

Abstract
Hedgehog (HH) signaling participates in hepatobiliary repair after injury and is activated in patients with cholangiopathies. Cholangiopathies are associated with bile duct (BD) hyperplasia, including expansion of peribiliary glands, the niche for biliary progenitor cells. The inflammation‐associated cytokine interleukin (IL)‐33 is also up‐regulated in cholangiopathies, including cholangiocarcinoma. We hypothesized that HH signaling synergizes with IL‐33 in acute inflammation‐induced BD hyperplasia. We measured extrahepatic BD (EHBD) thickness and cell proliferation with and without an IL‐33 challenge in wild‐type mice, mice overexpressing Sonic HH (pCMV‐Shh), and mice with loss of the HH pathway effector glioma‐associated oncogene 1 (Gli1lacZ/lacZ). LacZ reporter mice were used to map the expression of HH effector genes in mouse EHBDs. An EHBD organoid (BDO) system was developed to study biliary progenitor cells in vitro. EHBDs from the HH overexpressing pCMV‐Shh mice showed increased epithelial cell proliferation and hyperplasia when challenged with IL‐33. In Gli1lacZ/lacZ mice, we observed a decreased proliferative response to IL‐33 and decreased expression of Il6. The HH ligands Shh and Indian HH (Ihh) were expressed in epithelial cells, whereas the transcriptional effectors Gli1, Gli2, and Gli3 and the HH receptor Patched1 (Ptch1) were expressed in stromal cells, as assessed by in situ hybridization and lacZ reporter mice. Although BDO cells lacked canonical HH signaling, they expressed the IL‐33 receptor suppression of tumorigenicity 2. Accordingly, IL‐33 treatment directly induced BDO cell proliferation in a nuclear factor κB‐dependent manner. Conclusion: HH ligand overexpression enhances EHBD epithelial cell proliferation induced by IL‐33. This proproliferative synergism of HH and IL‐33 involves crosstalk between HH ligand‐producing epithelial cells and HH‐responding stromal cells.
Abbreviations
- ANOVA
analysis of variance
- BD
bile duct
- BDO
bile duct organoid
- BEC
biliary epithelial cell
- BrdU
5‐bromo‐2′‐deoxyuridine
- CK19
cytokeratin 19
- DAPI
4′,6‐diamidino‐2‐phenylindole
- EdU
5‐ethynyl‐2′‐deoxyuridine
- EHBD
extrahepatic bile duct
- GLI
glioma‐associated oncogene
- H&E
hematoxylin and eosin
- HH
hedgehog
- Hprt
hypoxanthine guanine phosphoribosyl transferase
- IHH
Indian hedgehog
- IL
interleukin
- IL‐6R
interleukin 6 receptor
- mRNA
messenger RNA
- NF‐κB
nuclear factor κB
- PBG
peribiliary gland
- PBS
phosphate‐buffered saline
- pCMV
promoter for cytomegalovirus
- PFA
paraformaldehyde
- PTCH1
Patched1
- QNZ
N4‐[2‐(4‐phenoxyphenyl)ethyl]‐4,6‐quinazolinediamine
- qPCR
quantitative real‐time polymerase chain reaction
- RT
room temperature
- SHH
Sonic hedgehog
- ST2
suppression of tumorigenicity 2
- Veh
vehicle
- WT
wild type
The Hedgehog (HH) pathway plays a role in hepatobiliary inflammation and injury‐related cancers. HH signaling involves Sonic hedgehog (SHH) and Indian hedgehog (IHH) ligands, the receptor Patched‐1 (PTCH1), and their transcriptional effectors glioma‐associated oncogene 1 (GLI1), GLI2, and GLI3.1 In the canonical HH pathway, cells expressing HH ligand signal to stromal cells expressing PTCH1 and GLIs in a variety of gastrointestinal tissues.2, 3, 4 In the liver, HH ligands are expressed in both epithelial cells and myofibroblasts after injury, and HH signaling is responsible for the “reactive” phenotype of injured cholangiocytes.5, 6 Prior studies, including from our group, suggest that HH signaling contributes to the initiation and progression of cholangiocarcinoma.7, 8 However, most studies describing HH signaling in hepatobiliary pathology have focused on hepatocytes, intrahepatic bile ducts (BDs), and fully developed cancer. This work focuses on the effects of activated HH signaling on extrahepatic BDs (EHBDs) in acute inflammation.
Cholangiopathies represent a group of chronic progressive diseases affecting biliary epithelial cells (BECs). Cholangiopathies, which include primary sclerosing cholangitis and cholangiocarcinoma, are associated with inflammation and fibrosis.9 Peribiliary glands (PBGs) are a specialized BEC compartment that contains biliary progenitor cells and participates in the maintenance and repair of large BDs.10, 11 PBGs contain mature and immature cell types and proliferate in response to BD injury in experimental mouse models of biliary atresia and BD obstruction.10 In humans, PBG hyperplasia is observed in numerous hepatobiliary pathologies, including cholangitis, cirrhosis, and hepatic necrosis, likely representing a compensatory mechanism after biliary injury to replace damaged BD epithelium.12 In patients with primary sclerosing cholangitis, increased HH signaling is associated with hyperplastic PBGs, dysplastic BD lesions, and advanced fibrosis.13 The mechanisms underlying HH regulation of EHBD as well as BEC and PBG epithelial hyperplasia have not been well described.
In children with biliary atresia, messenger RNA (mRNA) expression of the inflammatory cytokine interleukin‐33 (Il33) and its receptor suppression of tumorigenicity 2 (St2) is increased in the liver and IL‐33 is increased in serum.14, 15 Overexpression of IL‐33 was described in patients with inflammatory cholangiopathies,16 including hepatobiliary parasitic infection clonorchiasis17 and visceral leishmaniasis,18 and viral hepatitis B19 and C.20 The pro‐oncogenic effect of IL‐33 was recently described in a mouse model of cholangiocarcinoma, where biliary injury‐induced IL‐33 cooperated with Kirsten rat sarcoma viral oncogene and transforming growth factor βR2 mutations in the development of cholangiocarcinoma originating from PBGs.21 Moreover, it was recently shown that IL‐33 is a potent biliary mitogen, and IL‐33‐mediated proliferation of cholangiocytes was found to occur in a paracrine manner through IL‐13 secretion from nearby type 2 innate lymphoid cells responding to IL‐33.15 Furthermore, when coupled with the oncogenes AKT and yes‐associated protein, IL‐33 promotes cholangiocarcinoma in mouse, which involves an IL‐6‐sensitive mechanism.22 In addition to IL‐6 regulation by IL‐33, HH signaling and specifically GLI1 were reported to modulate IL‐6 expression in pancreatic cancer and stomach preneoplastic lesions.2, 23
These studies suggest a potential synergism between cell survival pathways and inflammation‐induced cytokines in cholangiocyte proliferation. In this study, we tested whether activated HH ligand signaling accelerates epithelial cell proliferation in EHBDs after an inflammatory challenge with IL‐33.
Materials and Methods
Animal Experiments
All mouse experiments were approved by the University of Michigan’s Institutional Animal Care and Use Committee. Transgenic Shh mice (promoter for cytomegalovirus [pCVM]‐Shh) and Gli1lacZ/lacZ mice have been described.2, 4 The Gli1lacZ./+and Gli1lacZ./lacZmice contain a knock‐in lacZ, encoding a nuclear‐localized β‐galactosidase, into exon 2 of the mouse Gli1 gene, producing a null allele.24, 25 The pCMV‐Shh; Gli1lacZ/+ and pCMV‐Shh; Gli1lacZ/lacZ mice were generated by crossing pCMV‐Shh and Gli1lacZ/lacZ mice. The Gli2lacZ/+, Gli3lacZ/+, and Ptch1lacZ/+ reporter mice have been described.26, 27, 28, 29 All lacZ reporter mice were maintained on a mixed C57BL/6J; 129S4/SvJaeJ background. Mice were housed in a specific pathogen‐free environment with a 12‐hour:12‐hour light–dark cycle in ventilated caging and provided Enviro‐Dri absorbent, cotton squares, or cardboard tubing as enrichment. Animals were provided with free access to food (5L0D; Purina LabDiet, St Louis, MO) and water. Recombinant mouse carrier free IL‐33 (R&D Systems, Minneapolis, MN) was reconstituted at 1 μg/100 μL in sterile phosphate‐buffered saline (PBS). During the light cycle, adult male and female mice were given intraperitoneal injections of either PBS (100 μL) or IL‐33 (1 μg) daily for 4 days, and tissues were isolated on day 5. Animals were euthanized during the light cycle with isoflurane combined with the removal of a vital organ according to institutional guidelines. Experimental replicates were sex and age matched as well as littermate matched when possible.
Human Samples
Human EHBD tissue from cholangiocarcinoma and adjacent noncancerous BD was collected with the approval of the University of Michigan’s Institutional Review Board according to the principles embodied in the Declaration of Helsinki. Paraffin‐embedded tissue was sectioned at 4 μm for further studies.
Immunohistochemistry
Mouse EHBDs were isolated and fixed in 10% formalin overnight at 4°C and then transferred to PBS before paraffin embedding. Human and mouse tissue sections (4 μm) were deparaffinized and rehydrated in serial xylene and alcohol dilutions and incubated in boiling citrate buffer (10 mM, pH 6) for 30 minutes for antigen retrieval. After antigen retrieval, slides were incubated in a 1% bovine serum albumin/5% fetal bovine serum/0.1% Triton X‐100–PBS blocking solution for 1 hour at room temperature (RT). Primary and secondary antibodies (Table 1) were diluted in the blocking solution. Tissue was incubated in primary antibodies overnight at 4°C and secondary antibodies for 1 hour at RT. ProLong Gold antifade reagent with 4′,6‐diamidino‐2‐phenylindole (DAPI) (Life Technologies, Waltham, MA) was used for nuclear counterstaining. Eosin (Thermo Fisher Scientific) was used for cytoplasmic staining, hematoxylin (Thermo Fisher Scientific) was used as a nuclear counterstain, and Permount (Thermo Fisher Scientific) was used as a mounting medium for hematoxylin and eosin (H&E) staining of deparaffinized and rehydrated slides. Images were taken on Olympus BX53 and Nikon Eclipse E800 microscopes (Tokyo, Japan).
Table 1.
Antibody List
| Target | Primary Antibody | Secondary Antibody |
|---|---|---|
| ST2 | Rabbit, 1:200; ProSci (3363) | Biotinylated donkey, ImmunoResearch (712‐065‐153) |
| BrdU | Sheep, 1:200; Abcam (AB2284) | Alexa Fluor 594‐Streptavidin, Invitrogen (S11227) |
| CK19 | Rabbit; 1:250; Abcam (AB53119) | Alexa Fluor 488‐labeled donkey IgG (H+L), Invitrogen (A21206) |
| IL‐6Rα | ||
| Rabbit; 1:200; Santa Cruz (SC661) | Biotinylated donkey, ImmunoResearch (712‐065‐153) | |
| IL‐33 (human) | Goat; 1:20; R&D Systems (AF3625) | Biotinylated donkey, Abcam (AB6884) |
Abbreviation: H, heavy chain; IgG, immunoglobulin G; L, light chain.
Histological Stains
For X‐gal staining, mouse EHBD was dissected, washed in ice‐cold PBS, and fixed in cold 4% paraformaldehyde (PFA)‐PBS for 1 hour before being washed in X‐gal wash buffer (1 M MgCl2, 2% NP‐40, 0.1 M sodium phosphate buffer pH 7.3) 3 times at RT on a rocker. Tissue was then stained overnight at 37°C in the dark with 1 mg/mL of X‐gal substrate (Roche, Basel, Switzerland) in 5 mM K3Fe(CN)6, 5 mM K4Fe(CN)6 · 3H2O, 2 mM MgCl2, 0.02% NP‐40, and 0.1% deoxycholate buffer. After staining, samples were washed for 30 minutes, postfixed in 4% PFA at 4°C overnight, paraffin embedded, sectioned at 4 μm, deparaffinized, counterstained with Nuclear Fast Red (Sigma‐Aldrich, St Louis, MO) for 30 minutes, rinsed in tap water, dehydrated in alcohol dilutions, and mounted with Permount.
In Situ Hybridization
Mouse EHBD formalin‐fixed paraffin‐embedded sections (5 μm) were deparaffinized and rehydrated in Histoclear (National Diagnostics, Atlanta, GA) and 100% ethanol, followed by 10 minutes of incubation in H2O2 (RNAscope Pretreatment Reagents, catalog #322330; ACD, Newark, CA). Next, slides were incubated in RNAScope Target Retrieval Reagent (catalog #322000; ACD) for 15 minutes at 95°C, rinsed with water and 100% ethanol, and treated with RNAScope Protease Plus (ACD) for 30 minutes at 40°C. Slides were then incubated with RNAScope probes (negative control [catalog #310043; ACD], Mm‐Shh [catalog #314361; ACD], Mm‐Ihh [catalog #413091; ACD], or Mm‐Gli1 [catalog #311001; ACD]) for 2 hours, with signal amplification using RNAscope 2.5 HD Detection Reagent (ACD) for 60 minutes at 40°C with washes using RNAscope Wash Buffer (ACD). Slides were incubated with 3,3′‐diaminobenzidine for 10 minutes, counterstained with 1:8 Harris’ hematoxylin (Thermo Fisher Scientific), dehydrated in graded ethanol and Histoclear, and mounted with Permount.
Biliary Proliferation
Proliferating cells were labeled by 5‐bromo‐2′‐deoxyuridine (BrdU) incorporation (50 mg/kg intraperitoneally 2 hours before tissue collection). The PBG and BEC areas were identified by cytokeratin 19 (CK19) immunostaining. Epithelial versus stromal cell proliferation was measured using ImageJ software (National Institutes of Health, Bethesda, MD) by an author blinded to experimental conditions.
Morphometrics
Each EHBD section was analyzed for BD wall thickness (excluding lumen) at three points 100 µm apart. Only sections with well‐aligned BD lumen and wall were used for analysis. Organoid growth was monitored by taking pictures with an inverted Leica DM IRB microscope (Wetzlar, Germany) using an Olympus DP7 (Tokyo, Japan) camera. The area occupied by organoids was measured with ImageJ software.
Quantitation of SHH Ligand
Blood was collected by cardiac puncture, and serum was isolated. SHH ligand levels were measured with a spectrophotometric enzyme‐linked immunosorbent assay kit following the manufacturer’s instructions (Sigma‐Aldrich). Absorbance was measured at 450 nm using a VICTOR3 microplate reader (PerkinEimer, Inc., Waltham, MA).
Mouse BD Organoid Culture
EHBD was isolated from adult mice and dissociated in Accutase (STEMCELL, Cambridge, MA) at 37°C for 15 minutes, followed by filtration through a 70‐µm strainer (BD Bioscience, San Jose, CA) and centrifugation at 300g for 5 minutes. Cells were resuspended in Matrigel (Corning, Tewksbury, MA) and plated in prewarmed 12‐ or 24‐well plates, followed by overlying with 300 or 600 µL culture media (50% L‐WRN [CRL‐3276; ATCC, Manassas, VA] conditioned media,30 1X penicillin–streptomycin, 1X GlutaMAX, 10 mM 4‐(2‐hydroxyethyl)‐1‐piperazine ethanesulfonic acid, 1X Fungizone, 1X gentamicin, 1X B27, and 1X N2 [Thermo Fisher Scientific] in advanced Dulbecco’s modified Eagle’s medium/F12 [Invitrogen; Carlsbad, CA]). Fibroblast growth factor 10 (100 ng/mL; PeproTech, Rocky Hill, NJ) and epithelial growth factor (50 ng/mL; Invitrogen) were added to the culture media for the first 3 days. BD organoids (BDOs) were passaged every 7 to 10 days by mechanically dissociating organoids with a 25‐gauge needle and resuspending them in Matrigel at 1:3 to 1:4 ratios. BDO growth and proliferation were measured after administration of recombinant IL‐33 (100 ng/mL; R&D Systems) or nuclear factor κB (NF‐κB) inhibitor N4‐[2‐(4‐phenoxyphenyl)ethyl]‐4,6‐quinazolinediamine (QNZ; 1 µM, pretreatment for 1 hour; Cayman Chemical, Ann Arbor, MI).
Quantitative Real‐Time Polymerase Chain Reaction
EHBD was isolated and placed into RNAlater (Qiagen, Hilden, Germany). Tissue was transferred to RPE buffer (RNeasy Micro Kit; Qiagen) and homogenized using a bead mill homogenizer (VWR, Radnor, PA). Total RNA was isolated from whole EHBD tissue or organoids after deoxyribonuclease I digestion (Qiagen) using an RNeasy Mini Kit or RNeasy Micro Kit (Qiagen). Complementary DNA (cDNA) was generated from 500 ng total RNA using the iScript cDNA synthesis kit (Bio‐Rad, Hercules, CA). Quantitative real‐time polymerase chain reaction (qPCR) was performed in duplicate reactions of 20 μL total volume using 100 μM of forward and reverse primers (Table 2), 1 μL SYBR Green I nucleic acid stain (Lonza, Rockland, ME), and 0.1 μL platinum Taq DNA polymerase (Invitrogen) using a CFX96 real‐time thermocycler (C1000; Bio‐Rad). Target gene expression was normalized to hypoxanthine guanine phosphoribosyl transferase (Hprt) control mRNA abundance.
Table 2.
Primer List
| Gene | Accession Number | Primer Sequence | Product Size |
|---|---|---|---|
| Hprt | NM_013556 | Forward 5′‐AACTTGCGCTCATCTTAGGCTTTG‐3′ | 173 bp |
| Reverse 5′‐AGGACCTCTCGAAGTGTTGGATAC‐3′ | |||
| Shh | NM_009170 | Forward 5′‐TCATCACAGAGATGGCCAAG‐3′ | 122 bp |
| Reverse 5′‐GGAACTCACCCCCAATTACA‐3′ | |||
| Ihh | NM_010544 | Forward 5′‐TGACAGAGATGGCCAGTGAG‐3′ | 119 bp |
| Reverse 5′‐AGAGCTCACCCCCAACTACA‐3′ | |||
| Gli1 | NM_010296 | Forward 5′‐TATGTCAGGGTCCCAGGGTTATG‐3′ | 110 bp |
| Reverse 5′‐GAGCCCGCTTCTTAGTCAGTTTG‐3′ | |||
| Gli2 | NM_001081125 | Forward 5′‐CCATTATGACCCTCACTCTGTC‐3′ | 102 bp |
| Reverse 5′‐GGGTGTGGAGAAAGTCGTATC‐3′ | |||
| Ptch1 | NM_008957 | Forward 5′‐CTGCTGTGGTGGTGGTATTC‐3′ | 120 bp |
| Reverse 5′‐GGCTTGTGAAACAGCAGAAA‐3′ | |||
| St2 | NM_001025602 | Forward 5′‐ATTCAGGGGACCATCAAGTG‐3′ | 117 bp |
| Reverse 5′‐CGTCTTGGAGGCTCTTTCTG‐3′ | |||
| Il‐6 | NM_031168.2 | Forward 5’‐TTCCATCCAGTTGCCTTCTTGG‐3’ | 175 bp |
| Reverse 5’‐TTCTCATTTCCACGATTTCCCAG‐3’ |
Abbreviation: bp, base pair.
Cell Lines
The L‐WRN (CRL‐3276) cell line30 was purchased from ATCC on November 6, 2015. The third and fourth passages were used to generate conditioned media for organoid culture. The mycoplasma detection test (ATCC) was performed on all cultures before experiments (last check on May 18, 2018).
Organoid Proliferation
BDOs were incubated in 5‐ethynyl‐2′‐deoxyuridine (EdU) (10 µM for 9 hours; Thermo Fisher Scientific), washed with PBS, and fixed with 4% PFA for 10 minutes at RT. After washing with PBS and permeabilization with 0.5% PBS–Triton X‐100 for 20 minutes, BDOs were incubated in the EdU reaction cocktail from a Click‐iT Plus EdU kit (Thermo Fisher Scientific). BDOs were then washed, resuspended in ice‐cold PBS, placed on a slide, and mounted with ProLong Gold with DAPI (Life Technologies). A Nikon Eclipse E800 microscope (Nikon, Tokyo, Japan) was used to take images.
Statistical Analysis
GraphPad Prism 7 (La Jolla, CA) was used for one‐way analysis of variance (ANOVA) and unpaired Student t test statistical analyses. Quantitative values were expressed as actual numbers (mRNA expression, serum SHH expression, BD thickness or area) and compared with the baseline (wild‐type [WT] animal or vehicle [Veh]) where applicable. Significance was set at P < 0.05. Results are presented as mean ± SEM.
Results
HH Signals to Stromal Cells in EHBDs
We examined the cellular basis for HH signaling in EHBD (Fig. 1A) to identify HH‐responsive cells expressing the general HH pathway components.31, 32 Using Gli1lacZ/+, Gli2 lacZ/+, Gli3lacZ/+, and PtchlacZ/+ reporter mice, we identified putative sites of active HH response in mouse EHBDs (i.e., Gli1‐expressing and Ptch1‐expressing cells) as well as those sites that are competent to transduce HH signals through expression of the GLI transcriptional effectors (i.e., Gli‐expressing cells). As expected, WT mice lacked β‐galactosidase activity (Fig. 1B). HH target gene expression was observed in stromal cells in Gli1lacZ/+and PtchlacZ/+ mice (Fig. 1C,F); similar stromal expression was noted in Gli2lacZ/+ and Gli3lacZ/+ mice (Fig. 1D,E). We tested WT mouse EHBDs for mRNA abundance of the HH ligands Shh and Ihh. Although mouse EHBDs lacked detectable Shh expression at baseline, they readily expressed Ihh mRNA (Fig. 1G). These findings indicate that EHBD stromal cells are HH‐signaling cells and IHH is the dominant HH ligand in EHBDs.
Figure 1.
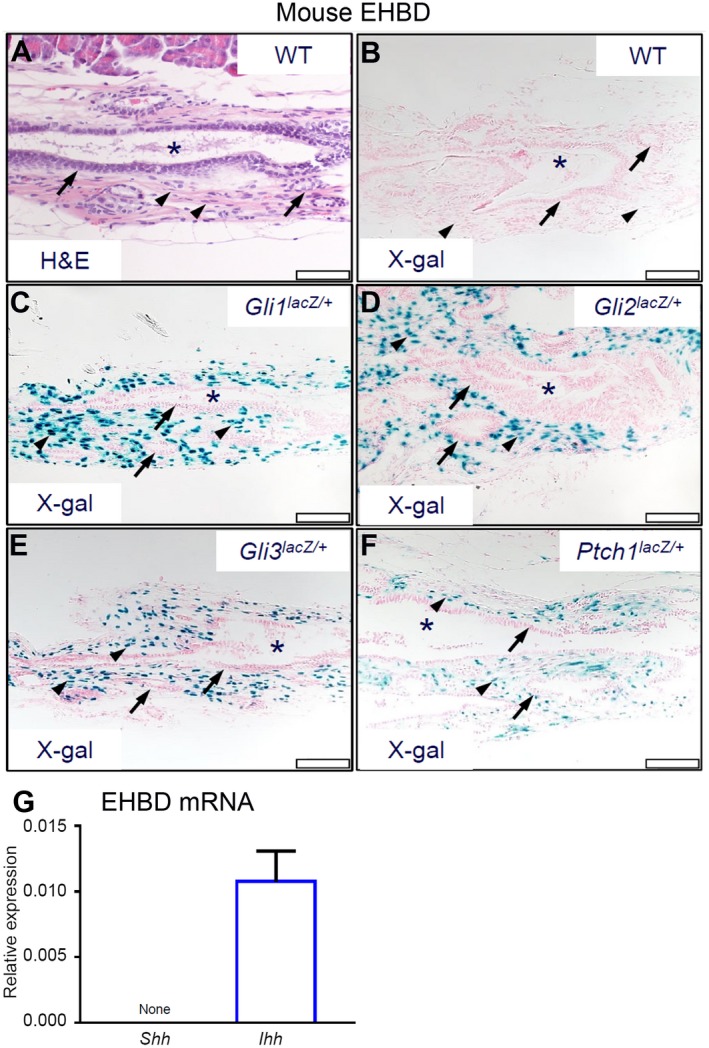
HH signaling receptive cells are located in the EHBD stroma. Mouse EHBDs were isolated from WT and reporter Gli1lacZ/+, Gli2lacZ/+, Gli3lacZ/+, and Ptch1lacZ/+ mice and stained by (A) H&E and (B‐F) for β‐galactosidase activity (asterisks, BD lumen; arrows, epithelial cells; arrowheads, stromal cells; scale bars, 50 µm) (representative images of two to five samples per genotype). (G) Total RNA was isolated from EHBDs of WT mice and analyzed for mRNA expression by qPCR of Shh and Ihh referenced to Hprt (n = 15‐16 animals per group). Results are expressed as mean ± SEM. Abbreviation: X‐gal, bromochloroindoxyl galactoside.
pCMV‐Shh and Gli1lacZ/lacZ Mice Model Chronic HH Ligand Overexpression and Gli1 Inhibition in EHBDs
We previously reported the use of genetically engineered murine models of HH ligand overexpression (pCMV‐Shh) and the loss of the transcriptional activator Gli1 (GlilacZ/lacZ) to investigate the consequences of modulating HH signaling on Helicobacter pylori‐induced stomach metaplasia.2, 4 We took advantage of these mouse models to examine the contribution of HH signaling in mouse EHBDs. We identified the site of HH ligands and effector Gli1 expression in EHBDs of pCMV‐Shh mice by in situ hybridization (Fig. 2B‐D). EHBDs from WT mice did not express Shh mRNA (Fig. 2A), consistent with the absence of detectable Shh mRNA expression in WT mouse EHBDs by qPCR (Fig. 1G). In pCMV‐Shh mice, Shh mRNA and Ihh mRNA were expressed and localized in epithelial cells (Fig. 2B,C). Consistent with findings in Gli1lacZ/+ mice, Gli1 mRNA was localized in stromal cells in EHBDs of pCMV‐Shh (Fig. 2D). Additionally, GLI1 was stromal in pCMV‐Shh; Gli1lacZ./+ and pCMV‐Shh; Gli1lacZ./lacZ mice (Fig. 2E,F). Further, pCMV‐Shh mice had an increased level of circulating SHH ligand in serum compared with WT and Gli1lacZ/lacZ mice (Fig. 2G). Gli1, Gli2, and Ptch1 mRNA abundance in pCMV‐Shh mice compared with WT and Gli1lacZ/lacZ mice was significantly increased (Fig. 2H‐J). Thus, we used the pCMV‐Shh mouse model to study the consequences of increased chronic HH pathway activity and Gli1lacZ/lacZ mice to study decreased canonical HH pathway activity in EHBD stroma in vivo.
Figure 2.
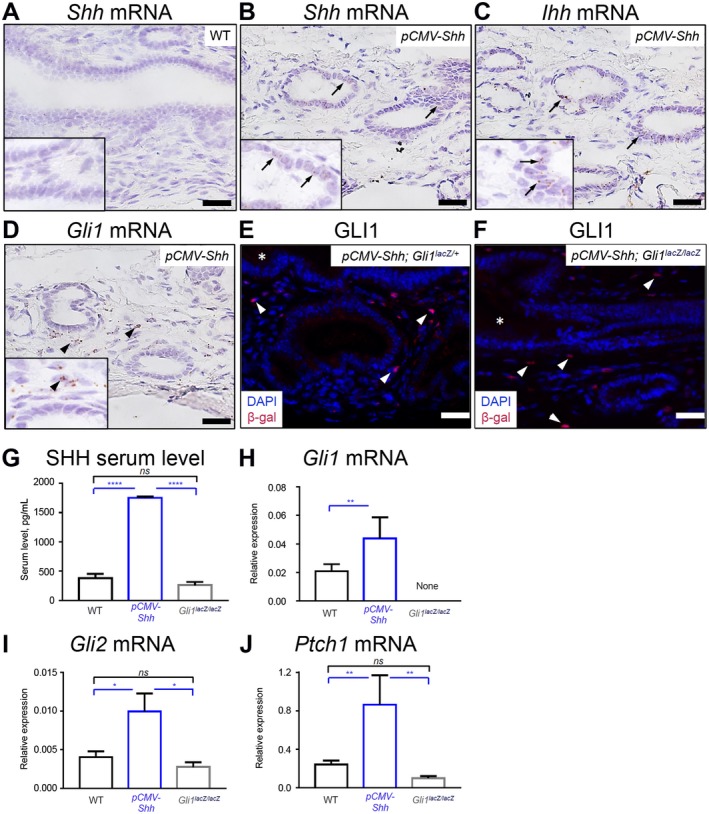
pCMV‐Shh and Gli1lacZ/lacZ mice model chronic HH overexpression and inhibition in EHBDs. EHBDs from WT, pCMV‐Shh, and Gli1lacZ/lacZ mice were studied for expression of HH ligands and signaling targets. (A‐D) We analyzed EHBDs from WT and pCMV‐Shh mice with in situ hybridization (RNAscope) for (A,B) expression of Shh mRNA (arrows) and EHBDs from pCMV‐Shh mice and for (C) expression of Ihh mRNA and (D) Gli1 mRNA (representative images of two samples). (E,F) Gli1 expression was also studied by immunofluorescence for β‐galactosidase (red; arrows) in pCMV‐Shh; Gli1lacZ/+ and pCMV‐Shh; Gli1lacZ/lacZ mice. Cell nuclei were marked with DAPI (blue; representative images of one and two samples, respectively; arrows, epithelial cells; arrowheads, stromal cells; asterisks, lumen; scale bars, 20 µm). (G) Serum level of the SHH ligand was measured by enzyme‐linked immunosorbent assay in WT, pCMV‐Shh, and Gli1lacZ/lacZ mice. (H‐J) Total RNA was isolated from EHBDs of the WT, pCMV‐Shh, and Gli1lacZ/lacZ mice and analyzed for mRNA expression by qPCR of (H) Gli1, (I) Gli2, and (J) Ptch1 referenced to Hprt. Results are expressed as mean ± SEM (n = 8‐19 animals per group; one‐way ANOVA); *P < 0.05, **P < 0.01, ****P < 0.0001. Abbreviation: ns, not significant.
HH Overexpression Amplifies IL‐33‐Induced Epithelial Cell Proliferation In Vivo
In chronic progressive fibroproliferative disorders, such as primary sclerosing cholangitis, chronic injury is associated with up‐regulation of inflammatory cytokines.33 Cholangiocarcinoma, which frequently complicates cholangiopathies, is associated with up‐regulation of HH signaling and also cytokines IL‐33 and IL‐6.7, 22, 34 Thus, we examined human EHBD cholangiocarcinoma and adjacent noncancerous tissue for expression of inflammatory cytokines with immunohistochemistry. We demonstrated that IL‐33, which is an alarmin and potent biliary mitogen,15, 21 was expressed in noncancerous EHBD and was overexpressed in cholangiocarcinoma (Fig. 3A). Further, normal adjacent bile duct and EHBD cholangiocarcinoma cells strongly expressed the IL‐6 receptor (IL‐6R) (Fig. 3B).
Figure 3.
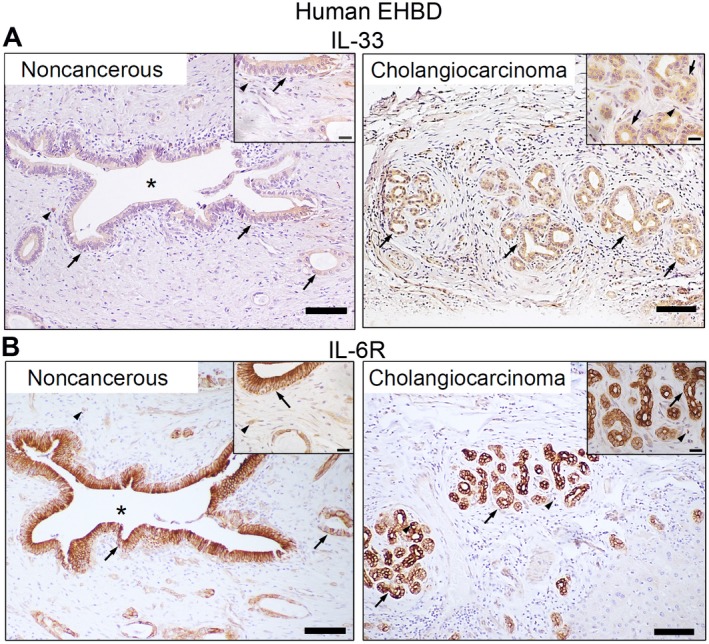
Cytokines IL‐33 and IL‐6 are expressed in human EHBDs. (A,B) Human extrahepatic cholangiocarcinoma and adjacent noncancerous tissue were examined for expression of (A) IL‐33 and (B) IL‐6R by immunohistochemistry (arrows, epithelial cells; arrowheads, stromal cells; asterisks, BD lumen; scale bars, 100 µm).
We tested the HH mouse models to determine whether increased HH signaling synergizes with inflammatory cytokines to promote cell proliferation. We showed that mouse EHBDs express the IL‐33 receptor ST2, which is primarily localized in epithelial cells at baseline and in both epithelial and stromal cells after IL‐33 treatment (Fig. 4A). We treated the pCMV‐Shh and Gli1lacZ/lacZ mouse models with IL‐33 (Fig. 4B, schema). We examined EHBDs of WT, pCMV‐Shh, and Gli1lacZ/lacZ mice macroscopically and histologically and observed no gross morphological differences between IL‐33 and Veh‐treated mice (Fig. 4C,D, upper panels). However, BD thickness, an indicator of hyperplasia, was increased in IL‐33‐treated pCMV‐Shh mice compared with WT controls and Gli1lacZ/lacZ mice (Fig. 4E), with increased cellularity observed by H&E staining (Fig. 4D).
Figure 4.
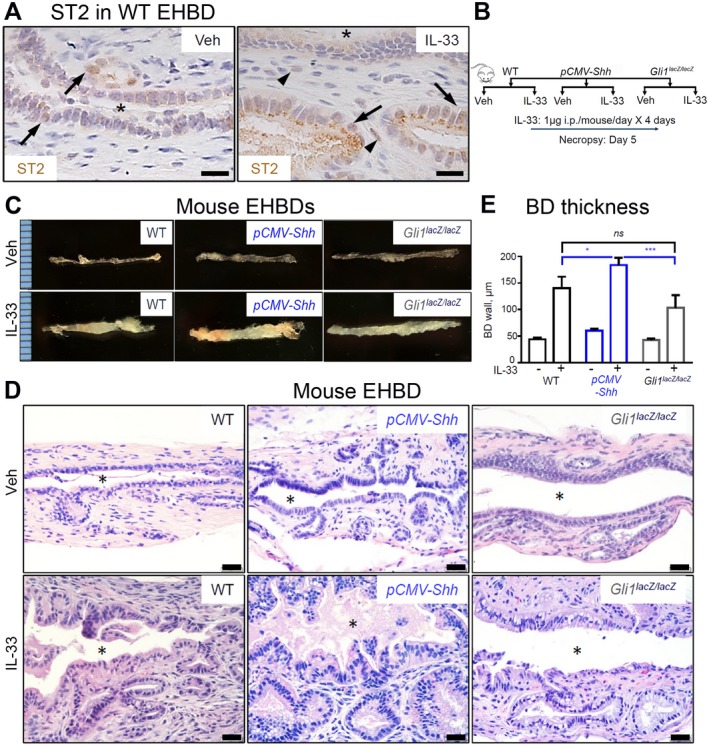
HH signaling synergizes with IL‐33 in EHBD hyperplasia. (A) EHBDs from WT mice treated with either Veh or IL‐33 (1 µg/mouse/day for 4 days; necropsy on day 5) were analyzed by immunohistochemistry for ST2 expression (representative images of three samples). (B,C) EHBDs from WT, pCMV‐Shh, and Gli1lacZ/lacZ mice treated with either Veh or mitogen IL‐33 intraperitoneally, as shown in (B) schema, were (C) examined macroscopically (representative photographs; ruler scale, 1 mm) and (D) histologically with H&E (scale bars, 20 µm; asterisks, BD lumen). (D) BD wall thickness from immunohistochemistry images was measured using ImageJ software and compared among WT, pCMV‐Shh, and Gli1lacZ/lacZ mice treated with either Veh or IL‐33 (n = 7‐11 animals per group). Results are expressed as mean ± SEM; one‐way ANOVA; *P < 0.05, ***P < 0.001. Abbreviations: i.p., intraperitoneally; ns, not significant.
Analysis of BrdU incorporation demonstrated no difference in epithelial and stromal cell proliferation in EHBDs of WT, pCMV‐Shh, and Gli1lacZ/lacZ Veh‐treated mice (Fig. 5A, upper panel; quantified in B). As expected, proliferation was very low because cholangiocytes are virtually mitotically dormant at baseline.5 IL‐33 induced proliferation of both epithelial and stromal cells (Fig. 5A‐C). However, HH overexpression amplified the proliferative effect of IL‐33 on epithelial cells but not on stromal cells (Fig. 5A‐C). Further, Gli1 ablation blunted the IL‐33‐induced epithelial cell proliferation (Fig. 5A,B). The results suggest that IL‐33 is the primary driver of EHBD cell proliferation and HH signaling enhances cytokine‐induced epithelial cell proliferation.
Figure 5.
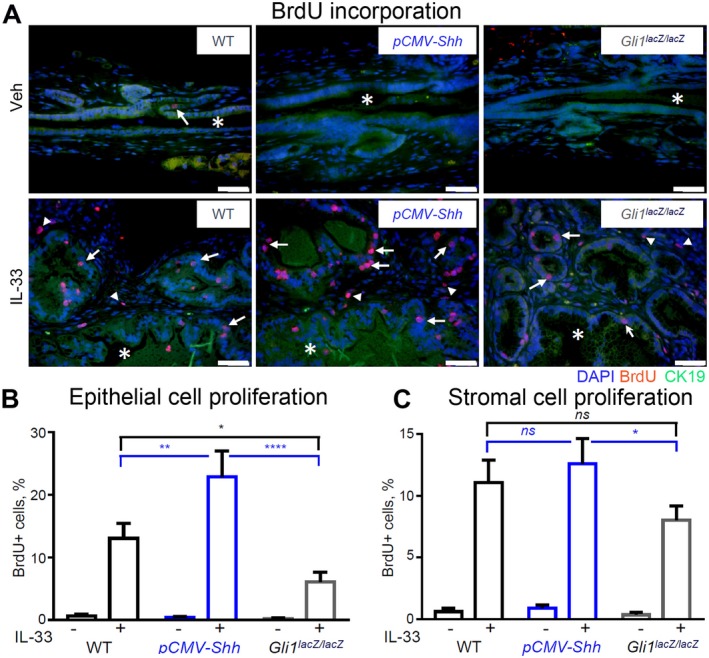
HH signaling synergizes with IL‐33 to promote biliary cell proliferation. EHBDs from the WT, pCMV‐Shh, and Gli1lacZ/lacZ mice treated with either Veh or IL‐33 (1 µg/mouse/day for 4 days; necropsy on day 5) were analyzed with immunofluorescence for cell proliferation. (A) Proliferating cells were labeled by BrdU incorporation (50 mg/kg, intraperitoneally, 2 hours before necropsy) (red). Cell nuclei were marked with DAPI (blue) and BECs with CK19 (green; original magnification ×400). (B,C) Proliferating cells were quantified in the epithelial (arrows) and stromal (arrowheads) cell compartments with ImageJ software (n = 5‐6 animals per group; five or more high‐power fields per BD; >500 cells/animal). Results are expressed as mean ± SEM; one‐way ANOVA; *P < 0.05, **P < 0.01, ****P < 0.0001. Abbreviation: ns, not significant.
IL‐6 Increase Is Associated With HH and IL‐33 Synergism
Both HH and IL‐33 signaling are associated with IL‐6 up‐regulation in human cholangiopathies. Therefore, we hypothesized that IL‐6 may contribute to the synergistic effect of HH and IL‐33 signaling on biliary proliferation in EHBDs. We demonstrated that IL‐33 increases IL‐6R expression in epithelial and stromal cells in WT mouse EHBDs (Fig. 6A). Although WT and pCMV‐Shh mice exhibited similar increases in Il6 mRNA expression after IL‐33 treatment, Gli1 ablation blunted the Il6 mRNA response to IL‐33lacZlacZ (Fig. 6B). These findings suggest that HH signaling is important for IL‐33‐induced Il6 expression.
Figure 6.
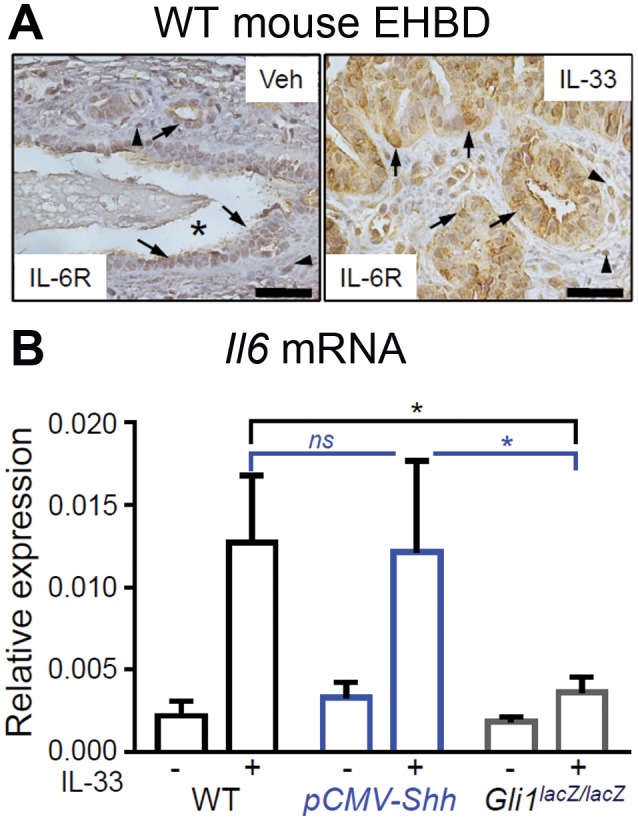
IL‐33‐induced IL‐6 expression in mouse EHBDs involves Gli1. (A) EHBDs from WT mice were analyzed by immunohistochemistry for IL‐6R expression after treatment with either Veh or recombinant IL‐33 intraperitoneally (representative images of two samples; asterisks, BD lumen; arrows, epithelial cells; arrowheads, stromal cells; scale bars, 10 µm). (B) Total RNA was isolated from BDs of the WT, pCMV‐Shh, and Gli1lacZ/lacZ mice treated with either Veh or IL‐33 and analyzed for mRNA expression by qPCR of Il6 referenced to Hprt. Results are expressed as mean ± SEM (n = 5‐8 animals per group; one‐way ANOVA; *P < 0.05).
We next examined the effect of IL‐33 on expression of HH components by immunohistochemistry and qPCR. We demonstrated that after treatment with IL‐33, Gli1 expression remained stromal (Supporting Fig. S1A‐C). We further showed that mRNA abundance of HH components was not significantly changed in various IL‐33‐treated mouse strains (Supporting Fig. S1D‐H). In particular, Shh mRNA still was not expressed in WT and Gli1lacZ/lacZ mice even after IL‐33 treatment (Supporting Fig. S1D), whereas Gli1 mRNA expression remained undetectable in Gli1lacZ/lacZ mice treated with IL‐33 (Supporting Fig. S1F). Collectively, these data indicated that IL‐33 does not have a direct effect on HH signaling modulator expression. However, HH signaling and specifically GLI1 primes EHBD for the proliferative response to IL‐33.
IL‐33 Induces Epithelial Cell Proliferation In Vitro
To study the direct effect of IL‐33 and HH on biliary cell proliferation, we examined WT mice‐derived BDOs. We examined organoids for mRNA abundance of St2 and the HH signaling effector Gli1. As expected from the mouse tissue expression patterns (cf. Fig. 1), BDOs expressed St2 but lacked Gli1 mRNA expression (Fig. 7A). Thus, BDOs, which comprise only epithelial cells, do not exhibit canonical GLI1‐mediated HH signaling. We investigated the effect of IL‐33 on epithelial cell proliferation in BDOs. Consistent with our in vivo data, we found that IL‐33 stimulated increased BDO growth (Fig. 7B,C) through increased epithelial cell proliferation, as measured by EdU incorporation (Fig. 7D,E). Further, there was no difference in the IL‐33‐induced proliferative response among WT, pCMV‐Shh, and Gli1lacZ/lacZ mice‐derived organoids (Supporting Fig. S2A,B). This result is consistent with the in vivo observation that HH signaling occurs in stromal cells.
Figure 7.
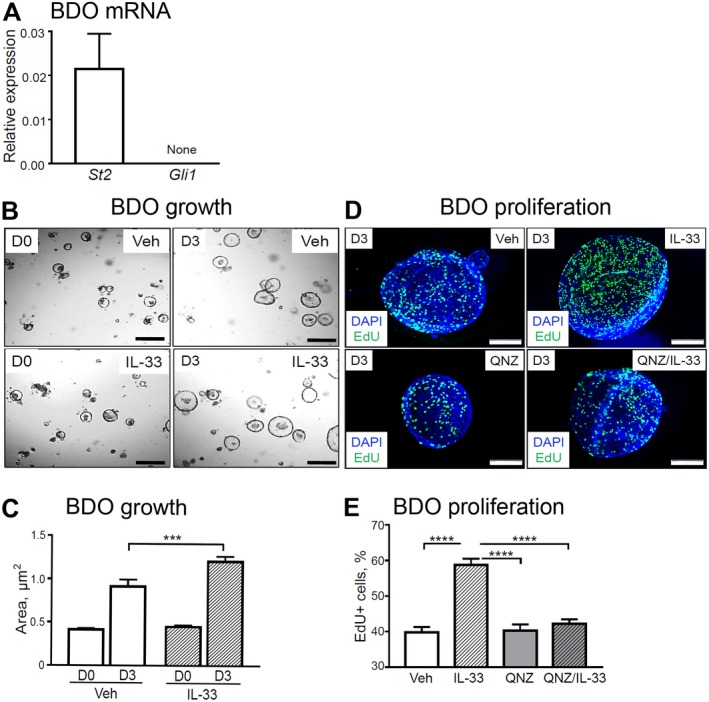
IL‐33‐induced BDO cell proliferation in vitro. (A) Total RNA was isolated from the WT mouse‐derived organoids and analyzed for mRNA expression of St2 and Gli1 (n = 3 organoid lines). (B,C) BDOs were treated with Veh or recombinant IL‐33 (100 ng/mL) for 72 hours (day 3) starting 24 hours after passage of established BDOs and analyzed for growth (n = 6 technical replicates per group). (D,E) In a separate experiment, BDOs were treated with either Veh or pretreated for 1 hour with the NF‐κB inhibitor QNZ (1 µM) and then treated with recombinant IL‐33 (100 ng/mL) for 72 hours (day 3) after passaging of established BDOs. BDOs were analyzed for proliferation (n = 3 technical replicates per group). (D) The organoids were incubated with EdU for 9 hours to label proliferating cells (green) among all BDO cells (cell nuclei, DAPI [blue]; original magnification ×600). (E) ImageJ software was used to enumerate proliferating cells. Results are expressed as mean ± SEM; one‐way ANOVA (C,E); ***P < 0.001, ****P < 0.0001. Abbreviation: D0/D3, day 0/day 3.
To identify the potential mechanism of IL‐33‐induced BDO proliferation, we used the NF‐κB inhibitor QNZ35 because IL‐33 has been reported to signal through NF‐κB.36 Pretreatment of BDOs with QNZ effectively prevented IL‐33‐induced BDO proliferation (Fig. 7D,E). Together, our findings suggest that IL‐33 can directly stimulate epithelial proliferation in an NF‐κB‐dependent manner and that the HH pathway indirectly supports IL‐33‐driven epithelial proliferation through activation of signaling in stromal cells.
Discussion
Our current results demonstrate that EHBD stromal cells express the HH transcriptional effectors GLI1, GLI2, and GLI3 as well as the receptor PTCH1 and thus appear to be the primary cells responding to the HH ligand. They also demonstrate that HH ligand expression is limited to the epithelium and that IHH is the predominant HH ligand expressed both under basal conditions and after inflammatory challenge with IL‐33. We used pCMV‐Shh mice as a model to study the consequences of HH ligand overexpression in EHBDs because IHH and SHH both signal through the same receptor and GLI effectors.37 Although the CMV promoter expressing the Shh transgene should be nonspecific, it has been demonstrated to preferentially drive expression in certain cells within different organ systems.38 For example, expression of SHH in pCMV‐Shh mice was enriched in gastric chief cells in the stomach.2 In our genetically engineered mouse model of HH ligand overexpression, Shh and Ihh were localized to epithelial cells. Therefore, our observation of HH ligand expression in epithelial cells and GLI1 in stromal cells is consistent with HH signaling from epithelial to stromal cells as observed in other organ systems, including the stomach and intestine.2, 39, 40
Our studies further suggest that increased HH signaling alone is insufficient to induce EHBD epithelial cell proliferation. However, HH overexpression augmented the proliferative effect of the inflammatory cytokine and alarmin IL‐33 on EHBD epithelial cells but not stromal cells. Interestingly, reduction in canonical HH signaling in receptive stromal cells attenuated the IL‐33 effect on epithelial proliferation, indicative that synergism between HH and IL‐33 signaling involves GLI1‐positive stromal cells. These findings suggest that BECs respond to HH up‐regulation differentially with no changes in cell proliferation at basal conditions and with an enhanced proliferative response under inflammatory challenge. However, the HH‐dependent factor that enhances epithelial cell response to IL‐33 is unknown.
As both HH and IL‐33 pathways are reported to signal through IL‐6,23, 41, 42 we explored the potential role of IL‐6 in the synergism between HH and IL‐33 signaling in IL‐33‐induced cell proliferation in EHBDs. We demonstrated that IL‐33 and IL‐6R are expressed in human cholangiocarcinoma and adjacent normal EHBD tissue. We also demonstrated that IL‐33 induces expression of Il6, which is in part dependent on GLI1‐positive stromal cells. Therefore, GLI1‐expressing stromal cells might be important in epithelial cell responses to inflammatory cytokines. It is possible that HH ligand overexpression “primes” GLI1‐positive stromal cells and activates GLI1 gene targets, which indirectly affects cytokine signaling in EHBD epithelial cells. Alternatively, IL‐33 signaling primes the epithelial cells to be more responsive to HH‐dependent stroma‐derived factors (Fig. 8).
Figure 8.
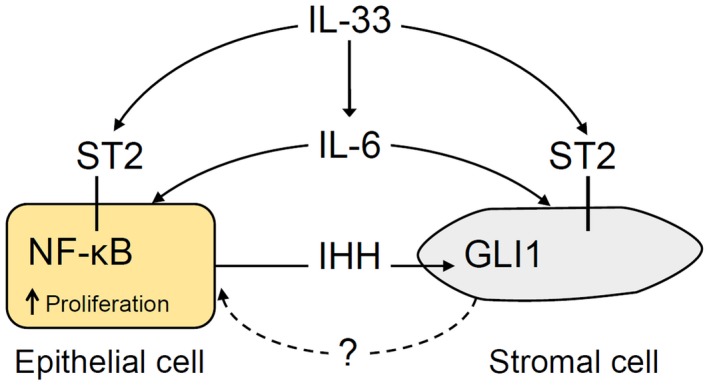
Model of HH and IL‐33 synergy in IL‐33‐induced EHBD epithelial cell proliferation. IHH is secreted by epithelial cells and activates GLI1‐positive stromal cells. After EHBD injury, IL‐33 signals to GLI1‐positive stromal and epithelial cells through the ST2 receptor. IL‐33 signaling involves the transcriptional effector NF‐κB in epithelial cells. IL‐33 induces IL‐6 expression, which can signal to both epithelial and stromal cells. Increased epithelial cell proliferation induced by IL‐33 is partially HH dependent through a yet uncharacterized stromal factor.
It was reported that IL‐33 promotes cholangiocyte proliferation indirectly through type 2 innate lymphoid cell IL‐13.15 However, in our biliary organoid culture, we demonstrated that IL‐33 also can directly induce proliferation in biliary progenitor cells. This IL‐33 proliferative effect on biliary progenitor cells is NF‐κB dependent. The latter observation is interesting as IL‐33 induces cytokine expression through NF‐κB.42 In addition, NF‐κB activation in cholangiocytes exposed to liver fluke Opisthorchis viverrini is associated with up‐regulation of IL‐6 and IL‐8,43 and IL‐33 overexpression occurs in hepatobiliary parasitic infection clonorchiasis17 and visceral leishmaniasis.18 Thus, IL‐33 can affect EHBD responses both directly and indirectly through regulating another cytokine function.
These data have relevance to human diseases as IL‐6 overexpression was recently described in cholangiocarcinomas arising in patients with primary sclerosing cholangitis associated with extensive PBG hyperplasia.44 HH signaling up‐regulation was also reported in patients with primary sclerosing cholangitis and PBG hyperplasia.13 An association between IL‐33 and IL‐6 has been shown in patients with cholangiocarcinoma, where IL‐33 overexpression was observed in cancer involving large versus small BDs, and was associated with IL‐6 overexpression.34
In summary, we report a previously unknown role for HH signaling in the regulation of EHBD homeostasis by promoting context‐dependent BEC proliferation contributing to EHBD hyperplasia. This involves crosstalk between HH ligand‐producing cells and receptive GLI1‐positive stromal cells (Fig. 8). We also showed that the alarmin IL‐33 can directly induce epithelial cell proliferation. Future studies identifying the proliferative signals from HH‐responding GLI1‐positive stromal cells toward epithelial cells and the mechanisms of HH signaling and inflammatory cytokine interactions will be needed. They could provide insight into how and, importantly, when HH and/or cytokine signaling can be targeted to prevent EHBD hyperplasia, which can be preneoplastic in chronic biliary conditions, such as primary sclerosing cholangitis.
Potential conflict of interest
Nothing to report.
Supporting information
Supported by the American Association for the Study of Liver Diseases Pinnacle award (to N.R.) and the National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases (awards P30 DK34933 to N.R., P01 DK062041 to J.L.M., and DK59427 to G.J.G.).
References
Author names in bold designate shared co‐first authorship.
- 1. Robbins DJ, Fei DL, Riobo NA. The Hedgehog signal transduction network. Sci Signal 2012;5:re6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. El‐Zaatari M, Kao JY, Tessier A, Bai L, Hayes MM, Fontaine C, et al. Gli1 deletion prevents Helicobacter‐induced gastric metaplasia and expansion of myeloid cell subsets. PLoS ONE 2013;8:e58935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Armas‐Lopez L, Zuniga J, Arrieta O, Avila‐Moreno F. The Hedgehog‐GLI pathway in embryonic development and cancer: implications for pulmonary oncology therapy. Oncotarget 2017;8:60684‐60703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ding L, Hayes MM, Photenhauer A, Eaton KA, Li Q, Ocadiz‐Ruiz R, et al. Schlafen 4‐expressing myeloid‐derived suppressor cells are induced during murine gastric metaplasia. J Clin Invest 2016;126:2867‐2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Omenetti A, Diehl AM. Hedgehog signaling in cholangiocytes. Curr Opin Gastroenterol 2011;27:268‐275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Maroni L, Haibo B, Ray D, Zhou T, Wan Y, Meng F, et al. Functional and structural features of cholangiocytes in health and disease. Cell Mol Gastroenterol Hepatol 2015;1:368‐380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Razumilava N, Bronk SF, Smoot RL, Fingas CD, Werneburg NW, Roberts LR, et al. miR‐25 targets TNF‐related apoptosis inducing ligand (TRAIL) death receptor‐4 and promotes apoptosis resistance in cholangiocarcinoma. Hepatology 2012;55:465‐475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Razumilava N, Gradilone SA, Smoot RL, Mertens JC, Bronk SF, Sirica AE, et al. Non‐canonical Hedgehog signaling contributes to chemotaxis in cholangiocarcinoma. J Hepatol 2014;60:599‐605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lazaridis KN, LaRusso NF. The cholangiopathies. Mayo Clin Proc 2015;90:791‐800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. DiPaola F, Shivakumar P, Pfister J, Walters S, Sabla G, Bezerra JA. Identification of intramural epithelial networks linked to peribiliary glands that express progenitor cell markers and proliferate after injury in mice. Hepatology 2013;58:1486‐1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Carpino G, Renzi A, Franchitto A, Cardinale V, Onori P, Reid L, et al. Stem/progenitor cell niches involved in hepatic and biliary regeneration. Stem Cells Int 2016;2016:3658013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Terada T, Nakanuma Y. Pathologic observations of intrahepatic peribiliary glands in 1,000 consecutive autopsy livers: IV. Hyperplasia of intramural and extramural glands. Hum Pathol 1992;23:483‐490. [DOI] [PubMed] [Google Scholar]
- 13. Carpino G, Cardinale V, Renzi A, Hov JR, Berloco PB, Rossi M, et al. Activation of biliary tree stem cells within peribiliary glands in primary sclerosing cholangitis. J Hepatol 2015;63:1220‐1228. [DOI] [PubMed] [Google Scholar]
- 14. Patman G. Biliary tract. IL‐33, innate lymphoid cells and IL‐13 are required for cholangiocyte proliferation. Nat Rev Gastroenterol Hepatol 2014;11:456. [DOI] [PubMed] [Google Scholar]
- 15. Li J, Razumilava N, Gores GJ, Walters S, Mizuochi T, Mourya R, et al. Biliary repair and carcinogenesis are mediated by IL‐33‐dependent cholangiocyte proliferation. J Clin Invest 2014;124:3241‐3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Baier JL, Mattner J. Mechanisms of autoimmune liver disease. Discov Med 2014;18:255‐263. [PubMed] [Google Scholar]
- 17. Yu Q, Li XY, Cheng XD, Shen LP, Fang F, Zhang B, et al. Expression and potential roles of IL‐33/ST2 in the immune regulation during Clonorchis sinensis infection. Parasitol Res 2016;115:2299‐2305. [DOI] [PubMed] [Google Scholar]
- 18. Rostan O, Gangneux JP, Piquet‐Pellorce C, Manuel C, McKenzie AN, Guiguen C, et al. The IL‐33/ST2 axis is associated with human visceral leishmaniasis and suppresses Th1 responses in the livers of BALB/c mice infected with Leishmania donovani. MBio 2013;4:e00383‐e00313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Huan SL, Zhao JG, Wang ZL, Gao S, Wang K. Relevance of serum interleukin‐33 and ST2 levels and the natural course of chronic hepatitis B virus infection. BMC Infect Dis 2016;16:200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang J, Zhao P, Guo H, Sun X, Jiang Z, Xu L, et al. Serum IL‐33 levels are associated with liver damage in patients with chronic hepatitis C. Mediators Inflamm 2012;2012:819636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nakagawa H, Suzuki N, Hirata Y, Hikiba Y, Hayakawa Y, Kinoshita H, et al. Biliary epithelial injury‐induced regenerative response by IL‐33 promotes cholangiocarcinogenesis from peribiliary glands. Proc Natl Acad Sci U S A 2017;114:E3806‐E3815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yamada D, Rizvi S, Razumilava N, Bronk SF, Davila JI, Champion MD, et al. IL‐33 facilitates oncogene‐induced cholangiocarcinoma in mice by an interleukin‐6‐sensitive mechanism. Hepatology 2015;61:1627‐1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mathew E, Collins MA, Fernandez‐Barrena MG, Holtz AM, Yan W, Hogan JO, et al. The transcription factor GLI1 modulates the inflammatory response during pancreatic tissue remodeling. J Biol Chem 2014;289:27727‐27743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ahn S, Joyner AL. Dynamic changes in the response of cells to positive hedgehog signaling during mouse limb patterning. Cell 2004;118:505‐516. [DOI] [PubMed] [Google Scholar]
- 25. Bai CB, Auerbach W, Lee JS, Stephen D, Joyner AL. Gli2, but not Gli1, is required for initial Shh signaling and ectopic activation of the Shh pathway. Development 2002;129:4753‐4761. [DOI] [PubMed] [Google Scholar]
- 26. Goodrich LV, Milenkovic L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science 1997;277:1109‐1113. [DOI] [PubMed] [Google Scholar]
- 27. Harfe BD, Scherz PJ, Nissim S, Tian H, McMahon AP, Tabin CJ. Evidence for an expansion‐based temporal Shh gradient in specifying vertebrate digit identities. Cell 2004;118:517‐528. [DOI] [PubMed] [Google Scholar]
- 28. Bai CB, Joyner AL. Gli1 can rescue the in vivo function of Gli2. Development 2001;128:5161‐5172. [DOI] [PubMed] [Google Scholar]
- 29. Garcia AD, Petrova R, Eng L, Joyner AL. Sonic hedgehog regulates discrete populations of astrocytes in the adult mouse forebrain. J Neurosci 2010;30:13597‐13608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Miyoshi H, Stappenbeck TS. In vitro expansion and genetic modification of gastrointestinal stem cells in spheroid culture. Nat Protoc 2013;8:2471‐2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Agren M, Kogerman P, Kleman MI, Wessling M, Toftgard R. Expression of the PTCH1 tumor suppressor gene is regulated by alternative promoters and a single functional Gli‐binding site. Gene 2004;330:101‐114. [DOI] [PubMed] [Google Scholar]
- 32. Dai P, Akimaru H, Tanaka Y, Maekawa T, Nakafuku M, Ishii S. Sonic Hedgehog‐induced activation of the Gli1 promoter is mediated by GLI3. J Biol Chem 1999;274:8143‐8152. [DOI] [PubMed] [Google Scholar]
- 33. Razumilava N, Gores GJ, Lindor KD. Cancer surveillance in patients with primary sclerosing cholangitis. Hepatology 2011;54:1842‐1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sawada R, Ku Y, Akita M, Otani K, Fujikura K, Itoh T, et al. Interleukin‐33 overexpression reflects less aggressive tumor features in large‐duct type cholangiocarcinomas. Histopathology 2018;73:259‐272. [DOI] [PubMed] [Google Scholar]
- 35. Tobe M, Isobe Y, Tomizawa H, Nagasaki T, Takahashi H, Fukazawa T, et al. Discovery of quinazolines as a novel structural class of potent inhibitors of NF‐kappa B activation. Bioorg Med Chem 2003;11:383‐391. [DOI] [PubMed] [Google Scholar]
- 36. Lin J, Zhang L, Zhao G, Su Z, Deng R, Pflugfelder SC, et al. A novel interleukin 33/ST2 signaling regulates inflammatory response in human corneal epithelium. PLoS ONE 2013;8:e60963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Varjosalo M, Taipale J. Hedgehog: functions and mechanisms. Genes Dev 2008;22:2454‐2472. [DOI] [PubMed] [Google Scholar]
- 38. Schmidt EV, Christoph G, Zeller R, Leder P. The cytomegalovirus enhancer: A pan‐active control element in transgenic mice. Mol Cell Biol 1990;10:4406‐4411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Razumilava N, Gumucio DL, Samuelson LC, Shah YM, Nusrat A, Merchant JL. Indian Hedgehog suppresses intestinal inflammation. Cell Mol Gastroenterol Hepatol 2017;5:63‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Westendorp BF, Buller N, Karpus ON, van Dop WA, Koster J, Versteeg R, et al. Indian Hedgehog suppresses a stromal cell‐driven intestinal immune response. Cell Mol Gastroenterol Hepatol 2017;5:67‐82.e61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hirsova P, Ibrahim SH, Bronk SF, Yagita H, Gores GJ. Vismodegib suppresses TRAIL‐mediated liver injury in a mouse model of nonalcoholic steatohepatitis. PLoS ONE 2013;8:e70599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fattori V, Hohmann MSN, Rossaneis AC, Manchope MF, Alves‐Filho JC, Cunha TM, et al. Targeting IL‐33/ST2 signaling: regulation of immune function and analgesia. Expert Opin Ther Targets 2017;21:1141‐1152. [DOI] [PubMed] [Google Scholar]
- 43. Ninlawan K, O’Hara SP, Splinter PL, Yongvanit P, Kaewkes S, Surapaitoon A, et al. Opisthorchis viverrini excretory/secretory products induce toll‐like receptor 4 upregulation and production of interleukin 6 and 8 in cholangiocyte. Parasitol Int 2010;59:616‐621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Carpino G, Cardinale V, Folseraas T, Overi D, Grzyb K, Costantini D, et al. Neoplastic transformation of peribiliary stem cell niche in cholangiocarcinoma arisen in primary sclerosing cholangitis. Hepatology 2018; 10.1002/hep.30210. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials