

Abstract
Background:
Historically, inflammatory periodontal diseases (gingivitis and periodontitis) have been recognized as being primarily of bacterial origin. Bacteria are necessary for disease development, but the presence of specific bacteria does not guarantee progression to periodontitis. Periodontitis is a multifactorial disease; specific bacteria are associated with disease, but may not be the target of treatment. Gingivitis and periodontitis are inflammatory conditions associated with bacterial overgrowth.
Aim:
To analyse evidence for established thought that specific bacteria directly participate in the pathogenesis of periodontitis and question the long-held tenet that penetration of the periodontal connective tissues by bacteria and their products is a significant phase in the initial development of periodontitis.
Methods:
The literature was searched for studies on initiation of gingivitis and periodontitis by specific pathogens. The search results were insufficient for a systematic review and have been summarized in a commentary instead.
Results:
There is very little evidence in the literature to support the commonly held concept that specific bacteria initiate periodontitis.
Conclusion:
We present evidence for a paradigm supporting the central role of inflammation, rather than specific microbiota, in the early pathogenesis of periodontitis, and discuss whether controlling the inflammation can influence the character and composition of the periodontal infection.
Keywords: periodontal infection, periodontal inflammation, periodontal pathogen, bacterial invasion, gingivitis, periodontitis
Introduction
Periodontitis, an inflammatory condition associated with bacterial infection, is modified by multiple host response genes in combination with lifestyle and environmental factors (Bartold and Van Dyke, 2013). The search for the causative periodontal pathogen(s) continues; however, emerging evidence suggests that it is the inflammatory response that drives changes in the periodontal microbiome and amplifies pathogenesis of progressive periodontitis.
Infection, Inflammation and Tissue Destruction
Plaque deposits account for only 20% of the risk for periodontitis (Grossi et al., 1994). Factors that contribute to the remaining 80% include environmental determinants and individual genetic variation that modify inflammation and its resolution (Kornman, 2008). Importantly, anti-infective therapy alone fails to effectively manage as many as 25% of periodontitis patients (Kornman et al., 2017). Most individuals with poor oral hygiene and little or no periodontal care develop only mild/moderate periodontitis (Loe et al., 1986, Baelum et al., 1986). The individual, not bacterial composition, determines risk for disease and adverse treatment outcomes (Socransky and Haffajee, 2005).
Hypotheses for the Development of Periodontitis
The ecological plaque hypothesis (Marsh, 1994), the keystone pathogen hypothesis (Hajishengallis et al., 2012, Hajishengallis and Lamont, 2012) and the polymicrobial synergy and dysbiosis model (Lamont and Hajishengallis, 2015) have widespread acceptance in contemporary periodontology. These hypotheses are consistent with observed reciprocal interactions between complexes of bacteria whereby gingival inflammation, in response to early colonising bacteria, changes the subgingival environment allowing increases in certain endogenous commensal bacteria (Socransky and Haffajee, 2002; Socransky and Haffajee, 2005).
However, these models fail to fully explain host-microbe interactions in health and disease. All three hypotheses place considerable emphasis on specific (keystone) bacteria being the driving force behind the development of periodontitis. While intuitively sound, the evidence is not conclusive. The oral microbiome in health is symbiotic (Dongari-Bagtzoglou, 2008). Putative pathogens are present in health, but are not pathogenic (Kolenbrander et al., 2010). We recognize that periodontitis is a continuum of inflammation, tissue destruction and microbial dysbiosis modified by factors such as smoking, systemic diseases and genetics. Further investigations into these complex interactions must determine how the metagenome of the host and microbes interact to result in disease.
Are specific bacteria the cause of periodontitis?
Early studies identified Aggregatibacter actinomycetemcomitans, Porphyromonas gingivalis, and Tannerella forsythia as “periodontal pathogens” (American Academy of Periodontology, 1996). The subgingival microbiota from periodontal health to periodontitis demonstrates ecologic succession, with emergence of dominant taxa in periodontitis without replacement of health-associated species (Oliveira et al., 2016), and an increase in bacterial load.
Recently, an additional 13 periodontal pathogens were identified (Oliveira et al., 2016). While these bacteria are present, we cannot infer that they cause periodontitis. It is just as likely they are selected for by inflammation-mediated environmental changes. To find “causative” periodontal pathogens, temporal data are required.
Can systemic inflammation modify periodontal destruction?
While not proven in humans, evidence from animal models shows that systemic inflammation results in exacerbation of periodontal inflammation and destruction. In three independent studies, induction of experimental arthritis had adverse actions on the periodontium in the absence of induction of experimental periodontitis (Ramamurthy et al., 2005; Park et al., 2010; Cantley et al., 2011).
Is bacterial invasion of the periodontal tissues an initiating event in periodontitis?
Penetration of periodontal tissues by bacteria and/or their metabolic products has long been considered a crucial step in the pathogenesis of periodontitis (Ji et al., 2015). However, this plausible assumption is based on very little supporting in vivo or in situ evidence. While studies report the presence of bacteria, or their products, in periodontal tissues (Ji et al., 2015), most, if not all, used biopsies from advanced periodontitis lesions (Guyodo et al., 2012, Kim et al., 2010, Baek et al., 2018). There are no publications concerning bacterial permeation in healthy, gingivitis or early periodontitis tissues. Invasion of epithelial cells by periodontal bacteria is reported in vitro, but in vivo demonstration has been limited (Tribble and Lamont, 2010). Furthermore, bacterial migration from within gingival epithelial cells across an intact basement membrane and into the gingival connective tissue has not been demonstrated in vivo. While bacterial/dendritic cell interaction has been demonstrated, this is confined to dendritic cells in the epithelium, not the underlying connective tissue (Cutler et al., 1999, Jotwani et al., 2001; El-Awady et al., 2015). No in vivo evidence exists for interaction/interface between fibroblasts, osteoblasts or osteoclasts and invading bacteria. In very advanced periodontitis, bacteria may be found in close approximation to CD-4 positive T cells (Guyodo et al., 2012).
What happens during the initial stages of development of the periodontal diseases?
It is essential to understand what happens at the during initiation of disease followed by definition of ensuing events. Several key factors should be considered:
1. Sulcular Epithelium: Barrier function and antimicrobial peptides
The epithelium provides a physical barrier between the external and internal environments (Bartold et al., 2000). In addition, the sulcular epithelium, in response to the accumulating bacterial load, releases potent antimicrobial peptides (alpha- and beta-defensins), as well as chemokines and cytokines responsible for neutrophil recruitment (Dale, 2002, Chung et al., 2007). Phagocytosis of bacteria and formation of a cellular barrier between the subgingival plaque and the epithelium ensues (Schroeder & Attstrom, 1980; Kornman et al., 1997). Given the critical role of innate immunity associated with the epithelium, it is time to consider the role periodontal epithelium plays in maintaining health and alerting the host of bacterial assault.
2. Epithelial Shedding and Epithelial cell renewal
The continuous shedding, turnover and replacement of gingival epithelial cells protects against invading bacteria. Non-keratinized tissues of the gingival sulcus have a 50% higher mitotic rate than the keratinized regions of attached gingiva that is increased in the presence of inflammation of the underlying connective tissue (Soni et al., 1965).
3. Gingival Crevicular Fluid
Gingival crevicular fluid (GCF) flow approximates 20 μL/h and increases dramatically with gingival inflammation. In a 5 mm pocket GCF is replaced 40 times/hour (Goodson, 2003). During transition from health to disease, GCF changes in composition, pH, oxygen levels, temperature, osmotic pressure and redox potential. Importantly, the rise in pH together with a rich source of nutrients from ongoing tissue destruction and bacterial metabolic activity favours the proliferation of acid-sensitive “pathogenic” bacteria (Barros et al., 2016).
4. Initial inflammatory response
Initially, the host contains the subgingival infection and tissue damage is limited to gingivitis with no ingress of specific bacteria or their metabolic products. If the inflammation cannot be resolved, the inflammatory exudate increases and alters the pocket environment allowing commensal bacteria, including P. gingivalis, to overgrow. The subgingival microbiome develops a disease-associated “dysbiotic” relationship dictated primarily by inflammation. With continuing bacterial challenge and uncontrolled gingival inflammation, the epithelial barrier is breached and new environment develops. It is conceivable that at this stage bacterial penetration of the tissues could occur. Continued inflammation leads to loss of periodontal ligament fibers, clinical loss of attachment and deepening of the periodontal pocket. Importantly, bone resorption is not directly microbial-induced; activation of osteoclasts is stimulated by inflammatory mediators.
Is there evidence in other systems for inflammation-mediated dysbiosis?
Dysbiosis, first described over 100 years ago in the gut, described disrupted symbiosis between bacteria and host (Plotnikoff and Riley, 2014). Dysbiosis of the gut is a useful model for understanding periodontal dysbiosis because, like periodontal dysbiosis, it is directly related to inflammation or immunodeficiency (Lupp et al., 2007, Kamada et al., 2013). It has been proposed that controlling gut inflammation is a good approach to limiting blooms of disease-associated bacteria (Zeng et al., 2017). Thus, evidence in other systems supports that it is inflammation that drives development of dysbiosis.
An inverted model for the pathogenesis of Periodontitis - inflammation exacerbates infection
The initial inflammatory response in periodontal disease(s) always precedes the emergence/overgrowth of periodontal pathogens (Lindhe et al., 1980, Listgarten, 1988, Tanner et al., 2007). Accordingly, an “inverted paradigm” is proposed whereby increasing inflammation selects specific bacteria based on growth requirements. Once overgrown, these bacteria promote inflammation to maintain their selective dominance (Figure 1).
Figure 1a.
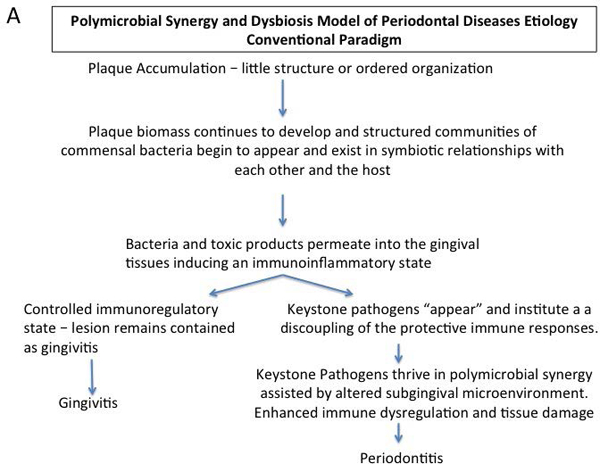
Conventional paradigm of polymicrobial synergy and dysbiosis model as a hypothesis for the pathogenesis of periodontitis
Management of periodontitis can be based on controlling the inflammation to control the infection (Bartold and Van Dyke, 2013, Bartold and Van Dyke, 2017). This inverted paradigm, which has been demonstrated experimentally in animal models (Hasturk et al. 2007, Lee et al. 2016), must be applied in the context of understanding that the ultimate successful clinical outcome of periodontal treatment will result from controlling, or resolving, the inflammatory response.
Future Research Directions
• Consider the validity of in vitro studies
There are innumerable examples of in vitro observations that are not reproducible in vivo. For example, direct interactions of bacteria with periodontal fibroblasts, bone cells, PMNs or lymphocytes are not supported by histopathological evidence. Studying such interactions in vitro is not warranted without in vivo confirmation.
• Investigate the continuum of disease
Descriptive studies of the progression of health to advanced periodontitis were performed in the 1970s. Mechanistic studies of the continuum are required.
• Modulate inflammation to modulate periodontal infection
Effective host response modulation is a crucial element missing in our current armamentarium of conventional periodontal therapy. Targeting inflammation reverses periodontitis and dysbiosis in animals. Studies are required in humans with active agents, not inhibitors of inflammation.
Conclusions
Current paradigms of initiation of periodontitis require reassessment. Traditional plaque control management approaches are only partially effective for periodontitis and fail in high-risk individuals. More detailed consideration of the host response, genetic and environmental factors is required. While the possibility exists that a hitherto unknown bacterial species may one day be identified as a true pathogen, this has eluded researchers for decades. Concepts focused on controlling the infection to control the inflammation should be reworked to consider controlling the inflammation to control the infection.
Figure 1b.
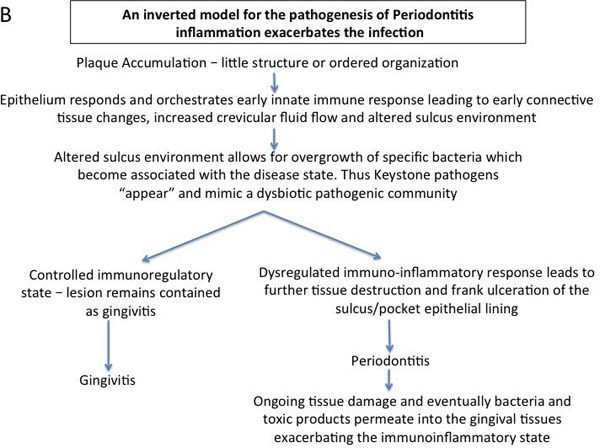
Inverted paradigm hypothesis for the pathogenesis of periodontitis whereby inflammation drives and exacerbated periodontal infection
Clinical Relevance:
Scientific Rationale.
Understanding disease pathogenesis requires knowing what initiates the disease followed by definition of ensuing events. There has been an overemphasis on the role of specific bacteria in the initiation of periodontitis at the expense of considering the host response, genetic and environmental factors.
Principle Findings.
The etiology of periodontitis is bacteria. However, evidence points to the pathogenesis being inflammatory. Data supporting in vivo invasion by bacteria has not been convincingly demonstrated; putative pathogens are part of the normal flora and overgrow after onset of disease, which is reversible by controlling inflammation; the ‘keystone pathogen’ hypothesis has no in vivo or in vitro evidence to support it; direct interactions of bacteria with fibroblasts or osteocytes is not supported by histopathological evidence.
Practical Implications.
Concepts focused solely on controlling the infection to control the inflammation should be reworked to consider controlling the inflammation to control the infection.
Acknowledgements
Disclosure: Boston University and the Forsyth Institute are assigned patents on lipoxins that are licensed for clinical development and are subject to consultant agreements for Dr. Thomas E. Van Dyke.
Research Support:
TE Van Dyke: USPHS grants DE025020, DE025383 and DE026934.
PM Bartold: NHMRC Grants 1027747 and 627143
Statement of Sources of Funding:
TE Van Dyke: USPHS grants DE025020, DE025383 and DE026934.
PM Bartold: NHMRC Grants 1027747 and 627143
Footnotes
Disclosure of any conflicts of interests:
Dr. Van Dyke is named in patents held by the Forsyth Institute that are subject to potential royalties or consulting fees.
Dr PM Bartold declares he has no conflicts of interest to declare.
References
- American Academy of Periodontology (1996). Consensus report. Periodontal diseases: epidemiology and diagnosis. Annals of Periodontology, 1, 216–222. [DOI] [PubMed] [Google Scholar]
- Baek K, Ji S & Choi Y (2018). Complex Intratissue Microbiota Forms Biofilms in Periodontal Lesions. Journal of Dental Research, 97, 192–200. doi: 10.1177/0022034517732754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baelum V, Fejerskov O & Karring T (1986). Oral hygiene, gingivitis and periodontal breakdown in adult Tanzanians. Journal of Periodontal Research, 21, 221–232. [DOI] [PubMed] [Google Scholar]
- Barros SP, Williams R, Offenbacher S & Morelli T (2016). Gingival crevicular fluid as a source of biomarkers for periodontitis. Periodontology 2000, 70, 53–64. doi: 10.1111/prd.12107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartold PM & Van Dyke TE (2013). Periodontitis: a host-mediated disruption of microbial homeostasis. Unlearning learned concepts. Periodontology 2000, 62, 203–217. doi: 10.1111/j.1600-0757.2012.00450.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartold PM & Van Dyke TE (2017). Host modulation: controlling the inflammation to control the infection. Periodontology 2000, 75, 317–329. doi: 10.1111/prd.12169. [DOI] [PubMed] [Google Scholar]
- Bartold PM, Walsh LJ & Narayanan AS (2000). Molecular and cell biology of the gingiva. Periodontology 2000, 24, 28–55. [DOI] [PubMed] [Google Scholar]
- Cantley MD, Haynes DR, Marino V & Bartold PM (2011) Pre-existing periodontitis exacerbates experimental arthritis in a mouse model. Journal of Clinical Periodontology, 38, 532–541. doi: 10.1111/j.1600-051X.2011.01714.x. [DOI] [PubMed] [Google Scholar]
- Chung WO, Dommisch H, Yin L & Dale BA (2007). Expression of defensins in gingiva and their role in periodontal health and disease. Current Pharmceutical Design, 13, 3073–3083. [DOI] [PubMed] [Google Scholar]
- Cutler CW, Jotwani R, Palucka KA, Davoust J, Bell D & Banchereau J (1999). Evidence and a novel hypothesis for the role of dendritic cells and Porphyromonas gingivalis in adult periodontitis. Journal of Periodontal Research, 34, 406–412. [DOI] [PubMed] [Google Scholar]
- Dale BA (2002). Periodontal epithelium: a newly recognized role in health and disease. Periodontology 2000 30, 70–78. [DOI] [PubMed] [Google Scholar]
- Dongari-Bagtzoglou A (2008) Pathogenesis of mucosal biofilm infections: challenges and progress. Expert Reviews in Anti Infective Therapy, 6, 201–208. doi: 10.1586/14787210.6.2.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Awady AR, Arce RM & Cutler CW (2015). Dendritic cells: microbial clearance via autophagy and potential immunobiological consequences for periodontal disease. Periodontology 2000, 69, 160–180. doi: 10.1111/prd.12096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodson JM (2003). Gingival crevice fluid flow. Periodontology 2000, 31, 43–54. [DOI] [PubMed] [Google Scholar]
- Grossi SG, Zambon JJ, Ho AW, Koch G, Dunford RG, Machtei EE, Norderyd OM & Genco RJ (1994). Assessment of risk for periodontal disease. I. Risk indicators for attachment loss. Journal of Periodontology, 65, 260–267. doi: 10.1902/jop.1994.65.3.260. [DOI] [PubMed] [Google Scholar]
- Guyodo H, Meuric V, Le Pottier L, Martin B, Faili A, Pers JO & Bonnaure-Mallet M (2012). Colocalization of Porphyromonas gingivalis with CD4+ T cells in periodontal disease. FEMS Immunology and Medical Microbiology, 64, 175–183. doi: 10.1111/j.1574-695X.2011.00877.x. [DOI] [PubMed] [Google Scholar]
- Hajishengallis G, Darveau RP & Curtis MA (2012). The keystone-pathogen hypothesis. Nature Reviews Microbiology, 10, 717–725. doi: 10.1038/nrmicro2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajishengallis G & Lamont RJ (2012). Beyond the red complex and into more complexity: the polymicrobial synergy and dysbiosis (PSD) model of periodontal disease etiology. Molecular and Oral Microbiology, 27, 409–419. doi: 10.1111/j.2041-1014.2012.00663.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasturk H, Kantarci A, Goguet-Surmenian E, Blackwood A, Andry C, Serhan CN & Van Dyke TE (2007). Resolvin E1 regulates inflammation at the cellular and tissue level and restores tissue homeostasis in vivo. Journal of Immunology, 179, 7021–7029. [DOI] [PubMed] [Google Scholar]
- Ji S, Choi YS & Choi Y (2015). Bacterial invasion and persistence: critical events in the pathogenesis of periodontitis? Journal of Periodontal Research, 50, 570–585. doi: 10.1111/jre.12248. [DOI] [PubMed] [Google Scholar]
- Jotwani R, Palucka AK, Al-Quotub M, Nouri-Shirazi M, Kim J, Bell D, Banchereau J & Cutler CW (2001). Mature dendritic cells infiltrate the T cell-rich region of oral mucosa in chronic periodontitis: in situ, in vivo, and in vitro studies. Journal of Immunology, 167, 4693–4700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada N, Seo SU, Chen GY & Nunez G (2013). Role of the gut microbiota in immunity and inflammatory disease. Nature Reviews Immunology, 13, 321–335. doi: 10.1038/nri3430. [DOI] [PubMed] [Google Scholar]
- Kim YC, Ko Y, Hong SD, Kim KY, Lee YH, Chae C & Choi Y (2010). Presence of Porphyromonas gingivalis and plasma cell dominance in gingival tissues with periodontitis. Oral Diseases, 16, 375–381. doi: 10.1111/j.1601-0825.2009.01649.x. [DOI] [PubMed] [Google Scholar]
- Kolenbrander PE, Palmer RJ Jr., Periasamy S & Jakubovics NS (2010). Oral multispecies biofilm development and the key role of cell-cell distance. Nature Reviews Microbiology, 8, 471–480. doi: 10.1038/nrmicro2381. [DOI] [PubMed] [Google Scholar]
- Kornman KS (2008). Mapping the pathogenesis of periodontitis: a new look. Journal of Periodontology, 79, 1560–1568. doi: 10.1902/jop.2008.080213. [DOI] [PubMed] [Google Scholar]
- Kornman KS, Giannobile WV & Duff GW (2017). Quo vadis: what is the future of periodontics? How will we get there? Periodontology 2000, 75, 353–371. doi: 10.1111/prd.12217. [DOI] [PubMed] [Google Scholar]
- Kornman KS, Page RC & Tonetti MS (1997). The host response to the microbial challenge in periodontitis: assembling the players. Periodontology 2000, 14, 33–53. [DOI] [PubMed] [Google Scholar]
- Lamont RJ & Hajishengallis G (2015). Polymicrobial synergy and dysbiosis in inflammatory disease. Trends in Molecular Medicine, 21, 172–183. doi: 10.1016/j.molmed.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CT, Teles R, Kantarci A, Chen T, McCafferty J, Starr JR, Brito LC, Paster BJ & Van Dyke TE. (2016). Resolvin E1 reverses experimental periodontitis and dysbiosis. Journal of Immunology, 197, 2796–2806. doi: 10.4049/jimmunol.1600859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindhe J, Liljenberg B & Listgarten M (1980). Some microbiological and histopathological features of periodontal disease in man. Journal of Periodontology, 51, 264–269. doi: 10.1902/jop.1980.51.5.264. [DOI] [PubMed] [Google Scholar]
- Listgarten MA (1988). The role of dental plaque in gingivitis and periodontitis. Journal of Clinical Periodontology, 15, 485–487. [DOI] [PubMed] [Google Scholar]
- Loe H, Anerud A, Boysen H & Morrison E (1986). Natural history of periodontal disease in man. Rapid, moderate and no loss of attachment in Sri Lankan laborers 14 to 46 years of age. Journal of Clinical Periodontology, 13, 431–445. [DOI] [PubMed] [Google Scholar]
- Lupp C, Robertson ML, Wickham ME, Sekirov I, Champion OL, Gaynor EC & Finlay BB (2007). Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe, 2, 119–129. doi: 10.1016/j.chom.2007.06.010. [DOI] [PubMed] [Google Scholar]
- Marsh PD (1994). Microbial ecology of dental plaque and its significance in health and disease. Advances in Dental Research, 8, 263–271. doi: 10.1177/08959374940080022001. [DOI] [PubMed] [Google Scholar]
- Oliveira RR, Fermiano D, Feres M, Figueiredo LC, Teles FR, Soares GM & Faveri M (2016). Levels of Candidate Periodontal Pathogens in Subgingival Biofilm. Journal of Dental Research, 95, 711–718. doi: 10.1177/0022034516634619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J-C, Su C, Jung I-H, Choi S-H, Cho K-S, Kim C-K, Park Y-B, Lee S-K & Kim C-S (2010) Mechanism of alveolar bone loss in a collagen-induced arthritis model in mice. Journal of Clinical Periodontology, 38, 122–130. doi: 10.1111/j.1600-051X.2010.01645.x [DOI] [PubMed] [Google Scholar]
- Plotnikoff GA & Riley D (2014). The human microbiome. Global Advances in Health and Medicine, 3, 4–5. doi: 10.7453/gahmj.2014.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramamurthy NS, Greenwald RA, Celiker MY & Shi EY (2005) Experimental arthritis in rats induces biomarkers of periodontitis which are ameliorated by gene therapy with tissue inhibitor of matrix metalloproteinases. Journal of Periodontology, 76: 229–233. [DOI] [PubMed] [Google Scholar]
- Schroeder HE, Attström R. (1980). Pocket formation: an hypothesis In: Lehner T, Cimasoni G (eds). The borderland between caries and periodontal disease (pp 99–123). London: II Academic/Grune & Stratton. [Google Scholar]
- Socransky SS & Haffajee AD (2002). Dental biofilms: difficult therapeutic targets. Periodontology 2000, 28, 12–55. [DOI] [PubMed] [Google Scholar]
- Socransky SS & Haffajee AD (2005). Periodontal microbial ecology. Periodontology 2000, 38, 135–187. doi: 10.1111/j.1600-0757.2005.00107.x. [DOI] [PubMed] [Google Scholar]
- Soni NN, Silberkweit M & Hayes RL (1965). Pattern of mitotic activity and cell densities in human gingival epithelium. Journal of Periodontology, 36, 15–21. [DOI] [PubMed] [Google Scholar]
- Tanner AC, Kent R Jr., Kanasi E, Lu SC, Paster BJ, Sonis ST, Murray LA & Van Dyke TE (2007). Clinical characteristics and microbiota of progressing slight chronic periodontitis in adults. Journal of Clinical Periodontology, 34, 917–930. doi: 10.1111/j.1600-051X.2007.01126.x. [DOI] [PubMed] [Google Scholar]
- Tribble GD & Lamont RJ (2010). Bacterial invasion of epithelial cells and spreading in periodontal tissue. Periodontology 2000, 52, 68–83. doi: 10.1111/j.1600-0757.2009.00323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng MY, Inohara N & Nunez G (2017). Mechanisms of inflammation-driven bacterial dysbiosis in the gut. Mucosal Immunology, 10, 18–26. doi: 10.1038/mi.2016.75. [DOI] [PMC free article] [PubMed] [Google Scholar]