

A single microscale system integrates multiplexed subset purification and gene expression profiling.
Abstract
Specialized immune cell subsets are involved in autoimmune disease, cancer immunity, and infectious disease through a diverse range of functions mediated by overlapping pathways and signals. However, subset-specific responses may not be detectable in analyses of whole blood samples, and no efficient approach for profiling cell subsets at high throughput from small samples is available. We present a low-input microfluidic system for sorting immune cells into subsets and profiling their gene expression. We validate the system’s technical performance against standard subset isolation and library construction protocols and demonstrate the importance of subset-specific profiling through in vitro stimulation experiments. We show the ability of this integrated platform to identify subset-specific disease signatures by profiling four immune cell subsets in blood from patients with systemic lupus erythematosus (SLE) and matched control subjects. The platform has the potential to make multiplexed subset-specific analysis routine in many research laboratories and clinical settings.
INTRODUCTION
Millions of immune cells can be obtained from a small blood draw, yet most methods for immune profiling from clinical samples fail to resolve the biological information contained within these cells. Recently, profiling the immune state of individuals using gene expression analysis of total peripheral blood mononuclear cells (PBMCs) has become instrumental in defining immune signatures and disease states in humans. These studies provide insight into the mechanisms of complex immune responses that occur in infection (1, 2) and autoimmunity (3–5), which are difficult to recapitulate in murine models (6–8). Furthermore, expression signatures can be used to stratify individuals into different disease subtypes (9–13) or to predict individualized clinical prognoses (14–16). However, profiling total PBMCs has a severely limited potential to resolve immune status since PBMCs constitute a complex mixture of specific cell types or cell “subsets” with distinct functions. Furthermore, no effective methods exist to resolve the underlying signatures of immune subsets from a total PBMC dataset. Only recently, gene expression profiles from purified subset samples have been shown to be better discriminants of immune status than total PBMC profiles due to the diversity of leukocyte responses (17–20). In addition, new immune subsets and cellular states, some of which are indicative of impaired immune function, have been discovered through gene expression profiling of PBMCs at the single-cell level (21–24). These observations have stirred interest in probing the gene expression and monitoring the activity of these subsets in particular. As a whole, this developing body of work suggests that molecular profiling of PBMC subsets is poised to become an important tool in basic studies of immune disease as well as a clinical tool useful for predicting and monitoring patient outcomes.
Despite its potential as a tool for immunomonitoring, available methods for subset-specific expression profiling are ill-suited for large studies and clinical translation. First, technologies for cell subset enrichment such as fluorescence-activated cell sorting (FACS) are capital intensive and require substantial attention from highly trained staff. As a result, FACS is challenging to scale for large clinical studies with many samples (e.g., multiple cell subsets across many patients at different time points). In addition, FACS requires a minimum sample input to establish gates for each target subset, which can frustrate its application to low-quantity samples and projects targeting many subsets from each sample. Another popular approach, magnetic affinity cell sorting (MACS), is most often practiced manually or on proprietary instrumentation with a capacity to run batches of 6 to 10 enrichments. Although the potential to automate MACS at higher throughput using pipetting robots exists, this also requires large samples for good results (typically 1 million cells per subset), would be capital intensive, requires extensive custom programming, and is only effective for central processing centers with a large steady supply of samples. Second, the throughput of complex RNA sequencing (RNA-seq) library construction protocols is generally limited by reagent cost and labor. Implementing library construction at high throughput on custom liquid handling systems has been widely demonstrated but suffers the same drawbacks described above in reference to a hypothetical automated MACS approach. Last, single-cell analysis obviates the need for subset purification since each cell is profiled individually. However, many thousands of cells would need to be analyzed from each sample, which is prohibitively expensive today for large studies and particularly inefficient where rare subsets are of interest or immune cells make up a small fraction of the total sample (25).
Because of these limitations, most clinical gene expression studies are currently limited to whole-blood or total PBMC profiling (9–11, 14, 15), which fail to resolve expression signatures from most cell subsets due to confounding signals from more abundant cell populations. To efficiently identify and monitor important disease signatures in lower-abundance subsets, we developed a multiplexed microfluidic system that integrates both human PBMC subset enrichment and sequence library construction procedures for subset-specific genome-wide expression measurements by RNA-seq. Our approach has the unique advantage of integrating the subset enrichment and complex multistep RNA-seq library construction protocols to provide an end-to-end solution in a single device that is not provided by any other approach. By integrating the key functionality into a single microfluidic device, we eliminate the need to acquire, program, and monitor complex liquid-handling robots or integrate a constellation of single-purpose proprietary instruments. Here, we show how this integrated workflow can be multiplexed to handle multiple cell subsets and sequence libraries in parallel. Because no comparable integrated instrumentation exists, we benchmark the technical performance of our system against the gold standard approaches for each step that are commonly implemented manually by expert immunology research laboratories.
The microfluidic system carries out multiplexed enrichment of target cell subsets based on affinity for cell surface markers by MACS and high-sensitivity sequence library construction for full-length RNA-seq using Smart-seq chemistry. From an input of 50,000 cells, the device can purify multiple PBMC subsets with high purity and produce highly quantitative gene expression data covering about 10,000 genes in each subset. In testing immune stimulation and challenge in vitro, we highlight the importance of subset-specific profiling by showing differential responses across four selected subsets. Last, we applied the microfluidic device to profile PBMCs of patients with systemic lupus erythematosus (SLE) and identified clear differences in the transcriptomic states of healthy individuals and patients with SLE in multiple immune cell subsets. By integrating multiplexed enrichment and library construction workflows in a single device, our platform has the potential to enable scalable PBMC sample preparation for large clinical studies and allow for both high-resolution and high-throughput profiling of the immune system. We foresee the routine application of this system as a tool to monitor immune responses in clinical studies and help diagnose patients with complex and/or critical immune conditions.
RESULTS
Microfluidic device design
We designed a custom two-layer microfluidic device capable of semiautomated cell isolation, cell disruption, and sequence library construction protocols. This system integrates microfluidic liquid handling with magnetic affinity purification and capability for onboard polymerase chain reaction (PCR). The device is fabricated using established methods for two-layer soft lithography (26), has “large” microliter-scale internal volumes to handle mammalian cell samples, and contains 39 micromechanical valve sets controlled by an external pneumatic valve controller (27). Each operating unit on the device consists of three main chambers, each having different capacities (1, 2, and 4 μl), that are partitioned by microvalves (Fig. 1A). The largest chamber is rectangular in shape and is used for cell isolation (Supplementary Note), while the two smaller “rotary reactor” chambers are used for library construction (28). These reactors are fitted with internal microvalves that are used to formulate sample and reagent combinations and to mix these by peristalsis around the circular channel (Fig. 1B). Bead resuspension is achieved by peristalsis in the rotary reactors and by a moving magnetic field in the large rectangular chamber. A 675-μm-thick silicon wafer was used as the substrate for these microfluidic devices to allow rapid heat transfer during temperature changes called for in the protocol, particularly for PCR (due to its thinness and high thermal conductivity). The substrate thinness also enables small external permanent magnets to closely approach magnetic beads in the device chambers and subject these to strong magnetic forces. The magnets are used to move beads between chambers and hold beads in place during buffer exchange steps. With such device functionality, we are able to automate many steps in the complex protocol for cell sorting, cell disruption, and RNA-seq library construction in a simple microarchitecture (fig. S1). The three-chamber operating unit is modular and constitutes a scalable microarchitecture for devices with variable sample multiplexing capacity. The data presented here were produced using 10-channel devices, although we have fabricated devices with 6 to 30 channels. The devices can also be reused following a simple washing procedure (particular devices were used up to four times in this study).
Fig. 1. Microfluidic chip design and integrated workflows.

(A) Photograph of a 10-channel chip filled with yellow, blue, and red dye to highlight compartments; control valves are filled with green dye. Scale bar, 2 cm. The inset shows the diagram of one channel, where compartments and control lines are colored similar to the photograph. Red and black arrows indicate sample input/output and reagent input ports, respectively. Blue arrows indicate control valves that control flow through the channels. (B) Schematic showing key device capabilities that enable various sample preparation steps. Black arrows indicate mixing valves that alternately open and close to generate flow within a compartment, allowing for reagent mixing and bead resuspension without external fluid input. Permanent magnets are used for moving magnetic beads across different compartments or preventing their flow during buffer exchange and washes. (C) Full and partial (cell sorting and RNA-seq) sample preparation workflows implemented in the microfluidic chip.
Microfluidic cell sorting and low-input RNA-seq
To validate the performance of our microfluidic device, we independently benchmarked the subset enrichment and RNA-seq workflows (Fig. 1C) against standard protocols for cell sorting and RNA-seq library construction. We tested our workflows using adult PBMC samples from healthy subjects obtained from a commercial supplier (Research Blood Components) at an input level of 50,000 cells per enrichment. We implemented MACS on the microfluidic device and configured a high-resolution eight-color flow cytometry analysis to read out the purity and yield of the resulting enriched cell subset samples (fig. S2). We optimized the conditions for microfluidic cell subset isolation by testing different reagents, incubation times, and washing procedures and compared the results of the optimized microfluidic protocol to conventional benchtop MACS (fig. S3 and Methods).
We tested positive selection of target cells, negative depletion of nontarget cells (where we recover cells that do not bind to the beads), and sequential isolation using both modalities in tandem. The total time required for isolation is about 1 hour. The CD4 and CD8 subsets were isolated using the tandem procedure. First, total T cells were isolated by depleting cells expressing markers for lineages other than T cells. The total T cell population was then positively selected for either CD4 or CD8 to isolate helper and cytotoxic T cell subsets separately. The previous negative selection reduced contamination from nontarget lineages that express CD4 or CD8. B cells and monocytes, on the other hand, were effectively isolated using single positive selection for CD19 and CD14, respectively. The device consistently achieved good purity (80 ± 8%) and excellent yield (76 ± 21%) for multiple targets and modes of isolation (Fig. 2, A and B), leading to 2- to 13-fold enrichment of the selected cell types. While the device workflow does not result in perfect enrichment of the target cell type, we found that the purity of the subsets isolated using the optimized microfluidic protocol was similar to those obtained by benchtop MACS, suggesting that our workflow is on par with current implementations used to process clinical immune cell samples (fig. S3A). In addition, our device achieved better yield with lower inputs than benchtop MACS (fig. S3B). These results show that microfluidic cell sorting with magnetic beads is a viable alternative to conventional sorting approaches and demonstrates the feasibility of subset-specific enrichment with limited quantity samples.
Fig. 2. Chip performance and validation.
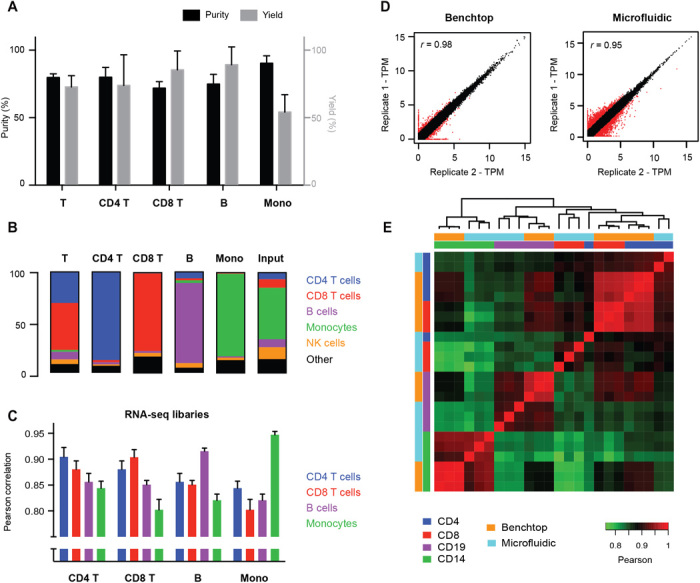
(A and B) Representative purity (black bars), yield (gray bars), and composition of immune cell types after microfluidic sorting. Yield is determined relative to fraction of the target subset in the input sample. (C) Pearson correlations between RNA-seq libraries of the four cell subsets processed through the microfluidic chip. (D) Scatterplot showing technical replicability of standard and microfluidic RNA-seq. Red points indicate genes with greater than twofold change between replicates. (E) Correlation matrix between standard and microfluidic RNA-seq libraries for four FACS-sorted cell lysates with single positive markers (CD4, CD8, CD14, and CD19). Error bars indicate SD (n = 3).
On the basis of the results of our cell isolation testing, we expected to capture thousands of cells in each subset using our microfluidic device. With these relatively low numbers in mind, we implemented a sensitive RNA-seq protocol (Smart-seq2) (29) in the chip with minor modifications. Instead of solid-phase reversible immobilization (SPRI)–based cleanup for RNA extraction, we used custom-prepared poly-dT capture beads (Methods) that captured mRNA molecules in lysate by direct hybridization to enable purification and subsequent solid-phase reverse transcription. Our protocol calls for amplifying the resulting complementary DNA (cDNA) molecules by PCR, purifying the products with SPRI, and subsequently recovering the samples from the device for transposase-based fragmentation and adaptation with subsequent enrichment PCR closely following the standard Smart-seq2 method. The timing for this procedure closely follows the Smart-seq2 protocol (about 2 hours hands-on time plus 5 hours for incubations). The cDNA amplicons from the microfluidic device showed the expected size distribution, and the RNA-seq datasets resulting from these samples show high technical reproducibility (Pearson correlation of 0.88 ± 0.04) and correlate well with libraries produced using the standard Smart-seq2 protocol on the benchtop across four different cell subsets (0.90 ± 0.03) (Fig. 2D, fig. S4, and Table 1). Despite the overall similarity between the gene expression profiles of the four subsets (Fig. 2C), the sequence libraries produced in our workflow can distinguish the subsets based on simple correlation and clustering procedures (Fig. 2E). In addition, the enrichment of polyadenylated RNA in the microfluidic protocol reduced the number of ribosomal RNA reads and improved transcript mapping rates over the standard Smart-seq2 protocol (Table 1 and fig. S5). Combining RNA-seq with cell isolation in an integrated workflow yields libraries of similar quality as standard processing approaches (Table 1 and figs. S6 and S7). These results demonstrate that full-length cDNA synthesis and amplification by PCR can be implemented in a microfluidic device with input from on-device–enriched cell subsets to support RNA-seq and that reduction in the reaction volume and reagent consumption (from 25 to 2 μl) does not negatively affect the quality of libraries obtained.
Table 1. RNA-seq library statistics for the samples generated in this study.
Two technical replicates are performed for each sample and isolated subset; n represents the total number of RNA-seq libraries generated for each column. Values are shown as means ± SD. rRNA, ribosomal RNA.
| Benchtop | Microfluidic | |||||
| Lysates (n = 12) | Lysates (n = 12) | PBMCs (n = 10) |
Cultured PBMCs (n = 24) |
SLE PBMCs (clinical study) (n = 32) |
Healthy PBMCs (clinical study) (n = 34) |
|
| Estimated library size (millions) |
16.1 ± 1.7 | 12.4 ± 5.5 | 7.7 ± 5.47 | 13.0 ± 8.7 | 6.3 ± 4.6 | 8.9 ± 3.6 |
| Genome map rate (%) | 91.6 ± 8.7 | 88.9 ± 1.7 | 86.8 ± 2.5 | 77.0 ± 16.3 | 79.8 ± 6.3 | 82.5 ± 5.7 |
| Transcript map rate (%) | 48.0 ± 7.0 | 63.0 ± 10.0 | 45.0 ± 15.0 | 65.0 ± 13.0 | 61.9 ± 10.5 | 63.1 ± 8.7 |
| Gene count | 13,592 ± 512 | 10,850 ± 481 | 10,336 ± 848 | 8899 ± 1379 | 9091 ± 1956 | 9817 ± 1757 |
| rRNA (%) | 8.67 ± 1.76 | 0.22 ± 0.06 | 2.48 ± 1.17 | 0.56 ± 0.37 | 1.29 ± 1.07 | 1.34 ± 1.08 |
Gene expression signatures of PBMCs stimulated in vitro
We assessed how accurately and reproducibly our system can profile the dynamic immune responses of different cell subsets and to what extent the responses were stereotypic or subset specific. We cultured healthy PBMCs and applied three distinct treatments known to affect immune cells [lipopolysaccharide (LPS), interferon-α (IFN-α), and dexamethasone (DEX)] in duplicate. Using the microfluidic device to carry out multiplexed subset enrichments and RNA-seq library construction, we profiled the treatment response of three different subsets (CD4+ T cells, B cells, and CD14+ monocytes). The device-processed libraries again showed strong reproducibility, coclustering the duplicates based on differential gene expression responses (Fig. 3A and fig. S8) and accurately recording differences in responses to the three treatments (Fig. 3B and fig. S9).
Fig. 3. Genomic characterization of PBMCs treated in vitro.
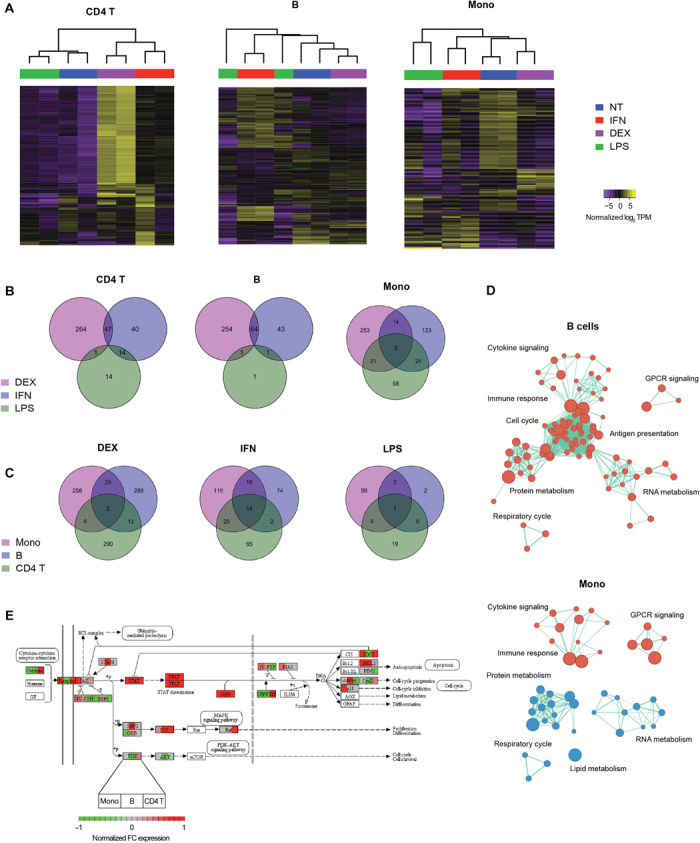
(A) Unsupervised clustering of untreated (NT) and treated (DEX, IFN, and LPS) PBMC subsets based on differentially expressed genes (false discovery rate < 0.05). Venn diagrams showing numbers of (B) treatment-specific differentially expressed genes for each subset and (C) subset-specific differentially expressed genes for each treatment. (D) Gene set enrichment analysis (Reactome sets, FDR < 0.01) of IFN-treated B cells and monocytes. Red nodes indicate up-regulation, while blue nodes indicate down-regulation. Node sizes are proportional to the number of genes in the gene set, while edge lengths are inversely proportional to the number of overlapping genes between the sets. (E) Normalized fold change (FC) in expression of Jak-STAT pathway genes in IFN-treated samples over untreated controls. Each node shows the fold-change expression of each gene in the three subsets profiled (left block, monocytes; middle block, B cells; right block, CD4 T cells).
Furthermore, our results highlight the heterogeneity in response between different cell subsets (Fig. 3, C and D), as evidenced by the minimal overlap in differentially expressed genes and enriched pathways among the three subsets profiled. Even in the canonical Jak-STAT (signal transducer and activator of transcription) signaling pathway, which is known to be directly activated by IFN-α, the pattern of downstream responses varied substantially across the three subtypes studied here (Fig. 3E). These results are consistent with previous reports that type I IFNs can have either proliferative or suppressive effects on lymphocytes, depending on the relative timing of receptor coactivation (30). In addition, these responses are greatly affected by cell-to-cell communication and the interplay between innate and adaptive immunity (31–33). After 24 hours of stimulation, IFN-α induces a strong proliferative response in B cells as shown by the up-regulation of cell cycle and metabolism pathways, while for monocytes, this effect is not observed (Fig. 3D). This finding is in line with previous studies (34–40) showing that IFN-α directly stimulates B cells to induce a strong antiviral response and a commitment to an effector phenotype, while in monocytes, the effect of IFN-α is costimulatory and primes monocytes to differentiate into dendritic cells after further stimulation. Together, these results emphasize the importance of subset-specific profiling to resolve differences that would be obscured in total PBMC expression profiling.
Transcriptomic profiling of SLE patients
To demonstrate the utility of the device for disease studies, we profiled the immune state of five patients with SLE and five healthy control individuals by isolating CD4+ T cells, CD8+ T cells, B cells, and CD14+ monocytes (plus a negative control) in duplicate from cryopreserved PBMC samples (for a total of 100 subset isolations and RNA-seq libraries including controls; table S1). For each sample, 0.5 million cells were split into eight channels on devices to isolate four subsets in duplicate and prepare RNA-seq libraries (Supplementary Methods). While we detected only a few differentially expressed genes when performing single-gene analyses (fig. S10), we found that gene sets with targets of IFN are up-regulated in patients with SLE compared to matched healthy control subjects based on gene set enrichment analyses (41), in line with our expectation that gene set enrichment analysis would have higher statistical power than analysis of single gene expression (Fig. 4A). This finding is in agreement with other studies that show the role of type I IFNs in the pathogenesis of SLE (42, 43) and validates the robustness of our method in identifying disease-relevant signatures. We found that IFN targets are enriched across all four subsets, which is expected based on previous profiling studies of cells from SLE patients (20, 44–46). Gene targets of the fusion protein NUP98-HOXA9, a potent driver of myeloid leukemia, were also enriched in all subsets. This supports previously published evidence that dysregulated lymphocyte proliferation is associated with both cancer and autoimmune disease and could explain the increased risk of malignancy in patients with SLE (47).
Fig. 4. Transcriptional immune profiling of SLE patients.
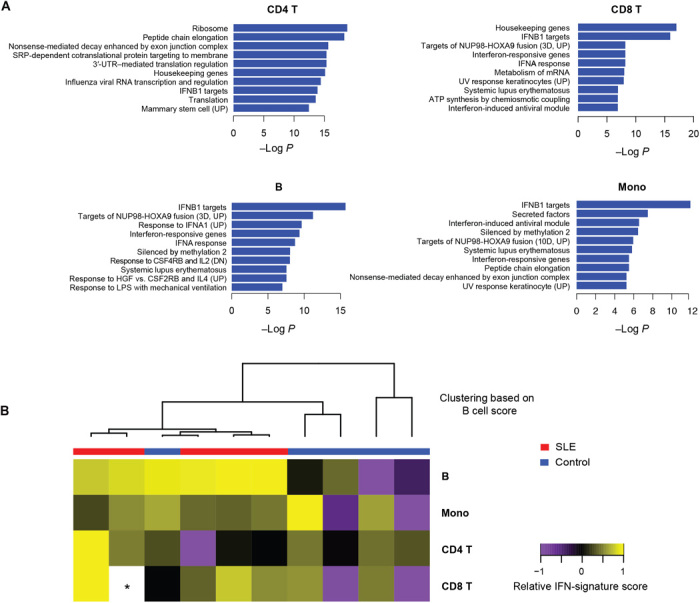
(A) Enriched gene sets (MSigDB C2) in SLE samples compared to healthy controls. P values are adjusted for multiple gene set testing (Benjamini-Hochberg). (B) Heat map showing relative IFN-signature scores across different cell types of 10 patients. Scores (transcripts-per-million sum for 37 genes; Supplementary Methods) are mean-centered across each subset. The dendrogram shows clustering of patients based on IFN-signature scores for B cells. The asterisk indicates missing data due to technical dropout.
Last, to further compare our results with previous studies, we generated an IFN gene score based on a panel of SLE signature genes established from previous studies that were not cell subset–resolved (Supplementary Methods and fig. S11) (48). Our data show that while this signature can be found across all the subsets we profiled, the difference between healthy and SLE signature scores is much more pronounced in B cells (P = 0.05 for B cells, P ≥ 0.2 for other subsets; with Bonferroni correction for testing multiple subsets) (Fig. 4B). This suggests that the diagnostic sensitivity and predictive power of the IFN signature for SLE may be improved by specifically profiling B cells instead of total PBMCs. Together, these initial findings show that gene expression responses in SLE differ across immune cell subsets and highlight the importance of subset-specific profiling in identifying disease signatures.
DISCUSSION
Through our multiplexed microfluidic workflow, we demonstrate the utility of subset-specific profiling of immune cells and its advantages over conventional total PBMC or total blood transcriptomics. Subset-specific analysis allows ready detection of biological signals from minority subsets by reducing confounding effects from abundant cell populations such as the monocytes that dominated our test samples. Our method is complementary to the application of single-cell transcriptomics approaches. For example, single-cell studies could reveal pathogenic subsets that can be enriched using the microfluidic device for large-scale research studies or clinical diagnostics, even for rare subsets. With this framework, single-cell RNA-seq (scRNA-seq) can be initially applied to a small cohort at a single time point to identify clinically relevant subsets, after which, the integrated subset-specific microfluidic workflow can be used to scale up to a larger cohort with multiple time points, increasing the study’s resolution and statistical power while lowering its cost. Another example would be the application of cell subset enrichment to target cells of interest ahead of scRNA-seq. This type of workflow could markedly improve the efficiency of scRNA-seq studies that target rare cell subsets by reducing the number of nontarget cells that need to be processed and sequenced to gain access to information about rare cells of interest.
Here, we describe the design and operation of a single microfluidic device that integrates both cell subset isolation and transcriptomic profiling, benchmark its performance, and demonstrate its applicability to clinical samples. The device can reliably isolate cell subsets of interest and reproducibly construct RNA-seq libraries for next-generation sequencing. The sample multiplexing capability and free scaling of our microfluidic MACS implementation to low numbers of input cells while maintaining good enrichment performance are key advantages over conventional MACS approaches. Furthermore, the microfluidic device can be readily repurposed for other library preparation techniques, such as chromatin accessibility and DNA methylation profiling. We highlighted the importance of subset-specific profiling through in vitro treatment of healthy PBMCs and the clinical utility of the workflow by profiling cell subsets in multiplex from patient samples with SLE. While our protocols do not yet reach the purity levels of FACS, the on-device enrichment approach boosts the signal from target subsets relative to total PBMC profiles by a significant 2- to 13-fold, enabling robust detection of weaker signals. This platform will enable high-resolution monitoring of immune responses in clinical studies, especially in applications where blood samples or other inputs bear limited numbers of target cells and large-scale immunomonitoring studies where significant sample throughput is required.
METHODS
Study samples
Human blood samples were obtained either from Research Blood Components (MA, USA) for technical validation experiments or from collections at the Brigham and Women’s Hospital (MA USA) (table S1). Research on the samples was approved by Institutional Review Boards at the Broad Institute of Massachusets Institute of Technology (MIT) and Harvard (MA, USA) and Brigham and Women’s Hospital (MA, USA). Blood samples from patients with SLE and healthy control donors were drawn with EDTA Vacutainer tubes (BD Biosciences) and processed within 3 hours of collection.
Isolation of PBMCs from whole blood
Cells were isolated from whole blood samples using density gradient centrifugation. Whole blood was diluted 1:1 with 1× phosphate-buffered saline, layered on top of Ficoll-Paque PLUS (GE Healthcare), and centrifuged at 1200g for 20 min. The PBMC layer was retrieved, resuspended in 10 ml of RPMI-1640 (Gibco), and centrifuged again at 300g for 10 min. The cells were counted using a manual hemocytometer, resuspended in fetal bovine serum (FBS) (Gibco) with 10% dimethyl sulfoxide (Sigma), and aliquoted in 1-ml cryopreservation tubes at a concentration of 5 million cells/ml. The tubes were kept at −80°C overnight and then transferred to liquid nitrogen for long-term storage. Before processing, cells were thawed at 37°C for 3 min, resuspended in 10 ml of RPMI-1640 supplemented with 10% FBS (Gibco), and centrifuged at 300g for 5 min. The cells were then resuspended in the desired concentration or buffer, depending on the experiment.
Microfluidic device design and fabrication
The microfluidic device was fabricated using a previously published protocol (27) with minor modifications. Flow layer molds were patterned in three steps: (i) rectangular (75 μm), (ii) rectangular (200 μm), and (iii) rounded (60 μm). All silicon wafers were precoated with hexamethyldisilazane (Sigma) before spin coating. Rectangular features were prepared by spin-coating SU-8 2075 (MicroChem) on a silicon wafer. The coated wafers were patterned by ultraviolet (UV) exposure (OAI 206 mask aligner) through a mask printed at 20,000 dots per inch (Fineline Imaging, design files are included in the Supplementary Materials). The features were then developed using an SU-8 developer (MicroChem). The rounded features were produced by spin-coating an AZ-40XT photoresist (MicroChem), patterning the wafer with UV exposure and a mask, and developing with an AZ 400K developer (MicroChem). After development, the wafer was subjected to an additional curing step (105°C for 10 min) to round the features. The control layer mold was patterned in one step: rectangular (40 μm), using methods similar for the flow layer with an SU-8 2015 photoresist (MicroChem). Device production was carried out using standard soft lithography, following the same published protocol, with the exception of final bonding to a silicon wafer substrate.
Magnetic affinity cell isolation and microfluidic implementation
Magnetic affinity cell sorting was done using commercially available EasySep kits (CD14, CD19, CD4, and CD8 positive isolation II and T cell negative isolation) from STEMCELL Technologies. To implement the isolation protocols on the device, the buffers were modified and volumes were scaled accordingly. EasySep buffer (STEMCELL Technologies) was supplemented with 10% FBS (Gibco) and 0.2% Pluronic-F127 (Sigma), to reduce nonspecific cell adhesion in the polydimethylsiloxane channels. The microchannels were also preincubated with 1% Pluronic-F127 (Sigma) before cell isolation. Neodynium magnets (Grainger) with a 43-lb pull were used for all magnetic capture steps.
Flow cytometry and FACS
For assessment of isolation purities, flow cytometry was conducted using the Cytoflex system (Beckman Coulter). For RNA-seq library validation experiments and benchtop comparisons, PBMCs were sorted using the MoFlo Astrios (Beckman Coulter). Lysate pools were generated by sorting 5000 cells into 20 μl of TCL buffer (Qiagen) and 1 μl proteinase K (20 mg/ml; Qiagen) and stored at −80°C to maintain RNA integrity. The following panel was used for both purity assessment and sorting: DAPI, CD45 BV605, CD3 AF700, CD4 FITC, CD8 PE, CD14 APC, CD19 PE-Cy7, and CD56 BV650 (all IgG1κ, BioLegend). Flow cytometry data were analyzed using FlowJo v10.1.
Low-input RNA-seq, microfluidic implementation, and sequencing
RNA-seq was performed using Smart-Seq2 (29) with minor modifications. Cells were sorted into 19 μl of TCL buffer and 1 μl proteinase K (20 mg/ml; Qiagen), and their RNA was purified by a 2.2× SPRI cleanup with RNAClean XP magnetic beads (Agencourt) before reverse transcription. For the microfluidic implementation of the protocol, Tween 20 (Teknova) was added to all reactions at a final concentration of 0.5%. For mRNA capture, a biotinylated oligo (/5BiosG/ - AAGCAGTGGTATCAACGCAGAGTAC-30T-VN) (Integrated DNA Technologies) was attached to streptavidin magnetic beads (New England Biolabs) following the manufacturer’s protocol. The beads were then used to capture mRNA from the lysates and were washed with 10 mM tris-HCl (pH 7.5), 0.15 M LiCl, 1 mM EDTA, and 0.5% Tween 20. The beads were then resuspended in the reverse transcription mix, following the same steps as the published protocol. cDNA processed on the benchtop and microfluidic device were amplified for 18 and 22 cycles, respectively. After amplification and cleanup, libraries were quantified using a Qubit fluorometer (Invitrogen), and their size distributions were determined using the Agilent Bioanalyzer 2100. After normalizing the amplicon concentrations to 0.1 to 0.2 ng/ml, sequencing libraries were constructed using the Nextera XT DNA Library Prep Kit (Illumina), following the manufacturer’s protocol. All RNA-seq libraries were sequenced with 38 × 37 paired-end reads using a MiniSeq or NextSeq (Illumina).
In vitro stimulation of PBMCs
Healthy PBMCs were resuspended in RPMI-1640 supplemented with 10% FBS and 1× penicillin-streptomycin (Gibco). Cells were cultured at a density of 1 M/ml and stimulated with LPS (5 μg/ml) (eBioscience), DEX (100 nM) (Millipore), IFN-α (250 U/ml) (Abcam), or no treatment. The cells were cultured for 24 hours at 37°C in a 5% CO2 environment before processing through the microfluidic device.
RNA-seq data analysis
RNA-seq libraries were sequenced to a depth of 5 to 15 million reads per sample. All technical validation libraries were subsampled to 10 million reads to remove potential confounding effects of sequencing depth. Sequencing reads were aligned to the UCSC hg19 transcriptome using STAR (49) and used as input to generate QC statistics with RNA-SeQC (50). RSEM (51) was used to generate an expression matrix for all samples. Both raw count and transcripts per million data were analyzed using edgeR and custom R scripts. Lowly expressed genes with log2 (counts per million) less than 5 were filtered out before analysis. Gene set analyses were performed using the Kolmogorov-Smirnov test implementation in GAGE (52). Cytoscape and the enrichMap (53) module extension were used to visualize pathway-specific differential expression data.
Supplementary Material
Acknowledgments
We thank S. Kim, G. Lagoudas, A. Kulesa, N. Ranu, D. Lieb, A. Arazi, and all the members of the Blainey and Hacohen Laboratories (Broad Institute) for helpful discussions. We also thank S. Leff for assistance in device fabrication and the Broad Flow Cytometry core for assistance in the FACS experiments. Funding: This work was supported by grant U24 AI118668 from the National Institute of Allergy and Infectious Disease (to N.H. and P.C.B.). P.C.B. was supported by a Career Award at the Scientific Interface from the Burroughs Wellcome Fund. N.H. was supported by the David P. Ryan, MD Endowed Chair in Cancer Research. Author contributions: D.V. and P.C.B. designed the device. M.R., D.V., and K.B. fabricated microfluidic devices, implemented and optimized the microfluidic implementations of the MACS and RNA-seq protocols, and performed all device experiments. M.R. and T.E. performed experiments for benchtop protocol comparisons. M.R. analyzed the RNA-seq data and performed the in vitro experiments. T.E., P.H., and D.A.R. aided in clinical sample processing. N.H. and P.C.B. conceived and supervised the study. M.R. and P.C.B. prepared the manuscript. All authors reviewed and edited the final manuscript. Competing interests: P.C.B. is an extramural faculty member of MIT's Koch Institute for Integrative Cancer Research and a consultant to and equity holder in two companies in the microfluidics industry, 10X Genomics (Pleasanton, CA) and GALT (San Carlos, CA). The Broad Institute and MIT may seek to commercialize aspects of this work, and related applications for intellectual property have been filed: N.H., P.C.B., and D.V. are inventors on these applications (USSN 16/075,258, filed on 3 August 2018; EP17706351.8, filed on 5 September 2018). The authors declare that they have no other competing interests. Data and materials availability: All gene expression results are available as Supplementary Data. Raw data are available from authors upon request. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/1/eaau9223/DC1
Fig. S1. Detailed schematic of integrated workflow implemented on microfluidic chip.
Fig. S2. Flow cytometry gating strategy.
Fig. S3. Chip sorting optimization and comparison with benchtop magnetic isolation.
Fig. S4. Representative Bioanalyzer traces for conventional benchtop and microfluidic device whole transcriptome amplicons (WTA) for four different lysate samples from the four indicated FACS-sorted cell subsets.
Fig. S5. Differential gene expression analysis between microfluidic and benchtop RNA-seq libraries.
Fig. S6. Chip enrichment validation by gene expression profiles.
Fig. S7. Comparison between conventional and device-processed RNA-seq libraries (full workflow).
Fig. S8. RNA-seq correlation plots for in vitro–treated PBMCs.
Fig. S9. Gene set enrichment analysis (Reactome sets) comparing treated PBMCs versus control.
Fig. S10. Volcano plots showing differentially expressed genes between patients with SLE and matched healthy controls for four different subsets.
Fig. S11. Heat map showing relative IFN-signature scores across different cell types of 10 patients.
Supplementary Note
Supplementary Methods
Table S1. Ex vivo treatment differential expression (DE) results.
Table S2. Patient information for clinical study.
Table S3. SLE DE and gene set enrichment analysis results.
Data file S1. Device design.
Data file S2. Gene expression matrices.
REFERENCES AND NOTES
- 1.Ko E. R., Yang W. E., McClain M. T., Woods C. W., Ginsburg G. S., Tsalik E. L., What was old is new again: Using the host response to diagnose infectious disease. Expert Rev. Mol. Diagn. 15, 1143–1158 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Tang B. M., Huang S. J., McLean A. S., Genome-wide transcription profiling of human sepsis: A systematic review. Crit. Care 14, R237 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pascual V., Chaussabel D., Banchereau J., A genomic approach to human autoimmune diseases. Annu. Rev. Immunol. 28, 535–571 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ermann J., Rao D. A., Teslovich N. C., Brenner M. B., Raychaudhuri S., Immune cell profiling to guide therapeutic decisions in rheumatic diseases. Nat. Rev. Rheumatol. 11, 541–551 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Banchereau R., Cepika A.-M., Banchereau J., Pascual V., Understanding human autoimmunity and autoinflammation through transcriptomics. Annu. Rev. Immunol. 35, 337–370 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seok J., Warren H. S., Cuenca A. G., Mindrinos M. N., Baker H. V., Xu W., Richards D. R., McDonald-Smith G. P., Gao H., Hennessy L., Finnerty C. C., López C. M., Honari S., Moore E. E., Minei J. P., Cuschieri J., Bankey P. E., Johnson J. L., Sperry J., Nathens A. B., Billiar T. R., West M. A., Jeschke M. G., Klein M. B., Gamelli R. L., Gibran N. S., Brownstein B. H., Miller-Graziano C., Calvano S. E., Mason P. H., Cobb J. P., Rahme L. G., Lowry S. F., Maier R. V., Moldawer L. L., Herndon D. N., Davis R. W., Xiao W., Tompkins R. G., the Inflammation and Host Response to Injury, Large Scale Collaborative Research Program , Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc. Natl. Acad. Sci. U.S.A. 110, 3507–3512 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee B. H., Gauna A. E., Pauley K. M., Park Y.-J., Cha S., Animal models in autoimmune diseases: Lessons learned from mouse models for Sjögren’s syndrome. Clin. Rev. Allergy Immunol. 42, 35–44 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Godec J., Tan Y., Liberzon A., Tamayo P., Bhattacharya S., Butte A. J., Mesirov J. P., Haining W. N., Compendium of immune signatures identifies conserved and species-specific biology in response to inflammation. Immunity 44, 194–206 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Banchereau R., Hong S., Cantarel B., Baldwin N., Baisch J., Edens M., Cepika A.-M., Acs P., Turner J., Anguiano E., Vinod P., Khan S., Obermoser G., Blankenship D., Wakeland E., Nassi L., Gotte A., Punaro M., Liu Y.-J., Banchereau J., Rossello-Urgell J., Wright T., Pascual V., Personalized immunomonitoring uncovers molecular networks that stratify lupus patients. Cell 165, 551–565 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davenport E. E., Burnham K. L., Radhakrishnan J., Humburg P., Hutton P., Mills T. C., Rautanen A., Gordon A. C., Garrard C., Hill A. V. S., Hinds C. J., Knight J. C., Genomic landscape of the individual host response and outcomes in sepsis: A prospective cohort study. Lancet Respir. Med. 4, 259–271 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsalik E. L., Henao R., Nichols M., Burke T., Ko E. R., McClain M. T., Hudson L. L., Mazur A., Freeman D. H., Veldman T., Langley R. J., Quackenbush E. B., Glickman S. W., Cairns C. B., Jaehne A. K., Rivers E. P., Otero R. M., Zaas A. K., Kingsmore S. F., Lucas J., Fowler V. G. Jr., Carin L., Ginsburg G. S., Woods C. W., Host gene expression classifiers diagnose acute respiratory illness etiology. Sci. Transl. Med. 8, 322ra11 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ramilo O., Allman W., Chung W., Mejias A., Ardura M., Glaser C., Wittkowski K. M., Piqueras B., Banchereau J., Palucka A. K., Chaussabel D., Gene expression patterns in blood leukocytes discriminate patients with acute infections. Blood 109, 2066–2077 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sweeney T. E., Wong H. R., Khatri P., Robust classification of bacterial and viral infections via integrated host gene expression diagnostics. Sci. Transl. Med. 8, 346ra91 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zaas A. K., Chen M., Varkey J., Veldman T., Hero A. O. III, Lucas J., Huang Y., Turner R., Gilbert A., Lambkin-Williams R., Øien N. C., Nicholson B., Kingsmore S., Carin L., Woods C. W., Ginsburg G. S., Gene expression signatures diagnose influenza and other symptomatic respiratory viral infections in humans. Cell Host Microbe 6, 207–217 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu X., Speranza E., Muñoz-Fontela C., Haldenby S., Rickett N. Y., Garcia-Dorival I., Fang Y., Hall Y., Zekeng E. G., Lüdtke A., Xia D., Kerber R., Krumkamp R., Duraffour S., Sissoko D., Kenny J., Rockliffe N., Williamson E. D., Laws T. R., N’Faly M., Matthews D. A., Günther S., Cossins A. R., Sprecher A., Connor J. H., Carroll M. W., Hiscox J. A., Transcriptomic signatures differentiate survival from fatal outcomes in humans infected with Ebola virus. Genome Biol. 18, 4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sweeney T. E., Shidham A., Wong H. R., Khatri P., A comprehensive time-course–based multicohort analysis of sepsis and sterile inflammation reveals a robust diagnostic gene set. Sci. Transl. Med. 7, 287ra71 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McKinney E. F., Lee J. C., Jayne D. R. W., Lyons P. A., Smith K. G. C., T-cell exhaustion, co-stimulation and clinical outcome in autoimmunity and infection. Nature 523, 612–616 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McKinney E. F., Lyons P. A., Carr E. J., Hollis J. L., Jayne D. R. W., Willcocks L. C., Koukoulaki M., Brazma A., Jovanovic V., Kemeny D. M., Pollard A. J., MacAry P. A., Chaudhry A. N., Smith K. G. C., A CD8+ T cell transcription signature predicts prognosis in autoimmune disease. Nat. Med. 16, 586–591 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Becker A. M., Dao K. H., Han B. K., Kornu R., Lakhanpal S., Mobley A. B., Li Q.-Z., Lian Y., Wu T., Reimold A. M., Olsen N. J., Karp D. R., Chowdhury F. Z., Farrar J. D., Satterthwaite A. B., Mohan C., Lipsky P. E., Wakeland E. K., Davis L. S., SLE peripheral blood B cell, T cell and myeloid cell transcriptomes display unique profiles and each subset contributes to the interferon signature. PLOS ONE 8, e67003 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Flint S. M., Jovanovic V., Teo B. W., Mak A., Thumboo J., McKinney E. F., Lee J. C., MacAry P., Kemeny D. M., Jayne D. R. W., Fong K. Y., Lyons P. A., Smith K. G. C., Leucocyte subset-specific type 1 interferon signatures in SLE and other immune-mediated diseases. RMD Open 2, e000183 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Villani A.-C., Satija R., Reynolds G., Sarkizova S., Shekhar K., Fletcher J., Griesbeck M., Butler A., Zheng S., Lazo S., Jardine L., Dixon D., Stephenson E., Nilsson E., Grundberg I., McDonald D., Filby A., Li W., de Jager P. L., Rozenblatt-Rosen O., Lane A. A., Haniffa M., Regev A., Hacohen N., Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 356, eaah4573 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.See P., Dutertre C.-A., Chen J., Günther P., McGovern N., Irac S. E., Gunawan M., Beyer M., Händler K., Duan K., Sumatoh H. R. B., Ruffin N., Jouve M., Gea-Mallorquí E., Hennekam R. C. M., Lim T., Yip C. C., Wen M., Malleret B., Low I., Shadan N. B., Fen C. F. S., Tay A., Lum J., Zolezzi F., Larbi A., Poidinger M., Chan J. K. Y., Chen Q., Rénia L., Haniffa M., Benaroch P., Schlitzer A., Schultze J. L., Newell E. W., Ginhoux F., Mapping the human DC lineage through the integration of high-dimensional techniques. Science 356, eaag3009 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gaublomme J. T., Yosef N., Lee Y., Gertner R. S., Yang L. V., Wu C., Pandolfi P. P., Mak T., Satija R., Shalek A. K., Kuchroo V. K., Park H., Regev A., Single-cell genomics unveils critical regulators of Th17 cell pathogenicity. Cell 163, 1400–1412 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Singer M., Wang C., Cong L., Marjanovic N. D., Kowalczyk M. S., Zhang H., Nyman J., Sakuishi K., Kurtulus S., Gennert D., Xia J., Kwon J. Y. H., Nevin J., Herbst R. H., Yanai I., Rozenblatt-Rosen O., Kuchroo V. K., Regev A., Anderson A. C., A distinct gene module for dysfunction uncoupled from activation in tumor-infiltrating T cells. Cell 166, 1500–1511.e9 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ranu N., Villani A.-C., Hacohen N., Blainey P. C., Targeting individual cells by barcode in pooled sequence libraries. Nucleic Acids Res., gky856 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Melin J., Quake S. R., Microfluidic large-scale integration: The evolution of design rules for biological automation. Annu. Rev. Biophys. Biomol. Struct. 36, 213–231 (2007). [DOI] [PubMed] [Google Scholar]
- 27.Kim S., De Jonghe J., Kulesa A. B., Feldman D., Vatanen T., Bhattacharyya R. P., Berdy B., Gomez J., Nolan J., Epstein S., Blainey P. C., High-throughput automated microfluidic sample preparation for accurate microbial genomics. Nat. Commun. 8, 13919 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hong J. W., Studer V., Hang G., Anderson W. F., Quake S. R., A nanoliter-scale nucleic acid processor with parallel architecture. Nat. Biotechnol. 22, 435–439 (2004). [DOI] [PubMed] [Google Scholar]
- 29.Picelli S., Faridani O. R., Björklund Å. K., Winberg G., Sagasser S., Sandberg R., Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 9, 171–181 (2014). [DOI] [PubMed] [Google Scholar]
- 30.Crouse J., Kalinke U., Oxenius A., Regulation of antiviral T cell responses by type I interferons. Nat. Rev. Immunol. 15, 231–242 (2015). [DOI] [PubMed] [Google Scholar]
- 31.González-Navajas J. M., Lee J., David M., Raz E., Immunomodulatory functions of type I interferons. Nat. Rev. Immunol. 12, 125–135 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moseman E. A., Wu T., de la Torre J. C., Schwartzberg P. L., McGavern D. B., Type I interferon suppresses virus-specific B cell responses by modulating CD8+ T cell differentiation. Sci. Immunol. 1, eaah3565 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sammicheli S., Kuka M., Di Lucia P., de Oya N. J., De Giovanni M., Fioravanti J., Cristofani C., Maganuco C. G., Fallet B., Ganzer L., Sironi L., Mainetti M., Ostuni R., Larimore K., Greenberg P. D., de la Torre J. C., Guidotti L. G., Iannacone M., Inflammatory monocytes hinder antiviral B cell responses. Sci. Immunol. 1, eaah6789 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hervas-Stubbs S., Perez-Gracia J. L., Rouzaut A., Sanmamed M. F., Le Bon A., Melero I., Direct effects of type I interferons on cells of the immune system. Clin. Cancer Res. 17, 2619–2627 (2011). [DOI] [PubMed] [Google Scholar]
- 35.Bogdan C., Mattner J., Schleicher U., The role of type I interferons in non-viral infections. Immunol. Rev. 202, 33–48 (2004). [DOI] [PubMed] [Google Scholar]
- 36.Le Bon A., Thompson C., Kamphuis E., Durand V., Rossmann C., Kalinke U., Tough D. F., Cutting edge: Enhancement of antibody responses through direct stimulation of B and T cells by type I IFN. J. Immunol. 176, 2074–2078 (2006). [DOI] [PubMed] [Google Scholar]
- 37.Hermann P., Rubio M., Nakajima T., Delespesse G., Sarfati M., IFN-α priming of human monocytes differentially regulates gram-positive and gram-negative bacteria-induced IL-10 release and selectively enhances IL-12p70, CD80, and MHC class I expression. J. Immunol. 161, 2011–2018 (1998). [PubMed] [Google Scholar]
- 38.Gujer C., Sandgren K. J., Douagi I., Adams W. C., Sundling C., Smed-Sörensen A., Seder R. A., Hedestam G. B. K., Loré K., IFN-α produced by human plasmacytoid dendritic cells enhances T cell-dependent naïve B cell differentiation. J. Leukoc. Biol. 89, 811–821 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Luft T., Luetjens P., Hochrein H., Toy T., Masterman K.-A., Rizkalla M., Maliszewski C., Shortman K., Cebon J., Maraskovsky E., IFN-α enhances CD40 ligand-mediated activation of immature monocyte-derived dendritic cells. Int. Immunol. 14, 367–380 (2002). [DOI] [PubMed] [Google Scholar]
- 40.Santodonato L., D'Agostino G., Nisini R., Mariotti S., Monque D. M., Spada M., Lattanzi L., Perrone M. P., Andreotti M., Belardelli F., Ferrantini M., Monocyte-derived dendritic cells generated after a short-term culture with IFN-α and granulocyte-macrophage colony-stimulating factor stimulate a potent Epstein-Barr virus-specific CD8+ T cell response. J. Immunol. 170, 5195–5202 (2003). [DOI] [PubMed] [Google Scholar]
- 41.Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S., Mesirov J. P., Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eloranta M.-L., Rönnblom L., Cause and consequences of the activated type I interferon system in SLE. J. Mol. Med. 94, 1103–1110 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Obermoser G., Pascual V., The interferon-α signature of systemic lupus erythematosus. Lupus 19, 1012–1019 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.O’Gorman W. E., Kong D. S., Balboni I. M., Rudra P., Bolen C. R., Ghosh D., Davis M. M., Nolan G. P., Hsieh E. W. Y., Mass cytometry identifies a distinct monocyte cytokine signature shared by clinically heterogeneous pediatric SLE patients. J. Autoimmun. 81, 74–89 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lyons P. A., McKinney E. F., Rayner T. F., Hatton A., Woffendin H. B., Koukoulaki M., Freeman T. C., Jayne D. R. W., Chaudhry A. N., Smith K. G. C., Novel expression signatures identified by transcriptional analysis of separated leucocyte subsets in systemic lupus erythematosus and vasculitis. Ann. Rheum. Dis. 69, 1208–1213 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Abbas A. R., Wolslegel K., Seshasayee D., Modrusan Z., Clark H. F., Deconvolution of blood microarray data identifies cellular activation patterns in systemic lupus erythematosus. PLOS ONE 4, e6098 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gayed M., Bernatsky S., Ramsey-Goldman R., Clarke A. E., Gordon C., Lupus and cancer. Lupus 18, 479–485 (2009). [DOI] [PubMed] [Google Scholar]
- 48.Arasappan D., Tong W., Mummaneni P., Fang H., Amur S., Meta-analysis of microarray data using a pathway-based approach identifies a 37-gene expression signature for systemic lupus erythematosus in human peripheral blood mononuclear cells. BMC Med. 9, 65 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dobin A., Davis C. A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T. R., STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.DeLuca D. S., Levin J. Z., Sivachenko A., Fennell T., Nazaire M.-D., Williams C., Reich M., Winckler W., Getz G., RNA-SeQC: RNA-seq metrics for quality control and process optimization. Bioinformatics 28, 1530–1532 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li B., Dewey C. N., RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 12, 323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Luo W., Friedman M. S., Shedden K., Hankenson K. D., Woolf P. J., GAGE: Generally applicable gene set enrichment for pathway analysis. BMC Bioinformatics. 10, 161 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Merico D., Isserlin R., Stueker O., Emili A., Bader G. D., Enrichment map: A network-based method for gene-set enrichment visualization and interpretation. PLOS ONE 5, e13984 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/1/eaau9223/DC1
Fig. S1. Detailed schematic of integrated workflow implemented on microfluidic chip.
Fig. S2. Flow cytometry gating strategy.
Fig. S3. Chip sorting optimization and comparison with benchtop magnetic isolation.
Fig. S4. Representative Bioanalyzer traces for conventional benchtop and microfluidic device whole transcriptome amplicons (WTA) for four different lysate samples from the four indicated FACS-sorted cell subsets.
Fig. S5. Differential gene expression analysis between microfluidic and benchtop RNA-seq libraries.
Fig. S6. Chip enrichment validation by gene expression profiles.
Fig. S7. Comparison between conventional and device-processed RNA-seq libraries (full workflow).
Fig. S8. RNA-seq correlation plots for in vitro–treated PBMCs.
Fig. S9. Gene set enrichment analysis (Reactome sets) comparing treated PBMCs versus control.
Fig. S10. Volcano plots showing differentially expressed genes between patients with SLE and matched healthy controls for four different subsets.
Fig. S11. Heat map showing relative IFN-signature scores across different cell types of 10 patients.
Supplementary Note
Supplementary Methods
Table S1. Ex vivo treatment differential expression (DE) results.
Table S2. Patient information for clinical study.
Table S3. SLE DE and gene set enrichment analysis results.
Data file S1. Device design.
Data file S2. Gene expression matrices.