

Abstract
Epimetabolites distinct from canonical metabolisms are identified by integrating three cheminformatics tools: BinVestigate, querying the BinBase GC-MS metabolome database to match unknowns with biological metadata across over 110,000 samples; MS-DIAL 2.0, a universal software for chromatographic deconvolution of high resolution GC- or LC-mass spectrometry; and MS-FINDER 2.0, a structure elucidation program with searching against an enzyme promiscuity library. The discoveries are showcased by N-methyl-alanine, N-methyl-UMP, lyso-monogalactosyl-monopalmitin, and two propofol derivatives.
Untargeted metabolomics detects many more unknown peaks than identified compounds because publicly available mass spectra libraries are still very small in comparison to the chemical sphere of more than 68 million known compounds1. Even in GC-MS where the spectra have been collected systematically and in a standardized manner in the NIST and Wiley libraries for more than 30 years with over 267,000 unique compounds, only about 40% of the reliably detectable peaks are identified in metabolomic profiles. This ‘dark matter of metabolomics’2 can be explained by at least five routes: (a) lack of knowledge of enzymatic transformations3, including substrate promiscuity4; (b) metabolic damage by spontaneous reactions or enzyme errors5; (c) signatures of exogenous compounds, for example from environmental sources6; (d) combined metabolic impact of a community of species, for example by gut microbiota7; and (e) formation of chemical artifacts during analytical protocols8. Recently, the new term ‘epimetabolite9’ was suggested to encompass such modified metabolites from the routes of (a) to (d) that gain physiological functions, similar to post-translational modifications of proteins. The purpose of this study is to identify novel epimetabolites from unknown chromatographic peaks, and the best strategy aims at reducing the number of important (functional) unknowns by investigating multiple studies simultaneously, including cross species analyses10. Once the origin, relevance and specificity of these unknowns have been asserted, accurate mass spectrometry and cheminformatics tools can be used to annotate and validate chemical structures.
We here present a unified method for functional and structural annotation of unknown epimetabolites (Fig. 1). BinBase is a large GC-MS based untargeted metabolomics database encompassing 1,561 studies with 114,795 samples for various species, organs, matrices, and experimental conditions that have been acquired over the past 13 years11. In BinBase, 9,563 unique metabolites have been discovered so far, where 1,020 have been identified by mass spectral libraries of authentic standards12, in addition to 256 known chemical artifacts. To query biological metadata for each metabolite, BinVestigate (http://binvestigate.fiehnlab.ucdavis.edu/) yields open access information about the abundance, frequency, species and organ origin. Once the importance of unknowns is evaluated, MS-DIAL 2.0 (http://prime.psc.riken.jp/) is utilized to obtain the deconvoluted spectra from high resolution GC-MS data as prerequisite for compound identification. MS-DIAL was previously developed for LC-MS data processing13 but now enables processing both LC-MS/MS and GC-MS data. Finally, again for either accurate mass GC-MS or LC-MS/MS, unknowns are annotated by their elemental formulas and in silico mass spectral fragmentation through MS-FINDER14 2.0 (http://prime.psc.riken.jp/). MS-FINDER integrates structures and formulas for 224,622 known metabolites and now also includes 643,307 hypothetical compounds from the enzyme promiscuity database – MINE-DB (http://minedatabase.mcs.anl.gov/)15. Notably, MS-DIAL 2.0 links mass spectra directly to BinVestigate and MS-FINDER 2.0 while the tools are also available as stand-alone software. We give five successful examples for this strategy, ranging from new methylation products in mammalian and microbial cells to plant-specific metabolites and transformations of exposome compounds. The performances of three programs, including false discovery rates in BinBase, are discussed in Online Methods.
Figure 1. Summary for functional and structural identification of unknown metabolites.
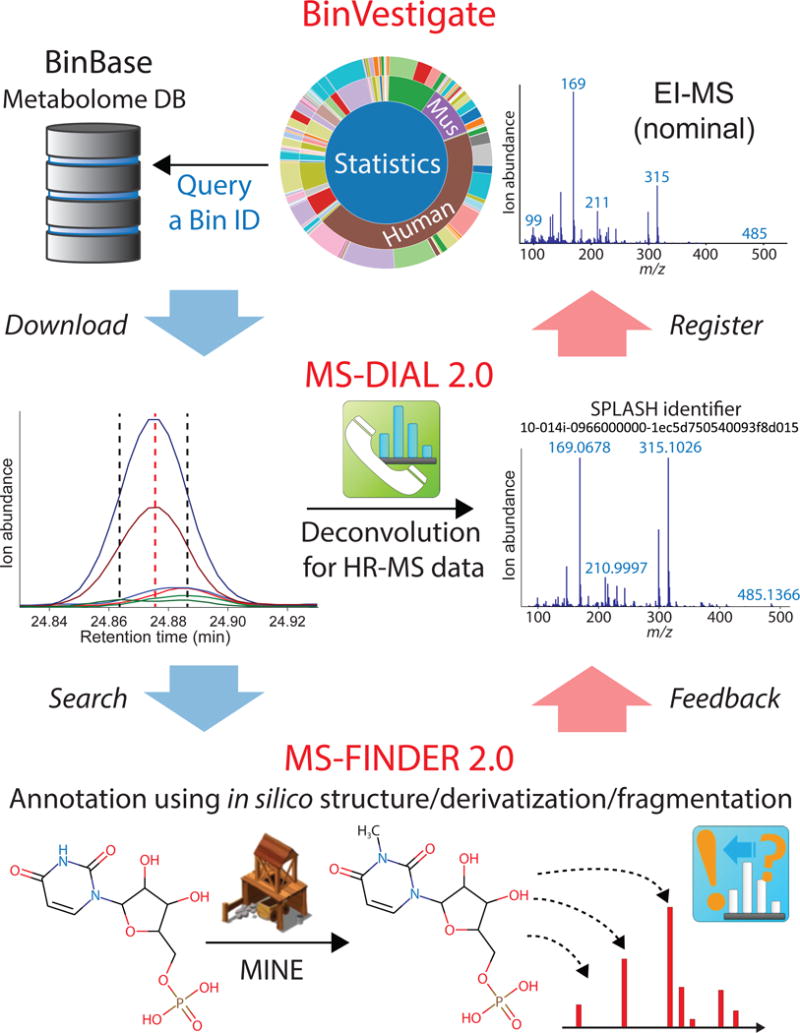
(a) BinVestigate to search unknown compounds for metabolomics study metadata and (nominal) EI-MS spectra in BinBase, with results shown as sunburst diagrams to illustrate the biological origin (species, organs, cell types) of unknowns. (b) MS-DIAL 2.0 for universal GC-MS or LC-MS/MS deconvolution with high resolution (HR) mass spectrometry analytics to obtain the deconvoluted HR-MS spectra of unknowns needed for compound identification. (c) MS-FINDER 2.0 for universal GC-EI-MS and LC-ESI-MS/MS spectral interpretation to annotate unknowns in combination with the enzyme promiscuity structure database (MINE), resulted in the discovery of biologically significant chemical structure. The tools are fully connected in MS-DIAL. Each tool is also available as standalone program.
BinVestigate is used to query unknowns from different West Coast Metabolomics Center (WCMC) metabolomics studies and to prioritize and select targeted unknowns for structural identification based on their cross-study specificity and relevance (Online Methods). The WCMC quality controls keep absolute signal intensities within two-fold deviations from the mean and avoid detector saturations, making intensities comparable across species and sample types. Therefore, besides frequency of detection in specific organs and species, BinVestigate uses average signal intensities to highlight relevance across studies. As the first case, unknown BinBase metabolite (BB160842) was detected in 44,128 samples (Fig. 2a), 90% of which were from microbial, fecal, or plasma studies. It was found at 5–10 times higher signal intensity in human or animal fecal matter compared to microbial cells, and up to 20-fold higher than in body fluids or tissues. It suggests a compound of microbial origin that is excreted into human plasma. The second example, BB106699 was detected in 7,228 samples, most abundantly in diverse cancer cell lines and cancer tissues (Fig. 2b). It showed up to 100-fold higher signal intensity in myeloma cancer cell lines compared to other cell types, such as mouse kidney cells. This compound was never found in fecal matter or bacterial samples, supporting the notion that it might be exclusive for eukaryotes and might have a specific role in cancer. Similarly, BB21735 (Supplementary Fig. 1) was found exclusively in 765 samples of algae, marine- and cyanobacteria, and plants but never in human or animal samples, suggesting a dedicated role in the biochemistry of photosynthetic organisms. Finally, BB171284 and BB118961 were found in only two clinical cohort studies in plasma and urine of 405 and 242 samples, respectively. As both studies involved pharmaceutical treatments, these compounds appeared to be significant for phase 2 drug clearance.
Figure 2. Metabolomic meta-analysis for origin exploration by BinVestigate.
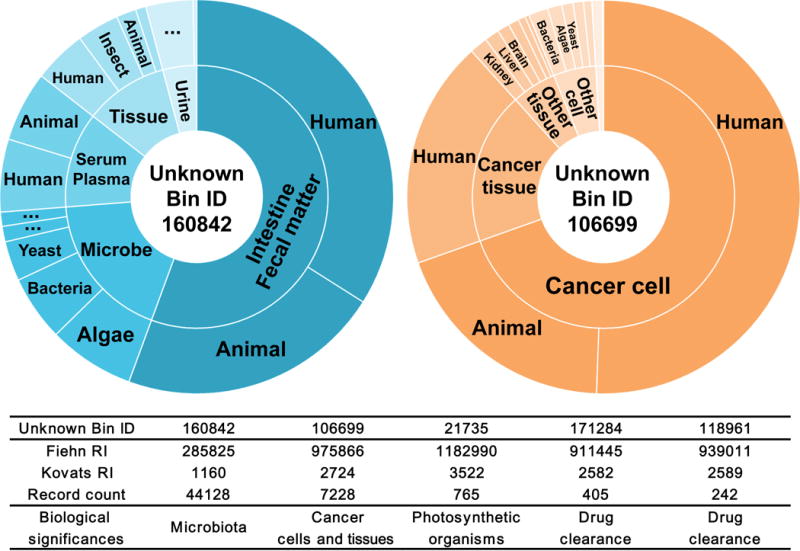
Bin IDs were queried in over 114,000 samples to show cross-study specificity and relevance of unknown BinBase ID 160842 (left) and unknown BinBase ID 106699 (right). In the sunburst diagrams, the area of the circular sector for each organ (inner cycle) or species (outer cycle) was mathematically determined by the average signal intensity of the unknown compound when present in such origin. Bin ID, Fiehn RI, Kovats RI, number of annotation records, and conclusion of biological significance for the five unknowns discussed in this paper were summarized in the table.
For identifying these unknown compounds, we first obtained high resolution (HR) accurate mass GC-MS data with different ionization techniques, and employed LC-MS/MS for validation. Unlike LC-MS/MS metabolomics, GC-MS based analyses lead to extensive fragmentation right at the source of ionization, even under soft chemical ionization. We have therefore developed a new version of our data processing software MS-DIAL. MS-DIAL 2.0 is the first universal program for processing untargeted metabolomics data including spectral deconvolution and compound identification (Supplementary Figs. S2, S3, Supplementary Data 1, and Online Methods), supporting multiple MS data types (low or high resolution MS, and GC-MS, LC-MS, or LC-MS/MS) of any major vendor- or open data formats (Supplementary Fig. S4). By using MS-DIAL 2.0, the deconvoluted spectra from GC-HR-MS and LC-HR-MS/MS were extracted from the representative biological samples containing the unknown metabolites discovered in BinVestigate.
The next step is MS-FINDER 2.0 for the structure elucidation of unknown HR-MS spectra, and we use BB106699 as a showcase (Fig. 3, Supplementary Figs. S5, S6, and Online Methods). First, the molecular adduct ion was identified to pursue the chemical formula of the unknown compound. As often observed for trimethylsilylated (TMS)-metabolites in GC-MS, the molecular ion ([M] +•) was absent from the hard electron ionization spectrum. Its initial methyl cleavage fragment ion ([M-CH3]+) was found to be very low abundant, impeding the calculation of the elemental formula for this unknown (Fig. 3a). We have therefore used softer methane chemical ionization GC-HR-MS that yielded a pattern of additional highly characteristic molecular adduct ions. For BB106699, the molecular mass was calculated as 626.212 Da based on the alignment of ions at m/z 611.118 ([M-CH3]+), m/z 627.214 ([M+H]+), m/z 655.244 ([M+C2H5]+), and m/z 667.245 ([M+C3H5]+). MS-FINDER 2.0 calculated C10H15N2O9P as most probable elemental formula by the optimized algorithm for TMS-derivatives, using both heuristic and chemical rules14 (Fig. 3b). The formula was further validated by comparing regular TMS derivatization to stable isotope labeled d9-TMS. This comparison directly yielded the number of TMS groups, i.e., the number of acidic protons in the unknown molecule (Fig. 3b left). Hence, the fact that GC-MS based metabolomics needs derivatization gave us the advantage to omit isomeric structures of C10H15N2O9P with less than four acidic protons. Moreover, comparing the chemical derivatization results by methoximation versus ethoximation showed that the unknown compound had no aldehyde or ketone functional groups16. In subsequent LC-HR MS/MS analysis, the formula was validated using both accurate mass and natural isotope abundance information (Fig. 3b right). In an analogous manner, formulas were confirmed as C4H9NO2 for BB160842 with 2 acidic protons, C25H48O9 for BB21735 with 5 acidic protons, and BB171284 and BB118961 as isomers of C18H26O8 with 5 acidic protons (Supplementary Table 1).
Figure 3. Identification of N-methyl-UMP by MS-DIAL 2.0 and MS-FINDER 2.0.
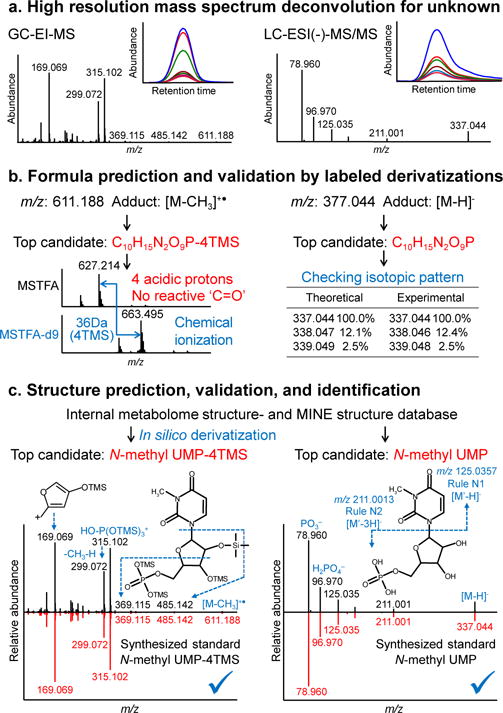
High resolution GC-MS analytics was first used for structure elucidation (left), then LC-MS/MS was applied as additional evidence line to validate the discovery (right). (a) Spectral deconvolution: fragment ions and molecular adduct ions of BinBase ID 106699 were deconvoluted and confirmed through MS-DIAL 2.0. (b) Formula prediction and validation: C10H15N2O9P was scored and ranked at 1st in MS-FINDER 2.0 based on mass errors, isotope ratio errors, and subformula assignments. For GC-MS flow, chemical ionization data with different derivatization methods (MSTFA vs. MSTFAd9) were obtained to verify the formula as well as to yield the number of acidic protons; for LC-MS flow, between theoretical values and experimental values, the mass errors were only 1 mDa, and the isotopic ratio errors were within 1%. (c) Structure prediction, validation, and identification: structure candidates were retrieved from MINE DB in addition to internal metabolome database, and in silico fragmented based on hydrogen rearrangement rules, bond dissociation energy, and comprehensive fragmentation rule library (including GC-EI-MS and LC-ESI-MS/MS). N-methyl-UMP was ranked at the most likely structure in MS-FINDER 2.0 with computational assigned substructures. The mass spectra and retention times in GC-MS (left) and LC-MS/MS (right) were matched between BinBase ID 106699 in cancer cell sample with chemically synthesized N-methyl-UMP standard for final validation.
Using the elemental formulas, all potential isomer structures were retrieved from databases for these five unknown BinBase metabolites. For retrieving structure candidates, MS-FINDER 2.0 uses a combination of 14 metabolomics databases comprising 47,311 formulas and 224,622 unique known structures. Yet, enzyme promiscuity and general lack of knowledge about enzyme reactions may be the reason of many unknown compounds. Therefore, MS-FINDER 2.0 also incorporates all MINE-DB structures15, a collection of 643,307 virtual metabolites that are predicted based on generalized enzymatic transformations as applied to KEGG pathway metabolites.
For BB106699, C10H15N2O9P yielded 6 isomers from the 14 metabolomic databases in MS-FINDER 2.0, and 33 isomers in MINE-DB. All structures were subsequently ranked by matching the experimental spectrum against predicted spectra for all isomers, considering chemical substructures recognized from the mass spectra as well as biochemical likelihood. For annotating chemical substructures from GC-MS spectra, MS-FINDER 2.0 exploits 228 true-positive fragmentation patterns from 80 reports that were published over the past 50 years17. These rules confirmed the presence of hydroxyl groups and a phosphate moiety in BB106699, a secondary amine and a carboxylic acid in BB160842, glycosylation of BB21735, and glucuronidation for BB171284 and BB118961 (Supplementary Table 1).
The unknown BB106699 was finally identified as N-methyl-UMP, a MINE predicted metabolite that has never been reported from biological samples. It ranked as the most likely structure in MS-FINDER 2.0 (Fig. 3c top) with all fragment ions rationalized by substructure annotations (Fig. 3c bottom). We validated the identification of N-methyl-UMP by synthesizing an authentic standard (Online Methods) and compared retention times and mass spectra in both GC-MS and LC-MS/MS to alternative O-methyl-UMP isomers (Supplementary Fig. S7). In the same manner, we annotated BB160842 as N-methyl-alanine (Supplementary Fig. S8), BB21735 as lyso-monogalactosylmonopalmitin (Supplementary Fig. S9), BB171284 as 4-hydroxypropofol-1-glucuronide (Supplementary Fig. S10), and BB118961 as 4-hydroxypropofol-4-glucuronide (Supplementary Fig. S11).
In summary, open access metabolomics repositories such as the NIH MetabolomicsWorkbench18 and EBI MetaboLights19 are important for comparing metabolomic results with respect to identified compounds. However, for comparing unknown metabolites across different biological studies, it is critical to standardize data acquisition methods and data processing parameters. At current, only GC-MS (and, in principle, NMR) data fulfill this criterion. Our strategy fully utilizes this advantage, using MS-DIAL 2.0 with BinVestigate and MS-FINDER 2.0 that outperformed alternative deconvolution and compound identification software in untargeted metabolomics (Supplementary Tables 1 and 2). Moreover, our approach found e.g. N-methyl-UMP to be highly upregulated in cancer cells and cancer tissues, in comparison to any other cell type or tissue. Recently, methylation of small molecules has been shown to directly regulate cellular progression in stem cells20, raising the possibility of related mechanisms in cancer cells or utilizing methylated metabolites as cancer biomarkers. More broadly, it has been shown quite regularly that small chemical alterations of metabolites may remove these compounds from primary biochemistry pathways, and that such modified metabolites, i.e. epimetabolites, subsequently gain regulatory functions, for example oxylipins. For the discovery of epimetabolites, the integration of BinVestigate, MS-DIAL, and MS-FINDER provides a systematic strategy to utilize the complete set of mass spectral information as well as biochemical metadata to successfully find and rank the most likely chemical structures, and it would form the basis for a unified and standardized input for a comprehensive metabolomics repository.
Online Methods
BinBase
BinBase is a large GC-TOF MS based metabolomics database encompassing 1,561 studies with 114,795 samples for various species, organs, matrices, and experimental conditions. By the physics of GC-MS, analysis is restricted to thermostable small molecules that range up to 650 Da in size, even if using derivatization by trimethylsilylation to reduce boiling points. Molecules profiled by trimethylsilylation GC-MS based metabolomics include amino acids, di- and tripeptides, hydroxyl acids, organic phosphates, fatty acids, alcohols, sugar acids, mono-, di- and trisaccharides including sugar acids and sugar alcohols, aromatic acids, nucleosides and mononucleotides (but not di- or trinucleotides), sterols, polyamines, and a large variety of miscellaneous compounds.
BinBase uses a retention index- and mass spectral quality filtering system based on GC-TOF based mass spectral deconvolution results as input21 to store and report unique metabolite signals that are detected in metabolomic studies. Through the connected MiniX system22, all studies in BinBase are associated with metadata such as species, organs, cell types, and treatments. The BinBase algorithm has been published previously11,23 and is used over the past 13 years. It relies on mass spectral deconvolution of GC-TOF MS data by the Leco ChromaTOF software and utilizes a multi-tiered filter system with different settings to annotate deconvoluted instrument peak spectra as unique database entries (“bins”). For typical studies on mammalian plasma with about 50–60 samples, about 1,000 peaks would be detected by ChromaTOF software at least in one chromatogram at signal/noise ratios s/n>5. BinBase removes low abundant, inconsistent and noisy peaks that cannot be assigned to existing bins in BinBase and that have too low spectra quality to generate a new bin in BinBase, resulting in datasets that typically report 400-500 peaks for mammalian plasma samples. Compound identifications within BinBase are managed by the administrator using spectral libraries and retention index information from the Fiehnlib libraries12 and NIST mass spectra. In a typical final BinBase report such as on mammalian plasma, about 30-40% of the reported bins are noted as identified metabolites, i.e. about 150 compounds, including database identifiers such as KEGG, PubChem and InChI keys.
BinVestigate
BinVestigate is an open-access query tool (http://binvestigate.fiehnlab.ucdavis.edu) to obtain information on known/unknown compounds present in BinBase. BinVestigate used data from trimethylsilyl-derivatized GC-MS based metabolomics with respect to the frequency, intensity and origin of such metabolites. Unknowns can be queried in two ways in BinVestigate: (a) users obtain result data from the West Coast Metabolomics Center (WCMC) or download public WCMC data from the free NIH database http://www.metabolomicsworkbench.org18; (b) alternatively, users can match EI-MS spectra obtained from own GC-MS datasets against BinBase within narrow Kovats retention index windows to gain a Bin ID for cross study analysis. BinVestigate result data are downloaded as CSV files and represented by sunburst diagrams. Some information, such as cell line genotypes and specific treatments is currently withheld to maintain confidentiality of study specifics of WCMC user data. For that reason, the WCMC uploads public data with more specific biological details to the NIH MetabolomicsWorkbench.
BinVestigate utilizes MongoDB for data storage and retrieval. The database is accessible and extendable by utilizing its REST services and the RSQL query language. To populate the MongoDB database, a Spring-based integration workflow is utilized to associate the study design information from our in-house study design database MiniX with metabolomic information. The metabolome data are contributed by the in-house data processing system BinBase that is based in PostgreSQL. Metabolite abundance data are normalized to the intensities of the sum of the internal standards (fatty acid methyl esters) in order to level the absolute differences between analyses over time. For comparing abundances in the BinVestigate sunburst diagrams, it is important to note that a mere 2-fold difference in normalized abundances between organs or species should be ignored because such values reflect average intensities across biological studies, and hence very much dependent on the conditions of such biological studies (e.g. mutants, stress conditions, and other factors that may greatly influence metabolite abundances). On the other hand, a 10-fold or 50-fold difference in relative intensities certainly qualifies for a high likelihood of different metabolite concentrations across different organs or species.
For users who query their own GC-MS mass spectra, Kovats retention indices are computed inside the integration workflow to enable access to BinVestigate for the general metabolomic community. Mass spectral similarity scores are calculated by the composite measure of the NIST algorithm. BinVestigate uses Java and Scala programming languages for data processing, and JavaScript for graphic user interface. Query results are available as JSON based documents or XLS compatible csv files. D3JS is utilized for data visualization.
In order to test the usability of BinVestigate, we tested one statistically significant unknown that was published by a European group investigating human cytomegalovirus infection24. Using similarity search with mass spectrum and retention index, the author’s unknown U180424 matched our BinBase unknown ID 8270 (Supplementary Fig. 12), demonstrating that BinVestigate is widely applicable for the metabolomics community.
False Discovery Rate (FDR) testing of BinBase
We have tested the BinBase data processing accuracy by determining false positive (FP), true positive (TP), false negative (FN) and true negative (TN) spectra annotation rates for the five unknown biomarkers highlighted in this research report. We have used the following equations:
| (1) |
| (2) |
| (3) |
| (4) |
For BB106699 (N-methyl-UMP), BinBase stored a total of 324,471 experimental mass spectra within the retention index search range. We determined a total 7,363 true positive spectra for N-methyl-UMP. For determining false positives, we chose spectra with an ion abundance ratio of m/z (352 / 315) > 0.3. We chose this ratio because UMP elutes within the retention index window (about 2 seconds later than N-methyl-UMP) and shares most fragment ions with N-methyl-UMP, except for m/z 352 that is abundant in UMP but absent from N-methyl-UMP (Supplementary Fig. 13). With this criterion, 3 false positive spectra were found, a FDR of 0.04%. The number of false negatives should be a lot higher than the number of false positives because BinBase was designed to assign ‘known peaks’ in a conservative way. Yet, very low abundant spectra as well as very complex chromatograms may lead to mass spectral deconvolution errors, leading to false negative peak reports. False negatives were defined as spectra in the retention index search range that were not annotated as N-methyl-UMP but had m/z ion ratios 169/315 between 10:1 and 1:1 and 169/299 between 10:1 and 1:1 (i.e. 1.0 to 10.0) and m/z 169 > 30% base peak intensity. With these criteria, 1,472 spectra were possible false negatives, yielding an overall sensitivity of 83.3%, specificity of 100% and predictive accuracy of 99.4%. We used similar detailed analyses for the other four BinBase spectra (Supplementary Table 3).
A close investigation of histograms and raw chromatograms for BB160842, however, yielded a co-elution of N-methyl-alanine and the isomer 2-aminobutyric acid for many sample chromatograms. For false positives we used the criterion m/z 218>2% base peak intensity, which is an ion that is a typical fragment for alpha-amino acids17. Unfortunately, N-methyl-alanine also shows low abundant m/z 218 ions, albeit much less abundant than in 2-aminobutyric acid. 2-aminobutyric acid shows also most fragments that occur in N-methyl-alanine (base peak m/z 130, m/z 100, m/z 114, m/z 147, and m/z 204). N-methyl-alanine presents low abundant diagnostic ions (m/z 144, m/z 142 and m/z 175) that are even lower abundant and in different ratios in pure 2-aminobutyric acid. Using these diagnostic ions we have validated the detection of pure N-methyl-alanine in biological samples, as well as detection of pure 2-aminobutyric acid in other samples. In most chromatograms, however, total peak intensities were too small to deconvolute the quantities of both co-eluting compounds due to the low abundance of the diagnostic ions. Hence, BB118961 should be regarded to reflect a mixture of both compounds in BinVestigate.
MS-DIAL 2.0
MS-DIAL 2.0 is designed as a universal program for MS data processing that supports any mass spectrometer, including GC-MS, GC-MS/MS, LC-MS, and LC-MS/MS. It is vendor-independent by supporting data conversion from file formats of many instrument manufactures, namely Agilent, Bruker, Leco, Sciex, Shimadzu, Thermo, and Waters. This software also supports any data acquisition method, from nominal or accurate mass analysis to data-dependent or data-independent MS/MS. It runs with a user-friendly graphical user interface on Windows system (.NET Framework 4.0 or later with at least 4GB RAM memory). MS-DIAL 2.0 is freely downloadable at the PRIMe website (http://prime.psc.riken.jp/) and as Supplementary Software 1.
The summary for processing high resolution GC-MS (GC-HR-MS) data is shown in Supplementary Fig. 2, using three primary metabolites as example – glycerol, phosphate, and leucine. Peak maxima of these metabolites co-elute within 1.02 s with 3 s peak widths. MS-DIAL 2.0 spots all m/z peaks and determines peak spot properties (Supplementary Fig. 2a) followed by constructing peak groups on the basis of local maxima of the second Gaussian filtered array of sharpness values (Supplementary Fig. 2b). The most important part is the subsequent chromatogram deconvolution to assign m/z spots, and fractions of shared m/z intensities to specific peak groups (Supplementary Fig. 2c). The deconvolution follows a least-square regression model based on unique ions, similar to the original MS-DIAL algorithm13 implemented for data independent MS/MS chromatogram deconvolution. The program substantially improved the spectral similarities of all co-eluting metabolites in the example data, greatly increasing the number of positively identified metabolites. For compound identification, a total of 15,302 GC-MS spectra and 21,770 LC-MS/MS spectra are currently available as internal mass spectral database in MS-DIAL 2.0.
Raw data handling and MS-DIAL scalability
The data stream including file formats and converters for MS-DIAL are summarized in Supplementary Fig. 4. MS-DIAL can import mzML, netCDF, and Analysis Base Framework (ABF) format, while ABF format is recommended for rapid data retrieval and for efficient data access. The ABF file converter is freely available at http://www.reifycs.com/AbfConverter/index.html. ABF file conversion and compatibility to MS-DIAL have been validated for open-access formats like mzML and netCDF, as well as vendor formats from Agilent Technologies (.D), Bruker Daltonics (.D), Leco (.netCDF), Sciex (.WIFF), Shimadzu (.LCD), Thermo Fisher Scientific (.RAW), and Waters (.RAW).
Peak detection
Profile data are centroided in MS-DIAL 2.0 before peak detection. First, data points are smoothed using a linearly weighted smoothing average as default setting. Noise is defined by ion amplitude and first and second derivatives. Peak start and end retention times are first approximated by noise levels. Then, the local minima within adjacent 5-point windows are explored to determine optimal peak start and end times by forward and back tracing. In order to avoid defining peak starts and ends too far away from the peak maxima, users can define an average peak width (APW) parameter. APW is utilized as a clamp for peak width definition within a maximal of ± 2 APW. MS-DIAL 2.0 involves background subtraction for filtering out chemical noise (Supplementary Fig. 14). After the initial peak detection program finished, the unsmoothed raw chromatogram is retrieved as control. Peaks are excluded if the ion abundance of one neighbor point from the peak top is zero, because smoothing algorithm may construct signal resembling actual peaks even for chromatographic noise. A secondary filter is used to exclude baseline noise arising from a sequence of ‘peak-like’ spike noise that may occur when too many peaks are detected in the initial peak picking algorithm. Here, the amplitude of spike noise is defined as the difference of two adjacent scan points. The current filter will exclude peaks if 4 spike noise signals are programmatically detected within a ± 5 APW region of a peak top.
MS1Dec deconvolution
MS-DIAL 2.0 deconvolution, named as MS1Dec, starts with MS1 fragment ions. The peak spotting program is first executed over the entire retention time and m/z ranges. In MS-DIAL, the detected m/z – retention time features are termed ‘peak spots’. The peak quality value is defined for each spot by comparing ideal slope values that evaluate the peak smoothness, i.e. whether the peak contains any spike noise within its peak width. There are three quality levels: high if the ideal slope value is higher than 0.999, middle if the value is between 0.9-0.999, low if it is less than 0.9. The peak sharpness value evaluates peak symmetry in combination with absolute intensity. The definitions of ideal slope- and peak sharpness values followed the previous work13.
Subsequently, all peak spots that have identical peak widths and peak top retention times are combined into single arrays. For each array, peak sharpness values are summed up and a second Gaussian derivative filter is applied in order to construct ‘peak groups’. The smoother is defined by a default sigma value of 0.5 in order to join m/z peak maxima even in case of small derivations. In practice, MS-DIAL 2.0 requires at least two scan differences in the peak tops of co-eluting metabolites to be distinguished because local maxima of the smoothed sharpness arrays are recognized as peak maxima and ignore neighboring peaks.
The main purpose of deconvolution is to estimate the peak abundance of m/z traces that are shared by two or more co-eluting metabolites. This is achieved by defining model peak m/z traces in the retention time region of each peak group. High quality m/z traces are utilized to construct model peak fitting for each m/z trace by least-square regression. Middle quality m/z traces will be used if there are no high quality traces in a focused peak group. Peak groups that only consist of low quality traces are recognized as ‘not detected’. In order to form the model peak for a peak group, the peak intensities that are above 90% of their base peaks are summed for modelling to increase the model accuracy for low ion signals. Peak maxima and peak start and end points are determined by tracing the local maximum, left local minimum, and right local minimum, respectively.
To deconvolute local model peaks Mt(n), co-eluting adjacent model peaks are considered if peak start and end points of the two (or more) model peaks are overlapping. For practical reasons, the current program considers up to four co-eluting metabolites (Mt−2(n) and Mt−1(n) for the left side of target compound; Mt+1(n) and Mt+2(n) for the right side of target compound) for the chromatogram deconvolution of targeted compound Mt(n). Therefore, the raw m/z trace C(n) will be decomposed to the model peaks as follows:
Retention time and full mass spectral similarity
Retention time and mass spectral similarities are used for compound identification and peak alignment in the data processing. In order to determine retention time (or index) similarity, a Gaussian function is utilized under the assumption that the potential retention time drifts between sets of chromatograms will follow Gaussian distribution. MS-DIAL 2.0 uses a combined value as ‘full mass spectral similarity’ with weight factor of 2:2:1 for dot product, reverse dot product and matched fragment ratio. For calculating overall similarities of chromatogram alignments, the software sums up values of retention time and mass spectral similarity.
Compound identification
Deconvoluted spectra are matched against mass spectral libraries that are imported as NIST MSP format. Library match hits are ranked against experimental data by the total retention time (or index) and mass spectral similarities across all samples that are processed in a batch. Users can define cut-off thresholds for both parameters. MS-DIAL 2.0 supports two retention indices: Kovats RI based on alkanes and Fiehn RI based on fatty acid methyl esters.
MS-DIAL aligner
The alignment algorithm for detecting peak groups across all samples of a data processing batch was optimized for GC-MS data. The aligner runs in the following procedures: (a) creating a reference table; (b) fitting each sample peak table to reference peak table; (c) filtering aligned peaks; and (d) missing value interpolation. For LC-MS/MS data, the MS-DIAL aligner focuses on MS1 precursor ions. While for GC-MS data, the MS-DIAL aligner determines the unique ion used for peak quantification, termed as ‘quant mass’. The m/z of highest ion abundance in high quality trace is defined as quant mass in the program, and the m/z from middle quality trace will be used if no high quality trace is present. A user-defined sample will serve for creating a starting reference table of all deconvoluted peak groups. Additional peak groups from further samples are inserted if the total retention and spectral similarity between the sample peak groups is lower than a user-defined cut-off compared to the existing peak groups in the reference table. This insertion routine is repeated for all peaks of all samples. The final table is utilized as the reference peak table for peak alignment. Each sample peak table is assigned into the reference peak table as the following criterion:
The coefficient a and b can be set by users. After all peaks of all samples are fitted to the reference peak table, alignment peak table including RT, quant mass and intensity is constructed with each row termed as ‘alignment spot’. The representative quant mass for each aligned spot is defined by the consideration of ion abundances- and frequencies of quant mass among samples and used for peak height and peak area determination. Average retention time (or index) and average quant mass (for accurate mass data) are calculated. A ‘fill percentage’ with respect to the positive detected sample number in a peak group is obtained. The results of compound identification by matching the reference database against experimental peak with best retention time and mass spectral similarity are stored. The corresponding spectrum for each aligned peak group is retrieved from the imported samples for each identified sample peak, or the spectrum with highest total ion intensities for unidentified peaks.
The interpolation program for missing values is executed as follows: (1) for the quantification mass of each aligned peak group, maximum and minimum retention times (or indices) are recorded with maximum and minimum peak widths; (2) for gap filling, local maxima and minima signal intensities are determined for the quant mass within the retention time (index) window of (minimum RT, maximum RT) ∩ (quant mass average – sigma mass, quant mass average + sigma mass); (3) peak height is defined by the amplitude different of local maxima and minima, while the corresponding estimated peak area is calculated on the basis of average peak width and actual peak maxima.
MS-FINDER 2.0
MS-FINDER 2.0 is launched as a universal program for structure elucidation of unknown mass spectra in LC-MS/MS (as previously reported14) and GC-MS. While vendors provide specific GC-MS/MS instruments, in practice, we have determined a high degree of in-source fragmentation in GC-MS (both hard electron ionization and soft chemical ionization) spectra, making the distinction of GC-MS and GC-MS/MS spectra unnecessary here. MS-FINDER 2.0 is compatible on Windows system (.NET Framework 4.0 or later with at least 8 GB RAM memory). It is freely downloadable at the PRIMe website (http://prime.psc.riken.jp/) and as Supplementary Software 2. MS-FINDER 2.0 employs a conventional spectral database search function based on dot product, reverse dot product and matched fragment ratio. More importantly, this program features a computational mass spectral fragmentation (in silico fragmenter search) for structure annotation. Here, the technical detail of in silico fragmenter search for full GC-MS spectra of trimethylsilylated (TMS) compounds is described. Additionally, the molecular adduct ions, often the [M]+• or [M-CH3]+• radical ions, have to be manually determined by peak alignments in MS-DIAL 2.0.
Molecular formula generator
Structure elucidation in MS-FINDER 2.0 is started by formula prediction. Full GC-MS spectra are mostly ionized compounds with odd electron (radical) ions, denoted as ‘+•’ (Supplementary Fig. 5a). This program supports eleven elements for formula generation, including carbon (C), hydrogen (H), oxygen (O), nitrogen (N), sulfur (S), phosphorus (P), fluorine (F), chlorine (Cl), bromine (Br), iodine (I), and silicon (Si). Atoms CHONSPSi are used for program evaluation presented in this paper.
Elemental formulas are computationally generated with valence rules and elemental ratio checks. Next, the number of trimethylsilyl (TMS) and methoxy (MeOX) moieties are simulated as follows. Here we use formula C22H47N2O9PSi4 as an example (Supplementary Fig. 5b). MS-FINDER 2.0 recognizes the origin of all Si elements as TMS moieties. Therefore, C22H47N2O9PSi4 is converted to C10H15N2O9P with each C3H8Si subtracted as one TMS from the derivatized formula. The number of MeOX moieties is simulated by the number of N atoms. All simulated candidates are used as the results of formula generation, and hence, C10H15N2O9P (0 MeOX), C9H12NO9P (1 MeOX) and C8H9O9P (2 MeOX) are obtained from the original C10H15N2O9P where CH3N is deleted per one N atom.
Molecular formula ranking
Formulas are ranked based on the sum of five diagnostic scores, specifically mass error, isotopic ratio error, formula assignment to fragment ions, neutral loss matching, and presence of the formula in the combined internal metabolome database. Our ‘existing formula database’ is an integrated database to retrieve biologically reported formulas in 15 repositories (total 90,227 unique formulas) including BMDB, ChEBI, DrugBank, ECMDB, FooDB, HMDB, KNApSAcK, PlantCyc, PubChem (Biomolecules), SMPDB, T3DB, UNPD, YMDB, STOFF, and MINE. The virtual enzyme expansion database MINE is evaluated separately from other repositories. If a formula candidate is present in one of the 14 databases (except for MINE), the evaluation score is 0.5, otherwise 0. To this value, the number of databases that include this formula, standardized by 0.5, is added. After this, 0.2 is added if the formula is also present in MINE database. The formula database is stored in ‘.EFD’ file of MS-FINDER folder as ASCII file format.
Searching of structure candidates and in silico derivatization
Currently, MS-FINDER 2.0 has three options to retrieve structural isomers: the internal combined metabolome database of 14 repositories with 224,622 unique structures, the MINE database with 643,307 unique structures, and the PubChem REST service for approximately 70 million structures (Supplementary Fig. 5c). Each repository in the combined metabolome database can also be selected by itself. The combined metabolome database and MINE database are stored in ‘.ESD’ and ‘.MSD’ files of MS-FINDER folder as ASCII file format.
After the structural data for a given formula is retrieved, the structure is computationally derivatized on the basis of the simulated TMS and MeOX numbers using the following procedures: (1) the acidic protons attached with heteroatoms ONSP are recognized as the reactive protons amenable to TMS derivatization; (2) the carbonyl groups as ketones or aldehydes are recognized as reactive ‘C=O’ for MeOX derivatization unless further heteroatoms ONSP are attached like carboxylic acids; (3) candidate structures are excluded if the number of acidic protons and carbonyl groups is less than the number of simulated TMS and MeOX; (4) TMS derivatization is prioritized by OH > COOH > NH2 > SH > NHR; (5) MeOX derivatization is prioritized by R1(C=O)R2 > R(C=O)H; (6) identical functional groups are derivatized in the same priority, i.e. the order of derivatization is determined by the order of atomic numbering.
Ranking of structure candidates
Hydrogen rearrangement (HR) rules (rules P1-P5 and N1-N4 for positive and negative ion mode, respectively) have been established to interpret mass spectra of LC-MS/MS with collision induced dissociation (CID) as previously reported14. Structure candidates are ranked by the integrated score of HR rules, fragment linkage and bond dissociation energy. Now, the updated rule-based mass spectral fragmentation library also includes structure elucidation for GC-MS spectra. A total of 533 fragment ions are rationalized by m/z, formula and SMILES code, and stored in ‘.EIF’ file of MS-FINDER folder as ASCII file format. MS-FINDER 2.0 utilizes this rule-based fragmentation library for substructure assignments because GC-MS spectra produce intense fragmentation schemes including electron shifts, hydrogen rearrangements, homolytic or heterolytic bond cleavages, and intramolecular rearrangements, rather than stabilizing fragments by aromatization. This program excludes aromatic fragment ions unless the aromatic substructures are detected in the original molecules. The likelihood of fragment ion with assigned substructure is evaluated by a Gaussian function based on experimental mass errors. Advanced likelihood by molecular fingerprints in combination with similarity calculation methods, such as Jaccard, will be used in the next version.
For fragment ion without substructure assignment by the rule-based library, in silico spectral annotation is performed by simulating an α-cleavage process for up to two bonds with a consideration of ± 2 hydrogen rearrangements. To specify the appropriate fragments within mass tolerance, computational ions are assigned to observed ions by the following priorities: (1) fragments from the first cleavage that differ up to two hydrogens from a neutralized substructure; (2) fragments from the second cleavage with assigned precursors in higher m/z area; (3) fragments of minimum mass errors. This scoring system is identical to that of LC-CID-MS/MS spectra except the penalty of HR rules: at current, the HR rules are always considered as ‘TRUE’ for GC-MS spectra.
Performance validation of MS-DIAL 2.0
The scalability and functionality of MS-DIAL 2.0 for GC-MS data processing were validated by six raw data files from five major MS vendors (Supplementary Fig. 3 and Supplementary Data 1). All raw data files (except Bruker and Thermo) are available at the PRIMe website (http://prime.psc.riken.jp/?action=drop_index) with the following data sources:
LECO GC-TOF(MS): The biological sample was Euglena gracilis; analysis procedures were performed according to the ‘LECO GC-TOF MS’ protocol of the previous report25.
Agilent GC-Q(MS): The biological sample was NIST standard human plasma; analysis procedures were performed according to the ‘Agilent GC-Quadrupole MS’ protocol of the previous report25.
Agilent GC-QTOF(MS): The biological sample was Chlamydomonas reinhardtii; analysis procedures were described in ‘Reagents and Sample Preparation’ section of this paper.
Shimadzu GC-Q(MS): The raw data was distributed from previously reported data26.
Bruker GC-Q(MS): The raw data was kindly distributed from Bruker Daltonics.
Thermo GC-QE(MS): The raw data was kindly distributed from Thermo Fisher Scientific.
The MS-DIAL 2.0 data processing procedures were followed by the software tutorial (http://prime.psc.riken.jp/Metabolomics_Software/MS-DIAL/). The analyzing parameters and MS libraries can be downloaded at http://prime.psc.riken.jp/?action=drop_index. The actual processing time for nominal and accurate mass GC-MS data were 20–30 sec and 1-2 min, respectively. The identification results were manually confirmed by the MS-DIAL 2.0 graphical user interface.
Performance validation of MS-FINDER 2.0
The performance of MS-FINDER 2.0 for structure elucidation was tested by the accurate mass GC-EI-MS spectra of total 441 trimethylsilylated compounds. The sample preparation and analytical conditions were described below. For each compound, the molecular mass as well as TMS and MeOX number of the derivatized form were determined by manual investigation, while the formula, SMILES and InChIKey of the non-derivatized form were generated by ChemAxon MolConverter and Calculator (www.chemaxon.com).
The mass tolerance, relative abundance cut off, and isotopic ratio tolerance were set to 0.01 Da, 1%, and 20%, respectively. The filtering of ‘LEWIS and SENIOR check’ and ‘common range for element ratio check’ was activated. The targeted atoms were set to C, H, O, N, S, and P as the option of ‘TMS-MeOX derivatized compound’. Tree depth was set to 2, and the ‘use of fragmentation library for electron ionization’ was applied. For batch process, the top 100 formula candidates were transferred to the structure searching procedure. The internal metabolome database including BMDB, ChEBI, DrugBank, ECMDB, FooDB, HMDB, KNApSAcK, PlantCyc, PubChem (Biomolecules), SMPDB, T3DB, UNPD, YMDB, and STOFF were selected for structure searching. PubChem ‘Biomolecules’ were retrieved from the ca. 70 million compounds in PubChem by restricting to ‘Biomolecular and interaction pathway’, then restricting to ‘Biosystems and pathways’. Currently, 12,400 compounds are retrievable from PubChem this way.
MS-FINDER 2.0 was tested by three structure resource sets. The first set is the internal metabolome database with 14 repositories as mentioned above, denoted as ‘FINDMetDB’. In order to spread search space, MINE and PubChem databases were also included for the second and the third set, which were denoted as ‘FINDMetDB+MINE’ and ‘FINDMetDB+PubChem’. Therefore, the number of total unique structures for the software accuracy test were 13,869, 92,628, and 280,245 in ‘FINDMetDB’, ‘FINDMetDB+MINE’, and ‘FINDMetDB+PubChem’, respectively. The performance test results of MS-FINDER 2.0 and of ‘random sampling method’ were shown in Supplementary Fig. 6. The logP and natural product likeness values were calculated by ChemAxon Calculator (www.chemaxon.com) and Natural Product Likeness Calculator (https://sourceforge.net/projects/np-likeness/). With a mass tolerance of 10 mDa for spectral annotation of CHNOSP elements, the probability of finding the correct structure for the top hit, or among the top 3, top 5, top 10 hits were 49.2%, 72.1%, 82.1%, and 91.8%, respectively.
Software comparison for MS-DIAL 2.0 and MS-FINDER 2.0
We compared the analyzing results obtained by MS-DIAL 2.0 (version 2.52) and MS-FINDER 2.0 (version 2.10) against other alternative programs, respectively. For the performance of GC-MS chromatogram deconvolution, MS-DIAL 2.0, AMDIS27, AnalyzerPro, and ChromaTOF were tested using identical raw data (http://prime.psc.riken.jp/?action=drop_index). Deconvoluted mass spectra were exported from the data processing software and imported to NIST MS Search program (http://chemdata.nist.gov/mass-spc/ms-search/) to obtain match scores for compound annotation. Based on the results of nine primary metabolites from four coeluting peak groups, MS-DIAL 2.0 outperformed AMDIS, AnalyzerPro and ChromaTOF for most individual spectral similarity matches and for the average match scores (Supplementary Table 2).
For the functionality of in silico GC-MS mass spectral annotation, MS-FINDER 2.0, CFM-ID28, MetFrag29, Molecular Structure Correlator (MSC), and Mass Frontier were evaluated using the mass spectra (http://prime.psc.riken.jp/?action=drop_index) of our five BinBase unknowns highlighted in this paper with same structure candidate lists that were downloaded from PubChem by formula query, computational derivatized and generated in ChemAxon Instant JChem (www.chemaxon.com) (Supplementary Table 1). For calculating, scoring and ranking 25–59 different isomers, MS-FINDER 2.0 required 2-12 s processing time, similar to MetFrag and MSC programs, while CFM-ID and Mass Frontier needed significantly more time (5-108 min). More importantly, MS-FINDER 2.0 was the only software that could confidently identify the five unknown compounds presented here as top hit; other programs had an average ranking of 3.8 (CFM-ID) to 9.6 (Mass Frontier).
The computer condition for data processing was an Intel (R) Core (TM) i7 with 4 GHz CPU and 8 GB RAM as Windows 7 System. The settings of MS-DIAL 2.0 and MS-FINDER 2.0 were default values as mentioned above. The parameters of other programs are listed as follows:
AMDIS: The software application was downloaded from http://chemdata.nist.gov/. Match Factor Penalty Level was ‘Very Strong’. Scan Direction was ‘Low to High’. Adjacent Peak Subtraction was ‘None’. Resolution, Sensitivity, and Shape Requirement were all ‘Medium’.
AnalyzerPro: The software application was purchased from Spectral Works. Data processing settings were vendor-suggested default parameters.
ChromaTOF: The software application was purchased from LECO Corporation. Data processing settings were vendor-suggested default parameters.
CFM-ID: The web application was performed in http://cfmid.wishartlab.com/. Spectra Type was ‘EI’. Number of Results, Mass Tolerance, and Scoring Function were 20, 0.01 Da, and Dot Product, respectively.
MetFrag: The web application was performed in http://msbi.ipb-halle.de/MetFrag/. Process Mode was ‘[M]’. MZABS and MZPPM were 0.01 Da and 20, respectively.
MSC: The software application was purchased from Agilent Technologies. Data processing settings were vendor-suggested default parameters.
Mass Frontier: The software application was purchased from Thermo Fisher Scientific. Data processing settings were vendor-suggested default parameters.
Reagents and sample preparation
The following reagents and authentic standard compounds were obtained from (suppliers): water, isopropanol, and acetonitrile (Fisher Optima); pyridine (Acros Organics); C8 – C30 fatty acid methyl esters [FAMEs], methoxyamine hydrochloride [MeOX], ethoxyamine hydrochloride [EtOX], N-methyl-N-(trimethylsilyl)-trifluoroacetamide [MSTFA], N-methyl-N-(trimethyl-d9-silyl)-trifluoroacetamide [MSTFA-d9], ammonium formate, formic acid, and N-methyl-L-alanine (Sigma-Aldrich); 2′-O-methyluridine-5′-triphosphate, 3′-O-methyluridine-5′-triphosphate, 5-methyluridine-5′-triphosphate (TriLink BioTechnologies); 4-hydroxypropofol-1-O-β-D-glucuronide, and 4-hydroxypropofol-4-O-β-D-glucuronide (Toronto Research Chemicals).
All metabolites extraction procedures are kept on ice, the quantities for sample aliquots were 25 μL for blood plasma, 5×106 for cells, 5 mg for tissues, 2 mL for algae cultures. Metabolites were extracted with 1,000 μL degassed acetonitrile:isopropanol:water (3:3:2, v/v/v), and then homogenized, centrifuged, decanted, and evaporated. Extracts were cleaned by 500 μL degassed acetonitrile:water (1:1, v/v) to remove triglycerides and membrane lipids, and evaporated again. For GC-MS analysis, internal standards C8 – C30 FAMEs were added to determine the retention index. The dried samples were derivatized with 10 μL MeOX (or EtOX) in pyridine and subsequently by 90 μL MSTFA (or MSTFA-d9) for trimethylsilylation of acidic protons. For LC-MS analysis, the extracted samples were resuspended in 50 μL acetonitrile:water (4:1, v/v) and submitted to instrument.
Analytical conditions
For gas chromatography – mass spectrometry analysis, the instrumentation used an Agilent 7890A GC system (Agilent Technologies, Santa Clara, CA, USA) and an Agilent 7200 accurate mass Q-TOF mass spectrometer (Agilent Technologies, Santa Clara, CA, USA), with transfer line temperature maintained at 290 °C. Chromatography was performed on a Rxi-5Sil MS column (30m×0.25mm, 0.25μm; Restek Corporation, Bellefonte, PA, USA) with Helium (99.999%; Airgas, Radnor, PA, USA) at a constant flow of 1 mL/min. The GC temperature program was set as follows: initial temperature of 60 °C with a hold time of 1 min, a temperature ramp of 10 °C/min to 325 °C, and a final hold time of 9.5 min at 325 °C. Injection volume was 1 μL in splitless mode at 250 °C. Mass spectra were acquired from m/z 50 to m/z 800 at 5 Hz scan rate and 750 V detector voltage in both electron ionization (EI) mode and chemical ionization (CI) mode. Other data acquisition parameters were EI ion source temperature 230 °C; EI electron energy, 70 eV; CI ion source temperature 300 °C; CI electron energy, 135 eV; CI gas flow rate, 20%; CI gas, Methane (99.999%; Airgas, Radnor, PA, USA).
For liquid chromatography – mass spectrometry analysis, the initial separation was achieved on an Agilent 1290 infinity LC system (Agilent Technologies, Santa Clara, CA, USA) with an Acquity UPLC BEH Amide column (150mm×2.1mm, 1.7μm; Waters Corporation, Milford, MA, USA). The mobile phases consisted of (A) 10 mM ammonium formate and 0.125% formic acid in water and (B) acetonitrile:water (95:5, v/v) with 10 mM ammonium formate and 0.125% formic acid. The gradient was 0 min, 100% B; 2 min, 100% B; 7.7 min, 70% B; 9.5 min, 40% B; 10.3 min, 30% B; 12.8 min, 100% B; 16.8 min, 100% B. A sample volume of 2 μL and 5 μL was used for the injection in ESI (+) and ESI (-) respectively, with the flow rate of 0.4 mL/min. The autosampler temperature was 4 °C, the column temperature was 45 °C. The mass spectrometry was equipped with an Agilent 6530 accurate mass Q-TOF system (Agilent Technologies, Santa Clara, CA, USA). MS and MS/MS data were collected in 4 Hz scan rate and m/z 50–800 mass range. Collision energy was applied at 20 eV. Mass calibration was maintained at constant infusion of reference ions at m/z 121.0509, m/z 922.0098 for positive mode and m/z 119.0363, m/z 966.0007 for negative mode.
Synthetic procedures
Glassware was oven dried at 100 °C overnight prior to the reaction. All reagents were purchased from commercial sources (Sigma-Aldrich or Fisher Scientific) and were used without further purification unless noted otherwise. Reactions were carried out under an atmosphere of dry argon. Liquid reagents were introduced by disposable syringes. Thin layer chromatography (TLC) was performed with EMD silica gel 60, F254 precoated TLC plates. Short and long wave visualization were performed with a Mineralight multiband ultraviolet lamp at 254 and 365 nm, respectively. Flash column chromatography was performed with Merck silica gel (Sorbent technologies, 60–200 mesh). Purification of nucleotide mono-phosphate was performed on a column of Sephadex DEAE-A25. The resin was swollen in 1 M NaHCO3 at 4°C for 1 day and washed with deionized water before use, unless noted otherwise. The fractions containing nucleotide mono-phosphate were identified by Beckman DU-7400 UV-Vis scanning spectrophotometer and Applied Biosystems QTrap Mass spectrometry.
N3-methyl uridine synthesis
N3-methyl uridine was synthesized as previously described30,31 with minor modifications. In brief, uridine (1.504 g, 6.16 mmol) and K2CO3 (1.704 g, 12.33 mmol) were added to a mixture of DMF (7.5 mL) and acetone (7.5 mL). Methyl iodide (383 μL, 6.16 mmol) was added dropwise to the suspension. The system was then refluxed for 5 hours. The solvent was removed in vacuo. The residue was purified by chromatography on a flash silica column. Eluting with (5-10% (v/v) MeOH in CH2Cl2). Fractions containing the product were dried in vacuo. Product was recrystallized in MeOH, which yield N3-methyl uridine as white crystal.
N3-methyl uridine 5′-mono-phosphate synthesis
N3-methyl uridine 5′-mono-phosphate was synthesized as previously described32 with minor modifications. N3-methyl uridine and proton sponge were dried overnight in a vacuum oven. Nucleoside (180 mg, 0.69 mmol) was dissolved in freshly distilled trimethyl phosphate (8 mL) by heating the solution, and the solution was cooled to −15 °C. Dry proton sponge (444 mg, 2.07 mmol) was then added to the solution and the solution was stirred at −15 °C for 20 minutes. Distilled phosphorus oxychoride (64 μL, 0.69 mmol) was added dropwise under argon with a micro syringe. The reaction solution was then stirred at −15 °C. After 2 hours at −15 °C, a solution of 1 M triethylammonium bicarbonate (30 mL, pH 8) was added. The clear solution was stirred at room temperature for 45 minutes and freeze dried. The crude mixture was dissolved in water and purified using a Sephadex DEAE-A25 column with a linear gradient of 0.01-0.10 M triethylammonium bicarbonate buffer. Fractions containing N3-methyl uridine 5′-mono-phosphate were identified by UV spectrophotometer and mass spectrometry. Combined fractions were evaporated under reduced pressure yielding N3-methyl uridine 5′-mono-phosphate as white solid.
Data availability
Any Supplementary Information and Source Data files are available in the online version of the paper.
Supplementary Material
Acknowledgments
This work was supported by the U.S. National Science Foundation (NSF) – Japan Science and Technology Agency (JST) Strategic International Collaborative Research Program (SICORP) for Japan – United States metabolomics. We appreciate funding by the U.S. National Science Foundation projects MCB 113944 and MCB 1611846, by the U.S. National Institutes of Health U24 DK097154, and as well as by the AMED-Core Research for Evolutionary Science and Technology (AMED-CREST) and JSPS KAKENHI Grant Numbers 15K01812, 15H05897, 15H05898, 17H03621.
Footnotes
Author Contributions
Z.L., H.T., M.A., and O.F. designed the research. G.W. and S.M. developed the BinVestigate program. H.T. developed the MS-DIAL 2.0 and MS-FINDER 2.0 program. Z.L. performed the sample preparation, instrumental analysis, and data processing for unknown identification. M.S. contributed biological and LC-MS studies for the identification of N-methyl-UMP. Z.L. and H.T. performed performance validation, and program comparison for MS-DIAL 2.0 and MS-FINDER 2.0. Y.Z. and P.B. synthesized the N-methyl-UMP standard compound. A.O. improved the raw data file reader in ABF conversion. J.M., K.T., O.F. and W.W. contributed to the identification of Lyso-MGMP and propofol derivatives. Z.L., H.T., M.A, and O.F. thoroughly discussed this project and wrote the manuscript.
Competing Financial Interests
The authors declare competing financial interests. Atsushi Ogiwara is a developer in Reifycs Inc., which provides the ABF converter of mass spectral data for free at http://www.reifycs.com/AbfConverter/.
References
- 1.Kim S, et al. Nucleic Acids Res. 2015;44:1202–1213. [Google Scholar]
- 2.Silva RR, Dorrestein PC, Quinn RA. Proc Natl Acad Sci. 2015;112:12549–12550. doi: 10.1073/pnas.1516878112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hanson AD, Pribat A, Waller JC, de Crécy-Lagard V. Biochem J. 2010;425:1–11. doi: 10.1042/BJ20091328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khersonsky O, Tawfik DS. Annu Rev Biochem. 2010;79:471–505. doi: 10.1146/annurev-biochem-030409-143718. [DOI] [PubMed] [Google Scholar]
- 5.Linster CL, Van Schaftingen E, Hanson AD. Nat Chem Biol. 2013;9:72–80. doi: 10.1038/nchembio.1141. [DOI] [PubMed] [Google Scholar]
- 6.Rappaport SM, Barupal DK, Wishart D, Vineis P, Scalbert A. Environ Health Perspect. 2014;122:769–774. doi: 10.1289/ehp.1308015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wikoff WR, et al. Proc Natl Acad Sci. 2009;106:3698–3703. doi: 10.1073/pnas.0812874106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kumari S, Stevens D, Kind T, Denkert C, Fiehn O. Anal Chem. 2011;83:5895–5902. doi: 10.1021/ac2006137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Showalter MR, Cajka T, Fiehn O. Curr Opin Chem Biol. 2017;36:70–76. doi: 10.1016/j.cbpa.2017.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Patti GJ, et al. Metabolomics. 2014;10:737–743. doi: 10.1007/s11306-013-0608-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fiehn O, Wohlgemuth G, Scholz M. Lect Notes Bioinformatics. 2005;3615:224–239. [Google Scholar]
- 12.Kind T, et al. Anal Chem. 2009;81:10038–10048. doi: 10.1021/ac9019522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsugawa H, et al. Nat Methods. 2015;12:523–526. doi: 10.1038/nmeth.3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsugawa H, et al. Anal Chem. 2016;88:7946–7958. doi: 10.1021/acs.analchem.6b00770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jeffryes JG, et al. J Cheminform. 2015;7:1. doi: 10.1186/s13321-015-0087-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fiehn O. Trends Anal Chem. 2008;27:261–269. doi: 10.1016/j.trac.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lai Z, Fiehn O. Mass Spectrom Rev. 2016;9999:1–13. doi: 10.1002/mas.21518. [DOI] [PubMed] [Google Scholar]
- 18.Sud M, et al. Nucleic Acids Res. 2015;44:463–470. [Google Scholar]
- 19.Haug K, et al. Nucleic Acids Res. 2013;41:781–786. [Google Scholar]
- 20.Sperber H, et al. Nat Cell Biol. 2015;17:1523–1535. doi: 10.1038/ncb3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Styczynski MP, et al. Anal Chem. 2007;79:966–973. doi: 10.1021/ac0614846. [DOI] [PubMed] [Google Scholar]
- 22.Scholz M, Fiehn O. in Pacific Symposium on Biocomputing Pacific Symposium on Biocomputing. 2006:169–180. [PubMed] [Google Scholar]
- 23.Fiehn O, et al. Plant J. 2008;53:691–704. doi: 10.1111/j.1365-313X.2007.03387.x. [DOI] [PubMed] [Google Scholar]
- 24.Fattuoni C, et al. Clin Chim Acta. 2016;460:23–32. doi: 10.1016/j.cca.2016.06.005. [DOI] [PubMed] [Google Scholar]
- 25.Fiehn O. Curr Protoc Mol Biol. 2016;114:30.34. 31–30.34. 32. doi: 10.1002/0471142727.mb3004s114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsugawa H, et al. J Biosci Bioeng. 2011;112:292–298. doi: 10.1016/j.jbiosc.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 27.Stein SE. J Am Soc Mass Spectrom. 1999;10:770–781. [Google Scholar]
- 28.Allen F, Pon A, Greiner R, Wishart D. Anal Chem. 2016;88:7689–7697. doi: 10.1021/acs.analchem.6b01622. [DOI] [PubMed] [Google Scholar]
- 29.Ruttkies C, Strehmel N, Scheel D, Neumann S. Rapid Commun Mass Spectrom. 2015;29:1521–1529. doi: 10.1002/rcm.7244. [DOI] [PubMed] [Google Scholar]
- 30.Flosadóttir HD, Jonsson H, Sigurdsson ST, Ingolfsson O. Phys Chem Chem Phys. 2011;13:15283–15290. doi: 10.1039/c1cp21298b. [DOI] [PubMed] [Google Scholar]
- 31.Yamamoto I, Kimura T, Tateoka Y, Watanabe K, Ho IK. J Med Chem. 1987;30:2227–2231. doi: 10.1021/jm00395a009. [DOI] [PubMed] [Google Scholar]
- 32.El-Tayeb A, Qi A, Müller CE. J Med Chem. 2006;49:7076–7087. doi: 10.1021/jm060848j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Any Supplementary Information and Source Data files are available in the online version of the paper.