

Abstract
Background & Aims
Hepatitis B virus (HBV) infection is a major health concern worldwide. Although currently used nucleos(t)ide analogs efficiently inhibit viral replication, viral proteins transcribed from the episomal viral covalently closed circular DNA (cccDNA) minichromosome continue to be expressed long-term. Because high viral RNA or antigen loads may play a biological role during this chronicity, the elimination of viral products is an ultimate goal of HBV treatment. HBV regulatory protein X (HBx) was recently found to promote transcription of cccDNA with degradation of Smc5/6 through the interaction of HBx with the host protein DDB1. Here, this protein–protein interaction was considered as a new molecular target of HBV treatment.
Methods
To identify candidate compounds that target the HBx–DDB1 interaction, a newly constructed split luciferase assay system was applied to comprehensive compound screening. The effects of the identified compounds on HBV transcription and cccDNA maintenance were determined using HBV minicircle DNA, which mimics HBV cccDNA, and the natural HBV infection model of human primary hepatocytes.
Results
We show that nitazoxanide (NTZ), a thiazolide anti-infective agent that has been approved by the FDA for protozoan enteritis, efficiently inhibits the HBx–DDB1 protein interaction. NTZ significantly restores Smc5 protein levels and suppresses viral transcription and viral protein production in the HBV minicircle system and in human primary hepatocytes naturally infected with HBV.
Conclusions
These results indicate that NTZ, which targets an HBV-related viral–host protein interaction, may be a promising new therapeutic agent and a step toward a functional HBV cure.
Keywords: Drug Screening, Minicircle, Primary Hepatocyte Infection
Abbreviations used in this paper: cccDNA, covalently closed circular DNA; Cluc, Cypridina luciferase; CMV, cytomegalovirus; DDB1, damage-specific DNA-binding protein 1; ddPCR, droplet digital polymerase chain reaction; DMEM, Dulbecco's modified Eagle's medium; DMSO, dimethyl sulfoxide; FDA, Food and Drug Administration; Flag-HBx, Flag-tagged hepatitis B virus regulatory protein X; Gluc, Gaussia luciferase; HBs, hepatitis B surface antibody; HBx, hepatitis B virus regulatory protein X; HBV, hepatitis B virus; IFNα, interferon alfa; IP, immunoprecipitation; LgBit, Large Bit; mRNA, messenger RNA; NTZ, nitazoxianide; mcHBV, minicircle hepatitis B virus; pgRNA, pregenomic RNA; PSAD, plasmid-safe DNase; qPCR, quantitative polymerase chain reaction; SDS, sodium dodecyl sulfate; SmBit, Small Bit; Smc5/6, structural maintenance of chromosomes 5/6
Graphical abstract

See editorial on page 289.
Summary.
We identified nitazoxanide as a novel inhibitor against the hepatitis B virus regulatory protein X–damage-specific DNA-binding protein 1 interaction via compound screening for drug repositioning. The inhibition of the hepatitis B virus regulatory protein X–damage-specific DNA-binding protein 1 interaction leads to the significant reduction of viral transcription and subsequent viral products.
Hepatitis B virus (HBV) is a major global health problem, although current antiviral drugs, such as nucleos(t)ide analogs, effectively reduce HBV-DNA levels. Despite the existence of a prophylactic vaccine, an estimated 240 million people are infected worldwide.1, 2 and are therefore at high risk of cirrhosis and hepatocellular carcinoma. To reduce these risks, reducing HBV-DNA levels is needed, as is elimination of hepatitis B surface antibody (HBs) antigens, referred to as a “functional cure,” which is a major clinical goal for chronic hepatitis B.3, 4 However, the HBV therapeutics that are currently available, such as interferon alfa (IFNα) and antiviral drugs, rarely achieve this goal.1
HBV virions contain a 3.2-kb genome, in the form of partially double-stranded, relaxed circular DNA, from which covalently closed circular DNA (cccDNA) is formed. HBV cccDNA is persistent in the hepatocyte nucleus, functioning as a minichromosome and as a transcriptional template for all HBV viral RNAs.5
Transcription of the viral genome is promoted by HBV regulatory X (HBx) protein.6, 7 Previous studies have suggested that this process requires the binding of HBx with the host protein damage-specific DNA-binding protein 1 (DDB1),8, 9 but the underlying mechanisms of the interaction of HBx with DDB1 and the promotion of viral transcription have long remained unknown. Recently, HBx was found to assemble an HBx-DDB1-CUL4-ROC1 E3 ligase complex to target structural maintenance of chromosomes 5/6 (Smc5/6), a host restriction factor that blocks viral transcription, for ubiquitination and degradation, resulting in enhanced viral transcription from cccDNA.10, 11 These results indicate that a primary function of the HBx–DDB1 interaction is the degradation of Smc5/6 to promote viral transcription.
Based on these findings, we hypothesized that compounds that inhibit the binding of HBx and DDB1 would block viral transcription, resulting in inhibition of the production of viral RNAs and proteins, including HBV antigens. In this study, we established a convenient screening system using a split luciferase for identification of inhibitors of the HBx–DDB1 interaction. Using this system, we identified a compound that significantly inhibits HBx–DDB1 binding and discovered that this compound reduces viral transcription and the levels of viral protein products, and therefore may provide a new therapeutic option for HBV.
Results
Establishment of a Screening System for Inhibitors of the HBX–DDB1 Interaction
To screen compounds for inhibition of the HBx–DDB1 interaction, we established a screening system using a split luciferase (NanoBiT, Promega, Madison, WI). The separated NanoBiT subunits, Large Bit (LgBit) (17.6 kDa) and Small Bit (SmBit) (11 amino acids), only associate weakly, so their assembly into a luminescent complex is dictated by interaction characteristics of the target proteins onto which they are tagged.12 By constructing NanoBiT-fused HBx and DDB1 proteins, we established a screening system for the HBx–DDB1 interaction (Figure 1A). Because the positions of the luciferase fragments relative to the target proteins are crucial for reflecting the interaction of the target proteins accurately,12 we first tested all 8 possible combinations of expression constructs to determine the optimal orientation of the LgBit or SmBit fusion to the target proteins (Figure 1B). Because the combination of HBx fused to LgBit at the C terminal of HBx (HBx–LgBit) and DDB1 fused to SmBit at the N terminal of DDB1 (SmBit–DDB1) provided the best results, ie, the brightest luciferase signals (Figure 1C) (P < .001), we used these combinations for subsequent screening.
Figure 1.

High-throughput screening system to monitor HBx–DDB1 binding. (A) The principle underlying detection of the HBx–DDB1 interaction using split luciferase. The separated NanoBiT subunits, LgBit and SmBit, associate weakly. However, once the target proteins to which the NanoBiT subunits are tagged interact, the luminescent complex is assembled and easily detected with the luciferase assay. (B) All possible combinations of the established constructs, which were used to determine the optimal tag positions and usage of LgBit or SmBit. (C) HBx fused to LgBit at its C terminus (HBx-LgBit) and DDB1 fused to SmBit at its N terminus (SmBit-DDB1) made up the best pair, which produced the brightest relative luciferase signal after transfection. Halo-tag-SmBit was included as a negative control. Data represent the mean (n = 3) ± SD of triplicate experiments. ***P < .0001 (t test). (D) Z’ scores at each time point during the course of the assays. (E, F) Time course of luciferase activity in (C) HEK293T and (D) HepG2 cells after addition of 5 candidate compounds selected from the initial screening. Data represent the mean (n = 3) ± SD of triplicate experiments. LOP, loperamide; PMZ, pimozide; TRF, toremifene; VBL, vinblastine. (G, H) Dose dependency of the inhibitory effect of NTZ on the HBx–DDB1 interaction in (E) HEK293T and (F) HepG2 cells. Data show the percentage of control results for luciferase activity 30 minutes after addition of NTZ. Experiments were performed in triplicate (n = 3). (I) Cell toxicity was determined with different doses of NTZ. HepG2 or HepG2 cells that constitutively express Flag-HBx protein (HepG2Flag-HBx) cells were treated with the indicated doses of NTZ for 24 hours. Cell viability was determined through a cell counting assay. Data show the results of triplicate experiments (n = 3).
Identification of Inhibitors of the HBx–DDB1 Interaction
We tested an Food and Drug Administration (FDA)–approved drug library of 817 compounds to identify inhibitors of the HBx–DDB1 interaction. Luciferase activity was measured every 30 minutes for 2 hours following the addition of compounds and we calculated the inhibitory effects by comparison with dimethyl sulfoxide (DMSO) treatment after normalization to baseline signals. Before compound screening, we examined the screening quality by calculating the Z’ scores using all wells of the plate containing DMSO, confirming that the screening quality was satisfactory, with Z’ scores above 0.6 at every time point (Figure 1D). When we set the threshold to >40% inhibition compared with DMSO in the first screening, we identified 5 candidate drugs: toremifene, loperamide, pimozide, vinblastine, and NTZ. We confirmed that these 5 candidate compounds have not been reported to be pan-assay interference compounds,13 suggesting that the results obtained here were not due to nonspecific interference. While some compounds did not show significant effects in different cell lines, NTZ had the most reproducible and strongest inhibitory function among these compounds in HEK293T and HepG2 cells (Figure 1E and F). We confirmed that its inhibitory effects occur in a dose-dependent manner with an IC50 below 10 μM (Figure 1G and H). Because NTZ exhibited minimal cell toxicity in both normal HepG2 cells and HepG2 cells that constitutively express Flag-tagged HBx (Flag-HBx) protein (HepG2Flag-HBx), with a cytotoxic concentration (CC50) of 57 μM and 60 μM, respectively (Figure 1I), the inhibitory effects observed here were not due to cytotoxicity.
To further confirm the inhibitory effects of NTZ on the HBx–DDB1 interaction, we performed immunoprecipitation (IP)-Western blot analysis using Flag-HBx. Endogenous DDB1 binding to Flag-HBx was significantly decreased following the addition of NTZ in the HEK293T cell line (Figure 2A). Among the 5 candidate compounds, NTZ showed the strongest inhibitory effect on the HBx–DDB1 interaction (Figure 2B), consistent with the results of the NanoBiT assay. Similar results were observed in the HepG2 cell line in a dose-dependent manner (Figure 2C). To determine whether NTZ inhibits the HBx–DDB1 interaction directly or indirectly, we performed an in vitro Glutathione S-transferase (GST) pull-down assay using GST-DDB1 and untagged HBx recombinant protein. Because NTZ significantly decreased the binding of HBx to GST-DDB1 in vitro (Figure 3), it appears to directly inhibit the HBx–DDB1 interaction. Based on these results, we focused on the inhibitory effects of NTZ on the HBx–DDB1 interaction in subsequent analyses.
Figure 2.

NTZ inhibits the HBx–DDB1 interaction. (A) HEK293T cells were transfected with plasmid expressing Flag-HBx, and treated with NTZ (10 μM) or DMSO (as a control) for 24 hours. Inhibition of the HBx–DDB1 interaction was examined through IP using anti-FLAG antibody, followed by Western blot analysis. Five percent of the total cell lysate was used as “input.” Representative results from 3 independent experiments are shown. Summarized results (n = 3) of relative band intensity are shown below the panels. *P = 6.8 × 10–5 (t test). (B) Inhibitory effects of the 5 candidate compounds on the HBx–DDB1 interaction. HEK293T cells were transfected with a plasmid expressing Flag-HBx, and treated with the indicated compounds (10 μM) for 24 hours. Inhibition of the HBx–DDB1 interaction was examined by IP using anti-FLAG antibody, followed by Western blot analysis. Representative results of 3 independent experiments are shown. Summarized results of relative band intensity are shown below the panels (n = 3). *P = 1.2× 10–3; **P = 2.1 × 10–3; ***P = 1.5 × 10–4 (t test). (C) HepG2 cells were transfected with a plasmid expressing Flag-HBx, and treated with NTZ (10 or 20 μM) or DMSO (as a control) for 24 hours. IP and Western blotting were performed as described in panel A. Representative results from 3 independent experiments are shown. Summarized results of relative band intensities are shown below the panels (n = 3). NS, not significant; *P = 1.3 × 10–3; **P = 1.4 × 10–4 (t test).
Figure 3.

Inhibitory effects of NTZ on the HBx–DDB1 interaction in vitro. GST-tagged recombinant DDB1 protein and untagged recombinant HBx protein were mixed in vitro. NTZ or DMSO was added to the mixture and incubated for 20 minutes, followed by pull-down using anti-GST antibody and Western blotting to determine the levels of DDB1 and HBx in the pulled-down samples. Representative results of 3 independent experiments are shown. Summarized results of relative band intensities are shown below the panels (n = 3). NS, not significant; *P = 7.1 × 10–5 (t test).
NTZ Inhibits Degradation of Smc5/6 Induced by HBx Protein
Next, we tested whether NTZ inhibits the degradation of Smc5/6 by HBx protein, using HepG2Flag-HBx cells. We first confirmed that NTZ had no effect on the expression of Smc5 in HepG2 cells without HBx expression (Figure 4A). While Smc5 was significantly degraded in HepG2Flag-HBx, which expresses the HBx protein, consistent with previous reports (Figure 4B),10, 11 NTZ restored the expression of Smc5 in these cells at least partially (Figure 4B). Furthermore, the effect was observed in a dose-dependent manner (Figure 4B). These results suggested that NTZ inhibits the degradation of Smc5/6 by HBx protein, which may subsequently result in the suppression of viral transcription.
Figure 4.

NTZ reverses the degradation of Smc5 induced by HBx. (A) HepG2 and (B) HepG2FlagHBx cells, which stably express Flag-HBx protein, were treated with NTZ or DMSO for 48 hours. The expression level of Smc5 protein was determined by Western blotting. Representative results from 3 independent experiments are shown. Summarized results of relative band intensities (n = 3) are shown below the panels. NS, not significant; *P = 5.4 × 10–3; **P = 1.1 × 10–5 (t test).
NTZ Inhibits the Transcription of HBV RNA From cccDNA
Next, to investigate whether NTZ could reduce transcription of HBV cccDNA, we measured HBV RNA through quantitative polymerase chain reaction (qPCR) with in vitro models of HBV persistence. As in the in vitro model of HBV persistence, we used a recently reported minicircle DNA that mimics HBV cccDNA.14, 15 Because minicircle DNA does not contain antibiotic resistance markers or the bacterial backbone, it provides long-term expression of HBV RNAs and proteins, resembling HBV cccDNA. We used the construct minicircle HBV with Gaussia luciferase (mcHBV-Gluc) (HBV genotype C), in which the Gluc construct was inserted into the core region of the HBV genome.14 This construct secretes Gluc into the culture supernatant, reflecting transcriptional activity of the HBV pregenomic RNA (pgRNA) promoter.14 While NTZ did not significantly affect activity of the internal control, cytomegalovirus (CMV) promoter–driven Cypridina luciferase (Cluc) (CMV-Cluc), NTZ downregulated the relative Gluc activity in a dose-dependent manner (Figure 5A). To further confirm the specificity of this effect, we examined Gluc activity combined with measurement of intracellular mcHBV-Gluc DNA construct levels rather than Cluc activity, which also showed specific inhibition of Gluc activity from the mcHBV construct with NTZ (Figure 5B). Notably, the effects of NTZ were stronger than those of IFNα2a (Figure 5C). Moreover, NTZ had no effect on HBx-depletion mutant of mcHBV-Gluc (mcHBV-Gluc-ΔX), while it suppressed Gluc activity when mcHBV-Gluc-ΔX was transfected into HepG2Flag-HBx cells (Figure 5D). These results suggested that NTZ significantly suppresses pgRNA promoter activity of HBV cccDNA in an HBx-dependent manner.
Figure 5.

NTZ inhibits viral RNA transcription. (A) HepG2 cells were transfected with mcHBV-Gluc and a control plasmid, pCMV-Cluc. After 3 days, NTZ (10 or 20 μM) or DMSO (as a control) was added. Five days after transfection, the culture medium was collected for analysis of Gluc and Cluc activities. Relative Gluc levels were calculated after normalization to Cluc values. Data represent the mean (n = 3) + SD of triplicate experiments. *P = 2.0 × 10–3; **P = 9.1 × 10–4 (t test). (B) HepG2 cells were transfected with mcHBV-Gluc. Four days after transfection, NTZ (10 μM) or control DMSO treatment was initiated. Three days later, Gluc activity in the culture media was assayed (left panel). To confirm equal Gluc minicircle DNA levels, levels in the cells were determined through qPCR and relative levels are shown. Data represent the mean (n = 3) + SD of triplicate experiments. *P = 8.5 × 10–6; NS, not significant (t test). (C) HepG2 cells were transfected with mcHBV-Gluc. Four days after transfection, NTZ (10 μM), IFNα2a (1.0 × 103 or 5.0 × 103 units/mL), or control DMSO treatment was started. Two days later, relative Gluc activities in the culture media were calculated after normalization using intracellular Gluc DNA levels. Data represent the mean (n = 3) + SD of triplicate experiments. *P = 2.8 × 10–3; **P = .016; ***P = 1.0 × 10–3; ****P = 1.2 × 10–3; *****P = 8.2 × 10–4; NS, not significant (t test). (D) HepG2 or HepG2Flag-HBx cells were transfected with mcHBV-Gluc (WT) or mcHBV-Gluc-ΔX (ΔX) as indicated and a control plasmid, pCMV-Cluc. After 3 days, NTZ (10 μM) or DMSO (as a control) was added. Five days after transfection, the culture medium was collected for analysis of Gluc and Cluc activities. Relative Gluc levels were calculated after normalization to Cluc values. Data represent the mean (n = 3) + SD of triplicate experiments. *P = 1.1 × 10–3; NS, not significant (t test). (E, F) HepG2 cells were transfected with mcHBV-Gluc. Six days after transfection, NTZ (10 μM) or DMSO was added. Culture medium was renewed every 2 days. Twelve days after transfection, total RNA was extracted and qPCR was performed to measure (E) total HBV mRNA levels and (F) 3.5-kb mRNA levels. Data represent the mean + SD of triplicate experiments. *P = 2.7 × 10–4, **P = 1.2 × 10–2. (G, H) HepG2FlagHBx cells were transfected with mcHBV-Gluc. Three days after transfection, cells were passaged and treated with NTZ (10 μM) or DMSO for another 2 days. (G) Five days after transfection, Gluc activities in the culture media were assayed. (H) Simultaneously, total RNA was extracted and qPCR was performed to measure 3.5-kb mRNA levels. Data represent the (G) mean (n = 3) ± SD or (H) mean (n = 3) + SD of triplicate experiments. *P = 3.7 × 10–6; **P = 7.0 × 10–4 (t test). (I, J) HepG2 cells were transfected with minicircle HBV (genotype D). Five days after transfection, NTZ (at the indicated doses) or DMSO (as a control) was added. Culture medium was renewed every 2 days. Eleven days after transfection, total RNA was extracted and qPCR was performed to measure (I) total HBV mRNA and (J) 3.5-kb mRNA levels. Data represent the mean (n = 3) + SD of triplicate experiments. *P = 9.7 × 10–3; ∗∗P = 2.2 × 10–3; ∗∗∗P = 1.1 × 10–2; ∗∗∗∗P = 2.4 × 10–4 (t test).
Next, to directly examine viral transcription, we next determined the viral RNA levels by qPCR using HepG2 cells transfected with mcHBV-Gluc. As expected, NTZ significantly suppressed HBV-derived total messenger RNA (mRNA) levels (Figure 5E) and 3.5-kb mRNA (pgRNA plus pre-core mRNA) levels (Figure 5F), as determined using specific primers. Similar results were observed with constitutive HBx expression in HepG2Flag-HBx cells (Figure 5G and H).
To further confirm these results in other constructs and genotypes, we used minicircle HBV genotype D without the luciferase construct and determined viral RNA levels transcribed from this construct through qPCR. NTZ significantly reduced both HBV total and 3.5-kb mRNA levels from this construct in HepG2 cells (Figure 5I and J). In addition, we used HepAD cells, which support tetracycline-regulated HBV production.16 Two days after disabling pgRNA transcription from the genome following the addition of tetracycline, NTZ was added for 5 days. The levels of 3.5-kb mRNA, which was theoretically produced from accumulated viral cccDNAs, decreased significantly in NTZ-treated cells (Figure 6). Together, these results suggest that NTZ significantly suppresses viral transcription of HBV cccDNA.
Figure 6.
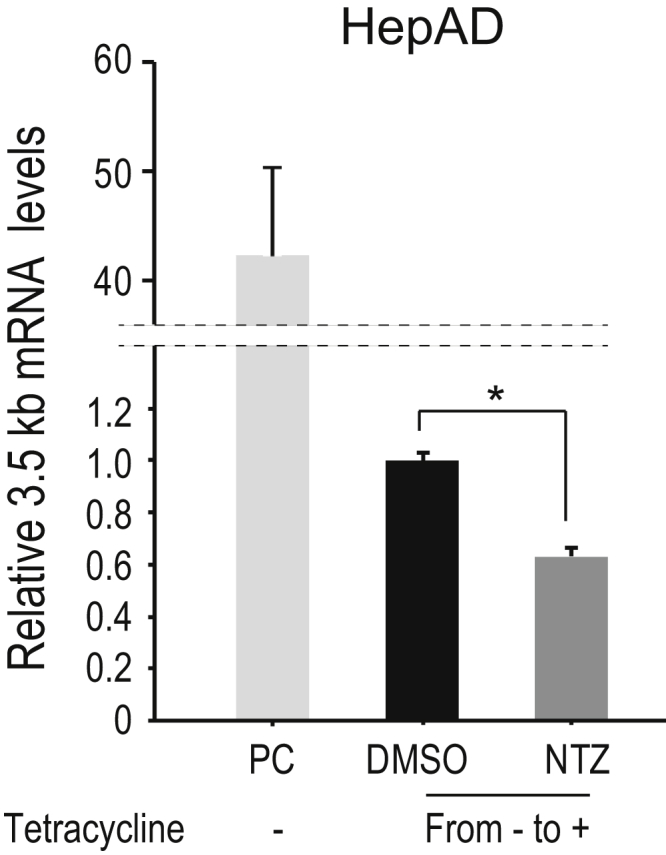
NTZ inhibits HBV mRNA transcription in HepAD38 cells. HepAD38 cells were cultured for 2 days after addition of tetracycline to stop HBV transcription from the genome. NTZ (10 μM) or DMSO was added, and culture media containing tetracycline was renewed every 2 days. After 5 days, qPCR was conducted to measure HBV 3.5-kb mRNA levels. Data represent the mean (n = 3) + SD of triplicate experiments. *P = 7.3 × 10–5 (t test). A positive control (PC) included HepAD38 cells cultured without tetracycline for a long time, allowing for constitutive HBV RNA transcription from the genome.
NTZ Decreases Viral Protein and cccDNA Levels
Next, we examined changes in viral protein levels caused by NTZ in an in vitro model of HBV persistence. Beginning 6 days after transfection with minicircle HBV genotype D, NTZ was added to the culture medium every 2 days. Twelve days after transfection, we observed that NTZ significantly reduced HB protein levels in a dose-dependent manner, along with significant restoration of Smc5 expression (Figure 7A).
Figure 7.

NTZ decreases viral protein and cccDNA levels. (A) HepG2 cells were transfected with minicircle HBV (genotype D). Four days after transfection, NTZ (at the indicated doses) or control DMSO was added. Culture medium was renewed every 2 days. Eleven days after transfection, cell lysates were subjected to Western blotting to determine the indicated protein levels. Representative results of 3 independent experiments are shown. Summarized data (n = 3) of relative band intensities are shown below the panels. NS, not significant: *P = 1.6 × 10–3; **P = 1.1 × 10–3; ***P = 1.6 × 10–3; ****P = 1.1 × 10–3 (t test). (B) HBV cccDNA levels were determined through ddPCR and Southern blotting. HepG2 cells were transfected with minicircle HBV (genotype D). Three days after transfection, NTZ (10 μM) or control DMSO was added. Five days after transfection, DNA extracted using the Hirt method was subjected to Southern blotting and ddPCR. For Southern blotting, control DNA from HBV transfected- and DMSO-treated cells was subjected to heat denaturing or heat denaturing with EcoRI digestion. Heat denaturing turns relaxed circular DNA and double-stranded linear DNA into single-stranded DNA, but does not affect cccDNA. EcoRI after heat denature cuts cccDNA into double-stranded linear DNA, but does not affect single-stranded DNA, confirming the integrities of the corresponding bands. Images of the cccDNA bands acquired with a longer exposure are shown in the lower panel. For ddPCR, the same samples subjected to Southern blotting were used for quantitation of cccDNA levels. Data represent the mean (n = 3) + SD of triplicate experiments. *P = 8.5 × 10–4 (t test). (C) Correlation between cccDNA levels in Southern blotting and ddPCR results. HepG2 cells were transfected with minicircle HBV (genotype D). Five days after transfection, DNA extracted using the Hirt method was subjected to Southern blotting and ddPCR with the indicated amounts of plasmid-safe digested DNA. The results of Southern blotting and ddPCR confirmed similar trends.
Because HBV viral proteins have been reported to support maintenance of HBV cccDNA,5 we next determined changes in HBV cccDNA levels after NTZ treatment. We specifically extracted cccDNA from cells using the Hirt extraction method,17, 18 and measured their cccDNA levels precisely through droplet digital PCR (ddPCR)19 and Southern blotting. The copy number of cccDNA decreased slightly but significantly with NTZ treatment (Figure 7B), which may be partly due to the decreased pgRNA levels as templates for the newly synthesized cccDNA as well as the destabilization of cccDNA due to decreased viral protein levels. To confirm that ddPCR has the specificity and accuracy to measure cccDNA levels, we visualized cccDNA levels via Southern blotting, which were generally consistent with those determined through ddPCR, confirming the validity of ddPCR measurement of cccDNA levels in this study (Figure 7B and C). These results suggest that NTZ silences transcription of cccDNA and decreases cccDNA levels slightly.
NTZ Inhibits Viral Protein, RNA, and DNA Production in a Natural HBV Infection Model
Finally, we tested the effects of NTZ in a more natural infection model. We used primary human hepatocytes, which provide a robust in vitro HBV infection model that can support the complete HBV life cycle.20 Following HBV (genotype C) infection, NTZ or DMSO treatment was initiated, and after 5 days of treatment, we quantified Smc5 protein, viral RNA, viral protein, and cccDNA levels. At the start of treatment, the baseline HBsAg and HBV-DNA levels in the culture media were comparable (Figure 8A and B), indicating that HBV infection levels were similar and viral product levels were similar in those cells. We confirmed that NTZ did not affect primary human hepatocyte viability at the doses used here (Figure 8C). Five days after NTZ treatment, degradation of Smc5 was significantly inhibited, although the degree of Smc5 degradation was not great, likely due to the low proportion of HBV-infected cells (Figure 8D). Viral RNA levels (total viral mRNA and 3.5-kb mRNA levels) were significantly downregulated with NTZ treatment (Figure 8E and F). HBs antigen levels in the culture media and intracellular HBV protein levels also decreased (Figure 8G and H), as did cccDNA levels in NTZ-treated cells (Figure 8I). Moreover, the degree of the viral product reduction was slightly increased, when NTZ treatment was started earlier after infection (Figure 9), indicating that NTZ inhibited the viral replication at early stages after infection. These results suggest that NTZ significantly reduces viral transcript and protein levels, leading to decreased cccDNA levels.
Figure 8.

Effects of NTZ in a natural HBV infection model. (A, B) Human primary hepatocytes were infected with HBV (genotype C). At 8 days after infection and before NTZ or DMSO were added, 1 mL of culture medium was collected for measurement of (A) HBsAg levels and (B) HBV-DNA) to confirm that the established infection load was equally distributed. Data represent the mean (n = 3) + SD of triplicate experiments. NS, not significant; NC, negative control without HBV infection. (C) Human primary hepatocytes were treated with the indicated doses of NTZ for 72 hours. Cell viability was determined through a cell counting assay. Data (n = 3) show the results of triplicate experiments. (D) Human primary hepatocytes were infected with HBV (genotype C). Eight days after infection, NTZ (20 μM) or DMSO was added. Culture medium was renewed every 24 hours. After treatment for 5 days, Smc5 protein levels in the cell lysates were determined through Western blotting. Representative results of 3 independent experiments are shown. Summarized results (n = 3) of relative band intensities are shown below the panels. *P = 7.3 × 10–5 (t test). (E, F) HBV infection and NTZ addition were performed as described in panel D. (E) Total viral mRNA and (F) 3.5-kb mRNA levels were determined by qPCR. Data represent the mean (n = 3) + SD of triplicate experiments. *P = 3.3 × 10–3; **P = 3.8 × 10–4 (t test). (G, H) After HBV infection and NTZ treatment as described in panel D, HBsAg levels in 1 mL of culture media were measured using (G) an enzyme-linked immunosorbent assay and (H) preS2 protein levels in cell lysates were determined through Western blotting. Data represent the mean (n = 3) + SD of triplicate experiments. *P = 3.4 × 10–3 (t test). (I) After HBV infection and NTZ treatment as described in panel D, cccDNA was isolated using plasmid-safe DNase treatment of total DNA extracted from the cells. cccDNA levels were determined via ddPCR. n.d., not detected in noninfected cells (NC). Data represent the mean (n = 3) + SD of triplicate experiments. *P = 6.7 × 10–4 (t test).
Figure 9.

Effects of NTZ in the early stage of natural HBV infection model. Human primary hepatocytes were infected with HBV (genotype C). Two days after infection, NTZ (20 μM) or DMSO was added. Culture medium was replaced every 24 hours. After treatment for 5 days, culture medium and cell lysates were collected for the following analyses. (A) Total viral mRNA and (B) 3.5-kb mRNA levels were determined by qPCR. Data represent the mean (n = 3) + SD of triplicate experiments. *P = 2.7 × 10–5; **P = 1.7 × 10–8 (t test). (C) HBs antigen levels in 1 mL of culture media were measured using an enzyme-linked immunosorbent assay. Data represent the mean (n = 3) + SD of triplicate experiments. *P = 3.6 × 10–4 (t test).
Discussion
We identified NTZ as a novel inhibitor of the HBx–DDB1 interaction through compound screening for drug repositioning. We showed that NTZ significantly inhibits the HBx–DDB1 interaction, resulting in suppression of viral transcription, protein expression, and cccDNA levels.
HBx binds to the DDB1 subunit of the DDB1-containing E3 ubiquitin ligase to target Smc5/6 for degradation. Smc5/6 was recently found to act as a restriction factor, selectively blocking transcription of HBV cccDNA.10, 11 Therefore, binding of HBx to DDB1 is a crucial factor for HBV viral transcription and subsequent viral protein production. Restoration of Smc5 protein levels with NTZ occurred only when HBx was expressed, suggesting that the effects of NTZ on Smc5 protein levels were HBx-dependent, most likely through inhibition of the HBx–DDB1 interaction.
We developed a novel and convenient screening method using split luciferase to identify inhibitors of the HBx–DDB1 interaction. Central to establishing an efficient screening system with this technology is identification of the optimal positions of the split luciferase fused to the target proteins. The optimal conditions identified here, HBx-LgBit and SmBit-DDB1, provide an optimal tool for screening inhibitors of the HBx–DDB1 interaction. Although we identified NTZ here through screening of a relatively small-scale compound library, screening a much larger compound library would be easy, and could result in the identification of novel compounds that may inhibit the HBx–DDB1 interaction more efficiently in the future.
NTZ [2-acetyloxy-N-(5-nitro-2-thiazolyl) benzamide] is an orally available thiazolide anti-infective agent, originally approved for the treatment of enteritis induced by Cryptosporidium parvum and Giardia lamblia in children and adults.21 Recently, NTZ has been reported to exhibit antiviral effects against various viruses, including influenza virus,22 rotavirus,21 norovirus,23 Japanese encephalitis virus,24 rubella virus,25 Zika virus,26 hepatitis C virus,27 and HBV.28 However, in most cases, including that of HBV, the molecular mechanisms of these effects have not been determined, except that blocking the viral NS2B–NS3 interaction and inhibition of viral hemagglutinin maturation have been proposed as possible mechanisms in the case of Zika26 and influenza22 viruses, respectively. It remains to be elucidated whether the molecular mechanism identified here, through which NTZ inhibits the interaction between viral and host cellular proteins, can be applied to other viral types.
Drug repositioning is an efficient strategy to bring laboratory discovery into the clinical setting, because repositioned drugs have already passed a large number of toxicity and other tests for clinical application, especially in the current situation in which more than 90% of drugs fail during development.29 Because we identified NTZ through screening of an FDA-approved compound library, and NTZ is already in clinical use for enteritis, NTZ may be applied to HBV cases without costly and time-consuming basic tests. Although the effects of NTZ against HBV observed in this study were modest but significant, further chemical modification or in silico analyses modifying the NTZ backbone may improve its antiviral effects. Although additional studies are necessary, NTZ, which targets the interaction between HBx and host protein, may provide a new strategy to work toward a functional cure for HBV.
Methods
Cells
The human embryonic kidney cell line, HEK293T, and human hepatocellular carcinoma cell line, HepG2, were purchased from the American Type Culture Collection (Manassas, VA) and cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum. HepAD38 cells, which express pgRNA under the control of the inducible tetracycline promoter, were kindly provided by Professor C. Seeger (Institute for Cancer Research, Fox Chase Cancer Center, Philadelphia, PA).16 The cells were maintained in Ham's F-10/DMEM culture medium, which was supplemented with 10% doxycycline-free fetal bovine serum with or without 1 μg/mL doxycycline to maintain repression of HBV expression or to maintain HBV replication, respectively. Primary human hepatocytes freshly isolated using the collagenase perfusion method from chimeric uPA/SCID mice with humanized livers20 were obtained from Phoenix Bio (Hiroshima, Japan). The purity of human hepatocytes was >95%. The cells were seeded onto a type I collagen-coated plate and maintained in dHCGM (DMEM with 10% fetal bovine serum, 20 mM HEPES, 44 mM NaHCO3, 100 U/mL penicillin, 100 μg/mL streptomycin, 15 μg/mL L-proline, 0.25 μg/mL insulin, 5 × 10–8 M dexamethasone, 5 ng/mL epidermal growth factor, 0.1 mM ascorbic acid-2-phosphate, and 2% DMSO).20 These cells were able to support long-term replication of HBV infection in vitro. All cells were incubated at 37°C in 20% O2 and 5% CO2.
Plasmids
TL7 plasmid,30 which expresses HBV pgRNA (genotype D: GenBank accession number; V01460), was kindly provided by Professor Loeb (McArdle Laboratory for Cancer Research, University of Wisconsin School of Medicine and Public Health, Madison, WI).
The Flexi Vector System (Promega) was used for cloning of split luciferase expression constructs according to the manufacturer’s instructions. Briefly, we amplified the cDNAs of HBx and DDB1 via PCR using the TL7 plasmid and Halo-tagged DDB1 plasmid (Promega) as templates, respectively. Primers used were as follows: HBx, 5′-GCGATCGCCATGGCTGCTAGGCTGTGCTGC-3′ (sense) and 5′-GTTTAAACGGCAGAGGTGAAAAAGTTGCA-3′ (antisense); and DDB1, 5′-ATGTCGTACAACTACGTGGTA-3′ (sense) and 5′-ATGGATCCGAGTTAGCTCCTC-3′ (antisense). Amplified PCR products were purified using the QIAquick PCR Purification Kit (Qiagen, Hilden, Germany). After digesting the terminal sequences with SgfI and PmeI, the cDNAs were ligated into the pF4A CMV Flexi Vector at the SgfI and PmeI sites. Then, pF4A CMV Flexi Vectors containing HBx or DDB1 cDNAs were mixed with acceptor Flexi Vectors and the cDNAs were subcloned into acceptor vectors using restriction enzymes. For N-terminal fusion, the pFN33K LgBit TK-neo and pFN35K SmBit TK-neo Flexi Vectors were used, while the pFC34K LgBit TK-neo and pFC36K SmBit TK-neo Flexi Vectors were used for C-terminal fusion. The NanoBiT Negative Control Vector encoding HaloTag-SmBit was used as a negative control.
The Flag-HBx–expressing plasmid was constructed by subcloning the Flag-HBx sequences using an Infusion method (Clontech, Mountain View, CA). After amplifying the complementary DNA (cDNA) using TL7 as a template with primers containing FLAG sequences, the PCR products were cloned into a pCDH-CMV-puro lentivirus vector (System Biosciences, Palo Alto, CA). The primer used was 5′-ATTTAAATCGGATCCACCATGGACTACAAAGACGATGACG-3′ (sense) and 5′-GATCGCAGATCCTTCTTAGGCAGAGGTGAAAAAGTTGC-3′ (antisense).
The pCMV-Cluc 2 Control Plasmid, which constitutively expresses the luciferase secreted by the ostracod Cypridina noctiluca (Cluc) under the control of the CMV promoter, was used as a transfection control for measurement of Gluc in HBV minicircles and was purchased from New England Biolabs (Ipswich, MA).
Minicircle HBV cccDNA with a Gluc reporter–containing plasmid (pre–mcHBV-Gluc) was kindly provided by Prof Su (Lineberger Comprehensive Cancer Center, Department of Microbiology and Immunology, University of North Carolina at Chapel Hill, Chapel Hill, NC). This plasmid harbors sequences of HBV genotype C.14 pre-HBV circle genotype D 15 was kindly provided by Dr. Gao (Roche Innovation Center Shanghai, Shanghai, China).
To construct pre-mcHBV-Gluc-ΔX, a QuikChange II XL Site-Directed Mutagenesis Kit (Agilent, Santa Clara, CA) was used to change the eighth amino acid in the X protein sequence to a stop codon, as Gln (CAA) to stop (TAA) in the pre-mcHBV-Gluc plasmid, which does not affect other viral protein sequences.14 Mutant strand synthesis was achieved using following primers: 5′-CGCAGGATCCAGTTAGCAGCACACCCGAG-3′ (sense) and 5′-CTCGGGTGTGCTGCTAACTGGATCCTGCG-3′ (antisense).
Compounds
Screen-Well FDA-approved drug library V2 version 1.0 was purchased from Enzo Life Sciences (Farmingdale, NY). Nitazoxanide (NTZ) was purchased from Toronto Research Chemicals Inc. (Toronto, Ontario, Canada) and IFNα2a was from Sigma-Aldrich (St. Louis, MO).
Compound Screening
The NanoBiT Protein:Protein Interaction system (Promega) was used to establish a screening assay for inhibition of the HBx–DDB1 interaction. Briefly, we transiently transfected 1 μg of HBx and DDB1 with split luciferases into HEK293T or HepG2 cells, at 80% confluence in a 10-cm dish. After 10 hours, cells were reseeded onto a 96-well plate with 5 × 104 cells/well and 50 μL medium/well. After 10 hours of incubation at 37°C, 12.5 μL of Nano-Glo live Cell Assay reagent was added to each well, and baseline luminescence was measured using a luminometer. Immediately after this initial measurement, compounds were added to a final concentration of 10 μM, and luminescence values were measured every 30 minutes using the GloMax Detection System (Promega). To calculate Z’-scores, used as a screening quality indicator, blank wells containing only media were set up with luciferase assay reagents in the first and last columns to determine the Average 0 and standard deviation (SD) 0. Then, transfected cells were seeded into all other columns with media, and DMSO was added followed by luciferase reagents to determine the Average DMSO and SD DMSO. The Z’ score was defined as 1 − ((3 × SD DMSO) + (3 × SD 0)/ (Average DMSO – Average 0)), and a screening system was considered acceptable if it was over 0.5.31
HBV Minicircle Extraction
Minicircle DNA was produced as previously described32 from HBV plasmids for minicircle DNA, as outlined previously.14, 15 Briefly, on day 1, parental plasmids were transformed into a ZYCY10P3S2T Escherichia coli minicircle-producing strain (System Biosciences), which expresses inducible ƟC31 integrase and I-SceI endonuclease to eliminate the bacterial plasmid DNA backbone. On day 2, we inoculated cells from 1 transformed colony to 5 mL of Terrific Broth (pH 7.0) with kanamycin (50 μg/mL), which was incubated at 37°C with shaking at 250 rpm. Later the same day, we amplified the bacteria by inoculating 50 μL of culture per 200 mL of Terrific Broth containing kanamycin (50 μg/mL) and continued incubation for 16 hours. On day 3, minicircle induction mixture (200-mL Luria-Bertani broth, 8-mL 1 N sodium hydroxide, and 0.2-mL 20% l-arabinose) was combined with the 200-mL overnight culture, and incubated at 32°C with shaking at 250 rpm for an additional 5 hours. Then, the minicircle was isolated using the Qiagen plasmid purification kit according to the manufacturer’s instructions with minor modifications. For every 200-mL overnight culture, we used 50 mL each of buffers P1, P2, and P3 to ensure complete resuspension and lysis of the bacteria and a high yield of the minicircle DNA vector. Agarose gel electrophoresis was used to confirm the absence of contamination with genomic and parental DNA.
Transfection and Lentiviral Transduction
Transient transfections were performed using Effectene Transfection Reagent (Qiagen). mcHBV-Gluc or mcHBV-Gluc-ΔX was co-transfected with a control plasmid, pCMV-Cluc, to normalize Gluc activities as indicated. To generate polyclonal cells with stable Flag-HBx expression, the Lentivirus Packaging System (System Biosciences) was used according to the manufacturer’s instructions. Briefly, 1.0 μg of Flag-HBx overexpressing vector and 5.0 μg of pPACKH1 packaging plasmid mix were transfected into HEK293T cells. After 24 hours, the collected culture media was mixed with one-fifth its volume of PEG-it Reagent (System Biosciences) and incubated overnight at 4°C to concentrate the viruses. The centrifuged pellet was resuspended in 1× phosphate-buffered saline and aliquots were stored at –80°C. The viruses were transduced into HEK293T and HepG2 cells using polybrene (Santa Cruz Biotechnology, Dallas, TX), followed by selection with 6 mg/mL puromycin to obtain polyclonal cells stably expressing Flag-HBx protein.
Western Blotting Analyses and Antibodies
Western blotting was performed as previously described.33 Briefly, lysate samples were separated on a 10%–20% gradient polyacrylamide gel by sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis following electrical transfer to polyvinylidene difluoride membranes (Merck Millipore, Burlington, MA). After blocking with 5% dry milk, membranes were probed overnight at 4°C with the appropriate primary antibodies diluted in Immunoshot Reagent 1 (Cosmo Bio Co, Tokyo, Japan). Horseradish peroxidase–conjugated corresponding secondary antibodies (GE Healthcare, Little Chalfont, United Kingdom) were subsequently applied. Bound antibodies were detected using Immunostar LD reagents (Wako Pure Chemical Industries, Osaka, Japan). The following antibodies were used: Smc5 (#ab18038, 1:10,000 [Abcam, Cambridge, United Kingdom]), β-actin (#A1978, 1:10,000 [Sigma-Aldrich, St. Louis, MO] or #5125, 1:10,000 [Cell Signaling Technology, Danvers, MA]), Hepatitis B preS2 (#NB100-93580, 1:1000 [Novus Biologicals, Littleton, CO]), DDB1 and GST tag ((#5428, 1:1000 and #2625S, 1:1000, respectively [Cell Signaling Technology]), and Flag (DYKDDDDK) tag (#018-22381, 1:1000 [Wako Pure Chemical Industries]). Protein band intensities were quantified through densitometric analysis using ImageJ software version 1.49v (National Institutes of Health, Bethesda, MD).
Immunoprecipitation
Anti-DYKDDDDK tag antibody magnetic beads (Wako Pure Chemical Industries) were used for IP. In brief, cell lysate samples were mixed with beads that had been prewashed in IP buffer (50-mM Tris-HCl, pH 7.5, 150-mM NaCl, 0.1% NP-40, 1-mM EDTA, pH 8.0, 0.25% gelatin, and 0.02% sodium azide), and incubated with rotation overnight at 4°C. The beads were separated with a magnetic separator and mixed with 4× SDS sample buffer (0.25 mol/L Tris-HCl, 8% SDS, 40% glycerol, and 0.02% bromophenol blue, pH 6.8), then boiled in a heat block at 95°C for 3 minutes to elute the proteins. For detection of the DYKDDDK tag fusion protein, Western blotting was performed as described previously. TrueBlot anti-mouse IgG horseradish peroxidase (1:1000; Rockland, Limerick, PA) was used as the secondary antibody to avoid interference of immunoprecipitated immunoglobulin heavy and light chains.
In Vitro Pull-Down Assay
GST-DDB1 protein (Abcam) and untagged HBx recombinant protein (Abcam) were used for the in vitro pull-down assay. After mixing the proteins in IP buffer with NTZ or DMSO for 20 minutes, GST-DDB1 and bound HBx proteins were pulled down with anti-GST antibody (#2625S, 1:200; Cell Signaling Technology) and protein A/G agarose (Santa Cruz Biotechnology). Western blotting was performed to visualize the precipitated proteins.
Measurement of Gluc and Cluc Activities
The Gluc signal from mcHBV-Gluc arises from pgRNA, providing a surrogate for cccDNA activity, as it is secreted from cells into the media. Gluc activity in aliquots of media was measured using the reagent for measurement of Renilla luciferase activity in the Dual Reporter Assay system (Promega) because Gaussia and Renilla catalyze their light-producing reactions using the same substrate. To measure Gluc, 10 μL of culture medium was added to 50 μL Renilla luciferase assay reagent (Promega), and luminescence was measured with a luminometer (Lumat LB 9507; Berthold Technologies, Bad Wildbad, Germany) with a 10-second integration. For Cluc, 1.0 μL of culture medium was added to 10 μL Cluc assay solution (BioLux Cypridina Luciferase Assay Kit, New England BioLabs), and luminescence was measured. The relative Gluc activity was normalized to Cluc unless otherwise specified. When using the endpoint mcHBV DNA copy number for normalization, the quantitation of mcHBV-Gluc DNA was conducted through qPCR using primers for the Gluc lesion as follows: 5′-ATGGTGAATGGCGTGAAG-3′ (sense) and 5′-TAGGTGTCATCGCCGCCAGC-3′ (antisense).34
Cytotoxicity Test
Cell Counting Kit-8 (Dojindo Molecular Technologies, Inc., Kumamoto, Japan) was used to test cytotoxicity according to the manufacturer’s instructions. Briefly, 100 μL of HepG2 or HepG2Flag-HBx cell suspension (5000 cells/well) was dispensed into a 96-well plate and incubated for 24 hours at 37°C under 5% CO2. Then, the culture medium was renewed with or without NTZ at the following concentrations: 1.0, 5.0, 10, 20, 40, 60, 80, 100, and 200 μM. After 24 hours, 10 μL of CCK-8 solution was added to each well, the plate was incubated for 3 hours, and the absorbance at 450 nm was measured using a microplate reader (Thermo Fisher Scientific, Waltham, MA). Cell viability was calculated according to the formula: [(ANTZ – Ablank)/(Acontrol – Ablank)] × 100, where ANTZ is the absorbance at 450 nm of wells with NTZ; Ablank is the absorbance at 450 nm of wells without cells; and Acontrol is the absorbance at 450 nm of wells without NTZ.
For primary human hepatocytes, cells grown in a 48-well plate were used. The cells were incubated for 3 days with or without NTZ at the following concentrations: 1.0, 5.0, 10, 20, 50, 80, and 100 μM. The medium was renewed every 24 hours. Cell viability was calculated using CCK-8 solution as described previously.
HBV Infection in Primary Human Hepatocytes
HBV infection of primary human hepatocytes has been reported previously.20 Briefly, the cells were inoculated with serum from HBV genotype C-infected humanized liver chimeric mice (PhoenixBio Co) at 5 viral genome equivalents per cell in dHCGM containing 4% PEG 8000 for 24 hours at 37°C. Then, the cells were washed with phosphate-buffered saline and cultured in dHCGM. The culture medium was renewed at 2 and 7 days after infection when compounds were added at day 8 after infection. NTZ (20 μM) or DMSO treatment was initiated 2 and 8 days after infection. The culture medium containing NTZ or DMSO was renewed every 24 hours and collected after 5 days of treatment.
Quantification of HBV RNA Levels
Total RNA was extracted from cells using RNeasy with DNase treatment (Qiagen). The extracted RNA was then reverse transcribed using SuperScript III First-Strand Synthesis SuperMix (Thermo Fisher Scientific). qPCR was carried out with THUNDERBIRD SYBR qPCR Mix (Toyobo Co, Osaka, Japan) using StepOnePlus real-time PCR system (Life Technologies, Carlsbad, CA). Concentrations of HBV 3.5-kb mRNA and total HBV mRNA were determined relative to the mRNA of β-actin or glyceraldehyde 3-phosphate dehydrogenase using the following primers: HBV 3.5-kb mRNA, 5′-GAGTGTGGATTCGCACTCC-3′ (sense) and 5′-GAGGCGAGGGAGTTCTTCT-3′ (antisense), which amplified the region from nucleotide (nt) 2268 to 2390 in the genotype D (HE815465.1) and genotype C (AB246345) HBV sequences; total HBV mRNA, 5′-TCACCAGCACCATGCAAC-3′ (sense for genotype C) or 5′-GCACCAGCACCATGCAAC-3′ (sense for genotype D) and 5′-AAGCCACCCAAGGCACAG-3′ (antisense), which amplified the region from nt 1803 to 1894 in genotype D (HE815465.1) and genotype C (AB246345) HBV sequences, covering all HBV transcripts (3.5-, 2.4-, 2.1-, and 0.7-kb mRNAs); β-actin, 5′-CTGTGCTACGTCGCCCTGG-3′ (sense) and 5′-GCCACAGGACTCCATGCCC-3′ (antisense); and glyceraldehyde 3-phosphate dehydrogenase, 5′-AGGGCTGCTTTTAACTCTGGT-3′ (sense) and 5′-CCCCACTTGATTTTGGAGGGA-3′ (antisense).
Quantification of HBV DNA Levels
HBV DNA levels in the culture media were measured using qPCR in a clinical laboratory, SRL (Tokyo, Japan).
Isolation of HBV cccDNA
HepG2 cells with and without transfection of mcHBV genotype D were harvested and HBV DNA including cccDNA was extracted using the Hirt method17, 18 to obtain protein-free DNA. Briefly, SDS is first applied to break down the lipid membranes and viral capsids and release all viral nucleic acids. A high concentration of salt is then added to precipitate high-molecular-weight cellular chromatin and protein covalently bonded to DNA in SDS-protein complexes. HBV cccDNA, a protein-free DNA, is one of the major viral products in the supernatant, which can be purified with an organic phenol method. Another protein free viral DNA, relaxed circular DNA, which is a precursor of cccDNA, can be extracted simultaneously. The concentration of extracted DNA was adjusted to 0.5 μg/μL using double-distilled water for ddPCR.
For primary human hepatocytes, cccDNA was isolated using the QIAamp DNA Mini Kit (Qiagen) and treated with plasmid-safe DNase (PSAD) (Epicentre, Madison, WI). Briefly, total cellular DNA was extracted with the QIAamp DNA Mini Kit according to the manufacturer’s instructions. Then, 200 ng of extracted DNA was mixed with 2 μL of 10× PSAD buffer, 1 μL of PSAD, and 0.8-μL 25-mM adenosine triphosphate, and double-distilled water was added to reach 20 μL. This mixture was incubated at 37°C for 2 hours followed by 70°C for 30 minutes to inactivate digestion.
Quantification of cccDNA Levels Through ddPCR
ddPCR was performed with the QX200 Droplet Digital PCR system (Bio-Rad, Hercules, CA) according to the manufacturer’s instructions. Briefly, 20 μL of ddPCR mixture (1-μL template DNA, 10-μL 2× ddPCR Supermix for Probe [no dUTP], 900-nM forward and reverse primers, and 250 nM of the corresponding probe) and 70 μL of droplet generation oil were added to the DG8 cartridge to produce droplets. Next, the droplets were carefully transferred to a PCR plate for amplification using a T100 Thermal Cycler (Bio-Rad) with a cycle starting at 95°C, followed by 35 cycles of 94°C for 30 seconds and 57°C for 60 second, and a final cycle of 98°C for 10 minutes. The droplets were analyzed on a QX200 droplet reader using QuantaSoft software for absolute counting of template DNA. As previously described,19 cccDNA primers were designed for specific amplification of cccDNA, including a forward primer, nt 1561–1579 5′-CTTCTCATCTGCCGGACC-3′ and reverse primer, nt 1865–1883 5′-CACAGCTTGGAGGCTTGA-3′. To detect PCR amplification signals, an HBV cccDNA-specific probe was used, which covers the HBV DNA minus-strand gap (nt 1838–1861 FAM-5′-AGGCTGTAGGCATAAATTGGTCT-3′-BHQ).
Southern Blotting
For Southern blotting, 10 μg of DNA was subjected to 1.2% agarose gel electrophoresis at 25 V overnight. Separated DNA on the gel was transferred to a Hybond-N+ membrane (GE Healthcare) in 20× saline sodium citrate (SSC) transfer buffer overnight. After transfer, the DNA was crosslinked using 120 mJ/cm2 UV in a UV crosslinker (Stratalinker, Stratagene, La Jolla, CA). To generate a full-length HBV RNA probe for hybridization, the DIG Northern starter kit (Roche, Basel, Switzerland) was used according to the manufacturer’s recommendations.
The template DNA was prepared from mcHBV genotype D plasmid through PCR using the following primers with the sequences of the T7 RNA polymerase promoter: 5′-TAATACGACTCACTATAGGGGGTGCGCAGACCAATTTATGC-3′ (sense) and 5′-GCACCATGCAACTTTTTCAC-3′ (antisense). Amplification of the correct product was confirmed via gel electrophoresis. The RNA probe was synthesized through an in vitro transcription reaction with digoxigenin-11-UTP using a labeling mixture. The probe was hybridized to the membrane using DIG Easy Hyb (Roche) and detected using the DIG Wash and Block Buffer Set (Roche) according to the manufacturer’s recommendations. Heat-denatured Hirt-extracted DNA that had been incubated at 85°C for 5 minutes and subsequently heat-denatured DNAs subjected to EcoRI (Thermo Fisher Scientific) digestion were applied for identification of the bands obtained.
Measurement of HBs Antigen Levels in Culture Media
HBs antigen levels in culture media were measured using an enzyme-linked immunosorbent assay at a clinical laboratory (SRL).
Statistical Analysis
All statistical analyses were conducted using the program R version 3.3.2 (R Foundation for Statistical Computing, Vienna, Austria). Continuous variables were reported as mean ± SD. The Welch t test was used for group comparisons of continuous variables. P values of <.05 were considered statistically significant.
Footnotes
Conflicts of interest The authors disclose no conflicts.
Funding This work was supported by Grants-in-Aid from the Ministry of Education, Culture, Sports, Science and Technology, Japan (#16H05149 [to Motoyuki Otsuka], #16KT0109 [to Motoyuki Otsuka], #18H05024 [to Motoyuki Otsuka], and #15H04807 [to Kazuhiko Koike]), by the Research Program on Hepatitis from Japan Agency for Medical Research and Development, AMED (#JP18fk0210214 [to Motoyuki Otsuka]), and by the Project for Cancer Research And Therapeutic Evolution (P-CREATE) from AMED (#JP19cm0106602 [to Motoyuki Otsuka]).
References
- 1.Tang L.S.Y., Covert E., Wilson E., Kottilil S. Chronic hepatitis B infection. JAMA. 2018;319:1802–1813. doi: 10.1001/jama.2018.3795. [DOI] [PubMed] [Google Scholar]
- 2.World Health Organization . WHO Press; Geneva, Switzerland: 2017. Global Hepatitis Report. [Google Scholar]
- 3.Fung J., Cheung K.S., Wong D.K., Mak L.Y., To W.P., Seto W.K., Lai C.L., Yuen M.F. Long Term outcomes and predictive scores for hepatocellular carcinoma and HBsAg seroclearance after HBeAg seroclearance. Hepatology. 2018;68:462–472. doi: 10.1002/hep.29874. [DOI] [PubMed] [Google Scholar]
- 4.Kim G.A., Lim Y.S., An J., Lee D., Shim J.H., Kim K.M., Lee H.C., Chung Y.H., Lee Y.S., Suh D.J. HBsAg seroclearance after nucleoside analogue therapy in patients with chronic hepatitis B: Clinical outcomes and durability. Gut. 2014;63:1325–1332. doi: 10.1136/gutjnl-2013-305517. [DOI] [PubMed] [Google Scholar]
- 5.Levrero M., Pollicino T., Petersen J., Belloni L., Raimondo G., Dandri M. Control of cccDNA function in hepatitis B virus infection. J Hepatol. 2009;51:581–592. doi: 10.1016/j.jhep.2009.05.022. [DOI] [PubMed] [Google Scholar]
- 6.Keasler V.V., Hodgson A.J., Madden C.R., Slagle B.L. Enhancement of hepatitis B virus replication by the regulatory X protein in vitro and in vivo. J Virol. 2007;81:2656–2662. doi: 10.1128/JVI.02020-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lucifora J., Arzberger S., Durantel D., Belloni L., Strubin M., Levrero M., Zoulim F., Hantz O., Protzer U. Hepatitis B virus X protein is essential to initiate and maintain virus replication after infection. J Hepatol. 2011;55:996–1003. doi: 10.1016/j.jhep.2011.02.015. [DOI] [PubMed] [Google Scholar]
- 8.Sitterlin D., Lee T.H., Prigent S., Tiollais P., Butel J.S., Transy C. Interaction of the UV-damaged DNA-binding protein with hepatitis B virus X protein is conserved among mammalian hepadnaviruses and restricted to transactivation-proficient X-insertion mutants. J Virol. 1997;71:6194–6199. doi: 10.1128/jvi.71.8.6194-6199.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Breugel P.C., Robert E.I., Mueller H., Decorsière A., Zoulim F., Hantz O., Strubin M. Hepatitis B virus X protein stimulates gene expression selectively from extrachromosomal DNA templates. Hepatology. 2012;56:2116–2124. doi: 10.1002/hep.25928. [DOI] [PubMed] [Google Scholar]
- 10.Decorsière A., Mueller H., van Breugel P.C., Abdul F., Gerossier L., Beran R.K., Livingston C.M., Niu C., Fletcher S.P., Hantz O., Strubin M. Hepatitis B virus X protein identifies the Smc5/6 complex as a host restriction factor. Nature. 2016;531:386–389. doi: 10.1038/nature17170. [DOI] [PubMed] [Google Scholar]
- 11.Murphy C.M., Xu Y., Li F., Nio K., Reszka-Blanco N., Li X., Wu Y., Yu Y., Xiong Y., Su L. Hepatitis B virus X protein promotes degradation of SMC5/6 to enhance HBV replication. Cell Rep. 2016;16:2846–2854. doi: 10.1016/j.celrep.2016.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dixon A.S., Schwinn M.K., Hall M.P., Zimmerman K., Otto P., Lubben T.H., Butler B.L., Binkowski B.F., Machleidt T., Kirkland T.A., Wood M.G., Eggers C.T., Encell L.P., Wood K.V. NanoLuc complementation reporter optimized for accurate measurement of protein interactions in cells. ACS Chem Biol. 2016;11:400–408. doi: 10.1021/acschembio.5b00753. [DOI] [PubMed] [Google Scholar]
- 13.Dahlin J.L., Nissink J.W., Strasser J.M., Francis S., Higgins L., Zhou H., Zhang Z., Walters M.A. PAINS in the assay: chemical mechanisms of assay interference and promiscuous enzymatic inhibition observed during a sulfhydryl-scavenging HTS. J Med Chem. 2015;58:2091–2113. doi: 10.1021/jm5019093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li F., Cheng L., Murphy C.M., Reszka-Blanco N.J., Wu Y., Chi L., Hu J., Su L. Minicircle HBV cccDNA with a Gaussia luciferase reporter for investigating HBV cccDNA biology and developing cccDNA-targeting drugs. Sci Rep. 2016;6:36483. doi: 10.1038/srep36483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yan Z., Zeng J., Yu Y., Xiang K., Hu H., Zhou X., Gu L., Wang L., Zhao J., Young J.A.T., HBVcircle Gao L. A novel tool to investigate hepatitis B virus covalently closed circular DNA. J Hepatol. 2017;66:1149–1157. doi: 10.1016/j.jhep.2017.02.004. [DOI] [PubMed] [Google Scholar]
- 16.Ladner S.K., Otto M.J., Barker C.S., Zaifert K., Wang G.H., Guo J.T., Seeger C., King R.W. Inducible expression of human hepatitis b virus (HBV) in stably transfected hepatoblastoma cells: a novel system for screening potential inhibitors of HBV replication. Antimicrob Agents Chemother. 1997;41:1715–1720. doi: 10.1128/aac.41.8.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hirt B. Selective extraction of polyoma DNA from infected mouse cell cultures. J Mol Biol. 1967;26:365–369. doi: 10.1016/0022-2836(67)90307-5. [DOI] [PubMed] [Google Scholar]
- 18.Cai D., Nie H., Yan R., Guo J.-T., Block T.M., Guo H. A Southern blot assay for detection of hepatitis B virus covalently closed circular DNA from cell cultures. Methods Mol Biol. 2013;1030:151–161. doi: 10.1007/978-1-62703-484-5_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mu D., Yan L., Tang H., Liao Y. A sensitive and accurate quantification method for the detection of hepatitis B virus covalently closed circular DNA by the application of a droplet digital polymerase chain reaction amplification system. Biotechnol Lett. 2015;37:2063–2073. doi: 10.1007/s10529-015-1890-5. [DOI] [PubMed] [Google Scholar]
- 20.Ishida Y., Yamasaki C., Yanagi A., Yoshizane Y., Fujikawa K., Watashi K., Abe H., Wakita T., Hayes C.N., Chayama K., Tateno C. Novel robust in vitro hepatitis B virus infection model using fresh human hepatocytes isolated from humanized mice. Am J Pathol. 2015;185:1275–1285. doi: 10.1016/j.ajpath.2015.01.028. [DOI] [PubMed] [Google Scholar]
- 21.Rossignol J.F., Abu-Zekry M., Hussein A., Santoro M.G. Effect of nitazoxanide for treatment of severe rotavirus diarrhoea: randomised double-blind placebo-controlled trial. Lancet. 2006;368:124–129. doi: 10.1016/S0140-6736(06)68852-1. [DOI] [PubMed] [Google Scholar]
- 22.Rossignol J.F., La Frazia S., Chiappa L., Ciucci A., Santoro M.G. Thiazolides, a new class of anti-influenza molecules targeting viral hemagglutinin at the post-translational level. J Biol Chem. 2009;284:29798–29808. doi: 10.1074/jbc.M109.029470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rossignol J.F., El-Gohary Y.M. Nitazoxanide in the treatment of viral gastroenteritis: A randomized double-blind placebo-controlled clinical trial. Aliment Pharmacol Ther. 2006;24:1423–1430. doi: 10.1111/j.1365-2036.2006.03128.x. [DOI] [PubMed] [Google Scholar]
- 24.Shi Z., Wei J., Deng X., Li S., Qiu Y., Shao D., Li B., Zhang K., Xue F., Wang X., Ma Z. Nitazoxanide inhibits the replication of Japanese encephalitis virus in cultured cells and in a mouse model. Virol J. 2014;11:10. doi: 10.1186/1743-422X-11-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perelygina L., Hautala T., Seppänen M., Adebayo A., Sullivan K.E., Icenogle J. Inhibition of rubella virus replication by the broad-spectrum drug nitazoxanide in cell culture and in a patient with a primary immune deficiency. Antiviral Res. 2017;147:58–66. doi: 10.1016/j.antiviral.2017.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Z., Brecher M., Deng Y.Q., Zhang J., Sakamuru S., Liu B., Huang R., Koetzner C.A., Allen C.A., Jones S.A., Chen H., Zhang N.N., Tian M., Gao F., Lin Q., Banavali N., Zhou J., Boles N., Xia M., Kramer L.D., Qin C.F., Li H. Existing drugs as broad-spectrum and potent inhibitors for Zika virus by targeting NS2B-NS3 interaction. Cell Res. 2017;27:1046–1064. doi: 10.1038/cr.2017.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rossignol J.-F., Elfert A., El-Gohary Y., Keeffe E.B. Improved virologic response in chronic hepatitis C genotype 4 treated with nitazoxanide, peginterferon, and ribavirin. Gastroenterology. 2009;136:856–862. doi: 10.1053/j.gastro.2008.11.037. [DOI] [PubMed] [Google Scholar]
- 28.Korba B.E., Montero A.B., Farrar K., Gaye K., Mukerjee S., Ayers M.S., Rossignol J.F. Nitazoxanide, tizoxanide and other thiazolides are potent inhibitors of hepatitis B virus and hepatitis C virus replication. Antiviral Res. 2008;77:56–63. doi: 10.1016/j.antiviral.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 29.Hay M., Thomas D.W., Craighead J.L., Economides C., Rosenthal J. Clinical development success rates for investigational drugs. Nat Biotechnol. 2014;32:40–51. doi: 10.1038/nbt.2786. [DOI] [PubMed] [Google Scholar]
- 30.Lentz T.B., Loeb D.D. Development of cell cultures that express hepatitis B virus to high levels and accumulate cccDNA. J Virol Methods. 2010;169:52–60. doi: 10.1016/j.jviromet.2010.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang J.H., Chung T.D.Y., Oldenburg K.R. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 32.Kay M.A., He C.Y., Chen Z.Y. A robust system for production of minicircle DNA vectors. Nat Biotechnol. 2010;28:1287–1289. doi: 10.1038/nbt.1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoshikawa T., Wu J., Otsuka M., Kishikawa T., Suzuki N., Takata A., Ohno M., Ishibashi R., Yamagami M., Nakagawa R., Kato N., Miyazawa M., Han J., Koike K. Repression of microRNA function mediates inflammation-associated colon tumorigenesis. Gastroenterology. 2017;152:631–643. doi: 10.1053/j.gastro.2016.10.043. [DOI] [PubMed] [Google Scholar]
- 34.Pan W., Dong Z., Li F., Meng W., Feng L., Niu X., Li C., Luo Q., Li Z., Sun C., Chen L. Visualizing influenza virus infection in living mice. Nat Commun. 2013;4:2369. doi: 10.1038/ncomms3369. [DOI] [PMC free article] [PubMed] [Google Scholar]