

Abstract
The acute hepatic porphyrias (AHPs) are a group of four inherited diseases of heme biosynthesis that present with episodic, acute neurovisceral symptoms. The four types are 5‐aminolevulinic acid (ALA) dehydratase deficiency porphyria, acute intermittent porphyria, hereditary coproporphyria, and variegate porphyria. Their diagnoses are often missed or delayed because the clinical symptoms mimic other more common disorders. Recent results indicate that acute intermittent porphyria, the most severe of the more common types of AHP, is more prevalent than previously thought, occurring in about 1 in 1600 Caucasians, but with low clinical penetrance (approximately 2%‐3%). Here we provide an updated review of relevant literature and discuss recent and emerging advances in treatment of these disorders. Symptomatic attacks occur primarily in females between 14 and 45 years of age. AHP is diagnosed by finding significantly elevated levels of porphyrin precursors ALA and porphobilinogen in urine. Acute attacks should be treated promptly with intravenous heme therapy to avoid the development of potentially irreversible neurologic sequelae. All patients should be counseled about avoiding potential triggers for acute attacks and monitored regularly for the development of long‐term complications. Their first‐degree relatives should undergo targeted gene testing. Patients who suffer recurrent acute attacks can be particularly challenging to manage. Approximately 20% of patients with recurrent symptoms develop chronic and ongoing pain and other symptoms. We discuss newer treatment options in development, including small interfering RNA, to down‐regulate ALA synthase‐1 and/or wild‐type messenger RNA of defective genes delivered selectively to hepatocytes for these patients. We expect that the newer treatments will diminish and perhaps obviate the need for liver transplantation as treatment of these inborn metabolic disorders.
Abbreviations
- AHPs
acute hepatic porphyrias
- AIP
acute intermittent porphyria
- ALA
aminolevulinic acid
- ALADP
ALA dehydratase deficient porphyria
- ALAS
ALA synthase
- CEP
congenital erythropoietic porphyria
- ED
emergency department
- EPP
erythropoietic protoporphyria
- HCC
hepatocellular carcinoma
- HCP
hereditary coproporphyria
- HEP
hepatoerythropoietic porphyria
- HMBS
hydroxymethylbilane synthase
- mRNA
messenger RNA
- PBG
porphobilinogen
- PCT
porphyria cutanea tarda
- PPOX
protoporphyrinogen oxidase
- siRNA
small inhibitory RNA
- VP
variegate porphyria
- XLP
X‐linked protoporphyria
Overview of Heme Metabolism and the Porphyrias
Heme is a primordial molecule that is essential to aerobic life on earth. It carries out an astonishing and still growing array of functions. It is an essential cofactor for a myriad of hemoproteins, including hemoglobin, myoglobin, cytochromes P‐450, mitochondrial cytochromes, catalase, peroxidase, and many others. In eukaryotes, biosynthesis of heme occurs in the mitochondria and cytoplasm, and the formation of the heme molecule is a multistep process involving eight enzymes (Fig. 1). Although most tissues in the human body synthesize heme, it is predominantly formed by erythroblasts in the bone marrow (75%‐80%) and hepatocytes in the liver (15%‐20%). The metabolic pathways of heme synthesis are essentially the same in both tissues, although the regulation of the rate‐limiting enzymes 5‐aminolevulinic acid synthase 1 (ALAS‐1) (hepatic, housekeeping) and ALAS‐2 (erythroid) is quite different. Porphyrias are inborn errors of metabolism that cause deficient activity within the eight‐step heme synthetic pathway. These deficiencies can lead to a build‐up of heme precursors, resulting in the clinical manifestations of the porphyrias.
Figure 1.
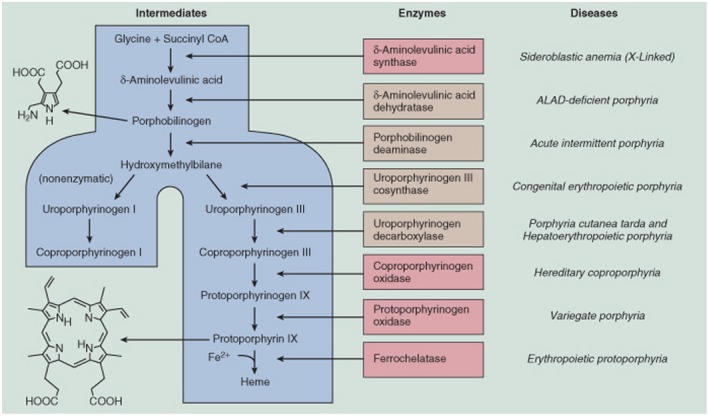
Summary of the heme synthetic pathway, highlighting the enzymatic defects associated with the porphyrias. The heme synthetic pathway involves eight enzymes, four of which are active in the mitochondria and four of which are active in the cytoplasm. The pathway is initiated and completed in the mitochondria. Intermediate steps in the cytoplasm begin with the activity of ALA dehydratase, also known as PBG synthase. Open arrows indicate progression through the pathway. Deficiency (indicated by blocked red arrows) in any of the eight enzymes involved in the pathway may contribute to the development of acute or chronic hepatic porphyrias or erythropoietic porphyrias, as shown in red. Abbreviations: Ac, acetate; CoA, coenzyme A; Copro’gen, coproporphyrinogen; Pr, propionate; proto’gen, protoporphyrinogen; Uro’gen, uroporphyrinogen; and Vi, vinyl. (From 43, used with permission of the authors and publisher.)
The conversion of glycine and succinyl coenzyme A to ALA by the mitochondrial enzyme ALAS is the rate‐limiting step in heme synthesis. In the liver and most other tissues in which ALAS‐1 is the dominant form of the enzyme, heme, the end product of the pathway, exerts negative feedback regulation by several mechanisms that tightly regulate ALAS‐1 activity (Fig. 2). Activity of ALAS‐1 is also down‐regulated by high levels of glucose or other metabolizable carbohydrates, the so‐called glucose effect,1 acting through modulation of peroxisome proliferator–activated receptor‐gamma coactivator 1 (PGC‐1) and other transcription factors.2 ALAS‐1 is increased by many substances that induce cytochromes P450, which are the major users of heme synthesized in hepatocytes, as implied by the thick arrow in Fig. 2.
Figure 2.
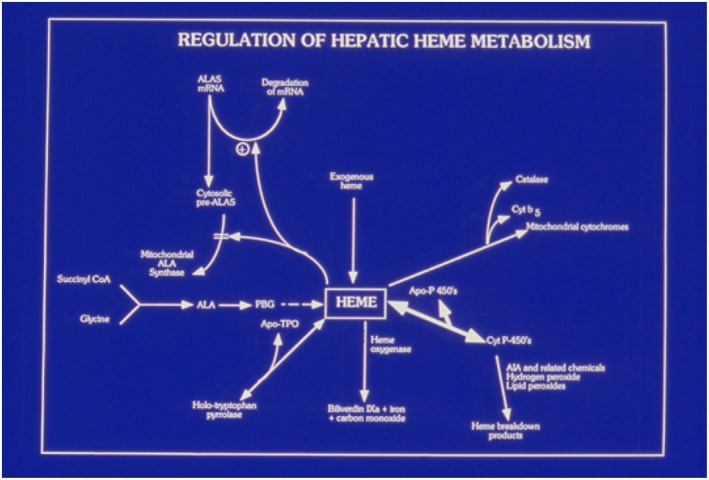
Key role of heme in the regulation of hepatic ALAS‐1 and heme metabolism. The figure shows the central role of the regulatory heme pool in regulating the activity of hepatic ALAS‐1 at the level of mRNA stability and uptake of the pre‐enzyme into mitochondria. Heme also down‐regulates transcription of ALAS‐1 and increases the rate of breakdown of the mature enzyme in mitochondria.3 Abbreviation: CoA, coenzyme A.
Because of the great demand for heme synthesis in developing red blood cells (approximately 80% of the total heme synthesized each day), there is a second form of ALAS‐2 that predominates in such cells. It shares considerable homology with ALAS‐1, but it is a separate gene that is located on the X chromosome. Its regulation is quite different from that of its housekeeping counterpart. ALAS‐2 is not down‐regulated by the end‐product heme, but rather is under the regulation of erythroid transcriptions factors, such as GATA binding factor‐1 (GATA‐1), also known as erythroid transcription factor, and the iron regulatory element–iron regulatory protein system. Thus, when there is deficiency of iron, activity of erythroid ALAS‐2 is restricted, with the eventual effect of restricting production of excess protoporphyrin, which is toxic to cells and would otherwise pile up in the absence of sufficient iron to fuel its conversion to heme. It is worthy of note that gain‐of‐function mutations of ALAS‐2 are the cause of the often‐severe cutaneous porphyria known as X‐linked protoporphyria (XLP), the phenotype of which closely resembles that of erythropoietic protoporphyria (EPP).
In both hepatocytes and erythroblasts, there are six additional steps required to convert ALA to protoporphyrin IX (Fig. 1). In the final step, ferrochelatase inserts ferrous iron into the protoporphyrin macrocycle to form heme. Enzyme deficiencies in any of the eight‐step heme synthetic pathway can cause clinically apparent porphyria.3 In acute porphyrias, the respective enzyme deficiencies predispose patients to a variety of triggering factors, especially drugs and female sex hormones, which induce the accumulation of the neurotoxin ALA and porphobilinogen (PBG).
The eight porphyrias are generally categorized in one of two ways: according to the main site of overproduction of heme precursors (hepatic or erythropoietic) or according to the cardinal clinical features (acute or cutaneous) (Table 1). The AHPs, which are the focus of this review, are a group of four inherited disorders: acute intermittent porphyria (AIP), hereditary coproporphyria (HCP), variegate porphyria (VP), and ALA dehydratase deficient porphyria (ALADP). AIP, HCP, and VP arise from autosomal dominant mutations of the genes that control normal hepatic heme biosynthesis, resulting in approximately 50% deficiencies of the enzymes hydroxymethylbilane synthase (HMBS, also known as PBG deaminase), coproporphyrinogen oxidase (CPOX), and protoporphyrinogen oxidase (PPOX), respectively. These enzyme deficiencies, in combination with other environmental factors (nutritional, hormonal, or other genetic factors), can lead to a critical deficiency in heme within a small but critical “regulatory pool” within hepatocytes (Fig. 2.) In the United States and most other countries of the world, AIP is the most common and most symptomatic of the three autosomal dominant conditions, occurring in 5 to 10 per 100,000 patients, and the most clinically severe. This is likely due primarily to the fact that, even under normal circumstances, activity of hepatic HMBS is relatively low and only slightly above that of ALAS‐1. Therefore, a 50% decrease in activity of HMBS is more likely to critically limit hepatic heme production and lead to uncontrolled up‐regulation of hepatic ALAS‐1, which is the sine qua non of acute porphyric attacks.3 Because of a strong founder effect among Afrikaners, VP is more common in South Africa than other locations. Although VP and HCP can give rise to neurologic symptoms similar to AIP, cutaneous manifestations may rarely also occur in HCP and often predominate in VP. ALA dehydratase deficiency is a rare autosomal recessive disorder in which enzymatic activity is less than 3% of normal (compared with asymptomatic carriers who have 50% of normal enzyme activity).
Table 1.
Main Schemes of Classification and Selected Aspects of the Porphyrias
| Acute Hepatic Porphyrias | Chronic Hepatic Porphyrias | Erythropoietic Porphyrias | |
|---|---|---|---|
| Types of porphyria | 1. ADP | 1. PCT | 1. CEP |
| 2. AIP | 2. HEP | 2. EPP | |
| 3. HCP | 3. XLP | ||
| 4. VP | |||
| Gender frequency | Female > male (~4 > 1) | Male > female | Female = male |
| Main site of overproduction of heme precursors | Liver | Liver | Erythroblasts |
| Inheritance | AIP, HCP, VP (autosomal dominant); ALADP (autosomal recessive) | 1. Usually acquired | 1. CEP |
| 2. Autosomal recessive or codominant | 2. EPP (autosomal recessive) | ||
| 3. XLP‐X‐linked | |||
| Key clinical features | Neurovisceral symptoms (photocutaneous disease in HCP, VP) | Cutaneous lesions on sun‐exposed areas, especially after minor mechanical trauma (painless blisters, skin fragility, sclerodermatous changes, cheeks and temples, hypertrichosis) | 1. CEP (cutaneous lesions on sun‐exposed areas [painless blisters, skin fragility, sclerodermatous changes, hypertrichosis]) |
| 2. EPP | |||
| 3. XLP (acute burning, pain, pruritus, swelling after sun exposure) | |||
| Key biochemical findings in active disease | 1. ALADP (increased urinary ALA, CP 3) | Increased urinary uroporphyrins and heptacarboxyl‐porphyrins; increased stool isocoproporphyrins | 1. CEP (increased urinary uroporphyrin 1, CP 1) |
| 2. AIP | 2. EPP | ||
| 3. HCP | 3. XLP (urine is normal unless hepatopathy has developed); increased PP in plasma and RBCs | ||
| 4. VP (increased urinary ALA, PBG, uroporphyrin 1, CP 3) HCP (increased stool CP) | |||
| VP (increased stool PP) |
Abbreviations: CP, coproporphyrin; PP, protoporphyrin; and RBC, red blood count.
The cutaneous porphyrias include congenital erythropoietic porphyria (CEP), EPP, XLP, hepatoerythropoietic porphyria (HEP), and porphyria cutanea tarda (PCT). The cutaneous porphyrias usually exhibit no neurologic symptoms. A rare exception is EPP complicated by serious hepatopathy, in which case a few patients have developed neurological deficits, especially after liver transplantation.4 In these diseases, excess porphyrins and porphyrinogens are deposited in the upper dermal capillary walls. These compounds are potent photoreactants that cause tissue damage, manifesting as cutaneous vesicles and bullae and increased skin fragility (CEP, HEP, PCT) or as acute pain, burning, and swelling (EPP, XLP) in skin exposed to sunlight. Over time, in CEP, HEP and PCT scarring, pigment changes, infection, and excessive hair growth (hypertrichosis) can occur.
Major Clinical Features of the AHPs
We first present a clinical vignette of a typical patient with AIP to emphasize the key aspects of presentation and diagnosis. A 28‐year‐old woman carrying a diagnosis of irritable bowel syndrome, constipation predominant, for 10 years (after previous imaging and endoscopic evaluation by a gastroenterologist) presented to the emergency department (ED) for the fourth time in 12 months for evaluation of severe abdominal pain, constipation, nausea, vomiting, and weakness. Surgical history is notable for an appendectomy, carried out at age 20, after an earlier episode of severe abdominal pain, despite a lack of computed tomography (CT) findings to suggest acute appendicitis. The appendix was said to have shown “mild chronic appendicitis.” The patient underwent a cholecystectomy 2 years ago for similar symptoms and a diagnosis of acalculous “chronic cholecystitis.”
Since the time of cholecystectomy, she has continued to have intermittent severe abdominal pain requiring repeated ED visits. Opioid pain medications provided minimal relief. On presentation, vital signs were notable for tachycardia (heart rate = 112/minute), systemic arterial hypertension (175/100 mm Hg), and normal oxygen saturation and respiratory rate. Physical exam was otherwise unremarkable, including a benign abdominal exam, despite the patient complaining loudly of severe stabbing lower abdominal pain. The CT of the abdomen/pelvis was unremarkable, noting only the previous appendectomy and cholecystectomy with expected mild common bile duct dilation and much stool in the colon. (The patient described chronic constipation with usual bowel movements only once or twice per week.) Laboratory evaluation was notable for minor elevations of serum aminotransferases (aspartate aminotransferase = 42 U/L; alanine aminotransferase = 48 U/L), hyponatremia (Na+ = 128 mEq/L), and hypomagnesemia (Mg++ = 1 .1 mg/dL). Urinalysis showed dark amber to brownish urine that is negative for blood or bilirubin. The urinalysis was otherwise unremarkable. A test for pregnancy was negative. An astute medical resident considered the diagnosis of an AHP, based on the constellation of symptoms and signs, and obtained a random urine sample for ALA, PBG, and creatinine. This was sent to a reference laboratory. The patient was treated with intravenous fluids that included 5% dextrose and 0.154 M NaCl and with promethazine, ondansetron, and narcotic analgesics, with partial gradual improvement over the ensuing 24 hours. She was discharged from the ED. After 5 days, the results of the spot urine test was returned (ALA = 38 mg/g creatinine [ref 0‐7]; PBG = 85 mg/g creatinine [ref 0‐4]), establishing a diagnosis of AHP. There was no history of cutaneous lesions. Genetic testing for mutations in all four of the genes involved in the AHPs revealed that the patient had a single missense mutation in the HMBS gene (p. R167Q), which is known to be associated with AIP. Genetic testing of all first‐degree relatives was recommended (Table 2).
Table 2.
Common Symptoms and Signs of AHPs in Subjects With Clinically Active Disease
| Symptoms | Signs |
|---|---|
| Pain | Systemic arterial hypertension (43%) |
| Abdominal (74%) | Chronic kidney disease (29%) |
| Back (56%) | Palpitations/tachycardia (19%) |
| Chest (58%) | Hyponatremia (20%) |
| Nausea/vomiting (73%) | Fever (18%) |
| Weakness (63%) | Hypomagnesemia (11%) |
| Constipation (60%) | |
| Anxiety/depression (55%) | |
| Diarrhea (29%) | |
| Sun sensitivity (20%) | |
| Chronic fatigue (11%) | |
| Seizures (9%) |
Adapted from 7.
As already described, the more common AHPs are inherited in an autosomal dominant pattern.3, 5 The most common and generally most severe type is AIP. Although previously thought to be quite rare, more recent population‐based data indicate that potential disease‐causing mutations in HMBS occur in approximately 1 in 1700 Europeans (Nordmann et al., approximately 1996). Thus, newer evidence indicates that disease penetrance is low (estimated to be 1%‐2%), with more than 90% of heterozygotes with disease‐causing mutations remaining asymptomatic for most or all of their lives, highlighting the facts that other factors are important in pathogenesis and that patients with severe disease represent only a small proportion of those carrying disease‐causing mutations.6
The preceding vignette describes a typical case history of AHP. Attacks are characterized by the development of acute neurovisceral pain. Attacks occur most frequently in young women (83%) compared with males (17%).7 Abdominal pain with nausea, vomiting, and accompanied weakness are the most common presenting symptoms. Pain tends to be localized to the lower abdomen and is often described as colicky in nature. It generally lasts hours to days and is poorly responsive to narcotic pain medications. Neurologic manifestations such as weakness and altered affect may be subtle, requiring a high index of suspicion. Signs often existing at presentation include tachycardia, hypertension, and dark‐colored urine, as in the patient described. Seizures occur during acute attacks in approximately 20% of patients, and if they are treated with hydantoins, barbiturates or valproic acid, the severity of the attacks is often amplified many‐fold.
The clinical history usually reveals recurrent symptoms with repeated, frequently nondiagnostic medical evaluations. The nonspecific symptoms may result in inaccurate diagnoses and unnecessary and ineffective surgical interventions (e.g., appendectomy, cholecystectomy, hysterectomy), as in the case described. The nonspecific signs/symptoms, coupled with nondiagnostic evaluations, result in mean delay in diagnosis of approximately 15 years.7
The neurological manifestations of acute porphyrias deserve specific attention, as they can be some of the most life threatening. Paresis/weakness generally begins proximally. This can progress to respiratory paralysis in some cases.8 Convulsions/seizures are not uncommonly seen (10%‐20%), and careful attention to choice of anti‐epileptic medication for treatment is required (see “Management of Acute Attacks” section).
Common laboratory abnormalities observed in the setting of acute attacks of porphyria include hyponatremia (most common). In some circumstances, hyponatremia may be severe and contribute to the development of seizures. Hypomagnesemia is also common. Mild aminotransferase elevations can also be observed. During acute attacks, the passage of reddish‐brown urine may be noted. If left exposed to air and light at room temperature, urine may darken further, which should prompt consideration of the diagnosis.
Keys to Diagnosis and Differential Diagnosis
Contrary to urban legend, it is not difficult to establish or rule out a diagnosis of AHP, provided that physicians or other providers think of the diagnosis and perform the appropriate diagnostic testing. As in the described clinical vignette, AHP should be considered in any patient, but especially in women in their child‐bearing years who present with recurrent, severe episodes of abdominal pain. The pain may occur elsewhere as well, including the back, extremities or chest, but is first and foremost in the abdomen. The first test of choice for diagnosis is a single random urine screening for ALA, PBG, and creatinine—not porphyrins. In our experience, the most frequent errors lie in (1) not considering the diagnosis in a timely fashion, leading to the average 15‐year delay in the United States from time of first symptoms to eventual correct diagnosis,7 and (2) ordering a random urine screening for porphyrins rather than for ALA and PBG. As described subsequently (in the “Secondary Porphyrinurias” section), many patients without any form of porphyria will show mild to moderate increases in urinary porphyrins, especially in coproporphyrins I and III. When these are found in patients with diverse symptoms, including abdominal pain, joint pain, muscle pain, fibromyalgia, anxiety and chronic fatigue syndrome, such patients, unfortunately, are labeled as having porphyria when they do not. Too often, this leads to the patients and their physicians ascribing a plethora of symptoms to an inaccurate diagnosis. Unfortunately, some of these patients have had central venous ports inserted and have been treated with recurrent infusions of intravenous heme, sometimes with disastrous results, such as recurrent infections and/or thrombosis of ports and central veins, thrombophlebitis, sepsis, and development of secondary iron overload (heme is 9% iron by weight).
Another misconception is that the pains and other features of AHP are transient and fleeting; they are not. Rather, they occur with a prodrome that is typical for the patient, with gradual crescendo. The pain typically lasts for several days and only gradually abates. Many patients try to treat their recurrent, often monthly attacks during the luteal phase of their menstrual cycles (when progesterone levels are at maximal) with narcotics or other analgesics and with increases in glucose intake. They try to avoid having to present to the ED or urgent care, because, too often, prompt and appropriate specific treatment with intravenous dextrose and heme and adequate narcotic analgesics is delayed or not provided at all. Such unfortunate patients are too often labeled as “drug seeking” or malingering. The latter has become much worse since the advent of the opioid overdose epidemic that has swept the United States, and shows no sign of abating.
As described in the clinical vignette, the key to diagnosis is measurement of ALA, PBG, and creatinine in a single spot urine test collected during or within a few days of an acute attack. Levels of ALA and/or PBG, expressed as per gram or per millimole creatinine in the urine, that are greater than four times the upper limit of normal establish the diagnosis of AHP. Most such patients will prove to have AIP, which is the most common and most severe form of AHP throughout the world, with the exception of South Africa, where, due to a founder effect, there are thousands of persons with the genetic defect in PPOX associated with VP. After an acute attack of AIP, urinary ALA and PBG remain elevated for many months to years.9 Thus, in bona fide AIP, collection of urine does not need to take place during the peak of clinical symptoms; unless the patient has been treated with a 4‐day to 5‐day course of intravenous heme, the urine can be collected days or weeks after the acute attack. This is less so in HCP or VP, in which levels of urinary ALA and PBG more often fall quickly after an acute attack. In these disorders, however, stool porphyrins are usually markedly elevated chronically and for years, and/or the fluorescence peak emission of serum points to the correct diagnosis (see subsequently).
The specific type of acute porphyria that is present now is usually established by genetic testing, with sequencing of the four genes that are defective in the acute porphyrias (i.e., ALAD, HMBS, CPOX, and PPOX) (Fig. 1). Such testing is available commercially at several labs, including Invitae (San Francisco, CA), Mayo Medical Labs (Rochester, MN), and Department of Genetics, Icahn School of Medicine (New York, NY). Another test of considerable use for diagnosis of VP is fluorescence of diluted sera at physiologic pH. The sera from subjects with VP, especially with biochemical activity, have a unique porphyrin‐peptide in plasma that has peak fluorescence at approximately 626 nm, following excitation by light of 410 nm (the Soret band). In contrast, the sera of subjects with biochemically active AIP, CEP, HCP, HEP, or PCT have emission peaks at approximately 619 nm to 620 nm, whereas those with EPP or XLP have peak emission peaks of protoporphyrin at approximately 634 nm.3
Management of Acute Attacks
Initial management includes discontinuation of any potential precipitants and management of symptoms. A thorough history to identify possible precipitating factors, specifically medications/chemicals, should be performed. If culprit agents are identified, these should be discontinued immediately. Common precipitants include excess alcohol intake, estrogen/progesterone, barbiturates, sulfonamides, and, more generally, any known inducers or suicide substrates of hepatic cytochromes (Table 3). Consultation with the American Porphyria Foundation website (www.porphyriafoundation.com/drug-database) and a newer mobile app (available at porphyriadrugs.com) is advised, as multiple medications commonly used in symptom management may have unanticipated deleterious effects. Other drug databases available on the Internet include one developed especially for European countries (www/drugs‐porphyria.org), a safe drug list produced jointly by the UK Porphyrias Medicines Information Service and the Cardiff Porphyria Service (www.wmic.wales.nhs.uk). These lists do not always agree, but, in general, we recommend caution in the use of drugs and herbals and dietary supplements for all persons, especially those with AHP.
Table 3.
Common Precipitants of Acute Attacks
| Drugs/Chemicals | Other Causes |
|---|---|
| Excess alcohol | Rapid weight loss |
| Anticonvulsants | Crash dieting, starvation |
| Barbiturates | Bariatric surgery |
| Carbamazepine | Acute illness/infections |
| Phenytoin | Stress/exhaustion |
| Primidone | Emotional/physical |
| Valproic acid | Surgery/anesthesia |
| Oral contraceptives | Luteal phase of menstrual cycle |
| Sulfonamides | Pregnancy |
| Cytochrome P450 inducers | Postpartum period |
| Suicide substrates of Cyp P450s |
Initial treatment in patients with confirmed acute porphyria without evidence of infection includes high carbohydrate intake (10% dextrose in 0.45% saline). Narcotic analgesics and anti‐emetics (chlorpromazine or promethazine and ondansetron, which have been found to be “safe” drugs in AHPs) are preferred.
At the pathophysiologic level, the goal of treatment in acute attacks is to reduce the activity of hepatic ALAS‐1. Carbohydrate/glucose administration works to down‐regulate ALAS‐1, thus resulting in decreased production of ALA and PBG, and at least 300 g of glucose or similar should be given daily. However, the most effective therapy in decreasing ALAS‐1 activity is the administration of intravenous heme (in the United States, Panhematin; in Europe, Normosang [Recordati Rare Chemicals]), which results in a decrease in urine and plasma ALA and PBG after two to three infusions,3, 10 with improvement in symptoms generally observed within 4 days.11
Given its instability in aqueous solution, it is recommended that Panhematin be reconstituted in human albumin and given at a dose of 3‐4 mg/kg/day.12, 13 There is potential risk for painful phlebitis when intravenous heme is administered into peripheral veins; thus, administration into a high‐flow, central vein is recommended.12 This risk is likely decreased when heme is administered as the heme‐albumin complex, which not only stabilizes the heme but also appears to diminish its toxicity to vascular endothelium. Other potential adverse effects of intravenous heme include transient thrombocytopenia and prolongation of prothrombin time (without resultant bleeding diathesis), hepatic iron overload (with repeated, chronic administration), and development of tachyphylaxis.14, 15
Monitoring for and treatment of expected electrolyte abnormalities, including hyponatremia and hypomagnesemia, is recommended. In addition to electrolyte replacement, if associated seizures are present, gabapentin and vigabatrin are the preferred anticonvulsant agents. Of note, commonly used anticonvulsants, including barbiturates, phenytoin, valproic acid and carbamazepine, should be avoided in patients with acute porphyria, as they may result in severe exacerbation of the attack and rapid development of profound weakness and acute respiratory compromise that requires mechanical ventilation.
The incapacitating pain associated with acute attacks nearly universally requires use of parenteral narcotic analgesics. If appropriate therapies (intravenous glucose and heme, safe analgesics) are administered without delay, improvement in pain should result within 3 to 5 days. Tachycardia and systemic arterial hypertension in acute attacks result from sympathetic hyperactivity. The preferred agents include beta blockers, angiotensin‐converting enzyme inhibitors, and calcium channel blockers (diltiazem is preferred over nifedipine, which is porphyrogenic in model systems).16
Some patients have predictable triggers for acute attacks that should be avoided and minimized as much as possible. There are multiple possible inciting factors that physicians should address (Table 3). Medications including but not limited to estrogens, progestogens, barbiturates, sulfonamides, and other inducers of cytochromes P450 and ALAS‐1 should be avoided, and websites with recommendations based on existing evidence should be consulted before starting any new medications. Patients should avoid smoking, including marijuana, and avoid heavy alcohol intake, as each can induce cytochromes P450, and alcohol is known to induce hepatic ALAS‐1.17 We recommend in general that men should adhere to no more than two, and women to no more than one, drink per day.
Sources of physical, psychological, and emotional stress should be avoided as much as possible. Infection or other illness that can cause metabolic stress can incite an attack; thus, prompt treatment of intercurrent infections or other illnesses with safe antibiotics and hydration is essential. All appropriate vaccinations to prevent infections that may trigger an attack should be administered. Consultation with a dietitian should be considered to promote and maintain a well‐balanced diet somewhat high in carbohydrates (60%‐70% of total calories). Extreme dieting, severe caloric restriction, and starvation should be avoided. First acute attacks have been reported following weight loss surgery.18
Some women suffer monthly recurrent attacks during the luteal phase of their menstrual cycles and can have symptomatic improvement with a gonadotropin‐releasing hormone analogue (leuprolide or histrelin) to suppress ovulation. Others report decreased symptoms when they take low‐dose oral contraceptives. Most women with AIP tolerate pregnancy well despite increased serum‐circulating progesterone. Some women have more frequent attacks while pregnant; such attacks are treated in the same manner as for nonpregnant women, and experience has shown intravenous heme to be safe and effective during pregnancy.
In VP, dietary supplementation with vitamins C and E may mitigate oxidative damage in plasma and neutrophils.19 In patients with cutaneous manifestations, skin trauma should be minimized as much as possible, as excessive mechanical manipulation can increase the damaging effects of photoreactive compounds. Usual sunscreens are of little help in the cutaneous porphyrias, as they do not block light in the Soret region (about 400 nm‐420 nm). Opaque sunscreens with zinc or titanium oxide paste are of more benefit, although they leave white pigment on the skin. Special filters, such as CLS‐200‐X or TA‐81 (Madico, Woburn, MA), can be used to protect against strong indoor lighting (particularly fluorescent lights or lights used in operating rooms).
Hematin infusions inhibit the stimulus for heme production, thereby diminishing or eliminating manifestations of the disease. A minority of patients require prophylactic intravenous heme, typically once or twice per week. In select patients, this may markedly improve quality of life and decrease total health care costs.20
Natural History: Recommendations for Long‐Term Follow‐up
Chronic Sequelae
Although most symptomatic patients with AHP have complete resolution of their symptoms between attacks, those with multiple recurrent attacks may develop chronic pain. The pathogenesis of chronic pain in these patients is poorly understood, but nerve conduction and electromyogram studies often reveal chronic neuropathy, typically an axonal motor polyneuropathy.8, 21 Referral to a neurologist is recommended for any patient with residual neurologic deficits. These chronic symptoms may not respond to administration of heme, and referral to a pain management specialist may be helpful for management of chronic neuropathic pain. Patients with AHP with persistent chronic pain symptoms can develop severe depression and anxiety, which are associated with an increased risk for suicide, and may require psychiatric monitoring and care.22
Of patients with AHP, 13% have increased serum aminotransferases during an acute attack.23 Asymptomatic patients may also show increased aminotransferases, although to a lesser degree.24 Persistent elevations in serum aminotransferases are common in patients who have recurrent attacks. The risk for developing cirrhosis is greater in patients with AHP than the general population. Regardless of cirrhosis status, patients with AHP have an increased risk for hepatocellular carcinoma (HCC).25, 26 HCC develops more commonly in symptomatic patients over 60 years of age.
Patients with AHP are also at increased risk for developing chronic renal disease.27, 28 The renal pathology is typically chronic tubulo‐interstitial nephropathy or focal cortical atrophy.28 Asymptomatic patients without elevated porphyrin precursors and porphyrins are not at risk of chronic renal disease.29 Although systemic arterial hypertension usually resolves after resolution of an acute attack, some patients may develop chronic hypertension, which may be a cause of chronic renal damage. Referral to a nephrologist is recommended if hypertension is not controlled by first‐line treatment or when renal dysfunction is first recognized. A common variant of peptide transporter 2, a transporter for ALA in the kidney and brain, may predispose patients with AHP to develop chronic renal disease.30 A number of patients with AHP have required dialysis or renal transplantation.
Long‐Term Management
Symptomatic patients with AHP should be followed at least annually and more frequently if they are receiving prophylactic treatment or continue to have acute attacks. Asymptomatic patients who have elevated porphyrin precursor levels should also be followed annually. Liver function tests should be checked in all patients at baseline. An initial abdominal ultrasound and Fibroscan to screen for the presence of advanced liver disease should be performed. Patients found to have elevated liver enzymes or abnormal liver imaging should be evaluated for concomitant liver disease. Those with evidence of chronic liver injury should have cirrhosis and HCC screening starting at age 50 with Fibroscan, ultrasound, and serum alpha‐fetoprotein annually. For those without evidence of chronic liver injury, annual screening is recommended starting at age 60.
Due to the high prevalence of chronic kidney disease, serum creatinine and estimated glomerular filtration rate should be monitored annually for all symptomatic patients. Hypertension can be difficult to manage, particularly during acute attacks, and blood pressure should be actively managed to prevent further decline in renal function. Hyponatremia may be chronic in some patients and may require careful Na+ replacement therapy and/or limitation of free water intake. Although the hyponatremia has often been ascribed to the syndrome of inappropriate antidiuretic hormone (ADH) secretion, when blood volumes were measured in patients with AIP, they were often found to be decreased.31 Thus, they do not truly fulfill the diagnostic criteria for the syndrome of inappropriate secretion of ADH. More detailed recommendations regarding long‐term management and follow‐up of subjects with AHP have been published recently by the Porphyrias Consortium.32
Prognosis
Data scarcely regard the prognosis of individuals with AHP due to the rarity of the conditions. Earlier studies showed increased mortality in patients with severe clinical manifestations.22 However, since the introduction of heme therapy, mortality has improved in these patients. Overall, patients can have a good prognosis, especially if their disease remains latent. When the diagnosis is made in a timely fashion, acute attacks are managed rapidly and future attacks prevented.
Patients with severe, intractable, and disabling attacks that are refractory to heme can be considered for orthotopic liver transplantation (OLT), which has been shown to be curative for these patients.33 However, due to the high morbidity and mortality associated with OLT, it is considered a treatment of last resort. Moreover, it has not been successful in patients with advanced neuropathy. Effects are also less clear for patients with ADP or severe homozygous deficiency of HMBS.34 Some patients with both intractable recurrent attacks and end‐stage renal disease have benefitted from combined liver and kidney transplantation.35
Secondary Porphyrinurias: Common Causes of Misdiagnosis
In the experience of the senior author, now stretching back 50 years, the most common cause of overdiagnosis (and erroneous diagnosis) of AHP is related to mild or moderate increases in urinary porphyrins, usually primarily coproporphyrins I and III. This occurs because providers fail to order the correct urinary test (i.e., ALA, PBG, and creatinine) and instead order only spot or 24‐hour urine tests for porphyrins. There are many reasons for levels of such porphyrins to be mildly or moderately elevated, and most of them are not porphyrias. The most common are excess alcohol and/or fatty liver disease. However, any condition that produces mild cholestasis or adversely affects the function of organic ion transport activity from hepatocytes into bile canaliculi may lead to a shift of coproporphyrin excretion from the bile and stool to the urine. In addition, many drugs, especially inducers of hepatic cytochromes P450, lead to mild induction of ALAS‐1 and to a mild increase in hepatic coproporphyrin production and/or to competitive inhibition of the organic anion transporters. In fact, elevations in urinary coproporphyrin levels have been proposed as biomarkers for such drug effects.36 Other causes of mild porphyrinuria without porphyria include a variety of anemias and dyserythropoietic states, heavy metal exposures, diabetes mellitus, chronic heart diseases, and many others.3, 37, 38 Regrettably, many patients with increases in urinary porphyrins have been mislabeled as having porphyria, when, in fact, they do not. Such overdiagnosis can lead to inappropriate, expensive, and potentially dangerous overtreatment with intravenous heme and other therapies. The importance of early and correct diagnosis—and of ruling out a diagnosis of AHP—cannot be overemphasized.
Emerging New Therapies for AHP
There has been no new effective therapies for AHP since the demonstration of the efficacy of intravenous heme10 and the development of Panhematin (hydroxyheme) and Normosang (heme arginate). Intravenous heme has withstood the test of time, and, with high glucose/carbohydrate intake, it continues to be the treatment of choice for acute porphyric attacks. Unfortunately, it has potential adverse effects, and there are patients for whom, with repeated use, tachyphylaxis develops (diminishing beneficial and increasing adverse effects). Thus, additional approaches to therapy, short of liver transplantation, are needed. Another approach for the down‐regulation of ALAS‐1 is to introduce a small inhibitory RNA (siRNA) specifically into hepatocytes. In general, the use of siRNAs as therapeutic agents is an exciting new field in therapeutics. A complementary approach may be to selectively introduce wild‐type messenger RNA (mRNA) for deficient genes and proteins into hepatocytes, to increase wild‐type protein levels sufficiently to overcome critical deficiencies in metabolic pathways. Both of these approaches are being pursued for therapy of AHPs.
Thus, because uncontrolled up‐regulation of ALAS‐1 in hepatocytes is the sine qua non for acute porphyric attacks, a novel treatment might be to introduce siRNA against ALAS‐1, specifically targeted to hepatocytes, by binding to asialoglycoproteins, which are taken up preferentially by the asialoglycoprotein receptors of hepatocytes. The specific drug that has shown good effect in preclinical studies in murine models39 and in phase I and phase IIa clinical trials in humans is Givosiran (Alnylam Pharma, Cambridge, MA) (Fig. 3). Givosiran, which is given once every month or two by subcutaneous injection, has been selectively taken up by hepatocytes and has led to marked and sustained decrements in urinary ALA and PBG in subjects with AHP and chronic high secretion of ALA and PBG, and to significant and clinically meaningful decreases in frequency and severity of acute attacks, hospitalizations, and intravenous heme therapy.40 Givosiran has had a favorable safety profile overall in these initial studies, which has led to the design and implementation of a pivotal phase III, multicenter, placebo‐controlled, double‐blind randomized prospective clinical trial that is currently in progress. This trial involves 6 months of treatment with Givosiran or matching placebo, followed by 18 months of open‐label therapy. It is being performed at centers throughout the world, and initial interim results will be available in the spring of 2019.
Figure 3.
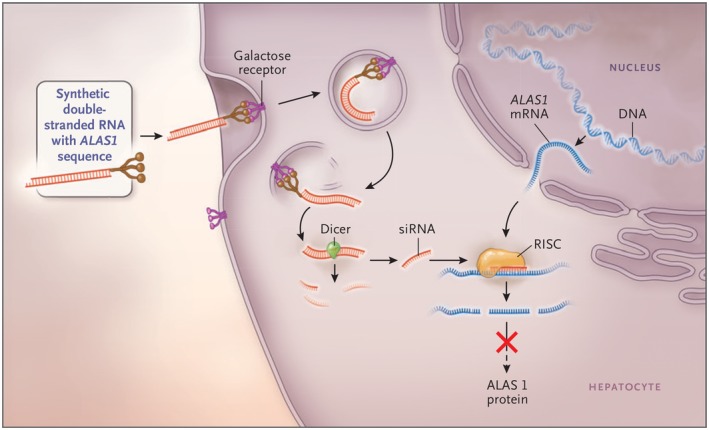
The mechanism of siRNA therapy. Synthetic double‐stranded RNA containing an ALAS‐specific sequence is derivatized with N‐acetylgalactosamine to target the asialoorosomucoid (galactose) receptor, which is expressed nearly exclusively on hepatocytes. Within the hepatocytes, the RNA is processed into approximately 20 base pair (bp) fragments by a cellular enzyme (dicer) and then separated into single strands. The strand that is complementary to ALAS‐1 (the guide strand) binds to cellular ALAS‐1 mRNA and enters the RNA‐induced silencing complex, where the new double‐stranded RNA is cleaved by a group of factors that include argonaute, a ribonuclease. The result is a reduction in the level of ALAS‐1 protein and decreased production of ALA. Abbreviations: RISC, RNA‐induced silencing complex. (From 5, used by permission of the authors and publisher.)
Another recent approach that was tried in Spain was to administer recombinant human wild‐type HMBS DNA packaged in adeno‐associated virus 8. The idea was that enough of the wild‐type DNA would get into hepatocytes and increase HMBS activity sufficient to decrease the overproduction of ALA and PBG and ameliorate symptoms of AIP. Unfortunately, this trial did not show any efficacy.41 Perhaps more promising is to introduce mRNA for HMBS selectively into hepatocytes, an approach being pursued by Moderna Therapeutics (Cambridge, MA). They have encouraging positive biochemical and clinical effects in murine models of AIP,42 and phase I and IIa studies in human subjects are anticipated to start within the next year.
In summary, the AHPs are more common than previously thought, although the clinical disease is rare. The keys to diagnosis are to think of the possibility of AHP early in the evaluation of persons, especially women between the ages of 14 and 45 years, and to obtain a random urine for measurement of ALA, PBG, and creatinine. Any with elevations of ALA and/or PBG above 4‐fold the upper limit of normal should be assumed to have an AHP, treated appropriately, and undergo genetic testing to pinpoint the affected gene and locus. First‐degree relatives of affected persons should undergo targeted genetic testing for the same mutation. Intravenous heme remains the time‐honored treatment of first choice, but in the future, there is the promise of siRNA and/or mRNA of defective genes delivered selectively to hepatocytes. We expect that the newer treatments will diminish and perhaps obviate the need for liver transplantation as treatment of these inborn metabolic disorders.
Potential conflict of interest
Dr. Bonkovsky received grants and consults for Alnylam and Mitsubishi Tanabe; he consults for Moderna Therapeutics and Recordati; he received grants from Gilead. Dr. Rudnick consults for Alnylam. Dr. Wang consults for Alnylam, Recordati, and Mitsubishi Tanabe.
Supported by National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Diseases (U54 DK 083909) and by Protect the Future funding provided by the American Porphyria Foundation.
References
- 1. Bonkowsky HL, Tschudy DP, Collins A, Doherty JM. Control of delta‐aminolevulinic acid synthetase and tyrosine aminotransferase in tumors and livers of tumor‐bearing rats. J Natl Cancer Inst 1973;50:1215‐1225. [DOI] [PubMed] [Google Scholar]
- 2. Handschin C, Lin J, Rhee J, Peyer AK, Chin S, Wu PH, et al. Nutritional regulation of hepatic heme biosynthesis and porphyria through PGC‐1alpha. Cell 2005;122:505‐515. [DOI] [PubMed] [Google Scholar]
- 3. Bonkovsky HL, Guo JT, Hou W, Li T, Narang T, Thapar M. Porphyrin and heme metabolism and the porphyrias. Compr Physiol 2013;3:365‐401. [DOI] [PubMed] [Google Scholar]
- 4. McGuire BM, Bonkovsky HL, Carithers RL Jr, Chung RT, Goldstein LI, Lake JR, et al. Liver transplantation for erythropoietic protoporphyria liver disease. Liver Transpl 2005;11:1590‐1596. [DOI] [PubMed] [Google Scholar]
- 5. Bissell DM, Anderson KE, Bonkovsky HL. Porphyria. N Engl J Med 2017;377:862‐872. [DOI] [PubMed] [Google Scholar]
- 6. Chen B, Solis‐Villa C, Hakenberg J, Qiao W, Srinivasan RR, Yasuda M, et al. Acute intermittent porphyria: predicted pathogenicity of HMBS variants indicates extremely low penetrance of the autosomal dominant disease. Hum Mutat 2016;37:1215‐1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bonkovsky HL, Maddukuri VC, Yazici C, Anderson KE, Bissell DM, Bloomer JR, et al. Acute porphyrias in the USA: features of 108 subjects from Porphyrias Consortium. Am J Med 2014;127:1233‐1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bonkowsky HL, Schady W. Neurologic manifestations of acute porphyria. Semin Liver Dis 1982;2:108‐124. [DOI] [PubMed] [Google Scholar]
- 9. Marsden JT, Rees DC. Urinary excretion of porphyrins, porphobilinogen and delta‐aminolaevulinic acid following an attack of acute intermittent porphyria. J Clin Pathol 2014;67:60‐65. [DOI] [PubMed] [Google Scholar]
- 10. Bonkowsky HL, Tschudy DP, Collins A, Doherty J, Bossenmaier I, Cardinal R, et al. Repression of the overproduction of porphyrin precursors in acute intermittent porphyria by intravenous infusions of hematin. Proc Natl Acad Sci U S A 1971;68:2725‐2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bissell DM. Treatment of acute hepatic porphyria with hematin. J Hepatol 1988;6:1‐7. [DOI] [PubMed] [Google Scholar]
- 12. Bonkovsky HL, Healey JF, Lourie AN, Gerron GG. Intravenous heme‐albumin in acute intermittent porphyria: evidence for repletion of hepatic hemoproteins and regulatory heme pools. Am J Gastroenterol 1991;86:1050‐1056. [PubMed] [Google Scholar]
- 13. Anderson KE, Bloomer JR, Bonkovsky HL, Kushner JP, Pierach CA, Pimstone NR, et al. Recommendations for the diagnosis and treatment of the acute porphyrias. Ann Intern Med 2005;142:439‐450. [DOI] [PubMed] [Google Scholar]
- 14. Glueck R, Green D, Cohen I, Ts'ao CH. Hematin: unique effects of hemostasis. Blood 1983;61:243‐249. [PubMed] [Google Scholar]
- 15. Willandt B, Langendonk JG, Biermann K, Meersseman W, D'Heygere F, George C, et al. Liver fibrosis associated with iron accumulation due to long‐term heme‐arginate treatment in acute intermittent porphyria: a case series. JIMD Rep 2016;25:77‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lambrecht RW, Gildemeister OS, Williams A, Pepe JA, Tortorelli KD, Bonkovsky HL. Effects of selected antihypertensives and analgesics on hepatic porphyrin accumulation: implications for clinical porphyria. Biochem Pharmacol 1999;58:887‐896. [DOI] [PubMed] [Google Scholar]
- 17. Badawy AA, Morgan CJ, Davis NR. Effects of acute ethanol administration on rat liver 5‐aminolaevulinate synthase activity. Biochem J 1989;262:491‐496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bonkovsky HL, Siao P, Roig Z, Hedley‐Whyte ET, Flotte TJ. Case records of the Massachusetts General Hospital. Case 20–2008. A 57‐year‐old woman with abdominal pain and weakness after gastric bypass surgery. N Engl J Med 2008;358:2813‐2825. [DOI] [PubMed] [Google Scholar]
- 19. Ferrer MD, Tauler P, Sureda A, Palacin C, Tur JA, Pons A. Variegate porphyria induces plasma and neutrophil oxidative stress: effects of dietary supplementation with vitamins E and C. Br J Nutr 2010;103:69‐76. [DOI] [PubMed] [Google Scholar]
- 20. Yarra P, Faust D, Bennett M, Rudnick S, Bonkovsky H. Benefits of prophylacticheme therapy in severe acute intermittent porphyria. Mol Genet Metab. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wu CL, Ro LS, Jung SM, Tsai TC, Chu CC, Lyu RK, et al. Clinical presentation and electrophysiological findings of porphyric neuropathies: a follow‐up study. Muscle Nerve 2015;51:363‐369. [DOI] [PubMed] [Google Scholar]
- 22. Jeans JB, Savik K, Gross CR, Weimer MK, Bossenmaier IC, Pierach CA, et al. Mortality in patients with acute intermittent porphyria requiring hospitalization: a United States case series. Am J Med Genet 1996;65:269‐273. [DOI] [PubMed] [Google Scholar]
- 23. Stein JA, Tschudy DP. Acute intermittent porphyria. A clinical and biochemical study of 46 patients. Medicine (Baltimore) 1970;49:1‐16. [PubMed] [Google Scholar]
- 24. Bylesjo I, Wikberg A, Andersson C. Clinical aspects of acute intermittent porphyria in northern Sweden: a population‐based study. Scand J Clin Lab Invest 2009;69:612‐618. [DOI] [PubMed] [Google Scholar]
- 25. Sardh E, Wahlin S, Bjornstedt M, Harper P, Andersson DE. High risk of primary liver cancer in a cohort of 179 patients with acute hepatic porphyria. J Inherit Metab Dis 2013;36:1063‐1071. [DOI] [PubMed] [Google Scholar]
- 26. Andant C, Puy H, Bogard C, Faivre J, Soule JC, Nordmann Y, et al. Hepatocellular carcinoma in patients with acute hepatic porphyria: frequency of occurrence and related factors. J Hepatol 2000;32:933‐939. [DOI] [PubMed] [Google Scholar]
- 27. Andersson C, Wikberg A, Stegmayr B, Lithner F. Renal symptomatology in patients with acute intermittent porphyria. A population‐based study. J Intern Med 2000;248:319‐325. [DOI] [PubMed] [Google Scholar]
- 28. Pallet N, Mami I, Schmitt C, Karim Z, Francois A, Rabant M, et al. High prevalence of and potential mechanisms for chronic kidney disease in patients with acute intermittent porphyria. Kidney Int 2015;88:386‐395. [DOI] [PubMed] [Google Scholar]
- 29. Stewart MF. Review of hepatocellular cancer, hypertension and renal impairment as late complications of acute porphyria and recommendations for patient follow‐up. J Clin Pathol 2012;65:976‐980. [DOI] [PubMed] [Google Scholar]
- 30. Tchernitchko D, Tavernier Q, Lamoril J, Schmitt C, Talbi N, Lyoumi S, et al. A variant of peptide transporter 2 predicts the severity of porphyria‐associated kidney disease. J Am Soc Nephrol 2017;28:1924‐1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bloomer JR, Berk PD, Bonkowsky HL, Stein JA, Berlin NI, Tschudy DP. Blood volume and bilirubin production in acute intermittent porphyria. N Engl J Med 1971;284:17‐20. [DOI] [PubMed] [Google Scholar]
- 32. Balwani M, Wang B, Anderson KE, Bloomer JR, Bissell DM, Bonkovsky HL, et al. Acute hepatic porphyrias: recommendations for evaluation and long‐term management. Hepatology 2017;66:1314‐1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Singal AK, Parker C, Bowden C, Thapar M, Liu L, McGuire BM. Liver transplantation in the management of porphyria. Hepatology 2014;60:1082‐1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dixon N, Li T, Marion B, Faust D, Dozier S, Molina A, et al. Pilot study of mitochondrial bioenergetics in subjects with acute porphyrias. Mol Genet Metab. In press. [DOI] [PMC free article] [PubMed]
- 35. Wahlin S, Harper P, Sardh E, Andersson C, Andersson DE, Ericzon BG. Combined liver and kidney transplantation in acute intermittent porphyria. Transpl Int 2010;23:e18‐e21. [DOI] [PubMed] [Google Scholar]
- 36. Shen H, Chen W, Drexler DM, Mandlekar S, Holenarsipur VK, Shields EE, et al. Comparative evaluation of plasma bile acids, dehydroepiandrosterone sulfate, hexadecanedioate, and tetradecanedioate with coproporphyrins I and III as markers of OATP inhibition in healthy subjects. Drug Metab Dispos 2017;45:908‐919. [DOI] [PubMed] [Google Scholar]
- 37. Watson CJ, Schwartz S, Schulze W, Jacobson LO, Zagaria R. Studies of coproporphyrin. III. Idiopathic coproporphyrinuria; a hitherto unrecognized form characterized by lack of symptoms in spite of the excretion of large amounts of coproporphyrin. J Clin Invest 1949;28:465‐468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Watson CJ, Hawkinson V, Capps RB, Rappaport EM. Studies of coproporphyrin. IV. The per diem excretion and isomer distribution in the urine in infectious hepatitis, infectious mononucleosis, and mechanical jaundice. J Clin Invest 1949;28:621‐637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yasuda M, Gan L, Chen B, Kadirvel S, Yu C, Phillips JD, et al. RNAi‐mediated silencing of hepatic Alas1 effectively prevents and treats the induced acute attacks in acute intermittent porphyria mice. Proc Natl Acad Sci U S A 2014;111:7777‐7782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sardh E, Harper P, Balwani M, Stein P, Rees D, Bloomer J, et al. Phase 1 randomized, placebo‐controlled study of Givosiran, an investigational RNA interference (RNAi) therapeutic, in patients with acute intermittent porphyria: interim study results. Hepatology 2017;66:427A. [Google Scholar]
- 41. D’Avola D, Lopez‐Franco E, Sangro B, Paneda A, Grossios N, Gil‐Farina I, et al. Phase I open label liver‐directed gene therapy clinical trial for acute intermittent porphyria. J Hepatol 2016;65:776‐783. [DOI] [PubMed] [Google Scholar]
- 42. Fontanellas A, Jiang L, Berraondo P, Guey L, Sampedro A, Frassetto A, et al. Efficacy of systemic messenger RNA therapy to treat and prevent porphyria attacks in a mouse model of acute intermittent porphyria. ICPP2017;Abstract.
- 43. Lane AM, McKay JT, Bonkovsky HL. Advances in the management of erythropoietic protoporphyria—role of afamelanotide. Appl Clin Genet 2016;9:179‐189. [DOI] [PMC free article] [PubMed] [Google Scholar]