

Abstract
The presence and stage of liver fibrosis in patients with nonalcoholic steatohepatitis (NASH) is strongly associated with mortality. Thus, both preventing and reversing fibrosis are critically important approaches to prevent death or the need for liver transplantation from NASH. Recently, fibrosis in several mouse models of organ injury was shown to be prevented and reversed with the potent small molecule, arginine‐glycine‐aspartic acid tripeptide (RGD)‐binding, integrin antagonist (3S)‐3‐(3‐bromo‐5‐(tert‐butyl)phenyl)‐3‐(2‐(3‐hydroxy‐5‐((5‐hydroxy‐1,4,5,6‐tetrahydropyrimidin‐2‐yl)amino)benzamido)acetamido)propanoic acid (Center for World Health and Medicine [CWHM]‐12). We hypothesized that RGD‐binding integrins may play an important role in fibrosis progression in NASH. We assessed the efficacy of CWHM‐12 in a choline deficient, amino‐acid defined, high‐fat diet (CDAHFD) mouse model of NASH. Mice were kept on the CDAHFD or a control diet for 10 weeks, and CWHM‐12 was delivered by continuous infusion for the final 4 weeks. The parameters of NASH and liver fibrosis were evaluated before and after drug treatment. Hepatic steatosis, liver injury, and inflammation were significantly induced by the CDAHFD at week 6 and did not change by week 10. Hepatic profibrogenic gene expression was induced by the CDAHFD at week 6, further increased at week 10, and decreased by CWHM‐12. Fibrosis measured by analysis of liver collagen was reduced by CWHM‐12 to levels significantly less than found at 6 weeks, demonstrating the possibility of reversing already established fibrosis despite ongoing injury. Demonstrated mechanisms of the antifibrotic effect of CWHM‐12 included loss of activated hepatic stellate cells through apoptosis and suppression of hepatic profibrotic signal transduction by transforming growth factor β. Conclusion: RGD‐binding integrins may be critical in the development of fibrosis in NASH and may represent potential targets for treating patients with NASH to reverse advanced liver fibrosis.
Abbreviations
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- CCL2
chemokine (C‐C motif) ligand 2
- CD
clusters of differentiation
- CDAHFD
choline‐deficient, amino‐acid defined, high‐fat diet
- CTGF
connective tissue growth factor
- CWHM
Center for World Health and Medicine
- ECM
extracellular matrix
- GAPDH
glyceraldehyde 3‐phosphate dehydrogenase
- H&E
hematoxylin and eosin
- HSC
hepatic stellate cell
- MCD
methionine‐choline‐deficient
- mRNA
messenger RNA
- NASH
nonalcoholic steatohepatitis
- p‐SMAD3
phosphorylated mothers against decapentaplegic homolog 3
- qPCR
quantitative reverse‐transcription polymerase chain reaction
- RGD
arginine‐glycine‐aspartic acid tripeptide
- α‐SMA
α‐smooth muscle actin
- TGFβ
transforming growth factor β
- TNFα
tumor necrosis factor α
- TUNEL
terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick‐end labeling
Nonalcoholic fatty liver disease (NAFLD) has become the major cause of chronic liver disease in developed countries.1 NAFLD includes a range of related liver disorders characterized by deposition of triglyceride as lipid droplets in the cytoplasm of hepatocytes. Nonalcoholic steatohepatitis (NASH) is distinguished by the additional presence of substantial hepatocyte injury (ballooning and apoptosis), inflammatory infiltrates, and varying degrees of fibrosis.2 Patients with NASH have an increased risk of disease progression to advanced fibrosis, cirrhosis, and hepatocellular carcinoma.3 The pathogenesis of NASH is based on an oversupply of metabolic substrates resulting in hepatocyte endoplasmic reticulum stress, inflammasome activation, apoptosis, and a dysfunctional repair response that promotes fibrosis.4 The treatment options for NASH are highly limited.4, 5 Recent longitudinal studies have demonstrated that the presence and stage of liver fibrosis are associated with all‐cause and liver‐related mortality in patients with NASH.6, 7, 8 Thus, it is critically important to identify drivers of liver fibrogenesis in NASH. Although early diagnosis and effective treatment of NASH would likely prevent fibrosis by diminishing the stimulus for fibrogenesis, many patients continue to come to initial medical attention with established advanced fibrosis; thus, developing therapies that can directly reverse fibrosis are needed.
Fibrosis is a pathologic process characterized by excessive accumulation of extracellular matrix (ECM) proteins that can lead to progressive tissue distortion and loss of organ function. Following activation, hepatic stellate cells (HSCs) give rise to myofibroblasts, which are the major source of excess ECM during liver fibrogenesis.9 The most potent inducer of ECM expression by HSCs is transforming growth factor β (TGFβ).10 TGFβ is initially synthesized and secreted by cells in an inactive state due to a noncovalent association with latency‐associated peptide.11 Recent data indicate that members of the arginine‐glycine‐aspartic acid tripeptide (RGD)‐binding integrin receptor subfamily appear to be the primary mediators of latent TGFβ activation in vivo in many organ injury states that lead to fibrosis. 12, 13, 14, 15 Integrins are large family of transmembrane cell‐adhesion and signaling receptors consisting of α and β subunits that together connect the inner cytoskeleton with outer ECM.16 Of the 24 integrin heterodimers, eight (αvβ1, αvβ3, αvβ5, αvβ6, αvβ8, α5β1, α8β1, and αIIbβ3) belong to the group of RGD‐binding integrins. In addition to activating TGFβ, RGD‐binding integrins are involved in other cell functions that are closely linked with ECM remodeling and the migration of different cell types during wound repair and angiogenesis.17, 18, 19 Thus, RGD‐binding integrins are attractive targets for antifibrotic therapies. Recently, a small molecule integrin antagonist compound, (3S)‐3‐(3‐bromo‐5‐(tert‐butyl)phenyl)‐3‐(2‐(3‐hydroxy‐5‐((5‐hydroxy‐1,4,5,6‐tetrahydropyrimidin‐2‐yl)amino)benzamido)acetamido)propanoic acid (Center for World Health and Medicine [CWHM]‐12), that simultaneously and selectively targets several of the profibrotic integrins was discovered by the CWHM at Saint Louis University. Treatment with this compound was demonstrated to alleviate liver, lung, muscle, and pancreatic fibrosis in mouse injury models.14, 20, 21 However, the role of RGD‐binding integrins in the development of NASH fibrosis and their pharmacologic inhibition to prevent or reverse NASH fibrosis have not been assessed.
In the present study, by targeting RGD‐binding integrins in a mouse model of NASH that closely resembles a human liver fibrosis phenotype in NASH, we identified a key role for these integrins in promoting NASH fibrosis and demonstrated the possibility of decreasing and even reversing already established fibrosis.
Materials and Methods
Mice
C57BL/6J 5‐week‐old male mice were obtained from the Jackson Laboratory (Bar Harbor, ME) and housed in standard facilities under controlled conditions of temperature, humidity, and a 12‐hour/12‐hour light/dark cycle with free access to water. Animal care and all animal procedures were approved by the Institutional Animal Care and Use Committee of Saint Louis University.
Choline Deficient, L‐Amino‐Acid Defined, High‐Fat Mouse Model of NASH
The choline deficient, amino‐acid defined, high‐fat diet (CDAHFD)22 was purchased from Research Diets (A06071309; New Brunswick, NJ). This diet was formulated as a high‐fat, choline‐deficient diet that includes 0.1% methionine and 45% Kcal% fat (20% lard, 25% soybean oil). The control diet was a standard rodent chow containing 13.6% of calories from fat (Harlan Teklad, Madison, WI). Starting from the age of 6 weeks, 30 mice were placed on the CDAHFD and 30 mice were placed on the standard diet. At the end of 6 weeks, 10 mice from each dietary group were fasted for 5 hours and killed. Blood and liver samples were collected. Livers were divided into sections that were fixed in 10% phosphate‐buffered formalin, frozen in liquid nitrogen, or placed in an RNA stabilization solution (RNAlater; Ambion, Austin, TX) for future evaluation. The remaining mice were kept on the diets for 4 more weeks for a total of 10 weeks and then fasted, killed, and samples collected as described above.
Administration of the RGD Integrin Antagonist CWHM‐12
The small molecular weight integrin antagonist compound CWHM‐12 was synthesized by the CWHM (Saint Louis University, St. Louis, MO). The structure and synthesis of this compound have been described.14 This prior report also showed excellent potency (50% inhibitory concentration [IC50] in the low nanomolar range) of CWHM‐12 against five RGD‐binding integrins (αvβ1, αvβ3, αvβ6, αvβ8, and α5β1) and good potency (IC50 <100 nm/L) against RGD‐binding αvβ5 integrin using in vitro ligand‐binding assays. It was also demonstrated that CWHM‐12 does not inhibit the function of αIIβ3 integrin, which is essential for platelet aggregation, nor does it affect non‐RGD‐binding integrins.14 CWHM‐12 was delivered by continuous infusion at 100 mg/kg/day (in 50% dimethyl sulfoxide [DMSO], 50% H2O) using Alzet mini‐osmotic pumps (Durect, Cupertino, CA) implanted subcutaneously in mice starting at the end of week 6 for the duration of the final 4 weeks. This dosing regimen has been demonstrated to be efficient in reducing fibrosis in other injury models.14, 20 Minipumps with vehicle (50% DMSO, 50% H2O) were implanted in CDAHFD and chow diet control groups. Ten mice from each group, CDAHFD and chow, received the CWHM‐12 compound, and 10 additional mice from these groups received the vehicle.
Biochemical Analysis
Hepatic triglyceride content was measured using the Triglyceride Colorimetric Assay kit (Cayman Chemicals, Ann Arbor, MI) according to the manufacturer’s instructions.
Plasma levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), glucose, insulin, triglyceride, and cholesterol were measured commercially by Advanced Veterinary Laboratory (Saint Louis, MO).
Real‐Time Quantitative Reverse‐Transcription Polymerase Chain Reaction
Isolation of total RNA from mouse liver tissues and quantitative reverse‐transcription polymerase chain reaction (qPCR) was conducted as described for mouse pancreatic tissues.23 The primer sequences of transcripts evaluated by qPCR are listed in Supporting Table S1. Results were normalized to ribosomal protein large P0 messenger RNA (mRNA).24 The comparative threshold cycle method25 was used to calculate changes in mRNA abundance.
Histopathology
Formalin‐fixed liver sections were embedded in paraffin, sectioned at 5 μm, and stained by hematoxylin and eosin (H&E) using a standard protocol for microscopic evaluation. Slides were graded by an experienced liver pathologist (K.O.) masked to treatment groups, using a semiquantitative scoring system similar to that described.26 Steatosis was scored similarly as follows: 0 (no steatosis or steatosis occupying less than 5% of the hepatic parenchyma), 1 (steatosis occupying 5%‐33% of the parenchyma), 2 (33%‐66% of hepatic parenchyma), 3 (>66% of hepatic parenchyma); lobular and portal inflammatory cell infiltration were scored as follows: 0 (none), 1 (mild, one to two foci per 200× field), 2 (moderate, three to four foci per 200× field), 3 (severe, more than four foci per 200× field); hepatocellular ballooning was scored as follows: 0 (ballooned cells are absent), 1 (ballooned cells are present). To evaluate liver collagen content, paraffin‐embedded liver sections were stained with sirius red/fast green dyes. Sirius red binds to all types of collagens, whereas fast green stains noncollagenous proteins.27 Briefly, liver sections were pretreated to remove paraffin, and nuclei were stained using Weigert’s iron hematoxylin solution. After washing, tissues were stained with 0.1% sirius red (Direct Red 80; Sigma, Saint Louis, MO), 0.1% fast green FCF certified (Sigma) in saturated picric acid for 2 hours. Slides were then washed in water, dehydrated with ethanol and xylene, and finally mounted in Permaslip (Alban Scientific, Inc., Saint Louis, MO). The degree of collagen accumulation was assessed by morphometric analysis.28 About 10 nonoverlapping images randomly selected from lobular areas of each liver section were captured by an investigator masked to treatment, using a Leica DM4000 B microscope (Wetzlar, Germany) equipped with a Leica DFC7000T camera using the 20× objective. To measure sirius red‐positive staining, the same threshold was applied to all images; positive sirius red staining was quantified by digital image analysis using ImageJ software (version 1.37; National Institutes of Health, Bethesda, MD). The amount of collagen was expressed relative to the amount of collagen in the 6‐week chow control group.
Determination of Hepatic Hydroxyproline Content
Hydroxyproline content was determined as a measure of the amount of total collagen present in the liver. Liver tissues were homogenized in distilled water, precipitated with trichloroacetic acid, and hydrolyzed for 48 hours in 12 N HCl at 105°C. Samples were evaporated, and dry pellets were reconstituted in distilled water. Reconstituted samples were centrifuged for 10 minutes at 13,000g, and supernatants were diluted with 12 N HCl to achieve 4 N HCl concentration in the final samples. Liver hydroxyproline content was determined using the Sensitive Tissue Hydroxyproline Assay (QuickZyme Biosciences, Leiden, the Netherlands) according to the manufacturer’s protocol.
Immunohistochemistry
Formalin‐fixed liver tissues were pretreated to remove paraffin by standard methods. Endogenous peroxidase activity was blocked by incubation in 3.3% hydrogen peroxide in methanol for 1 hour at room temperature. Antigen retrieval was performed by a high‐temperature unmasking procedure using citrate‐based Antigen Unmasking Solution (Vector Laboratories, Inc., Burlingame, CA). The antibody to F4/80 (AbD Serotec MCA497GA, clone CI: A3‐1) was obtained from Thermo Fisher Scientific (Waltham, MA), antibody to clusters of differentiation (CD)11b (EPR1344) was obtained from Abcam (Cambridge, MA), antibody to α‐smooth muscle actin (α‐SMA; alternative name ACTA2) clone 1A4 was obtained from Sigma, and antibody to desmin (clone Y66) was obtained from Abcam. F4/80 and CD11b antibodies were detected using the Elite Universal Vectastain ABC Kit (Vector Laboratories). α‐SMA antibody was detected using anti‐mouse immunoglobulin (Ig)2A‐biotin secondary antibody obtained from Thermo Fisher Scientific. Desmin antibody was detected using ImmPRESS Horseradish Peroxidase Anti‐Rabbit IgG (Peroxidase) Polymer Detection kit (Vector Laboratories). Secondary antibody peroxidase activity was detected with diaminobenzidine stain (ImmPACT DAB Peroxidase Substrate kit; Vector Laboratories). Tissues were counterstained with hematoxylin. Multiple slides from each group were immunostained, and about 15 images from each slide at magnification ×200 were captured by an investigator masked to treatment, using a Leica DM4000 B microscope equipped with a Leica DFC7000T camera (Wetzlar, Germany); positive antibody‐stained areas were quantified by digital image analysis using ImageJ software (version 1.37). CD11b‐positive cells were counted on 15 images at magnification ×400 per tissue slide by an investigator masked to the treatment groups.
Apoptotic Cell Detection in Liver Tissues
Apoptotic cells in liver tissues were identified by labeling and detecting DNA strand breaks by the terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick‐end labeling method (TUNEL) using the ApopTag Peroxidase Detection kit (Millipore, Temecula, CA) according to the manufacturer’s instructions. Stained apoptotic cells with hepatocyte morphology were counted by an experienced pathologist masked to the treatment groups. Fifteen high‐power fields on each tissue section were used for counting. To detect apoptotic cells with HSC morphology, TUNEL‐stained liver tissues were subsequently stained with the antibody to desmin (a marker of HSC) as described in Materials and Methods, except that secondary antibody peroxidase activity was detected with the ImmPACT VIP Peroxidase Substrate kit (Vector Laboratories). This kit produces a purple product that is easily distinguished from the brown product of the TUNEL staining. Tissues were counterstained with Contrast Green (SeraCare Life Sciences, Milford, MA). Cells costained for TUNEL and desmin were counted on fifteen 200× fields per each tissue slide by an investigator masked to treatment groups.
Western Blot
Western blotting was performed as described23 using the following primary antibodies: anti‐phosphorylated mothers against decapentaplegic homolog 3 (p‐SMAD3), ab52903, 1:1,000 (Abcam); anti‐glyceraldehyde 3‐phosphate dehydrogenase (anti‐GAPDH), sc‐25778 (Santa Cruz Biotechnology, Inc., Dallas, TX). Protein band intensities were quantified using ImageJ software (version 1.46).
Statistical Analysis
The nonparametric U test was used for statistical analysis of histologic slides. Statistical analysis of data for all other experiments was performed using one‐way analysis of variance followed by a two‐tailed t test (software version 3.1; SigmaStat, San Jose, CA). For qPCR results, ΔCT values were used for statistical analysis, as recommended.29 Data were expressed as mean ± SEM.
Results
Expression of Most Subunits of RGD‐Binding Integrins is Induced in Livers of Mice on CDAHFD Compared to Chow Diet Controls
Expression of RGD‐binding integrin subunits was analyzed in mouse livers by qPCR in mice kept on the CDAHFD and chow diet for 6 and 10 weeks. mRNA expression for all RGD‐binding integrin subunits except β6 was detected in the livers (Fig. 1). High mRNA expression of αv, β1, and β5 subunits detected in the livers (Fig. 1A) is consistent with the possible expression of αvβ1 and αvβ5 integrin heterodimers. Relatively low levels of liver mRNA expression of α5, α8, β3, and β8 (Fig. 1B) are consistent with the possible expression of α5β1, α8β1, αvβ3, and αvβ8 integrin heterodimers, respectively. The expression of the β6 integrin subunit was below the detection level of our experiment with the exception of the CDAHFD group of mice at 10 weeks; this group demonstrated a very low level of liver β6 subunit expression. The mRNA expression of all integrin subunits except β1 was significantly induced in the mice that received the NASH‐inducing CDAHFD.
Figure 1.

Expression of RGD‐binding integrin subunits in mouse livers. Mice were subjected to the NASH‐inducing CDAHFD (gray column) or control chow diet (white column) for 6 and 10 weeks. At the end of the indicated periods, mice were killed and the expression of RGD‐binding subunits was measured by qPCR as described in Materials and Methods. Results were normalized to the ribosomal protein large P0 and depicted as fold difference over the level of the lowest discreetly detected integrin subunit, β8, in the chow control group of mice at week 10. Data are expressed as mean ± SEM; n = 10; ***P < 0.001, **P < 0.01, *P < 0.05. (A) RGD‐binding integrin subunits with relatively high level of expression. (B) RGD‐binding integrin subunits with relatively lower level of expression. Abbreviation: N/D, not detected.
Treatment with the RGD‐Binding Integrin Antagonist CWHM‐12 does not Significantly Affect Hepatic Steatosis or Metabolic Parameters in the CDAHFD‐Fed Group
To assess the effect of CWHM‐12 on the development of hepatic steatosis, we quantified liver fat droplet accumulation by examining H&E‐stained liver sections and measured liver triglyceride content as described in Materials and Methods. In agreement with published data,22 mice on the CDAHFD gained significantly less weight than mice on the chow diet at 6 and 10 weeks (Table 1; the average weight of the mice at the start date was 21.6 g). In contrast, liver weight as well as the ratio of liver to body weight of mice on the CDAHFD was significantly increased at week 6 compared to the chow diet group, consistent with the development of fatty liver. The CWHM‐12 treatment group did not have any significant differences in body or liver weight in the control chow or CDAHFD groups and showed only a slight increase in the ratio of liver to body weight in the CDAHFD group of mice compared with the chow group at week 10 (Table 1). Histologic examination of H&E‐stained liver sections revealed significant lipid droplet accumulation at week 6 that was not further increased by week 10. CWHM‐12 administration did not have any effect on the liver lipid droplet accumulation in any evaluated group of mice (Fig. 2; Table 2). As expected, hepatic triglyceride content dramatically increased in the CDAHFD mouse groups at weeks 6 and 10. CWHM‐12 did not affect liver triglyceride accumulation (Table 1). Similar to the methionine‐choline‐deficient (MCD) diet,30 the CDAHFD caused a decrease in plasma levels of glucose, insulin, triglyceride, and cholesterol (Table 1). CWHM‐12 treatment did not further change any of these metabolic parameters in the CDAHFD‐fed groups of mice, but the compound treatment group did have lower glucose, insulin, and triglyceride levels in the control chow‐fed mice at the end of the study (Table 1).
Table 1.
Effect of CWHM‐12 Administration on Mouse and Liver Weights, Liver Injury, Plasma Metabolic Parameters, and Liver Triglyceride Content
| Chow Week 6 | CDAHFD Week 6 | Chow Week 10 | Chow CWHM‐12 Week 10 | CDAHFD Week 10 | CDAHFD CWHM‐12 Week 10 | |
|---|---|---|---|---|---|---|
| Body weight (g) | 27.9 ± 0.4 | 23.4 ± 0.2‡ | 28.1 ± 0.7 | 28.7 ± 0.8 | 23.6 ± 0.4‡ | 23.7 ± 0.4‡ |
| Liver weight (g) | 1.3 ± 0.03 | 1.6 ± 0.04‡ | 1.3 ± 0.05 | 1.3 ± 0.04 | 1.6 ± 0.1‡ | 1.8 ± 0.1‡ |
| Liver/body weight (%) | 4.5 ± 0.1 | 7.0 ± 0.1‡ | 4.5 ± 0.1 | 4.6 ± 0.1 | 6.9 ± 0.3‡ | 7.8 ± 0.2‡,§ |
| Liver triglyceride (µg/mg) | 6.1 ± 0.6 | 68.2 ± 4.9‡ | 7.1 ± 0.6 | 8.8 ± 1.0 | 70.9 ± 4.1‡ | 71.7 ± 6.1‡ |
| ALT (U/L) | 35 ± 4 | 282 ± 25‡ | 37 ± 23 | 89 ± 18|| | 262 ± 21‡ | 284 ± 15‡ |
| AST (U/L) | 153 ± 13 | 288 ± 16‡ | 190 ± 23 | 228 ± 24 | 282 ± 17† | 345 ± 22‡ |
| Glucose (mg/dL) | 228 ± 7 | 134 ± 4‡ | 214 ± 11 | 148 ± 7¶ | 137 ± 6‡ | 122 ± 11‡ |
| Insulin (ng/mL) | 0.81 ± 0.08 | 0.31 ± 0.02‡ | 0.96 ± 0.13 | 0.52 ± 0.05|| | 0.44 ± 0.07‡ | 0.41 ± 0.05‡ |
| Triglyceride (mg/dL) | 78 ± 3 | 72 ± 2 | 98 ± 6# | 73 ± 3|| | 78 ± 4* | 72 ± 2† |
| Cholesterol (mg/dL) | 94 ± 2 | 48 ± 3‡ | 96 ± 3 | 98 ± 3 | 57 ± 3‡ | 51 ± 4‡ |
Values are expressed as mean ± SEM; n = 10. *P < 0.05, † P < 0.01, ‡ P < 0.001, compared with the chow control group of the corresponding week. § P < 0.05, || P < 0.01, ¶ P < 0.001, compared with the corresponding diet group without CWHM‐12 administration. # P < 0.001, compared with the 6‐week corresponding diet group.
Figure 2.
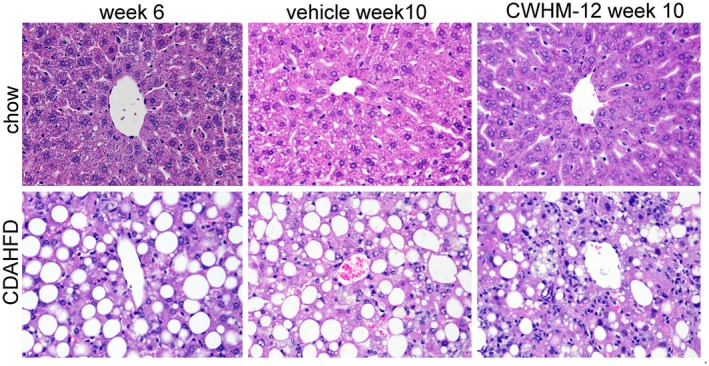
Effect of CWHM‐12 administration on mouse liver histology. Mice were subjected to the NASH‐inducing CDAHFD or control chow diet for 6 and 10 weeks. During the last 4 weeks of the 10‐week period, mice were treated with CWHM‐12. Representative microscopic images of H&E‐stained mouse liver sections (magnification ×200) are depicted with the central vein from the centrilobular area present at the center. Histologic evaluation of these sections is presented in Table 2.
Table 2.
Histologic Analysis of H&E‐Stained Liver Sections
| Histopathology | Chow Week 6 | CDAHFD Week 6 | Chow Week 10 | Chow CWHM‐12 Week 10 | CDAHFD Week 10 | CDAHFD CWHM‐12 Week 10 |
|---|---|---|---|---|---|---|
| Steatosis | 0 ± 0 | 3 ± 0† | 0 ± 0 | 0 ± 0 | 2.89 ± 0.11† | 3 ± 0† |
| Lobular inflammation | 0.20 ± 0.13 | 2.40 ± 0.16† | 0 ± 0 | 0.44 ± 0.18 | 1.33 ± 0.17†,‡ | 2.0 ± 0.29† |
| Portal inflammation | 0.10 ± 0.10 | 0.9 ± 0.10† | 0 ± 0 | 0.22 ± 0.15 | 0.11 ± 0.11‡ | 0.56 ± 0.18* |
| Ballooning | 0 ± 0 | 0.6 ± 0.16* | 0 ± 0 | 0 ± 0 | 0.22 ± 0.15 | 0.56 ± 0.18* |
Histopathology was scored as described in Materials and Methods. Higher values indicate greater severity. Data are expressed as mean ± SEM; n = 10. *P < 0.01, † P < 0.001, compared with the chow control group of the corresponding week. ‡ P < 0.001, compared with the 6‐week corresponding diet group.
Effects of Treatment with the RGD‐Binding Integrin Antagonist CWHM‐12 on Liver Injury, Inflammation, and Hepatocyte Apoptosis
Mice fed the CDAHFD demonstrated a significant increase in the markers of liver injury (plasma ALT and AST) by week 6, and the level of these markers remained similarly elevated at week 10 (Table 1). The group of chow‐fed mice treated with CWHM‐12 showed a mild increase in ALT at 10 weeks, but there were no significant ALT or AST differences with drug treatment in the CDAHFD groups. Hepatocyte ballooning, a structural manifestation of hepatic injury in NASH, was mildly induced by week 6 in CDHAFD mice (Table 2). The CDAHFD also caused substantial lobular and portal inflammation by week 6 as assessed by histopathologic evaluation of H&E‐stained slides (Fig. 2A; Table 2). By week 10 on the CDAHFD, hepatocyte ballooning and inflammatory cell infiltration decreased in all groups, although these reductions were smaller in the CWHM‐12‐treated groups (Table 2). This observation together with the appearance of mild infiltration in the chow‐fed animals treated with high‐dose CWHM‐12 is consistent with the expected pharmacologic effect of inhibiting TGFβ activity. The mRNA expression level of mouse liver proinflammatory cytokines tumor necrosis factor α (TNFα) and chemokine (C‐C motif) ligand 2 (CCL2; also known as macrophage chemoattractant protein 1) were greatly increased with the CDAHFD by week 6 and increased still further by week 10. CWHM‐12‐treated groups had slightly higher mRNA levels of these cytokines compared to vehicle‐treated groups, but the increase did not reach statistical significance (Fig. 3A,B).
Figure 3.
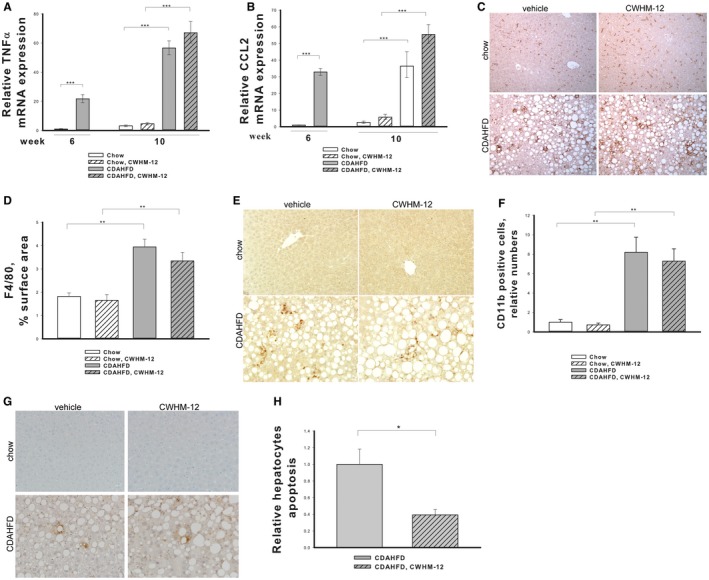
Effect of CWHM‐12 administration on CDAHFD‐induced liver inflammation and hepatocyte apoptosis. Mice were subjected to the NASH‐inducing CDAHFD or control diet for 6 and 10 weeks. During the last 4 weeks, mice were treated with CWHM‐12. Expression of the proinflammatory cytokines mRNAs for (A) TNFα and (B) CCL2 was measured by qPCR and normalized to the acidic ribosomal protein large P0. Data are depicted as fold increase over the level of corresponding genes in the chow control group of mice at week 6 and expressed as mean ± SEM; n = 10; ***P < 0.001. (C) Representative microscopic images of macrophage‐specific F4/80 antibody‐stained liver sections after 10 weeks of CAHFD or control chow diet (magnification ×200). (D) Macrophage‐specific F4/80 immunochistochemical staining was quantified in 15 random macroscopic fields per animal by morphometry as described in Materials and Methods. Data are presented as average percentage of F4/80‐stained areas ± SEM; n = 4; **P < 0.01. (E) Representative microscopic images of macrophage precursor‐specific CD11b antibody‐stained liver seactions after 10 weeks of CDAHFD or control chow diet (magnification ×400). (F) CD11b‐positive cells were counted as described in Materials and Methods. Data are presented as fold increase over the amount of CD11b‐positive cells in the chow control group ± SEM; n = 4; **P < 0.01. (G) Representative images of apoptotic TUNEL‐stained liver sections after 10 weeks of CDAHFD or chow diet (magnification ×200). (H) Apoptotic TUNEL staining of hepatocytes was quantified in 15 randon microscopic fields (magnification ×400) as described in Materials and Methods. Data are presented as fold decrease over the staining in the CDAHFD group ± SEM; n = 8; *P < 0.05.
To further evaluate the effects of CWHM‐12 treatment on liver inflammation in the CDAHFD model, we assessed the infiltration of macrophages in the livers of studied groups of mice at week 10 by immunohistochemistry using the macrophage‐specific markers F4/80 and CD11b. F4/80 marks the majority of tissue‐resident macrophages, while CD11b is expressed by newly migrated macrophage precursors.31 There was an increase in both these macrophage populations in livers of CDAHFD groups of mice (Fig. 3C,E), and the macrophages formed crown‐like structures,32 which are a unique histologic feature of NASH in mice and humans (Fig. 3C). CWHM‐12 treatment did not influence the amount or structural organization of macrophages (Fig. 3D,F).
Increased hepatocyte apoptosis is a key feature in the pathogenesis of NASH. To evaluate the effect of CWHM‐12 on hepatocyte apoptosis, we performed TUNEL staining of apoptotic cells in liver tissues (Fig. 3G). We did not detect any apoptotic cells in livers of the mice on the chow diet, whereas the CDAHFD led to noticeable TUNEL staining. Counting apoptotic cells with hepatocyte morphology demonstrated a significant decrease in apoptotic cells in the CWHM‐12‐treated group (Fig. 3H).
The RGD‐Binding Integrin Antagonist CWHM‐12 Significantly Reverses Liver Fibrosis Induced by the CDAHFD
At week 6, collagen deposition assessed by sirius red staining was already observed in the CDAHFD group of mice, and this increased in severity by week 10 (Fig. 4A). CWHM‐12 treatment significantly decreased collagen deposition in the livers as assessed by morphometric analysis of sirius red‐stained liver tissues and by determination of liver hydroxyproline content (Fig. 4B,C), even to a level below that observed at the start of treatment at week 6. Increased liver collagen deposition was mostly detected in lobular areas. At the transcriptional level, the CDAHFD induced increases in gene expression of type I and type III collagens that were significantly reduced by CWHM‐12 administration (Fig. 4D,E).
Figure 4.

Effect of CWHM‐12 administration on CDAHFD‐induced liver collagen accumulation. Mice were subjected to NASH‐inducing CDAHFD or control diet for 6 and 10 weeks. During the last 4 weeks of the 10‐week period, mice were treated with CWHM‐12. (A) Representative microscopic images of sirius red‐stained liver sections (magnification ×200). (B) Sirius red staining in lobular areas of the livers was quantified by morphometric analysis and depicted as fold increase over the 6‐week chow diet group. Data are presented as mean ± SEM; n = 10; *P < 0.05, **P < 0.01, ***P < 0.001. (C) Hydroxyproline was measured in the livers as described in Materials and Methods. Data are presented as mean ± SEM; n = 4; **P < 0.01. (D,E) Expression of Col1α1 and Col3α1 mRNA was measured by qPCR and normalized to the acidic ribosomal protein large P0. Data are depicted as fold increase over the level of corresponding genes in the chow control group of mice at week 6 and expressed as mean ± SEM; n = 10; ***P < 0.001.
Treatment with the RGD‐Binding Integrin Antagonist CWHM‐12 Reduces CDAHFD‐Activated HSCs by Inducing HSC Apoptosis
Activated HSCs are the main source of ECM in hepatic fibrogenesis. To gain insight into the antifibrotic effect of CWHM‐12, we examined the effect of CWHM‐12 on α‐SMA, a marker of HSC activation, in the CDAHFD model. The CDAHFD caused significant induction in α‐SMA‐positive staining in the parenchyma, indicating the presence of activated HSCs (Fig. 5A). CWHM‐12 treatment significantly decreased α‐SMA staining, indicating a reduction in the abundance of activated HSCs (Fig. 5A,B). Consistent with the current understanding of HSCs, the decrease in α‐SMA staining could be caused by HSC senescence and apoptosis as well as by reversal of activated HSCs to a quiescent phenotype. To gain insights into the mechanisms of the CWHM‐12‐induced decrease in α‐SMA‐positive HSCs, we analyzed the effect of CWHM‐12 on total HSCs by measuring changes in the expression of desmin, which is expressed in both activated and quiescent HSCs,33 at weeks 6 and 10. The expression of desmin was induced by the CDAHFD by week 6 and further induced by week 10 (Fig. 5C,D). Livers from CDAHFD‐treated mice had significantly reduced levels of desmin expression (below the starting point at week 6), suggesting that HSC loss may account for the treatment‐associated decrease in activated HSCs. To determine if HSC loss might be responsible for the decrease in fibrosis after CWHM‐12 administration, we quantified apoptotic HSCs in our model by double staining liver tissues to detect apoptosis (TUNEL) in HSCs identified by staining for desmin (Fig. 5E). On masked histologic assessment, the abundance of apoptotic cells with HSC morphology significantly decreased after CWHM‐12 administration (Fig. 5F).
Figure 5.
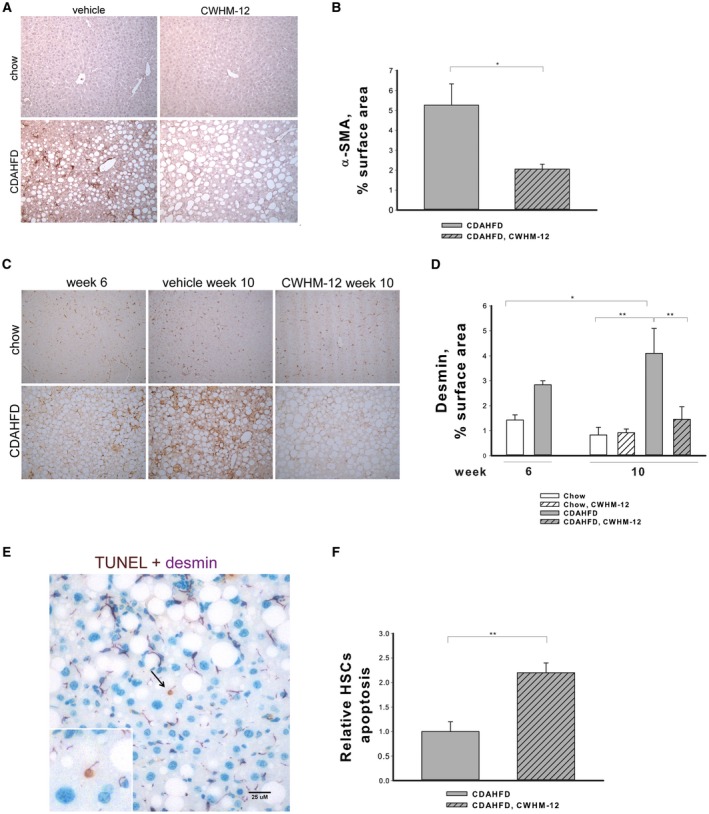
Effect of CWHM‐12 administration on CDAHFD‐induced HSC activation. Mice were subjected to the NASH‐inducing CDAHFD or control diet for 6 and 10 weeks. During the last 4 weeks of the 10‐week period, mice were treated with CWHM‐12. (A) Representative microscopic images of activated HSCs detected by α‐SMA staining of liver sections at 10 weeks (magnification ×200). (B) Activated HSC‐specific α‐SMA immunochistochemical staining was quantified in 15 random microscopic fields per liver tissue slide by morphometry as described in Materials and Methods. Data are presented as the average percentage of α‐SMA‐stained areas ± SEM; n = 4; *P < 0.05. (C) Representative microscopic images of HSC‐specific desmin antibody‐stained liver sections (magnification ×200). (D) Desmin staining was quantified in 15 random microscopic fields per liver tissue slide by morphometry as described in Materials and Methods. Data are presented as the average percentage of desmin‐stained areas ± SEM; n =3; *P < 0.05, **P < 0.01. (E) Representative microscopic image of liver tissue from CDAHFD‐treated mice stained with the apoptotic TUNEL stain (brown) and with desmin (purple), the marker of HSC (magnification ×200). Magnified image of doubly stained apoptotic HSC is depicted in the left corner. (F) Doubly stained apoptotic HSCs were quantified in 15 random microscopic fields (magnification ×200) as described in Materials and Methods. Data are presented as fold increase over the staining in the CDAHFD group ± SEM; n = 4; **P < 0.01.
Treatment with the RGD‐Binding Integrin Antagonist CWHM‐12 Decreases CDAHFD‐Induced TGFβ Activation in the Liver
As a measure of TGFβ activation, p‐SMAD3 was evaluated in the liver extracts by western blotting. p‐SMAD3 was barely detectible in the livers of mice on the chow diet, while the CDAHFD greatly increased the liver expression of p‐SMAD3 (Fig. 6A). Treatment with CWHM‐12 significantly reduced expression of p‐SMAD (Fig. 6B), suggesting reduced TGFβ‐mediated signal transduction. As a second measure of TGFβ activation, we assessed the expression of connective tissue growth factor (CTGF) because it is a downstream target of TGFβ and is a regulator of many profibrogenic actions of TGFβ in liver fibrosis.34 Analysis by qPCR showed that CWHM‐12 administration significantly decreased CDAHFD‐induced expression of this cytokine (Fig. 6C).
Figure 6.
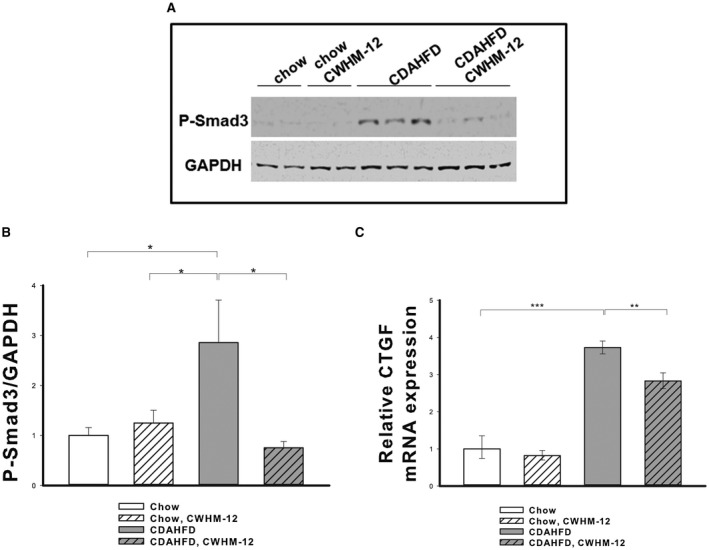
Effect of CWHM‐12 administration on CDAHFD‐induced liver TGFβ signaling. Mice were subjected to the NASH‐inducing CDAHFD or control diet for 10 weeks. During the last 4 weeks, 10 mice were treated with CWHM‐12. (A) Representative western blot of liver extracts with p‐SMAD3‐specific antibodies. GAPDH was used as a loading control. (B) Densitometric analysis of p‐SMAD expression normalized to GAPDH epression. Data are expressed as mean ± SEM; n = 10; *P < 0.05. (C) CTGF expression was measured by qPCR, normalized to the ribosomal protein large P0, and depicted as fold increase over the level of CTGF expression in the chow control group of mice. **P < 0.01, ***P < 0.001.
Discussion
NASH is a common disease that can lead to progressive liver fibrosis, cirrhosis, liver cancer, and death.5 Treatment options for NASH are currently being evaluated in clinical trials and are focused on NASH and NASH‐induced fibrosis. Because the severity of NASH‐induced liver fibrosis is the main predictor of liver‐related mortality in NASH,7 it is critically important to identify the mechanisms responsible for fibrosis development in NASH and to develop treatments that are effective in the prevention and reversal of fibrosis. Recently, RGD‐binding integrins were found to play an important role in the development of organ fibrosis,35 and several studies, including a study from our laboratory, demonstrated the efficacy of a potent small‐molecule synthetic analog of the RGD peptide (CWHM‐12) in mouse‐injury models of liver, lung, muscle, and pancreatic fibrosis.14, 20, 21 Here, by assessing CWHM‐12 in a mouse model of NASH, we provide evidence supporting the role of RGD‐binding integrins in the pathogenesis of NASH‐induced liver fibrosis.
There are many mouse models of NASH that reproduce different aspects of the human disease.36 We chose the CDAHFD mouse model of NASH22 for this study because together with excessive liver fat accumulation and liver injury, the mice rapidly develop progressive hepatic fibrosis without the weight loss seen in the classic MCD model. Although they do not fully recapitulate the metabolic changes in human NASH, the MCD‐based diets have been shown to be efficient and reproducible for studying severe liver damage and progressive fibrosis with mechanisms similar to human NASH progression.37 Here, we demonstrated that most of the RGD‐binding integrin subunits were expressed in the mouse liver at the transcript level and that the expression of all but integrin α6/β1 (Itgb1) was induced by the CDAHFD. The αv subunit forms heterodimers with multiple β subunits; α5 and α8 subunits form heterodimers only with β1. The expression of liver Itgb6 mRNA was not detectable in our experimental conditions in the groups of mice except the CDAHFD group at 10 weeks; this group demonstrated a low level of expression. Because the β6 subunit only forms a heterodimer with αv integrin, this suggests that αvβ6 is not widely expressed in either the normal liver or CDAHFD‐induced NASH. However, these data do not preclude the possibility of its significant expression in small subpopulations of the total cells in mouse liver. The CWHM‐12 compound has the potential to affect functions of all the expressed RGD‐binding integrins simultaneously,14 allowing direct functional evaluation of the importance of the whole group in NASH.
Steatohepatitis, or the presence of excess liver fat associated with hepatocellular injury and inflammation, is the driver of liver fibrogenesis in NASH. CWHM‐12 was not expected to alter the underlying steatosis in this model because there is no prior evidence of integrin involvement in this process. Indeed, we observed that the CDAHFD caused significant liver steatosis that was not affected by CWHM‐12 treatment.
The CDAHFD increased liver injury and inflammatory markers, which were highest by week 6. After an additional 4 weeks on the CDAHFD, liver portal and lobular inflammation and ballooning had decreased as assessed by pathology evaluation. This reduction from the peak of mouse liver inflammatory scores with additional time on the CDAHFD has been reported.22 Inflammatory scores for the CWHM‐12‐treated mice on the CDAHFD were also reduced following the peak observed at week 6, although to a lesser extent than in the vehicle‐treated control CDAHFD group, and ballooning was not decreased after 6 weeks on the CDAHFD. Thus, at the time of sacrifice at 10 weeks, livers from the CWHM‐12‐treated group of mice on the CDAHFD may have retained more inflammation and ballooning than the CDAHFD vehicle control, although the difference did not reach statistical significance. Because TGFβ is a pleotropic cytokine that participates in all stages of liver disease progression38 and has well‐known inflammation‐suppressing properties, it seems possible that this difference may be attributable to the blockade by CWHM‐12 of RGD‐binding integrin‐induced TGFβ activation.11, 12, 13 Additional analysis of liver inflammatory parameters induced by the CDAHFD, including macrophage infiltration and expression of the proinflammatory cytokines TNF‐α and CCL2, did not identify any statistically significant differences between control and CWHM‐12‐treated groups.
Interestingly, the very high dose of continuous CWHM‐12 administration used in this proof‐of‐concept study for the role of RGD‐binding integrins in liver fibrosis decreased blood glucose, insulin, and triglyceride levels in the chow‐feeding group of mice but not in the CDAHFD‐feeding group. It is noteworthy that the CDAHFD mouse model, being a derivative from the MCD model, is not the best model to study metabolic aspects of NASH because this diet does not cause the insulin resistance seen in most patients with NASH.30, 37 Similar to the MCD model, CDAHFD‐feeding groups have decreased evidence of metabolic disease, and in contrast to the chow diet group, CWHM‐12 administration did not decrease them further. The role of RGD‐binding integrins in metabolic diseases is largely unknown, and future studies, including use of a range of treatment doses, are needed to further validate and determine whether our observation has not just statistical but also biological significance.
In accordance with the accumulating evidence for the critical importance of RGD‐binding integrins in tissue fibrogenesis across a range of mouse models, inhibiting the RGD‐binding integrins with CWHM‐12 dramatically reversed established liver fibrosis that developed in the NASH‐inducing diet. Liver collagen content measured by morphometric analysis of sirius red collagen staining and by liver hydroxyproline content was significantly increased by the CDAHFD by week 6 and increased further by week 10. CWHM‐12 administration from week 6 to week 10 of diet treatment decreased collagen content to a level significantly less than the amount of collagen found at week 6, demonstrating the possibility not only of preventing continued collagen deposition but also reversal of established fibrosis despite ongoing injury. The expression of major fibrotic collagen types I and III genes in the liver39 was also substantially reduced by CWHM‐12, supporting this conclusion.
Activated HSCs are the main source of ECM in hepatic fibrogenesis.40 We therefore examined the effect of CWHM‐12 on HSC activation in this model by evaluating α‐SMA staining, an established marker of HSC activation.10 We found it to be markedly reduced. The decrease in HSC activation could be attributed either to the loss of activated HSCs, the reversal of activated HSCs to a quiescent phenotype, or a combination of these mechanisms.41 Assessment with a marker of total (activated and nonactivated) HSCs, desmin,33 revealed significantly lower abundance of HSCs after CWHM‐12 administration in diet‐fed animals. Apoptosis is considered to be the main mechanism for the clearance of HSCs during resolution of liver fibrogenesis.42 Because RGD‐binding integrins play an important role in TGFβ activation15, 43, 44, 45, 46 and TGFβ has been demonstrated to prevent apoptosis of HSCs,47, 48 blocking TGFβ activation by CWHM‐12 could lead to an increase in HSC apoptosis. In support of this hypothesis, we found that CWHM‐12 significantly increased the number of apoptotic HSCs (cells that were both TUNEL and desmin positive).
TGFβ is considered to be the central cytokine responsible for many aspects of fibrotic liver diseases,49 and SMAD3 phosphorylation is a major downstream mediator of TGFβ signaling. Our data demonstrate that diet‐induced SMAD3 phosphorylation, together with transcription of the TGFβ‐inducible genes collagen 1a1 (Col1a1) and CTGF, were significantly decreased by CWHM‐12 administration. These findings are consistent with TGFβ activation as a probable primary mechanism through which RGD‐binding integrins regulate liver fibrogenesis. RGD‐binding integrins not only play an important role in mediating the release of latency‐associated peptide from the latent complex but are also important mechanotransducing sensors that convey information to the cell regarding stiffness of the ECM.50 Interference with these signals from the fibrotic matrix may modulate the ability of the HSCs to generate the force needed for effective TGFβ activation and may promote the apoptosis we observed in activated HSCs if these cells have become dependent on sustained integrin signaling for survival.
The decrease in hepatocyte apoptosis observed in our experiments in the CWHM‐12‐treated group is also consistent with the decrease in TGFβ signaling because, in contrast to its effect on HSCs, TGFβ promotes hepatocyte apoptosis.51 In addition to activation of TGFβ, RGD‐binding integrins mediate other functions that we speculate are also likely to be important at various stages of initiation, development, maintenance, and resolution of fibrosis. RGD‐binding integrins are the principal regulators of ECM remodeling and migration of many different cell types so that modulating their function may alter trafficking of fibroblasts, fibroblast precursors, and immune cells to sites of developing fibrosis.52
A limitation of the present study is that it was conducted on young animals (3 and 4 months old at the time of sacrifice). Young animals have a different ECM composition, higher regenerative capacity, and higher collagen turnover rate compared to old animals.53, 54 To address this issue, future studies using aged mice for evaluation of the RGD inhibitors are warranted.
CWHM‐12 inhibits multiple RGD integrins, a property that may be beneficial for efficacy due to activity against redundant or complementary profibrotic mechanisms. Various RGD integrins have been implicated in latent TGFβ activation,12 mechanosensing of matrix stiffness controlling myofibroblast function and survival,50, 55 recruitment of myofibroblast precursors to sites of organ injury,56 binding of profibrotic growth factors (such as CTGF),57 and the fibronectin organization necessary for collagen deposition and maintenance.58 However, an important question that must be addressed in future studies is the potential for RGD integrin treatments to affect not only the targeted pathologic process of fibrosis but also nontargeted physiologic processes in which these integrins participate. They have other diverse normal and disease‐specific biological roles affecting osteoclast function and bone resorption,59 autoimmunity and colitis,60 senescence,61 cell migration, proliferation, apoptosis, and angiogenesis.62 The proof‐of‐concept study described here was limited to testing a single high‐dose treatment, 100 mg/kg/day, delivered by continuous infusion. Studies using various other dosing regiments in this model and other NASH models will be critical to evaluate the net benefits and safety of agents, such as CWHM‐12, for possible clinical applications. It is encouraging that other inhibitors targeting multiple RGD integrins have progressed to clinical studies for cancer indications and have generally been shown to have acceptable safety profiles.63
In conclusion, this study provides evidence that RGD‐binding integrins play an important role in NASH‐associated liver fibrosis. The small molecule, RGD peptidomimetic, integrin‐antagonist CWHM‐12 demonstrated efficacy in decreasing already established fibrosis and reducing hepatocyte apoptosis in a NASH mouse model. The data support as possible mechanisms suppression of TGFβ activation and induction of HSC apoptosis. Targeting multiple specific integrins that are up‐regulated in a temporospatially specific manner during the course of NASH progression could be a highly promising approach to treating patients with NASH with advanced fibrosis.
Potential conflict of interest
Dr. Griggs owns intellectual property rights and stock in, received grants from, and consults for Indalo Therapeutics. Dr. Neuschwander‐Tetri consults for and advises Allegran, Arrowhead, Blade, Boehringer Ingleheim, Bristol‐Myer Squibb, Coherus, Consynance, Cymabay, Enanta, Gelesis, Gilead, Intercept, Karos, Lexicon, Madrigal, Merck, Medimmune, Metacrine, NGM, pH‐Pharma, and Prometheus. The other authors have nothing to report.
Supporting information
Supported by a Saint Louis University Liver Center Grant and H.N. was supported by a grant from Japan Research Foundation for Clinical Pharmacology.
References
- 1. Younossi ZM, Stepanova M, Afendy M, Fang Y, Younossi Y, Mir H, et al. Changes in the prevalence of the most common causes of chronic liver diseases in the United States from 1988 to 2008. Clin Gastroenterol Hepatol 2011;9:524‐530. [DOI] [PubMed] [Google Scholar]
- 2. Ahmed A, Wong RJ, Harrison SA. Nonalcoholic fatty liver disease review: diagnosis, treatment, and outcomes. Clin Gastroenterol Hepatol 2015;13:2062‐2070. [DOI] [PubMed] [Google Scholar]
- 3. Singh S, Allen AM, Wang Z, Prokop LJ, Murad MH, Loomba R. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: a systematic review and meta‐analysis of paired‐biopsy studies. Clin Gastroenterol Hepatol 2015;13:643‐654.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Neuschwander‐Tetri BA. Non‐alcoholic fatty liver disease. BMC Med 2017;15:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Younossi ZM, Loomba R, Rinella ME, Bugianesi E, Marchesini G, Neuschwander‐Tetri BA, et al. Current and future therapeutic regimens for nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology 2018;68:361‐371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ekstedt M, Hagstrom H, Nasr P, Fredrikson M, Stal P, Kechagias S, et al. Fibrosis stage is the strongest predictor for disease‐specific mortality in NAFLD after up to 33 years of follow‐up. Hepatology 2015;61:1547‐1554. [DOI] [PubMed] [Google Scholar]
- 7. Dulai PS, Singh S, Patel J, Soni M, Prokop LJ, Younossi Z, et al. Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: systematic review and meta‐analysis. Hepatology 2017;65:1557‐1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hagstrom H, Nasr P, Ekstedt M, Hammar U, Stal P, Hultcrantz R, et al. Fibrosis stage but not NASH predicts mortality and time to development of severe liver disease in biopsy‐proven NAFLD. J Hepatol 2017;67:1265‐1273. [DOI] [PubMed] [Google Scholar]
- 9. Puche JE, Saiman Y, Friedman SL. Hepatic stellate cells and liver fibrosis. Compr Physiol 2013;3:1473‐1492. [DOI] [PubMed] [Google Scholar]
- 10. Bataller R, Brenner DA. Liver fibrosis. J Clin Invest 2005;115:209‐218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shi M, Zhu J, Wang R, Chen X, Mi L, Walz T, et al. Latent TGF‐beta structure and activation. Nature 2011;474:343‐349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Worthington JJ, Klementowicz JE, Travis MA. TGFbeta: a sleeping giant awoken by integrins. Trends Biochem Sci 2011;36:47‐54. [DOI] [PubMed] [Google Scholar]
- 13. Nishimura SL. Integrin‐mediated transforming growth factor‐beta activation, a potential therapeutic target in fibrogenic disorders. Am J Pathol 2009;175:1362‐1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Henderson NC, Arnold TD, Katamura Y, Giacomini MM, Rodriguez JD, McCarty JH, et al. Targeting of alphav integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat Med 2013;19:1617‐1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Reed NI, Jo H, Chen C, Tsujino K, Arnold TD, DeGrado WF, et al. The alphavbeta1 integrin plays a critical in vivo role in tissue fibrosis. Sci Transl Med 2015;7:288ra79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Takada Y, Ye X, Simon S. The integrins. Genome Biol 2007;8:215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Weis SM, Cheresh DA. alphaV integrins in angiogenesis and cancer. Cold Spring Harb Perspect Med 2011;1:a006478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Larsen M, Artym VV, Green JA, Yamada KM. The matrix reorganized: extracellular matrix remodeling and integrin signaling. Curr Opin Cell Biol 2006;18:463‐471. [DOI] [PubMed] [Google Scholar]
- 19. De Franceschi N, Hamidi H, Alanko J, Sahgal P, Ivaska J. Integrin traffic ‐ the update. J Cell Sci 2015;128:839‐852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ulmasov B, Neuschwander‐Tetri BA, Lai J, Monastyrskiy V, Bhat T, Yates MP, et al. Inhibitors of Arg‐Gly‐Asp‐binding integrins reduce development of pancreatic fibrosis in mice. Cell Mol Gastroenterol Hepatol 2016;2:495‐518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Murray IR, Gonzalez ZN, Baily J, Dobie R, Wallace RJ, Mackinnon AC, et al. alphav integrins on mesenchymal cells regulate skeletal and cardiac muscle fibrosis. Nat Commun 2017;8:1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Matsumoto M, Hada N, Sakamaki Y, Uno A, Shiga T, Tanaka C, et al. An improved mouse model that rapidly develops fibrosis in non‐alcoholic steatohepatitis. Int J Exp Pathol 2013;94:93‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ulmasov B, Oshima K, Rodriguez MG, Cox RD, Neuschwander‐Tetri BA. Differences in the degree of cerulein‐induced chronic pancreatitis in C57BL/6 mouse substrains lead to new insights in identification of potential risk factors in the development of chronic pancreatitis. Am J Pathol 2013;183:692‐708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Akamine R, Yamamoto T, Watanabe M, Yamazaki N, Kataoka M, Ishikawa M, et al. Usefulness of the 5' region of the cDNA encoding acidic ribosomal phosphoprotein P0 conserved among rats, mice, and humans as a standard probe for gene expression analysis in different tissues and animal species. J Biochem Biophys Methods 2007;70:481‐486. [DOI] [PubMed] [Google Scholar]
- 25. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) Method. Methods 2001;25:402‐408. [DOI] [PubMed] [Google Scholar]
- 26. Liang W, Menke AL, Driessen A, Koek GH, Lindeman JH, Stoop R, et al. Establishment of a general NAFLD scoring system for rodent models and comparison to human liver pathology. PLoS One 2014;9:e115922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lopez‐De Leon A, Rojkind M. A simple micromethod for collagen and total protein determination in formalin‐fixed paraffin‐embedded sections. J Histochem Cytochem 1985;33:737‐743. [DOI] [PubMed] [Google Scholar]
- 28. French SW, Miyamoto K, Wong K, Jui L, Briere L. Role of the Ito cell in liver parenchymal fibrosis in rats fed alcohol and a high fat‐low protein diet. Am J Pathol 1988;132:73‐85. [PMC free article] [PubMed] [Google Scholar]
- 29. Yuan JS, Reed A, Chen F, Stewart CN Jr. Statistical analysis of real‐time PCR data. BMC Bioinformatics 2006;7:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rinella ME, Green RM. The methionine‐choline deficient dietary model of steatohepatitis does not exhibit insulin resistance. J Hepatol 2004;40:47‐51. [DOI] [PubMed] [Google Scholar]
- 31. Lloyd CM, Phillips AR, Cooper GJ, Dunbar PR. Three‐colour fluorescence immunohistochemistry reveals the diversity of cells staining for macrophage markers in murine spleen and liver. J Immunol Methods 2008;334:70‐81. [DOI] [PubMed] [Google Scholar]
- 32. Itoh M, Kato H, Suganami T, Konuma K, Marumoto Y, Terai S, et al. Hepatic crown‐like structure: a unique histological feature in non‐alcoholic steatohepatitis in mice and humans. PLoS One 2013;8:e82163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev 2008;88:125‐172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rachfal AW, Brigstock DR. Connective tissue growth factor (CTGF/CCN2) in hepatic fibrosis. Hepatol Res 2003;26:1‐9. [DOI] [PubMed] [Google Scholar]
- 35. Conroy KP, Kitto LJ, Henderson NC. alphav integrins: key regulators of tissue fibrosis. Cell Tissue Res 2016;365:511‐519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ibrahim SH, Hirsova P, Malhi H, Gores GJ. Animal models of nonalcoholic steatohepatitis: eat, delete, and inflame. Dig Dis Sci 2016;61:1325‐1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Machado MV, Michelotti GA, Xie G, Almeida Pereira T, Boursier J, Bohnic B, et al. Mouse models of diet‐induced nonalcoholic steatohepatitis reproduce the heterogeneity of the human disease. PLoS One 2015;10:e0127991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fabregat I, Moreno‐Caceres J, Sanchez A, Dooley S, Dewidar B, Giannelli G, et al. IT‐LIVER Consortium. TGF‐beta signalling and liver disease. FEBS J 2016;283:2219‐2232. [DOI] [PubMed] [Google Scholar]
- 39. Maher JJ, McGuire RF. Extracellular matrix gene expression increases preferentially in rat lipocytes and sinusoidal endothelial cells during hepatic fibrosis in vivo. J Clin Invest 1990;86:1641‐1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mederacke I. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun 2013;4:2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liu X, Xu J, Brenner DA, Kisseleva T. Reversibility of liver fibrosis and inactivation of fibrogenic myofibroblasts. Curr Pathobiol Rep 2013;1:209‐214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Elsharkawy AM, Oakley F, Mann DA. The role and regulation of hepatic stellate cell apoptosis in reversal of liver fibrosis. Apoptosis 2005;10:927‐939. [DOI] [PubMed] [Google Scholar]
- 43. Asano Y, Ihn H, Yamane K, Jinnin M, Mimura Y, Tamaki K. Increased expression of integrin alpha(v)beta3 contributes to the establishment of autocrine TGF‐beta signaling in scleroderma fibroblasts. J Immunol 2005;175:7708‐7718. [DOI] [PubMed] [Google Scholar]
- 44. Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell 1999;96:319‐328. [DOI] [PubMed] [Google Scholar]
- 45. Asano Y, Ihn H, Yamane K, Jinnin M, Mimura Y, Tamaki K. Involvement of alphavbeta5 integrin‐mediated activation of latent transforming growth factor beta1 in autocrine transforming growth factor beta signaling in systemic sclerosis fibroblasts. Arthritis Rheum 2005;52:2897‐2905. [DOI] [PubMed] [Google Scholar]
- 46. Mu D, Cambier S, Fjellbirkeland L, Baron JL, Munger JS, Kawakatsu H, et al. The integrin alpha(v)beta8 mediates epithelial homeostasis through MT1‐MMP‐dependent activation of TGF‐beta1. J Cell Biol 2002;157:493‐507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Saile B, Matthes N, Knittel T, Ramadori G. Transforming growth factor beta and tumor necrosis factor alpha inhibit both apoptosis and proliferation of activated rat hepatic stellate cells. Hepatology 1999;30:196‐202. [DOI] [PubMed] [Google Scholar]
- 48. Saile B, Matthes N, El Armouche H, Neubauer K, Ramadori G. The bcl, NFkappaB and p53/p21WAF1 systems are involved in spontaneous apoptosis and in the anti‐apoptotic effect of TGF‐beta or TNF‐alpha on activated hepatic stellate cells. Eur J Cell Biol 2001;80:554‐561. [DOI] [PubMed] [Google Scholar]
- 49. Dooley S, ten Dijke P. TGF‐beta in progression of liver disease. Cell Tissue Res 2012;347:245‐256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lampi MC, Reinhart‐King CA. Targeting extracellular matrix stiffness to attenuate disease: from molecular mechanisms to clinical trials. Sci Transl Med 2018;10.pii:eaao0475. [DOI] [PubMed] [Google Scholar]
- 51. Black D, Lyman S, Qian T, Lemasters JJ, Rippe RA, Nitta T, et al. Transforming growth factor beta mediates hepatocyte apoptosis through Smad3 generation of reactive oxygen species. Biochimie 2007;89:1464‐1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Theocharis AD, Skandalis SS, Gialeli C, Karamanos NK. Extracellular matrix structure. Adv Drug Deliv Rev 2016;97:4‐27. [DOI] [PubMed] [Google Scholar]
- 53. Karsdal MA, Genovese F, Madsen EA, Manon‐Jensen T, Schuppan D. Collagen and tissue turnover as a function of age: implications for fibrosis. J Hepatol 2016;64:103‐109. [DOI] [PubMed] [Google Scholar]
- 54. Delire B, Lebrun V, Selvais C, Henriet P, Bertrand A, Horsmans Y, et al. Aging enhances liver fibrotic response in mice through hampering extracellular matrix remodeling. Aging (Albany NY) 2016;9:98‐113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Santos A, Lagares D. Matrix stiffness: the conductor of organ fibrosis. Curr Rheumatol Rep 2018;20:2. [DOI] [PubMed] [Google Scholar]
- 56. McCurley A, Alimperti S, Campos‐Bilderback SB, Sandoval RM, Calvino JE, Reynolds TL, et al. Inhibition of alphavbeta5 integrin attenuates vascular permeability and protects against renal ischemia‐reperfusion injury. J Am Soc Nephrol 2017;28:1741‐1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gao R, Brigstock DR. Connective tissue growth factor (CCN2) induces adhesion of rat activated hepatic stellate cells by binding of its C‐terminal domain to integrin alpha(v)beta(3) and heparan sulfate proteoglycan. J Biol Chem 2004;279:8848‐8855. [DOI] [PubMed] [Google Scholar]
- 58. Zollinger AJ, Smith ML. Fibronectin, the extracellular glue. Matrix Biol 2017;60‐61:27‐37. [DOI] [PubMed] [Google Scholar]
- 59. Nakamura I, Duong LT, Rodan SB, Rodan GA. Involvement of alpha(v)beta3 integrins in osteoclast function. J Bone Miner Metab 2007;25:337‐344. [DOI] [PubMed] [Google Scholar]
- 60. Travis MA, Reizis B, Melton AC, Masteller E, Tang Q, Proctor JM, et al. Loss of integrin alpha(v)beta8 on dendritic cells causes autoimmunity and colitis in mice. Nature 2007;449:361‐365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rapisarda V, Borghesan M, Miguela V, Encheva V, Snijders AP, Lujambio A, et al. Integrin beta 3 regulates cellular senescence by activating the TGF‐beta pathway. Cell Rep 2017;18:2480‐2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nieberler M, Reuning U, Reichart F, Notni J, Wester HJ, Schwaiger M, et al. Exploring the role of RGD‐recognizing integrins in cancer. Cancers (Basel) 2017;9,pii.E116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ley K, Rivera‐Nieves J, Sandborn WJ, Shattil S. Integrin‐based therapeutics: biological basis, clinical use and new drugs. Nat Rev Drug Discov 2016;15:173‐183. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials