether group with a phenyl group abolished activity (36), consistent with the presence of a narrow pocket in this region. The effect of these derivatives was also evaluated with in vitro CK2 assays. Their CK2 inhibitory activities (fig. S5B) were well correlated with the effects on cellular circadian rhythms (Fig. 6D). Therefore, the CK2α-GO289 interactions from the crystal structure are remarkably consistent with the activity in cells.
We then compared the binding pocket of CK2α-GO289 to that of other kinases, PIM1, PIM2, CLK2, DYRK2, and HIPK1 (Fig. 6E and fig. S6A). The CK2 residues V66 and H160 that made crucial interactions with GO289 are replaced by Ala (A65 in PIM1) and Glu (E171 in PIM1), respectively, in all other kinases. Furthermore, the methyl in the methoxy group of bromoguaiacol pointed outward and was spatially close to the inward bent Phe (F49 in PIM1) in other kinases, while this residue is replaced by Tyr (Y50) and pointed outward in CK2α (fig. S6A). The steric hindrance caused by the Phe, along with the loss of hydrophobic interactions and π stacking contributed by V66 and H160, respectively, provides a clear explanation for the highly selective nature of GO289 for CK2α compared to other kinases.
We further compared the binding mode of GO289 to that of TBB, DMAT, and CX-4945 in complex with CK2α (Fig. 6F). TBB shows polar interactions with K68 and mostly hydrophobic interactions with the rest of the binding pocket. DMAT, a derivative of TBB, makes halogen bond interactions with the hinge region but does not make any direct polar interactions with K68. GO289 had many more interactions with the ATP-binding pocket of CK2 than these compounds, explaining its higher potency. Selectivity of GO289 over TBB and DMAT can be attributed to its unique interactions with residues specific to CK2α (V66, M163, and H160) (Fig. 6F). Furthermore, TBB adopts different modes of binding depending on the kinase (42), consistent with its low selectivity. In contrast, the binding mode of GO289 was similar to that of CX-4945. In both complexes, K68 made a hydrogen bond with the methoxy (GO289) or carbonyl (CX-4945) group, the W1 crystal water occupied the same position to interact with the hydroxy group, V66 made hydrophobic interactions with the methyl thioether group (GO289) or tricyclic scaffold (CX-4945), and H160 was in the up conformation to make a stacking interaction with the phenyl ring of the two compounds (Fig. 6F). These shared interactions highlight the high potency of both compounds against CK2α. The most significant difference between GO289 and CX-4945 was the absence of a direct interaction of GO289 with the CK2 hinge region. Because this region is highly conserved among kinases and is a common binding site for kinase inhibitors, we propose that a combination of several factors, including the lack of a direct interaction with the hinge region, could be important for the selectivity of GO289 (see Discussion). These results together revealed unique features of the GO289-CK2α interaction that contribute to the high potency and selectivity of GO289.
DISCUSSION
Our cell-based phenotypic screen for circadian clock modifiers and affinity-based strategy for target identification led to the discovery of a novel CK2 inhibitor, GO289, that caused strong period lengthening. The chemical structure of GO289 was highly unusual compared to other kinase inhibitors, making its identification by target-based drug design difficult. A SAR study together with x-ray crystal structural analysis revealed tight interactions between GO289 and CK2α that result in strong inhibition. Furthermore, GO289 showed remarkable specificity for CK2, and we revealed structural features for this selectivity. These results demonstrate that an unbiased chemical biology approach enabled identification of unique compounds, even against well-studied protein kinases, such as CK2.
Small-molecule inhibitors of CK2 have been studied extensively. Among them, CX-4945 is a highly potent and selective inhibitor of CK2, but it has also been shown to inhibit DYRK, HIPK, PIM, and CLK family kinases from profiling of more than 200 kinases (30, 31, 35). In our proteome-wide affinity purification assay, GO289 interacted with only few kinases (CK2α, CK2α′, CK2β, and GSK-3β) among hundreds of kinases expressed in U2OS cells. These data, together with profiling of 60 diverse kinases, as well as DYRK, HIPK, PIM, and CLK family kinases, indicate much higher selectivity of GO289 than CX-4945. Most kinase inhibitors, including CX-4945, interact with the hinge region by forming hydrogen bonds similar to the adenine moiety of ATP. This interaction is thought to be important for potency among molecules targeting the ATP-binding pocket (43). By using CX-4945 derivatives, it was suggested that the loss of hydrophobic interactions caused by the CK2α V66A mutation can be compensated for by increasing interactions with the hinge region (35). This V66 of CK2α is replaced by Ala in DYRK, HIPK, PIM, and CLK family kinases. Because GO289 showed no direct interactions with the hinge region, the added loss of an interaction with the hydrophobic residue (V66A) may reduce binding of GO289 to DYRK, HIPK, PIM, and CLK family kinases compared to CK2α, resulting in its high selectivity. Another characteristic of GO289 contributing to its high selectivity could be its planar structure. CX-4945 has rotational freedom around the amine function, possibly causing unfavorable entropy to the free energy of binding, which is compensated for by the enthalpy contribution from polar interactions with the hinge region (35). In contrast, the presence of an imine function connecting the bromoguaiacol and triazole groups limits rotational freedom of GO289 along this bond. Thus, the planar structure of GO289 is likely to achieve better shape complementarity to the narrow binding pocket of CK2α. This possibility was supported by modification of the imine function to amine (26), methyl group addition (27), or amide group addition (28), which caused complete loss of CK2 inhibition.
In addition to the ATP-binding pocket, previous studies have identified five regions in the CK2α structure that show conformational variability (44, 45). The p-loop and β4/β5 loop of the CK2α-GO289 complex showed “up” and “bent” conformations, respectively (fig. S6B), similar to the CX-4945–bound structure (35). The β3/αC loop and the αGH2 helix showed slightly different conformations, and the hinge/αD region showed remarkably different conformations between the GO289- and CX-4945–bound forms. The hinge/αD region has been intensely explored owing to its proximity to the ATP-binding pocket and its role in CK2α function and inhibition (46). In previous CK2α crystal structures, the hinge/αD region was proposed to exist in two conformations, “open” and “closed,” depending primarily on the orientation of F121. However, recent computational and experimental analyses found that multiple conformations of the hinge region can exist in solution or in complex with different molecules (45, 47). The hinge/αD region of the CK2α-GO289 complex moved outward with a maximum displacement of 4.3 Å from that of CK2α-CX-4945 (fig. S6C). Furthermore, the Y125 side chain in CK2α-GO289 pointed outward, leaving a cavity behind the αD helix that contained three molecules of ethylene glycol from the crystallization buffer, whereas Y125 in CK2α-CX-4945 is buried in the pocket behind the αD helix. This unique cavity has been the subject of particular interest for targeting CK2α using fragments (47) and may form the basis for the design of more potent and selective derivatives of GO289 through a fragment-based approach.
The high potency and selectivity of GO289 enabled specific manipulation of cellular CK2 activity. Using GO289, we identified cellular phosphorylation sites of core clock proteins, whose modification was reduced upon CK2 inhibition. PER2 contained the largest number of GO289 target sites, including S693 and T998, which showed overlap with CK2 phosphorylation sites previously identified in in vitro studies of full-length PER2 ([S12 or T13], [S693 or S697], S971, [S980 or S981], [S997 or T998], and S1231) (33). In in vitro assays, however, S693/S697 are also phosphorylated by CKIδ, and S997/T998 are phosphorylated by CKIδ and GSK-3β (33). Our peptide-based assay unambiguously revealed that CK2, but not CKIδ, phosphorylated S693. Recent phosphoproteomic analysis of mouse liver identified rhythmic phosphorylation of S693/S697, with a peak at ZT18-21 (48). This rhythm is coincident with a CK2 activity rhythm that peaks at CT18-22, as measured by phosphorylation of CDC37 at S13 (36). These findings together indicate that CK2 is a rhythmic kinase responsible for PER2 S693 phosphorylation. In addition, GO289 reduced phosphorylation of PER2 S10 (Fig. 4A), whose mutation to Ala in combination with T12, S13, and T15 affected PER2 stability and period length (33). Because S10 is not phosphorylated by CK2 in vitro (33), it might be affected by a kinase or phosphatase downstream of CK2 in cells. Despite these results, Per2 knockout had only a marginal effect on GO289-dependent regulation of circadian period. Unlike the disruption of behavioral rhythms in Per2 knockout mice (49), Per2 knockout had a minimal effect on period in mesodermal cells differentiated from ES cells (period lengths of wild-type and Per2 knockout cells were 22.15 ± 0.27 hours and 22.00 ± 0.40 hours, respectively). It is possible that the contribution of PER2 to period is different among cell types, and multiple target proteins are involved in CK2-dependent period regulation. Drosophila CK2 phosphorylates PER and TIM, leading to nuclear accumulation (50, 51), and also phosphorylates CLOCK, resulting in protein stabilization (52). Our results in mammalian cells form the basis of future studies to further dissect the complex phosphorylation network of the circadian clock.
It has also been reported that CK2 is associated with a wide range of cancers, including RCC and AML (53). RCC is frequently accompanied by mutation of the Von Hippel–Lindau (VHL) gene encoding an E3 ubiquitin ligase for hypoxia-inducible factor (HIF) that activates transcription of genes involved in cancer progression. CK2 phosphorylates VHL that leads to cleavage of the N-terminal region, followed by proteasomal degradation of VHL. Inhibition of this cleavage stabilizes VHL and leads to HIF degradation (54), suggesting a role of the VHL-HIF pathway in CK2-dependent regulation of cancer. Unexpectedly, however, the response of the RCC cell lines to GO289 was not consistent with the level of HIF-1α, since only Caki-2, RCC4+vec, and A704 cells showed detectable expression (40). Instead, we found a strong correlation between growth inhibition by GO289 and Bmal1 reporter induction. The RCC lines had a disrupted circadian clock but still showed one circadian cycle of reporter expression (40). This may reflect partial clock function, since the Bmal1 promoter is regulated by REV-ERB and ROR, as part of the interconnected feedback loop. It is possible that REV-ERB/ROR, CLOCK-BMAL1, or other clock proteins are co-opted to drive downstream genes necessary for tumor growth, even in the absence of robust rhythmicity. Therefore, cancer cells with partial clock function might be more sensitive to GO289. Consistently, we found that MLL-AF9 leukemia cells showed profiles similar to Caki-2 cells with higher Bmal1 induction and greater GO289 sensitivity among RCC lines. Growth of MLL-AF9 leukemia cells was strongly inhibited by GO289, and one cycle of rhythmic Per2::Luc expression was observed in spleen and bone marrow, although MLL-AF9–driven AML caused severe disruption of the circadian clock. Together, these results suggest a possible connection between the circadian clock and CK2-dependent inhibition of cancer cell growth. Disruption of circadian rhythms in RCC and AML is consistent with the findings from a comprehensive systems analysis of clock genes across multiple human cancers (55). GO289 showed much less of an effect on the growth of a normal hematopoietic counterpart, hematopoietic progenitor LSK cells. Recent studies revealed an important role of CK2 in the regulation of leukemia stem cells (LSCs) in a mouse AML model (41) and in patients with AML (56). Because targeted regulation of slowly proliferating LSCs is a key challenge in AML therapies, GO289 and its derivatives will provide a useful tool to inhibit CK2 and control the circadian clock with high selectivity.
In conclusion, GO289 belongs to a new class of highly specific CK2 inhibitors that strongly regulates circadian rhythms and cancer cell growth. Its unique mode of action will likely form the basis of new design strategies for highly selective kinase inhibitors, a critical issue in drug discovery. Optimization of GO289 to increase its drug-likeness will enable selective and conditional control of CK2 function in vivo and further our understanding of the molecular connections between the circadian clock and cancer.
MATERIALS AND METHODS
Compound screen and cell-based circadian assays
An in-house collection of drug-like compounds with diverse chemical scaffolds was screened at a final concentration of 7 μM by using a high-throughput circadian assay system, as described previously (15, 16, 22). In follow-up studies, stable U2OS reporter cells harboring Bmal1-dLuc or Per2-dLuc reporters (34) were suspended in culture medium [Dulbecco’s modified Eagle medium (DMEM; Gibco, 11995-073) supplemented with 10% fetal bovine serum (FBS), l-glutamine (0.29 mg/ml), penicillin (100 U/ml), and streptomycin (100 μg/ml)] and plated onto a white, solid-bottom 384-well plate at 30 μl (3000 cells) per well. After 2 days, 40 μl of explant medium [DMEM (Gibco, 12800-017) supplemented with 2% B27 (Gibco), 10 mM Hepes, sodium bicarbonate (0.38 mg/ml), l-glutamine (0.29 mg/ml), penicillin (100 U/ml), streptomycin (100 μg/ml), and 0.2 or 1 mM luciferin (pH 7.2)] was dispensed into each well, followed by the application of 500 nl of compounds [dissolved in dimethyl sulfoxide (DMSO); final 0.7% DMSO]. The plate was covered with an optically clear film, and luminescence was recorded every 100 min for 5 days in a microplate reader [Infinite M200 PRO (Tecan) or Synergy2 (BioTek)].
Lung tissue was dissected from Per2::Luc knock-in mice (57). Spleen and bone marrow tissues were dissected from Per2::Luc MLL-AF9 leukemia mice and control transplant mice (see below). For leukemic mice, tissues were collected when mice exhibited typical symptoms due to leukemia (e.g., weakness and slow movements) at 3 to 4 weeks after transplantation with leukemic cells. The tissue pieces were cultured in explant medium containing compounds (final 0.24% DMSO) in a black, clear-bottom 24-well plate. The plate was covered with an optically clear film, and luminescence was recorded every 30 min for 5 days in a LumiCEC luminometer (Churitsu Electric Corporation).
Circadian period was determined from luminescence rhythms by a curve-fitting program, MultiCycle (Actimetrics). The luminescence intensity was calculated by averaging the intensity during the entire experiment. Data from the first day were excluded from analysis, because of transient changes in luminescence upon medium change. LogEC50 was obtained by sigmoidal dose-response fitting of dilution series data (threefold, 12 points) with Prism software (GraphPad Software).
Circadian assay in differentiated ES cells
Per2::LucSV knock-in ES cells (17) and Bmal1-Luc ES cells (58) were maintained on mouse embryonic fibroblast feeder cells in ES cell medium [Glasgow minimum essential medium supplemented with 15% FBS (HyClone), 0.1 mM minimum essential medium (MEM) nonessential amino acids, 0.1 mM 2-mercaptoethanol, leukemia inhibitory factor (1000 U/ml), and penicillin-streptomycin (100 U/ml)]. Per2 was disrupted using the CRISPR-Cas9 system with the target sequence of 5′-GGAACACACTGACGTCATGAGG-3′ where “AGG” is the PAM. Frameshift deletions in the targeted exon were confirmed by DNA sequencing (fig. S7) (59). For the in vitro differentiation of ES cells, cells were dissociated and seeded in low-attachment 96-well plates in differentiation medium [DMEM supplemented with 10% FBS, 1 mM sodium pyruvate, 0.1 mM nonessential amino acids, GlutaMAX-I (Invitrogen), 0.1 mM 2-mercaptoethanol, and penicillin-streptomycin (100 U/ml)]. Two days later, the embryoid bodies were plated onto gelatin-coated 24-well plates and cultured for 26 days in differentiation medium, which was exchanged every other day. On differentiation day 28, the cells were treated with 100 nM dexamethasone for 1 hour, and then, the medium was replaced with differentiation medium containing 0.2 mM luciferin and the indicated concentrations of the compound. Luminescence was measured and integrated for 1 min at 20-min intervals with photomultiplier tube–based equipment. Period analysis was performed by sine-wave fitting, as previously described (59).
In vitro kinase assay
Effects of compounds on CKIδ and CKIα activity in vitro were analyzed, as described previously (15). The CK2, PIM2, and CLK2 kinase assays were performed in a 384-well plate (10-μl volume). The reaction mixture contained the following: for CK2, CK2 (1.5 ng/μl; New England Biolabs, P6010), 50 μM CK2 substrate peptide (Millipore, 12-330), and CK2 buffer [40 mM tris, 10 mM MgCl2, 0.5 mM dithiothreitol (DTT), and 150 mM NaCl (pH 7.5)]; for PIM2, PIM2 (3 ng/μl; Sigma-Aldrich, K3518), 100 μM S6K substrate peptide (SignalChem; S05-58), and PIM2 buffer [25 mM tris, 10 mM MgCl2, 2 mM DTT, and 6.25 mM NaCl (pH 7.5)]; for CLK2, CLK2 (3 ng/μl; Invitrogen, PV4201), 100 μM S6K substrate peptide (SignalChem, S05-58), and CLK2 buffer [25 mM tris, 10 mM MgCl2, 0.5 mM EGTA, and 2.5 mM DTT (pH 7.5)]. Five hundred nanoliters of compound was added to the mixture (final 5% DMSO), and the reaction was started by adding ATP (final 3 μM). After incubation at 37°C for 1.5 hours (CK2) or 30°C for 3 hours (PIM2 and CLK2), 10 μl of Kinase-Glo Luminescent Kinase Assay reagent (Promega) was added, and luminescence was detected to determine the amount of the remaining ATP. None of the tested compounds inhibited luciferase activity directly. IC50 values were obtained by sigmoidal dose-response fitting with Prism software (GraphPad Software).
Profiling of 60 kinases, as well as DYRK, HIPK, and PIM family kinases, was done using KinaseProfiler (Millipore). PER2(680-705) peptide RKKKPQPELETVEDMASGPESLDGAAGGL (underlined residues were mutated to Ala where indicated) and CKI control peptide RKKKAEpSVASLTSQCSYSS (pS represents phosphorylated Ser), corresponding to human PER2(659-674), were synthesized and used for CK2 and CKIδ assays.
CRY1 degradation assay
Effects of compounds on CRY1 stability were analyzed in CRY1-LUC and LUC stable HEK293 cells, as described previously (22).
Compound synthesis
Methods for compound synthesis are described in data file S1.
Pull-down assay
Compound-interacting proteins were affinity-purified as described previously (22) with modifications. Unsynchronized, confluent U2OS cells (1.6 × 108 cells) were homogenized with a Dounce homogenizer and 5 ml of lysis buffer 1 [25 mM Mops, 15 mM EGTA, 15 mM MgCl2, 1 mM DTT, and cOmplete Protease Inhibitor Cocktail (Roche) (pH 7.2)]. After sonication, the homogenate was supplemented with NP-40 (final 0.5%) and incubated on ice for 5 min, followed by centrifugation (14,000g) at 4°C for 20 min. The resulting supernatant was split into two, and each portion was incubated with 0 or 50 μM GO289 (final 0.5% DMSO) at 4°C for 30 min with rotation. Then, 120 μl of agarose-conjugated GO457 [50% slurry in bead buffer 1 (50 mM tris, 250 mM NaCl, 5 mM EDTA, 5 mM EGTA, 0.1% NP-40, and cOmplete Protease Inhibitor Cocktail (pH 7.4)] was added to the mixture and incubated at 4°C for 1 hour with rotation. Agarose beads were washed six times with 1 ml of bead buffer 1 and boiled with SDS sample buffer to elute bound proteins. Proteins were partially separated (~2 cm) by SDS–polyacrylamide gel electrophoresis (PAGE). The gel lane for each condition was cut horizontally into six pieces (~0.2 cm3 for each piece) and subjected to protein mass spectrometric analysis. For recombinant CK2, 300 ng of CK2 (P6010, New England Biolabs) was diluted with 300 μl of lysis buffer 1 and pulled down with 25 μl of agarose-conjugated GO457.
Protein MS
In-gel digestion was performed, and samples were analyzed by nanoflow reverse-phase LC followed by MS/MS. A capillary reverse-phase high-performance LC (HPLC)–MS/MS system composed of an Eksigent ekspert nanoLC 400 HPLC system (SCIEX) was directly connected to a tandem quadrupole time-of-flight (QqTOF) TripleTOF 5600+ mass spectrometer (SCIEX) in Trap and Elute mode. Samples were automatically injected into a peptide trap column [ChromeXP; C18-CL, 200-μm inner diameter (I.D.) × 0.5 mm, 3-μm particle size, 120-Å pore size; SCIEX] attached to a cHiPLC system (SCIEX) for desalting and concentrating peptides. After washing the trap with MS-grade water containing 0.1% trifluoroacetic acid and 2% acetonitrile (solvent C), the peptides were loaded into a separation capillary reverse-phase column (ChromeXP; C18-CL, 75 μm I.D. × 150 mm, 3-μm particle size, 120-Å pore size; SCIEX) by switching the valve. The eluents used were as follows: A, 100% water containing 0.1% formic acid, and B, 100% acetonitrile containing 0.1% formic acid. The column was developed at a flow rate of 0.3 μl/min with a concentration gradient of acetonitrile: from 2% B to 32% B in 100 min, 32% B to 80% B in 1 min, sustaining 80% B for 10 min, from 80% B to 2% B in 1 min, and finally re-equilibrating with 2% B for 15 min. Mass spectra and tandem mass spectra were recorded in positive-ion and “high-sensitivity” mode with a resolution of ~35,000 full width at half maximum. The nanospray needle voltage was typically 2300 V in HPLC-MS mode. After acquisition of ~5 samples, TOF MS spectra and TOF MS/MS spectra were automatically calibrated during dynamic LC-MS and MS/MS autocalibration acquisitions injecting 50 fmol bovine serum albumin. Analyst TF1.6 system (SCIEX) was used to record peptide spectra over the mass range of mass/charge ratio (m/z) 400 to 1250 and MS/MS spectra in information-dependent data acquisition over the mass range of m/z 100 to 1600. For collision-induced dissociation (CID)–MS/MS, the mass window for precursor ion selection of the quadrupole mass analyzer was set to 0.7 ± 0.1 Da. The precursor ions were fragmented in a collision cell using nitrogen as the collision gas. Advanced information-dependent acquisition was used for MS/MS collection on the TripleTOF 5600+ to obtain MS/MS spectra for the 10 most abundant parent ions following each survey MS1 scan (250-ms acquisition time per MS1 scan and typically 100-ms acquisition time per MS/MS). Dynamic exclusion features were based on the m/z value and were set to an exclusion mass width of 50 mDa and an exclusion duration of 12 s.
Searches were performed by using the Mascot server (ver. 2.4.0, Matrix Science) against the latest Homo sapiens (human; TaxID = 9609) SwissProt database for protein identification. Searching parameters were set as follows: enzyme selected as used with three maximum missing cleavage sites, species limited to H. sapiens, a mass tolerance of 45 parts per million for peptide tolerance, 0.1 Da for MS/MS tolerance, fixed modification of carbamidomethyl (C), and variable modification of oxidation (M). The maximum expectation value for accepting individual peptide ion scores [−10*log(p)] was set to ≤0.05, where p is the probability that the observed match is a random event. Protein identification and modification information returned from Mascot were manually inspected and filtered to obtain confirmed protein identification and modification lists of CID-MS/MS.
Protein immunoblot
Protein samples were boiled in SDS sample buffer, separated by SDS-PAGE, and analyzed by immunoblotting with anti-CK2α (Santa Cruz Biotechnology, sc-6479), anti-CK2β (Calbiochem, 218712), anti-phospho CK2 substrate (Cell Signaling Technology, 8738), anti-phospho PKA substrate (Cell Signaling Technology, 9624), and anti–β-actin (Wako Pure Chemical, 017-24573) antibodies. For detection of BMAL1 S90-phosphorylation, an NIH-3T3 cell lysate was subjected to immunoprecipitation with anti-BMAL1 antibody followed by immunoblotting with anti-P-BMAL1-S90 and anti-BMAL1 antibodies (37). The protein concentration of each sample was measured by the Bradford method.
Transient transfection and immunoprecipitation
HEK293T cells (2 × 107 cells) were reverse-transfected on 15-cm dishes with 32 μg of expression vector by Lipofectamine 2000. After 24 hours, 0 or 8 μM GO289 was applied. After 24 hours, the cells were collected in ice-cold phosphate-buffered saline (PBS) and resuspended in 1.5 ml of incubation buffer [50 mM tris, 50 mM NaCl, 2 mM EDTA, 10% glycerol, 1 mM DTT, cOmplete Protease Inhibitor Cocktail, and Phosphatase Inhibitor Cocktail 2 and 3 (Sigma-Aldrich) (pH 8.0)]. The mixture was supplemented with NP-40 (final 1%) and incubated on ice for 15 min, followed by centrifugation (14,000g) at 4°C for 10 min. The supernatant was used for immunoprecipitation. The following expression vectors were used: C-terminal 3XFlag-tagged PER2, CRY1, and BMAL1 in p3XFLAG-CMV-14 (Sigma-Aldrich); C-terminal 3XFlag-tagged CLOCK in pcDNA3-intron (22); and C-terminal 3XHA-tagged CK2α in pRC-CMV (Addgene #27086).
Before purification, 30 μg of anti-Flag antibody (F1804, Sigma-Aldrich) was cross-linked to 167 μl of Dynabeads Protein G (Invitrogen, 100-09D). Cell lysates (500 μl) were subjected to immunoprecipitation for 1 hour at 4°C. The beads were washed three times with 1 ml of incubation buffer and boiled with SDS sample buffer to elute bound proteins. The proteins were separated by SDS-PAGE and stained with Coomassie Brilliant Blue. The band for each protein was cut out and subjected to phosphoproteomics.
Phosphoproteomics
In-gel digestion was performed by using chymotrypsin/AspN, AspN/typsin/Lys-C, or chymotrypsin/trypsin/LysC mix (Promega). Obtained peptides were analyzed by nanoflow reverse-phase LC followed by MS, using an LTQ XL ion trap mass spectrometer (Thermo Fisher). A capillary reverse-phase HPLC-MS/MS system is composed of an Agilent 1100 series gradient pump equipped with Valco C2 valves with 150-μm ports and an LTQ XL ion trap mass spectrometer equipped with an XYZ nanoelectrospray ionization source (AMR). Samples were automatically injected using the PAL system (CTC analytics) directly using a peptide L-trap column (Trap and Elute mode, Chemical Evaluation Research Institute) attached to an injector valve for desalting and concentrating peptides. After washing the trap with solvent C, the peptides were loaded into a separation capillary reverse-phase column (MAGIC C18 packed with gel particles of 3 μm in diameter and 200-Å pore size, 150 mm by 0.2 mm; Michrom BioResources) by switching the valve. The eluents used were as follows: A, 100% water containing 0.1% formic acid, and B, 100% acetonitrile containing 0.1% formic acid. The column was developed at a flow rate of 1 μl/min with a concentration gradient of acetonitrile: from 5% B to 40% B in 20 min, then from 40% B to 95% B in 1 min, sustaining 95% B for 3 min, from 95% B to 5% B in 1 min, and finally re-equilibrating with 5% B for 20 min. Xcalibur 2.2 system (Thermo) was used to record peptide spectra over the mass range of m/z 350 to 1500 and MS/MS spectra in information-dependent data acquisition over the mass range of m/z 150 to 2000. Repeatedly, MS spectra were recorded followed by three data-dependent CID-MS/MS spectra generated from three highest intensity precursor ions. Multiple charged peptides were chosen for MS/MS experiments because of their excellent fragmentation characteristics.
Searches were performed by using the Mascot server (ver. 2.4.0, Matrix Science) against mouse PER2, CRY1, CLOCK, or BMAL1 peptide sequence for phosphorylation site identification. Search parameters were set as follows: enzyme selected as used with three maximum missing cleavage sites, a mass tolerance of 2 Da for peptide tolerance, 0.8 Da for MS/MS tolerance, fixed modification of carbamidomethyl (C), and variable modification of oxidation (M) and phosphorylation (S, T, Y). The maximum expectation value for accepting individual peptide ion scores [−10*log(p)] was set to ≤0.05. Phosphorylation site information returned from Mascot was manually inspected and filtered to obtain confirmed phosphorylation lists of CID-MS/MS.
Proliferation assays of RCC lines
Human RCC cell lines (Caki-2, 786-O, A498, A704, 769-P, ACHN, RCC4+vec, and RCC4+VHL) were maintained in RPMI-1640 medium (Sigma-Aldrich) supplemented with 10% FBS, penicillin (24 U/ml), and streptomycin (25 μg/ml). Caki-2 (2.0 × 104 cells per well), 786-O (1.0 × 104 cells per well), A498 (1.0 × 104 cells per well), A704 (2.0 × 104 cells per well), 769-P (1.0 × 104 cells per well), ACHN (1.0 × 104 cells per well), RCC4+vec (0.7 × 104 cells per well), and RCC4+VHL (0.8 × 104 cells per well) cells were plated into 96-well plates 1 day before GO289 treatments. Two days after treatment, cells were stained with Hoechst 33342 (2 μg/ml; Invitrogen) for 1 hour and counted with ArrayScan XTI (Thermo Scientific).
Generation of Per2::Luc MLL-AF9 mice
Bone marrow cells were isolated from femurs and tibias of Per2::Luc knock-in mice by flushing with PBS. Freshly isolated LSK CD48−CD150+ cells were sorted with a fluorescence-activated cell sorter FACSAria (BD Biosciences) and precultured for 1 day in SF-O3 medium (EIDIA) containing stem cell factor (SCF; 100 ng/ml) (PeproTech) and thrombopoietin (TPO; 100 ng/ml) (PeproTech). The cells were then infected with MLL-AF9–expressing retroviral particles using the Magnetofection technology (OZ Biosciences). After 2 days of infection, green fluorescent protein (GFP)–positive cells were sorted into single cells using FACSAria and cultured for at least six passages to leukemic granulocyte/megakaryocytes progenitor clones. The leukemia cells were subjected to retro-orbital transplantation (0.5 × 106 to 1.0 × 106 cells per mouse) into sublethally irradiated (6 Gy) C57BL/6 J mice to develop AML. Serial transplantations of the leukemia cells caused AML in nonirradiated recipients, which were used for experiments. As controls for the circadian assay, bone marrow cells from Per2::Luc knock-in mice were transplanted into C57BL/6 J mice and used 3 months after transplantation.
Proliferation assays of MLL-AF9 leukemia cells and OCI-AML3 cells
GFP-positive leukemia cells were isolated from the bone marrow of MLL-AF9 leukemia mice. The cells were maintained in Iscove’s modified Dulbecco’s medium supplemented with 10% FBS, SCF (20 ng/ml), mIL-3 (10 ng/ml; TONBO), mIL-6 (10 ng/ml; TONBO), and granulocyte-macrophage colony-stimulating factor (10 ng/ml; TONBO). The cells were plated into 96-well plates (2.0 × 104 cells per well) in the presence of GO289, incubated at 37°C for 2 days, and counted with FACSAria.
LSK hematopoietic progenitor cells as a normal counterpart were maintained in StemSpan medium (STEMCELL Technologies) supplemented with SCF (100 ng/ml) and TPO (100 ng/ml). The cells were plated into 96-well plates (1.0 × 104 cells per well) in the presence of GO289, incubated at 37°C for 2 days, and counted with FACSAria.
The human myeloid leukemia cell line, OCI-AML3, was maintained in MEM alpha medium supplemented with 20% FBS. The cells were plated into 96-well plates (1.0 × 104 cells per well) in the presence of GO289, incubated at 37°C for 4 days, and analyzed with a Cell Counting Kit-8 (Dojindo).
Structure determination
Recombinant human CK2α was prepared according to a reported protocol (60). The C-terminal truncated form of CK2α was cloned into the pGEX-6P-1 expression vector (GE Healthcare) and expressed in Escherichia coli strain HMS174 (DE3) (Merck) as an N-terminal glutathione S-transferase (GST)–fusion protein. GST-CK2α protein was purified by glutathione Sepharose 4B resin (GE Healthcare) and digested with PreScission Protease (80 U/ml; GE Healthcare). The GST-free CK2α protein was further purified by anion-exchange chromatography with a Mono Q column (GE Healthcare) using an AKTA purification system (GE Healthcare). The CK2α-GO289 complex was cocrystallized using 20 to 25% ethylene glycol as a precipitant, similar to our previous studies (60). Crystallographic data were collected by the MX-325HE CCD detector (Rayonix) at the beamline BL44XU of SPring-8. The dataset was processed with the program HKL2000. The initial phase was determined by the molecular replacement method using the highest-resolution CK2α structure (3WAR in the Protein Data Bank) (60) as a starting model with program MolRep. Structural modification and refinement were performed by Coot and Refmac5. Data collection and refinement statistics are shown in table S2. Maestro (Release 2017-1, Schrödinger) was used for molecular visualization.
Statistical analysis
Statistical significance was evaluated using one-way or two-way analysis of variance (ANOVA), followed by a Tukey’s multiple comparisons test using Prism software (GraphPad Software).
Supplementary Material
Acknowledgments
We thank Y. Nagai, K. Goto, D. Yokogawa, and T. Suzuki for technical assistance; J. W. Lee and P. G. Schultz for help during the initial stage of the project; J. S. Takahashi for Per2::Luc knock-in mice; and M. Hatori and K. Tamai for critical reading of the manuscript. Funding: This work was supported in part by PRESTO Grant JPMJPR14LA from JST (to T.Hi.); Grant-in-Aid for Research Activity start-up 26891011, Young Scientists (A) 15H05590, and Scientific Research (B) 18H02402 from JSPS (to T.Hi.); the Naito Foundation (to T.Hi.); the Inamori Foundation (to T.Hi.); the Takeda Science Foundation (to T.Hi.); Grant-in-Aid for JSPS Fellows 16J04435 (to T.O.) and 15J05509 (to Y.N.) from JSPS; Grant-in-Aid for Scientific Research (B) 18H02841 from JSPS (to Y.K.); and Grant-in-Aid for Scientific Research 26119006 and 15K21711 from JSPS (to F.T.). Preliminary experiments and diffraction data collection were carried out at the beamline BL17A of the Photon Factory (proposal no. 2016G665) and at the Osaka University beamline BL44XU of SPring-8 (proposal no. 2015A6518). Ethics statement: All animal studies were approved by the Animal Experiment Committee of Nagoya University and Kyushu University and performed in accordance with these guidelines. Author contributions: T.Hi. and K.I. designed the research. T.O., Y.N., K.K., T.Hy., Y.T., M.K., T.T., A.Su., N.O., N.Z., S.O., A.N., T.K., and T.Hi. performed the experiments. T.O., A.Sr., T.Hy., Y.T., M.T., T.T., A.Su., Y.A., F.A., S.H., J.Y., F.T., Y.K., K.Y., M.I., T.K., and T.Hi. analyzed the data. T.Hi., K.I., and S.A.K. supervised the research. T.Hi., A.Sr., and T.O. wrote the paper. All authors discussed the results and commented on the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: The final coordinates of the CK2α-GO289 complex were deposited into the Protein Data Bank with accession number 6A1C. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/1/eaau9060/DC1
Fig. S1. Effect of GO289 on circadian period, CKI activity, and CRY stability.
Fig. S2. Effect of GO289 on kinase activity.
Fig. S3. Effect of CKI inhibitor LH846 and GSK-3 inhibitor CHIR-99021 on the immunoreactivity of an anti-phospho CK2 substrate antibody.
Fig. S4. Effect of CK2 inhibition on the growth and circadian rhythms of cancer cells.
Fig. S5. Effect of GO289 derivatives on CK2 activity.
Fig. S6. Structural features of the CK2α-GO289 complex.
Fig. S7. Mouse Per2 gene knockout with CRISPR-Cas9.
Table S1. Effect of GO289 on the phosphorylation of PER2 residues previously reported to be phosphorylated by CK2.
Table S2. Data collection and refinement statistics.
Table S3. Statistical analysis of Figs. 3E and 5A.
Data file S1. Compound synthesis.
Data file S2. Compound charts.
REFERENCES AND NOTES
- 1.Bass J., Lazar M. A., Circadian time signatures of fitness and disease. Science 354, 994–999 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Panda S., Circadian physiology of metabolism. Science 354, 1008–1015 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Takahashi J. S., Transcriptional architecture of the mammalian circadian clock. Nat. Rev. Genet. 18, 164–179 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee C., Etchegaray J.-P., Cagampang F. R. A., Loudon A. S. I., Reppert S. M., Posttranslational mechanisms regulate the mammalian circadian clock. Cell 107, 855–867 (2001). [DOI] [PubMed] [Google Scholar]
- 5.Lowrey P. L., Shimomura K., Antoch M. P., Yamazaki S., Zemenides P. D., Ralph M. R., Menaker M., Takahashi J. S., Positional syntenic cloning and functional characterization of the mammalian circadian mutation tau. Science 288, 483–492 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Toh K. L., Jones C. R., He Y., Eide E. J., Hinz W. A., Virshup D. M., Ptáček L. J., Fu Y.-H., An hPer2 phosphorylation site mutation in familial advanced sleep phase syndrome. Science 291, 1040–1043 (2001). [DOI] [PubMed] [Google Scholar]
- 7.Xu Y., Padiath Q. S., Shapiro R. E., Jones C. R., Wu S. C., Saigoh N., Saigoh K., Ptáček L. J., Fu Y.-H., Functional consequences of a CKIδ mutation causing familial advanced sleep phase syndrome. Nature 434, 640–644 (2005). [DOI] [PubMed] [Google Scholar]
- 8.Gallego M., Eide E. J., Woolf M. F., Virshup D. M., Forger D. B., An opposite role for tau in circadian rhythms revealed by mathematical modeling. Proc. Natl. Acad. Sci. U.S.A. 103, 10618–10623 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vanselow K., Vanselow J. T., Westermark P. O., Reischl S., Maier B., Korte T., Herrmann A., Herzel H., Schlosser A., Kramer A., Differential effects of PER2 phosphorylation: Molecular basis for the human familial advanced sleep phase syndrome (FASPS). Genes Dev. 20, 2660–2672 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ode K. L., Ukai H., Susaki E. A., Narumi R., Matsumoto K., Hara J., Koide N., Abe T., Kanemaki M. T., Kiyonari H., Ueda H. R., Knockout-rescue embryonic stem cell-derived mouse reveals circadian-period control by quality and quantity of CRY1. Mol. Cell 65, 176–190 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Hirota T., Kay S. A., Identification of small-molecule modulators of the circadian clock. Methods Enzymol. 551, 267–282 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Chen Z., Yoo S.-H., Takahashi J. S., Development and therapeutic potential of small-molecule modulators of circadian systems. Annu. Rev. Pharmacol. Toxicol. 58, 231–252 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hirota T., Lewis W. G., Liu A. C., Lee J. W., Schultz P. G., Kay S. A., A chemical biology approach reveals period shortening of the mammalian circadian clock by specific inhibition of GSK-3β. Proc. Natl. Acad. Sci. U.S.A. 105, 20746–20751 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Isojima Y., Nakajima M., Ukai H., Fujishima H., Yamada R. G., Masumoto K.-h., Kiuchi R., Ishida M., Ukai-Tadenuma M., Minami Y., Kito R., Nakao K., Kishimoto W., Yoo S.-H., Shimomura K., Takao T., Takano A., Kojima T., Nagai K., Sakaki Y., Takahashi J. S., Ueda H. R., CKIε/δ-dependent phosphorylation is a temperature-insensitive, period-determining process in the mammalian circadian clock. Proc. Natl. Acad. Sci. U.S.A. 106, 15744–15749 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hirota T., Lee J. W., Lewis W. G., Zhang E. E., Breton G., Liu X., Garcia M., Peters E. C., Etchegaray J.-P., Traver D., Schultz P. G., Kay S. A., High-throughput chemical screen identifies a novel potent modulator of cellular circadian rhythms and reveals CKIα as a clock regulatory kinase. PLOS Biol. 8, e1000559 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee J. W., Hirota T., Peters E. C., Garcia M., Gonzalez R., Cho C. Y., Wu X., Schultz P. G., Kay S. A., A small molecule modulates circadian rhythms through phosphorylation of the period protein. Angew. Chem. Int. Ed. Engl. 50, 10608–10611 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen Z., Yoo S.-H., Park Y.-S., Kim K.-H., Wei S., Buhr E., Ye Z.-Y., Pan H.-L., Takahashi J. S., Identification of diverse modulators of central and peripheral circadian clocks by high-throughput chemical screening. Proc. Natl. Acad. Sci. U.S.A. 109, 101–106 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fabian M. A., Biggs W. H. III, Treiber D. K., Atteridge C. E., Azimioara M. D., Benedetti M. G., Carter T. A., Ciceri P., Edeen P. T., Floyd M., Ford J. M., Galvin M., Gerlach J. L., Grotzfeld R. M., Herrgard S., Insko D. E., Insko M. A., Lai A. G., Lélias J.-M., Mehta S. A., Milanov Z. V., Velasco A. M., Wodicka L. M., Patel H. K., Zarrinkar P. P., Lockhart D. J., A small molecule–kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol. 23, 329–336 (2005). [DOI] [PubMed] [Google Scholar]
- 19.Badura L., Swanson T., Adamowicz W., Adams J., Cianfrogna J., Fisher K., Holland J., Kleiman R., Nelson F., Reynolds L., St. Germain K., Schaeffer E., Tate B., Sprouse J., An inhibitor of casein kinase Iϵ induces phase delays in circadian rhythms under free-running and entrained conditions. J. Pharmacol. Exp. Ther. 322, 730–738 (2007). [DOI] [PubMed] [Google Scholar]
- 20.Walton K. M., Fisher K., Rubitski D., Marconi M., Meng Q.-J., Sládek M., Adams J., Bass M., Chandrasekaran R., Butler T., Griffor M., Rajamohan F., Serpa M., Chen Y., Claffey M., Hastings M., Loudon A., Maywood E., Ohren J., Doran A., Wager T. T., Selective inhibition of casein kinase 1ϵ minimally alters circadian clock period. J. Pharmacol. Exp. Ther. 330, 430–439 (2009). [DOI] [PubMed] [Google Scholar]
- 21.Meng Q.-J., Maywood E. S., Bechtold D. A., Lu W.-Q., Li J., Gibbs J. E., Dupré S. M., Chesham J. E., Rajamohan F., Knafels J., Sneed B., Zawadzke L. E., Ohren J. F., Walton K. M., Wager T. T., Hastings M. H., Loudon A. S. I., Entrainment of disrupted circadian behavior through inhibition of casein kinase 1 (CK1) enzymes. Proc. Natl. Acad. Sci. U.S.A. 107, 15240–15245 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hirota T., Lee J. W., St. John P. C., Sawa M., Iwaisako K., Noguchi T., Pongsawakul P. Y., Sonntag T., Welsh D. K., Brenner D. A., Doyle F. J. III, Schultz P. G., Kay S. A., Identification of small molecule activators of cryptochrome. Science 337, 1094–1097 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Humphries P. S., Bersot R., Kincaid J., Mabery E., McCluskie K., Park T., Renner T., Riegler E., Steinfeld T., Turtle E. D., Wei Z.-L., Willis E., Carbazole-containing amides and ureas: Discovery of cryptochrome modulators as antihyperglycemic agents. Bioorg. Med. Chem. Lett. 28, 293–297 (2017). [DOI] [PubMed] [Google Scholar]
- 24.Solt L. A., Wang Y., Banerjee S., Hughes T., Kojetin D. J., Lundasen T., Shin Y., Liu J., Cameron M. D., Noel R., Yoo S.-H., Takahashi J. S., Butler A. A., Kamenecka T. M., Burris T. P., Regulation of circadian behaviour and metabolism by synthetic REV-ERB agonists. Nature 485, 62–68 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.He B., Nohara K., Park N., Park Y.-S., Guillory B., Zhao Z., Garcia J. M., Koike N., Lee C. C., Takahashi J. S., Yoo S.-H., Chen Z., The small molecule nobiletin targets the molecular oscillator to enhance circadian rhythms and protect against metabolic syndrome. Cell Metab. 23, 610–621 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sulli G., Rommel A., Wang X., Kolar M. J., Puca F., Saghatelian A., Plikus M. V., Verma I. M., Panda S., Pharmacological activation of REV-ERBs is lethal in cancer and oncogene-induced senescence. Nature 553, 351–355 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duncan J. S., Litchfield D. W., Too much of a good thing: The role of protein kinase CK2 in tumorigenesis and prospects for therapeutic inhibition of CK2. Biochim. Biophys. Acta 1784, 33–47 (2008). [DOI] [PubMed] [Google Scholar]
- 28.Ruzzene M., Pinna L. A., Addiction to protein kinase CK2: A common denominator of diverse cancer cells? Biochim. Biophys. Acta 1804, 499–504 (2010). [DOI] [PubMed] [Google Scholar]
- 29.Pagano M. A., Bain J., Kazimierczuk Z., Sarno S., Ruzzene M., Di Maira G., Elliott M., Orzeszko A., Cozza G., Meggio F., Pinna L. A., The selectivity of inhibitors of protein kinase CK2: An update. Biochem. J. 415, 353–365 (2008). [DOI] [PubMed] [Google Scholar]
- 30.Siddiqui-Jain A., Drygin D., Streiner N., Chua P., Pierre F., O'Brien S. E., Bliesath J., Omori M., Huser N., Ho C., Proffitt C., Schwaebe M. K., Ryckman D. M., Rice W. G., Anderes K., CX-4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits prosurvival and angiogenic signaling and exhibits antitumor efficacy. Cancer Res. 70, 10288–10298 (2010). [DOI] [PubMed] [Google Scholar]
- 31.Kim H., Choi K., Kang H., Lee S.-Y., Chi S.-W., Lee M.-S., Song J., Im D., Choi Y., Cho S., Identification of a novel function of CX-4945 as a splicing regulator. PLOS ONE 9, e94978 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao P.-L., Duan A.-N., Zou M., Yang H.-K., You W.-W., Wu S.-G., Synthesis and cytotoxicity of 3,4-disubstituted-5-(3,4,5-trimethoxyphenyl)-4H-1,2,4-triazoles and novel 5,6-dihydro-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazole derivatives bearing 3,4,5-trimethoxyphenyl moiety. Bioorg. Med. Chem. Lett. 22, 4471–4474 (2012). [DOI] [PubMed] [Google Scholar]
- 33.Maier B., Wendt S., Vanselow J. T., Wallach T., Reischl S., Oehmke S., Schlosser A., Kramer A., A large-scale functional RNAi screen reveals a role for CK2 in the mammalian circadian clock. Genes Dev. 23, 708–718 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang E. E., Liu A. C., Hirota T., Miraglia L. J., Welch G., Pongsawakul P. Y., Liu X., Atwood A., Huss J. W. III, Janes J., Su A. I., Hogenesch J. B., Kay S. A., A genome-wide RNAi screen for modifiers of the circadian clock in human cells. Cell 139, 199–210 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Battistutta R., Cozza G., Pierre F., Papinutto E., Lolli G., Sarno S., O’Brien S. E., Siddiqui-Jain A., Haddach M., Anderes K., Ryckman D. M., Meggio F., Pinna L. A., Unprecedented selectivity and structural determinants of a new class of protein kinase CK2 inhibitors in clinical trials for the treatment of cancer. Biochemistry 50, 8478–8488 (2011). [DOI] [PubMed] [Google Scholar]
- 36.Tsuchiya Y., Akashi M., Matsuda M., Goto K., Miyata Y., Node K., Nishida E., Involvement of the protein kinase CK2 in the regulation of mammalian circadian rhythms. Sci. Signal. 2, ra26 (2009). [DOI] [PubMed] [Google Scholar]
- 37.Tamaru T., Hirayama J., Isojima Y., Nagai K., Norioka S., Takamatsu K., Sassone-Corsi P., CK2α phosphorylates BMAL1 to regulate the mammalian clock. Nat. Struct. Mol. Biol. 16, 446–448 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tamaru T., Takamatsu K., Circadian modification network of a core clock driver BMAL1 to harmonize physiology from brain to peripheral tissues. Neurochem. Int. 119, 11–16 (2018). [DOI] [PubMed] [Google Scholar]
- 39.Lamia K. A., Ticking time bombs: Connections between circadian clocks and cancer. F1000Res 6, 1910 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Okabe T., Kumagai M., Nakajima Y., Shirotake S., Kodaira K., Oyama M., Ueno M., Ikeda M., The impact of HIF1α on the Per2 circadian rhythm in renal cancer cell lines. PLOS ONE 9, e109693 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Puram R. V., Kowalczyk M. S., de Boer C. G., Schneider R. K., Miller P. G., McConkey M., Tothova Z., Tejero H., Heckl D., Järås M., Chen M. C., Li H., Tamayo A., Cowley G. S., Rozenblatt-Rosen O., Al-Shahrour F., Regev A., Ebert B. L., Core circadian clock genes regulate leukemia stem cells in AML. Cell 165, 303–316 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.De Moliner E., Brown N. R., Johnson L. N., Alternative binding modes of an inhibitor to two different kinases. Eur. J. Biochem. 270, 3174–3181 (2003). [DOI] [PubMed] [Google Scholar]
- 43.Xing L., Klug-Mcleod J., Rai B., Lunney E. A., Kinase hinge binding scaffolds and their hydrogen bond patterns. Bioorg. Med. Chem. 23, 6520–6527 (2015). [DOI] [PubMed] [Google Scholar]
- 44.Niefind K., Raaf J., Issinger O.-G., Protein kinase CK2 in health and disease: Protein kinase CK2: From structures to insights. Cell. Mol. Life Sci. 66, 1800–1816 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Srivastava A., Hirota T., Irle S., Tama F., Conformational dynamics of human protein kinase CK2α and its effect on function and inhibition. Proteins 86, 344–353 (2018). [DOI] [PubMed] [Google Scholar]
- 46.Klopffleisch K., Issinger O.-G., Niefind K., Low-density crystal packing of human protein kinase CK2 catalytic subunit in complex with resorufin or other ligands: A tool to study the unique hinge-region plasticity of the enzyme without packing bias. Acta Crystallogr. D Biol. Crystallogr. 68, 883–892 (2012). [DOI] [PubMed] [Google Scholar]
- 47.Brear P., De Fusco C., Hadje Georgiou K., Francis-Newton N. J., Stubbs C. J., Sore H. F., Venkitaraman A. R., Abell C., Spring D. R., Hyvönen M., Specific inhibition of CK2α from an anchor outside the active site. Chem. Sci. 7, 6839–6845 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Robles M. S., Humphrey S. J., Mann M., Phosphorylation is a central mechanism for circadian control of metabolism and physiology. Cell Metab. 25, 118–127 (2017). [DOI] [PubMed] [Google Scholar]
- 49.Zheng B., Larkin D. W., Albrecht U., Sun Z. S., Sage M., Eichele G., Lee C. C., Bradley A., The mPer2 gene encodes a functional component of the mammalian circadian clock. Nature 400, 169–173 (1999). [DOI] [PubMed] [Google Scholar]
- 50.Lin J.-M., Kilman V. L., Keegan K., Paddock B., Emery-le M., Rosbash M., Allada R., A role for casein kinase 2α in the Drosophila circadian clock. Nature 420, 816–820 (2002). [DOI] [PubMed] [Google Scholar]
- 51.Top D., Harms E., Syed S., Adams E. L., Saez L., GSK-3 and CK2 kinases converge on timeless to regulate the master clock. Cell Rep. 16, 357–367 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Szabó Á., Papin C., Zorn D., Ponien P., Weber F., Raabe T., Rouyer F., The CK2 kinase stabilizes CLOCK and represses its activity in the Drosophila circadian oscillator. PLOS Biol. 11, e1001645 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chua M. M. J., Ortega C. E., Sheikh A., Lee M., Abdul-Rassoul H., Hartshorn K. L., Dominguez I., CK2 in cancer: Cellular and biochemical mechanisms and potential therapeutic target. Pharmaceuticals (Basel) 10, E18 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.German P., Bai S., Liu X.-D., Sun M., Zhou L., Kalra S., Zhang X., Minelli R., Scott K. L., Mills G. B., Jonasch E., Ding Z., Phosphorylation-dependent cleavage regulates von Hippel Lindau proteostasis and function. Oncogene 35, 4973–4980 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ye Y., Ozguc F. M., Kim Y., Liu C.-J., Park P. K., Hu Q., Diao L., Lou Y., Lin C., Guo A.-Y., Zhou B., Wang L., Chen Z., Takahashi J. S., Mills G. B., Yoo S.-H., Han L., The genomic landscape and pharmacogenomic interactions of clock genes in cancer chronotherapy. Cell Syst. 6, 314–328.e2 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Quotti Tubi L., Canovas Nunes S., Brancalion A., Doriguzzi Breatta E., Manni S., Mandato E., Zaffino F., Macaccaro P., Carrino M., Gianesin K., Trentin L., Binotto G., Zambello R., Semenzato G., Gurrieri C., Piazza F., Protein kinase CK2 regulates AKT, NF-κB and STAT3 activation, stem cell viability and proliferation in acute myeloid leukemia. Leukemia 31, 292–300 (2017). [DOI] [PubMed] [Google Scholar]
- 57.Yoo S.-H., Yamazaki S., Lowrey P. L., Shimomura K., Ko C. H., Buhr E. D., Siepka S. M., Hong H.-K., Oh W. J., Yoo O. J., Menaker M., Takahashi J. S., PERIOD2::LUCIFERASE real-time reporting of circadian dynamics reveals persistent circadian oscillations in mouse peripheral tissues. Proc. Natl. Acad. Sci. U.S.A. 101, 5339–5346 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yagita K., Horie K., Koinuma S., Nakamura W., Yamanaka I., Urasaki A., Shigeyoshi Y., Kawakami K., Shimada S., Takeda J., Uchiyama Y., Development of the circadian oscillator during differentiation of mouse embryonic stem cells in vitro. Proc. Natl. Acad. Sci. U.S.A. 107, 3846–3851 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tsuchiya Y., Umemura Y., Minami Y., Koike N., Hosokawa T., Hara M., Ito H., Inokawa H., Yagita K., Effect of multiple clock gene ablations on the circadian period length and temperature compensation in mammalian cells. J. Biol. Rhythms 31, 48–56 (2016). [DOI] [PubMed] [Google Scholar]
- 60.Kinoshita T., Nakaniwa T., Sekiguchi Y., Sogabe Y., Sakurai A., Nakamura S., Nakanishi I., Crystal structure of human CK2α at 1.06 Å resolution. J. Synchrotron Radiat. 20, 974–979 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/1/eaau9060/DC1
Fig. S1. Effect of GO289 on circadian period, CKI activity, and CRY stability.
Fig. S2. Effect of GO289 on kinase activity.
Fig. S3. Effect of CKI inhibitor LH846 and GSK-3 inhibitor CHIR-99021 on the immunoreactivity of an anti-phospho CK2 substrate antibody.
Fig. S4. Effect of CK2 inhibition on the growth and circadian rhythms of cancer cells.
Fig. S5. Effect of GO289 derivatives on CK2 activity.
Fig. S6. Structural features of the CK2α-GO289 complex.
Fig. S7. Mouse Per2 gene knockout with CRISPR-Cas9.
Table S1. Effect of GO289 on the phosphorylation of PER2 residues previously reported to be phosphorylated by CK2.
Table S2. Data collection and refinement statistics.
Table S3. Statistical analysis of Figs. 3E and 5A.
Data file S1. Compound synthesis.
Data file S2. Compound charts.