

Abstract
Novel biocompatible nanoparticles based on a calcium analogue of Prussian blue were designed and synthesized to take advantage of their ability to penetrate the cell membrane in Staphylococcus aureus and to undergo selective ion exchange with intracellular iron to disrupt iron metabolism in such pathegenic bacteria for antibacterial applications.
Keywords: iron metabolism, Prussian blue analogue, biocompatible nanoparticles, iron depletion, antimicrobial
Graphical Abstract:
An ironclad guarantee for defeating bacterial infections! KCa(H2O)2[FeIII(CN)6]·H2O nanoparticles can penetrate the bacterial cell membrane and sequester intracellular iron by ion exchange to form insoluble Prussian blue, and thus inhibiting bacterial growth.
Iron is an essential element for nearly all forms of life. In the battle between the invading pathogenic microorganisms and the host vertebrates for this strategic micronutrient, evolution seems to have granted vertebrates the upper hand over pathogenic bacteria. For instance, nature has developed sophisticated machinery in vertebrates to sequester, transport and store iron with an extremely high efficiency to tightly regulate the amount of free iron at both the systemic and cellular level, which significantly decreases the chance for the invading pathogenic bacteria to multiply and cause disease.[1-6] In contrast, pathogenic microorganisms totally depend on the iron pool of the host vertebrate for iron supply and acquire iron by means of theft and robbery.[7,8] At the time of infection, one of the host defense mechanisms is the withholding of iron from invading pathogens.[9,10] This process, often referred to as nutritional immunity, mobilizes a score of iron transport and storage proteins to further restrict the availability of free iron.[11,12] In response, pathogenic bacteria, after encountering iron starvation upon entering the host, will deploy a variety of equally sophisticated mechanisms with multiple redundant uptake routes that use siderophores and heme-uptake systems to evade nutritional immunity in order to gain access to the host iron during infection, resulting in a constant arms race for iron between the host tissues and the invading pathogenic microorganisms.[13-17] It seems logical that external interventions to disrupt iron acquisition and metabolism in pathogenic bacterial cells will shift the balance of power in the battle, and may constitute a novel approach to treating bacterial infections.[18-21] However, use of synthetic iron chelators for intracellular iron depletion to exploit this vulnerability has thus far met with limited success as the naturally occurring siderophores form iron chelates with higher thermodynamic stability than the synthetic iron-chelating ligands,[22-26] which makes it difficult for the latter to compete with siderophores and to displace iron from them. Furthermore, the typical synthetic iron chelators lack the ability to penetrate the bacterial cell membrane, which renders iron chelation therapy feasible only in lowering the systemic iron level in host vertebrates, but not in targeting the intracellular iron in bacteria for depletion.[27-29]
In this work, we report on the design, synthesis, characterization and investigation of antimicrobial properties of the novel nanoparticles (NPs) based on a calcium analogue of Prussian blue KCa[FeIII(CN)6] (hereafter abbreviated as CaPB).[30] We demonstrate that such NPs can be readily internalized by the bacterial cell and selectively deplete the intracellular iron by an ion-exchange reaction, thus inhibiting bacterial growth. To the best of our knowledge, the CaPB NPs are the first example of any known NP system to exhibit such striking antibacterial activity by disrupting iron acquisition and metabolism in bacteria.
The simplistic approach to selectively depleting iron from the bacterial cell would be to react K3[FeIII(CN)6] directly with intracellular Fe(II) in bacteria to form the insoluble Prussian blue FeIII4[FeII(CN)6]3 in which both Fe(II) and Fe(III) are tightly bound by the CN- ligands and completely locked up in the 3D coordination polymer network (Ksp = 3.0 × 10−41 for dissociation into Fe3+ and [FeII(CN)6]4– ions in aqueous solution), which diminishes the bioavailability of iron inside the cell. However, due to its high negative charge and a large hydration shell in solution, the [FeIII(CN)6]3– anion lacks the ability to penetrate the bacterial cell membrane on its own because the surface of cell membrane happens to carry a negative charge, as demonstrated by our comparison studies using the K3[FeIII(CN)6] salt to treat bacterial cells (vide infra). We then endeavored to assemble an electrically neutral 3D framework between this anion and a judiciously selected cation in the hope that the nanoparticles of such network compound can undergo rapid ion-exchange reaction with intracellular Fe(II) after entering bacterial cells. The metal analysis of this product using the atomic absorption spectrometric (AAS) method showed that the molar ratio of K: Ca: Fe is close to unity, confirming that the composition of the nanoparticle core has the expected formula KCa[FeIII(CN)6]. The transmission electron microscopic (TEM) imaging studies revealed that the PVP-coated KCa[FeIII(CN)6] nanoparticles have near spherical shape with an average diameter of 5.7 ± 0.2 nm as determined by counting and averaging the size of 64 NPs in the TEM picture frame (Figure 1). Furthermore, the energy dispersive X-ray spectroscopic (EDX) measurements showed the characteristic signals of Ca, Fe and K from several individual NPs randomly selected from the TEM grid (Supporting information, Figure S1).
Figure 1.

TEM image of the PVP-coated CaPB NPs (left) and a histogram of the size distribution (right).
The Fourier transform infrared (FTIR) spectra of the PVP-coated CaPB NPs exhibited a characteristic C≡N stretching vibration at 2094 cm−1, a peak also observed in the bulk sample, in addition to the other characteristic stretching and bending vibrations attributable to PVP (Supporting information, Figure S2). To investigate the structure of this novel coordination polymer, we made numerous attempts to grow single crystals using the slow evaporation method under various conditions. For example, to decrease the nucleation rate and to prevent the formation of a precipitate, we attempted to crystallize the product in the acidic conditions with pH ≈ 1, as this approach had been successfully used to crystallize many Prussian blue analogue compounds including the prototypical Prussian blue itself.[31] Thus far, all of the attempted experiments have yielded powdered product with poor crystallinity or a precipitate of Prussian blue. However, when the [CoIII(CN)6]3– anion was used as the surrogate for [FeIII(CN)6]3–, we obtained pale-yellow single crystals of (H3O)Ca(H2O)2[CoIII(CN)6] (hereafter abbreviated as CaCoPB). The X-ray single crystal analysis showed that CaCoPB crystallizes in the orthorhombic crystal system (space group Pnma) with a = 12.940(3) Å, b = 13.697(3) Å, c = 7.3605(2) Å and V = 1304.6(5) Å3 (CCDC 1530204). The details of structure refinement including the crystallographic data and selected interatomic distances and bond angles are given in Tables S1-S3 (Supporting Information). The structure can be best viewed as consisting of Co3+ octahedra and Ca2+ biface-capped by six C atoms from trigonal prisms linked to form a 3D framework by the CN- groups. The Co3+ ion is coordinated by the CN- groups while the Ca2+ ion is coordinated by six N atoms and further capped by two O atoms of water molecules (Figure 2). Previously, we reported on the synthesis and MRI contrast properties of KGd(H2O)2[FeII(CN)6]·H2O, an isostructural compound with the current CaCoPB phase.[32] In KGd(H2O)2[FeII(CN)6]·H2O, The K+ ion and a water molecule are located in the framework cavity, showing site-occupancy disorder on the same crystallographic position. In (H3O)Ca(H2O)2[CoIII(CN)6], this zeolitic water cavity is occupied by the hydronium H3O+ ion. It is reasonable to assume that CaPB forms the same type of coordination polymer with a 3D network structure in which the Ca2+ ion is found in a similar biface-capped trigonal prism environment. The presence of calcium-coordinated water was readily confirmed by the TGA measurements of the bulk CaPB sample (Supporting information, Figure S3), which clearly showed the presence of two coordinative water molecules and a zeolitic water molecule from the structural cavity, and hence indicating that KCa(H2O)2[FeIII(CN)6]·H2O is a more appropriate formula for the NP core.
Figure 2.

Single-crystal X-ray structure of (H3O)Ca(H2O)2[CoIII(CN)6], showing the 3D coordination polymer framework and the coordination environment of the Ca2+ ion. The H3O+ cations are omitted for clarity.
We studied the selectivity of Fe2+ depletion by CaPB NPs in the presence of other biologically relevant divalent metal ions including Cu2+, Mn2+, Zn2+, Mg2+ and Ca2+ each at the concentration level of 100 ppm in the same aqueous solution to compete with Fe2+ for ion exchange with the NPs over a time period of 24 hours. The resulting selectivity for each ion was normalized against the removal of Fe2+ ions being set as 100 %. The results were expressed as the percent removal for each ion. As depicted in Figure 3a, CaPB NPs appeared to be the most selective toward the Fe2+ ion in the present of Cu2+, Mn2+ and Zn2+, Mg2+ and Zn2+ ions. The observed selectivity is more or less consistent with that predicted by the Irving-Williams series, i.e. Mn(II) < Co(II) < Zn(II) > Cu(II), except for Fe(II) that forms the exceptionally stable Prussian blue from this ion-exchange reaction.
Figure 3.

a) Selectivity of metal-ion removal. b) Kinetics of ion exchange between the Fe2+ ion and CaPB NPs in aqueous solution.
The kinetics of Fe2+ removal was followed by measuring the decrease of Fe2+ ion concentration in aqueous solution where the dialysis bag containing the PVP-coated CaPB NPs was immersed. The simultaneous increase of Ca2+ ion concentration in the same solution was also determined quantitatively by AAS to verify the kinetics (Figure 3b). The kinetics data can be fitted to two separate rate laws, i.e., a pseudo-first-order reaction up to the time point of 100 min with a rate constant of k1 = 2.1×10−4 s−1 and a half-life of T1/2 = 55 min, and a second-order reaction with a rate constant of k2 = 9.2×10−2 M−1 s−1 (Supporting information, Table S4 and Figures S4 - S5). Interestingly, such ion exchange can be visually followed by the color change of the solution in the dialysis bag that turned from pale yellow to blue in accordance one the same time scale as determined by the kinetic measurements, indicating an ion-exchange reaction between CaPB NPs and Fe2+ ions to give Prussian blue (Supporting information, Figure S6).
To investigate cellular uptake, intracellular iron depletion, and antimicrobial properties of the PVP-coated CaPB NPs, we used the opportunistic pathogen Staphylococcus aureus (S. aureus) as an in vitro bacterial cellular model. S. aureus is a Gram-positive coccoid bacterium that causes a spectrum of ailments ranging from wound infections to more severe diseases such as respiratory infections and food poisoning. In these studies, colony-forming unit (CFU) values of bacterial cells treated with CaPB NPs were first determined, followed by cellular lysis performed by the use of concentrated nitric acid in order to quantitatively analyze the metal concentrations. Both calcium and iron in cellular lysates were then measured by AAS to monitor the cellular uptake of CaPB NPs. The calcium and iron concentrations were expressed as concentration per 108 cells in microgram. As shown in Figure 4a, there was a significant increase of both the calcium (i.e. ca. 5 times higher than the control) and iron (i.e. ca. 4 times higher than the control) concentration. It should be noted that the results of the total iron quantification in this study cannot distinguish the existing intracellular iron that was immobilized by CaPB NPs via ion exchange or the inorganic iron from [FeIII(CN)6]3– that was introduced into the cell by cellular uptake of the NPs. Nonetheless, the increase of both calcium and iron in cellular lysates provided sufficiently strong evidence to confirm the internalization of CaPB NPs in S. aureus cells.
Figure 4.

Effect of CaPB NPs on S. aureus cells. a) Results of metal concentration analysis from the S. aureus cellular lysates after 24 hours of incubation. *Mean value significantly different from that for the control group. P < 0.05, n = 3. b) Inhibiting curves of S. aureus cells incubated with various amounts of CaPB NPs (red), CaCl2 (black) and K3[Fe(CN)6] (blue). Experiments were performed in TSB medium at 37 °C for 24 hours. *Mean value significantly different from that for the untreated CaPB NPs groups. P < 0.05, n = 3. c) Results of time-dependent CaPB NPs growth inhibition of S. aureus with different concentrations of CaPB NPs. Experiments were performed in TSB medium at 37 °C. *Mean value significantly different from that for the control group. P < 0.05, n = 3. d) OD600 values of S. aureus cells incubated with CaPB NPs alone (black) and with CaPB NPs followed by addition of an equivalent amount of Fe to the cell culture after 2 hours incubation (red). *Mean value significantly different from that for the iron added groups. P < 0.05, n = 3.
For bacterial cytotoxicity studies, we incubated bacteria with the TSB medium containing various amounts of CaPB NPs in comparison with the bacteria cultured with the corresponding amounts of either Ca2+ ions or [FeIII(CN)6]3– ions, the two constituent components in the CaPB NPs, as two separate controls. As shown in Figure 4b, CaPB NPs inhibited the growth of S. aureus in a dose-dependent manner at concentrations greater than 10 μM (supporting information, Figure S7), while either CaCl2 or K3[Fe(CN)6] almost has no effect on growth at the concentration range of 10 μM to 400 μM. In addition, we also tested the time-dependent growth inhibition curve for CaPB NPs (Figure 4c). There is a pronounced effect on the bacterial growth after 9 hours of incubation.
To ascertain that such a striking antibacterial action of CaPB NPs was attributable to the depletion or sequestration of intracellular iron by the internalized NPs, we carried out a rescue test by replenishing the cell culture with iron two hours after the cells were treated with CaPB NPs. The amount of iron in the form of FeCl2 added to each cell culture corresponds to the concentration of CaPB NPs that was used to deplete the intracellular iron these cells before the rescue test. As shown in Figure 4d, iron addition to the cell cultures somewhat reversed the growth-inhibitory effect by the NPs. However, for the cells treated with higher concentrations of NPs (i.e. > 50 μM), full restoration of growth becomes unachievable, suggesting that some irreversible damage might have been done to the iron metabolism of the cells after they were treated with higher concentrations of NPs for 2 hours. We also tested the possibility of rescuing the CaPB NPs-treated cells using higher amounts of iron (i.e. 2- and 5-equivalent amounts). The results from these studies are thus far inconclusive because a higher amount of iron itself in the cell culture caused cells to die probably via the production of reactive oxygen species (ROS) triggered by the Fe2+ ion acting as the Fenton catalyst (Supporting information, Figure S8).
To probe whether CaPB NPs can cause any physical damage to the bacterial cell membrane, we carried out a LIVE/DEAD bacterial viability assay to probe the membrane integrity of the cell. Bacteria were cultured with CaPB NPs at the concentration of 200-μM to ensure that both live and dead cells would be present. The cultured cell suspensions were stained with the SYTO 9 green-fluorescent nucleic acid and propidium iodide red-fluorescent nucleic acid stains. Hence, cells with a compromised membrane that were dead or dying would stain red, whereas live or dead cells with an intact membrane would stain green. As revealed by the presence of strong green fluorescent signals in Figure 5, the cell membranes of both live (upper) and dead (lower) cells remained intact under the experimental conditions (see Supporting Information for experimental details).
Figure 5.

The fluorescence images of S. aureus bacterial cells treated with 200-μM CaPB NPs and stained with the LIVE/DEAD assay kit consisting of SYTO 9 and propidium iodide stains for monitoring the membrane integrity of both live (upper) and dead (lower) cells.
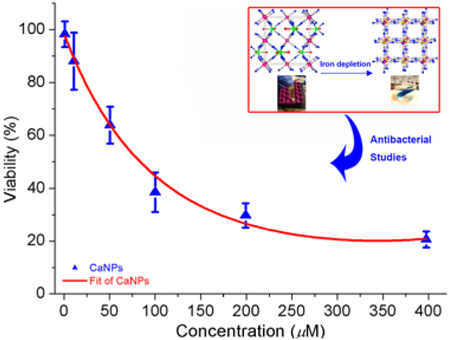
To evaluate the cytotoxicity of CaPB NPs in mammalian cells, we incubated murine RAW 264.7 macrophage-like cells, normal human fibroblast cells and human umbilical endothelial cells with various concentrations of NPs in the range of 10 μM to 500 μM. The cell viability was all assayed using the MTT method (see Supporting Information for experimental details). As seen in Figure 6, after incubation for 24 hours with 500 μM PVP-coated CaPB NPs, i.e. approximately ten times of the LD50 value in S. aureus, more than 80 % of the cells remained viable for all the three mammalian cell lines, indicating that such NPs are less toxic in mammalian cells. More importantly, when examined under the microscope, RAW cells and human fibroblast cells treated with such NPs all appeared to be healthy and viable with no morphological change discernable as shown in Figure S9 (Supporting Information), which indicates that a wide therapeutic window may exist for the use of such NPs to treat bacterial infections). In other words, the therapeutic index of CaPB NPs should be relatively large (i.e. TI >10).
Figure 6.

Results of cell viability studies in murine RAW 264.7 cells (black), human fibroblast cells (red), and endothelial cells (blue) incubated with different concentrations of CaPB NPs. Experiments were performed using MTT assay and incubated for 24 hours. *Mean value significantly different from that for the untreated CaPB NPs groups. P < 0.05, n = 3.
In summary, our approach to using PVP-coated CaPB NPs to selectively deplete intracellular iron in bacteria offers a unique opportunity to design and develop novel antimicrobial agents for treating bacterial infections. The currently known mechanisms of bacterial resistance to antibiotics include: (1) the enzymatic degradation of antibacterial drug molecules, (2) an alteration of bacterial proteins targeted by antibacterial drugs to reduce their binding capacity, and (3) a change in membrane permeability to antibacterial drugs by either decreasing permeability or increasing active efflux of antibacterial drugs. However, the current iron-depleting strategy may allow us to circumvent all of these classical mechanisms, and thus opening up a new avenue to defeat drug resistance in the MDROs. In other words, the ability to target iron acquisition and metabolism in pathogenic bacterial cells using NPs such as those demonstrated in this communication may represent a paradigm shift in treating bacterial infections. Work is under way in this laboratory to evaluate the antimicrobial activity of PVP-coated CaPB NPs in several strains of multidrug-resistant S. aureus. On the other hand, our preliminary results from the studies of antimicrobial properties of PVP-coated CaPB NPs in Pseudomonas aeruginosa (P. aeruginosa), a Gram-negative rod-shaped bacterium that is associated with a number of serious hospital-acquired infections, indicated that the PVP-coated NPs exhibit a low cell-membrane permeability, and thus modest antimicrobial activity in P. aeruginosa. We are investigating the use of different polymers and targeting agents in replacement of PVP to enhance the cell-membrane permeability of CaPB NPs in P. aeruginosa. The results of this research will be published in due course.
Supplementary Material
Acknowledgements
This study was supported in part by a grant from NIH-NINR (Grant R01NR015674). The TEM data were obtained using the Cryo-TEM Facility at Liquid Crystal Institute, KSU supported by the Ohio Research Scholars Program.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- [1].Bothwell TH and Finch CA, Iron Metabolism, Little, Brown, Boston, 1962. [Google Scholar]
- [2].Hentze MW, Muckenthaler MU and Andrews NC, Cell, 2004, 117, 285–297. [DOI] [PubMed] [Google Scholar]
- [3].Ganz T and Nemeth E, Biochim. Biophys. Acta, 2006, 1763, 690–699. [DOI] [PubMed] [Google Scholar]
- [4].Silva B and Faustino P, Biochim. Biophys. Acta, 2015, 1852, 1347–1359. [DOI] [PubMed] [Google Scholar]
- [5].Papanikolaou G and Pantopoulos K, Toxicol. Appl. Pharmacol, 2005, 202, 199–211. [DOI] [PubMed] [Google Scholar]
- [6].Arosio P and Levi S, Free Radic. Biol. Med, 2002, 33, 457–463. [DOI] [PubMed] [Google Scholar]
- [7].Crosa JH, Mey AR and Payne SM, ed., Iron transport in bacteria, 2004, Washington D.C.: ASM Press; p499. [Google Scholar]
- [8].Jurado RL, Clin. Infect. Dis, 1997, 25, 888–895. [DOI] [PubMed] [Google Scholar]
- [9].Bullen JJ and Griffiths E, Iron and infection: molecular, physiological and clinical aspects. New York: John Wiley and Sons, 1999. [Google Scholar]
- [10].Schaible UE and Kaufmann SH, Nat. Rev. Microbiol, 2004, 12, 946–953. [DOI] [PubMed] [Google Scholar]
- [11].Kehl-Fie TE and Skaar EP, Curr Opin Chem Biol, 2010, 14, 218–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Flo TH, Smith KD, Sato S, Rodriguez DJ, Holmes MA, Strong RK, Akira S and Aderem A, Nature, 2004, 432, 917–921. [DOI] [PubMed] [Google Scholar]
- [13].Skaar EP, Humayun M, Bae T, DeBord KL and Schneewind O, Science, 2004, 305, 1626–1628. [DOI] [PubMed] [Google Scholar]
- [14].Bullen JJ, Rogers HJ, Spalding PB and Ward CG, FEMS Immunol. Med. Microbiol, 2005, 43, 325–330. [DOI] [PubMed] [Google Scholar]
- [15].Wandersman C and Delepelaire P, Annu. Rev. Microbiol, 2004, 58, 611–647. [DOI] [PubMed] [Google Scholar]
- [16].Ratledge C and Dover LG, Annu. Rev. Microbiol, 2000, 54, 881–941. [DOI] [PubMed] [Google Scholar]
- [17].Letoffe S, Heuck G, Delepelaire P, Lange N, Wandersman C, Proc. Natl. Acad. Sci. U S A, 2009, 106, 11719–11724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Watanabe NA, Nagasu T, Katsu K, and Kitoh K, Antimicrob. Agents Chemother, 1987, 31, 497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Minnick AA, McKee JA, Dolence EK and Miller MJ, Antimicrob. Agents Chemother, 1992, 36, 840–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Zheng T, Bullock JL and Nolan EM, J. Am. Chem. Soc, 2012, 134, 18388–18400. [DOI] [PubMed] [Google Scholar]
- [21].Sassone-Corsi M, Chairatana P, Zheng T, Perez-Lopez A, Edwards RA, George MD, Nolan EM and Raffatellu M, Proc. Natl. Acad. Sci. U. S. A, 2016, 113, 13462–13467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Neilands JB, J. Biol. Chem, 1995, 270, 26723–26726. [DOI] [PubMed] [Google Scholar]
- [23].Königsberger L-C, Königsberger E, May PM and Hefter GT, J. Inorg. Biochem, 2000, 78, 175–184. [DOI] [PubMed] [Google Scholar]
- [24].Boukhalfa H and Crumbliss AL, BioMetals, 2002, 15, 325–339. [DOI] [PubMed] [Google Scholar]
- [25].Raymond KN, Dertz EA and Kim SS, Proc. Natl. Acad. Sci. U. S. A, 2003, 100, 3584–3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Hider RC and Kong X, Nat. Prod. Rep, 2010, 27, 637–657. [DOI] [PubMed] [Google Scholar]
- [27].Yang Nicole J. and Hinner Marlon J., Methods Mol. Biol, 2015, 1266, 29–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Engelman DM, Nature, 2005, 438, 578–580. [DOI] [PubMed] [Google Scholar]
- [29].Sugano K, Kansy M, Artursson P , Avdeef A , Bendels S , Di L , Ecker GF , Faller B , Fischer H , Gerebtzoff G , Lennernaes H and Senner F, Nat. Rev. Drug Discov, 2010, 9, 597–614. [DOI] [PubMed] [Google Scholar]
- [30].Dunbar KR and Heintz RA, Prog. Inorg. Chem, 1997, 45, 283–391. [Google Scholar]
- [31].Buser HJ, Schwarzenbach D, Petter W, Ludi A, Inorg. Chem, 1977, 16, 2704–2710. [Google Scholar]
- [32].Perera VS, Yang LD, Hao J, Chen G, Erokwu BO, Flask CA, Zavalij PY, Basilion JP and Huang SD, Langmuir, 2014, 30, 12018–12026. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.