

Supplemental Digital Content is Available in the Text.
Key Words: LC-MS/MS, antibiotics, TDM
Abstract
Background:
Adequate antibiotic treatment is a prerequisite for the successful treatment of systemic infections. Based on accumulating scientific evidence, a fixed dosage regimen can lead to insufficient and ineffective antibiotic therapy. Thus, the aim of this study was to develop and validate a simplified, but sensitive method for the simultaneous quantification of antimicrobials by using liquid chromatography with tandem mass spectrometry (LC-MS/MS) for the development of personalized therapy regimens using therapeutic drug monitoring.
Methods:
A method was developed for the simultaneous quantification of 9 antimicrobials (aciclovir, ampicillin, cefuroxime, ciprofloxacin, meropenem, metronidazole, piperacillin, rifampicin, and tazobactam) in lithium–heparin plasma. A simple sample preparation method and a chromatographic run time of 10 minutes enabled the quick processing of the samples. The method was validated according to the guidelines for bioanalytical method validation of the European Medicines Agency and addressed sensitivity, specificity, linearity, accuracy, precision, dilution integrity, carry-over, recovery, matrix effects, and stability.
Results:
The chromatographic run time was 10 minutes and antimicrobials eluted at retention times ranging from 1.1 to 2.2 minutes. Calibration curve for all antimicrobials was linear over a range of 1–100 mg/L, and a 2-fold or 5-fold dilution of the samples was possible. The method accuracy ranged from 85.1% to 114.9% for all measured antimicrobials, and the within- and between-run precision values were <11.9% and <16.5% for the lower limit of quantification. No interferences and carry-over were observed. The samples were stable for at least 5 hours at room temperature or in the autosampler (10°C).
Conclusions:
The LC-MS/MS method developed in this study is appropriate and practical for the therapeutic drug monitoring of antimicrobials in the daily clinical laboratory practice because of its short analysis time, the need for a small amount of plasma, high specificity, and accuracy.
INTRODUCTION
In addition to the timely administration of appropriate antimicrobials, the effective treatment of infectious diseases in the critically ill patients requires a sufficient drug concentration at the site of infection. Current data from the Medusa (medical education for sepsis source control and antibiotics) study showed that only 58.1% of the patients were administered with an adequate antibiotic treatment exhibiting a 10.6% increase in survival.1 Inadequate drug exposure from fixed dosage regimens based on noncritically ill patients may be one reason for antimicrobial treatment failure.2 Traditionally, therapeutic drug monitoring (TDM) is used for drugs with a narrow therapeutic index to prevent toxicity. However, accumulating evidence stresses the importance of maintaining free drug concentrations that exceed the bacterial minimum inhibitory concentration for a specified time.3–5 Moreover, the emergence of multidrug resistant microorganisms has dramatically increased in the last few years,6–8 together with a lack of new antimicrobial substances emphasize the need for an optimized use of antimicrobials.9 Currently, numerous indications render an individual dosage meaningful, at least in critically ill patients. In the critically ill patients, antibiotic pharmacokinetics are variably altered by fluid resuscitation using large volumes, body weight, organ dysfunction, augmented renal elimination, comorbidity, hypoproteinemia, and drug interactions,10,11 leading to changes in volume of distribution and drug clearance, and thus, the individual pharmacokinetics of ß-lactam antibiotics are unpredictable.12
Therefore, TDM represents a meaningful possibility for optimizing therapy.13–15 Nevertheless, TDM of various classes of antibiotics is largely limited by the lack of robust and rapid methods, particularly commercial procedures for their quantification in human plasma. Thus, several methods have been developed and validated in-house. High-performance liquid chromatography (HPLC) using ultraviolet detection has frequently been used.16,17 More sensitive and selective methods have been developed using HPLC coupled with tandem mass spectrometry (LC-MS/MS),18,19 however, only for the simultaneous measurement of one class of antibiotics, such as β-lactam antibiotics. Thus, a sensitive, specific, fast, and reliable method for the simultaneous quantification of antimicrobials of different classes in small sample volumes in a relatively short time is urgently needed to identify the under-dosing of the at-risk patients particularly in the case of parallel administration of different antimicrobials and to perform individual adjustments of treatment algorithms. Moreover, simultaneous quantification is also advantageous with regard to practical reasons in the laboratory workflow to save resources.
In this study, we aimed to develop and validate an LC-MS/MS procedure for the simultaneous measurement of 9 clinically relevant antimicrobials for use in daily clinical laboratory practice: aciclovir (ACV), ampicillin (AMP), cefuroxime (CFX), ciprofloxacin (CIP), meropenem (MEM), metronidazole (MTZ), piperacillin (PIP), rifampicin (RIF), and tazobactam (TAZ).
MATERIALS AND METHODS
Chemicals and Reagents
Analytical standards of ACV (purity of 99.7%), AMP (purity of 99.7%), CFX (purity of 95.1%), CIP (purity of 99.5%), MEM trihydrate (purity of 99.7%), MTZ (purity of 99.9%), PIP (purity of 94.4%), RIF (purity of 97.6%), TAZ (purity of 99.6%), and the internal standard dicloxacillin sodium salt monohydrate (purity of 100%) were purchased from Sigma-Aldrich (St. Louis, MO). The labeled internal standard piperacillin-d5 (purity of 99.6%, IS for PIP) was supplied by Toronto Research Chemicals (Ontario, Canada). LC-MS-grade methanol was purchased from Carl Roth (Karlsruhe, Germany). Formic acid was supplied by Sigma-Aldrich (St. Louis, MO). Pooled drug-free human lithium–heparin human plasma (collection tubes without a gel separator) was obtained from 8 healthy, untreated volunteers. Written informed consent was obtained from all participants. The study was approved by the local ethics committee (no. 4619/-11/15).
Calibration Standards, Quality Control Samples, and Internal Standards
Two stock solutions were prepared at a concentration of 1 g/L to prepare calibration standards and quality controls. The stock solutions were prepared by weighing an appropriate amount of each antimicrobial and dissolving it in drug-free lithium–heparin plasma. The 100 mg/L working solution was prepared as a composite of all 9 antimicrobials. Further working calibration solutions were prepared by diluting the compound in drug-free lithium–heparin plasma at concentrations of 75, 50, 20, 10, 5, and 1 mg/L. Working quality controls were similarly prepared from the 100 mg/L working solution at concentrations of 2.5, 25, and 60 mg/L. The solutions were stored in the dark in brown tubes for up to 3 months at −80°C as 50 µL aliquots.
Dicloxacillin and piperacillin-d5 were dissolved in HPLC-grade water to achieve a final concentration of 1 g/L (stock solution) and 75 mg/L (working solution), respectively.
Sample Preparation
For protein precipitation, 50 µL of the internal standard piperacillin-d5 (75 mg/L) and 700 µL deproteinization solution (10 mg/L dicloxacillin in methanol) were added to 50 µL of calibration standard, quality control, or plasma samples. Samples were vortexed for 1 minute, and the tubes were then centrifuged at 16,000g for 5 minutes at room temperature. Approximately 200 µL of the supernatant was transferred to 2-mL screw-top glass vial with 250 µL glass inserts and silicon septa caps (Agilent, Santa Clara, CA) and 5 µL was injected for analysis.
LC-MS/MS Analyses
Analyses were performed using an Agilent 1200 series system (Santa Clara, CA) consisting of a thermostatic autosampler, a binary solvent delivery manager, and a thermostated column compartment. Chromatographic separation was achieved using a Kinetex F5 core–shell reverse-phase column (50 × 2.1 mm, 100 Å pore size, 2.6 µm particle size; Phenomenex, Aschaffenburg, Germany) and a corresponding F5 precolumn with a 2.1-mm internal diameter (Phenomenex, Aschaffenburg, Germany). The column compartment was maintained at a temperature of 40 ± 1°C. Mobile phase A consisted of 0.1% formic acid in water, and mobile phase B consisted of 0.1% formic acid in methanol. After every injection, the needle was washed in a flush port for 15 seconds in 50% methanol. Furthermore, the main pass and bypass of the sample loop were washed 5 times between the injections.
The 9 antimicrobials were separated in 2 consecutive chromatographic runs with different gradients for positive and negative ionization. The quantification of ACV, AMP, CIP, MEM, MTZ, PIP, and RIF was performed in positive mode (hold at 100% B for 0.5 minutes with a flow rate of 200 µL/min, 100%–0% B from 0.5 to 1 minute with a flow rate of 400 µL/min, 0%–50% B from 1 to 2 minutes with a flow rate of 400 µL/min, 50%–0% B from 2 to 3 minutes with a flow rate of 400 µL/min, hold at 0% B until 3.5 minutes with a flow rate of 400 µL/min, 0%–100% B from 3.5-4 minutes with a flow rate of 400 µL/min, hold at 100% B hold until 4.5 minutes with a flow rate of 400 µL/min, and hold at 100% B until 5 minutes with a flow rate of 200 µL/min). Negative mode was used to measure the CFX and TAZ concentrations (hold at 100% B for 0.5 minutes with a flow rate of 200 µL/min, 100%–0% B from 0.5 to 1 minute with a flow rate of 400 µL/min, 0%–30% B from 1 to 2 minutes with a flow rate of 400 µL/min, 30%–0% B from 2 to 2.5 minutes with a flow rate of 400 µL/min, 0%–100% B from 2.5 to 4 minutes with a flow rate of 400 µL/min, hold at 100% B until 4.5 minutes with a flow rate of 400 µL/min, and hold at 100% B until 5 minutes with a flow rate of 200 µL/min).
Detection was conducted with an API4000 LC-MS/MS System (AB SCIEX, Framingham, MA) equipped with an electrospray ionization source. The instrument was operated in multiple reaction monitoring (MRM) mode with the following instrument settings: collision gas: 8 psi, curtain gas: 10 psi, nebulizer gas: 55 psi, heater gas: 55 psi, ion spay voltage: 5500 V/-4500 V, heater temperature: 550°C, and entrance potential: 10 V. Nitrogen was used as the collision gas. For each antimicrobial, 2 transitions, 1 for quantification (quantifier) and 1 for confirmation (qualifier), were detected. Compound-specific instrument parameters were optimized for each transition to obtain the most robust and intense signal (Table 1).
TABLE 1.
Mass Spectrometry Parameters for Antimicrobials

Eight-level calibration was performed daily, and concentrations were calculated using Analyst 1.5.1 software (AB SCIEX, Framingham, MA). The calibration curves were generated by a quadratic fit of the antimicrobial/IS standard area response ratio plotted against the nominal concentration of the standard sample (1/x weighting).
Validation
Validation was performed according to the current Food and Drug Administration (FDA) and European Medicines Agency (EMA) guidelines.20,21 The developed method was validated in terms of selectivity, specificity, sensitivity, lower limit of quantification (LLOQ), carry-over, linearity, accuracy, precision, dilution integrity, matrix effect, and stability.
Selectivity
Six individual sources of blank matrix were individually analyzed and evaluated for interferences from endogenous components with the antimicrobials of interest. The absence of interferences was accepted as a response of less than 20% of the LLOQ for the analytes and 5% for the internal standards.
Specificity, LOD, and LLOQ
About 100 mg/L solution of each antimicrobial was separately analyzed to determine specificity. Interferences between the antimicrobials should be less than 20% of the LLOQ for the analyte. The LLOQ was defined as the lowest concentration with a precision of ±20% and accuracy of 80%–120%. Therefore, samples with concentrations that were approximately the same as the expected LLOQ (0.25, 0.5, 0.75, and 1 mg/L) were analyzed. The limit of detection (LOD) was calculated by multiplying the background of the mean of 10 measurements of a blank sample of pooled lithium–heparin matrix by 3.
Carry-Over
Carry-over was assessed by injecting 3 blank samples after the highest calibration standard (100 mg/L). In the first blank, the carry-over should be less than 20% of the LLOQ for the analytes and 5% for the internal standards.
Linearity
Linearity was assessed by measuring triplicates 2-fold serial dilutions of the highest calibration standard (100 mg/L) within ranges of the LOD in drug-free lithium–heparin plasma.
Accuracy and Precision
The accuracy and precision of the method were repeatedly quantified using 4 concentrations of quality control samples (LLOQ, 2.5, 25, and 60 mg/L) 10 times within a run (intraday) and in a single series per day on 10 different days (interday). The mean value should be within 15% (20% for LLOQ) of the nominal values for the quality control samples. The determined precision should not exceed 15% (20% for LLOQ) of the coefficient of variation.
Dilution Integrity
Dilution integrity was validated to quantify drug concentrations greater than the calibration interval, which may occur during the analysis of samples from actual patients. The blank matrix was spiked with a concentration exceeding the highest calibration standard (150 mg/L) and diluted 2-fold or 5-fold with blank matrix after sample processing. The accuracy and precision should be within 85%–115% and ±15% in 5 determinations per dilution factor, respectively.
Recovery
For the recovery study, several concentrations of the compounds (2.5, 25, and 60 mg/L) were prepared in 6 different blank matrices. Recovery was calculated as the mean ratio between the peak area of spiked samples after extraction and before extraction. The variation in recovery among all concentrations should be less than 15%.
Matrix Effect
The matrix factor was determined using the blank matrix from 6 individual donors by calculating the ratio of the peak area of spiked blank matrix after sample processing to the peak area in the absence of matrix (50% methanol) for 3 different concentrations of antimicrobial (2.5, 25, and 60 mg/L). The internal standard–normalized matrix factor (the matrix factor of the analyte divided by the matrix factor of the internal standard) should not show a variation greater than 15%. In addition, the effects of increased hemolysis, icterus, and lipemia on each analyte were examined in 2 different donors compared with normal levels. Therefore, samples were spiked with 100 mg/L antimicrobials, and accuracy was determined.
Stability
Stability studies included analyses of the stability of the stock solution and working solution, freeze and thaw stability, and the stability of extracted samples at room temperature and in the autosampler (10°C). Concentrations of analytes in solutions stored for up to 3 months at −80°C were compared with fresh samples to evaluate the stability of the stock and working solutions. The short-term stability of the processed sample at room temperature and the processing temperature (autosampler at 10°C) was analyzed at 3 concentrations (2.5, 25, and 60 mg/L) for 5 and 24 hours.
Acceptance Criteria for an Analytical Run
Acceptance criteria for each analytical run were defined by considering the following issues: the precision of the internal standard, accuracy, calibration range, and reinjection of study samples. The precision of the internal standard during the analytical run was determined as the variation of each sample from the mean of the peak area of the calibration standards. This variation should be less than 12.5%. The back-calculated concentrations of the calibration standards should show an accuracy ranging from 85% to 115%. At least 75% of the calibration standards (at least 6) should fulfill these criteria. Calibration standards that did not meet these criteria were excluded from the calibration curve. If the excluded calibration standard was the LLOQ or the highest standard, the calibration range was revised to the next lowest or highest calibration standard, respectively. Furthermore, at least 2 of the 3 quality controls should also fulfill the internal standard and accuracy criteria. Criteria for the reanalysis of study samples were predefined as not fulfilling the internal standard criteria, a concentration was obtained that exceeded the calibration range, and implausible results.
Clinical Samples
We evaluated the applicability of the newly developed LC-MS/MS method by processing plasma samples from 1 patient who was treated with a standard dosage (13.5 g/24 hours) and 1 patient who was treated with an individual dosage of PIP/TAZ. Approval was obtained from the leading institutional review board at Jena University Hospital (ref: 4825-06/16), all relevant institutional review boards of participating study sites, and the Federal Institute for Drugs and Medical Devices (EudraCT: 2016-000136-17, ref: 4041358). Blood samples were collected in lithium–heparin–containing tubes. All samples were centrifuged at 2000g for 10 minutes and immediately analyzed.
RESULTS
Method Development
Antimicrobials eluted at retention times ranging from 1.1 to 2.2 minutes under the chromatographic conditions described above. Representative MRM chromatograms for ACV, AMP, CFX, CIP, MEM, MTZ, PIP, RIF, and TAZ, as well as dicloxacillin and piperacillin-d5, are shown in Figure 1. The analytical run time was 5 minutes for each ionization mode, including the time needed to equilibrate the column to baseline conditions before the next injection.
FIGURE 1.

Representative MRM chromatograms. The peaks are shown in an overall overview of positive and negative ionization, including retention times in a sample MRM chromatogram for 10 mg/L of antimicrobials in lithium–heparin plasma. A, MRM chromatogram of all antimicrobials (quantifier) with positive ionization, (B) MRM chromatogram of all antimicrobials (quantifier) with negative ionization.
Validation Results
Selectivity
Endogenous compounds in the 6 individual donors of blank matrix did not show interferences greater than 20% of the LLOQ for the analytes and 5% for the internal standards (Supplemental Digital Content 1, http://links.lww.com/TDM/A269, which illustrates the selectivity of the developed method).
Specificity, LOD, and LLOQ
No interferences greater than 20% of LLOQ of the analyte were observed for the analyzed antimicrobials (Supplemental Digital Content 2, http://links.lww.com/TDM/A270, which illustrates the specificity of the developed method). The LOD for ACV, AMP, CFX, CIP, MEM, MTZ, PIP, RIF, and TAZ was 0.02, 0.67, 0.001, 0.17, 0.14, 0.008, 0.02, 0.05, and 0.005 mg/L, respectively, and the LLOQs for all antimicrobials were 1 mg/L. Data for accuracy and precision at the LLOQ are summarized in Table 2.
TABLE 2.
Accuracy and Precision

Carry-Over
Peak areas observed in a blank sample measured after the highest calibration standard were 18.2, 19.9, 12.7, 19.8, 8.9, 19.9, 18.7, 17.9, and 11.4% of the LLOQ for ACV, AMP, CFX, CIP, MEM, MTZ, PIP, RIF, and TAZ, respectively. The internal standards dicloxacillin and piperacillin-d5 showed a carry-over of 1.1% (positive ionization)/0.2% (negative ionization) and 0.3% of the LLOQ, respectively.
Linearity
The calibration curves were calculated by determining the peak area ratio from the antimicrobial and the internal standard at 8 standard concentrations (including a zero sample). Quadratic regression analyses with a weighting scheme of 1/x provided the best description of the generated data set. The regression coefficient for all calibration curves was greater than 0.99. Linearity was established between 0.1 and 100 mg/L for ACV, CFX, and TAZ, between 0.2 and 100 mg/L for MEM and PIP, between 0.39 and 100 mg/L for AMP and RIF, and between 0.78 and 100 mg/L for CIP and MTZ.
Accuracy and Precision
Data for intraday and interday accuracy and precision are summarized in Table 2. Within-day accuracy ranged from 85.1% to 114.9%, with a mean of 103.4%. Between-day accuracy ranged from 94.5% to 114.9% (mean: 102.1%). Intraday and interday coefficients of variation ranged from 1.3% to 5.5% and 1.8% to 11.9%, respectively. Overall, the results indicate the establishment of a reproducible method for each compound.
Dilution Integrity
Accuracy values for dilution integrity at 2-fold and 5-fold dilutions were −1.6% and −0.9% for ACV, −4.1% and 8.0% for AMP, 0.6% and 2.3% for CFX, −14.3% and −6.3% for CIP, −9.1% and −7.8% for MEM, 9.9% and 8.9% for MTZ, −14.8% and −14.9% for PIP, −1.1% and −3.9% for RIF, and 4.8% and −0.7% for TAZ, respectively. Precision was found to be 1.3% and 3.5% for ACV, 3.1% and 1.3% for AMP, 2.7% and 3.6% for CFX, 3.2% and 3.3% for CIP, 1.6% and 2.2% for MEM, 6.0% and 3.9% for MTZ, 1.8% and 2.7% for PIP, 1.6% and 3.5% for RIF, and 3.4% and 2.4% for TAZ at these 2 dilutions, respectively.
Recovery and Matrix Effect
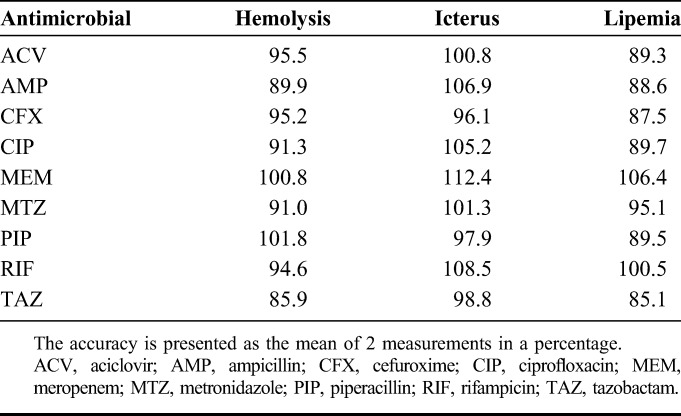
Recovery, matrix factor, internal standard–normalized matrix factor, and variability of matrix effect at different concentrations are summarized in Table 3. The recovery of all compounds at all concentrations was greater than 88.9%. The variation in the matrix effect among all concentrations was less than 15%. Increased hemolysis, icterus, and lipemia had no effects on the measurement of ACV, AMP, CFX, CIP, MEM, MTZ, PIP, RIF, and TAZ concentrations (Table 4).
TABLE 3.
Recovery, Matrix Factors, and Internal Standard–Normalized Matrix Factors

TABLE 4.
Effects of Increased Hemolysis, Icterus, and Lipemia
Stability
The concentrations of antimicrobials in extracted samples were stable in the autosampler (10°C) for 5 hours for CIP and MEM and for 24 hours for ACV, AMP, CFX, MTZ, PIP, RIF, and TAZ. At room temperature, ACV, AMP, CFX, CIP, MEM, MTZ, RIF, and TAZ concentrations were stable in processed samples for 5 hours, and PIP for 24 hours. Stock solutions and working solutions of the antimicrobials and internal standards were stable at −80°C for 3 months.
Clinical Application
Antimicrobial concentrations in plasma obtained from 2 selected patients are illustrated in Figure 2. The control patient treated with a standard dosage showed high intraindividual differences in antimicrobial concentrations over treatment time. At some time points, this patient nearly exhibited antimicrobial concentrations (>150 mg/L) associated with a risk of side effects. The target concentration for the patient treated with an individual dosage was 80 mg/L for an unknown minimum inhibitory concentration (MIC) within the first 2 days of treatment. On day 3, the MIC was determined to be ≤4 mg/L, and the target concentration was adjusted to 20 mg/L. Therefore, with a very high starting concentration of 269 mg/L piperacillin, the antimicrobial dose and the risk of side effects were remarkably reduced.
FIGURE 2.
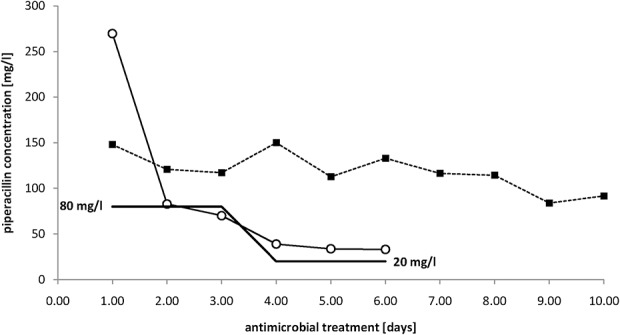
Antimicrobial concentrations in selected patients over time. Antimicrobial concentrations in a patient treated with standard dosage of 13.5 g of PIP/TAZ for 24 hours (square) and a patient treated with individualized dosing according to the minimum inhibitory concentration (MIC) measured throughout the treatment period are shown. The target concentrations (100% f T >4MIC-percentage of time during a dosing interval that the free (f) drug concentration exceeded 4 times the MIC) for the second patient are highlighted as a black line (80 mg/L for unknown pathogen, and 20 mg/L for Escherichia coli with a MIC <4 mg/L).
DISCUSSION
In this study, we developed and validated a LC-MS/MS-based method that simultaneously measured the concentrations of 9 antimicrobials (ACV, AMP, CFX, CIP, MEM, MTZ, PIP, RIF, and TAZ) accurately and precisely in small amount (50 µL) of plasma. Other investigators have also determined drug concentrations in low plasma volumes. However, most studies included antimicrobials of the same drug class and no antivirals.16,18 The ability to simultaneously determine the concentrations of antimicrobials from different substance classes, including antivirals, is a milestone in the dose adjustment of antimicrobials, particularly in critically ill patients with unpredictable pharmacokinetics.
Method Development
Several combinations of different mobile phases and reverse-phase HPLC columns were tested to achieve the simultaneous measurements of the concentrations of the selected antimicrobials with a short retention time and a high response. Of all possible combinations of mobile phases consisting of water and acetonitrile or methanol as organic phase, a mobile phase composed of water and methanol with 0.1% formic acid offered the highest MS response. Other parameters such as column oven temperature and injection volume were also optimized for a fast and reliable separation. The best results were observed when 40°C (versus 30, 50, or 60°C) was used as the column oven temperature and 5 µL (versus 10, or 20 µL) were injected.
The simple and straightforward extraction process (protein precipitation) provided excellent recovery (>88%) of all antimicrobials. Thus, the extraction procedure used in this method exhibits a high level of efficiency in sample preparation and high-throughput processing of patient samples. In addition to the simplicity of the sample processing procedure, the major advantage of our method is the simultaneous measurement of 9 antimicrobials in just 2 runs. The short chromatographic run time of 10 minutes is another advantage, which is comparable with previously described methods ranging from 3.5 to 11.8 minutes.18,19,22,23 Methods with significantly shorter chromatography times have been reported, but only one ESI mode was used for detection.24–27
Validation
Carry-over should be addressed and minimized during method validation. In contrast to other published procedures,17,22,23 a carry-over study was conducted here, and no carry-over was detected.
The LLOQ for all antimicrobials for the developed quantification method was 1 mg/L. Considering the breakpoint minimum inhibitory concentrations for many pathogens, it is particularly difficult to treat pathogens observed in critically ill patients with Staphylococcus aureus (0.03–4 mg/L) and Pseudomonas aeruginosa (0.5–16 mg/L),28 an LLOQ of 1 mg/L meets the clinical needs for adequate TDM. Furthermore, similar results for LLOQs ranging from 0.5 to 0.96 mg/L were in agreement with other studies,18,25 while methods using cleaner but more complex extraction procedures (solid–phase extraction or protein precipitation combined with liquid–liquid extraction) not surprisingly produced better results (LLOQ: 0.1–0.5 mg/L).22,23
The accuracy and precision of the concentration ranges for the developed method meet the requirements listed in the guidelines of EMA and FDA and were similar to or better than values reported in other publications.16,24,25 These results indicate a robust and reliable quantification.
We used a compromise for calibration ranges because of the wide range of concentrations of antimicrobials used during the different stages of treatment. Thus, some samples required further dilution.
In contrast to methods described in other previous publications,17,22–24 a dilution integrity study was conducted in this study with an imprecision and bias of 15%, according to the EMA and FDA criteria.
Matrix effects of lithium–heparin plasma are attributed to ion suppression or ion enhancement effects. The use of standards diluted in the blank matrix compensates for these observed matrix effects, and importantly, the signal was consistent across the 6 tested plasma matrices. The variations in the accuracy and precision of these different plasma batches were within the limits of the guidelines, indicating that no further sources of variation were introduced by the use of different plasma sources. Therefore, the measurement of samples from different patients should be considered as reliable and comparable.
Antimicrobials, particularly piperacillin, have been shown to absorb to the gel barrier in collection tubes with a gel separator.29 Thus, antimicrobial concentrations should be measured in plasma collected in tubes without gel separators as performed in this study. The poor stability of antimicrobials, particularly β-lactam antibiotics, at room temperature and under refrigeration is well described. Thus, precautions should be taken, for example, the use of a cooled autosampler, extraction of samples immediately before the measurement, and a minimal storage time at room temperature, to prevent the decomposition of antimicrobials in the processed samples. The stability of some antimicrobials, such as piperacillin, meropenem, and tazobactam, is reduced (not more than 20 days) in plasma samples stored at −20°C or −30°C.22,25,30 Consequently, stock solutions, working solutions, and samples should be stored at −80°C. Under these conditions, our solutions were stable for up to 3 months, confirming other observations of stability for 2 to 8 months.16,30,31 According to the literature, plasma samples and samples extracted in methanol are stable at room temperature from 4 to 12 hours16,17,19,23 and less than 6 hours.30 Regarding previous published data and our results, the time for which plasma and processed samples are stored at room temperature and in the autosampler should be minimized.
Although the validation criteria were fulfilled, the developed method had some limitations, with regard to the LC method. The 9 analyzed antimicrobials display differences in their chemical characteristics, and thus, the separation as well as peak shape of some drugs is limited, particularly ACV and MEM. Measurements of the antimicrobials without matrix excluded artifacts (see Figure, Supplemental Digital Content 3, http://links.lww.com/TDM/A271, which shows a representative chromatogram without matrix).
CONCLUSIONS
A short, robust, and reliable LC-MS/MS procedure for the quantification of 9 antimicrobials in small samples of human plasma was developed in this study. The assay was successfully validated in terms of selectivity, specificity, sensitivity, carry-over, linearity, accuracy, precision, dilution integrity, recovery, matrix effect, and stability. The simple sample preparation procedure and short chromatographic run time saves time. Therefore, this method can be applied daily to meet all requests in one sequence run.
Supplementary Material
ACKNOWLEDGMENTS
The authors gratefully acknowledge the financial support from the BMBF through the integrated research and treatment center “Center for Sepsis Control and Care” (grant 01EO1502).
Footnotes
This work was supported within the framework of the German Center for Sepsis Control and Care (CSCC) by the Ministry of Education and Research (BMBF), Grant-No. 01EO1502.
S. Neugebauer: method development, method validation, drafting the manuscript, critical review, and final approval of the manuscript. C. Wichmann: method development, method validation, critical review, and final approval of the manuscript. S. Bremer-Streck: method validation, critical review, and final approval of the manuscript. S. Hagel: clinical data acquisition, critical review, and final approval of the manuscript. M. Kiehntopf: project management, critical review, and final approval of the manuscript
The authors declare no conflict of interest.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Web site (www.drug-monitoring.com).
REFERENCES
- 1.Bloos F, Thomas-Ruddel D, Ruddel H, et al. Impact of compliance with infection management guidelines on outcome in patients with severe sepsis: a prospective observational multi-center study. Crit Care. 2014;18:R42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Waele JJ, Carrette S, Carlier M, et al. Therapeutic drug monitoring-based dose optimisation of piperacillin and meropenem: a randomised controlled trial. Intensive Care Med. 2014;40:380–387. [DOI] [PubMed] [Google Scholar]
- 3.Gonzalez D, Schmidt S, Derendorf H. Importance of relating efficacy measures to unbound drug concentrations for anti-infective agents. Clin Microbiol Rev. 2013;26:274–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roberts JA, Norris R, Paterson DL, et al. Therapeutic drug monitoring of antimicrobials. Br J Clin Pharmacol. 2012;73:27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Craig WA. Basic pharmacodynamics of antibacterials with clinical applications to the use of beta-lactams, glycopeptides, and linezolid. Infect Dis Clin North America. 2003;17:479–501. [DOI] [PubMed] [Google Scholar]
- 6.World Health Organizsation. Antimicrobial resistance: Global Report on Surveillance 2014 [web site] 2014. Available at: http://www.who.int/antimicrobial-resistance/publications/surveillancereport/en/. Accessed March 03, 2017.
- 7.European Centre for Disease Prevention and Control. Antimicrobial Resistance Surveillance in Europe 2015 [Web Site] 2015. Stockholm: European Centre for Disease Prevention and Control; 2017. Available at: http://ecdc.europa.eu/en/publications/Publications/antimicrobial-resistance-europe-2015.pdf. Accessed March 23, 2017. [Google Scholar]
- 8.Carlet J, Collignon P, Goldmann D, et al. Society's failure to protect a precious resource: antibiotics. Lancet. 2011;378:369–371. [DOI] [PubMed] [Google Scholar]
- 9.Boucher HW, Talbot GH, Bradley JS, et al. Bad bugs, no drugs: no ESKAPE! an update from the Infectious Diseases Society of America. Clin Infect Dis. 2009;48:1–12. [DOI] [PubMed] [Google Scholar]
- 10.Roberts JA, Taccone FS, Lipman J. Understanding PK/PD. Intensive Care Med. 2016;42:1797–1800. [DOI] [PubMed] [Google Scholar]
- 11.Udy AA, Roberts JA, Lipman J. Clinical implications of antibiotic pharmacokinetic principles in the critically ill. Intensive Care Med. 2013;39:2070–2082. [DOI] [PubMed] [Google Scholar]
- 12.Goncalves-Pereira J, Povoa P. Antibiotics in critically ill patients: a systematic review of the pharmacokinetics of beta-lactams. Crit Care. 2011;15:R206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huttner A, Harbarth S, Hope WW, et al. Therapeutic drug monitoring of the beta-lactam antibiotics: what is the evidence and which patients should we be using it for? J Antimicrob Chemother. 2015;70:3178–3183. [DOI] [PubMed] [Google Scholar]
- 14.Roberts JA, Paul SK, Akova M, et al. DALI: defining antibiotic levels in intensive care unit patients: are current beta-lactam antibiotic doses sufficient for critically ill patients?. Clin Infect Dis. 2014;58:1072–1083. [DOI] [PubMed] [Google Scholar]
- 15.Wong G, Sime FB, Lipman J, et al. How do we use therapeutic drug monitoring to improve outcomes from severe infections in critically ill patients? BMC Infect Dis. 2014;14:288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Legrand T, Vodovar D, Tournier N, et al. Simultaneous determination of eight beta-lactam antibiotics, amoxicillin, cefazolin, cefepime, cefotaxime, ceftazidime, cloxacillin, oxacillin, and piperacillin, in human plasma by using ultra-high-performance liquid chromatography with ultraviolet detection. Antimicrob Agents Chemother. 2016;60:4734–4742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Verdier MC, Tribut O, Tattevin P, et al. Simultaneous determination of 12 beta-lactam antibiotics in human plasma by high-performance liquid chromatography with UV detection: application to therapeutic drug monitoring. Antimicrob Agents Chemother. 2011;55:4873–4879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rigo-Bonnin R, Ribera A, Arbiol-Roca A, et al. Development and validation of a measurement procedure based on ultra-high performance liquid chromatography-tandem mass spectrometry for simultaneous measurement of beta-lactam antibiotic concentration in human plasma. Clin Chim Acta. 2017;468:215–224. [DOI] [PubMed] [Google Scholar]
- 19.Cohen-Wolkowiez M, White NR, Bridges A, et al. Development of a liquid chromatography-tandem mass spectrometry assay of six antimicrobials in plasma for pharmacokinetic studies in premature infants. J Chromatogr B Analyt Technol Biomed Life Sci. 2011;879:3497–3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.European Medicines Agency (EMA). Guideline on bioanalytical method validation [web site] 2015. Available at: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2011/08/WC500109686.pdf. Accessed March 28, 2017.
- 21.Food and Drug Administration (FDA). Bioanalytical method validation [web site] 2018. Available at: https://www.fda.gov/downloads/drugs/guidances/ucm070107.Pdf. Accessed August 10, 2018.
- 22.Cazorla-Reyes R, Romero-Gonzalez R, Frenich AG, et al. Simultaneous analysis of antibiotics in biological samples by ultra high performance liquid chromatography-tandem mass spectrometry. J Pharm Biomed Anal. 2014;89:203–212. [DOI] [PubMed] [Google Scholar]
- 23.Sime FB, Roberts MS, Roberts JA, et al. Simultaneous determination of seven beta-lactam antibiotics in human plasma for therapeutic drug monitoring and pharmacokinetic studies. J Chromatogr B Analyt Technol Biomed Life Sci. 2014;960:134–144. [DOI] [PubMed] [Google Scholar]
- 24.Abdulla A, Bahmany S, Wijma RA, et al. Simultaneous determination of nine beta-lactam antibiotics in human plasma by an ultrafast hydrophilic-interaction chromatography-tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2017;1060:138–143. [DOI] [PubMed] [Google Scholar]
- 25.Lefeuvre S, Bois-Maublanc J, Hocqueloux L, et al. A simple ultra-high-performance liquid chromatography-high resolution mass spectrometry assay for the simultaneous quantification of 15 antibiotics in plasma. J Chromatogr B Analyt Technol Biomed Life Sci. 2017;1065-1066:50–58. [DOI] [PubMed] [Google Scholar]
- 26.Ahsman MJ, Wildschut ED, Tibboel D, et al. Microanalysis of beta-lactam antibiotics and vancomycin in plasma for pharmacokinetic studies in neonates. Antimicrob Agents Chemother. 2009;53:75–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carlier M, Stove V, De Waele JJ, et al. Ultrafast quantification of beta-lactam antibiotics in human plasma using UPLC-MS/MS. J Chromatogr B Analyt Technol Biomed Life Sci. 2015;978-979:89–94. [DOI] [PubMed] [Google Scholar]
- 28.European Committee on Antimicrobial Susceptibility Testing. EUCAST MIC distribution website [web site]. Available at: https://mic.eucast.org/Eucast2/SearchController/search.jsp?action=init. Accessed October 10, 2017.
- 29.Carlier M, De Waele JJ, Verstraete AG, et al. Exploration of the pre-analytical stability of beta-lactam antibiotics in plasma and blood—implications for therapeutic drug monitoring and pharmacokinetic studies. Clin Chem Lab Med. 2015;53:e227–230. [DOI] [PubMed] [Google Scholar]
- 30.Zander J, Maier B, Zoller M, et al. Effects of biobanking conditions on six antibiotic substances in human serum assessed by a novel evaluation protocol. Clin Chem Lab Med. 2016;54:265–274. [DOI] [PubMed] [Google Scholar]
- 31.McWhinney BC, Wallis SC, Hillister T, et al. Analysis of 12 beta-lactam antibiotics in human plasma by HPLC with ultraviolet detection. J Chromatogr B Analyt Technol Biomed Life Sci. 2010;878:2039–2043. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.