

Site-selective blocking by ALD disentangles the geometric and electronic effects of catalysts beyond the selectivity volcano.
Abstract
The prominent size effect of metal nanoparticles shapes decisively nanocatalysis, but entanglement of the corresponding geometric and electronic effects prevents exploiting their distinct functionalities. In this work, we demonstrate that in palladium (Pd)–catalyzed aerobic oxidation of benzyl alcohol, the geometric and electronic effects interplay and compete so intensively that both activity and selectivity showed in volcano trends on the Pd particle size unprecedentedly. By developing a strategy of site-selective blocking via atomic layer deposition along with first principles calculations, we disentangle these two effects and unveil that the geometric effect dominates the right side of the volcano with larger-size Pd particles, whereas the electronic effect directs the left of the volcano with smaller-size Pd particles substantially. Selective blocking of the low-coordination sites prevents formation of the undesired by-product beyond the volcano relationship, achieving a remarkable benzaldehyde selectivity and activity at the same time for 4-nm Pd. Disentangling the geometric and electronic effects of metal nanoparticles opens a new dimension for rational design of catalysts.
INTRODUCTION
Metal nanoparticles (NPs) are one of the most important materials in a broad application of heterogeneous catalysis, sensors, batteries, biology and medicine, etc., because of the notable size effects from their bulk counterparts (1, 2). Geometrically, in addition to high surface area, the smaller NPs expose more low-coordination sites (LCSs) instead of high-coordination sites (HCSs), and their different local geometries considerably change chemical bond breaking and making in catalysis (3–9). Electronically, quantum size effects (QSEs) also alter significantly the energy levels of the entire particle, thereby affecting the orbital hybridization and overall charge transfer between metal and reactants (10, 11). The above two distinct functionalities endow precise tailoring of the particle size, in addition to the particle composition (12, 13), to be a valuable methodology to optimize catalysts (14–18). Nevertheless, HCSs and LCSs might have very different electronic structures (19), whereas QSEs depend sensitively on geometric structures (20). This strong entanglement of the geometric and electronic properties with the particle size makes identification of their distinct size-dependent functionalities nearly impossible. Disentangling the geometric and electronic properties of metal NPs and identifying when one or the other starts to dominate the reactivity would go beyond the established structure-activity relationship and open a new dimension for rational design of the catalysts (21, 22). However, the above demand has never been met and represents a grand challenge.
Selective oxidation of alcohols to the corresponding carbonyl compounds, the key intermediates for fine chemical synthesis, is one of the most fundamental and useful industrial processes (23). Supported palladium (Pd)–based nanocatalysts have attracted significant attention because of their superior catalytic performance under mild conditions with molecular oxygen as the oxidant (23–25). However, the effects of the structural and electronic properties of Pd NPs on activity and selectivity remain controversial (17, 23, 25, 26).
In this work, we report that the strong entanglement of the electronic and geometric structures of Pd NPs in Pd/Al2O3 catalysts prompts the activity and selectivity both in volcano relationships with the Pd particle size unprecedentedly in aerobic oxidation of benzyl alcohol. To manipulate solely the geometric structure without interfering with the electronic structures, we developed a new strategy of selectively blocking the LCSs and HCSs of Pd NPs using atomic layer deposition (ALD) of Al2O3 and FeOx, respectively. Combining with density functional theory (DFT) calculations, we disentangle unambiguously the geometric and electronic effects of Pd NPs on the activity and selectivity of oxidation of benzyl alcohol. We find that it is the geometric effect that controls the right side of the volcano peak at ~4 nm, whereas the electronic effect controls the left side. Selectively blocking the Pd LCSs by Al2O3 ALD goes beyond the selectivity volcano and results in a remarkable benzaldehyde selectivity and intrinsic activity on the 4-nm Pd NP catalyst at the same time.
RESULTS AND DISCUSSION
Solvent-free aerobic benzyl alcohol oxidation using O2 as the oxidant for benzaldehyde was conducted at 120°C over various Pd/Al2O3 catalysts, with the Pd particle size carefully tuned from ~2.1 to 19.1 nm (figs. S1 and S2 and table S1). Typically, there are two major reaction pathways: the dehydrogenation path to benzaldehyde (the desired product) (Eq. 1) and the hydrogenolysis path to toluene (the major by-product at this reaction temperature) (Eq. 2) (27).
| (1) |
| (2) |
The H* in Eq. 2 is the intermediate surface hydrogen species generated during the dehydrogenation process. Strikingly, an inverse volcano relationship for the selectivity to benzaldehyde on the Pd particle size was found (Fig. 1A). On the left and right sides of this volcano, selectivity to benzaldehyde was as high as 87% (2.1 nm) and 93% (19.1 nm) at 50% conversion, while a lowest benzaldehyde selectivity of 72% occurred at the valley of the inverse volcano on the 4.2-nm Pd/Al2O3 sample. Meanwhile, the activity in terms of turnover frequencies (TOFs) and specific rates also showed a traditional volcano shape with a maximum of 4.3 × 104 hour−1 and 1.2 × 104 hour−1 on the 4.2-nm Pd/Al2O3 sample, respectively (Fig. 1B). At the volcano peak, corresponding apparent activation energies for formation of benzaldehyde and toluene were measured with values of 0.36 and 0.83 eV, respectively (fig. S3). Note that the volcano relationship of TOFs on the Pd particle size is in good agreement with the literature (17).
Fig. 1. Catalytic performance of Pd/Al2O3 catalysts with different particle sizes in aerobic oxidation of benzyl alcohol.
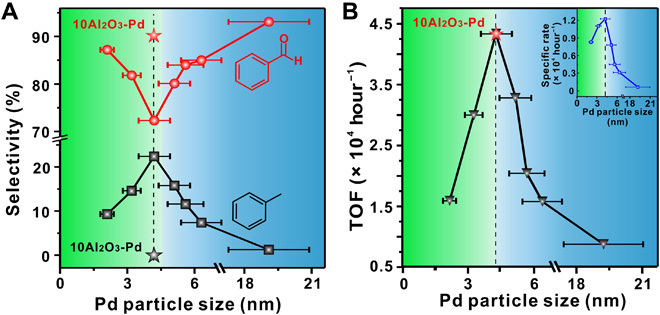
(A) Benzaldehyde and toluene selectivity as a function of Pd particle size at 50% benzyl alcohol conversion. (B) TOFs as a function of Pd particle size. The inset in (B) shows the specific rates as a function of Pd particle size. The selectivity and TOF of the 4.2-nm Pd/Al2O3 catalyst with 10 cycles of ALD alumina overcoat (10Al2O3-Pd) under the same reaction conditions were also presented in (A) and (B), respectively, for comparison (star). Reaction conditions: normalized Pd content, 1.6 μmol Pd; substrate, 48 mmol benzyl alcohol; reaction temperature, 120°C; O2 flow rate, 15 ml/min.
In situ x-ray photoelectron spectroscopy (XPS) measurements showed that the Pd 3d binding energy gradually shifted to a higher position in a magnitude of 1.3 eV as Pd particle size decreased from 19.1 to 2.1 nm (Fig. 2A). A sharp increase in Pd 3d binding energy was noticed below 4.2 nm (Fig. 2B), indicating a dramatic change in the corresponding electronic structure. To see the change in geometric structure, we estimated the HCS-to-LCS ratio by assuming all Pd NPs synthesized in a cubooctahedral shape, as often adopted in the literature (25). It is found that the resulting HCS-to-LCS ratio increases significantly from 0.93 to 16.7, with an increase in the Pd particle size from ~2.1 to 19.1 nm (Fig. 2B). These results show that both the geometric (the HCS-to-LCS ratio) and electronic structures change largely with size. However, the entanglement between the geometric and electronic structures prevents differentiation of their individual contributions to the observed volcano relationships.
Fig. 2. The electronic and geometric properties of Pd/Al2O3 catalysts on Pd particle size.
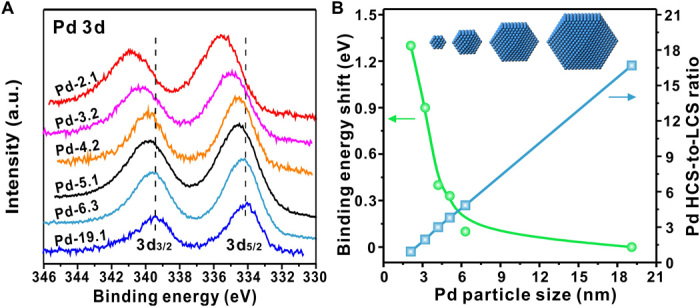
(A) In situ XPS spectra of the Pd/Al2O3 catalysts with various particle sizes in the Pd 3d region. a.u., arbitrary units. (B) The Pd 3d5/2 binding energy shift compared with the 19.1-nm Pd NP sample as a function of Pd particle size, and the calculated ratios of Pd HCSs to LCSs for the Pd/Al2O3 catalysts with various particle sizes based on the cubooctahedral cluster model, as illustrated by the inset in (B).
To disentangle the geometric and electronic effects, ALD of Al2O3 and FeOx was performed on the 4.2-nm Pd/Al2O3 sample to selectively block the LCSs and HCSs of Pd NPs, respectively (herein nAl2O3-Pd and nFeOx-Pd for n cycles of Al2O3 and FeOx ALD, respectively) (28, 29). Transmission electron microscopy (TEM) and energy-dispersive x-ray spectroscopy (EDS) elemental mapping confirmed the Al2O3 and FeOx coating layers on Pd NPs (figs. S4 and S5). It was found that both the average Pd particle size (fig. S6) and the corresponding electronic properties, measured by in situ XPS (fig. S7), were essentially intact after overcoating. Diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) of CO chemisorption showed that the uncoated sample exhibited three peaks centered at about 1922, 1966, and 2088 cm−1 (Fig. 3A). They can be assigned to the μ2 bridge–bonded CO on Pd(111) facets (HCSs), μ2 bridge–bonded CO on the Pd NPs edges/corners (LCSs), and linear CO on HCSs, respectively (28, 30, 31). When ALD of Al2O3 was applied, the peak at 1966 cm−1 decreased much more quickly compared with the peak at 1922 cm−1 with increase in ALD cycles and vanished on 10Al2O3-Pd. The above evidence suggests firmly that the LCSs of Pd NPs in 10Al2O3-Pd were selectively blocked by the Al2O3 overcoat, leaving the HCSs accessible for catalysis (inset of Fig. 3A), according to the literature (32, 33). In contrast, FeOx coating showed an opposite trend by attenuating the CO peak at 1922 cm−1 much more quickly with ALD cycles (Fig. 3B), suggesting a preferential decoration of the HCSs. Semiquantitative analysis of CO DRIFT chemisorption showed that the HCS-to-LCS ratio was ~4.3 for uncoated Pd/Al2O3, close to estimation of 3.0 by assuming corresponding Pd NPs in a cubooctahedral shape (Fig. 2B and fig. S8) (25). Although this semiquantification has certain error bars, it could still properly represent the evolution of the HCS-to-LCS ratio with oxide overcoating. We found that the HCS-to-LCS ratio increased rapidly with Al2O3 overcoating and became about 55 on 10Al2O3-Pd, with negligible LCSs exposed. On the contrary, the FeOx overcoating decreased this ratio considerably to ~0.8 for 8FeOx-Pd.
Fig. 3. Structural characterization of Al2O3- and FeOx-overcoated 4.2-nm Pd/Al2O3 samples and their catalytic performances.
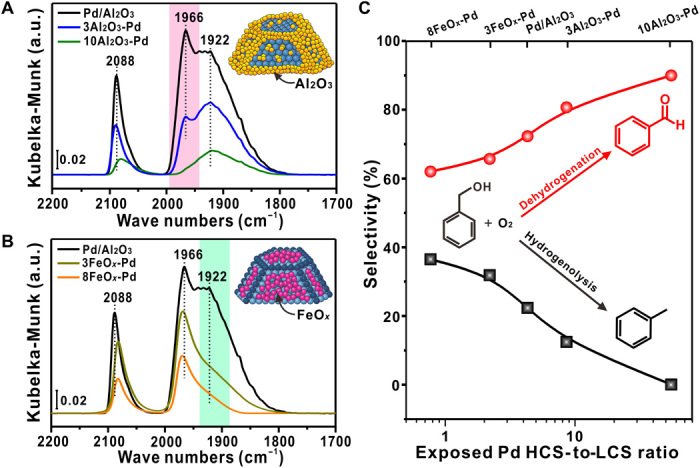
DRIFT spectra of CO chemisorption on the (A) ALD Al2O3- and (B) FeOx-overcoated Pd/Al2O3 samples at the CO saturated coverage. The insets illustrate selective blocking of the LCSs (A) or HCSs (B) of Pd NPs with Al2O3 and FeOx ALD overcoat, respectively. (C) Tuned selectivity to benzaldehyde and toluene in benzyl alcohol oxidation on Pd/Al2O3 by oxide overcoats as a function of exposed Pd HCS-to-LCS ratio, measured at 50% benzyl alcohol conversion; the lines in (C) are only for guiding the eyes. Reaction conditions: normalized Pd content, 1.6 μmol Pd; substrate, 48 mmol benzyl alcohol; reaction temperature, 120°C; O2 flow rate, 15 ml/min.
The above results show that selective blocking of Pd NPs can tune effectively the HCS-to-LCS ratio from ~0.8 to ~55 without changing the particle size necessarily (fig. S9), thus providing an ideal platform for disentangling the geometric and electronic effects. With the increase of Al2O3 ALD cycles, benzaldehyde selectivity increased gradually from only 72 to almost 90% (fig. S10A), whereas the toluene selectivity decreased from about 24 to only 0.1% (fig. S10B). In contrast, with the increase of the FeOx coverage, benzaldehyde selectivity decreased from 72 to 62%, along with an increase in toluene selectivity from 22 to 37%. According to the strong support effect observed by Hutchings and co-workers (34), the Pd-FeOx interface might also influence the selectivity. However, we found that a Pd/Fe2O3 catalyst with a Pd particle size of 3.5 ± 0.4 nm exhibited a benzaldehyde selectivity of 90% at 50% conversion, even higher than the Pd/Al2O3 catalysts with similar particle sizes (Fig. 1A). Therefore, it turns out that the Pd-FeOx interface favors the benzaldehyde formation (35) and that the decreased benzaldehyde selectivity through FeOx overcoating is mainly due to the geometric effect through “site-selective blocking” of the HCSs of Pd NPs, rather than the formation of the Pd-FeOx interface. Plotting the selectivity of the two products together showed compellingly that with increase of the HCS-to-LCS ratio, benzaldehyde selectivity increased but the toluene selectivity decreased monotonously (Fig. 3C). At the HCS-to-LCS ratio of ~50, the corresponding TOF of 4.3 × 104 hour−1 remained similar with the volcano peak of uncoated 4.2-nm Pd/Al2O3 (Fig. 1B and fig. S11). Notably, toluene selectivity was nearly completely suppressed. This remarkable selectivity manipulation by oxide overcoating was also observed at different reaction temperatures and other Pd catalysts with different particle sizes and supports (figs. S12 to S17). As shown in Fig. 1, site-selective blockages of the LCSs broke down the selectivity volcano on the particle size and achieved a great performance in both activity and selectivity on 4.2-nm Pd/Al2O3. It is also worth noting that the 10Al-Pd/Al2O3 catalyst demonstrated excellent recyclability and metal stability against sintering and leaching (fig. S18).
Selective blocking of Pd NPs by Al2O3 and FeOx provides unambiguous evidence that dehydrogenation to benzaldehyde in benzyl alcohol oxidation takes place mainly at the HCSs, whereas hydrogenolysis to toluene occurs at the LCSs. Therefore, the geometric effect, namely, the decreased HCS-to-LCS ratio, controls the observed decrease in benzaldehyde selectivity as a decrease in the Pd particle size from ~19.1 to 4.2 nm (Fig. 1A). However, when the particle size was smaller than 4.2 nm, the trend of the benzaldehyde selectivity was reversed. The resulting inverse volcano implies that the electronic effect starts to control the selectivity, as supported by a significant increase in the Pd 3d energy level (Fig. 2).
DFT calculations were performed to shed more light on the geometric and electronic effects of Pd NPs in benzyl alcohol oxidation. First, terraced Pd(111) and stepped Pd(211) were used to simulate the HCSs and LCSs of Pd NPs, respectively (figs. S19 to S21). For aerobic oxidation of benzyl alcohol to benzaldehyde and water, oxygen is mainly for promoting dehydrogenation and removing surface hydrogen. Oxygen-assisted dehydrogenation of benzyl alcohol to benzyloxy and hydroxyl (Fig. 4A, figs. S22 to S24, and table S2) is facile, with barriers of 0.38 eV on Pd(111) and 0.44 eV on Pd(211). Further dehydrogenation of benzyloxy to benzaldehyde and hydrogen without assistance by oxygen necessarily has modest barriers of 0.51 eV on Pd(111) and 0.44 eV on Pd(211). In the following, reaction between hydroxyl and hydrogen to generate water closes the catalytic cycle. Calculated barriers are 0.75 eV on Pd(111) and 1.12 eV on Pd(211) (Fig. 4B); therein, the lower barrier for the former comes from its weaker binding toward hydroxyl, compared with the latter (−2.68 eV versus −3.04 eV) (fig. S20), which is in agreement with previous result (36). In this cycle, benzaldehyde formation is facile at both the HCSs and LCSs, and hydrogen removal by hydroxyl is demanding and more efficient at the HCSs.
Fig. 4. Theoretical calculations of Pd-catalyzed benzyl alcohol oxidation.
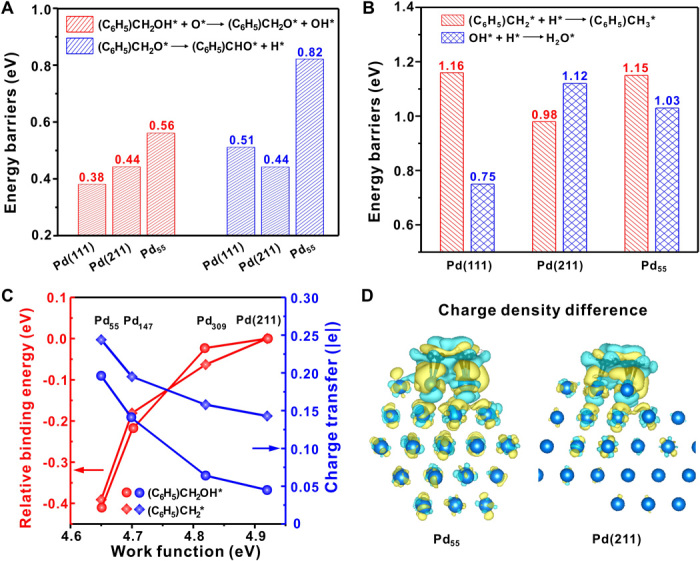
(A) Calculated energy barriers in the oxidative dehydrogenation from benzyl alcohol to benzyloxy and OH and from benzyloxy to benzaldehyde and hydrogen on Pd(111), Pd(211), and Pd55. (B) Calculated energy barriers for competing hydrogenation of benzyl and hydroxyl to toluene and water on Pd(111), Pd(211), and Pd55. (C) Relative binding energies of benzyl alcohol and benzyl on Pd55, Pd147, and Pd309 with respect to Pd(211) surface, as well as calculated charge transfer toward these species and corresponding work function. The absolute binding energies of (C6H5)CH2OH and (C6H5)CH2 on Pd(211) are −2.05 and −2.51 eV, respectively (see fig. S19). (D) Charge density difference upon adsorption of benzyl on Pd55 and Pd(211). The blue balls are Pd atoms, while the areas of cyan and yellow contours represent the depletion and accumulation of electrons in a unit of 0.001e/Å3, respectively.
For toluene formation, the scission of the C–O bond in benzyl alcohol or its derived intermediates is the prerequisite. Compared with the scission of the C–O bond in benzyl alcohol–derived intermediates, we found that direct scission of the C–O bond of benzyl alcohol to form benzyl and hydroxyl is preferential with barriers of 1.02 eV on Pd(111) and 0.95 eV on Pd(211) (figs. S25 and S26 and table S2). Hydrogen’s reaction with benzyl produces toluene with barriers of 1.16 eV on Pd(111) and 0.98 eV on Pd(211) (Fig. 4B). Compared with hydrogen’s reaction with hydroxyl, hydrogen’s reaction with benzyl is kinetically more favorable on Pd(211), but not on Pd(111). Namely, LCSs prefers to hydrogenate benzyl for toluene, and HCSs prefers to hydrogenate hydroxyl for water. It is therefore the competitive removal of hydrogen governing the reaction paths on HCSs and LCSs. With an increase in the HCS-to-LCS ratio, the benzaldehyde selectivity would increase while the toluene decreases, as found exactly on the right side of the volcano relationship (Fig. 1A) and the site-selective blocking experiments (Fig. 3C).
To see the electronic effect on the small particles and rationalize the left side of the volcano relationship, we investigated the adsorption of benzyl alcohol, benzyl, and hydroxyl on freestanding Pdn clusters (n = 55, 147, and 309; figs. S27 and S28) with cubooctahedral structures (11, 37). Although the change of hydroxyl binding is negligible, the binding strength of benzyl alcohol and benzyl at favorable sites increases pronouncedly with a decrease in the size (Fig. 4C), ~0.40 eV stronger on Pd55 than on Pd(211), where the calculated binding energies of benzyl alcohol and benzyl are −2.05 and −2.51 eV, respectively (fig. S19). Changes in binding energies cannot come from the geometric effect, since the corresponding adsorption sites (fig. S28) are similar. On the other hand, the clusters constructed have different electronic structures from Pd(211); in particular, the work function decreases systematically by ~0.3 eV for Pd55. This indicates that the corresponding Fermi level shifts upward with a decrease in particle size, in line with upshifting the Pd 3d level to higher positions observed experimentally (Fig. 2). Accordingly, there is more electron transfer toward the adsorbates (Fig. 4C), enhancing eventually the corresponding binding strengths, a fact that was also found previously in Pt and Au (11, 38). As shown in Fig. 4D and figs. S29 and S30, increasing electron transfer toward the adsorbates with a decrease in the particle size comes not only from the coordinated metal atoms contributing more electrons but also from the fact that there are more metal atoms contributing electrons. This is understandable since a change in Fermi level affects the entire particle, in contrast to the HCSs and LCSs differing only in their local geometries of the particle.
Enhanced binding strengths on small particles change significantly the activity and selectivity. Taking Pd55 as an example, the barriers for the O–H (0.56 eV) bond and subsequent C–H (0.82 eV) bond cleavages of benzyl alcohol are higher than those on Pd(111) and Pd(211), respectively (Fig. 4A and figs. S31 and S32). Moreover, a barrier of 1.03 eV for hydrogen removal by hydroxyl is lower than that of 1.15 eV by benzyl (Fig. 4B), opposite to the order on Pd(211). This indicates that although the toluene formation is more favorable than water at the LCSs, stronger binding of benzyl due to the pronounced electronic effect of small particles leads to subsequent hydrogenation less favorable than water formation. These rationalize well the low activity and the high selectivity to benzaldehyde on small particles on the left side of volcanos (Fig. 1). In the moderate particle size, the electronic effect is weakened, the geometric effect on the HCS-to-LCS ratio increases, and the interplay between these two effects results in the volcano of activity and selectivity on the particle size.
In summary, we have demonstrated that in aerobic oxidation of benzyl alcohol, both the activity and selectivity over Pd/Al2O3 catalysts showed volcano relationships with the Pd particle size unprecedentedly, due to the strong entanglement of electronic and geometric structures. Selective blocking of Pd NPs by Al2O3 and FeOx, according to DRIFTS CO chemisorption, provides a powerful tool to manipulate solely the geometric structure, which unambiguously confirms that the geometric effect controls the right side of the volcanos. On the smaller Pd particles, the electronic effect is involved significantly as indicated by XPS, thus reversing the trend of the benzaldehyde selectivity and decreasing the activity. First principles calculations further unveiled that the competitive removal of surface hydrogen via reaction with hydroxyl and benzyl intermediates on HCSs and LCSs makes the reaction structure sensitive and selective. Decreasing the particle size lowers the work function and increases the binding strength of benzyl alcohol and benzyl and the reaction barriers. These effects, in turn, alter the reaction path of hydrogen removal and significantly change the corresponding activity and selectivity, consistent with the experimental observation. Given the essential roles of the geometric and electronic effects in metal NPs, disentangling the geometric and electronic effects of metal NPs opens a new dimension for metal catalyst rational design.
MATERIALS AND METHODS
Catalyst preparation
Pd/Al2O3 catalysts were synthesized using the incipient wetness impregnation (WI) method (28). For the synthesis of the 4.2-nm Pd/Al2O3 catalyst, 0.1 g of Pd(NO3)2·2H2O (>97.7%; Aladdin Chemicals) and 0.43 g of citric acid were first dissolved in 2.6 ml of deionized water. Then, 4.4 g of spherical Al2O3 powder (99.5%; NANODUR, Alfa Aesar) was added into the solution and mixed thoroughly. The mixture was dried overnight at 125°C. Last, the dried material was calcined in 10% O2 in Ar (Nanjing Special Gases) at 300°C for 5 hours and then reduced at 250°C for another 1 hour in 10% H2 in Ar (Nanjing Special Gases) to obtain the 4.2-nm Pd/Al2O3 catalyst. Tuning the ratio of the amount of the Pd precursor to citric acid, as well as the calcination temperature, would allow precise control of the Pd particle size. Details can be seen in table S1.
A Pd/C catalyst was also prepared for excluding the support effect according to a procedure reported previously (39). Briefly, 0.1 mmol PdCl2 (Aladdin Chemicals) and 0.8 mmol sodium citrate (Sinopharm Chemical Reagent Co. Ltd.) were dissolved into 150 ml of water. Carbon black (400 mg; Vulcan XC72R, Carbot Corp.) was then added. After stirring the mixture for 20 min, followed by a 30-min sonication, 15 ml of 0.1 M sodium borohydride (96%; Sinopharm Chemical Reagent Co. Ltd.) solution was added dropwise into the suspension under vigorous stirring and then aged at 25°C for 8 hours. Next, the precipitate was filtered and washed with deionized water thoroughly. The obtained materials were then dried overnight in a vacuum oven at 45°C. Last, the dried sample was calcined in 10% O2 in Ar at 250°C for 1 hour and then reduced at 250°C for 1 hour in 10% H2 in Ar to obtain the Pd/C catalyst.
Alumina and FeOx ALD were carried out in a viscous flow reactor (GEMStar-6 Benchtop ALD, Arradiance). Ultrahigh purity N2 (99.999%) was used as a carrier gas at a flow rate of 200 ml/min. Al2O3 ALD was performed by alternatively exposing the sample to trimethylaluminum (TMA; 99.999%; Nanjing MO Yuan Scientific Instruments & Materials Co., Ltd.) and deionized water at 200°C at different cycles (28). The timing sequence was 3, 200, 10, and 200 s for TMA exposure, N2 purge, water exposure, and N2 purge, respectively. Besides the 4.2-nm Pd/Al2O3 samples, alumina overcoating was also applied on 5.1-nm Pd/Al2O3 and Pd/C samples. The obtained alumina-coated 5.1-nm Pd/Al2O3 and Pd/C samples were denoted as nAl2O3-Pd-5.1 and nAl2O3-Pd/C, respectively, where n represents the number of ALD cycles (n = 1, 3, 5, and 10). All catalysts were calcined in 10% O2 in Ar at 300°C for 2 hours and then reduced at 200°C for 1 hour in 10% H2 in Ar before any characterization and reaction tests.
FeOx ALD was performed by alternatively exposing the sample to ferrocene (98%; Sigma-Aldrich) and ultrahigh purity O2 (99.999%) at 150°C at different cycles (29). The timing sequence was 60, 200, 60, and 200 s for ferrocene exposure, N2 purge, O2 exposure, and N2 purge, respectively. Besides the 4.2-nm Pd/Al2O3 samples, FeOx overcoating was also applied on 5.1-nm Pd/Al2O3 and Pd/C samples. The obtained FeOx-coated 5.1-nm Pd/Al2O3 and Pd/C samples were denoted as nFeOx-Pd-5.1 and nFeOx-Pd/C, respectively, where n represents the number of ALD cycles (n = 1, 3, 5, and 10). All catalysts were calcined in 10% O2 in Ar at 150°C for 1 hour and then reduced at 150°C for 1 hour in 10% H2 in Ar before any characterization and reaction tests.
Catalyst characterization
TEM measurements were performed on a JEM-2100F instrument operated at 200 kV to characterize the morphology of the 10Al2O3-Pd catalyst. Scanning TEM (STEM) characterization of 8FeOx-Pd catalysts and Pd/Al2O3 samples with various particle sizes was carried out on an aberration-corrected high-angle annular dark-field STEM instrument at 200 kV (JEM-ARM200F, University of Science and Technology of China). Elemental mapping using EDS was performed on the same equipment. The Pd contents in various catalysts were analyzed by an inductively coupled plasma atomic emission spectrometer.
The DRIFTS CO chemisorption measurements were performed on a Nicolet iS10 spectrometer equipped with a mercury-cadmium-telluride detector and a low-temperature reaction cell (Praying Mantis Harrick). After loading a sample into the cell, it was first calcined in 10% O2 in Ar at 150°C for 1 hour followed by a reduction in 10% H2 in Ar at 150°C for another 1 hour. After cooling the sample to room temperature under a continuous flow of Ar, a background spectrum was collected. Subsequently, the sample was exposed to 10% CO in Ar at a flow rate of 20 ml/min for about 30 min until saturation. Next, the sample was purged with Ar at a flow rate of 20 ml/min for another 30 min to remove the gas-phase CO, and then, the DRIFT spectrum was collected with 256 scans at a resolution of 4 cm−1.
In situ x-ray photoemission spectroscopy (XPS) measurements were conducted at the BL10B beamline photoemission end station at the National Synchrotron Radiation Laboratory (NSRL) in Hefei, China. Briefly, the beamline is connected to a bending magnet and covers photon energies from 100 to 1000 eV. The end-station consists of four chambers, i.e., analysis chamber, preparation chamber, quick sample load lock chamber, and a high pressure reactor. The analysis chamber, with a base pressure of <5 × 10−10 torr, is connected to the beamline and equipped with a VG Scienta R3000 electron energy analyzer and a twin anode x-ray source. The high pressure reactor houses a reaction cell where the samples can be treated with different gases up to 20 bars and simultaneously heated up to 650°C. After sample treatment, the reactor can be pumped down to high vacuum (<10−8 torr) for sample transfer. In the current work, the samples were first treated with the flowing 10% H2 in Ar (20 ml/min) at 150°C for 0.5 hour at ambient pressure in the high pressure reactor. Next, the samples were transferred to the analysis chamber for XPS measurements in the Pd 3d region without exposing to air. The Al 2p binding energy at 74.3 eV was used as internal reference (40).
The Pd dispersions were determined by CO pulse chemisorption, which were conducted using a Micromeritics AutoChem II chemisorption analyzer at room temperature. Before CO pulse, the catalysts were first calcined in 10% O2 in He at 200°C for 1 hour followed by a reduction in 10% H2 in He at 200°C for 2 hours. Then, the catalysts were cooled to room temperature in He, and CO pulses were introduced to the catalyst surface using 10% CO in He (41). A stoichiometry of CO:Pd = 1 for dispersion calculations was assumed according to literature.
Catalyst evaluation
Solvent-free aerobic oxidation of benzyl alcohol using molecular O2 was conducted in a batch-type reactor, as described in our previous work (42). The amount of catalyst used for the reaction test was normalized to have the same Pd content of 1.6 μmol. Typically, 5 ml of benzyl alcohol (48 mmol) and the normalized Pd catalyst were co-added into a 25-ml three-necked glass flask equipped with a reflux condenser. Before reaction, the system was first charged with O2 by bubbling ultrahigh purity O2 (99.999%; Nanjing Special Gases) at a flow rate of 15 ml/min for 20 min to remove air. Under the continuous flow of O2, the reactor was quickly immersed into a silicon oil bath at the desired temperature (90, 120, or 140°C) to initiate the reaction. During each reaction, the mixture was vigorously stirred at a rate of 1500 rpm to exclude any mass transfer limitation. The effect of oxygen flow rates on the possible selectivity change was also ruled out. Last, the reaction products were analyzed using a Shimadzu GC-2014 gas chromatograph, equipped with an Rtx-1 capillary column and an autoinjector. The TOFs were calculated using Eq. 3
| (3) |
Here, the moles of active sites were normalized to the number of exposed surface Pd sites calculated by the cubooctahedral “magic cluster” model. On alumina-coated samples, the number of exposed surface Pd sites was determined by the Pd dispersion data (table S3). The TOFs were calculated from the rate of the intitial 60-min reaction, unless otherwise noted. The specific rates were also calculated on the basis of Eq. 4
| (4) |
Theoretical calculations
DFT calculations were performed with the Vienna ab initio Simulation Package (43, 44). The exchange-correlation interaction was described by optPBE functional with van der Waals correction (45–47). The Kohn-Sham equations were solved in a plane wave basis set with a kinetic energy cutoff of 400 eV. To model Pd(111) and Pd(211) surfaces, the four-layer slab models were used with (4 by 4) and (2 by 4) unit cells, respectively. The freestanding Pd55, Pd147, and Pd309 clusters were modeled using the cubeoctahedral structure based on the literature (11, 38). A (3 by 3 by 1) k-point mesh and Γ-point were used to sample the surface Brillouin zone for the surface and cluster, respectively. A 12-Å vacuum was introduced with correction of dipole moment between the repeated slabs/clusters. During optimization, the bottom two layers of the slab were fixed, while the remaining atoms of the slab, all the atoms of the clusters and the adsorbates were relaxed until the residual force was less than 0.02 eV/Å. The barriers of the elementary steps were calculated by the climbing image nudged elastic band (CI-NEB) method (48), and the transition states were confirmed by single imaginary frequency.
Supplementary Material
Acknowledgments
We thank the BL10B beamlines at NSRL, China, and Y. Lin for electron microscopy study. Funding: This work was supported by the National Natural Science Foundation of China (21673215, 91645202, and 21473169), National Key R&D Program of China (2017YFB0602205 and 2018YFA0208603), the Chinese Academy of Sciences (QYZDJ-SSW-SLH054), the Fundamental Research Funds for the Central Universities (WK6030000015 and WK2060030017), and the Max-Planck Partner Group. X.-K.G. thanks the partial support of the Early Career Program (DE-SC0014347) from the Department of Energy, Basic Energy Science. Author contributions: J.L. received the project and designed the experiments. H.W. performed all catalyst synthesis, characterization, and reaction tests. W.-X.L. and X.-K.G. designed the calculations. X.-K.G. performed the DFT calculations. H.W., X.Z., H.P., and J.Z. performed the in situ XPS measurements. S.C. and L.C. assisted in other catalyst characterization. H.W., X.-K.G., W.-X.L., and J.L. co-wrote the paper, and all the other authors contributed to the overall scientific interpretation and editing of the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/1/eaat6413/DC1
Supplementary Text
Fig. S1. Morphologies of Pd/Al2O3 catalysts with various particle sizes.
Fig. S2. The corresponding Pd size distributions in the Pd/Al2O3 catalysts.
Fig. S3. The apparent activation energies for benzaldehyde and toluene formation.
Fig. S4. A representative TEM image of the 10Al2O3-Pd catalyst.
Fig. S5. A representative STEM image of the 8FeOx-Pd catalyst.
Fig. S6. The Pd particle size distributions of uncoated and oxide-coated samples.
Fig. S7. In situ XPS spectra of Pd/Al2O3, 10Al2O3-Pd, and 8FeOx-Pd samples in the Pd 3d region.
Fig. S8. Semiquantitative analysis of the DRIFT spectra.
Fig. S9. The ratios of exposed Pd HCSs to LCSs on oxide-coated and uncoated Pd/Al2O3 samples determined by semiquantitative analysis of the DRIFT CO chemisorption spectra.
Fig. S10. Catalytic performance of 4.2-nm Pd/Al2O3 catalysts with different cycles of alumina and FeOx overcoating in aerobic oxidation of benzyl alcohol at 120°C.
Fig. S11. Catalytic performance of 4.2-nm Pd/Al2O3 catalysts with different cycles of alumina overcoating in aerobic oxidation of benzyl alcohol at 120°C.
Fig. S12. Catalytic performance of 4.2-nm Pd/Al2O3 and 10Al2O3-Pd catalysts in aerobic oxidation of benzyl alcohol at different temperatures.
Fig. S13. Catalytic performance of 4.2-nm Pd/Al2O3 and 10Al2O3-Pd catalysts in aerobic oxidation of benzyl alcohol at 90°C.
Fig. S14. Catalytic performance of 4.2-nm Pd/Al2O3 and 10Al2O3-Pd catalysts in aerobic oxidation of benzyl alcohol at 140°C.
Fig. S15. Structural characterization of Al2O3- and FeOx-overcoated 5.1-nm Pd/Al2O3 samples and their catalytic performances.
Fig. S16. Morphology of the 2.9-nm Pd/C catalyst.
Fig. S17. Catalytic performance of the 2.9-nm Pd/C, 10Al2O3-Pd/C, and 8FeOx-Pd/C catalysts in aerobic oxidation of benzyl alcohol at 120°C.
Fig. S18. Recycling stability of the 10Al2O3-Pd catalyst.
Fig. S19. Optimized geometries and calculated binding energies of benzyl alcohol and its derived key intermediates on Pd surfaces.
Fig. S20. Optimized geometries and calculated binding energies of O2, O, H, OH, and H2O intermediates on Pd surfaces with respect to the corresponding species in the gas phase.
Fig. S21. The side views of optimized geometries of benzyl alcohol and its derived key intermediates on Pd surfaces.
Fig. S22. The transition states of elementary steps involved in benzyl alcohol oxidation on Pd(111).
Fig. S23. The transition states of elementary steps involved in benzyl alcohol oxidation on Pd(211).
Fig. S24. Energy profiles of the direct and oxidative dehydrogenation paths on Pd surfaces.
Fig. S25. Energy profiles of the hydrogenolysis path to toluene formation on Pd surfaces.
Fig. S26. Energy profiles of C–O scission of (C6H5)CH2OH*, (C6H5)CHOH*, and (C6H5)CH2O* on Pd surfaces.
Fig. S27. Models of freestanding Pd55, Pd147, and Pd309 clusters.
Fig. S28. The optimized most stable geometries of (C6H5)CH2OH*, (C6H5)CH2*, and OH* on freestanding Pd55, Pd147, and Pd309 clusters.
Fig. S29. Comparison of the side view of charge density difference upon adsorption of benzyl on Pd55, Pd147, Pd309, and Pd(211) surfaces.
Fig. S30. Comparison of the top view of charge density difference upon adsorption of benzyl on Pd55, Pd147, Pd309, and Pd(211) surfaces.
Fig. S31. Optimized transition state structures of the key steps involved in benzaldehyde and toluene formations on Pd55 cluster.
Fig. S32. Energy profiles of the oxidative dehydrogenation and hydrogenolysis paths on Pd55 cluster.
Table S1. Conditions for synthesis of Pd/Al2O3 samples with various Pd particle sizes using the WI method.
Table S2. Calculated activation barriers (Ea) and reaction energies (Er) of elementary steps in oxidation of benzyl alcohol on Pd(111) and Pd(211) surfaces.
Table S3. Pd dispersion determined by CO pulse chemisorption for various Pd catalysts.
REFERENCES AND NOTES
- 1.G. Bergeret, P. Gallezot, G. Ertl, H. Knözinger, J. Weitkamp, Handbook of Heterogeneous Catalysis (VCH, 1997), 2, 439 pp. [Google Scholar]
- 2.Jain P. K., Huang X. H., El-Sayed I. H., El-Sayed M. A., Noble metals on the nanoscale: Optical and photothermal properties and some applications in imaging, sensing, biology, and medicine. Acc. Chem. Res. 41, 1578–1586 (2008). [DOI] [PubMed] [Google Scholar]
- 3.Calle-Vallejo F., Tymoczko J., Colic V., Vu Q. H., Pohl M. D., Morgenstern K., Loffreda D., Sautet P., Schuhmann W., Bandarenka A. S., Finding optimal surface sites on heterogeneous catalysts by counting nearest neighbors. Science 350, 185–189 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Calle-Vallejo F., Loffreda D., Koper M. T. M., Sautet P., Introducing structural sensitivity into adsorption–energy scaling relations by means of coordination numbers. Nat. Chem. 7, 403–410 (2015). [DOI] [PubMed] [Google Scholar]
- 5.den Breejen J. P., Radstake P. B., Bezemer G. L., Bitter J. H., Frøseth V., Holmen A., de Jong K. P., On the origin of the cobalt particle size effects in Fischer–Tropsch catalysis. J. Am. Chem. Soc. 131, 7197–7203 (2009). [DOI] [PubMed] [Google Scholar]
- 6.Kuhn J. N., Huang W. Y., Tsung C.-K., Zhang Y. W., Somorjai G. A., Structure sensitivity of carbon–nitrogen ring opening: Impact of platinum particle size from below 1 to 5 nm upon pyrrole hydrogenation product selectivity over monodisperse platinum nanoparticles loaded onto mesoporous silica. J. Am. Chem. Soc. 130, 14026–14027 (2008). [DOI] [PubMed] [Google Scholar]
- 7.Reske R., Mistry H., Behafarid F., Cuenya B. R., Strasser P., Particle size effects in the catalytic electroreduction of CO2 on Cu nanoparticles. J. Am. Chem. Soc. 136, 6978–6986 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Englisch M., Jentys A., Lercher J. A., Structure sensitivity of the hydrogenation of crotonaldehyde over Pt/SiO2 and Pt/TiO2. J. Catal. 166, 25–35 (1997). [Google Scholar]
- 9.Cao S. W., Tao F., Tang Y., Li Y. T., Yu J. G., Size- and shape-dependent catalytic performances of oxidation and reduction reactions on nanocatalysts. Chem. Soc. Rev. 45, 4747–4765 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Halperin W. P., Quantum size effects in metal particles. Rev. Mod. Phys. 58, 533–606 (1986). [Google Scholar]
- 11.Li L., Larsen A. H., Romero N. A., Morozov V. A., Glinsvad C., Abild-Pedersen F., Greeley J., Jacobsen K. W., Nørskov J. K., Investigation of catalytic finite-size-effects of platinum metal clusters. J. Phys. Chem. Lett. 4, 222–226 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Nørskov J. K., Bligaard T., Rossmeisl J., Christensen C. H., Towards the computational design of solid catalysts. Nat. Chem. 1, 37–46 (2009). [DOI] [PubMed] [Google Scholar]
- 13.Ferrando R., Jellinek J., Johnston R. L., Nanoalloys: From theory to applications of alloy clusters and nanoparticles. Chem. Rev. 108, 845–910 (2008). [DOI] [PubMed] [Google Scholar]
- 14.Roduner E., Size matters: Why nanomaterials are different. Chem. Soc. Rev. 35, 583–592 (2006). [DOI] [PubMed] [Google Scholar]
- 15.Valden M., Lai X., Goodman D. W., Onset of catalytic activity of gold clusters on titania with the appearance of nonmetallic properties. Science 281, 1647–1650 (1998). [DOI] [PubMed] [Google Scholar]
- 16.Bai L., Wang X., Chen Q., Ye Y., Zheng H., Guo J., Yin Y., Gao C., Explaining the size dependence in platinum-nanoparticle-catalyzed hydrogenation reactions. Angew. Chem. Int. Ed. 55, 15656–15661 (2016). [DOI] [PubMed] [Google Scholar]
- 17.Chen J., Zhang Q., Wang Y., Wan H., Size-dependent catalytic activity of supported palladium nanoparticles for aerobic oxidation of alcohols. Adv. Synth. Catal. 350, 453–464 (2008). [Google Scholar]
- 18.Shao M., Peles A., Shoemaker K., Electrocatalysis on platinum nanoparticles: Particle size effect on oxygen reduction reaction activity. Nano Lett. 11, 3714–3719 (2011). [DOI] [PubMed] [Google Scholar]
- 19.Nørskov J. K., Abild-Pedersen F., Studt F., Bligaard T., Density functional theory in surface chemistry and catalysis. Proc. Natl. Acad. Sci. U.S.A. 108, 937–943 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bond G. C., Supported metal catalysts: Some unsolved problems. Chem. Soc. Rev. 20, 441–475 (1991). [Google Scholar]
- 21.Vojvodic A., Nørskov J. K., New design paradigm for heterogeneous catalysts. Natl. Sci. Rev. 2, 140–143 (2015). [Google Scholar]
- 22.Wang P., Chang F., Gao W., Guo J., Wu G., He T., Chen P., Breaking scaling relations to achieve low-temperature ammonia synthesis through LiH-mediated nitrogen transfer and hydrogenation. Nat. Chem. 9, 64–70 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Mallat T., Baiker A., Oxidation of alcohols with molecular oxygen on solid catalysts. Chem. Rev. 104, 3037–3058 (2004). [DOI] [PubMed] [Google Scholar]
- 24.Enache D. I., Edwards J. K., Landon P., Solsona-Espriu B., Carley A. F., Herzing A. A., Watanabe M., Kiely C. J., Knight D. W., Hutchings G. J., Solvent-free oxidation of primary alcohols to aldehydes using Au-Pd/TiO2 catalysts. Science 311, 362–365 (2006). [DOI] [PubMed] [Google Scholar]
- 25.Mori K., Hara T., Mizugaki T., Ebitani K., Kaneda K., Hydroxyapatite-supported palladium nanoclusters: A highly active heterogeneous catalyst for selective oxidation of alcohols by use of molecular oxygen. J. Am. Chem. Soc. 126, 10657–10666 (2004). [DOI] [PubMed] [Google Scholar]
- 26.Ferri D., Mondelli C., Krumeich F., Baiker A., Discrimination of active palladium sites in catalytic liquid-phase oxidation of benzyl alcohol. J. Phys. Chem. B 110, 22982–22986 (2006). [DOI] [PubMed] [Google Scholar]
- 27.Galvanin F., Sankar M., Cattaneo S., Bethell D., Dua V., Hutchings G. J., Gavriilidis A., On the development of kinetic models for solvent-free benzyl alcohol oxidation over a gold-palladium catalyst. Chem. Eng. J. 342, 196–210 (2018). [Google Scholar]
- 28.Lu J. L., Fu B., Kung M. C., Xiao G., Elam J. W., Kung H. H., Stair P. C., Coking- and sintering-resistant palladium catalysts achieved through atomic layer deposition. Science 335, 1205–1208 (2012). [DOI] [PubMed] [Google Scholar]
- 29.Rooth M., Johansson A., Kukli K., Aarik J., Boman M., Hårsta A., Atomic layer deposition of iron oxide thin films and nanotubes using ferrocene and oxygen as precursors. Chem. Vap. Deposition 14, 67–70 (2008). [Google Scholar]
- 30.Zeinalipour-Yazdi C. D., Willock D. J., Thomas L., Wilson K., Lee A. F., CO adsorption over Pd nanoparticles: A general framework for IR simulations on nanoparticles. Surf. Sci. 646, 210–220 (2016). [Google Scholar]
- 31.Lear T., Marshall R., Lopez-Sanchez J. A., Jackson S. D., Klapötke T. M., Baumer M., Rupprechter G., Freund H.-J., Lennon D., The application of infrared spectroscopy to probe the surface morphology of alumina-supported palladium catalysts. J. Chem. Phys. 123, 174706 (2005). [DOI] [PubMed] [Google Scholar]
- 32.Lu J., Liu B., Guisinger N. P., Stair P. C., Greeley J. P., Elam J. W., First-principles predictions and in situ experimental validation of alumina atomic layer deposition on metal surfaces. Chem. Mater. 26, 6752–6761 (2014). [Google Scholar]
- 33.Schauermann S., Hoffmann J., Johánek V., Hartmann J., Libuda J., Freund H.-J., Catalytic activity and poisoning of specific sites on supported metal nanoparticles. Angew. Chem. Int. Ed. 41, 2532–2535 (2002). [DOI] [PubMed] [Google Scholar]
- 34.Cao E., Sankar M., Nowicka E., He Q., Morad M., Miedziak P. J., Taylor S. H., Knight D. W., Bethell D., Kiely C. J., Gavriilidis A., Hutchings G. J., Selective suppression of disproportionation reaction in solvent-less benzyl alcohol oxidation catalysed by supported Au–Pd nanoparticles. Catal. Today 203, 146–152 (2013). [Google Scholar]
- 35.Zhao G., Yang F., Chen Z., Liu Q., Ji Y., Zhang Y., Niu Z., Mao J., Bao X., Hu P., Li Y., Metal/oxide interfacial effects on the selective oxidation of primary alcohols. Nat. Commun. 8, 10439 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Trinchero A., Hellman A., Grönbeck H., Methane oxidation over Pd and Pt studied by DFT and kinetic modeling. Surf. Sci. 616, 206–213 (2013). [Google Scholar]
- 37.Stenlid J. H., Brinck T., Extending the σ-hole concept to metals: An electrostatic interpretation of the effects of nanostructure in gold and platinum catalysis. J. Am. Chem. Soc. 139, 11012–11015 (2017). [DOI] [PubMed] [Google Scholar]
- 38.Kleis J., Greeley J., Romero N. A., Morozov V. A., Falsig H., Larsen A. H., Lu J., Mortensen J. J., Dułak M., Thygesen K. S., Nørskov J. K., Jacobsen K. W., Finite size effects in chemical bonding: From small clusters to solids. Catal. Lett. 141, 1067–1071 (2011). [Google Scholar]
- 39.Li J., Chen W., Zhao H., Zheng X., Wu L., Pan H., Zhu J., Chen Y., Lu J., Size-dependent catalytic activity over carbon-supported palladium nanoparticles in dehydrogenation of formic acid. J. Catal. 352, 371–381 (2017). [Google Scholar]
- 40.Elmasides C., Kondarides D. I., Grünert W., Verykios X. E., XPS and FTIR study of Ru/Al2O3 and Ru/TiO2 catalysts: Reduction characteristics and interaction with a methane–oxygen mixture. J. Phys. Chem. B 103, 5227–5239 (1999). [Google Scholar]
- 41.Narayanan S., Krishna K., Hydrotalcite-supported palladium catalysts part I: Preparation, characterization of hydrotalcites and palladium on uncalcined hydrotalcites for CO chemisorption and phenol hydrogenation. Appl. Catal. A. Gen. 174, 221–229 (1998). [Google Scholar]
- 42.Wang H., Wang C., Yan H., Yi H., Lu J., Precisely-controlled synthesis of Au@Pd core–shell bimetallic catalyst via atomic layer deposition for selective oxidation of benzyl alcohol. J. Catal. 324, 59–68 (2015). [Google Scholar]
- 43.Kresse G., Furthmüller J., Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996). [DOI] [PubMed] [Google Scholar]
- 44.Kresse G., Furthmüller J., Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996). [DOI] [PubMed] [Google Scholar]
- 45.Perdew J. P., Burke K., Ernzerhof M., Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996). [DOI] [PubMed] [Google Scholar]
- 46.Klimeš J., Bowler D. R., Michaelides A., Van der Waals density functionals applied to solids. Phys. Rev. B 83, 195131 (2011). [Google Scholar]
- 47.Klimeš J., Bowler D. R., Michaelides A., Chemical accuracy for the van der Waals density functional. J. Phys. Condens. Matter 22, 022201 (2010). [DOI] [PubMed] [Google Scholar]
- 48.Henkelman G., Uberuaga B. P., Jónsson H., A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 113, 9901–9904 (2000). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/1/eaat6413/DC1
Supplementary Text
Fig. S1. Morphologies of Pd/Al2O3 catalysts with various particle sizes.
Fig. S2. The corresponding Pd size distributions in the Pd/Al2O3 catalysts.
Fig. S3. The apparent activation energies for benzaldehyde and toluene formation.
Fig. S4. A representative TEM image of the 10Al2O3-Pd catalyst.
Fig. S5. A representative STEM image of the 8FeOx-Pd catalyst.
Fig. S6. The Pd particle size distributions of uncoated and oxide-coated samples.
Fig. S7. In situ XPS spectra of Pd/Al2O3, 10Al2O3-Pd, and 8FeOx-Pd samples in the Pd 3d region.
Fig. S8. Semiquantitative analysis of the DRIFT spectra.
Fig. S9. The ratios of exposed Pd HCSs to LCSs on oxide-coated and uncoated Pd/Al2O3 samples determined by semiquantitative analysis of the DRIFT CO chemisorption spectra.
Fig. S10. Catalytic performance of 4.2-nm Pd/Al2O3 catalysts with different cycles of alumina and FeOx overcoating in aerobic oxidation of benzyl alcohol at 120°C.
Fig. S11. Catalytic performance of 4.2-nm Pd/Al2O3 catalysts with different cycles of alumina overcoating in aerobic oxidation of benzyl alcohol at 120°C.
Fig. S12. Catalytic performance of 4.2-nm Pd/Al2O3 and 10Al2O3-Pd catalysts in aerobic oxidation of benzyl alcohol at different temperatures.
Fig. S13. Catalytic performance of 4.2-nm Pd/Al2O3 and 10Al2O3-Pd catalysts in aerobic oxidation of benzyl alcohol at 90°C.
Fig. S14. Catalytic performance of 4.2-nm Pd/Al2O3 and 10Al2O3-Pd catalysts in aerobic oxidation of benzyl alcohol at 140°C.
Fig. S15. Structural characterization of Al2O3- and FeOx-overcoated 5.1-nm Pd/Al2O3 samples and their catalytic performances.
Fig. S16. Morphology of the 2.9-nm Pd/C catalyst.
Fig. S17. Catalytic performance of the 2.9-nm Pd/C, 10Al2O3-Pd/C, and 8FeOx-Pd/C catalysts in aerobic oxidation of benzyl alcohol at 120°C.
Fig. S18. Recycling stability of the 10Al2O3-Pd catalyst.
Fig. S19. Optimized geometries and calculated binding energies of benzyl alcohol and its derived key intermediates on Pd surfaces.
Fig. S20. Optimized geometries and calculated binding energies of O2, O, H, OH, and H2O intermediates on Pd surfaces with respect to the corresponding species in the gas phase.
Fig. S21. The side views of optimized geometries of benzyl alcohol and its derived key intermediates on Pd surfaces.
Fig. S22. The transition states of elementary steps involved in benzyl alcohol oxidation on Pd(111).
Fig. S23. The transition states of elementary steps involved in benzyl alcohol oxidation on Pd(211).
Fig. S24. Energy profiles of the direct and oxidative dehydrogenation paths on Pd surfaces.
Fig. S25. Energy profiles of the hydrogenolysis path to toluene formation on Pd surfaces.
Fig. S26. Energy profiles of C–O scission of (C6H5)CH2OH*, (C6H5)CHOH*, and (C6H5)CH2O* on Pd surfaces.
Fig. S27. Models of freestanding Pd55, Pd147, and Pd309 clusters.
Fig. S28. The optimized most stable geometries of (C6H5)CH2OH*, (C6H5)CH2*, and OH* on freestanding Pd55, Pd147, and Pd309 clusters.
Fig. S29. Comparison of the side view of charge density difference upon adsorption of benzyl on Pd55, Pd147, Pd309, and Pd(211) surfaces.
Fig. S30. Comparison of the top view of charge density difference upon adsorption of benzyl on Pd55, Pd147, Pd309, and Pd(211) surfaces.
Fig. S31. Optimized transition state structures of the key steps involved in benzaldehyde and toluene formations on Pd55 cluster.
Fig. S32. Energy profiles of the oxidative dehydrogenation and hydrogenolysis paths on Pd55 cluster.
Table S1. Conditions for synthesis of Pd/Al2O3 samples with various Pd particle sizes using the WI method.
Table S2. Calculated activation barriers (Ea) and reaction energies (Er) of elementary steps in oxidation of benzyl alcohol on Pd(111) and Pd(211) surfaces.
Table S3. Pd dispersion determined by CO pulse chemisorption for various Pd catalysts.