

Abstract
Inhibition of the hedgehog (Hh) signaling pathway has been validated as a therapeutic strategy to treat basal cell carcinoma and holds potential for several other forms of human cancer. Itraconazole and posaconazole are clinically useful triazole anti-fungals that are being repurposed as anti-cancer agents based on their ability to inhibit the Hh pathway. We have previously demonstrated that removal of the triazole from itraconazole does not affect its ability to inhibit the Hh pathway while abolishing its primary side effect, potent inhibition of Cyp3A4. To develop structure-activity relationships for the related posaconazole scaffold, we synthesized and evaluated a series of des-triazole analogues designed through both ligand- and structure-based methods. These compounds demonstrated improved anti-Hh properties compared to posaconazole and enhanced stability without inhibiting Cyp3A4. In addition, we utilized a series of molecular dynamics and binding energy studies to probe specific interactions between the compounds and their proposed binding site on Smoothened. These studies strongly suggest that the tetrahydrofuran region of the scaffold projects out of the binding site and that π-π interactions between the compound and Smoothened play a key role in stabilizing the bound analogues.
Graphical Abstract:
1. Introduction
The hedgehog (Hh) signaling pathway plays an essential role in embryonic development by regulating the proliferation and differentiation of cells in a time-dependent manner [1-3]. Hh signaling is significantly less active in adults with the exception of maintaining stem cell homeostasis, predominately in the skin and central nervous system [2]. Aberrant activation of the Hh signaling pathway has been implicated as a driving force in several cancers; most notably, basal cell carcinoma (BCC) and medulloblastoma (MB) [4-5]. Constitutive activation of the Hh pathway in these cancers is primarily caused by mutations in several key members of the Hh signal cascade, including Patched (Ptch), Smoothened (Smo), and Suppressor of fused (Sufu) [6]. Current FDA approved drugs that target the Hh pathway (vismodegib and sonidegib) function through direct antagonism of Smo; however, resistance due to point mutations in the binding pocket of these drugs on Smo has been identified in both BCC and MB patients who used these drugs for initial treatment [7-11]. The need to develop potent inhibitors that overcome the resistance observed with vismodegib and sonidegib has led to the identification of multiple small-molecule scaffolds that target different points along the Hh signaling pathway. [12-16]
Posaconazole (PSZ) and itraconazole (ITZ) are clinically approved azole antifungals that have been identified as Hh pathway inhibitors in vitro and in vivo [17-20]. Previous studies strongly suggest that both of these drugs inhibit the Hh pathway by directly binding Smo [17-18, 21-22]. PSZ and ITZ retain their anti-Hh activity in the presence of mutant forms of Smo that confer resistance to vismodegib and sonidegib, highlighting their potential as improved anti-cancer drugs targeting the Hh pathway. This activity is presumably a result of their ability to bind Smo in a manner distinct from the clinically approved drugs. We have undertaken a series of studies to more fully explore the anti-cancer potential of the PSZ scaffold through targeting the Hh pathway. Herein, we report the synthesis and evaluation of PSZ analogues designed to enhance both their Hh inhibitory activity and their drug-like properties. In addition, we have generated a homology model of Smo that was utilized in an extensive series of computational studies to probe potential intermolecular interactions between PSZ and Smo and to examine the conformational changes in Smo that drive its inactivation following antagonist binding.
2. Chemistry
2.1. PSZ Analogue Design
Multiple strategies were employed to rationally design improved analogues of PSZ for evaluation as anti-cancer agents that target the Hh signaling pathway. Our previous studies with the ITZ scaffold demonstrated that the triazole moiety, which is responsible for the potent inhibition of Cyp3A4 associated with the ITZ scaffold, is not required for inhibition of Hh signaling [17]. With this in mind, we designed an initial proof-of-concept des-triazole PSZ analogue, 1, that removed the triazole while also incorporating a simplified alkyl side chain present in our most potent ITZ analogues (Figure 1, top). Based on our preliminary docking studies for PSZ in complex with Smo (vide infra), we incorporated modified side chains that contained either a primary alcohol or carboxylic acid to take advantage of enhance intermolecular interactions with the PSZ binding site on Smo (Figure 1, middle). Finally, we prepared several simplified PSZ analogues that replace the triazolone/side chain region with an aromatic moiety tethered to the central region of the scaffold through an amide bond (Figure 1, bottom). These compounds were prepared based on recent SAR results for the ITZ scaffold generated in our group [20].
Figure 1.

Structure of PSZ and design strategies for PSZ analogues.
2.2. Compound Synthesis
PSZ analogue 1 was prepared through modification of a previously published synthesis of PSZ (Scheme 1) [23]. Friedel-Craft acylation of 1,3-difluorobenzene (2) with succinic anhydride (3), followed by conversion of the resulting ketone to the alkene through standard Wittig conditions provided carboxylic acid 5. Amidation of 5 with (4R)-(+)−4-benzyl-2-oxazolidinone (OXZ) affords OXZ-protected intermediate 6, which undergoes selective hydroxymethylation at the opposite face in the presence of titanium tetrachloride to form intermediate 7. Iodocyclization of 7 results in selective formation of the 2S,4R conformation around the intact tetrahydrofuran (THF) moiety (8). Reduction of 8 with sodium borohydride removes both the iodine and reduces the OXZ to the primary alcohol, which is subsequently tosylated to provide key des-triazole THF intermediate 10. Intermediate 11 was prepared as described previously [17] and coupled directly to 10 to afford desired analogue 1 with the trans conformation in 5% overall yield.
Scheme 1.

Synthesis of THF Ring and final coupling to make Des-PSZ Analogue 1. Reagents and conditions: (a) AlCl3, DCM, 45 °C, 3-4 h, 65%; (b) NaOtBu, MePPh3Br, THF, 40 °C, 12 h, 100%; (C) i. DCM, TEA, pivaloyl chloride, ii. OXZ, DMAP, DMF, 45-50 °C, 12 h, 68%; (d) i. DCM, TiCl4, DIPEA, 0 °C, 1 h, ii. s-trioxane, TiCl4, DCM, 0 °C, 12 h, 50%; (e) I2, pyridine, CH3CN, R.T., 20 h, 61%; (f) NaBH4, DMSO, 110°C, 12 h, 62%; (g) Pyridine, TsCl, R.T., 12 h, 100%; (h) Cs2CO3, DMSO, 90 °C, 12 h, 63%.
Analogues incorporating either a carboxylic acid or primary alcohol in the side chain region of the PSZ scaffold were prepared by coupling either ethyl 3-bromopropanoate or ethyl bromoacetate to well-characterized intermediate 12 [17] under basic conditions, followed by demethylation with boron tribromide to afford esters 15 and 16, respectively (Scheme 2). Subsequent coupling of the tosylated THF intermediate 10 to both esters resulted in 17 and 18, which had been hydrolyzed to the carboxylic acid, presumably due to the basic conditions and elevated temperature required for the coupling. Alcohol 19 was prepared through reduction of 17 with lithium aluminum hydride. Surprisingly, attempts to reduce 18 to the corresponding alcohol were unsuccessful, resulting in multiple products that were difficult to characterize. Because 18 was significantly less active than 17 against the Hh signaling pathway (Table 2 below), we did not optimize the conditions necessary to reduce 18 to the corresponding alcohol. A similar synthetic route was also utilized to synthesize an ITZ analogue incorporating the propanoic acid side chain (21) to explore whether incorporation of this moiety affected the activity of the des-triazole ITZ scaffold (Scheme 3).
Scheme 2.

Synthesis of Hydroxylated Des-triazole PSZ Analogues. Reagents and conditions: (a) ethyl 3-bromopropanoate or ethyl bromoacetate, Cs2CO3, DMF, 60 °C – 100 °C, 12 h, quant; (b) BBr3 (1M), DCM, 0 °C - R.T., 4 h, 29-35%; (c) Cs2CO3, DMSO, 90 °C, 12 h, 36%; (d) LAH, THF, 0 °C to R.T., 63%.
Table 2.
Hh Inhibition of PSZ Analogues in ASZ cells.
| Cmpd | IC50 (μM)a, b |
|---|---|
| PSZ | 0.50 ± 0.1 |
| 1 | 0.19 ± 0.03 |
| 17 | 0.032 ± 0.01 |
| 18 | 0.15 ± 0.02 |
| 19 | 0.020 ± 0.01 |
| 21 | 0.012 ± 0.005 |
| 23 | 0.74 ± 0.15 |
| 24 | 0.33 ± 0.12 |
| 25 | >10 |
| 26 | 0.22 ± 0.08 |
| 27 | 0.19 ± 0.10 |
| 28 | 0.024 ± 0.009 |
| 29 | 0.19 ± 0.03 |
| 30 | >10 |
IC50 values represent the Mean ± SEM of at least two separate experiments performed in triplicate.
All analogues evaluated following 48 h incubation.
Scheme 3.

Synthesis of Hydroxylated Side Chain Des-triazole PSZ Analogues. Reagents and conditions: (a) Cs2CO3, DMSO, 90 °C, 12 h, 54%.
A series of ‘right side’ analogues based on previous ITZ SAR conducted in our lab were also synthesized (Scheme 4). They were prepared by coupling THF region 10 and the linker region precursor 22, which provides the nitro-containing intermediate 23. The nitro intermediate was reduced in the presence of 10% palladium on carbon and hydrazine monohydrate to produce aniline 24. The carbamate 25 was generated through the addition of phenyl chloroformate to aniline 24 and subsequent treatment with hydrazide monohydrate provided carboxamide intermediate 26. The final unsubstituted triazolone 27 was generated by heating carboxamide 26 with formamidine acetate. Finally, three analogues that replaced the triazolone side chain with an aromatic amide were prepared by directly coupling of aniline 24 to the requisite aromatic carboxylic acid (28-30, Scheme 5).
Scheme 4.

Synthesis of ‘Right-Side Analogues’– Linker Precursors. Reagents and conditions: (a) Cs2CO3, DMSO, 90 °C, 12 h, 67%; (b) 10% Pd/C, EtOH, NH2NH2, reflux, 12 h, 90%; (c) Pyr, ClCOOPh, 3 h, 40%; (d) NH2NH2-H2O, reflux, 3h, 30%; (e) formamidine acetate, acetic acid, reflux, 3 h, 46%.
Scheme 5.

Synthesis of ‘Right-Side Analogues’– Amide Linkage. Reagents and conditions: (a) EDCI, DMAP, DCM, RT, 12 h, 27 - 63% or HATU, NMM, DMF, RT, 12h, 80%.
3. Initial Biological and Computational Studies for PSZ and Analogue 1.
3.1. Hh inhibitory activity for PSZ and 1.
To determine whether removal of the triazole moiety had any effect on the ability of the PSZ scaffold to inhibit Hh signaling, we evaluated whether 1 could down-regulate mRNA expression of the Hh-dependent target gene Gli1 in ASZ cells, a well-characterized Hh-dependent murine BCC cell line [24]. Interestingly, des-triazole analogue 1 was approximately 2.5-fold more potent than PSZ (IC50 values 0.19 and 0.50 μM, respectively; Table 1). A similar trend was observed with regards to the ability of 1 to inhibit the growth of a primary Hh-dependent MB derived from a conditional Patched knockout (Math1-Cre-ER:Ptchfl/fl, termed Ptch-CKO) mouse [25].
Table 1.
In Vitro and Pharmacokinetic Activity of PSZ and initial Des-triazole PSZ Analogue.
| Cmpd | IC50 (μM)a | Ptch-CKO GI50 (μM)b |
CYP3A4 IC50 (μM)c |
P-gp Substratec |
T1/2 (min)c |
Clint (μL/min/mg)c |
|---|---|---|---|---|---|---|
| PSZ | 0.50 ± 0.1 | 1.5 ± 0.3 | 0.15 | Yes | 108 | 21.5 |
| 1 | 0.19 ± 0.03 | 0.61 ± 0.12 | >20 | No | 61.8 | 37.4 |
IC50 values from ASZ cells represent the Mean ± SEM of at least two separate experiments performed in triplicate following a 48 hr incubation.
Values represent the Mean ± SEM of at least two separate experiments performed in triplicate.
PK data generated by Pharmaron or Cyprotex as a CRO service via their standard protocols.
3.2. Computational Modeling of the Smoothened Receptor.
Several distinct lines of evidence strongly suggest that both PSZ and ITZ bind to the 7-TM domain of Smo in the same region where other Smo agonists and antagonists bind [17-18, 21-22]. Both analogues were active in Ptch−/− knockout mouse embryonic fibroblasts (MEFs), but neither compound was inhibited Hh signaling in the presence of a constitutively active Smo mutant, indicating that inhibition of pathway signaling as at the level of Smo [17,21]. In addition, ITZ significantly displaced a tritiated Smo antagonist from HEK293 cells that stably express human Smo [22]. Both azoles retain in vitro and in vivo activity in the presence of several mutant forms of Smo resistant to other Smo antagonists, suggesting that the PSZ/ITZ scaffold may adopt a distinct binding conformation when in complex with Smo [17, 21]. With this in mind, we had three primary goals for our computational studies. First, we sought to predict the conformation these compounds adopt when binding to Smo to determine how they maintain potent anti-Hh activity in the presence of mutant forms of Smo. Second, we sought to correlate inhibition of the Hh pathway with binding energy calculations. Finally, we explored the conformational changes that occur in Smo following compound binding to validate the recent models predicting whether a Smo ligand is an agonist or antagonist [26, 27]. Taken together, this information could aid in the future design of PSZ/ITZ analogues as improved Hh pathway inhibitors. Our first step in this process was to generate a homology model of SMO based on previously published Smo structures in complex with known Smo antagonists (PDB IDs: 4JKV [26], 5V56 [28], and 5L7I [5]). We chose to generate a new model rather than use the existing structures because the complexes described above either removed or mutated multiple residues to allow for improved crystallization. We replaced these altered residues with the natural amino acids and refined the overall structure through homology modeling (Modeller 9.16).
The three-dimensional structure of the SMO receptor generated through our homology modeling process is analogous to the experimentally-derived structures previously disclosed. Smo contains an extracellular domain (ECD), which is composed of the well-characterized cysteine rich domain (CRD) and a linker domain (ELD) (Figure 2A). Other key regions of the Smo receptor include the seven transmembrane domain (7TM), extracellular loop 2 (ECL2) that connects TM4 and TM5, and the KTXXXW motif (K539-T553) located in helix VIII of the intracellular domain. ECL2 contains a β-hairpin that encompasses the primary binding pocket for SMO ligands. Orientation of the intracellular KTXXXW motif is important for both stabilizing the entire Smo receptor in an inactive state and inducing a conformational change that results in Smo activation [28]. Stability of the KTXXXW motif in the inactive state is primarily governed by molecular interactions of T541, I544 and W545 in helix VIII with T251 and A254 in helix I. Most notably, the side chain hydroxyl of T541 forms a hydrogen bond with the backbone carbonyl of V536 in the intracellular end of helix VII and the indole hydrogen of W545 forms a hydrogen bond with the hydroxyl in the side chain of T251 in helix I (Supplemental Figure 1).
Figure 2.

Binding mode of PSZ and 1 in complex with Smo. (A) The binding orientations of PSZ (yellow) and 1 (blue) inside the binding pocket on Smo. The opening of the binding site is sandwiched between the ELD and ECL2. (B) The furan region of PSZ and 1 orient out of the binding pocket towards the ELD. (C) Compared to PSZ, 1 penetrates more deeply inside the binding pocket.
3.3. Binding modes for PSZ, 1, and 19
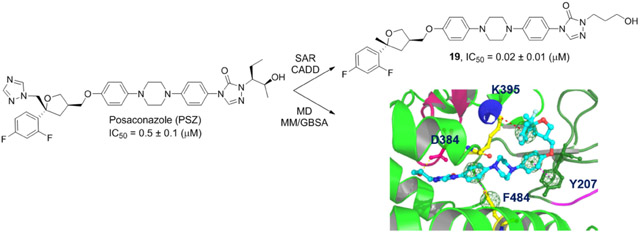
In order to explore the binding interaction of PSZ and 1 with Smo, a grid box was generated around the well-characterized Smo binding site. Both PSZ and 1 were docked into this grid on Smo utilizing the XP mode in Schrödinger Glide. The putative ligand binding site for these compounds is located at the extracellular surface of the β-hairpin in ECL2 and the α-helical extension of TM6 of the 7-TM domain. As shown in Figure 2, both PSZ and 1 adopt similar orientations and binding interactions inside the binding pocket. For both compounds, the triazole region is oriented towards the extracellular surface with the linker/side chain region penetrating deeply into the Smo binding pocket. The primary difference in the binding modes of the two compounds is in regards to the triazole ring of PSZ, which orients towards the solvent accessible surface of the binding pocket resulting in unfavorable polar binding interactions (Figure 2B and 2C). By contrast, removal of the triazole in analogue 1 results in reduced interactions with the water molecules present at the pocket surface, which may be a primary reason why analogue 1 demonstrates enhanced anti-Hh activity. Additional intermolecular interactions with Smo common to both compounds include a network of π–π and cation–π interactions between the phenyl ring adjacent to the ether and Y207, K395, R485, and/or F484 (Figure 3). The triazolone/side chain region of the scaffold forms two hydrogen bonds with polar residues (D384 and N219) while also exhibiting favorable non-polar interactions between the aliphatic side chain and several hydrophobic residues (Y394, E518 and P513).
Figure 3.

Two-dimensional Smo:ligand contact maps for PSZ (A), 1 (B), and 19 (C) in complex with our Smo structural model. Color code is as follows: red residue, negatively charged residue; purple, positively charged residue; green, hydrophobic residue; light blue, polar residue; gray, solvent exposed area; pink arrow, hydrogen bond; green line, pi-pi interactions.
Based on these predicted binding modes for PSZ and 1, we sought to design an improved analogue that focused on exploring whether an analogue of 1 that replaced the sec-butyl with a simplified hydrophilic side chain would maintain potent anti-Hh activity. In addition, the incorporation of the terminal alcohol could potentially improve the solubility of the scaffold. Prior to the synthesis of this analogue (19), the compound was docked into our Smo model. Overall, the orientation and binding mode of 19 was comparable to both PSZ and 1 (Figure 3). The difluorophenyl ring and tetrahydrofuran are oriented at the solvent accessible surface, while the triazolone/side chain penetrated deeply into the pocket. The primary alcohol on the side chain formed a hydrogen bond with D384, mimicking the interaction between this amino acid residue and the secondary alcohol in the side chain of PSZ (Figure 3). Finally, neither PSZ, 1, nor 19 demonstrated specific intermolecular interactions with the primary residue mutations that have been linked to vismodegib (D477G and E522K) and sonidegib resistance (N223D, L225R, and G457S), providing evidence as to why these compounds maintain activity in the presence of mutated forms of Smo.
4. Biological Activity of Second Generation PSZ Analogues.
4.1. Anti-Hh activity of PSZ analogues.
Preliminary docking studies with our Smo model encouraged the synthesis of 19 as well as two additional hydroxylated analogues, 17 and 18. When evaluated in ASZ cells for Hh inhibition, 19, which incorporates a propanol side chain, was most active amongst the hydroxylated analogues (IC50 = 0.020 μM). Overall, 17-19 had significantly increased anti-Hh activity when compared to PSZ and our initial proof-of-concept analogue 1, highlighting the potential of introducing a hydrophilic side chain in addition to the removal of the triazole ring when improving this scaffold. Curious about the impact a hydroxylated side chain would have on the Hh activity of the ITZ scaffold, the non-stereochemically defined propanoic acid side chain analogue, 21, was synthesized. Interestingly, 21 was more active than 19 by approximately 2-fold.
Based on our recent SAR for the ITZ scaffold [20], PSZ analogues 23-30 focused on incorporating modifications to the triazolone/side chain region or ‘right-side’ of the scaffold. Analogues 23-27 integrated the des-triazole THF region with the linker intermediates leading to the un-substituted triazolone ring while an amide linkage in analogues 28-30 introduced phenyl rings with hydrogen bonding potential. Evaluation in ASZ cells revealed the truncated nitro (23) and aniline (24) PSZ analogues had weakened Hh inhibition compared to 1 indicating an extended scaffold is required for optimal activity The carbamate (25), carboxamide (26), and triazolone (27) PSZ analogues had similar Hh activity as their trans-ITZ counterparts previously published from our group [20]. When investigating the amide linkage analogues, we found that the addition of a meta-acetyl phenyl ring (28), increased anti-Hh activity (IC50 = 0.024 μM) that was comparable to the hydroxylated analogues (17-19). A meta-hydroxylated phenyl ring (29) maintained Hh activity similar to 1 while the addition of a meta-pyridyl group (30) abolished Hh inhibition in ASZ cells.
4.2. Preliminary pharmacokinetic profiles of PSZ and analogues.
Based on the promising and improved potency of the PSZ analogues, we evaluated several pharmacokinetic parameters of representative compounds (Tables 1 and 3). All of the compounds evaluated were minimally soluble in PBS at pH 7.4. As expected, removal of the triazole moiety completely abolished the ability of PSZ analogues (1, 21, and 28) to inhibit Cyp3A4. Interestingly, while PSZ was identified as a substrate for P-glycoprotein (Pgp), 1 was not a substrate, suggesting the triazole also plays a role in efflux pump recognition for the PSZ scaffold (Table 1). In addition, none of the compounds inhibited the hERG channel at concentrations up to 25 μM. Overall, PSZ was more stable than ITZ; however, removal of the triazole from PSZ resulted in a significant decrease in metabolic stability for analogue 1 (T1/2 = 108 and 61.8 min, respectively). By contrast, analogues 28-30 had improved metabolic stability compared to both PSZ and 1 as evidenced by T1/2 values ranging from 115 to >180 min and internal clearance rates as low at 2.23 μl/min/mg.
Table 3.
Preliminary Pharmacokinetic Activity of PSZ and Analogues.
| Cmpd | Solubility (μM)a |
cCYP3A4 IC50 (μM) |
hERG Inhibition (%)b |
T1/2 (min)c |
Clint (μL/min/mg)c |
|---|---|---|---|---|---|
| PSZ | 0.781 | 0.15 | 14.7 | 108 ± 5.3 | 21.5 |
| ITZ | 0.8 | 0.04 | < 1 | 27.0 ± 6.2 | --- |
| 1 | --- | >20 | --- | 61.8 ± 2.1 | 37.4 |
| 19 | 1.56 | --- | --- | --- | --- |
| 21 | 0.781 | >20 | 3.6 | --- | --- |
| 28 | 3.13 | >20 | 4.2 | 115 ± 9.8 | 20.0 |
| 29 | 3.13 | --- | --- | > 180 | 2.23 |
| 30 | 1.56 | --- | --- | > 180 | 7.15 |
Kinetic solubility determined in PBS at pH 7.4.
Percent inhibition of the hERG channel at 25 μM.
T1/2 and Clint were measured in human liver microsomes at 1 μM.
---indicates values were not determined.
4.3. Molecular dynamics studies for PSZ, 1, and 19.
4.3.1. Structural conformation studies.
The molecular docking described above represents a single snapshot of the binding complex between Smo and our PSZ-based ligands. In order to more closely probe the binding interactions and conformational changes generated when PSZ or its analogues bind with Smo, we performed additional molecular dynamics (MD, 50 ns) studies on the Smo:PSZ, Smo:1, and Smo:19 docked structures. First, we utilized the root mean square deviations (RMSDs) of the heavy atoms in the Smo:ligand complexes to evaluate the dynamic structural changes that occurred during the MD simulations (Figure 4). The overall Smo:ligand complex was more stable for analogues 1 and 19 compared to PSZ, which suggests a tighter binding affinity (Table 3). A similar trend was observed for the RMSD values of the ligand, further highlighting that the two analogues are more stable in complex with Smo. The RMSD values of the 7-TM domain and the KTXXXW motif suggest that these regions of Smo were in a stable conformation throughout the simulation. By contrast, the CRD is more flexible, most likely due to fluctuation of the ELD. The KTXXXW motif, which drives downstream activation of the pathway following a conformational change [27], demonstrated stable RMSD values for each complex providing strong evidence that Smo adopts its inactive conformation upon binding to the three small molecules (Table 3). To further probe the activation/inactivation state of Smo in complex with our analogues, we evaluated the conformational stability of helix VI/TM6 by RMSD analysis of the backbone atoms in this region. The α-helical extension of TM6 remained stable throughout the 50 ns simulation time for two primary reasons. First, a cation-π interaction between R451 of TM6 and W535 in TM7 was maintained throughout the simulation. This specific interaction is known to play a critical role in stabilizing Smo in an inactive conformation [27]. Second, a network of hydrogen bonds between several water molecules and amino residues 475-479 stabilize the non-proline kink of the α-helical extension and TM6.
Figure 4.

RMSD for heavy atoms of the Smo:PSZ (A), Smo:1 (B), and SMO:19 complexes. 7-TM (black), ligand (blue), CRD (red), KTXXXW motif (green), TM6 (magenta), and R451-W535 (gray) with respect to the equilibrated structure are plotted as a function of MD simulation time.
4.3.2. Binding energy studies.
The RMSD values for all the heavy atoms of the ligands and the 7-TM domain of the Smo:PSZ and Smo:1 complex converged and equilibrated after 2 ns, which suggests that these binding conformations are appropriate to utilize for analysis of the binding energies of the complexes. By contrast, convergence and equilibration of the Smo:19 complex was achieved after 10 ns; therefore, the stable trajectories of the last 45 ns of the simulation were employed to calculate the average binding energies for the three complexes using the MM/GBSA method. The predicted binding energies of PSZ, 1, and 19 with Smo are in general agreement with experimental inhibition of Hh inhibition described above, with the ΔGbinding for the Smo:1 and Smo:19 complexes (−39.41 and −36.07 kcal/mol, respectively) significantly lower than the Smo:PSZ complex (−27.90 kcal/mol) (Table 4). To gain a clearer understanding of which individual energy terms have the greatest impact on the calculated binding affinities, four individual energy components were calculated and compared. Overall, the van der Waals (ΔEvdw) interactions served as the major contributor to the binding energies. The polar contributions (ΔGpolar) negatively affected compound binding, primarily through polar solvation effects that antagonized binding affinity.
Table 4.
MM/PBSA binding energies (kcal/mol) for Smo:ligand complexes.
| PSZ | 1 | 19 | |
|---|---|---|---|
| ΔGbinding | −27.90 ± 4.9a | −39.41 ± 4.3 | −36.07 ± 6.5 |
| ΔGvdw | −57.96 ± 5.2 | −67.79 ± 3.3 | −68.13 ± 4.2 |
| ΔGelect | −11.07 ± 3.2 | −8.08 ± 2.8 | −18.64 ± 3.2 |
| ΔGpolar | 47.67 ± 7.1 | 43.95 ± 7.5 | 58.11 ± 8.7 |
| ΔGnonpolar | −6.54 ± 0.4 | −7.48 ± 0.2 | −7.41 ± 0.3 |
All values are kcal/mol.
To evaluate the structural and energetic convergence of the complexes, averages of the MM/PBSA ΔGbinding values were plotted as a function of simulation time (Figure 5). These plots allowed us to determine the most stable structure across the entire simulation. For Smo:PSZ, the most energetically stable conformation occurred at 21 ns. In this complex, the difluorophenyl ring and the triazole moiety of PSZ are located between the ELD and ECL2 of Smo and have minimal interactions with the residues present at the surface groove adjacent to the binding site (Figure 6A). The central piperazine adopts a pseudo chair conformation within the binding site that is stabilized by two strong π –π interactions between the phenyl ring adjacent to the furan and Y207 and the phenyl ring adjacent to the triazolone ring with F484. In addition, the piperazine demonstrates strong hydrophobic interactions with multiple residues in the binding site (M301, L303, Y394, K395, E481, and R485). The triazolone adopts a unique orientation such that the carbonyl forms a hydrogen bond (52% occupancy, interatomic distance 2.37Å) with the backbone of S385. The hydroxyl group on the propyl side chain forms a hydrogen bond with the side chain of D384 (40% occupancy rate, interatomic distance 2.63Å) while the aliphatic regions of the side chain are stabilized by multiple van der Waals contacts.
Figure 5.

Binding energies for Smo:ligand complexes over time. The lowest energy conformation was utilized to explore intermolecular interactions between Smo and PSZ (blue), 1 (red), and 19 (green).
Figure 6.

Energetically stabilized structures of Smo in complex with PSZ (A), 1 (B), and 19 (C). Intermolecular interactions between Smo and the ligand are represented as dotted lines. Yellow, hydrogen bond; Dark blue, π-π interaction; Red, π-cation interaction.
In a similar manner, the binding conformation of Smo:1 at 35 ns was used to explore the key interactions between the ligand and receptor (Figure 6B). When the triazole moiety is removed, the conformation of the THF region of the scaffold reorients such that the difluorophenyl ring of 1 interacts with the same region previously occupied by the triazole of PSZ. The van der Waals contacts between the difluorophenyl ring and multiple residues at the surface of the binding site (L303, R296, K204, S205 and I215) are stronger than those for the triazole in PSZ, which provides insight into the improved activity of the des-triazole analogues. The biaryl piperazine in 1 also adopts a chair conformation that is stabilized by π–π and π–cation interactions between the phenyl ring adjacent to the furan and F484 and K395, respectively. Interestingly, in this structure, N1 of the triazolone moiety is protonated, resulting in the formation of a salt bridge with the carboxylate side chain of D384 (49% occupancy, interatomic distance 1.81Å). Additional stabilization of the triazolone ring is provided by a π–π interaction with Y394 and a hydrogen bond with the side chain of N219. Finally, the highly flexible aliphatic side chain forms multiple hydrophobic contacts with several amino acids at the bottom of the binding pocket.
The most energetically stable conformation of Smo:19 (20 ns) demonstrated that analogue 19 penetrates deeper into the binding site than 1, allowing for a π–cation interaction between the difluorophenyl ring and K395 that is not present for the other two compounds (Figure 6C). The slightly deeper positioning of 19 also allows for the phenoxy ether to form a hydrogen bond with the backbone of E208 (36% occupancy, interatomic distance 1.81Å). The central diaryl piperazine of 19 is stabilized in a chair orientation by the same π–π interactions between the two flanking phenyl rings and Y207 and F484 seen in the Smo:PSZ structure. Finally, the primary hydroxyl of the propanol side chain forms a hydrogen bond with D384 (58% occupancy with interatomic distance 1.85Å).
5. CONCLUSION
We have broadened our understanding of the optimal structural motifs required for potent inhibition of the Hh signaling pathway for small molecule analogues based on the general PSZ and ITZ scaffolds. Our studies continue to demonstrate that the triazole moiety present in the parent scaffold is not required for potent anti-Hh activity and that its removal abrogates the primary off-target side effect associated with PSZ/ITZ administration. Interestingly, removal of the triazole in the PSZ scaffold results in a significant increase in Hh pathway inhibition and additional modifications to the ‘right-side’ of the scaffold have resulted in analogues with low nanomolar IC50 values in Hh-dependent cell lines. To more fully understand our experimental SAR, we performed extensive computational studies, the results of which provide important context for further analogue preparation. The THF region extends out of the binding pocket and removal of the triazole decreases unfavorable polar interactions with water molecules at the binding surface, which supports the improved activity of the des-triazole analogues. Finally, the incorporation of a hydrophilic moiety in the triazolone/side chain region of the scaffold can increase activity through improved hydrogen bond interactions with key binding site residues.
6. Experimental section
6.1. General Information.
Starting materials and solvents were purchased from Sigma-Aldrich or Fisher Scientific. All reactions were performed under an argon atmosphere. Intermediates 11 and 20 were prepared as previously described [17]. NMR data was collected on a Bruker AVANCE 500 MHz spectrometer and analysis performed using MestReNova. HRMS data was obtained at the Mass Spectrometry Facility at the University of Connecticut. FT-IR analysis was performed on a Bruker Alpha Platinum ATR instrument using OPUS software (v 7.2). All PSZ analogues evaluated in the biological assays were greater than 95% purity. Purity was determined by injecting 25 μL of a 1 mg/mL solution each PSZ analogue dissolved in HPLC-grade MeCN into an Agilent Manual FL-Injection Valve (600 bar) on an Agilent 1100/1200 Series HPLC equipped with an Agilent Eclipse Plus C18 (4.6 × 100 mm) column and Agilent 1100 Series Photodiode Array Detector. The mobile phase consisted of 70% MeCN/30% H2O. All analogues were run at a flow rate of 1.0 mL/min for 12 min and purity was assessed at 254 nm.
6.2. Chemical synthesis.
6.2.1. Synthesis of des-triazole intermediates.
6.2.1.1. ((3S,5R)-5-(2,4-difluorophenyl)-5-methyltetrahydrofuran-3-yl)methanol (9).
To a solution of 8 (150 mg, 0.3 mmol) in anhydrous DMSO, NaBH4 (43 mg, 1.1 mmol, 4 equiv.) was added. The reaction was stirred at 100 °C overnight. The reaction is cooled to 0 °C before being quenched by the dropwise addition of NH4Cl (aqueous, saturated). Once bubbling ceased, the quenched reaction is extracted using EtOAc (4x). The organic layers were combined, dried over Na2SO4, filtered and concentrated. The crude was purified by column chromatography (SiO2, 10-30% EtOAc in Hexanes). The fractions containing product were collected and dried by rotary evaportation to obtain a clear, colorless oil (Yield = 40 mg, 62%, 40 mg). 1HNMR (500 MHz, CDCl3) δ 7.57 (ddd, J = 34.4, 15.6, 8.8 Hz, 1H), 6.91 – 6.78 (m, 2H), 4.23 – 4.18 (m, 1H), 4.04 (t, J = 8.1 Hz, 1H), 3.91 – 3.86 (m, 1H), 3.75 – 3.66 (m, 2H), 2.72 (dt, J = 14.6, 7.4 Hz, 1H), 2.58 – 2.51 (m, 1H), 2.48 – 2.35 (m, 1H), 1.87 (dd, J = 12.7, 8.4 Hz, 1H), 1.62 (s, 2H), 1.55 (s, 1H). 13CNMR (126 MHz, CDCl3) δ 163.04, 162.94, 161.07, 159.96, 158.08, 157.99, 127.68, 127.63, 127.60, 127.56, 110.77, 110.58, 104.55, 104.34, 104.13, 82.80, 82.77, 77.30, 77.04, 76.79, 69.89, 64.76, 42.31, 42.03, 41.86, 41.83, 41.59, 28.29. DART-HRMS: m/z [M+H]+ calcd. for [C12H13F2O2]+, 227.0883; found 227.0899.
6.2.1.2. ((3S, 5R)-5-(2,4-difluorophenyl)-5-methyltetrahydrofuran-3-yl)methyl 4-methylbenzenesulfonate (10).
Alcohol 9 (33 mg, 0.145 mmol, 1 equiv.), trimethylamine (0.29 mmol, 0.032 mL, 2 equiv.) and 4-DMAP (18 mg, 0.145 mmol, 1 equiv.) were dissolved in DCM at room temperature. Next, TsCl was added slowly. The reaction was stirred for 2 hours at room temperature. Upon completion, the reaction was diluted with EtOAc and washed with H2O and brine. The organic layer was collected, dried over Na2SO4, filtered and concentrated. The crude was purified by column chromatography (SiO2, 10-25% EtOAc in Hexanes). The fractions containing product were collected and dried by rotary evaportation to obtain a clear, colorless oil (Yield = 55 mg, ~100%). 1HNMR (500 MHz, CDCl3) δ 7.82 (d, J = 8.1 Hz, 1H), 7.74 (d, J = 8.1 Hz, 1H), 7.49 – 7.45 (m, 1H), 7.37 (d, J = 8.1 Hz, 1H), 7.36 (d, J = 8.1 Hz, 1H), 6.88 – 6.75 (m, 2H), 4.15 – 3.60 (m, 5H), 2.50 (m, 4H), 2.50 (m, 1H, hidden by CH3) 2.41 – 2.37 (m, 1H), 1.91 – 1.77 (m, 1H), 1.55 (s, 2H), 1.50 (s, 1H). 13CNMR (126 MHz, CDCl3) δ 163.07, 161.11, 159.91, 158.04, 145.02, 132.88, 129.96, 129.89, 129.60, 127.92, 127.87, 127.53, 127.45, 127.41, 127.07, 110.89, 110.72, 110.01, 109.98, 104.63, 104.42, 104.21, 82.74, 77.31, 77.05, 76.80, 71.29, 70.98, 69.75, 69.18, 42.02, 41.49, 41.46, 39.19, 39.00, 29.72, 28.26, 21.66. DART-HRMS: m/z [M+H]+ cald. for [C19H21F2O4S]+, 383.1050; found 383.1117. Also found [M–CH3]+ = 367.0803 and [M+NH4]+ = 400.1388.
6.2.2. Synthesis of proof-of-concept analogue 1.
6.2.2.1. General synthetic procedure for coupling the des-triazole THF region to linker intermediate.
CS2CO3 (10 equiv.) and the requisite linker (1 equiv.) were diluted in anhydrous DMSO and stirred for 30 minutes at room temperature before addition of the des-triazole THF region in DMSO. The mixture was heated to 90 °C and stirred for 12 h. The reaction was quenched with water and extracted using EtOAc (2x) and DCM (2x). For 17 and 18, the water layer was acidified to pH = 2 to ensure complete extraction of the carboxylic acid into the organic phase. The organic layers were combined, dried over Na2SO4, filtered and concentrated. The crude analogue was purified by column chromatography (SiO2, 10-30% Acetone in Hexanes).
6.2.2.2. 2-((R)-sec-butyl)-4-(4-(4-(4-(((3R,5R)-5-(2,4-difluorophenyl)-5-methyltetrahydrofuran-3-yl)methoxy)phenyl)-piperazin-1-yl)phenyl)-2,4-dihydro-3H-1,2,4-triazol-3-one (1).
Analogue 1 was prepared by coupling THF region intermediate 10 and linker/side chain intermediate 11 as described above. Dark grey solid. (Yield = 63%). 1HNMR (500 MHz, CDCl3) δ 7.66 (s, 1H), 7.59 – 7.54 (dd, J = 16.2, 8.2 Hz, 1H), 7.47 (d, J = 8.5 Hz, 2H), 7.07 (d, J = 8.5 Hz, 2H), 6.98 (m, 1H), 6.89 (d, J = 7.6 Hz, 3H), 6.85 – 6.80 (m, 1H), 4.34 (m, 1H), 4.14 – 4.11 (t, J = 7.7 Hz, 1H), 3.97 – 3.96 (m, 3H), 3.85 – 3.74 (m, 1H), 3.41 (m, 4H), 3.28 (m, 4H), 2.64 – 2.62 (m, 2H), 2.10 – 1.87 (m, 2H), 1.79 – 1.75 (m, 1H), 1.65 (s, 3H), 1.43 (d, J = 6.7 Hz, 3H), 0.95 (t, J = 7.3 Hz, 3H). 13CNMR (126 MHz, CDCl3) δ 152.08, 150.59, 145.72, 127.74, 127.66, 125.98, 123.59, 118.57, 116.70, 115.32, 104.50, 104.36, 104.15, 82.80, 70.16, 52.72, 50.75, 49.27, 42.07, 39.54, 29.72, 28.47, 28.39, 19.27, 10.81. DART-HRMS: m/z [M+H]+ cald. for [C34H40F2N5O3]+, 604.3021; found 604.3085. IR (solid) vmax: 2932, 2842, 1702, 1513, 1230, 1041, 966, 823. Purity = 95.1%.
6.2.3. Synthesis of des-triazole analogues containing a modified side chain.
6.2.3.1. ethyl 3-(4-(4-(4-(4-methoxyphenyl)piperazin-1-yl)phenyl)-5-oxo-4,5-dihydro-1H-1,2,4-triazol-1-yl)propanoate (13).
Ethyl-3-bromoproponate was coupled with linker region intermediate 10 via the general procedure described in 5.2.2.1. above to afford 13. Light tan solid. (Yield = quantitative) 1HNMR (500 MHz, CDCl3) δ 7.64 (s, 1H), 7.42 (d, J = 8.9 Hz, 2H), 7.05 (d, J = 9.0 Hz, 2H), 6.99 (d, J = 9.0 Hz, 3H), 6.90 (d, J = 9.0 Hz, 3H), 4.21 – 4.19 (m, 4H), 3.81 (s, 3H), 3.40 – 3.38 (m, 4H), 3.26 – 3.24 (m, 4H), 2.87 – 2.84 (t, J = 7.1 Hz, 2H), 1.31 – 1.28 (t, J = 7.1 Hz, 3H). 13CNMR (126 MHz, CDCl3) δ 170.83, 154.22, 151.99, 150.72, 145.49, 134.42, 125.60, 123.63, 121.26, 118.62, 116.78, 116.60, 114.56, 110.01, 109.98, 77.40, 77.15, 76.89, 60.81, 55.59, 50.78, 49.87, 49.18, 41.36, 33.36, 29.97, 14.20. DART-HRMS: m/z [M+H]+ cald. for [C24H30N5O4]+, 452.2220; found 452.2326.
6.2.3.2. ethyl 2-(4-(4-(4-(4-methoxyphenyl)piperazin-1-yl)phenyl)-5-oxo-4,5-dihydro-1H-1,2,4-triazol-1-yl)acetate (14).
3-bromoacetate was coupled with linker region intermediate 10 via the general procedure described in 5.2.2.1. above to afford 14. Light tan to white solid. (Yield = quantitative). 1HNMR (500 MHz, CDCl3) δ 7.71 (s, 1H), 7.46 (d, J = 9.0 Hz, 2H), 7.07 (d, J = 9.0 Hz, 2H), 7.00 (d, J = 9.0 Hz, 2H), 6.92 (d, J = 9.0 Hz, 2H), 4.67 (s, 2H), 4.47 – 4.28 (dd, J = 15.9, 8.8 Hz, 3H), 3.83 (s, 3H), 3.43 – 3.41 (m, 4H), 3.29 – 3.27 (m, 4H), 1.36 – 1.33 (t, J = 7.1 Hz, 3H). 13CNMR (126 MHz, CDCl3) δ 167.58, 167.05, 154.57, 154.27, 152.54, 150.78, 135.11, 125.94, 125.50, 123.67, 118.66, 116.61, 114.56, 110.01, 109.98, 77.34, 77.09, 76.83, 63.88, 61.83, 61.69, 55.60, 50.83, 49.16, 46.81, 14.16, 14.10. DART-HRMS: m/z [M+H]+ cald. for [C23H28N5O4]+, 437.2063; found 438.2149.
6.2.3.3. ethyl 3-(4-(4-(4-(4-hydroxyphenyl)piperazin-1-yl)phenyl)-5-oxo-4,5-dihydro-1H-1,2,4-triazol-1-yl)propanoate (15).
To a solution of 13 (1 equiv.) in anhydrous DCM as 0 °C was added 1M BBr3 in DCM (4-5 equiv.) dropwise. The mixture was stirred for 30 min at 0 °C before warming to RT and stirred for an additional 3 h. The reaction was quenched by pouring the mixture over ice, followed by washing with DCM (2x) and EtOAc (2x). The organic layers were combined, dried over Na2SO4, filtered and concentrated. Column chromatography (SiO2, 1-5% MeOH in DCM) afforded 15 as a light tan solid (Yield = 28%). 1HNMR (500 MHz, CDCl3) δ 7.64 (s, 1H), 7.40 (d, J = 8.9 Hz, 2H), 7.02 (d, J = 9.0 Hz, 2H), 6.88 (d, J = 8.8 Hz, 2H), 6.75 (d, J = 8.8 Hz, 2H), 4.25 – 4.22 (m, 4H), 3.40 – 3.38 (m, 4H), 3.25 – 3.13 (m, 4H), 2.89 – 2.86 (t, J = 7.1 Hz, 2H), 1.32 – 1.30 (t, J = 7.1 Hz, 3H). 13CNMR (126 MHz, CDCl3) δ 170.87, 152.24, 150.90, 150.69, 145.04, 134.66, 125.24, 124.16, 118.84, 116.53, 116.00, 110.01, 109.98, 77.31, 77.05, 76.80, 60.90, 51.01, 49.10, 41.46, 33.35, 14.18. DART-HRMS: m/z [M+H]+ cald. for [C23H28N5O4]+, 438.2063; found 438.2150.
6.2.3.4. ethyl 2-(4-(4-(4-(4-hydroxyphenyl)piperazin-1-yl)phenyl)-5-oxo-4,5-dihydro-1H-1,2,4-triazol-1-yl)acetate (16).
Alcohol 16 was prepared using precursor 14 and BBr3 as described above. Tan solid. (Yield = 23%). 1HNMR (500 MHz, CDCl3) δ 8.41 (s, 1H), 7.71 (s, 1H), 7.42 (d, J = 8.3 Hz, 1H), 7.20 (d, J = 8.5 Hz, 1H), 7.04 – 6.99(m, 2H), 6.93 – 6.90 (t, J = 9.1 Hz, 2H), 6.82 – 6.77 (m, 2H), 4.69 (d, J = 8.7 Hz, 1H), 4.47 (s, 1H), 4.30 – 4.22 (m, 2H), 3.38 −3.24 (m, 8H), 1.35 – 1.31 (m, 3H). 13CNMR (126 MHz, CDCl3) δ 168.51, 162.68, 150.85, 150.56, 150.10, 150.07, 145.56, 135.14, 132.85, 125.96, 123.79, 118.84, 116.70, 116.61, 115.96, 114.58, 61.86, 61.50, 55.61, 52.65, 50.89, 49.23, 49.17, 47.79, 46.83, 46.70, 40.99, 29.71, 14.14. DART-HRMS: m/z [M+H]+ cald. for [C22H26N5O4]+, 424.1907; found 424.1958.
6.2.3.5. 3-(4-(4-(4-(4-(((3R, 5R)-5-(2,4-difluorophenyl)-5-methyltetrahydrofuran-3- yl)methoxy)phenyl)piperazin-1-yl)phenyl)-5-oxo-4,5-dihydro-1H-1,2,4-triazol-1-yl)propanoic acid (17).
Analogue 17 was prepared by coupling des-triazole intermediate 10 and linker region intermediate 15 as described above. White solid. (Yield = 42%) 1HNMR (500 MHz, CDCl3) δ 7.63 (s, 1H), 7.59 – 7.51 (m, 2H), 7.44 (d, J = 8.9 Hz, 1H), 7.07 (d, J = 9.0 Hz, 2H), 7.00 – 6.97 (m, 3H), 6.90 – 6.79 (m, 5H), 4.29 – 4.21 (m, 1H), 4.14 – 4.07 (m, 1H), 4.03 – 3.91 (m, 4H), 3.8 – 3.72 (m, 1H), 3.42 – 3.40 (m, 3H), 3.32 – 3.31 (m, 1H), 3.28 – 3.27 (m, 4H), 2.71 – 2.51 (m, 3H), 2.11 – 1.97 (m, 2H), 1.66 (s, 1H), 1.65 (s, 2H). 13CNMR (126 MHz, CDCl3) δ 176.37, 162.98, 161.01, 153.50, 152.29, 150.77, 145.57, 134.42, 127.72, 125.49, 123.73, 118.69, 118.60, 118.04, 116.67, 116.50, 115.31, 110.79, 110.63, 110.01, 109.98, 104.57, 104.36, 104.16, 82.80, 77.32, 77.06, 76.81, 70.43, 70.16, 70.10, 50.77, 50.21, 49.12, 48.00, 42.99, 42.04, 39.51, 39.42, 36.79, 29.71, 28.36, 20.87. DART-HRMS: m/z [M+H]+ cald. for [C33H36F2N5O5]+, 620.2060; found [M – 114.9653]+, 505.2407 m/z. IR (solid) vmax: 2931, 1705, 1614, 1512, 1231.6, 1039, 966, 822. Purity = 95.1%.
6.2.3.6. 2-(4-(4-(4-(4-(((3R, 5R)-5-(2,4-difluorophenyl)-5-methyltetrahydrofuran-3-yl)methoxy)phenyl)piperazin-1-yl)phenyl)-5-oxo-4,5-dihydro-1H-1,2,4-triazol-1-yl)acetic acid (18).
Analogue 18 was prepared by coupling des-triazole intermediate 10 and linker region intermediate 16 as described above. Tan solid. (Yield = 27%) 1HNMR (500 MHz, CDCl3) δ 7.63 – 7.51 (m, 1H), 7.00 – 6.94 (m, 5H), 6.90 – 6.88 (m, 3H), 6.81 – 6.79 (m, 2H), 4.29 – 4.26 (m, 1H), 4.14 – 4.11 (m, 1H), 3.99 – 3.94 (m, 3H), 3.87 – 3.72 (m, 2H), 3.40 (s, 1H), 3.33 – 3.26 (m, 8H), 2.65 – 2.62 (m, 2H), 1.98 (d, J = 4.3 Hz, 1H), 1.65 (s, 2H), 1.58 (s, 1H), 1.30 (s, 3H). 13CNMR (126 MHz, CDCl3) δ 163.11, 161.01, 153.31, 148.19, 145.76, 129.69, 127.65, 118.49, 117.91, 116.63, 115.30, 110.79, 110.61, 104.57, 104.36, 104.15, 82.71, 70.12, 50.80, 50.08, 42.07, 39.54, 29.72, 28.36. DART-HRMS: m/z [M+H]+ cald. for [C32H34F2N5O5]+, 606.2450; found [M – 101.0026]+, 505.2424 m/z. IR (solid) vmax: 2928, 2854, 2229, 1614, 1511, 1230, 966, 823; Purity = 97.2%.
6.2.3.7. 4-(4-(4-(4-(((3R,5R)-5-(2,4-difluorophenyl)-5-methyltetrahydrofUran-3-yl)methoxy)phenyl)piperazin-1-yl)phenyl)-2-(3-hydroxypropyl)-2,4-dihydro-3H-1,2,4-triazol-3-one (19).
Carboxylic acid 17 (24 mg, 0.039 mmol) was dissolved in THF (5 mL) and cooled to 0 °C. LiAlH4 (2M in THF, 0.062 mmol, 0.031 mL) was added dropwise. The mixture was warmed to RT and stirred for 4 h. The mixture was cooled to 0 °C and saturated NH4Cl was added dropwise and the resulting solution was washed with EtOAc (3x). The organic layers were combined, dried over Na2SO4, filtered and concentrated. Column chromatography (SiO2, 1% MeOH in DCM) afforded alcohol 19 as a tan solid. (Yield = 17 mg, 87%). 1HNMR (500 MHz, CDCl3) δ 7.64 (s, 1H), 7.61 – 7.52 (m, 2H), 7.43 (d, J = 8.9 Hz, 1H), 7.06 (d, J = 9.1 Hz, 1H), 7.00 – 6.97 (m, 4H), 6.89 – 6.87 (m, 3H), 6.82 – 6.79 (m, 2H), 4.29 – 4.21 (m, 1H), 4.14 – 4.07 (m, 2H), 4.03 – 3.91 (m, 4H), 3.87 – 3.71 (m, 2H), 3.40 (m, 2H), 3.31 (m, 2H), 3.27 (s, 4H), 2.63 – 2.62 (m, 3H), 2.12 – 1.98 (m, 2H), 1.65 (s, 3H), 1.37 – 1.33 (dd, J = 13.6, 6.7 Hz, 2H). 13CNMR (126 MHz, CDCl3) δ 163.04, 161.11, 160.07, 158.01, 153.39, 152.22, 150.77, 148.03, 145.76, 134.29, 127.73, 123.64, 118.58, 118.50, 117.92, 116.65, 116.61, 115.31, 111.50, 110.78, 110.63, 110.01, 109.98, 104.57, 104.36, 104.15, 82.74, 77.29, 77.03, 76.78, 70.46, 70.19, 70.12, 69.90, 50.81, 50.73, 50.11, 49.19, 47.98, 43.01, 42.05, 39.54, 39.45, 28.38, 22.35, 14.07. DART-HRMS: m/z [M+H]+ cald. for [C33H38F2N5O4]+, 605.2814; found [M – 100.0.390]+, 505.2424. IR (solid) vmax: 2932, 2828, 2232, 1689, 1614, 1511, 1230, 1112, 1039, 966, 821. Purity = 96.2%.
6.2.3.8. 3-(4-(4-(4-(4-((2-(2,4-dichlorophenyl)-2-methyl-1,3-dioxolan-4-yl)methoxy)phenyl)piperazin-1-yl)phenyl)-5-oxo-4,5-dihydro-1H-1,2,4-triazol-1-yl)propanoic acid (21).
Analogue 21 was prepared by coupling des-triazole intermediate 20 and linker region intermediate 15 as described above. White solid. (Yield = 20 mg, 54%) 1H NMR (500 MHz, CDCl3) δ 7.77 (d, J = 8.4 Hz, 1H), 7.69 – 7.62 (m, 2H), 7.57 (s, 1H), 7.45 – 7.39 (m, 2H), 7.29 – 7.23 (m, 1H), 7.08 – 7.06 (m, 1H), 7.00 – 6.98 (s, 2H), 6.95 – 6.93 (m, 2H), 6.78 – 6.77 (m, 1H), 4.67 – 4.64 (m, 1H), 4.42 – 4.33 (m, 2H), 4.19 – 4.14 (m, 1H), 4.07 – 4.00 (m, 2H), 3.91 – 3.77 (m, 2H), 3.42 – 3.39 (m, 3H), 3.30 – 3.25 (m, 5H), 1.86 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 153.06, 153.06, 152.54, 152.54, 150.80, 150.75, 147.98, 145.94, 145.94, 139.35, 139.13, 138.14, 134.63, 134.58, 132.84, 132.77, 131.21, 131.16, 130.98, 129.00, 128.91, 128.59, 126.81, 126.75, 125.49, 123.70, 123.62, 118.50, 118.46, 118.42, 117.92, 116.66, 115.52, 115.27, 110.00, 109.17, 109.11, 77.30, 77.05, 76.79, 75.07, 75.07, 74.77, 74.77, 73.96, 73.96, 73.62, 73.62, 69.29, 69.29, 68.43, 68.43, 67.94, 67.37, 67.37, 67.31, 67.01, 67.01, 50.73, 50.64, 50.64, 50.14, 49.16, 49.16, 48.23, 48.23, 47.25, 31.61, 29.72, 25.90, 25.74, 25.74, 25.66, 22.67. DART-HRMS: m/z [M+H]+ cald. for [C32H34Cl2N5O6]+, 654.1808; found [M – 115.0225]+, 539.1583 m/z. IR (solid) vmax: 2937, 2881, 2826, 1705, 1509, 1225, 1193, 1034, 819, 731, 500. Purity = 30.1 % : 69.0% (cis: trans), total = 99.1%.
6.2.4. Synthesis of triazolone/side chain truncated analogues.
6.2.4.1. 1-(4-(((3R,5R)-5-(2,4-difluorophenyl)-5-methyltetrahydrofuran-3-yl)methoxy)phenyl)-4- (4-nitrophenyl)piperazine (23).
Analogue 23 was prepared by coupling des-triazole intermediate 10 and linker region intermediate 22 as described above. Orange oil. (Yield = 40 mg, 67%). 1HNMR (500 MHz, CDCl3) δ 8.21 – 8.19 (m, 2H), 7.65 – 7.51 (m, 1H), 6.98 – 6.80 (m, 8H), 4.29 – 4.26 (m, 1H), 4.19 – 4.10 (m, 1H), 4.00 – 3.94 (m, 2H), 3.87 – 3.72 (m, 1H), 3.63 – 3.60 (m, 4H), 3.28 – 3.26 (m, 4H), 2.98 – 2.95 (m, 1H), 2.66 – 2.52 (m, 2H), 2.07 – 1.95 (m, 1H), 1.65 (s, 1H), 1.58 (s, 2H). 13CNMR (126 MHz, CDCl3) δ 154.79, 145.38, 130.16, 127.72, 127.64, 126.99, 125.98, 118.68, 115.38, 115.33, 113.38, 112.92, 104.36, 104.15, 82.81, 70.19, 70.08, 69.90, 60.31, 50.47, 47.29, 42.03, 39.53, 28.37. DART-HRMS: m/z [M+H]+ cald. for [C28H30F2N3O4]+, 510.2126; found 510.2205. IR (solid) vmax: 2932, 2850, 1596, 1509, 1322, 1229, 1115, 1038, 944, 826, 754. Purity = 95.6%.
6.2.4.2. 4-(4-(4-(((3R,5R)-5-(2,4-difluorophenyl)-5-methyltetrahydrofuran-3-yl)methoxy)phenyl)piperazin-1-yl)aniline (24).
Pd/C (10 wt. %, 0.05 equiv.) and ethanol were added to a round bottom flask purged with argon. Intermediate 23 (20 mg, 0.04 mmol) was added followed by dropwise addition of hydrazine monohydrate (13 mg, 0.40 mmol, 0.013 mL) and the solution was refluxed for 12 h. The mixture was cooled, filtered through celite and washed with ethanol. The filtrate was dried and the resulting crude gray solid was purified by column chromatography (SiO2, 20-30% Acetone in Hexanes). White solid. (Yield =17 mg, 90%). 1HNMR (500 MHz, CDCl3) δ 7.65 – 7.54 (m, 1H), 6.99 – 6.97 (m, 2H), 6.92 – 6.85 (m, 5H), 6.83 – 6.79 (m, 2H), 6.73 – 6.71 (m, 2H), 4.29 – 4.26 (m, 1H), 4.14 – 4.11 (m, 1H), 3.97 – 3.94 (m, 3H), 3.81 – 3.72 (m, 1H), 3.49 (br, s, 1H), 3.27 – 3.23 (m, 8H), 2.63 – 2.62 (m, 2H), 2.00 – 1.95 (m, 1H), 1.65 (s, 3H), 1.58 (s, 1H). 13CNMR (126 MHz, CDCl3) δ 162.97, 158.04, 153.12, 145.98, 144.46, 140.40, 127.74, 127.67, 118.86, 118.34, 116.24, 115.27, 115.22, 110.80, 110.61, 104.56, 104.35, 104.15, 82.70, 70.20, 70.14, 69.92, 51.26, 50.92, 42.06, 39.77, 39.55, 29.72, 28.38. DART-HRMS: m/z [M+H]+ cald. for [C28H32F2N3O2]+, 480.2384; found 480.2450. IR (solid) vmax: 3459, 3364, 2974, 2821, 1614, 1510, 1496, 1221, 1038, 942, 819, 515. Purity = 98.1%.
6.2.4.3. phenyl (4-(4-(4-(((3R,5R)-5-(2,4-difluorophenyl)-5-methyltetrahydrofuran-3-yl)methoxy)phenyl)piperazin-1-yl)phenyl)carbamate (25).
Aniline 24 (100 mg, 0.42 mmol) was dissolved in CHCl3 (15 mL) under argon followed by the addition of pyridine (6.72 mmol, 0.27 mL). The solution was cooled to 0 °C and phenylchloroformate (33 mg, 0.42 mmol, 0.025 mL) was added dropwise. The mixture was stirred at room temperature for 3 h before being diluted with water and washed with DCM (3x). The crude mixture was purified by column chromatography (SiO2, 20-30% Acetone in Hexanes). White solid. (Yield = 50 mg, 40%). 1HNMR (500 MHz, CDCl3) δ 7.65 – 7.54 (m, 1H), 7.45 – 7.40 (m, 4H), 7.29 – 7.23 (m, 3H), 7.02 – 6.94 (m, 4H), 6.89 – 6.88(m, 3H), 6.86 – 6.79 (m, 1H), 4.30 – 4.27 (dd, J = 8.5, 7.5 Hz, 1H), 4.14 – 4.11 (m, 1H), 4.00 – 3.94 (m, 2H), 3.87 – 3.72 (m, 1H), 3.35 – 3.32 (m, 4H), 3.28 – 3.25 (m, 4H), 2.99 – 2.94 (m, 1H), 2.67 – 2.61 (m, 1H), 2.56 – 2.52 (m, 1H), 2.07 – 1.98 (m, 1H), 1.65 (s, 2H), 1.58 (s, 1H). 13CNMR (126 MHz, CDCl3) δ 159.99, 158.02, 153.26, 150.76, 145.82, 129.39, 127.69, 125.59, 121.67, 120.34, 118.46, 118.42, 117.16, 115.31, 115.31, 115.26, 110.81, 110.64, 104.36, 104.15, 82.71, 70.21, 70.13, 69.92, 50.81, 49.98, 42.06, 39.77, 39.56, 28.39. DART-HRMS: m/z [M+H]+ cald. for [C35H36F2N3O4]+, 600.2596; found 600.2595. IR (solid) vmax: 3334, 3295, 3044, 2966, 2831, 1707, 1534, 1488, 1197, 1030, 820, 687. Purity = 96.2%.
6.2.4.4. N-(4-(4-(4-(((3R,5R)-5-(2,4-difluorophenyl)-5-methyltetrahydrofuran-3-yl)methoxy)phenyl)piperazin-1-yl)phenyl)hydrazinecarboxamide (26).
Compound 25 (40 mg, 0.067 mmol) was dissolved in 1,4-dioxane (2 mL). Hydrazine monohydrate (12 mg, 0.36 mmol, 0.011 mL, 5.4 equiv.) was added and the solution was refluxed for 3 h. The solution was cooled to RT, diluted with water and washed with EtOAc (3x). The organic layers were combined, dried over Na2SO4, filtered and concentrated and purified with column chromatography (SiO2, 20-30% Acetone in Hexanes). Tan solid. (Yield = 10.4 mg, 30%). 1HNMR (500 MHz, CDCl3) δ 8.08 (s, 1H), 7.61 (s, 1H), 7.57 – 7.54 (m, 1H), 7.46 (d, J = 8.0 Hz, 2H), 7.01 – 6.94 (m, 3H), 6.89 – 6.79 (m, 3H), 4.29 – 4.26 (t, J = 7.3 Hz, 1H), 4.14 – 4.11 (t, J = 7.0 Hz, 1H), 3.97 (br, s, 2H), 3.88 – 3.72 (m, 1H), 3.40 (s, 1H), 3.32 – 3.27 (m, 7H), 2.98 – 2.95 (m, 1H), 2.63 – 2.62 (m, 1H), 2.56 – 2.52 (m, 1H), 1.65 (s, 2H), 1.57 (s, 1H). 13CNMR (126 MHz, CDCl3) δ 162.97, 153.71, 147.67, 147.11, 145.91, 131.06, 127.74, 121.17, 118.43, 117.25, 115.28, 115.24, 104.36, 104.15, 70.19, 70.13, 69.91, 50.84, 50.21, 42.05, 39.76, 39.54, 28.38, 25.26. DART-HRMS: m/z [M+H]+ cald. for [C29H34F2N5O3]+, 538.2551; found. IR (solid) vmax: 3349, 3206, 2927, 2874, 2819, 1677, 1508, 1223, 1014, 817, 625, 516. Purity = 96.7%.
6.2.4.5. 4-(4-(4-(4-(((3R, 5R)-5-(2,4-difluorophenyl)-5-methyltetrahydrofuran-3-yl)methoxy)phenyl)piperazin-1-yl)phenyl)-2,4-dihydro-3H-1,2,4-triazol-3-one (27).
A solution of formamidine acetate (8.7 mg, 0.084 mmol) and 26 (10 mg, 0.019 mmol) in anhydrous DMF (5 mL) was stirred at 130 °C for 5 h. The mixture was cooled to RT, diluted with water, and washed with EtOAc (3x). The organic layers were combined, dried over Na2SO4, filtered and concentrated. Purification by column chromatography (SiO2, 15-30% Acetone in Hexanes) afforded triazolone 27. The fractions containing product were collected and dried by rotary evaportation. Tan solid. (Yield = 4.7 mg, 46%). 1HNMR (500 MHz, CDCl3) δ 8.55 (d, J = 11.6 Hz, 1H), 8.38 (s, 1H), 7.63 – 7.48 (m, 2H), 7.24 (d, J = 11.5 Hz, 1H), 7.06 – 6.79 (m, 8H), 4.29 – 4.26 (m, 1H), 4.14 – 4.11 (m, 1H), 3.98 – 3.94 (m, 2H), 3.87 – 3.72 (m, 1H), 3.40 (s, 1H), 3.37 – 3.27 (m, 7H), 2.63 – 2.51 (m, 2H), 2.08 – 1.90 (m, 1H), 1.65 (s, 1H), 1.58 (s, 2H). 13CNMR (126 MHz, CDCl3) δ 162.54, 158.58, 153.28, 148.76, 129.35, 127.68, 121.57, 121.41, 118.48, 117.31, 116.85, 115.31, 104.36, 70.20, 50.77, 49.79, 49.65, 42.08, 39.55, 29.72, 28.38. DART-HRMS: m/z [M+H]+ cald. for [C30H32F2N5O3]+, 548.2395; found 548.2406. IR (solid) vmax: 2923, 2852, 1690, 1509, 1226, 1076, 1018, 966, 944, 816, 730, 516. Purity = 96.7%.
6.2.5. Synthesis of analogues containing an amide.
6.2.5.1. 3-acetyl-N-(4-(4-(4-(((3R, 5R)-5-(2,4-difluorophenyl)-5-methyltetrahydrofUran-3-yl)methoxy)phenyl)piperazin-1-yl)phenyl)benzamide (28).
To a dry round bottom flask purged with Ar(g) was added 24 (17 mg, 0.035 mmol), 3-acetyl benzoic acid (18 mg, 0.11 mmol), N-(3-dimethylaminopropyl)-N’-ethylcarbodiimide hydrochloride (EDCI) (21 mg, 0.11 mmol) and 4-dimethylaminopyridine (13 mg, 0.11 mmol). The solids were dissolved with anhydrous DCM (8 mL) and the mixture was stirred for 12 h at RT before diluting with water and washing with DCM (3x). The organic layers were combined, dried over Na2SO4, filtered and concentrated. The crude product was purified by column chromatography (SiO2, 30% Acetone in Hexanes). White solid. (Yield = 5.2 mg, 27%). 1HNMR (500 MHz, CDCl3) δ 8.48 (s, 1H), 8.18 – 8.16 (m, 2H), 7.85 (s, 1H), 7.68 – 7.54 (m, 4H), 7.06 – 6.80 (m, 7H), 4.30 – 4.27 (dd, J= 8.7, 7.3 Hz, 1H), 4.14 − 4.11 (m, 1H), 4.00 – 3.94 (m, 2H), 3.87 – 3.73 (m, 1H), 3.40 – 3.37 (m, 4H), 3.30 – 3.28 (m, 4H), 2.73 (s, 3H), 2.65 – 2.53 (m, 2H), 2.07 – 1.95 (m, 1H), 1.65 (s, 3H). 13CNMR (126 MHz, CDCl3) δ 197.40, 164.45, 162.98, 161.01, 160.08, 158.02, 153.28, 148.70, 145.85, 137.41, 135.64, 131.74, 131.38, 130.19, 129.28, 127.69, 126.29, 121.80, 118.49, 118.45, 116.87, 115.31, 115.26, 104.56, 104.36, 104.15, 82.71, 70.20, 70.13, 69.92, 50.80, 49.78, 42.05, 39.77, 39.55, 29.72, 28.38, 26.77. DART-HRMS: m/z[M+H]+ cald. for [C37H38F2N3O4]+, 626.2752; found 626.2807. IR (solid) vmax: 3305, 2944, 2830, 1642, 1591, 1511, 1497, 1227, 1023, 818, 706, 537, 515. Purity = 100%.
6.2.5.2. N-(4-(4-(4-(((3R,5R)-5-(2,4-difluorophenyl)-5-methyltetrahydrofuran-3-yl)methoxy)phenyl)piperazin-1-yl)phenyl)-3-hydroxybenzamide (29).
To a dry round bottom flask purged with argo, 3-hydroxybenzoic acid (4 mg, 0.03 mmol) and HATU (15 mg, 0.04 mmol) were dissolved in DMF (5 mL). Next, NMM (12 mg, 0.12 mmol, 0.013 mL) was added and the mixture was stirred at RT for 30 min followed by the addition of 24 (16 mg, 0.03 mmol). The mixture was stirred at RT for 12 h, poured over ice cold water (~5 mL) and washed with EtOAc (3x). The organic layers were combined, dried over Na2SO4, filtered and concentrated. The crude product was purified by column chromatography (SiO2, 30% Acetone in Hexanes). White solid. (Yield = 10 mg, 80%). 1HNMR (500 MHz, CDCl3) δ 8.18 – 8.10 (m, 1H), 7.83 – 7.54 (m, 5H), 7.44 – 7.39 (m, 2H), 7.18 (d, J = 7.3 Hz, 1H), 7.09 – 6.79 (m, 7H), 4.28 – 4.26 (m, 1H), 4.12 – 4.10 (t, J = 7.8 Hz, 1H), 3.97 (m, 2H), 3.81 – 3.74 (m, 1H), 3.39 – 3.27 (m, 8H), 3.19 – 2.93 (m, 2H), 2.66 – 2.53 (m, 2H), 1.65 (s, 2H), 1.57 (s, 1H). 13CNMR (126 MHz, CDCl3) δ 156.31, 155.97, 153.28, 151.10, 148.63, 145.86, 136.88, 130.03, 127.79, 122.69, 121.73, 121.19, 118.61, 118.49, 116.92, 115.31, 104.36, 104.15, 82.71, 70.20, 70.13, 50.79, 49.81, 42.05, 39.55, 29.72, 28.37. DART-HRMS: m/z [M+H]+ cald. for [C35H36F2N3O4]+, 600.2596; found 600.2681. IR (solid) vmax: 3269, 2925, 2825, 1676, 1642, 1511, 1225, 1020, 817, 731, 685, 590, 532, 516.Purity = 100%
6.2.5.3. N-(4-(4-(4-(((3R, 5R)-5-(2,4-difluorophenyl)-5-methyltetrahydrofuran-3-yl)methoxy)phenyl)piperazin-1-yl)phenyl)nicotinamide (30).
Analogue 30 was prepared utilizing the procedure described above for analogue 28. White solid. (Yield = 11.4 mg, 63%). 1HNMR (500 MHz, CDCl3) δ 9.13 (s, 1H), 8.81 (d, J = 3.3 Hz, 1H), 8.25 – 8.24 (d, J = 7.6 Hz, 1H), 7.87 (s, 1H), 7.60 – 7.47 (m, 4H), 7.05 – 6.79 (m, 7H), 4.29 – 4.26 (dd, J = 8.7, 7.3 Hz, 1H), 4.14 – 4.11 (dd, J = 8.6, 7.4 Hz, 1H), 3.99 – 3.94 (m, 2H), 3.85 – 3.74 (m, 1H), 3.39 – 3.36 (m, 4H), 3.29 – 3.27 (m, 4H), 2.99 – 2.94 (m, 1H), 2.67 – 2.52 (m, 2H), 2.07 – 1.95 (m, 1H), 1.65 (s, 2H), 1.58 (s, 1H). 13CNMR (126 MHz, CDCl3) δ 163.63, 162.98, 161.17, 153.29, 152.43, 148.83, 147.81, 145.82, 135.26, 130.97, 129.92, 127.62, 123.68, 121.93, 118.49, 116.82, 115.30, 110.78, 110.65, 104.57, 104.36, 104.15, 82.74, 70.20, 70.13, 50.79, 49.72, 42.05, 39.55, 28.39. DART-HRMS: m/z [M+H]+ cald. for [C34H35F2N4O3]+, 585.2599; found 585.2649. IR (solid) vmax: 3312, 2932, 2874, 2826, 1737, 1646, 1614, 1592, 1242, 966, 945, 849, 748. Purity = 97.4%.
6.3. Cell Culture Studies.
Protocols for general cell culture, qPCR, and Hh inhibition in ASZ cells have been previously described [17, 20]. All PSZ analogues were tested for Hh inhibitory activity by measuring their inhibition of endogenous Gli1 mRNA expression in a murine BCC cell line (Ptch+/−), ASZ. Cells at 80% confluence were plated in 154CF growth media (2% FBS, ThermoFisher) in a 96-well plate (10,000 cells/well). Cells were incubated for 24 hours (37°C, 5% CO2) before media without fetal bovine serum (FBS) was added. Cells were again incubated for 24 hours (37°C, 5% CO2) before being treated with DMSO (vehicle control), PSZ, or PSZ analoguea at varying concentrations (10 – 0.001 μM). Compounds were incubated with the cells for 48 hours (37°C, 5% CO2). Cells were visually examined to ensure there was no non-specific cytotoxicity present for the concentrations evaluated. Cells were lysed, mRNA was extracted, and Act B (internal control) and Glil expression levels were measured by qRT-PCR using the TaqMan Fast Cells-to-CT kit (ThermoFisher). Compound concentrations that significantly decreased actin B levels were excluded form our qRT-PCR calculations. Data was analyzed using GraphPad Prism 5 and IC50 values computed as mean ± SEM from at least three separate experiments performed in triplicate.
Supplementary Material
Highlights.
A des-triazole posaconazole (PSZ) analogue maintained potent inhibition of the hedgehog (Hh) signaling pathway with enhanced drug-like properties.
Docking studies provide insight into the binding orientation these compounds adopt when complexed with their molecular target Smoothened.
Structure-based design of PSZ analogues resulted in compounds with improved potency and stability.
Molecular dynamics simulations demonstrate that PSZ and analogues stabilize an inactive conformation of Smo upon binding.
Acknowledgements
The authors gratefully acknowledge support of this work by the National Institutes of Health/National Cancer Institute (CA190617). ASZ001 cells were kindly provided by Dr. Ervin Epstein (Children's Hospital Oakland Research Institute).
Abbreviations
- ITZ
itraconazole
- PSZ
posaconazole
- Hh
Hedgehog
- Gli
glioblastoma associated oncogene
- Ptch
patched
- BCC
Basal cell carcinoma
- MB
Medulloblastoma
- Smo
Smoothened
- THF
Tetrahydrofuran
- OXZ
(4R)-(+)-4-benzyl-2-oxazolidinone
- Ptch-CKO
Patched knockout
- ECD
Extracellular domain
- CRD
Cysteine rich domain
- ELD
Linker domain
- 7TM
Seven transmembrane domain
- ECL2
Extracellular loop 2
Footnotes
Appendix A. Supporting Information. Synthetic protocols and characterization of previously disclosed intermediates. Protocols for the computational modeling and PK studies. 1H, 13C and 2D NMR (HMBC and HSQC) for all new intermediates and final des-triazole PSZ analogues. Additional figures.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Ingham PW, McMahon AP, Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 15 (2001) 3059–3087. [DOI] [PubMed] [Google Scholar]
- [2].Briscoe J, Thérond PP, The mechanisms of hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol 14 (2013) 416–429. [DOI] [PubMed] [Google Scholar]
- [3].Amakye D, Jagani Z, Dorsch M, Unraveling the therapeutic potential of the hedgehog pathway in cance. Nat. Med 19 (2013) 1410–1422. [DOI] [PubMed] [Google Scholar]
- [4].Fecher LA, Sharfman WH, Advanced basal cell carcinoma, the hedgehog pathway, and treatment options – role of smoothened inhibitors. Biologics 9 (2015) 129–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kieran MW Targeted treatment for sonic hedgehog-dependent medulloblastoma. Neuro-Oncol. 16 (2014) 1037–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Wu F, Zhang Y, Sun B, McMahon AP, Wang Y, Hedgehog signaling: From basic biology to cancer therapy. Cell Chem. Biol 24 (2017) 252–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Rudin CM, Hann CL, Laterra J, Yauch RL, Callahan CA, Fu L, Holcomb T, Stinson J, Gould SE, Coleman B, LoRusso PM, Von Hoff DD, de Sauvage FJ, Low JA, Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N. Engl. J. Med 361 (2009) 1173–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Yauch RL, Dijkgraaf GJ, Alicke B, Januario T, Ahn CP, Holcomb T, Pujara K, Stinson J, Callahan CA, Tang T, Bazan JF, Kan Z, Seshagiri S, Hann CJ, Gould SE, Low JA, Rudin CM, de Sauvage FJ, Smoothened mutation confers resistance to a hedgehog pathway inhibitor in medulloblastoma. Science 326 (2009) 572–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Atwood SX, Sarin KY, Whitson RJ, Li JR, Kim G, Rezaee M, Ally MS, Kim J, Yao C, Chang ALS, Oro AE, Tang JY, Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell 27 (2015) 342–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sharpe HJ, Pau G, Dijkgraaf GJ, Basset-Seguin N, Modrusan Z, Januario T, Tsui V, Durham AB, Dlugosz AA, Haverty PM, Bourgon R, Tang JY, Sarin KY, Dirix L, Fisher DC, Rudin CM, Sofen H, Migden MR, Yauch RL, de Sauvage FJ, Genomic analysis of smoothened inhibitor resistance in basal cell carcinoma. Cancer Cell 27 (2015) 327–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Dijkgraaf GJ, Alicke B, Weinmann L, Januario T, West K, Modrusan Z, Burdick D, Goldsmith R, Robarge K, Sutherlin D, Scales SJ, Gould SE, Yauch RL, de Sauvage FJ, Small molecule inhibition of GDC-0449 refractory smoothened mutants and downstream mechanisms of drug resistance. Cancer Res. 71 (2011) 435–444. [DOI] [PubMed] [Google Scholar]
- [12].Wen J, Hadden MK, Structure-activity relationship studies of vitamin D3 analogues containing an ether or thioether linker as Hedgehog pathway inhibitors. ChemMedChem 13 (2018) 748–753. [DOI] [PubMed] [Google Scholar]
- [13].Carballo GB, Honorato JR, Farias de Lopes GP, Leite TC de Sampaio e Spohr, A highlight on sonic hedgehog pathway. Cell Communication and Signaling 16 (2018) 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Dong X, Wang C, Chen Z, Zhao W, Overcoming the resistance mechanisms of Smoothened inhibitors. Drug Discovery Today 23 (2018) 704–710. [DOI] [PubMed] [Google Scholar]
- [15].Ghirga F, Mori M, Infante P, Current trends in Hedgehog signaling pathway inhibition by small molecules. Bioor. Med. Chem. Lett 28 (2018) 3131–3140. [DOI] [PubMed] [Google Scholar]
- [16].Xin M, Ji X, De La Cruz LK, Thareja S, Wang B, Strategies to target the Hedgehog signaling pathway for cancer therapy. Med. Res. Rev 38 (2018) 870–913. [DOI] [PubMed] [Google Scholar]
- [17].Kim J, Tang JY, Gong R, Kim J, Lee JJ, Clemons KV, Chong CR, Chang KS, Fereshteh M, Reya T, Liu JO, Epstein EH, Stevens DA, Beachy PA, Itraconazole, a commonly used anti-fungal that inhibits Hedgehog pathway activity and cancer growth Cancer Cell 17 (2010) 388–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kim J, Aftab BT, Tang JY, Kim D, Lee AH, Rezaee M, Kim J, Chen B, King EM, Borodovsky A, Riggins GJ, Epstein EH Jr., Beachy PA, Rudin CM, Itraconazole and arsenic trioxide inhibit Hedgehog pathway activation and tumor growth associated with acquired resistance to smoothened antagonists. Cancer Cell 23 (2013) 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Pace JR, DeBerardinis AM, Sail V, Tacheva-Grigorova SK, Chan KA, Tran R, Raccuia DS, Wechsler-Reya RJ, Hadden MK, Repurposing the clinically efficacious antifungal agent itraconazole as an anticancer chemotherapeutic. J. Med. Chem 59 (2016) 3635–3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Pace JR; Teske KA; Chou LQ; Wechsler-Reya RJ; Hadden MK Structure-activity relationships for itraconazole-based triazolone analogues as hedgehog pathway inhibitors. J. Med. Chem Under Revision. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Chen B, Trang V, Lee A, Williams NS, Wilson AN, Epstein EH, Tang JY, Kim J, Posaconazole, a second-generation triazole antifungal drug, inhibits the hedgehog signaling and progression of basal cell carcinoma. Mol. Cancer Ther 15 (2016) 866–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Hoch L, Faure H, Roudaut H, Schoenfelder A, Mann A, Girard N, Bihannic L, Ayrault O, Petricci E, Taddei M, Rognan D, Raul M, MRT-92 inhibits hedgehog signaling by blocking overlapping binding sites in the transmembrane domain of the smoothened receptor. FASEB J. 29 (2015) 1817–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Saksena AK, Girijavallabhan VM, Wang H, Liu Y–T, Pike RE, Ganguly AK, Concise asymmetric routes to 2,2,4-trisubstituted tetrahydrofurans via chiral titanium imide enolates: Key intermediates towards synthesis of highly active azole antifungals SCH 51048 and SCH 56592. Tet. Lett 37 (1996) 5657–60. [Google Scholar]
- [24].So P–L, Langston AW, Daniallinia N, Hebert JL, Fujimoto MA, Khaimskiy Y, Asxterbaum M, Epstein EH, Long-term establishment, characterization and manipulation of cell lines from mouse basal cell carcinoma tumors. Exp. Dermatol 15 (2006) 742–750. [DOI] [PubMed] [Google Scholar]
- [25].Brun SN, Markant SL, Esparza LA, Garcia G, Terry D, Huang J–M, Pavlyukov MS, Li X–N, Grant GA, Crawford JR, Levy ML, Conway EM, Smith LH, Nakano I, Berezov A, Greene MI, Wang Q, Wechsler-Reya RJ, Survivin as a therapeutic target in Sonic hedgehog-driven medulloblastoma. Oncogene 34 (2015) 3770–3779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wang C, Wu H, Katritch V, Han GW, Huang X–P, Liu W, Siu FY, Roth BL, Cherezov V, Stevens RC, Structure of the human smoothened receptor bound to an antitumour agent. Nature 497 (2013) 338–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Huang P, Zheng S, Wierbowski BM, Kim Y, Nedelcu D, Aravena L, Liu J, Kruse AC and Salic A, Structural basis of smoothened activation in hedgehog signaling. Cell 174 (2018) 312–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zhang X, Zhao F, Wu Y, Yang J, Han GW, Zhao S, Ishchenko A, Ye L, Lin X, Ding K, Dharmarajan V, Griffin PR, Gati C, Nelson G, Hunter MS, Hanson MA, Cherezov V, Stevens RC, Tan W, Tao H and Xu F, Crystal structure of a multi-domain human smoothened receptor in complex with a super stabilizing ligand. Nat. Commun 8 (2017) 15383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Byrne EFX, Sircar R, Miller PS, Hedger G, Luchetti G, Nachtergaele S, Tully MD, Mydock-McGrane L, Covey DF, Rambo RP, Sansom MSP, Newstead S, Rohatgi R, Siebold C, Structural basis of Smoothened regulation by its extracellular domains. Nature 535 (2016) 517–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.