

Supplemental Digital Content is available in the text.
Keywords: blood-brain barrier, brain edema, brain ischemia, cell death, magnetic resonance imaging, neuroprotection
Abstract
Background and Purpose—
Acetylsalicylic acid and clopidogrel are the 2 main antithrombotic drugs for secondary prevention in patients with ischemic stroke (IS) without indication for anticoagulation. Because of their limited efficacy and potential side effects, novel antiplatelet agents are urgently needed. Cilostazol, a specific phosphodiesterase (PDE)-3 inhibitor, protected from IS in clinical studies comprising mainly Asian populations. Nevertheless, the detailed mechanistic role of PDE-3 inhibitors in IS pathophysiology is hardly understood. In this project, we analyzed the efficacy and pathophysiologic mechanisms of a novel and only recently described PDE-3 inhibitor (substance V) in a mouse model of focal cerebral ischemia.
Methods—
Focal cerebral ischemia was induced by transient middle cerebral artery occlusion in 6- to 8-week-old male C57Bl/6 wild-type mice receiving substance V or vehicle 1 hour after ischemia induction. Infarct volumes and functional outcomes were assessed between day 1 and day 7, and findings were validated by magnetic resonance imaging. Blood-brain barrier damage, as well as the extent of local inflammatory response and cell death, was determined.
Results—
Inhibition of PDE-3 by pharmacological blockade with substance V significantly reduced infarct volumes and improved neurological outcome on day 1 and 7 after experimental cerebral ischemia. Reduced blood-brain barrier damage, attenuated brain tissue inflammation, and decreased local cell death could be identified as potential mechanisms. PDE-3 inhibitor treatment did neither increase the number of intracerebral hemorrhages nor affect platelet function.
Conclusions—
The novel PDE-3 inhibitor substance V protected mice from IS independent from platelet function. Pharmaceutical inactivation of PDE-3 might become a promising therapeutic approach to combat IS via inhibition of thromboinflammatory mechanisms and stabilization of the blood-brain barrier.
Ischemic stroke (IS) is the second leading cause of mortality and a major contributor to long-term morbidity worldwide. Revascularization with r-tPA (recombinant tissue-type plasminogen activator) and mechanical thrombectomy for large vessel occlusions are the only acute treatment options currently available.1 In patients with stroke, the coadministration of platelet inhibitors during thrombolysis to improve microvascular patency is associated with an increased intracerebral bleeding risk.2 Moreover, the efficacy of currently approved platelet inhibitors is limited. Consequently, novel antithrombotic compounds with better benefit-risk ratios are urgently needed.
Novel antiplatelet agents currently investigated are phosphodiesterase (PDE) inhibitors.3,4 More than 60 different isoforms of PDEs are described in mammalian tissue, grouped into 11 broad families (PDE-1 to PDE-11). By catalyzing the hydrolysis of cAMP and cGMP, PDEs limit intracellular levels of cyclic nucleotides and thus regulate the amplitude, duration, and compartmentation of cyclic nucleotide signaling.3,5 cAMP and cGMP are 2 critical intracellular second messengers regulating fundamental platelet processes. Therefore, inhibition of platelet aggregation via PDE blockade has shown great benefit for the treatment of ischemic cardiovascular disease.3 Inhibitors of the PDE-3 subfamily (eg, cilostazol and milrinone) are nowadays used for peripheral artery disease.6 In addition to positive preclinical studies, large randomized controlled trials have been performed in Asia testing cilostazol versus acetylsalicylic acid in patients with acute IS.7–10 Cilostazol was noninferior in preventing further IS and, importantly, showed lower rates of intracranial hemorrhages.3,7,8 Because of potential side effects of the currently available PDE-3 inhibitors and also their limited efficacy, which is potentially related to their unspecific mode of action, recently, novel PDE-3 inhibitors with superior specificity, lower side effects, and increased inhibitory potency have been developed.11,12 So far these novel inhibitors have not yet been evaluated in an experimental stroke model.
The aim of this study was to characterize a prototypic member of this class of novel PDE-3 inhibitors (substance V) about safety and efficacy and to delineate the underlying in vivo mechanisms in a mouse model of brain ischemia/reperfusion injury.
Methods
This article adheres to the American Heart Association Journals implementation of the Transparency and Openness Promotion Guidelines. The detailed experimental description can be found in the online-only Data Supplement (including Tables I and II and Figures I and II in the online-only Data Supplement). Data that support the findings of this study are available from the corresponding author on reasonable request.
Animals
All animal experiments were approved by local animal care committees (Regierung von Unterfranken) and were conducted in accordance with the US National Institutes of Health Guide for the Care and Use of Laboratory Animals. The experiments were designed, performed, and reported according to the Animal Research: Reporting of In Vivo Experiments guidelines.13 All male C57Bl/6 mice were purchased from Charles River Laboratories (Sulzfeld, Germany). We randomized the mice and conducted transient middle cerebral artery occlusion (tMCAO) for 60 minutes as described previously.14
Animal Treatment
The PDE-3 inhibitor substance V (provided by G.J. Kumar and Dr Vaidya; 0.5 mg/kg body weight, dissolved in 0.5 % dimethyl sulfoxide [DMSO]), cilostazol (Sigma Aldrich, Germany; 10 mg/kg body weight, dissolved in 0.5 % DMSO), or vehicle (0.5 % DMSO) were given by an intraperitoneal injection before tMCAO or directly after reperfusion, that is, 60 minutes after inserting the occluding thread in the MCA.
Assessment of Functional Outcome
Global neurological deficits were quantified according to the Bederson score.15 To monitor motor function and coordination, the grip test was used.16 Neurological outcome was assessed 24 hours after stroke.
Analysis of Infarct Size by 2,3,5-Triphenyltetrazolium Chloride Staining and Magnetic Resonance Imaging
We analyzed infarct size by vital staining using 2% (wt/vol) 2,3,5-triphenyltetrazolium chloride (TTC) in phosphate buffer. Magnetic resonance imaging was performed by serial measurements on a 3.0T unit (MAGNETOM Trio, SIEMENS, Erlangen, Germany).
Determination of Blood-Brain Barrier Leakage and Brain Edema Formation
Blood-brain barrier (BBB) leakage after stroke by injecting the vascular tracer Evans blue and brain edema formation were determined as described previously.17
Protein Extraction, Western Blot Analysis, and Quantitative Real-Time Polymerase Chain Reaction
Protein extraction and Western blot analysis were performed according to standard procedures.17 Quantitative real-time polymerase chain reaction was performed as described previously.18
Histology, Immunohistochemistry, and TUNEL Assay
We stained mouse brains with antibodies against CD11b (Bio-Rad, MCA711), CD31 (Bio-Rad MCA2388GA), Claudin-5 (Thermo Fisher Scientific, 34–1600), Ly-6B (Bio-Rad, MCA771G), NeuN (Merck-Millipore, MAB377), and TUNEL (Sigma Aldrich, In situ Cell Death Detection Kit, TMR red, 12156792910).
Platelet Functional Assays
Determination of platelet counts, flow cytometry assays, preparation of washed platelets, in vitro aggregometry, and platelet adhesion under flow was performed as described previously.19,20
Statistical Analysis
All results are expressed as mean±SEM. For statistical analysis, the GraphPad Prism 6 software package was used. Data were tested for Gaussian distribution with the D’Agostino-Pearson omnibus normality test and then analyzed by 1-way ANOVA or in the case of measuring the effects of 2 factors simultaneously by 2-way ANOVA with post hoc Bonferroni adjustment for P values. Nonparametric functional outcome scores were compared by Kruskal-Wallis test with post hoc Dunn corrections. If only 2 groups were compared, an unpaired, 2-tailed Student t test, or in the case of nonparametric functional outcome, the Wilcoxon-Mann-Whitney U test, was applied. Probability values <0.05 were considered to indicate statistically significant results.
Results
Substance V Regulates Cyclic Nucleotide Signaling in the Brain and Improves Stroke Outcome in Wild-Type Mice
PDE-3 inhibitors mainly act by preventing the hydrolysis of cAMP.5 Therefore, we first assessed the total levels of cAMP in brain tissue after vehicle- or substance V treatment. In line with a previous report,3 substance V treatment showed significantly higher cAMP accumulation in the brain tissue (Figure 1A). We next analyzed whether substance V protects from acute IS in a clinically relevant setting. For this purpose, C57Bl/6 wild-type mice were subjected to 60 minutes of tMCAO and treated with substance V before tMCAO (Figure III in the online-only Data Supplement) or immediately after reperfusion (Figure 1B). In substance V–treated mice infarct volumes on day 1 were significantly reduced compared with vehicle treatment without an increase of cerebral bleeding complications as histologically assessed with the use of the TTC stainings. Accordingly, functional outcome was improved in the treatment group on day 1 using the Bederson score and the grip test (Figure 1C).
Figure 1.
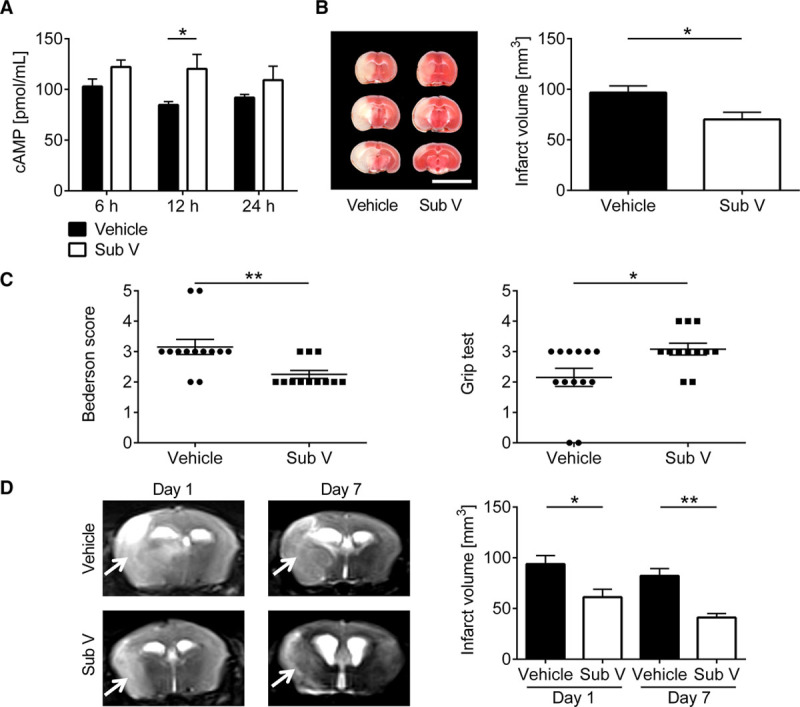
Therapeutic treatment with substance V (Sub V) reduces stroke severity without increasing the risk of intracerebral hemorrhages. A, Levels of cAMP in the brain tissue of vehicle-treated mice (0.5% dimethyl sulfoxide) or mice treated with 0.5 mg/kg Sub V at 6, 12, and 24 h after treatment (n=8–10). B, Left, Representative 2,3,5-triphenyltetrazolium chloride staining of 3 corresponding coronal brain sections of vehicle- or 0.5 mg/kg Sub V–treated mice euthanized 24 h after transient middle cerebral artery occlusion (tMCAO; scale bar=10 mm). The infarcts (white) appear smallest in the Sub V–treated mice and this could be confirmed by infarct volumetry (right; n=12–13). C, Bederson score (left) and grip test (right) on day 1 after tMCAO in the 2 mouse groups indicated above (n=12–13). D, Left, Serial coronal T2-weighted gradient echo magnetic resonance images (MRI) show extensive hyperintense (bright) ischemic lesions (arrows) in vehicle-treated mice on day 1 and 7 after tMCAO, whereas the infarctions were smaller in Sub V–treated mice. One representative imaging panel per group is depicted. Right, MRI based infarct volumetry (n=7–8). Statistical significances analyzed by (A) 2-way ANOVA with Bonferroni post hoc test, (B) Student t test, (C) Mann-Whitney U test, and (D) 1-way ANOVA with Bonferroni post hoc test. *P<0.05 and **P<0.01.
Feared complications of antithrombotic treatment after acute IS are intracranial hemorrhages.21 Therefore, we analyzed the consequences of substance V treatment on the dynamics of infarct development by serial magnetic resonance imaging in vivo (Figure 1D). Confirming the TTC-based planimetry (Figure 1B), the extension of cerebral infarction as delineated by the T2-weighted hyperintense lesion of substance V–treated mice was significantly smaller than in vehicle-treated mice 1 day after tMCAO. Furthermore, the size of infarctions of substance V–treated mice by sequential magnetic resonance imaging remained smaller compared with vehicle-treated mice until day 7 after tMCAO, excluding delayed infarct growth. In all animals hypointense areas, which typically indicate hemorrhages in T2-weighted images, were absent, supporting the observation that treatment with substance V does not increase the risk of cerebral hemorrhage compared with vehicle treatment, even at later stages of infarct development.
Substance V Stabilizes BBB After Stroke
BBB disruption and subsequent edema formation can cause clinical deterioration up to cerebral herniation. Given the known BBB stabilizing properties of PDE inhibitors, we first evaluated the stroke-related consequences of substance V on BBB structure and function.4,22 The extent of edema formation on day 1 after tMCAO, as reflected by the concentration of the vascular tracer Evans blue leaking into the brain parenchyma, was markedly reduced in the ipsilesional hemispheres of substance V–treated mice in comparison to vehicle-treated controls (Figure 2A). This finding correlated with a similar reduction in brain water content in mice receiving substance V after tMCAO (Figure 2B). In line with these findings, the expression of the tight junction protein claudin-3 was increased in cortices and basal ganglia of substance V–treated compared with vehicle-treated mice (Figure 2C). Accordingly, immunohistochemistry of infarcted brain tissue from substance V–treated animals consistently confirmed preserved tight junction expression, in this case claudin-5, at the cerebral microvessels 24 hours after tMCAO (Figure 2D).
Figure 2.
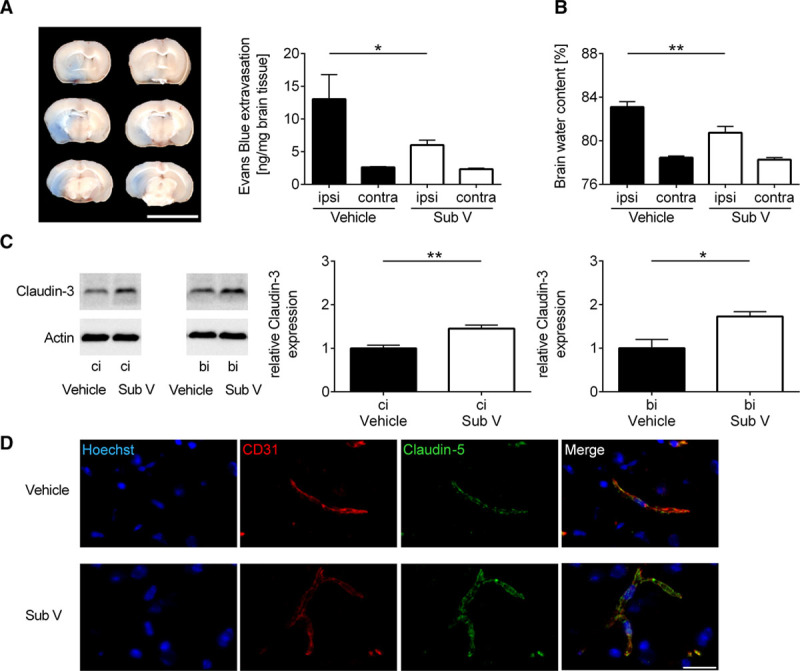
Substance V (Sub V) shows blood-brain barrier stabilization and antiedematous effects in ischemic stroke. A, Left, Representative corresponding coronal brain sections of vehicle-treated mice and mice treated with 0.5 mg/kg Sub V after the injection of the vascular tracer Evans blue. Brains were analyzed on day 1 after transient middle cerebral artery occlusion (tMCAO; scale bar=10 mm). Vascular leakage was decreased in the cortical and subcortical areas after Sub V treatment. Right, Tissue concentration of Evans blue in the ischemic (ipsi) and contralateral (contra) hemispheres of treated and untreated mice 24 h after tMCAO determined by photometry (n=9–10). B, Edema formation as measured by brain water content in the ischemic (ipsi) and contralateral (contra) hemispheres of vehicle- or Sub V–treated mice 24 h after tMCAO (n=8). C, Left, Claudin-3 protein expression in the ischemic hemispheres of vehicle-treated mice and mice treated with 0.5 mg/kg Sub V on day 1 after tMCAO as determined by immunoblot. Actin was used as loading control. Right, Densitometric quantification of claudin-3 immunoreactivity in the ipsilateral cortex (ci) and ipsilateral basal ganglia (bi) of controls and Sub V–treated mice (n=4). D, Immunohistochemistry confirmed that tight junction integrity (anticlaudin-5, green) between cerebral endothelial cells (anti-CD31, red) was markedly reduced in vehicle-treated mice compared with mice treated with Sub V on day 1 after tMCAO. Hoechst staining (blue) depicts DNA. One representative panel per group out of 3 independent experiments is shown. Scale bar=20 µm. Statistical significances analyzed by (A and B) 1-way ANOVA with Bonferroni post hoc test and (C) Student t test. *P<0.05 and **P<0.01.
Substance V Reduces Inflammation After Stroke
We further analyzed downstream signaling of cAMP accumulation in the brain tissue and found elevated levels of phosphorylated PKA (protein kinase A) in the ipsilesional cortex and basal ganglia of substance V–treated mice 24 hours after tMCAO (Figure 3A). This leads to a reduction of NF-κB (nuclear factor-κB) p65 protein expression (Figure 3A) caused by downregulation of p65 gene expression levels in the same tissue (Figure IV in the online-only Data Supplement). NF-κB is a central regulator of inflammatory response which is required for the transcriptional induction of many proinflammatory mediators involved in innate immunity.23 To assess whether this and the maintenance of the BBB in substance V–treated mice have an effect on the invasion of inflammatory cells,24 we determined the amount of macrophages/microglia and neutrophils in the ischemic hemisphere by immunocytochemistry (Figure 3B). Until day 1 after stroke, less immune cells entered the ipsilesional hemispheres of substance V–treated mice. Additionally, we analyzed the gene expression profiles of several prototypic proinflammatory cytokines in the ischemic cortices and basal ganglia of substance V– and vehicle-treated mice 24 hours after tMCAO (Figure 3C). The mRNA expression of all tested cytokines, TNF (tumor necrosis factor)-α, IL (interleukin)-1β, and IL-6, was significantly reduced in substance V–treated animals.
Figure 3.
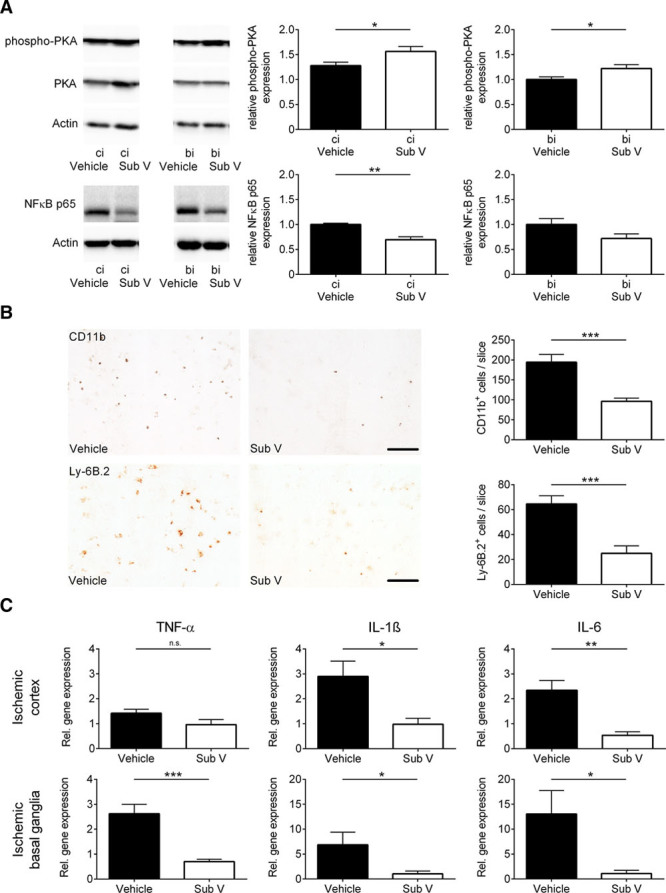
Substance V (Sub V) exerts anti-inflammatory effects in ischemic stroke. A, Intracellular downstream signaling of cAMP in the ipsilesional cortex (ci) and basal ganglia (bi) of vehicle- and Sub V–treated mice was analyzed by immunoblotting 24 h after transient middle cerebral artery occlusion (tMCAO) using antibodies against NF-κB (nuclear factor-κB) p65 and PKA (protein kinase A). The protein phosphorylation was expressed as a relative amount of phosphoprotein to total protein. Actin was used as loading control. In addition to the densitometric quantification, one representative immunoblot is shown (n=4–5). B, Left, Representative immunocytochemical staining against CD11b+ macrophages/microglia and against Ly-6B.2+ neutrophils in the ischemic hemisphere of vehicle-treated mice and mice treated with 0.5 mg/kg Sub V. Scale bar=100 µm. Right, Quantification of CD11b+ macrophages/microglia and Ly-6B.2+ neutrophils per slice in the infarcted hemispheres on day 1 after tMCAO (n=4). C, Relative gene expression of TNF (tumor necrosis factor)-α, IL (interleukin)-1β, and IL-6 in the ischemic cortices (top) and basal ganglia (bottom) of vehicle- and Sub V–treated mice 24 h after tMCAO (n=4–5). Statistical significances analyzed by (A, B, and C) Student t test and (B; Ly-6B.2+ cells) Mann-Whitney U test. *P<0.05, **P<0.01, and ***P<0.001.
Stroke Protective Effect of Substance V Depends Also on Enhanced Neuroprotection
Another well-established mechanism of tissue damage in IS is neuronal apoptosis. It has previously been shown that PDE inhibitors dampen neuronal damage in acute brain injuries.4,10,25 Therefore, we aimed to clarify the underlying mechanism of a possible neuroprotective effect of substance V. Looking for 2 cellular pathways which are known to be involved in neuroprotection, we found elevated expression levels of phosphorylated eNOS (endothelial nitric oxide synthase) and phosphorylated AKT (protein kinase B) in substance V–treated mice (Figure 4A). Furthermore, immunolabeling with NeuN and TUNEL indicated reduced neuronal apoptosis in substance V–treated mice 24 hours after tMCAO (Figure 4B). Additionally, we assessed the extent of neuronal apoptosis by Western blot against caspase-3 and cleaved caspase-3 (Figure 4C). The intensity of caspase-3 protein expression in the ipsilesional cortices and basal ganglia of substance V–treated animals was preserved in comparison to vehicle-treated mice, whereas the intensity of cleaved caspase-3 is elevated in vehicle-treated mice indicating less conversion of caspase-3 into its proapoptotic effector enzyme.26
Figure 4.
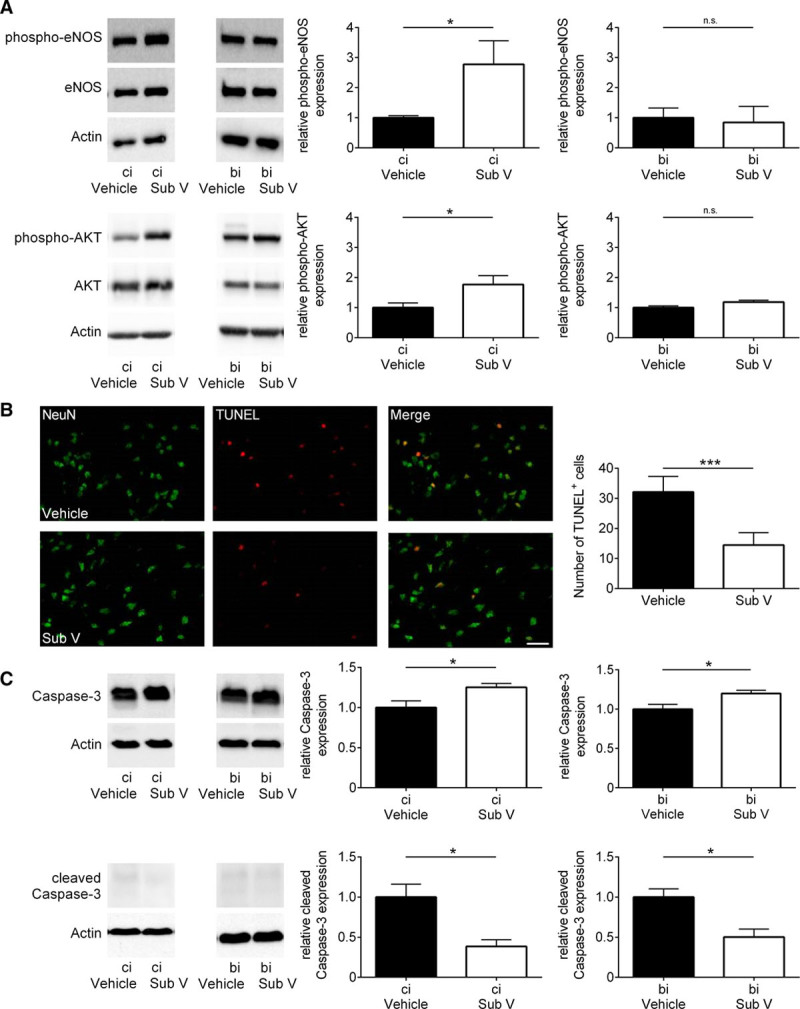
Substance V (Sub V) has an enhanced neuroprotective effect in ischemic stroke. A, Relative expression of the phosphorylated form of eNOS (endothelial nitric oxide synthase) and AKT (protein kinase B) in the ipsilesional cortex (ci) and basal ganglia (bi) of vehicle- and Sub V–treated mice 24 h after transient middle cerebral artery occlusion (tMCAO) determined by immunoblot and densitometric quantification of the bands (n=4–5). The protein phosphorylation was expressed as a relative amount of phosphoprotein to total protein. Actin was used as loading control. B, Left, Representative brain sections from vehicle-treated and Sub V (0.5 mg/kg)–treated mice 24 h after tMCAO immunolabeled for the neuronal marker NeuN and subjected to TUNEL to depict apoptosis (scale bar=25 µm). Right, The number of TUNEL-positive neurons in the ischemic hemispheres of controls and Sub V–treated mice on day 1 after tMCAO (n=4). C, Left, Caspase-3 and cleaved caspase-3 protein expression in the ipsilesional cortex (ci) and basal ganglia (bi) of vehicle-treated mice and mice treated with 0.5 mg/kg Sub V on day 1 after tMCAO as determined by immunoblot. Right, Densitometric quantification of caspase-3 and cleaved caspase-3 immunoreactivity in the ischemic regions of controls and Sub V–treated mice (n=4). Statistical significances analyzed by (A and C) Student t test and (B) Mann-Whitney U test. *P<0.05 and ***P<0.001.
Substance V Does Not Alter Platelet Function
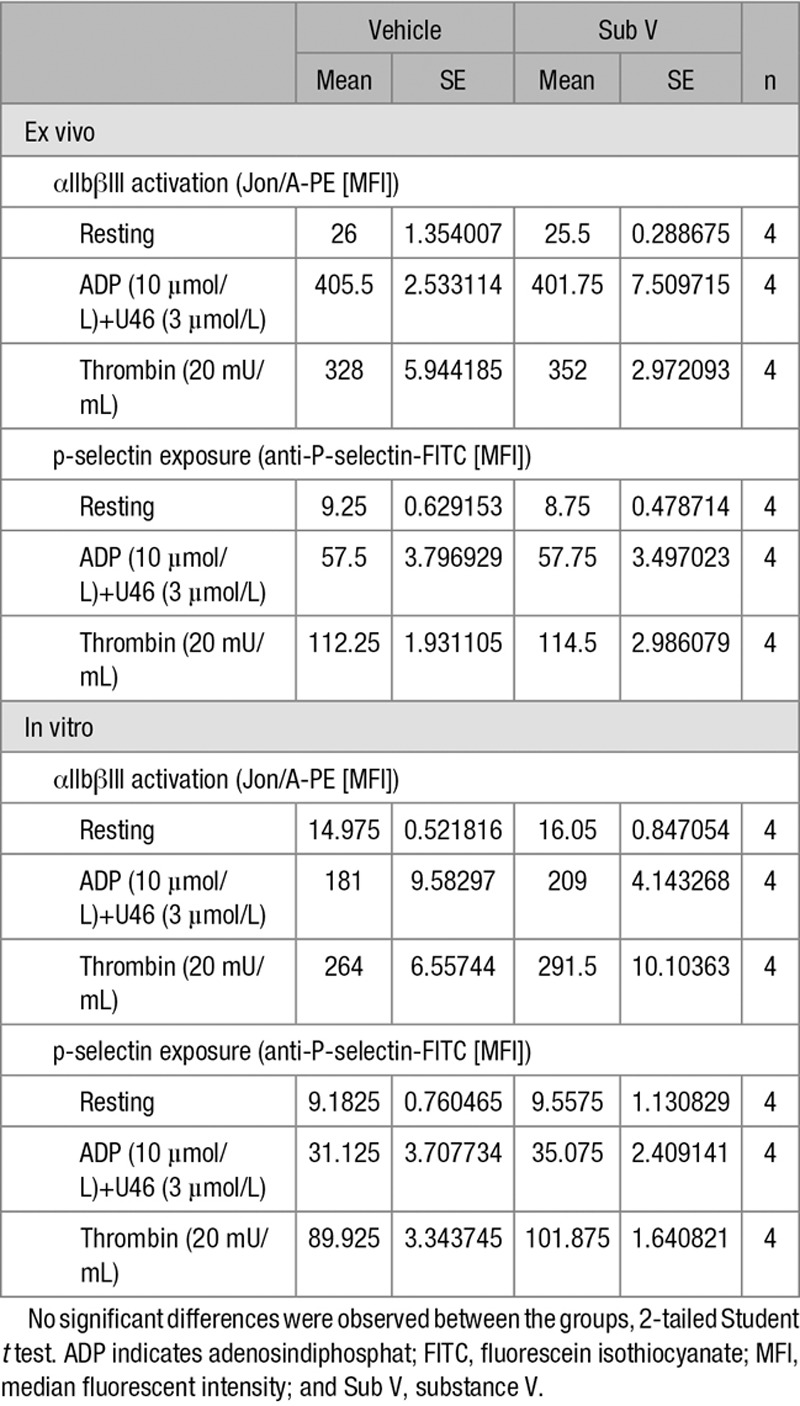
Given the fact that one main target of PDE inhibitors is platelets,3 we analyzed the direct effect of substance V treatment on platelets and their function (Table; Table III in the online-only Data Supplement). In addition to unchanged platelet counts, there was no significant difference in platelet activation reflected by αIIbβIII integrin activation and p-selectin exposure in ex vivo and in vitro analysis of substance V– and vehicle-treated platelets. Furthermore, we analyzed thrombin- and collagen-dependent platelet aggregation with in vitro aggregometry, and platelet adhesion on collagen underflow via surface coverage and relative thrombus volume determination. There was no difference between substance V and vehicle-treated animals.
Table.
Platelet Activation After Vehicle or Sub V Treatment
Superior Therapeutic Outcome of Substance V Treatment in Comparison to Cilostazol
Finally, we aimed to examine whether substance V treatment is as effective as treatment with cilostazol, which has been shown to effectively reduce stroke damage after experimental stroke.9,10 Administration of substance V reduced infarct volumes and also improved functional outcome on day 1 after tMCAO in comparison to control mice and also mice treated with cilostazol (Figure 5A and 5B).
Figure 5.
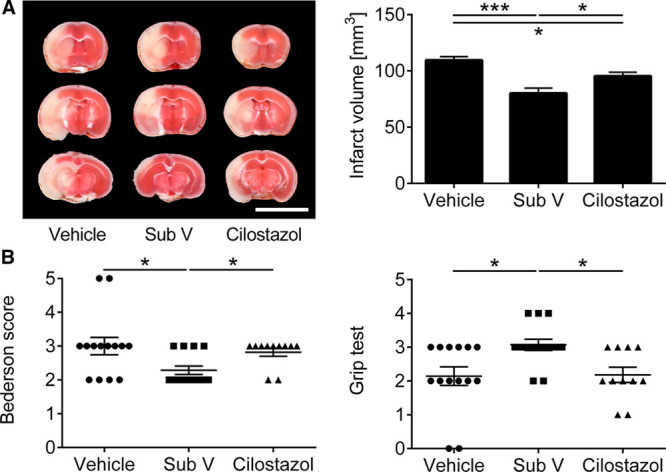
Superior therapeutic outcome of substance V (Sub V) treatment in comparison to Cilostazol. A, Left, Representative 2,3,5-triphenyltetrazolium chloride staining of 3 corresponding coronal brain sections of vehicle-, 0.5 mg/kg Sub V–, or 10 mg/kg Cilostazol-treated mice euthanized 24 h after transient middle cerebral artery occlusion (tMCAO; scale bar=10 mm). In comparison to the 2 other groups, the infarcts (white) appear smallest in the Sub V–treated mice and this could be confirmed by infarct volumetry (right; n=11–14). B, Bederson score (left) and grip test (right) on day 1 after tMCAO in the 3 mouse groups indicated above (n=11–14). Statistical significances analyzed by (A) 1-way ANOVA with Bonferroni post hoc test and (B) Kruskal-Wallis test with post hoc Dunn corrections. *P<0.05 and ***P<0.001.
Discussion
In the present study, we demonstrate that the novel PDE-3 inhibitor substance V protects from stroke in a clinically meaningful setting. Therapeutic administration after the onset of ischemia reduced infarct volume and improved neurological outcome. The protective effect was driven by a combination of BBB stabilization, the reduction of the local inflammatory response in the brain and reduced apoptotic mechanisms. Importantly, the protective effect seems to be independent of platelet function. Clinically relevant, the novel PDE-3 inhibitor substance V seems to have superior protective effects compared with cilostazol, a well-known PDE-3 inhibitor approved for peripheral artery disease and investigated in large clinical IS studies.8,9
Within their function as modulators of intracellular cAMP and their wide distribution in the brain, PDEs are viable therapeutic targets in neurological disorders of neuronal survival (Huntington disease), cognition and memory (Alzheimer disease), mood (anxiety, depression), and thought (Schizophrenia).27–31 Additionally, PDEs show beneficial effects after ischemia-reperfusion injury in different organs such as the lung, the kidney, the gut, and especially the brain.4,32–34 These largely consistent findings in CNS disorders of different causes support the principal therapeutic potential of PDE inhibitors in the field of neurology.
The observation of a substantial reduction in proinflammatory cytokine levels, edema formation, and immune cell invasion into the brain induced by application of a PDE-4 inhibitor (rolipram) supports our findings.4 Also, another PDE-3 inhibitor (cilostazol) had already been described as neuroprotective against brain ischemia.10,35 In these studies, cilostazol decreased ischemic brain infarction in association with inhibition of apoptotic and oxidative cell death,35 reduction of hemorrhagic transformation, and reduction in BBB permeability.22
In our experimental setup, treatment with substance V did not show direct effects on platelets. Therefore, it can be assumed that platelet-specific effects play at the most a minor role in the stroke protection observed in substance V–treated mice. In contrast, substance V treatment significantly reduced the numbers of neutrophils and macrophages/activated microglia invading the brain after tMCAO. Neutrophils and macrophages/microglia have been shown to be involved in stroke development by producing free radicals and other neurotoxic factors.36 Nevertheless, the pathophysiological role of neutrophils in IS development in rodents and humans has not fully been understood so far.37 The expression of soluble immune mediators was altered in substance V–treated animals: mice expressed lower amounts of proinflammatory TNF-α, IL-1β, and IL-6 in the ipsilateral cortices and basal ganglia. TNF-α, IL-1β, and IL-6 are prototypic cytokines that contribute to ischemia-reperfusion injury.38
Our observations in the tMCAO model are in good accordance with previous in vitro and in vivo studies describing PDE-3 inhibition dependent cAMP increase, downstream increase of phospho-PKA, phospho-eNOS, and phospho-AKT, and decrease of NF-κB. Therefore, PDE-3 can be considered as an important anti-inflammatory and antiapoptotic signaling mediator.39,40 The neuroprotective mechanisms of substance V in stroke have to be further investigated in different stroke models (eg, hypertensive animals) to increase translational validity.
Recently, a study showed that in mouse models of sciatic nerve crush and lung fibrosis low cAMP levels reduce the release of endogenous tPA leading to impaired fibrin degradation. This effect was partially reversible by forskolin which, similar to substance V, increases intracellular cAMP levels.4,41 Therefore, enhancement of fibrinolysis via PDE-3 inhibition could additionally contribute to the protective effects of substance V.
Taken together, substance V improves stroke outcome by modulating pivotal mechanisms of ischemic neurodegeneration such as BBB disruption, inflammation, and neuronal apoptosis. Given our finding that substance V protects mice better than cilostazol and given the fact that currently there are ongoing clinical trials employing cilostazol in IS, substance V might become the new lead compound within the class of PDE-3 inhibitors and could move forward towards clinical trials.42 One has to bear in mind, however, that the safety and efficacy of substance V in patients with stroke are unknown so far and findings from animal experiments cannot be easily transferred to the human situation as seen in PDE-3 inhibition in patients with multiple sclerosis.43 Another unresolved issue is the optimum dosage, the time point and delivery of substance V, that is, single versus continuous application, as well as its long-term effects in the setting of acute IS. In addition, based on our experiments it cannot be concluded that substance V reduces stroke risk. To address that question, a completely different experimental design including a model of spontaneous IS would be necessary. Furthermore, in future studies, potential interactions between substance V and r-tPA have to be investigated as it is known that parallel treatment with acetylsalicylic acid and r-tPA increases the risk for bleeding complications. In conclusion, our study demonstrates that the PDE-3 inhibitor substance V protects from IS in a clinically meaningful setting by a combined antiedematous, anti-inflammatory, and neuroprotective mechanism. Modulation of the PDE-3 cAMP signaling pathway might become a novel effective and safe strategy to combat IS and possibly other thromboembolic disorders in the future.
Acknowledgments
We thank L. Frieß, S. Braunschweig, G. Köllner, B. Martin, and S. Hellmig for excellent technical assistance. Dr Vaidya thanks CSIR, Delhi for Emeritus Scientist honor.
Sources of Funding
This study was supported by the Deutsche Forschungsgemeinschaft. The sponsor did not play any active role in the study.
Disclosures
Dr Pham reports a grant from the German Research Foundation outside the submitted work. The other authors report no conflicts.
Supplementary Material
{kind=link}
Footnotes
The online-only Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/STROKEAHA.118.023664.
References
- 1.Berkhemer OA, Fransen PS, Beumer D, van den Berg LA, Lingsma HF, Yoo AJ, et al. MR CLEAN Investigators. A randomized trial of intraarterial treatment for acute ischemic stroke. N Engl J Med. 2015;372:11–20. doi: 10.1056/NEJMoa1411587. doi: 10.1056/NEJMoa1411587. [DOI] [PubMed] [Google Scholar]
- 2.Zinkstok SM, Beenen LF, Majoie CB, Marquering HA, de Haan RJ, Roos YB. Early deterioration after thrombolysis plus aspirin in acute stroke: a post hoc analysis of the Antiplatelet Therapy in Combination with Recombinant t-PA Thrombolysis in Ischemic Stroke trial. Stroke. 2014;45:3080–3082. doi: 10.1161/STROKEAHA.114.006268. doi: 10.1161/STROKEAHA.114.006268. [DOI] [PubMed] [Google Scholar]
- 3.Gresele P, Momi S, Falcinelli E. Anti-platelet therapy: phosphodiesterase inhibitors. Br J Clin Pharmacol. 2011;72:634–646. doi: 10.1111/j.1365-2125.2011.04034.x. doi: 10.1111/j.1365-2125.2011.04034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kraft P, Schwarz T, Göb E, Heydenreich N, Brede M, Meuth SG, et al. The phosphodiesterase-4 inhibitor rolipram protects from ischemic stroke in mice by reducing blood-brain-barrier damage, inflammation and thrombosis. Exp Neurol. 2013;247:80–90. doi: 10.1016/j.expneurol.2013.03.026. doi: 10.1016/j.expneurol.2013.03.026. [DOI] [PubMed] [Google Scholar]
- 5.Maurice DH, Ke H, Ahmad F, Wang Y, Chung J, Manganiello VC. Advances in targeting cyclic nucleotide phosphodiesterases. Nat Rev Drug Discov. 2014;13:290–314. doi: 10.1038/nrd4228. doi: 10.1038/nrd4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morley RL, Sharma A, Horsch AD, Hinchliffe RJ. Peripheral artery disease. BMJ. 2018;360:j5842. doi: 10.1136/bmj.j5842. doi: 10.1136/bmj.j5842. [DOI] [PubMed] [Google Scholar]
- 7.Huang Y, Cheng Y, Wu J, Li Y, Xu E, Hong Z, et al. Cilostazol versus Aspirin for Secondary Ischaemic Stroke Prevention Cooperation; Investigators. Cilostazol as an alternative to aspirin after ischaemic stroke: a randomised, double-blind, pilot study. Lancet Neurol. 2008;7:494–499. doi: 10.1016/S1474-4422(08)70094-2. doi: 10.1016/S1474-4422(08)70094-2. [DOI] [PubMed] [Google Scholar]
- 8.Shinohara Y, Katayama Y, Uchiyama S, Yamaguchi T, Handa S, Matsuoka K, et al. CSPS 2 group. Cilostazol for prevention of secondary stroke (CSPS 2): an aspirin-controlled, double-blind, randomised non-inferiority trial. Lancet Neurol. 2010;9:959–968. doi: 10.1016/S1474-4422(10)70198-8. doi: 10.1016/S1474-4422(10)70198-8. [DOI] [PubMed] [Google Scholar]
- 9.Wakida K, Morimoto N, Shimazawa M, Hozumi I, Nagase H, Inuzuka T, et al. Cilostazol reduces ischemic brain damage partly by inducing metallothionein-1 and -2. Brain Res. 2006;1116:187–193. doi: 10.1016/j.brainres.2006.07.125. doi: 10.1016/j.brainres.2006.07.125. [DOI] [PubMed] [Google Scholar]
- 10.Ye YL, Shi WZ, Zhang WP, Wang ML, Zhou Y, Fang SH, et al. Cilostazol, a phosphodiesterase 3 inhibitor, protects mice against acute and late ischemic brain injuries. Eur J Pharmacol. 2007;557:23–31. doi: 10.1016/j.ejphar.2006.11.003. doi: 10.1016/j.ejphar.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 11.Ravinder M, Mahendar B, Mattapally S, Hamsini KV, Reddy TN, Rohit C, et al. Synthesis and evaluation of novel 2-pyridone derivatives as inhibitors of phosphodiesterase3 (PDE3): a target for heart failure and platelet aggregation. Bioorg Med Chem Lett. 2012;22:6010–6015. doi: 10.1016/j.bmcl.2012.05.019. doi: 10.1016/j.bmcl.2012.05.019. [DOI] [PubMed] [Google Scholar]
- 12.Sorkin EM, Markham A. Cilostazol. Drugs Aging. 1999;14:63; discussion 72–71; discussion 72. doi: 10.2165/00002512-199914010-00005. doi: 10.2165/00002512-199914010-00005. [DOI] [PubMed] [Google Scholar]
- 13.Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol. 2010;8:e1000412. doi: 10.1371/journal.pbio.1000412. doi: 10.1371/journal.pbio.1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schuhmann MK, Kraft P, Stoll G, Lorenz K, Meuth SG, Wiendl H, et al. CD28 superagonist-mediated boost of regulatory T cells increases thrombo-inflammation and ischemic neurodegeneration during the acute phase of experimental stroke. J Cereb Blood Flow Metab. 2015;35:6–10. doi: 10.1038/jcbfm.2014.175. doi: 10.1038/jcbfm.2014.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H. Rat middle cerebral artery occlusion: evaluation of the model and development of a neurologic examination. Stroke. 1986;17:472–476. doi: 10.1161/01.str.17.3.472. [DOI] [PubMed] [Google Scholar]
- 16.Moran PM, Higgins LS, Cordell B, Moser PC. Age-related learning deficits in transgenic mice expressing the 751-amino acid isoform of human beta-amyloid precursor protein. Proc Natl Acad Sci USA. 1995;92:5341–5345. doi: 10.1073/pnas.92.12.5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Langhauser F, Göb E, Kraft P, Geis C, Schmitt J, Brede M, et al. Kininogen deficiency protects from ischemic neurodegeneration in mice by reducing thrombosis, blood-brain barrier damage, and inflammation. Blood. 2012;120:4082–4092. doi: 10.1182/blood-2012-06-440057. doi: 10.1182/blood-2012-06-440057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bieber M, Werner RA, Tanai E, Hofmann U, Higuchi T, Schuh K, et al. Stroke-induced chronic systolic dysfunction driven by sympathetic overactivity. Ann Neurol. 2017;82:729–743. doi: 10.1002/ana.25073. doi: 10.1002/ana.25073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deppermann C, Cherpokova D, Nurden P, Schulz JN, Thielmann I, Kraft P, et al. Gray platelet syndrome and defective thrombo-inflammation in Nbeal2-deficient mice. J Clin Invest. 2013;123:3331–3342. doi: 10.1172/JCI69210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nieswandt B, Brakebusch C, Bergmeier W, Schulte V, Bouvard D, Mokhtari-Nejad R, et al. Glycoprotein VI but not alpha2beta1 integrin is essential for platelet interaction with collagen. EMBO J. 2001;20:2120–2130. doi: 10.1093/emboj/20.9.2120. doi: 10.1093/emboj/20.9.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Flaherty ML, Kissela B, Woo D, Kleindorfer D, Alwell K, Sekar P, et al. The increasing incidence of anticoagulant-associated intracerebral hemorrhage. Neurology. 2007;68:116–121. doi: 10.1212/01.wnl.0000250340.05202.8b. doi: 10.1212/01.wnl.0000250340.05202.8b. [DOI] [PubMed] [Google Scholar]
- 22.Nonaka Y, Tsuruma K, Shimazawa M, Yoshimura S, Iwama T, Hara H. Cilostazol protects against hemorrhagic transformation in mice transient focal cerebral ischemia-induced brain damage. Neurosci Lett. 2009;452:156–161. doi: 10.1016/j.neulet.2009.01.039. doi: 10.1016/j.neulet.2009.01.039. [DOI] [PubMed] [Google Scholar]
- 23.Harari OA, Liao JK. NF-κB and innate immunity in ischemic stroke. Ann N Y Acad Sci. 2010;1207:32–40. doi: 10.1111/j.1749-6632.2010.05735.x. doi: 10.1111/j.1749-6632.2010.05735.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Magnus T, Wiendl H, Kleinschnitz C. Immune mechanisms of stroke. Curr Opin Neurol. 2012;25:334–340. doi: 10.1097/WCO.0b013e328352ede6. doi: 10.1097/WCO.0b013e328352ede6. [DOI] [PubMed] [Google Scholar]
- 25.Li LX, Cheng YF, Lin HB, Wang C, Xu JP, Zhang HT. Prevention of cerebral ischemia-induced memory deficits by inhibition of phosphodiesterase-4 in rats. Metab Brain Dis. 2011;26:37–47. doi: 10.1007/s11011-011-9235-0. doi: 10.1007/s11011-011-9235-0. [DOI] [PubMed] [Google Scholar]
- 26.Djafarzadeh S, Vuda M, Takala J, Jakob SM. Effect of remifentanil on mitochondrial oxygen consumption of cultured human hepatocytes. PLoS One. 2012;7:e45195. doi: 10.1371/journal.pone.0045195. doi: 10.1371/journal.pone.0045195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.García-Osta A, Cuadrado-Tejedor M, García-Barroso C, Oyarzábal J, Franco R. Phosphodiesterases as therapeutic targets for Alzheimer’s disease. ACS Chem Neurosci. 2012;3:832–844. doi: 10.1021/cn3000907. doi: 10.1021/cn3000907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang HT, Huang Y, Masood A, Stolinski LR, Li Y, Zhang L, et al. Anxiogenic-like behavioral phenotype of mice deficient in phosphodiesterase 4B (PDE4B). Neuropsychopharmacology. 2008;33:1611–1623. doi: 10.1038/sj.npp.1301537. doi: 10.1038/sj.npp.1301537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wild EJ, Tabrizi SJ. Targets for future clinical trials in Huntington’s disease: what’s in the pipeline? Mov Disord. 2014;29:1434–1445. doi: 10.1002/mds.26007. doi: 10.1002/mds.26007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Siuciak JA. The role of phosphodiesterases in schizophrenia: therapeutic implications. CNS Drugs. 2008;22:983–993. doi: 10.2165/0023210-200822120-00002. doi: 10.2165/0023210-200822120-00002. [DOI] [PubMed] [Google Scholar]
- 31.O’Donnell JM, Xu Y. Evidence for global reduction in brain cyclic adenosine monophosphate signaling in depression. Biol Psychiatry. 2012;72:524–525. doi: 10.1016/j.biopsych.2012.07.017. doi: 10.1016/j.biopsych.2012.07.017. [DOI] [PubMed] [Google Scholar]
- 32.Souza DG, Cassali GD, Poole S, Teixeira MM. Effects of inhibition of PDE4 and TNF-alpha on local and remote injuries following ischaemia and reperfusion injury. Br J Pharmacol. 2001;134:985–994. doi: 10.1038/sj.bjp.0704336. doi: 10.1038/sj.bjp.0704336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Okusa MD, Linden J, Huang L, Rosin DL, Smith DF, Sullivan G. Enhanced protection from renal ischemia-reperfusion [correction of ischemia:reperfusion] injury with A(2A)-adenosine receptor activation and PDE 4 inhibition. Kidney Int. 2001;59:2114–2125. doi: 10.1046/j.1523-1755.2001.00726.x. doi: 10.1046/j.1523-1755.2001.00726.x. [DOI] [PubMed] [Google Scholar]
- 34.Kyoi T, Kitazawa S, Tajima K, Zhang X, Ukai Y. Phosphodiesterase type IV inhibitors prevent ischemia-reperfusion-induced gastric injury in rats. J Pharmacol Sci. 2004;95:321–328. doi: 10.1254/jphs.fpj04009x. [DOI] [PubMed] [Google Scholar]
- 35.Choi JM, Shin HK, Kim KY, Lee JH, Hong KW. Neuroprotective effect of cilostazol against focal cerebral ischemia via antiapoptotic action in rats. J Pharmacol Exp Ther. 2002;300:787–793. doi: 10.1124/jpet.300.3.787. [DOI] [PubMed] [Google Scholar]
- 36.Jin R, Yang G, Li G. Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J Leukoc Biol. 2010;87:779–789. doi: 10.1189/jlb.1109766. doi: 10.1189/jlb.1109766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Strecker JK, Schmidt A, Schäbitz WR, Minnerup J. Neutrophil granulocytes in cerebral ischemia - Evolution from killers to key players. Neurochem Int. 2017;107:117–126. doi: 10.1016/j.neuint.2016.11.006. doi: 10.1016/j.neuint.2016.11.006. [DOI] [PubMed] [Google Scholar]
- 38.Allan SM, Tyrrell PJ, Rothwell NJ. Interleukin-1 and neuronal injury. Nat Rev Immunol. 2005;5:629–640. doi: 10.1038/nri1664. doi: 10.1038/nri1664. [DOI] [PubMed] [Google Scholar]
- 39.Aizawa T, Wei H, Miano JM, Abe J, Berk BC, Yan C. Role of phosphodiesterase 3 in NO/cGMP-mediated antiinflammatory effects in vascular smooth muscle cells. Circ Res. 2003;93:406–413. doi: 10.1161/01.RES.0000091074.33584.F0. doi: 10.1161/01.RES.0000091074.33584.F0. [DOI] [PubMed] [Google Scholar]
- 40.Ding B, Abe J, Wei H, Xu H, Che W, Aizawa T, et al. A positive feedback loop of phosphodiesterase 3 (PDE3) and inducible cAMP early repressor (ICER) leads to cardiomyocyte apoptosis. Proc Natl Acad Sci USA. 2005;102:14771–14776. doi: 10.1073/pnas.0506489102. doi: 10.1073/pnas.0506489102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sachs BD, Baillie GS, McCall JR, Passino MA, Schachtrup C, Wallace DA, et al. p75 neurotrophin receptor regulates tissue fibrosis through inhibition of plasminogen activation via a PDE4/cAMP/PKA pathway. J Cell Biol. 2007;177:1119–1132. doi: 10.1083/jcb.200701040. doi: 10.1083/jcb.200701040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Knott EP, Assi M, Rao SN, Ghosh M, Pearse DD. Phosphodiesterase inhibitors as a therapeutic approach to neuroprotection and repair. Int J Mol Sci. 2017;18:696. doi: 10.3390/ijms18040696. doi: 10.3390/ijms18040696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bielekova B, Richert N, Howard T, Packer AN, Blevins G, Ohayon J, et al. Treatment with the phosphodiesterase type-4 inhibitor rolipram fails to inhibit blood–brain barrier disruption in multiple sclerosis. Mult Scler. 2009;15:1206–1214. doi: 10.1177/1352458509345903. doi: 10.1177/1352458509345903. [DOI] [PMC free article] [PubMed] [Google Scholar]