

Abstract
The engineering of microorganisms to monitor environmental chemicals or to produce desirable bioproducts is often reliant on the availability of a suitable biosensor. However, the conversion of a ligand-binding protein into a biosensor has been difficult. Here, we report a general strategy for generating biosensors in Escherichia coli that act by ligand-dependent stabilization of a transcriptional activator and mediate ligand concentration-dependent expression of a reporter gene. We constructed such a biosensor by using the lac repressor, LacI, as the ligand-binding domain and fusing it to the Zif268 DNA-binding domain and RNA polymerase omega subunit transcription-activating domain. Using error-prone PCR mutagenesis of lacI and selection, we identified a biosensor with multiple mutations, only one of which was essential for biosensor behavior. By tuning parameters of the assay, we obtained a response dependent on the ligand isopropyl β-D-1-thiogalactopyranoside (IPTG) of up to a seven-fold increase in the growth rate of E. coli. The single destabilizing mutation combined with a lacI mutation that expands ligand specificity to D-fucose generated a biosensor with improved response both to D-fucose and to IPTG. However, a mutation equivalent to the one that destabilized LacI in either of two structurally similar periplasmic binding proteins did not confer ligand-dependent stabilization. Finally, we demonstrated the generality of this method by using mutagenesis and selection to engineer another ligand-binding domain, MphR, to function as a biosensor. This strategy may allow many natural proteins that recognize and bind to ligands to be converted into biosensors.
Keywords: biosensors, ligand-dependent stabilization, directed evolution
Graphical Abstract:
Biosensors transmit ligand binding into an observable output signal, such as the activation of a gene whose product confers resistance (e.g. β-lactamase for ampicillin resistance) or that is directly observable (e.g. green fluorescent protein). They can be used for many different purposes, including environmental monitoring of chemicals, orthogonal control of gene expression, construction of genetic circuits, and detection of desired products in metabolic engineering experiments. Biosensors have been constructed that provide output at the transcriptional, translational, or protein level. At the transcriptional level, biosensors based on transcriptional repressors typically exploit naturally occurring allosteric transcription factors,1–3 whose ligand-specificity has in some cases been altered.4–6 However, challenges with maintaining allostery can make these efforts difficult. At the transcriptional and translational level, biosensors have been constructed from RNA devices that convert ligand-binding into increased translation,7 but identifying riboswitches that respond to new ligands can require many rounds of selection. At the protein level, biosensors have been constructed by inserting a protein that binds a ligand into a reporter protein, whose signal can be measured.8–14 This strategy, however, requires the identification of suitable protein insertion sites and can require optimization of amino acid linkers.
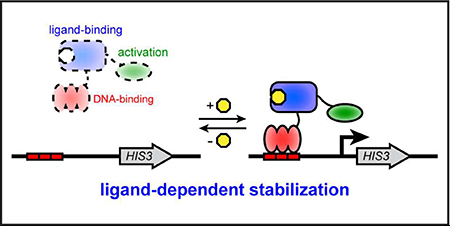
To rapidly engineer biosensors for new ligands, it would be advantageous to exploit the large number of proteins known to bind ligands, such as enzymes, transport proteins, and transcription factors. Many of these proteins have already evolved robust ligand recognition, with sensitive and selective binding. The key challenge is to convert the ligand binding by these natural proteins into an observable biosensor signal. One general strategy to meet this challenge employs ligand-dependent stability, which has been demonstrated in eukaryotic hosts.15, 16 It is an appealing strategy because protein stabilization upon ligand binding is an inherently general protein property, which should allow many protein domains that bind to a ligand to be converted to a biosensor. In this design, the biosensor is unstable in the absence of ligand and rapidly degraded by cellular protein degradation mechanisms; after binding ligand, the biosensor is stabilized and degraded more slowly. In a simple version of this design, the ligand-binding domain is part of a transcriptional activator such that its stability can be linked to reporter gene expression. Here, we develop this strategy in Escherichia coli, with increased biosensor stability due to ligand binding leading to increased expression of a metabolic reporter gene and hence increased growth rate.
Results and Discussion
A LacI-based biosensor
The biosensor design requires a destabilized ligand-binding domain fused to a DNA-binding domain and transcription-activating domain (Figure 1A). This protein fusion will localize to a DNA-binding site upstream of a reporter gene and activate its expression. We chose biosensor components that had been used in bacterial one-hybrid assays that also rely on reporter gene activation mediated by transcription factor binding,17, 18 namely Zif268 as the DNA-binding domain and the RNA polymerase omega subunit (encoded by the rpoZ gene) as the transcription-activating domain. For the reporter gene, we chose the HIS3 gene from Saccharomyces cerevisiae, which can be used as a selectable marker in E. coli and is required for growth in minimal media lacking histidine. As the ligand-binding domain, we chose the LacI transcription repressor because it binds to isopropyl β-D-1-thiogalactopyranoside (IPTG), which is neither toxic to nor metabolized by E. coli. A map of each plasmid is provided (Figure S1).
Figure 1.

Design and selection of E. coli biosensors based on ligand-dependent stability. A. A destabilized ligand-binding domain is fused between the Zif268 DNA-binding domain and RpoZ transcription-activating domain. In this biosensor scheme, the absence of ligand would lead to biosensor degradation by natural protein degradation processes, leading to weak transcriptional expression of HIS3. However, in the presence of ligand, binding to ligand would stabilize the biosensor, leading to increased transcriptional expression of HIS3. B. An error-prone PCR library of LacI is fused to Zif268 and RpoZ and subjected to positive selection in minimal media lacking histidine with IPTG ligand. E. coli with biosensor variants that activate expression of HIS3 grow in these selections. Individual biosensors are then screened for ligand-dependence by replica plating their E. coli host on minimal media lacking histidine with and without IPTG.
We used mutagenesis and selection to identify potentially destabilizing mutations in LacI (Figure 1B). We mutagenized lacI by error-prone PCR and inserted the pool of mutagenized fragments between RpoZ and Zif268 using linkers based on the 23 amino acids connecting RpoZ and Zif268 in a bacterial one-hybrid experiment.18 The 15 amino acids AAADYKDDDDKFRTG, which include a FLAG epitope for tracking accumulation of the biosensor, connect RpoZ and LacI, and the remaining eight amino acids, SKTPPHGT, connect LacI and Zif268. The resultant library, together with a reporter plasmid bearing the HIS3 gene downstream of a Zif268 binding site, was transformed into an E. coli host lacking the native hisB, pyrF, and rpoZ genes, to obtain approximately 12,000 variants. Twelve library members assessed by Sanger sequencing showed an average of 5.8 amino acid mutations across the lacI gene.
We first performed a positive selection to identify variants that grew in minimal media lacking histidine and supplemented with 1 mM IPTG and 3 mM 3-amino-1,2,4-triazole (3-AT), a competitive inhibitor of the His3 enzyme. In this positive selection, we expected to identify both the many variants containing mutations that are not destabilizing as well as the rare variants destabilized by mutation that can be restabilized by binding ligand. As LacI does not contact the Zif268 binding site, its normal allosteric properties in response to IPTG are not relevant in this context. We replica-plated approximately 1000 colonies that survived the positive selection on to minimal media lacking histidine with or without 1 mM IPTG, and with varying concentrations of 3-AT, to reveal colonies that showed improved growth with IPTG compared to without this ligand. We observed that many of the biosensor constructs did not contain an insert encoding a LacI variant, requiring us to screen colonies of interest by PCR to confirm that they contained the full-length lacI gene. Two colonies with full-length biosensors showed improved growth due to the presence of IPTG. Sequencing of lacI in these two colonies revealed the identical sequences of six mutations, which we assume arose via duplication of a single insert during the PCR or transformation steps. The mutations, numbered according to their LacI position,19 were: V4L, Q153L, V244I, G272R, S279R, and K290I. We named this biosensor L1.0. Because expression of the L1.0 biosensor construct itself was driven by a weak lacUV5 promoter, which is affected by induction with IPTG, we subsequently used the proA constitutively active promoter20 to drive expression of the L1.0 biosensor in subsequent experiments.
The six amino acid mutations are mapped to the LacI structure in Figure S2. These mutations are present on both the N-terminal domain of LacI that binds DNA, and the C-terminal domain that binds ligand. In particular, amino acid position 4 is intolerant to mutation because it directly contacts DNA,21 and thus it is likely that the V4L mutation prevents L1.0 from binding to the genomic binding site for LacI. Other mutations in L1.0, including Q153L and K290I, are present in spacer regions or solvent-exposed regions, and are tolerated in LacI repressor function.21 Finally, mutations V244I, G272R, and S279R are located in buried regions, and mutation of any of these generally results in a broken LacI protein that is unable to repress transcription.21
A single mutation in biosensor L1.0 is sufficient for biosensor activity
The plasmid encoding L1.0 was retransformed with the reporter plasmid into fresh E. coli cells. Growth assays in minimal media lacking histidine revealed that L1.0 increased growth rate in 30 mM 3-AT in a concentration-dependent manner, with a maximal three-fold increase at 100 mM IPTG compared to no ligand (Figure 2A). Deletion of rpoZ from L1.0 abolished growth in the absence of histidine, confirming that the increase in growth rate was due to RpoZ-mediated activation of HIS3 (Figure 2A).
Figure 2.

A. Response of L1.0 and L1.0 without RpoZ at increasing IPTG concentration in minimal media with 30 mM 3-AT. Each data point shows mean μmax (n = 3 biological replicates; error bars represent ± one standard deviation). Uninduced response is indicated with the horizontal gray line (n = 3 biological replicates; gray shading represents ± one standard deviation). B. Amino acid mutations present in each LacI-based biosensor. C. Response of L1.1-L1.6, relative to L1.0, when induced with 100 mM IPTG, in minimal media with 30 mM 3-AT. Each bar shows mean μmax relative to mean μmax of L1.0 (n = 3 biological replicates; error bars correspond to the quadratic sum of the fractional uncertainties in L1.1–1.6 and in L1.0). D. Response of L1.7 biosensor at increasing concentrations of IPTG in minimal media with 30 mM 3-AT. Each data point shows mean μmax (n = 3 biological replicates, error bars represent ± one standard deviation). Uninduced response is indicated with the horizontal gray line (n = 3 biological replicates; gray shading represents ± one standard deviation). E. Accumulation of L1.7 during exponential growth phase and stationary phase as assessed by Western blot. F. Structure of LacI dimer (taken from a co-crystal with DNA) showing amino acids 1–333 (PDB: 1EFA). G272 is highlighted in red.
We sought to compare the ligand-dependent response of L1.0 to a similar construct in which the wild type LacI served as the ligand-binding domain. However, with the wild type LacI, the protein fusion was unable to drive reporter gene expression regardless of IPTG concentration, and we observed no growth in media without histidine. Transformation of the plasmid encoding the wild type LacI fusion protein into the E. coli strain resulted in a slow-growth phenotype, independent of the presence of the reporter plasmid, suggesting that the fusion protein itself was toxic. The slow-growth phenotype was not observed in the E. coli DH10B strain used for cloning. The key difference between these strains is likely the native copy of rpoZ present in DH10B. We speculate that a fusion protein containing wild type LacI may be extremely stable, such that highly efficient recruitment of RNA polymerase by its RpoZ component limits availability of the polymerase for expression of essential E. coli genes. In the DH10B strain, the native copy of RpoZ may compete with the RpoZ in the fusion protein for the polymerase. Despite the toxicity of the construct containing the wild type LacI, the mutagenesis and selection strategy enabled us to identify mutations that convert this construct into a biosensor, highlighting the robustness of the method.
To determine if all six amino acid mutations were required for biosensor activity, we individually reverted each single amino acid mutation back to its wild type identity in biosensors named L1.1-L1.6 (Figure 2B). In all cases except G272R, reversion back to the wild type amino acid identity resulted in a growth rate similar to L1.0 when the cells were induced with 100 mM IPTG (Figure 2C). Reversion of G272R, in biosensor L1.4, abolished biosensor function, and the cells had a poor growth phenotype in media without histidine similar to that with the wild type LacI as the ligand-binding domain. We evaluated whether the G272R mutation alone when present in the biosensor construct was sufficient to confer biosensor activity. L1.7, containing only the G272R mutation (Figure 2B), resulted in a similar ligand-dependent response as L1.0, conferring an approximately 2.6-fold increase in growth rate from 1 mM to 100 mM IPTG (Figure 2D).
To confirm that L1.7 was indeed functioning by ligand-dependent stabilization, we assayed the level of L1.7 protein by Western blot using the FLAG epitope. Samples were grown in minimal media lacking histidine with various concentrations of IPTG. After reaching mid-log phase (OD600 of 0.4–0.5), cells were pelleted and lysed, and the proteins were fractionated and blotted. The level of L.1.7 protein at 100 mM IPTG was 5.2-fold greater than with no IPTG in the media (Figure 2E), confirming the effect of ligand concentration on biosensor accumulation. However, when these samples were allowed to reach stationary phase, no L1.7 protein was detected (Figure 2E). Similarly, when cells were cultured in media that does not require HIS3 expression for growth, such as LB media or minimal media supplemented with histidine, no L1.7 protein was detected (data not shown). These results suggest that the L1.7 biosensor is itself somewhat toxic, accumulating only when it provides a growth benefit and being degraded after growth has ceased.
LacI G272R, displayed on the LacI structure (Figure 2E), is within a β-sheet buried in the C-terminal globular domain of LacI, and this mutation likely prevents proper packing of this domain and reduces protein stability. Previous work showing that large amino acids at position 272 result in a LacI variant no longer able to repress transcription21 is consistent with the effect of this mutation in the biosensor context.
Transcriptional activation of the L1.7 biosensor
To directly assess transcriptional activation of the L1.7 biosensor, we measured its response using a GFP reporter gene. We constructed a reporter plasmid with HIS3 replaced by sfGFP, and the kanamycin resistance gene replaced by a chloramphenicol resistance gene, and used it to transform the selection strain of E. coli carrying the L1.7 biosensor plasmid and HIS3 reporter plasmid. Cultures were grown in M9 media with 10 mM 3-AT, a low concentration that did not yield a ligand-dependent effect on growth rate (Figure S3). We observed a maximal increase in fluorescence of 2.7-fold after 32 h with 100 mM IPTG compared to no IPTG (Figure S3). In nonselective media or when the HIS3 reporter plasmid was not present, fluorescence did not increase (data not shown), consistent with the finding that L1.7 was observed only in selective conditions and was degraded upon cells reaching stationary phase.
Tuning the biosensor response
A major advantage of the growth-based biosensors developed here is the ability to optimize biosensor response for different ligand concentrations through changes in growth conditions or properties of the reporter gene. One potential variable is the concentration of 3-AT, a competitive inhibitor of the His3, as more His3 production is needed for cells to grow when 3-AT is included in the media. We wanted to fully assess the relationship between varying 3-AT and IPTG concentrations and biosensor response. With the L1.7 biosensor at low 3-AT concentrations, no change in growth rate was observed regardless of IPTG concentration (Figure 3A), presumably because the biosensor even when not bound to ligand is stable enough to activate a low level of HIS3 expression sufficient for growth. At high 3-AT concentrations, robust growth was observed with 100 mM IPTG, resulting in a maximal 7.3-fold increase in growth rate at 40 mM 3-AT compared with the uninduced sample, although little growth was observed in IPTG concentrations below 100 mM at this 3-AT concentration. We did not observe improved sensitivity to lower concentration of IPTG at lower concentration of 3-AT, suggesting that the relatively high background signal of the biosensor limits this sensitivity.
Figure 3.

A. Heat map showing response of L1.7 at various concentrations of 3-AT and IPTG. Each condition shows mean μmax of two biological replicates. B. Response of L1.7 with mutated Zif268 binding sites. Each data point shows mean μmax (n = 3 biological replicates; error bars represent ± one standard deviation) in minimal media with 30 mM 3-AT.
In addition to varying ligand and His3 inhibitor concentration, we attempted to tune the biosensor response based on the affinity of the DNA-binding domain for its binding site. Reducing the affinity of the Zif268 domain for its cognate DNA may further reduce the growth rate of E. coli at low concentrations of ligand. The reduced affinity may thus improve the sensitivity and dynamic range of the biosensor, as the biosensor would be slower to localize to the reporter gene, providing more time for biosensor degradation. The Zif268 domain binds to a DNA site of nine base pairs. We assessed the ability of L1.7 to activate a reporter gene with three different mutated DNA-binding sites (Figure 3B); the wild type binding site has affinity for Zif268 that is ~4-fold higher than two of these mutated sites (those ending in GAG or GCA), and ~20-fold higher than the other (ending in GGG).22 The response of L1.7 with the reporter containing the mutated DNA-binding sites showed the expected trend, with the binding sites ending in GAG or GCA conferring a larger response compared to the binding site ending in GGG. However, the response of L1.7 with the binding site ending in GAG and GCA was similar in its dynamic range compared to with the wild type binding site, suggesting that the limited reduction in affinity is not sufficient to decrease biosensor background.
A mutation altering LacI ligand-specificity results in improved response to IPTG and D-fucose
Mutations that alter the ligand specificity of the LacI transcription repressor have been identified.4 Several of these mutations relax ligand specificity toward other sugars, and other mutations change ligand-specificity from IPTG to another sugar. One mutation, LacI Q291T, improves the response of LacI both to IPTG and D-fucose.4 We asked whether Q291T also altered the specificity of L1.7.
We incorporated the Q291T mutation into L1.7 and evaluated the response of the resulting biosensor to both IPTG and fucose (Figure 4). Compared to L1.7, the L1.7+Q291T biosensor showed a higher response to IPTG (3.4-fold increase in growth rate compared to no ligand) and to fucose (3.5-fold increase in growth rate compared to no ligand). We also observed a 2.8-fold increase in the growth rate of E. coli containing L1.7 when induced with fucose, which was unexpected given that the native LacI protein shows essentially no response with fucose.4 This result suggests that fucose may bind LacI but not induce its allosteric response, or that the binding specificity of L1.7 is relaxed as part of LacI destabilization. This result highlights the strong specificity of many allosteric transcription repressors for their native ligand. When engineering new biosensors, it is important to consider whether relaxed ligand specificity outweighs the increased challenge of maintaining allostery in transcriptional repressors.
Figure 4.

Response of L1.7 and L1.7+Q291T to induction with IPTG and fucose in minimal media with 30 mM 3-AT. Bar plots show mean μmax (n = 3 biological replicates; error bars represent ± one standard deviation).
Biosensors based on ligand-dependent stabilization is a general method for biosensor engineering
The core ligand-binding region of LacI is structurally similar to a large family of transport proteins called periplasmic binding proteins.23, 24 Phylogenetic analysis suggests that LacI, as well as several other common transcriptional repressors, evolved by fusion of a periplasmic binding protein and a domain for binding DNA.25 Analysis of the more than 500 periplasmic binding proteins with structural data showed that the core ligand-binding domain of LacI is similar to several other periplasmic binding proteins that bind sugars, sugar alcohols, and autoinducer-2.26 Because of this structural similarity, we speculated that destabilizing mutations in LacI might also be destabilizing in related periplasmic binding proteins.
We assessed the mutation analogous to G272R in two E. coli periplasmic binding proteins: AlsB, the D-allose binding protein,27 and RbsB, the D-ribose binding protein.28 We constructed the protein fusions RpoZ-AlsBZif268 and RpoZ-RbsB-Zif268, and introduced the equivalent mutation into each (G247R in AlsB and G238R in RbsB). However, neither the wild type nor mutated variants of AlsB or RbsB conferred a ligand-dependent increase in growth rate (Figure 5A, 5B and Figure S4). Comparing AlsB and RbsB, we observed that fusion proteins derived from RbsB were weaker activators of HIS3, resulting in much lower growth rate at 3-AT concentrations above 10 mM (Figure S4). This observation suggests that inherent differences in stability between AlsB and RbsB, despite their strong structural similarities, may explain why simply transferring a destabilizing protein mutation to a structurally similar protein is not sufficient to generate novel biosensors based on ligand-dependent stabilization.
Figure 5.

Construction of biosensors based on other ligand-binding domains. Response of protein fusions with A. AlsB and B. RbsB in minimal media with 40 mM 3-AT (AlsB) and 20 mM 3-AT (RbsB). Bar plots show mean μmax (n = 3 biological replicates; error bars represent ± one standard deviation). Response of each transcriptional activating protein fusion with 10–40 MM 3-AT is shown in Figure S4. C. Response of WT MphR protein fusion and biosensor M1.0 in minimal media with 40 mM 3-AT. Bar plot shows mean μmax (n = 4 biological replicates for wild type MphR, and n = 6 for M1.0; error bars represent ± one standard deviation; *indicates p < 0.05 as determined by a two-tailed t-test).
To demonstrate that this method of constructing biosensors could be generalized, we engineered a second ligand-binding domain. For this experiment, we chose the allosteric transcription repressor MphR,29, 30 which binds the macrolide antibiotic erythromycin A. We generated an error-prone PCR library of MphR and fused this library between RpoZ and Zif268. We employed an additional negative selection based on the URA3 gene, which converts 5-fluoroorotic acid into a toxic product, to reduce the number of variants that activate reporter expression in the absence of ligand. After selection and screening, we identified a biosensor (called M1.0) with two mutations in the MphR ligand-binding domain, A34S and V169D. We observed a 1.4-fold increase in growth rate of cells carrying M1.0 with 100 μM erythromycin compared to no erythromycin (Figure 5C). Although this response was modest, it supports the idea that the method to construct biosensors may be general. We anticipate that the response of the MphR biosensor could be improved with additional mutagenesis and selection.
Concluding comments
This directed evolution strategy used here to identify destabilizing mutations could be general, and it might be improved by increasing the throughput of the approach. To identify LacI-based biosensors, we used replica-plating to screen variants for ligand-dependent response, which limited the screening to between 103–104 variants. To identify MphR-based biosensors, we employed a negative selection to select against variants that are not destabilized, allowing the screening of libraries that are several orders of magnitude larger.
The biosensors we generated display a more limited dynamic range than many artificial and engineered allosteric transcription repressors, which frequently show more than ten-fold activation of reporter gene expression,1, 2, 4 or the eukaryotic biosensors based on ligand-dependent stabilization, which show nearly 100-fold activation of a reporter gene.16 Nonetheless, the magnitude of reporter response observed with the biosensors developed here, as assessed by growth rate or by activation of the GFP gene, is consistent with some engineered biosensors.3, 13, 14 This dynamic range might be improved by additional rounds of mutagenesis and selection, by beginning with a domain that has greater affinity for its ligand than LacI or MphR, or by decreasing the amount of background reporter expression, such as by increased degradation using a degradation tag.
Biosensors that function based on ligand-dependent stability allow the enormous diversity of natural ligand-binding proteins to be exploited; many bind their ligand with high affinity and specificity. Thus, this biosensor platform should facilitate the engineering of proteins and metabolic pathways with improved function.
Materials and methods
Enzymes and reagents
All oligonucleotides were purchased from Integrated DNA Technologies with standard desalting and used without further purification. All plasmids used in this study are listed in Table S1, and all oligonucleotides used in this study are listed in Table S2. PCR reactions were performed using Phusion polymerase purchased from New England Biolabs or with Kappa HiFi polymerase purchased from Roche. Error-prone PCR was performed using the Agilent GeneMorph II Random Mutagenesis kit. Cloning was performed using Gibson Assembly, and selection of plasmids was performed using chemically competent DH10B cells. The sequence of each plasmid was verified using Sanger sequencing. IPTG was obtained from Chem-Impex International and prepared as a 10x stock in water for all assays. Erythromycin was obtained from Sigma and prepared as a 100x stock in 0.1% acetic acid for all assays. All other chemicals were obtained from Sigma or Acros.
Strains and Media
Biosensor growth assays were performed using E. coli strain USO hisBΔ pyrFΔ rpoZΔ.18 Cloning was performed using laboratory strain E. coli DH10B from Invitrogen. Cells were grown at 37 °C. LB media was used as rich media for all experiments. NM selective media, prepared as described previously,18 was used as minimal media during selection experiments and in biosensor growth assays.
Preparation of Library
1 ng of LacI template DNA was amplified using the Agilent GeneMorph II Random Mutagenesis kit following the manufacturer’s instructions. The PCR product was purified by 1% agarose gel and isolated from the gel using the Qiagen QIAquick Gel Extraction kit.
Plasmid pB1H2ω2-zif268, which served as the backbone for the LacI library, was linearized by PCR and cleaned up using the Zymogen DNA Clean and Concentrator Kit.
70 ng of product from error-prone PCR of lacI and 240 ng of linearized pB1H2ω2-Zif268 were diluted to 10 μL volume with water, combined with 10 μL of NEB NEBuilder HiFi DNA Assembly 2x master mix, and incubated at 50 °C for 60 min. The assembly reaction was cleaned up using the Zymogen DNA Clean and Concentrator Kit, and the entire product was transformed into 100 μL of Invitrogen Electromax DH10B cells. Transformed cells were recovered by shaking in 1 mL of SOC media for 1 h at 37 °C. 1% of cells were plated to estimate library size, and the remaining transformation was added to 50 mL LB media supplemented with ampicillin, and incubated with shaking overnight at 37 °C. Plasmid containing the error-prone PCR library was isolated from 5 mL of culture using the Qiagen QIAprep Spin Miniprep kit.
The biosensor library based on the mphR ligand-binding domain was generated similarly (described in detail in the supporting information). Transformation of the MphR library yielded ~500,000 unique transformants. These were grown overnight in 50 mL of LB media supplemented with carbenicillin and incubated with shaking overnight at 37 °C. Plasmid containing the error-prone PCR library was isolated from 5 mL of culture using the Qiagen QIAprep Spin Miniprep kit.
E. coli transformation with library and positive selection for HIS3 expression
200 ng of plasmid containing the error-prone PCR library was cotransformed with 200 ng of reporter plasmid into 200 μL of electrocompetent USO hisBΔ pyrFΔ rpoZΔ cells in four separate 50 μL transformations. The cells from each transformation were recovered by shaking in 1 mL of SOC media for 1 h at 37 °C. 1% of cells were plated on LB plates supplemented with ampicillin and kanamycin to estimate the number of unique colonies from the library transformation. The rest of the transformation was pelleted, resuspended in NM minimal media supplemented with ampicillin and kanamycin, 3 mM 3-AT, and 1 mM IPTG and incubated with shaking at 37 °C for 48 h and directly used for replica plating.
Replica-plating to identify variants with ligand-dependent growth
Cells surviving positive selection were diluted to 1 × 103 cells/mL (based on OD600), and 100 μL of this dilution (100 cells) were plated on LB plates containing ampicillin and kanamycin. Plates were incubated overnight at 37 °C, then replica plated onto NM plates supplemented with ampicillin, kanamycin, 3, 10, or 30 mM 3-AT, either without IPTG or with 1 mM IPTG. Plates were incubated at 37 °C for 48 h. Plates with and without IPTG were compared to one another to identify colonies that showed a ligand-dependent difference in growth.
Measurement of biosensor growth rates
Single colonies from freshly transformed cells were used to inoculate 5 mL cultures of LB media containing the antibiotics required to maintain plasmids. Culture tubes were sealed with parafilm and incubated without shaking overnight at 37°C, usually growing to OD600 between 0.2 and 0.4. We observed more consistent growth assay results when starter cultures were not allowed to reach stationary phase.31 Approximately 1 × 108 cells (as measured by OD600) were removed from overnight culture and pelleted by spinning for 30 sec at 14 15,000 x g. LB media was removed, and cells resuspended in 200 μL of water.
For growth assays with LacI biosensors, 2 μL of resuspended cells (~1 × 106 cells) were used to inoculate each well of a 96-well plate containing 178 μL of NM media (supplemented ampicillin, kanamycin, and 3-AT) and 20 μL of ligand from a 10x stock. For growth assays with MphR biosensors, 2 μL of resuspended cells (~1 × 106 cells) were used to inoculate each well of a 96-well plate containing 196 μL of NM media (supplemented with ampicillin, kanamycin, and 3-AT) and 2 μL of ligand from a 100x stock. The plate was covered with transparent LightCycler480 Sealing Foil (Roche), and grown for 48 h in a Biotek Synergy H1 plate reader at 37 °C with double orbital shaking, measuring OD600 every 10 minutes.
Analysis of growth rates
The maximum growth rate (μmax) for each growth curve was determined using the R package Growthrates,32, 33 using the function that computes growth rates as described by Hall and coworkers.31 Growth curves were fit using 6 hours of growth curve data.
Assessing biosensor transcriptional activation
Single colonies from freshly transformed cells were used to inoculate 5 mL cultures of LB media containing the antibiotics required to maintain plasmids. Culture tubes were sealed with parafilm and incubated without shaking overnight at 37°C. Approximately 5 × 108 cells (as measured by OD600) were removed from overnight culture and pelleted by spinning for 30 sec at 15,000 x g. LB media was removed, and cells resuspended in 500 μL of water.
20 μL of resuspended cells (~2 × 107 cells) were used to inoculate each well of a 96-well plate containing 160 μL of M9 media (supplemented with ampicillin, kanamycin, and 3-AT) and 20 μL of ligand from a 10x stock. The plate was covered with transparent LightCycler480 Sealing Foil (Roche), and grown for 48 h in a Biotek Synergy H1 plate reader at 37 °C with double orbital shaking, measuring OD600 and fluorescence (excitation at 485 nm and emission at 515 nm) every 10 minutes.
Assessing biosensor level by Western blot
Single colonies from freshly transformed cells were used to inoculate 5 mL cultures of LB media containing the antibiotics required to maintain plasmids. Culture tubes were sealed with parafilm and incubated without shaking overnight at 37°C. Approximately 5 × 108 cells (as measured by OD600) were removed from overnight culture and pelleted by spinning for 30 sec at 15,000 x g. LB media was removed, and cells resuspended in 500 μL of water.
50 μL of resuspended cells (~5 × 107 cells) were used to inoculate a 5 mL culture of NM media supplemented with ampicillin, kanamycin, 3-AT, and IPTG. Cultures were grown at 37 °C with shaking until cell density reached mid-log phase (at OD600 of approximately 0.4–0.5). 1.5 mL of culture was pelleted by spinning for 30 sec at 15,000 x g, NM media was removed, and cells were resuspended in 75 uL of lysis buffer (12.5 mM Tris, pH 8, 4% SDS), heated at 95C for 10 min, and spun for 2 min at 15,000 x g. The remaining culture was allowed to reach stationary phase and left for approximately 12 h, after which 1.5 mL of culture was isolated and lysed by the same protocol.
Samples were separated on a 4–12% Tris-Glycine Novex SDS-PAGE gel in MOPS SDS-PAGE running buffer at 120 volts for 1.5 h. Samples were transferred to nitrocellulose membrane by electrophoresis in Towbin transfer buffer for 1.5 h at 50 volts. The nitrocellulose membrane was blocked for 1 h at room temperature with 3% nonfat milk in pH 7.5 TBS with 0.1% Tween, blotted with primary antibodies in 3% nonfat milk in pH 7.5 TBS with 0.1% Tween for 1 h at room temperature, and blotted with secondary antibody in 3% nonfat milk in pH 7.5 TBS with 0.1% Tween for 30 min at room temperature. Monoclonal Anti-FLAG M2 (Sigma) and monoclonal GA1R anti-GAPDH (Invitrogen) were used to detect the biosensor protein and the loading control, respectively. Primary antibodies were detected using GE ECL Mouse IgG, HRP-linked Whole Antibody secondary antibody. Blots were detected using West Dura Extended Duration HRP substrate (Thermo Scientific) and imaged with HyBlot Autoradiography film (Denville Scientific).
Positive and negative selection to identify MphR biosensors
For each round of selection, ten agar plates were used, allowing us to screen approximately 100,000 transformants. Although this number did not cover our entire biosensor library of ~500,000 transformants, we speculated that it would be a sufficient number of variants to identify some with biosensor activity.
Approximately 100,000 cells were plated on NM minimal media plates supplemented with ampicillin, kanamycin, 3 mM 3-AT, and 30 μM erythromycin A. Plates were incubated at 37 °C for 48 h, after which colonies were scraped, and the population expanded by growing overnight at 37 °C in LB media with ampicillin and kanamycin. From this population, 100,000 cells were then plated on NM minimal media plates supplemented with ampicillin, kanamycin, 0.2 g/L histidine, 0.2 g/L uracil, and 1 g/L 5-fluoorotic acid (5-FOA). Plates were incubated at 37 °C for 48 h, and colonies were scraped and grown overnight in at 37 °C in LB media with ampicillin and kanamycin. Two additional rounds of positive and negative selection were performed, after which colonies were screened for ligand-dependent growth by replica plating. Colonies that appeared to show ligand-dependent growth were subsequently screened in a liquid growth assay, after which biosensor plasmids were isolated and sequenced to identify the biosensor variant.
Supplementary Material
Acknowledgments
This work was supported by NIH grant 1P41 GM103533 (to S.F.). B.M.B. was supported by NIH T32HG000035 and NIH F32GM122202. S.F. is an investigator of the Howard Hughes Medical Institute. We thank members of the Fields lab for helpful suggestions on the manuscript. We thank Scot Wolfe and George Church for gifts of plasmids and the E. coli strain lacking hisB, pyrF, and rpoZ.
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website. Figure S1–S4, Tables S1–S2, and description of cloning procedures.
References:
- [1].Rogers JK, Guzman CD, Taylor ND, Raman S, Anderson K, and Church GM (2015) Synthetic biosensors for precise gene control and real-time monitoring of metabolites, Nucleic Acids Res. 43, 7648–7660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Dietrich JA, Shis DL, Alikhani A, and Keasling JD (2013) Transcription factor-based screens and synthetic selections for microbial small-molecule biosynthesis, ACS Synth. Biol 2, 47–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Ho JCH, Pawar SV, Hallam SJ, and Yadav VG (2018) An Improved Whole-Cell Biosensor for the Discovery of Lignin-Transforming Enzymes in Functional Metagenomic Screens, ACS Synth. Biol 7, 392–398. [DOI] [PubMed] [Google Scholar]
- [4].Taylor ND, Garruss AS, Moretti R, Chan S, Arbing MA, Cascio D, Rogers JK, Isaacs FJ, Kosuri S, Baker D, Fields S, Church GM, and Raman S (2016) Engineering an allosteric transcription factor to respond to new ligands, Nat. Methods 13, 177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Tang SY, Qian S, Akinterinwa O, Frei CS, Gredell JA, and Cirino PC (2013) Screening for enhanced triacetic acid lactone production by recombinant Escherichia coli expressing a designed triacetic acid lactone reporter, J. Am. Chem. Soc 135, 10099–10103. [DOI] [PubMed] [Google Scholar]
- [6].Kasey CM, Zerrad M, Li Y, Cropp TA, and Williams GJ (2018) Development of Transcription Factor-Based Designer Macrolide Biosensors for Metabolic Engineering and Synthetic Biology, ACS Synth. Biol 7, 227–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Serganov A, and Nudler E (2013) A decade of riboswitches, Cell 152, 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Tucker CL, and Fields S (2001) A yeast sensor of ligand binding, Nat. Biotechnol 19, 1042–1046. [DOI] [PubMed] [Google Scholar]
- [9].Guntas G, and Ostermeier M (2004) Creation of an allosteric enzyme by domain insertion, J. Mol. Biol 336, 263–273. [DOI] [PubMed] [Google Scholar]
- [10].Guntas G, Mansell TJ, Kim JR, and Ostermeier M (2005) Directed evolution of protein switches and their application to the creation of ligand-binding proteins, Proc. Natl. Acad. Sci. U.S.A 102, 11224–11229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Oakes BL, Nadler DC, Flamholz A, Fellmann C, Staahl BT, Doudna JA, and Savage DF (2016) Profiling of engineering hotspots identifies an allosteric CRISPR-Cas9 switch, Nat. Biotechnol 34, 646–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Nadler DC, Morgan SA, Flamholz A, Kortright KE, and Savage DF (2016) Rapid construction of metabolite biosensors using domain-insertion profiling, Nat. Commun 7, 12266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Younger AK, Dalvie NC, Rottinghaus AG, and Leonard JN (2017) Engineering Modular Biosensors to Confer Metabolite-Responsive Regulation of Transcription, ACS Synth. Biol 6, 311–325. [DOI] [PubMed] [Google Scholar]
- [14].Younger AKD, Su PY, Shepard AJ, Udani SV, Cybulski TR, Tyo KEJ, and Leonard JN (2018) Development of novel metabolite-responsive transcription factors via transposon-mediated protein fusion, Protein Eng. Des. Sel 31, 55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Banaszynski LA, Chen LC, Maynard-Smith LA, Ooi AG, and Wandless TJ (2006) A rapid, reversible, and tunable method to regulate protein function in living cells using synthetic small molecules, Cell 126, 995–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Feng J, Jester BW, Tinberg CE, Mandell DJ, Antunes MS, Chari R, Morey KJ, Rios X, Medford JI, Church GM, Fields S, and Baker D (2015) A general strategy to construct small molecule biosensors in eukaryotes, Elife 4, e10606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Meng X, Brodsky MH, and Wolfe SA (2005) A bacterial one-hybrid system for determining the DNA-binding specificity of transcription factors, Nat. Biotechnol 23, 988–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Noyes MB, Meng X, Wakabayashi A, Sinha S, Brodsky MH, and Wolfe SA (2008) A systematic characterization of factors that regulate Drosophila segmentation via a bacterial one-hybrid system, Nucleic Acids Res. 36, 2547–2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Farabaugh PJ (1978) Sequence of the lacI gene, Nature 274, 765–769. [DOI] [PubMed] [Google Scholar]
- [20].Davis JH, Rubin AJ, and Sauer RT (2011) Design, construction and characterization of a set of insulated bacterial promoters, Nucleic Acids Res. 39, 1131–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Suckow J, Markiewicz P, Kleina LG, Miller J, Kisters-Woike B, and Müller-Hill B (1996) Genetic studies of the Lac repressor. XV: 4000 single amino acid substitutions and analysis of the resulting phenotypes on the basis of the protein structure, J. Mol. Biol 261, 509–523. [DOI] [PubMed] [Google Scholar]
- [22].Miller JC, and Pabo CO (2001) Rearrangement of side-chains in a Zif268 mutant highlights the complexities of zinc finger-DNA recognition, J. Mol. Biol 313, 309–315. [DOI] [PubMed] [Google Scholar]
- [23].Tam R, and Saier MH Jr. (1993) Structural, functional, and evolutionary relationships among extracellular solute-binding receptors of bacteria, Microbiol. Rev 57, 320–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Berntsson RP, Smits SH, Schmitt L, Slotboom DJ, and Poolman B (2010) A structural classification of substrate-binding proteins, FEBS Lett. 584, 2606–2617. [DOI] [PubMed] [Google Scholar]
- [25].Fukami-Kobayashi K, Tateno Y, and Nishikawa K (2003) Parallel evolution of ligand specificity between LacI/GalR family repressors and periplasmic sugar-binding proteins, Mol. Biol. Evol 20, 267–277. [DOI] [PubMed] [Google Scholar]
- [26].Scheepers GH, Lycklama a Nijeholt JA, and Poolman B (2016) An updated structural classification of substrate-binding proteins, FEBS Lett. 590, 4393–4401. [DOI] [PubMed] [Google Scholar]
- [27].Chaudhuri BN, Ko J, Park C, Jones TA, and Mowbray SL (1999) Structure of D-allose binding protein from Escherichia coli bound to D-allose at 1.8 Å resolution, J Mol Biol 286, 1519–1531. [DOI] [PubMed] [Google Scholar]
- [28].Bjürkman AJ, Binnie RA, Zhang H, Cole LB, Hermodson MA, and Mowbray SL (1994) Probing protein-protein interactions. The ribose-binding protein in bacterial transport and chemotaxis, J. Biol. Chem 269, 30206–30211. [PubMed] [Google Scholar]
- [29].Noguchi N, Takada K, Katayama J, Emura A, and Sasatsu M (2000) Regulation of transcription of the mph(A) gene for macrolide 2’-phosphotransferase I in Escherichia coli: characterization of the regulatory gene mphR(A), J. Bacteriol 182, 5052–5058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Zheng J, Sagar V, Smolinsky A, Bourke C, LaRonde-LeBlanc N, and Cropp TA (2009) Structure and function of the macrolide biosensor protein, MphR(A), with and without erythromycin, J. Mol. Biol 387, 1250–1260. [DOI] [PubMed] [Google Scholar]
- [31].Hall BG, Acar H, Nandipati A, and Barlow M (2014) Growth rates made easy, Mol. Biol. Evol 31, 232–238. [DOI] [PubMed] [Google Scholar]
- [32].R Core Team. (2017) R: A Language and Environment for Statistical Computing, https://www.R-project.org.
- [33].Petzoldt T (2017) Estimate Growth Rates from Experimental Data, R Package Version 0.7.1.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.