

Abstract
The mechanism for the borylation of an aromatic substrate by a cobalt pincer complex was investigated by density functional theory calculations. Experimental observations identified trans-(iPrPNP)CoH2(BPin) as the resting state in the borylation of five-membered heteroarenes, and 4-BPin-(iPrPNP)Co(N2)BPin as the resting state in the catalytic borylation of arene substrates. The active species, 4-R-(iPrPNP)CoBPin (R=H, BPin), were generated by reductive elimination of H2 in the former, through Berry pseudorotation to the cis isomer, and N2 loss in the latter. The catalytic mechanism of the resulting Co(I) complex was computed to involve three main steps: C-H oxidative addition of the aromatic substrate (C6H6), reductive elimination of PhBPin, and regeneration of the active complex. The oxidative addition product formed through the most favorable pathway, where the breaking C-H bond of C6H6 is parallel to a line between the two phosphine atoms, leaves the complex with a distorted PNP ligand, which rearranges to a more stable complex via dissociation and re-association of HBPin. Alternative pathways, σ-bond metathesis and the oxidative addition in which the breaking C-H bond is parallel to the Co-B bond, are predicted to be unlikely for this Co(I) complex. The thermodynamically favorable formation of the product PhBPin via reductive elimination drives the reaction forward. The active species regenerates through the oxidative addition of B2Pin2 and reductive elimination of HBPin. In the overall reaction, the flipping (refolding) of the five-membered phosphine rings, which connects the species with two phosphine rings folded in the same direction and that with them folded in different directions, is found to play an important role in the catalytic process, as it relieves steric crowding within the PNP ligand and opens Co coordination space. Metal-ligand cooperation based on the ligand’s aromatization/dearomatization, a common mechanism for heavy-metal pincer complexes, and the dissociation of one phosphine ligand, do not apply in this system. This study provides guidance for understanding important features of pincer ligands with first-transition-row metals that differ from those in heavier metal complexes.
Keywords: cobalt pincer complex, catalytic C-H borylation, the flipping of the five-membered phosphine rings, DFT calculations, mechanistic studies
Graphical Abstract
1. Introduction
Catalytic C-H borylation is one of the most attractive methods for C-H functionalization, as the C-B products are versatile synthetic reagents for constructing new carbon-carbon and carbon-heteroatom bonds.1 Efforts by many researchers have developed catalytic borylations for C(sp3)-H, C(sp2)-H,1–6 C(sp)-H bonds,7–11 and chemo-, regio-, and stereo-selective C-H borylations.1–4,9–11 Among them, catalytic C-H borylation of arenes and heteroarenes is a particularly attractive approach to synthesize organoboronate esters, which are key synthetic intermediates for Suzuki-Miyaura cross-coupling reactions.1–6
Transition-metal complexes have been widely applied to catalyze the C-H borylations, for which the iridium-based complexes are most common.1–5,8 Furthermore, some iridium complexes have been reported to catalyze methane borylations,12,13 a major challenge. However, widespread applications for iridium and other noble metal catalysis suffer from their many disadvantages: expense, toxicity, scarcity, and difficulty in separation. These limitations have motivated researchers to develop catalysts with earth abundant first transition-row metals,14,15 paving the way for sustainable and environmentally friendly synthesis. Although Hartwig and coworkers reported several C-H borylations with CpFe(CO)2Bcat (Cp = η5-C5H5; cat = O2C6H4) under photochemical conditions in 1995, catalytic reactions were not observed in their systems.16 Significant progress in developing catalytic C-H borylations with first transition-row metal complexes has only been made recently. Although catalytic C-H borylations has been reported by using metal-free systems based on the concept of “Frustrated Lewis Pairs”,17–21 most of the work has involved bimetallic copper-iron,22–24 zinc-iron complexes,22,23 monometallic iron complexes,25–27 and recently a series of cobalt28–35 and nickel complexes.36,37
Important applications of C-H borylations in synthetic chemistry need new efficient catalysts, whose development can benefit from fully understanding the catalytic mechanism. Such mechanisms at various level of detail have been reported from both experiments and computations.20,23,33,34,38–45 Furthermore, efforts to elucidate the origin of chemo-, regio-, and stereo-selectivity have also had some success.4,46–54 Generally, catalytic borylations involve C-H bond cleavage by oxidative addition and C-B bond formation by reductive elimination, routes that utilize the inherent two-electron redox property of heavier transition metals. However, metal-assisted σ-bond metathesis mechanism has been suggested for the C-H bond cleavage and H transfer,39,42,45,47,49 a route that avoids the formation of higher oxidation-state intermediates. In addition, both steric and electronic effects, together with weak interactions between ligands and substrates, were found to govern the selectivity of some reactions.47,49,50,54
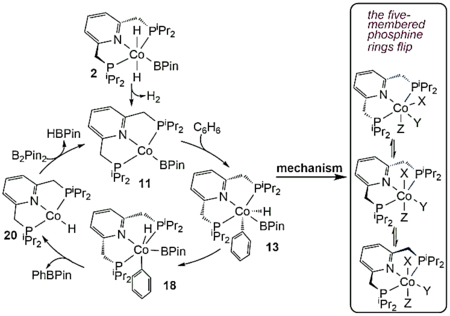
In spite of these studies, detailed mechanistic investigations of C-H borylations with first transition-row metals are still scarce.20,43 Unlike heavier transition metals with two-electron redox chemistry, the first-row metals usually demonstrate one-electron redox processes,14,15 a difference that suggest some new mechanistic pathways may be involved. Furthermore, variable coordination geometries and multiple spin states of the first transition-row metal complexes, especially those with weak field ligands, challenge characterizations of their electronic structures and catalytic reactivity. Thus, metal-metal cooperativity has been recognized to facilitate the catalytic photochemical C-H borylation with bimetallic Cu-Fe and Zn-Fe complexes, which may mimic heavier transition metal complexes.22–24 Herein, we are interested in understanding the catalytic mechanism for the C-H borylations of heterocycles and arenes with several pincer-ligated cobalt complexes reported by Chirik and coworkers.28 Among their tested cobalt complexes, the pyridine-based PNP pincer complex, (iPrPNP)Co(CH2SiMe3) (iPrPNP = 2,6-bis(di-iso-propyl)phosphinomethyl)pyridine, 1 in Scheme 1), was observed to perform best. With the catalyst 1, the borylation of methyl furan-2-carboxylate has achieved up to 5000 turnovers at 23 °C (eq 1 in Scheme 1), and the borylation of benzene has also been realized with excess benzene (benzene: B2Pin2 = 20:1, Pin = O2C2Me4) at 80 °C (eq 2 in Scheme 1). The trans-dihydride cobalt complex, trans-(iPrPNP)CoH2(BPin) (Pin = O2C2Me4, complex 2 in Figure 1), synthesized separately by adding two equivalents of HBPin to the cobalt alkyl compound 1 and releasing one equivalent of Me3SiCH2BPin, was identified as the resting state in the borylation of 2-methylfuran with HBPin. According to their proposed mechanism (Figure 1), complex 2 first isomerizes to a cis-dihydride cobalt complex, which then releases H2 to generate the active species (iPrPNP)CoBPin, followed by the oxidative addition of a C-H bond and the subsequent reductive elimination of the B-C bond to form the borylated product. Finally, addition of HBPin to (iPrPNP)CoH regenerates complex 2. Although several experiments have been conducted to explore the catalytic mechanism and the structures of cobalt intermediates,28–34,55 a complete catalytic cycle has not been established yet. Furthermore, related PNP pincer ligated metal complexes synthesized by Milstein and coworkers are well known to show a mode of metal-ligand cooperation based on the ligand’s aromatization/dearomatization.56–58 This gives rise to an interesting question that whether the trans-(iPrPNP)CoH2(BPin) complex shows similar reactivity or not during its catalytic cycle. Building on our previous mechanistic studies of the borylation with transition metal complexes.39,42,48 and studies of Milstein’s systems,58,59 we herein employed density functional theory (DFT) calculations to fully understand the catalytic mechanism by computing the detailed reaction pathways for the proposed mechanism as shown in Figure 1.
Scheme 1.

A cobalt pincer complex 1 and its catalyzed borylation reactions.
Figure 1.

A proposed mechanism for the catalytic borylation of 2-methylfuran with HBPin from the resting-state complex 2 in ref. 28.
2. Computational methods
Detailed DFT calculations of the catalytic mechanism were conducted by using benzene and B2Pin2 as representative substrates with catalyst 1 (eq 1 in Scheme 1), as all its C-H bonds are equal, benzene simplifies our explorations on the mechanism. With the ωB97XD functional,60 geometries of all intermediates and transition states were optimized in gas phase by employing the basis set BS1, where atoms of the iPrPNP ligand except N and P use cc-pVDZ61 and the others (including the Co) use Def2TZVP.62 On the basis of gas-phase optimized geometries, single point calculations were conducted in C6H6 solvent with the SMD solvent model63 at the level of ωB97XD/Def2TZVP, where all the atoms use Def2TZVP. In addition, an ‘ultrafine’ grid was used to make optimizations of large molecules with many soft modes such as methyl rotations more reliable. Furthermore, ‘5D’ option was used for all d functions in these calculations. Unless noted otherwise, free energies and enthalpies in C6H6 solvent together with the free energies in gas phase, which are represented by ΔGC6H6[ΔHC6H6](ΔGgas), respectively, are reported in this work.
In addition to calculating the experimentally reported system, we also computed a simplified model system to explore various alternative reaction pathways. In the simplified model system, we replaced all the iPr substituents of the iPrPNP ligand with Me, and further replaced B2Pin2 with B2eg2 (eg = ethyleneglycolato) to save computation time. The ωB97XD functional was also used to calculate the model system. Geometries of all intermediates and transition states were optimized in gas phase at the level of BS2, where Co employs LANL2DZ ECP-basis set64 and the others use cc-pVDZ. With the optimized geometries in gas phase, the energetic results were refined by single point calculations with the SMD solvent model at the level of ωB97XD/BS3, where Co employs SDD ECP-basis set65 and the others use cc-pVTZ. The THF solvent, which was used in the catalytic borylation of some substituted pyridines in experiments, was selected in the SMD single point calculations. Free energies and enthalpies in THF and the free energies in gas phase, which are represented by ΔGTHF[ΔHTHF](ΔGgas), were reported. Results for the model system are presented mainly in the Supporting Information.
Results reported in this work are based on optimized geometries for closed shell singlet states. However, triplet states or open shell singlet states may also be possible for cobalt complexes, especially for four-coordinate cobalt species. For example, the solid-state structure of the cobalt alkyl complex 1 was confirmed to be diamagnetic with a four-coordinate Co in a square-planar geometry, while its 1H NMR spectra appears to be somewhat paramagnetically shifted at high temperature, implying a low-lying triplet state.28 To verify this observation, several DFT functionals involving hybrid functionals (ωB97XD, M06,66 TPSSh67) and non-hybrid functionals (TPSS,67 TPSS-D3 (with D3 = Empirical Dispersion = GD3BJ,68), M06L,69 PBE70) were selected to calculate its possible structures. As expected, the relative energies between singlet and triplet states depend strongly on the functionals, where TPSS gives square-planar, singlet ground states and ωB97XD over stabilizes tetrahedral triplet states (SI1). To further confirm this, several experimentally isolated species, (iPrPNP)CoCl, (tBuPNP)CoCl, and (tBuPNP)CoH,55 were investigated by using TPSS and ωB97XD. Similarly, TPSS predicts singlet ground states consistent with experiment, while ωB97XD over stabilizes triplet states (SI1). Comparisons of the optimized geometries at TPSS and ωB97XD show that ωB97XD predicts longer Co-X bonds (X any coordinated atom) than TPSS, and these differences are larger for triplet states than for singlet states (SI1). With TPSS, singlet and triplet states of selected stationary points, especially the four-coordinate cobalt species involved in the reaction, such as (iPrPNP)CoBPin (11), (iPrPNP)CoPh (16), and (iPrPNP)CoH (20), were recalculated, and all are predicted to be singlet ground states (SI1). Thus, the singlet states are reported for the mechanism. The geometries and the energies reported for the reaction mechanism are from the ωB97XD calculations, as their predicted rate-determining barriers (around 25 kcal/mol) are more consistent with the experimental conditions at 80°C than the barriers of over 34 kcal/mol predicted by using the TPSS functional (see the following discussions). However, using TPSS to recalculate the rate-determining transition states and thermodynamics of key steps did not change the conclusions.
All the DFT calculations were performed with the Gaussian 09 program.71 The three-dimensional molecular structures reported in this work were drawn using the JIMP2 molecular visualizing and manipulating program.72–74
3. Results and discussion
Following the proposed mechanism of catalytic borylation of 2-methylfuran with HBPin from the trans-(iPrPNP)CoH2(BPin) complex (2, a resting state in the reaction) as in Figure 1,28,34 the active species (iPrPNP)CoBPin was generated from the trans-(iPrPNP)CoH2(BPin) complex by releasing H2. Furthermore, in the catalytic borylation of arene substrates, the 4-BPin-(iPrPNP)Co(N2)BPin complex (BPin substituted the 4-position of the pincer ligand) was identified as the resting state, which was proposed to form the related active species 4-BPin-(iPrPNP)CoBPin through the dissociation of N2.30 From the active species, the catalytic borylation mechanism involves similar steps: oxidative addition of the C-H bond, reductive elimination of B-C bond, and regeneration of the active species. We first examined the mechanism for the generation of the active species (iPrPNP)CoBPin from the trans-(iPrPNP)CoH2(BPin) complex to determine if aromatization/dearomatization of the pincer ligand is operative in this system or not. Then, we calculated the following three steps from the active species (iPrPNP)CoBPin by using C6H6 and B2Pin2 as substrates to reduce the computational cost: oxidative addition of C6H6, reductive elimination of B-C bond to form PhBPin, and regeneration of (iPrPNP)CoBPin. Considering that 4-BPin-(iPrPNP)CoBPin is the active species in the catalytic borylation of arene substrates, some selected transition states and intermediates involved in the oxidative addition and reductive elimination steps were recalculated by using the 4-BPin-(iPrPNP)CoBPin complex. Details for these steps will be reported separately in the following sections.
3.1. Generation of (iPrPNP)CoBPin from trans-(iPrPNP)CoH2(BPin)
In the experiments, the trans-(iPrPNP)CoH2(BPin) complex 2 was observed as a resting state in the catalytic reaction, and was proposed to release H2 to generate an active species (iPrPNP)CoBPin.28,34 However, direct release of H2 from 2 is unlikely as the two H atoms on Co are trans. Thus, an isomerization process from 2 to a cis-(iPrPNP)CoH2(BPin) complex was suggested.28 Our calculations support this suggestion, and the detailed pathway for this process is shown in Figure 2 with optimized geometries of some species in Figure 3.
Figure 2.

Calculated energy profiles for the release of H2 from trans-(iPrPNP)CoH2(BPin) complex 2, which isomerizes to cis-(iPrPNP)CoH2(BPin) before the rate determining reductive elimination.
Figure 3.

Optimized geometries of key species involved in the mechanism in Figure 2 (other species are in SI2). Some bond distances in Å, angles in °, and dihedral angles in ° are given in black, red, and green colors, respectively. Values in the parentheses in complex 2 are from its crystal structure. Geometries of 2, TS2–3, and 3 drawn in another perspective are also shown here, where iPr groups on P and BPin on Co are omitted for clarity.
The optimized geometry of complex 2 is close to its X-ray crystal structure, as the differences of bond lengths and angles between them are no more than 0.03 Å and 3.2°, respectively (see Figure 3 and their superimposed structures in SI3), an agreement that supports the methodology used here. In 2, two five-membered phosphine rings of the pincer ligand are folded in different directions; one sp3-C of the ring tips down (in blue), while the other tips up (in black). Via transition state TS2–3, the sp3-C of the phosphine ring in blue flips up, and a new complex 3 was formed. This ring-flipping process is clear from their optimized geometries and is especially obvious from the alternative perspective in Figure 3. The two hydrides at Co are trans in 3, and then move simultaneously towards the B atom via transition state TS3–4 where the Co-H1 bond lengthens and both B-H distances shorten, generating complex 4 with two unequally bridging H atoms (Co-H-B) (see its optimized geometry in Figure 3). In contrast, the stepwise pathway where the two hydrides move separately is unlikely; as optimizations of corresponding intermediates formed after moving one hydride repeatedly converge to 3 again. Complex 4 is higher than 2 by only 1.7 kcal/mol, and the formation of 4 from 2 is calculated to proceed via two low-barrier transition states, TS2–3 and TS3–4. In addition, an alternative pathway for the formation of 4 from 2, where the two hydrides at Co in 2 firstly move simultaneously toward the B atom and then the sp3-C of the phosphine ring in blue flips up to generate 4, was calculated to compete with the path in Figure 2 (see SI4). Furthermore, a dynamic process was suggested by experiments, which show a broad cobalt hydride signal in 1H NMR spectrum.28 Thus, this experimental observation can be ascribed to the rapidly transformation between 2 and 4 which represent a mer isomer and a fac isomer, respectively, the latter also displaying semibridging Hs.
In the optimized geometry of 4 (Figure 3), the N, Co, and B atoms are not in line with the BPin group bending upwards. In addition, the B-H1 bond is shorter than the B-H2 bond by 0.10 Å, implying that the B atom is bound to H1 more tightly than to H2. Correspondingly, the Co-H1 bond is longer than the Co-H2 bond by 0.19 Å. Furthermore, the B-H1 bond at 1.31 Å and the Co-H2 bond at 1.55 Å are in the normal range for these bonds. Thus, complex 4 could be considered the product formed by coordinating H1BPin to a cobalt-hydride complex (i.e., 20 in the following sections). Consistent with this proposal, a process for the dissociation of HBPin from an isomer of complex 4, where two five-membered phosphine rings of the pincer ligand are folded in different directions, has been located with a barrier of 12.4 kcal/mol higher than 4. Therefore, the H1BPin group in 4 is able to rotate in a counterclockwise direction along the B-Co axis towards P1 and simultaneously H2 moves far from B, as is described by TS4–5, to form complex 5, which adopts a distorted trigonal bipyramidal geometry in which H1BPin and phosphine ligands occupy the equatorial positions with H2 and pyridine ligands in the axial positions. Subsequently, the H1BPin group of 5 continues to rotate in an anticlockwise direction along the B-Co axis towards H2, and concurrently H2 moves downwards (i.e., far from the B atom) via transition state TS5–6, giving complex 6, which adopts a distorted trigonal bipyramidal geometry in which H2, H1BPin, and pyridine ligands occupy the equatorial positions with phosphine ligands lying almost in the axial positions. Here, we are considering H1BPin as a single σ-bonded ligand with H1-B bond occupying one coordination site, although both H1 and B are shown as coordinated to Co in Figure 3. Thus, the isomerization process from 4 to 6 via 5 proceeds like a Berry pseudorotation,75 which occurs widely in trigonal bipyramidal structures. Compared to 4, the B-H2 distance is longer and the P1-Co-P2 angle is smaller in 5 (Figure 3), thus, 5 is higher in energy due to the loss of the B-H2 bonding interaction and the steric effects of two phosphine ligands in 5. Furthermore, 6 is higher than 5 because of additional steric effects between H2, H1BPin, and pyridine ligands in the same plane. For the same reasons, the barrier of TS5–6 is also higher than that of TS4–5.
The two hydrides, which in 6 are cis with H1 bridging Co and B atoms, then simultaneously move away from B (via TS6–7) to generate 7. Complex 7 adopts an octahedral geometry with one H trans to N and the other trans to B, and the two five-membered phosphine rings in 7 are folded in the same direction. Next, one sp3-C of the phosphine ring (in blue) flips down via TS7–8 to form complex 8, cis-(iPrPNP)CoH2(BPin), which is more stable than 7 but less stable than the trans-(iPrPNP)CoH2(BPin) complex 2 by 7.8 kcal/mol. Then in TS8–9 the other phosphine ring (in black) is also flipping down to form 9, in which the BPin group is bending towards the trans position of the pyridine ligand because of steric clashes with the P substituents. The movement of BPin drives the H atoms closer and the H2-formation transition state, TS9–10, leads to an H2 σ-complex, 10, which then releases H2 to generate a four-coordinated Co(I) complex 11 with a planar geometry around Co. The formation of 11 has been confirmed in experiments by capturing it as a CO adduct, 11(CO), with CO occupying a vacant position on Co.30 It should be noted that the five-membered ring plane of BPin is perpendicular to the plane of the pyridine ring in the optimized geometry of 11. In contrast, the two planes are parallel to each other in the crystal structure of 11(CO). Comparisons of the Co-X bonds (X any coordinated atom) in the optimized geometry and crystal structure of 11(CO) show that the largest difference (0.023 Å) corresponds to the Co-N bond. When CO is added to the structure of 11, BPin rotates toward being planar, but there is a barrier to overcome to form the planar 11(CO); however, the planar structure is more stable than this intermediate structure. Furthermore, optimizations of the structure generated by removing CO from 11(CO) converge back to the geometry of 11 in Figure 3. In the process for the release of H2 from 2 (Figure 2), the rate-determining transition state is TS9–10 with a barrier of 18.5[19.8](17.6) kcal/mol, and the formation of 11 and H2 is uphill by 11.3[21.4](9.9) kcal/mol relative to complex 2. The TPSS functional predicts a more favorable process, where the barrier for TS9–10 and the products 11 and H2 are 15.2[16.2](16.2) and 6.5[16.8](16.2) kcal/mol, respectively.
In the reverse reaction complex 9 can be considered an H2-activation product that is formed from 11 and H2 via TS9–10, where the H2 bond-breaking H atoms are moving towards BPin and pyridine ligands, respectively. Alternatively, H2 can be also cleaved along a direction parallel to the line between two phosphine atoms to form an isomer of 9, which adopts an octahedral geometry in which two H atoms and two phosphine ligands occupy the equatorial positions, and the pyridine ligands and BPin occupy the axial positions. As shown in SI5, this isomer of 9 could be formed reversibly, which then releases H2 to regenerate complex 11.
For each species in Figure 2, several different geometries are possible by rotating iPr groups of the phosphine ligands, and the geometries shown in Figure 3 and SI2 correspond to the most stable ones. In addition, these geometries are considered to isomerize between each other very easily by rotating the iPr groups separately (see SI6 for an example of the isomerization process from 4 to its isomer). Thus, species with different orientations of iPr groups may exist, but that would not change the results obtained in the following discussions.
The mechanism shown in Figure 2 is generally similar to that for the model systems (SI7), in which all the iPr substituents of the iPrPNP ligand were replaced with Me and B2Pin2 was replaced with B2eg2 (superscript “m” is added to the reported complex’s number to denote their corresponding model complex). Due to less steric clashing between the substituents on the phosphine ligands in the model systems, the barriers for the ring “flipping” transition states TS2–3m, TS7–8m, and TS8–9m are 2.6, 8.0, and 16.5 kcal/mol relative to 2m (Figure S5 and S6), which are lower than the corresponding (full system) transition states TS2–3, TS7–8, and TS8–9 in Figure 2. By using the model systems, other possibilities were checked. For example, the isomerization between 11m and its isomer in which two five-membered phosphine rings are folded in different directions was calculated to occur very easily (Figure S6). Furthermore, isomers of 5m, 6m, and 10m in which two five-membered phosphine rings are folded in different directions are found to be unlikely, as optimizations of these isomers repeatedly converge to 5m, 6m, and 10m, respectively. In addition, other possible pathways are calculated to be unlikely or less favorable, which include the direct release of H2 from the cis-dihydride cobalt complex 8m, the formation of 8m from the trans-dihydride cobalt complex 2m via the dissociation and association of HBeg (Figure S7), the formation of 8m from 2m through dissociation of one phosphine (Figure S8), and the release of H2 via the dearomatization of the pyridine ligand (Figure S9). Our results, which involve reductive elimination (RE) of H2 from a Co(III) pincer complex, differ from a previous study of the C-C bond formation via RE from a cobalt(III) dimethyl complex, where dissociation of a phosphine ligand was required prior to RE.76 Maybe for future work, it is also interesting to compare the PNP system with all of this flexibility to terpyridine ones that also borylate but are more rigid.
Among these unfavorable pathways, the dearomatization of the pyridine ligand is quite interesting, as a series of PNP pincer ligated Ru complexes that are similar to complex 2 have been synthesized by Milstein and coworkers and reported to show a mode of metal-ligand cooperation based on the ligand’s aromatization/dearomatization.56–58 Pincer ligand benzylic deprotonations and hydrogen abstractions in related Co(I) complexes suggest that the metal-ligand cooperation in the Rh complexes may involve deprotonation at the benzylic site,55 consistent with calculations on the Ru mechanism.56–58 To confirm the conclusions obtained with model systems, we recalculated this pathway by using complex 2 with the full ligand (iPrPNP). Considering that the pyridine ligand and other atoms use different basis sets in the optimizations (see SI1 in Computational Details), we employed another basis set BS4 (LANL2DZ for Co and 6–31G* for the others) to optimize the species involved in this pathway. The rest of the methodology was the same (see Computational Details). Thus, the results for this pathway used in the following discussions are obtained at the level of ωB97XD/Def2TZVP(SMD)//ωB97XD/BS4. At this level, the barrier for TS9–10 and the product complex 11 with the release of H2 are 18.2[19.5] and 10.7[21.3] kcal/mol (ΔGC6H6[ΔHC6H6]), respectively, relative to 2. As shown in SI8, the level of ωB97XD/Def2TZVP(SMD)//ωB97XD/BS4 is suitable in the calculations of this system by giving reasonable optimized geometries and the results close to that at the level of ωB97XD/Def2TZVP(SMD)//ωB97XD/BS1.
With ωB97XD/BS4, the optimized geometries for transition state TS2–12 and the product complex 12 involved in the pathway via the dearomatization of the pyridine ligand are shown in Figure 4. TS2–12 corresponds to the formation of H2 by eliminating a hydride at Co and a proton from the sp3-C of the pyridine ligand. In contrast to the reductive elimination pathway to form the Co(I) complex 11 via TS9–10, complex 12 remains a Co(III) complex due to the dearomatization of the pyridine ligand. In addition, the hydride at Co is close to B, which could indicate an interaction between the hydride and the vacant orbital on B. At the level of ωB97XD/Def2TZVP(SMD)//ωB97XD/BS4, TS2–12 and complex 12 with the release of H2 are 31.5[32.4] and 6.1[17.1] kcal/mol (ΔGC6H6[ΔHC6H6]), respectively, relative to 2. In addition, the dissociation of HBPin from 12 is unfavorable, as this process is endothermic by 13.1[27.4] kcal/mol. The origin of the higher barrier for TS2–12 can be seen in the distance between two eliminating H atoms in their optimized geometries. The distance between the hydride at Co and the hydrogen atom at sp3-C of the pyridine ligand) decreases by 2.02 Å in forming TS2–12, the corresponding distance between the two eliminating H atoms decreases by only 0.82 Å in forming TS9–10. Thus, changing geometries from 2 to TS2–12 requires more energy. Moreover, the dearomatization of the pyridine ligand further destabilizes transition state TS2–12. Therefore, the mechanism for the release of H2 via the dearomatization of the pyridine ligand is less favorable than that via the reductive elimination mechanism shown in Figure 2. Furthermore, the mechanism via the reversible ligand dearomatization-aromatization was also ruled out based on the experimental observations.30
Figure 4.

Optimized geometries of transition state TS2–12 and complex 12. Some bond distances in Å and angles in ° are given in black and red colors, respectively.
To understand the effect of the metal, we investigated the release of H2 from the corresponding iridium complex 2Ir (trans-(iPrPNP)IrH2(BPin)) formed by replacing Co in 2 with Ir. The barriers for H2 formation in the iridium analogues are higher than those in the cobalt systems: TS9–10Ir and TS2–12Ir are higher than TS9–10 and TS2–12 by 14.3 and 5.3 kcal/mol, respectively (Table 1). To understand the effect of the ligand, we also calculated the release of H2 from their phenyl analogues (2Ph = trans-(iPrPNP)CoH2(Ph) and 2Ir_Ph = trans-(iPrPNP)IrH2(Ph)), which are generated by using Ph to replace BPin in 2 and 2Ir, respectively. The barriers for H2 formation for 2 and 2Ir are close to those for their phenyl analogues (2Ph and 2Ir_Ph) with the variations of less than 3 kcal/mol (Table 1). Thus, the choice of the metal affects the reactivity to a greater degree than the choice of the ligand. In particular, the barriers for H2 formation via the reductive elimination mechanism for iridium analogues (TS9–10Ir and TS9–10Ir_Ph) are higher by more than 14 kcal/mol than those for cobalt systems (TS9–10 and TS9–10Ph, respectively). In contrast, H2 formation via the dearomatization of the pyridine ligand for the iridium analogues (TS2–12Ir and TS2–12Ir_Ph) are higher by only 5.3 kcal/mol than those for the cobalt systems (TS2–12 and TS2–12Ph, respectively). Relative to 2Ir, the barriers for the H2 formation, TS9–10Ir and TS2–12Ir, are over 32 kcal/mol (Table 1). Consistent with these results, Chirik and coworkers observed that the iridium congener with a pyrrolidinyl substituent in 4-position of the pincer ligand is very stable under catalytic conditions, which can be ascribed to a more difficult reductive elimination from Ir(III) to Ir(I) than from Co(III) to Co(I), as proposed by Chirik and coworkers.30 Furthermore, Milstein and coworkers did not observe the release of H2 from an iridium complex 2Ir_Ph_tBu (trans-(tBuPNP)IrH2(Ph)),77 for which the predicted barriers for H2 formation are over 33 kcal/mol (Table 1). In addition, TS2–12Ir_Ph_tBu is very close to TS2–12Ir_Ph, while TS9–10Ir_Ph_tBu is much higher than TS9–10Ir_Ph, which can be ascribed to the steric effects between tBu groups in TS9–10Ir_Ph_tBu (see their optimized geometries in SI9).
Table 1.
Results for the release of H2 at the level of ωB97XD/Def2TZVP(SMD)//ωB97XD/BS4. Values in kcal/mol are ΔGC6H6[ΔHC6H6](ΔGgas), and are relative to 2R, respectively.
| R | TS9–10R | 11R + H2 | TS2–12R | 12R + H2 |
|---|---|---|---|---|
| 18.2[19.5](17.2) | 10.7[21.3](4.8) | 31.5[32.4](32.0) | 6.1[17.1](5.0) | |
| Ir | 32.5[34.4](31.8) | 17.4[28.8](16.3) | 36.8[36.8](39.0) | 14.3[24.3](18.1) |
| Ph | 19.1[20.5](18.0) | −0.6[11.6](−7.8) | 28.9[29.3](28.7) | 6.3[17.6](7.5) |
| Ir_Ph | 35.0[34.5](35.7) | 9.1[20.1](8.3) | 34.2[34.2](36.2) | 12.1[22.7](17.2) |
| Ir_Ph_tBu | 41.5[45.9](42.6) | 9.0[20.0](8.2) | 33.5[34.1](35.5) | 10.9[22.1](15.5) |
3.2. Oxidative addition of C6H6
The cobalt boryl complex (iPrPNP)CoBPin (11), which is generated from complex 2 via the H2 release mechanism (Figure 2), is the active species that catalyzes the borylation reactions of five-membered heteroarenes. This active species can also be formed in the presence of B2Pin2 directly from the catalyst precursor (iPrPNP)Co(CH2SiMe3) (1) by releasing Me3SiCH2BPin, as observed in experiments.28 This reaction was calculated to be thermodynamically very favorable (ΔGC6H6[ΔHC6H6](ΔGgas)=−25.1[−24.6](−27.3) kcal/mol), and a possible pathway with reasonable barriers was located by using the model systems (SI10). The calculated energy profiles for the oxidative addition of C6H6 by 11 together with their optimized geometries are shown in Figure 5. A search along the backward direction of the imaginary frequency from TS11–13 toward 11 failed to find either a Co σ-complex or a Co π-complex. In the forward direction of the oxidative-addition transition state TS11–13, the breaking C-H bond of C6H6 is parallel to a line between the two phosphine atoms. Thus, TS11–13 and the product complex 13 adopt a distorted octahedral geometry in which C6H5, H, and phosphine ligands occupy the equatorial positions, and the pyridine ligands and BPin occupy the axial positions. In this geometry, the pyridine ligand occupies one face of the octahedron around Co, i.e., the N and P atoms of the pyridine ligand are cis to each other, a facial isomeric form. Therefore, due to the distorted backbone of pyridine ligands and the steric effects between the substituents of phosphine ligands, complex 13 is very high in energy, 23.0 kcal/mol relative to separated 11 and C6H6. This step is also unfavorable under the TPSS functional, where TS11–13 and 13 are 34.8[18.9](19.5) and 33.6[18.2](18.8) kcal/mol, respectively. However, the next process, the reductive elimination of B-C bond to form PhBPin, can drive the overall reaction forward.
Figure 5.

Calculated energy profiles for the oxidative addition of C6H6 to 11. Some bond distances in Å, angles in °, and dihedral angles in ° are given in black, red, and green colors, respectively.
In addition to the pathway shown in Figure 5, alternative pathways for the oxidative addition of C6H6 to the cobalt boryl complex in the model systems were explored (SI11): (1) an oxidative-addition pathway to form another facial isomer of 13m in which C6H5 and Beg are switched (Figure S13(B) in SI11), (2) oxidative-addition pathways directly forming meridional complex 15m (see Figure 6 and Figure S13(C) in SI11) and its meridional isomer in which C6H5 and H are switched (Figure S13(D) in SI11), (3) σ-bond metathesis pathways, and (4) a pathway for the oxidative-addition of C6H6 to the cobalt complex with one phosphine ligand dissociated (Figure S14 in SI11). However, these alternative pathways were calculated to be unlikely or less favorable than that in Figure 5.
Figure 6.

Calculated energy profiles for the reductive elimination of B-C bond to form PhBPin. The energies for these species are relative to separate C6H6 and 11, and their optimized geometries are shown in SI14.
Some conclusions obtained from the model systems remain the same for the experimentally reported systems (SI12). For example, alternative σ-bond metathesis pathways remain unlikely relative to oxidative addition, because the Co(I) in 11 is easily oxidized to Co(III) via the oxidative addition. In contrast, previously reported σ-bond metathesis pathways are proposed for the reactions of complexes in high oxidation states, such as W(III),39,45 Rh(III),42 and Ir(III)47,49 complexes. In addition, alternative oxidative-addition pathways to form meridional complex 15 and its meridional isomer are still less favorable than that via TS11–13 in Figure 5 (Figure S15 in SI12). The lower barrier of TS11–13 can be ascribed to lower distortion energy of the Co catalyst and the more negative interaction energy (Table S6 in SI12).
Using model complexes, pathways for adding C6H6 to the corresponding Rh and Ir complexes (Rh11m and Ir11m) obtained by replacing Co in 11m with Rh and Ir, respectively, were examined (SI13). Like the Co complex, σ-bond metathesis pathways for the Rh and Ir complexes are still unlikely because of their low oxidation state. In addition, the oxidative-addition pathways via RhTS11–13m and IrTS11–13m (corresponding Rh and Ir transition states of TS11–13m) are still more favorable than that for directly forming Rh15m and Ir15m (corresponding Rh and Ir complexes of 15m) and their isomers.
With the active species 4-BPin-(iPrPNP)CoBPin, we calculated this process by optimizing corresponding species TS11–13BPin and 13BPin with ωB97XD/BS4 (LANL2DZ for Co and 6–31G* for the others). The same method was employed to recalculate TS11–13 and 13 for consistency. At the level of ωB97XD/Def2TZVP(SMD)//ωB97XD/BS4, TS11–13, 13, TS11–13BPin, and 13BPin are 24.2[8.2](23.0), 21.9[6.9](22.0), 23.8[8.2](22.8), and 21.6[7.1](21.7) kcal/mol, respectively. Although the barrier for TS11–13BPin is slightly lower than that for TS11–13 in free energy, they are the same in enthalpy, and the former is slightly higher than the latter by 0.05 kcal/mol in SCF energy. Furthermore, as shown by Table 2 and corresponding discussions in the following section, the relative energies of TS11–13BPin and TS11–13 depend on selected calculation methods. The experimental observations that the C-H oxidative addition with 4-BPin-(iPrPNP)CoBPin is slower than that with (iPrPNP)CoBPin30 may be explained by these differences.
Table 2.
Calculated barriers (in kcal/mol) for transition states TS11–13 and TS18–19 relative to separate 11 and C6H6 by using different functionals and basis sets.
| TS11–13 | TS18–19 | |
|---|---|---|
| ωB97XD/Def2TZVP(SMD)//ωB97XD/BS1 | 24.2 | 25.2 |
| M06/Def2TZVP(SMD)//M06/BS1 | 30.1 | 30.7 |
| B3LYP/Def2TZVP(SMD)//B3LYP/BS1 | 43.0 | 44.5 |
| B3LYP(GD3BJ)/Def2TZVP(SMD)//B3LYP(GD3BJ)/BS1 | 25.8 | 27.2 |
| M06L/Def2TZVP(SMD)//M06L/BS1 | 26.5 | 26.4 |
| ωB97XD/Def2TZVP(SMD)//ωB97XD/BS2 | 23.5 | 25.0 |
| ωB97XD/Def2TZVP(SMD)//ωB97XD/BS41 | 24.2 | 26.0 |
| ωB97XD/Def2TZVP(SMD)//ωB97XD/BS52 | 24.7 | 25.9 |
| M06/Def2TZVP(SMD)//M06/BS52 | 30.4 | 32.3 |
| B971/Def2TZVP(SMD)//B971/BS52 | 36.6 | 38.8 |
| TPSS/Def2TZVP(SMD)//TPSS/BS52 | 34.8 | 36.1 |
| TPSS/Def2TZVP(SMD)//TPSS/BS1 | 34.8 | 35.8 |
Co uses LANL2DZ and the others use 6–31G* in BS4;
Co uses 6–31+G* and the others use 6–31G* in BS5.
3.3. Reductive elimination of B-C bond to form PhBPin
From the C6H6 addition product 13, the calculated energy profiles for the reductive elimination of the B-C bond to generate the borylation product PhBPin are shown in Figure 6. By beginning at complex 13, where the pincer is folded and C6H5, H, and BPin are in the opposite fac site, the H and BPin groups rotate via transition state TS13–14 to form complex 14, which adopts a distorted octahedral geometry where C6H5, H, BPin, and the pyridine ligands are in the same plane, and the P-Co-P angle begins to open. Then, the P arm in blue tips up and the pincer unfolds into its mer site to form 15, where the Ph and BPin groups are trans. Subsequently, the HBPin group dissociates from 15 to form a four-coordinated Co-phenyl complex 16, followed by the re-association of HBPin to generate complex 17 where the Ph and BPin groups are in cis positions (alternative dissociation/re-association route are possible but were not investigated). For the vibrational modes of transition states TS15–16 and TS16–17, in addition to the oxidation and reductive elimination of HBPin, the Ph groups are also rotating around Co towards or away from the trans position of the N atom. From complex 17 where the two five-membered phosphine rings of the pincer ligand are folded in different directions, the P arm in blue then flips down to afford complex 18. In 18, the Ph and BPin groups are in cis positions, from which they proceed through a reductive elimination transition state TS18–19 to form complex 19, followed by the release of the product PhBPin to generate a Co-hydride complex 20. The transition state TS18–19 corresponds to the reductive elimination of PhBPin and the rotation of H atom around Co.
Due to the release of steric effects between the ligands, complex 14 is more stable than 13 by 3.1 kcal/mol, and complex 15 is more stable than 14 by 2.0 kcal/mol. Moreover, complex 17 is more stable than 15 by 6.8 kcal/mol. According to Figure 6, the reductive elimination processes are very favorable. The formations of Co(I) complexes 16 and 20 from their corresponding Co(III) complexes 15 and 18, respectively, are favorable by releasing more than 9 kcal/mol. Overall, the generated PhBPin and complex 20 are lower than complex 13 by 26.5 kcal/mol, and they are also lower than separate reactants (C6H6 and 11) by 3.5 kcal/mol. Thus, the reductive elimination process is thermodynamically favorable, which drives the reaction to produce PhBPin.
In comparison with the oxidative addition process in Figure 5, the reductive elimination process in Figure 6 appears to have the rate-determining step, as several transition states involved in Figure 6 (TS13–14, TS14–15, TS15–16, TS16–17, and TS18–19) are a little higher than the C-H oxidative-addition transition state TS11–13. The same conclusion was also obtained by using the experimentally proposed active species 4-BPin-(iPrPNP)CoBPin, as the barrier of TS11–13BPin (23.8[8.2](22.8) kcal/mol) for the C-H oxidative addition is still lower than that of TS18–19BPin (25.7[9.7](23.0) kcal/mol) for the reductive elimination at the level of ωB97XD/Def2TZVP(SMD)//ωB97XD/BS4. These results appear to be inconsistent with the experimental observations that the C-H oxidative addition was indicated to be the rate-determining step for the borylation of 2,6-lutidine with B2Pin2.30 Of course, the differences between these barriers are small and the transition states involved in Figure 6 could adopt other lower-barrier conformations due to the flexibility of ligands, in particular the rotations of iPr groups and the orientations of phosphine ligands. Here, we hesitate to try to find all possible conformations because of the large computation cost and because we could not trust such small differences. To support this postulation, a similar reductive elimination pathway was calculated by using model systems (Figure S17 in SI15). The results show that the reductive elimination transition state TS18–19m (corresponding to TS18–19) is the highest one among these transition states. However, TS18–19m is slightly lower by 0.5 kcal/mol than the C-H oxidative addition transition state TS11–13m. Thus, for the model system, the oxidative addition step is the rate-determining step. To further explore the effects of functionals and basis sets, calculations were conducted for these two important transition states TS11–13 (the oxidative addition transition state) and TS18–19 (the reductive elimination transition state) by employing several different DFT functionals and basis sets (Table 2). Although the barriers for TS18–19 are predicted to be slightly higher than that for TS11–13 with most of these tested methods, TS18–19 is lower than TS11–13 with M06L. In addition, the results at the level of M06/Def2TZVP(SMD)//M06/BS1 are very close with a difference of only 0.6 kcal/mol. Thus, the choice of functional could be a source of this small inconsistency. Considering that the C-H oxidative addition is the rate-determining step, the overall barrier for the reaction is 24.2 kcal/mol from 11 to TS11–13. Consistent with the calculated barrier, the reaction was observed to occur at 80°C for 24 h with the isolated yield of 87% by employing a 20:1 ration of arene to B2Pin2 in the reported experiments.28
From complex 13, the barriers for the reductive elimination of the C-H bond via TS11–13, the reductive elimination of the B-H bond via TS15–16, and the reductive elimination of the B-C bond via TS18–19 are 1.2, 5.8, and 2.2 kcal/mol, respectively. Furthermore, their corresponding thermodynamics are −23.0, −14.7, and −26.5 kcal/mol for the formations of 11 and C6H6, the formations of 16 and HBPin, and the formations of 20 and PhBPin. Thus, the reductive elimination of the B-C bond is the most favorable process, which drives the reaction forward to produce PhBPin, while the reductive eliminations of the C-H and B-H bond are considered to be reversible.
The barriers for the ring “flipping” transition states TS14–15m and TS17–18m, relative to separate C6H6 and 11m, are 12.7 and −2.3 kcal/mol (Figure S17), which are lower than the corresponding (full system) transition states TS14–15 and TS17–18 in Figure 6, as there is less steric clashing between the substituents on the phosphine ligands in the model systems. In addition to the pathway shown in Figure 6, several other possibilities were considered by using model systems and found to be less favorable (see SI15): direct reductive elimination of the B-C bond from the oxidative addition product 13m (Figure S18), reductive elimination through an isomer of the TS18–19m with two phosphine ligands in different directions (Figure S19(A)), reductive elimination with the assist of HBeg (Figure S19(B)), reductive elimination from the C6H6 oxidative addition product where the cleaved C6H5 and H are in cis positions (Figure S19(C)), reductive elimination via the dissociation of one phosphine ligand (Figure S19(D)), and reductive elimination from the oxidative addition product from B2eg2 and the Co(I)-Ph complex (Figure S20). With the experimentally reported systems, the process for the reductive elimination of the B-C bond to generate the borylated product PhBPin through the direct reductive elimination from 13 was further calculated to be unlikely, as the barrier for TS13–20 is very high, 38.4[23.6](36.3) kcal/mol relative to separated C6H6 and 11 (Figure S21). In addition, another possible pathway for the oxidative addition of C6H6 to the Co(I)-H complex 20m was also considered (Figure S22), but it was less favorable thermodynamically and kinetically than the oxidative addition of B2eg2 to regenerate the active species (Figure S24 in SI17).
At the level of ωB97XD/Def2TZVP(SMD)//ωB97XD/BS4, TS18–19, 20, TS18–19BPin, and 20BPin are 26.0[9.7](23.3), −3.2[−0.8](2.2), 25.7[9.7](23.0), and −5.2[−1.0](0.2) kcal/mol, respectively, where 20BPin and TS18–19BPin are the active species 4-BPin-(iPrPNP)CoBPin and its corresponding transition state. Accordingly, the reductive elimination process with 4-BPin-(iPrPNP)CoBPin is more favorable than that with (iPrPNP)CoBPin both kinetically and thermodynamically.
3.4. Regeneration of (iPrPNP)CoBPin
In the presence of B2Pin2, the active species (iPrPNP)CoBPin (11) can be regenerated from (iPrPNP)CoH (20) by following the pathway shown in Figure 7. Coordination of B2Pin2 to 20 gives complex 21, in which the B-B bond is parallel with N-Co-H axis, is followed by transition state TS21–22 corresponding to simultaneous cleavage of B-B bond and rotation of Co-H bond to generate an octahedral complex 22. Subsequently, the sp3-C of the phosphine ring in blue flips up via transition state TS22–23 to form complex 23. Like the dihydride cobalt complexes in Figure 2, complex 23, where the two five-membered rings are folded in different directions, is slightly more stable than complex 22 where the two five-membered rings are folded in the same direction. Then, through transition state TS23–24, in which the H and BPin on Co are moving towards each other, and transition state TS24–25, in which BPin rotated and forms the new H-B bond. Finally, HBPin dissociates from 25 via transition state TS25–11 to regenerate the active species 11. An alternative pathway for the generation of 11 from 25 via an intermediate that is formed from 25 by flipping up the sp3-C of the phosphine ring in blue is slightly less favorable, and the transition state for the dissociation of HBPin from this intermediate is higher than TS25–11 by 0.7 kcal/mol. As shown in Figure 7, the formation of 11 with releasing HBPin from 20 and B2Pin2 is favorable thermodynamically and kinetically; the rate-determining barrier for this process is only 14.1 kcal/mol (TS24–25 relative to 23).
Figure 7.

Calculated energy profiles for the regeneration of (iPrPNP)CoBPin (11) from (iPrPNP)CoH (20) and B2Pin2. Optimized geometries of these species are shown in SI16.
The pathways discussed above are consistent with those for the model systems (SI17). In the model system several less favorable alternative paths were explored: formation of an isomer of 21, in which the B-B bond is parallel with the P-Co-P axis, formation of 24 from 22, in which the H and Beg on Co firstly move towards each other, followed by the sp3-C of one phosphine ring flips up, formation of 25 from 24, in which the sp3-C of one phosphine ring firstly flips up to generate an intermediate with two phosphine rings bending in the same direction, and the pathway with one phosphine ligand dissociated (Figure S25 in SI17).
4. Conclusions
Density functional theory (DFT) calculations on C-H borylation catalyzed by cobalt pincer complexes with C6H6 and B2Pin2 as substrates predict a mechanism involving three distinct steps: oxidative addition of C6H6, reductive elimination PhBPin, and regeneration of (iPrPNP)CoBPin. The trans-(iPrPNP)CoH2(BPin) complex was experimentally observed as a resting state in the borylation of five-membered heteroarenes. Starting from this complex, the active species, (iPrPNP)CoBPin, is generated by rearrangement to the cis isomer and reductive elimination of H2. The isomerization pathway is similar to the well-known Berry pseudorotation mechanism with the H1BPin group acting as a unit in a 5-coordinate Co. The metal-ligand cooperative mechanism based on the ligand’s aromatization/dearomatization, which is widely proposed to explain the catalytic reactions by the PNP pincer ligated to heavier metals, is computed to be less favorable for this system. Additionally, the 4-BPin-(iPrPNP)Co(N2)BPin complex, which can easily release N2 to form the active species, was identified as the resting state in the catalytic borylation of arene substrates. The lowest barrier for oxidative addition of C6H6 forms a complex with a distorted PNP ligand, which rearranges to a more stable complex via dissociation and re-association of HBPin. Possible σ-bond metathesis pathways are predicted to be unlikely for this Co(I) complex. Reductive elimination of PhBPin is thermodynamically favorable and drives the reaction forward. The regeneration of the active species was found to proceed through the oxidative addition of B2Pin2 and reductive elimination of HBPin. In the overall reaction, the flipping up and down of the sp3-C of the five-membered phosphine rings, which connects the species with two phosphine rings folded in the same direction and that with them folded in different directions, is found to play important roles in the catalytic process, as this motion releases steric crowding within the PNP ligand and opens Co coordination space. This investigation provides some guidance for further understanding of important features of pincer ligands with first-transition-row metals that differ from those in heavier metal complexes.
Supplementary Material
ACKNOWLEDGMENT
We thank the National Science Foundation (CHE-1300787 and 1664866) and the Welch Foundation (A-0648) for financial support, and the Texas A&M High Performance Research Computing Center and the Laboratory for Molecular Simulation for providing computing resources. J.V.O. and P.J.C. would like to thank the National Institutes of Health (1R01GM121441–01).
Footnotes
SUPPORTING INFORMATION AVAILABLE:
Verification of ground states of four-coordinate cobalt species; optimized geometries of some species involved in Figure 2; the superimposed structures of the optimized geometry of 2 and its X-ray crystal structure; an alternative pathway for the formation of 4 from 2; formation of the isomer of 9; isomerization of 4 to its isomer; results for the release of H2 from the trans-dihydride Co-complex with model systems; verification of ωB97XD/Def2TZVP(SMD)//ωB97XD/BS4; optimized geometries of transition states for the release of H2 from 2Ir, 2Ph, 2Ir_Ph, and 2Ir_Ph_tBu; a possible pathway for the formation of the active species from the catalyst precursor with model systems; alternative pathways for the addition of C6H6 to the cobalt boryl species with model systems; comparisons of transition state TS11–13 and that for the formation of 15 and its isomer; addition of C6H6 to corresponding Rh and Ir complexes; optimized geometries for the species in Figure 6 in the text; results for the reductive elimination of the B-C bond with model systems; optimized geometries of the species in Figure 7; results for the regeneration of the active species with model systems; Cartesian coordinates and energies of the species involved in the text. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Mkhalid IAI; Barnard JH; Marder TB; Murphy JM; Hartwig JF C-H Activation for the Construction of C-B Bonds. Chem. Rev 2010, 110, 890–931. [DOI] [PubMed] [Google Scholar]
- (2).Hartwig JF Regioselectivity of the Borylation of Alkanes and Arenes. Chem. Soc. Rev 2011, 40, 1992–2002. [DOI] [PubMed] [Google Scholar]
- (3).Hartwig JF Borylation and Silylation of C-H Bonds: A Platform for Diverse C-H Bond Functionalizations. Acc. Chem. Res 2012, 45, 864–873. [DOI] [PubMed] [Google Scholar]
- (4).Li Q; Driess M; Hartwig JF Iridium-Catalyzed Regioselective Silylation of Aromatic and Benzylic C-H Bonds Directed by a Secondary Amine. Angew. Chem., Int. Ed 2014, 53, 8471–8474. [DOI] [PubMed] [Google Scholar]
- (5).Ishiyama T; Miyaura N Transition Metal-Catalyzed Borylation of Alkanes and Arenes via C-H Activation. J. Organomet. Chem 2003, 680, 3–11. [Google Scholar]
- (6).Ros A; Fernandez R; Lassaletta JM Functional Group Directed C-H Borylation. Chem. Soc. Rev 2014, 43, 3229–3243. [DOI] [PubMed] [Google Scholar]
- (7).Thomas RL; Souza FES; Marder TB Highly Efficient Monophosphine Platinum Catalysts for Alkyne Diboration. J. Chem. Soc.-Dalton Trans 2001, 1650–1656. [Google Scholar]
- (8).Lee CI; Shih WC; Zhou J; Reibenspies JH; Ozerov OV Synthesis of Triborylalkenes from Terminal Alkynes by Iridium-Catalyzed Tandem C-H Borylation and Diboration. Angew. Chem., Int. Ed 2015, 54, 14003–14007. [DOI] [PubMed] [Google Scholar]
- (9).Kubota K; Iwamoto H; Yamamoto E; Ito H Silicon-Tethered Strategy for Copper(I)-Catalyzed Stereo- and Regioselective Alkylboration of Alkynes. Org. Lett 2015, 17, 620–623. [DOI] [PubMed] [Google Scholar]
- (10).Nakagawa N; Hatakeyama T; Nakamura M Iron-Catalyzed Diboration and Carboboration of Alkynes. Chem.-Eur. J 2015, 21, 4257–4261. [DOI] [PubMed] [Google Scholar]
- (11).Itoh T; Shimizu Y; Kanai M Ligand-Enabled, Copper-Catalyzed Regio- and Stereoselective Synthesis of Trialkylsubstituted Alkenylboronates from Unactivated Internal Alkynes. J. Am. Chem. Soc 2016, 138, 7528–7531. [DOI] [PubMed] [Google Scholar]
- (12).Cook AK; Schimler SD; Matzger AJ; Sanford MS Catalyst-Controlled Selectivity in the C-H Borylation of Methane and Ethane. Science 2016, 351, 1421–1424. [DOI] [PubMed] [Google Scholar]
- (13).Smith KT; Berritt S; Gonzalez-Moreiras M; Ahn S; Smith MR; Baik MH; Mindiola DJ Catalytic Borylation of Methane. Science 2016, 351, 1424–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Chirik PJ Iron- and Cobalt-Catalyzed Alkene Hydrogenation: Catalysis with Both Redox-Active and Strong Field Ligands. Acc. Chem. Res 2015, 48, 1687–1695. [DOI] [PubMed] [Google Scholar]
- (15).Holland PL Distinctive Reaction Pathways at Base Metals in High-Spin Organometallic Catalysts. Acc. Chem. Res 2015, 48, 1696–1702. [DOI] [PubMed] [Google Scholar]
- (16).Waltz KM; He XM; Muhoro C; Hartwig JF Hydrocarbon Functionalization by Transition-Metal Boryls. J. Am. Chem. Soc 1995, 117, 11357–11358. [Google Scholar]
- (17).Légaré MA; Courtemanche MA; Rochette E; Fontaine FG Metal-free Catalytic C-H Bond Activation and Borylation of Heteroarenes. Science 2015, 349, 513–516. [DOI] [PubMed] [Google Scholar]
- (18).Bose SK; Marder TB A Leap Ahead for Activating C-H Bonds. Science 2015, 349, 473–474. [DOI] [PubMed] [Google Scholar]
- (19).Légaré MA; Rochette E; Lavergne JL; Bouchard N; Fontaine FG Bench-Stable Frustrated Lewis Pair Chemistry: Fluoroborate Salts as Precatalysts for the C-H Borylation of Heteroarenes. Chem. Commun 2016, 52, 5387–5390. [DOI] [PubMed] [Google Scholar]
- (20).Chernichenko K; Lindqvist M; Kótai B; Nieger M; Sorochkina K; Pápai I; Repo T Metal-Free sp(2)-C-H Borylation as a Common Reactivity Pattern of Frustrated 2-Aminophenylboranes. J. Am. Chem. Soc 2016, 138, 4860–4868. [DOI] [PubMed] [Google Scholar]
- (21).Iashin V; Chernichenko K; Pápai I; Repo T Atom-Efficient Synthesis of Alkynylfluoroborates Using BF3-Based Frustrated Lewis Pairs. Angew. Chem., Int. Ed 2016, 55, 14146–14150. [DOI] [PubMed] [Google Scholar]
- (22).Mazzacano TJ; Mankad NP Base Metal Catalysts for Photochemical C-H Borylation That Utilize Metal-Metal Cooperativity. J. Am. Chem. Soc 2013, 135, 17258–17261. [DOI] [PubMed] [Google Scholar]
- (23).Parmelee SR; Mazzacano TJ; Zhu YQ; Mankad NP; Keith JA A Heterobimetallic Mechanism for C-H Borylation Elucidated from Experimental and Computational Data. ACS Catal 2015, 5, 3689–3699. [Google Scholar]
- (24).Mankad NP Non-Precious Metal Catalysts for C-H Borylation Enabled by Metal-Metal Cooperativity. Synlett 2014, 25, 1197–1201. [Google Scholar]
- (25).Dombray T; Werncke CG; Jiang S; Grellier M; Vendier L; Bontemps S; Sortais JB; Sabo-Etienne S; Darcel C Iron-Catalyzed C-H Borylation of Arenes. J. Am. Chem. Soc 2015, 137, 4062–4065. [DOI] [PubMed] [Google Scholar]
- (26).Mazzacano TJ; Mankad NP Thermal C-H Borylation Using a CO-free Iron Boryl Complex. Chem. Commun 2015, 51, 5379–5382. [DOI] [PubMed] [Google Scholar]
- (27).Yoshigoe Y; Kuninobu Y Iron-Catalyzed Ortho-Selective C-H Borylation of 2-Phenylpyridines and Their Analogs. Org. Lett 2017, 19, 3450–3453. [DOI] [PubMed] [Google Scholar]
- (28).Obligacion JV; Semproni SP; Chirik PJ Cobalt-Catalyzed C-H Borylation. J. Am. Chem. Soc 2014, 136, 4133–4136. [DOI] [PubMed] [Google Scholar]
- (29).Schaefer BA; Margulieux GW; Small BL; Chirik PJ Evaluation of Cobalt Complexes Bearing Tridentate Pincer Ligands for Catalytic C-H Borylation. Organometallics 2015, 34, 1307–1320. [Google Scholar]
- (30).Obligacion JV; Semproni SP; Pappas I; Chirik PJ Cobalt-Catalyzed C(sp(2))-H Borylation: Mechanistic Insights Inspire Catalyst Design. J. Am. Chem. Soc 2016, 138, 10645–10653. [DOI] [PubMed] [Google Scholar]
- (31).Palmer WN; Obligacion JV; Pappas I; Chirik PJ Cobalt-Catalyzed Benzylic Borylation: Enabling Polyborylation and Functionalization of Remote, Unactivated C(sp(3))-H Bonds. J. Am. Chem. Soc 2016, 138, 766–769. [DOI] [PubMed] [Google Scholar]
- (32).Leonard NG; Bezdek MJ; Chirik PJ Cobalt-Catalyzed C(sp(2))-H Borylation with an Air-Stable, Readily Prepared Terpyridine Cobalt(II) Bis(acetate) Precatalyst. Organometallics 2017, 36, 142–150. [Google Scholar]
- (33).Obligacion JV; Bezdek MJ; Chirik PJC (sp(2))-H Borylation of Fluorinated Arenes Using an Air-Stable Cobalt Precatalyst: Electronically Enhanced Site Selectivity Enables Synthetic Opportunities. J. Am. Chem. Soc 2017, 139, 2825–2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Obligation JV; Chirik PJ Mechanistic Studies of Cobalt-Catalyzed C(sp(2))-H Borylation of Five-Membered Heteroarenes with Pinacolborane. ACS Catal 2017, 7, 4366–4371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Ren HL; Zhou YP; Bai YP; Cui CM; Driess M Cobalt-Catalyzed Regioselective Borylation of Arenes: N-Heterocyclic Silylene as an Electron Donor in the Metal-Mediated Activation of C-H Bonds. Chem.-Eur. J 2017, 23, 5663–5667. [DOI] [PubMed] [Google Scholar]
- (36).Furukawa T; Tobisu M; Chatani N Nickel-Catalyzed Borylation of Arenes and Indoles via C-H Bond Cleavage. Chem. Commun 2015, 51, 6508–6511. [DOI] [PubMed] [Google Scholar]
- (37) (a).Zhang H; Hagihara S; Itami K Aromatic C-H Borylation by Nickel Catalysis. Chem. Lett 2015, 44, 779–781. [Google Scholar]; (b) Furukawa T; Tobisu M; Chatani N Nickel-Catalyzed Borylation of Arenes and Indoles via C-H Bond Cleavage. Chem. Commun 2015, 51, 6508–6511. [DOI] [PubMed] [Google Scholar]
- (38).Tamura H; Yamazaki H; Sato H; Sakaki S Iridium-Catalyzed Borylation of Benzene with Diboron. Theoretical Elucidation of Catalytic Cycle Including Unusual Iridium(V) Intermediate. J. Am. Chem. Soc 2003, 125, 16114–16126. [DOI] [PubMed] [Google Scholar]
- (39) (a).Webster CE; Fan YB; Hall MB; Kunz D; Hartwig JF Experimental and Computational Evidence for a Boron-Assisted, Sigma-Bond Metathesis Pathway for Alkane Borylation. J. Am. Chem. Soc 2003, 125, 858–859. [DOI] [PubMed] [Google Scholar]; (b) Vastine B; Hall MB The Molecular and Electronic Structure of Carbon–Hydrogen Bond Activation and Transition Metal Assisted Hydrogen Transfer Coord. Chem. Rev 2009, 253, 1202–1218. [Google Scholar]
- (40).Sumimoto M; Iwane N; Takahama T; Sakaki S Theoretical Study of Trans-Metalation Process in Palladium-Catalyzed Borylation of Iodobenzene with Diboron. J. Am. Chem. Soc 2004, 126, 10457–10471. [DOI] [PubMed] [Google Scholar]
- (41).Boller TM; Murphy JM; Hapke M; Ishiyama T; Miyaura N; Hartwig JF Mechanism of the Mild Functionalization of Arenes by Diboron Reagents Catalyzed by Iridium Complexes. Intermediacy and Chemistry of Bipyridine-Ligated Iridium Trisboryl Complexes. J. Am. Chem. Soc 2005, 127, 14263–14278. [DOI] [PubMed] [Google Scholar]
- (42).Hartwig JF; Cook KS; Hapke M; Incarvito CD; Fan YB; Webster CE; Hall MB Rhodium Boryl Complexes in the Catalytic, Terminal Functionalization of Alkanes. J. Am. Chem. Soc 2005, 127, 2538–2552. [DOI] [PubMed] [Google Scholar]
- (43).Dang L; Lin ZY; Marder TB DFT Studies on the Borylation of Alpha, beta-Unsaturated Carbonyl Compounds Catalyzed by Phosphine Copper(I) Boryl Complexes and Observations on the Interconversions Between O- and C-Bound Enolates of Cu, B, and Si. Organometallics 2008, 27, 4443–4454. [Google Scholar]
- (44).Olsson VJ; Szabo KJ Functionalization of Unactivated Alkenes through Iridium-Catalyzed Borylation of Carbon-Hydrogen Bonds. Mechanism and Synthetic Applications. J. Org. Chem 2009, 74, 7715–7723. [DOI] [PubMed] [Google Scholar]
- (45).Sawyer KR; Cahoon JF; Shanoski JE; Glascoe EA; Kling MF; Schlegel JP; Zoerb MC; Hapke M; Hartwig JF; Webster CE; Harris CB Time-resolved IR Studies on the Mechanism for the Functionalization of Primary C-H Bonds by Photoactivated Cp*W(CO)(3)(Bpin). J. Am. Chem. Soc 2010, 132, 1848–1859. [DOI] [PubMed] [Google Scholar]
- (46).Lam WH; Lam KC; Lin ZY; Shimada S; Perutz RN; Marder TB Theoretical Study of Reaction Pathways for the Rhodium Phosphine-Catalysed Borylation of C-H Bonds with Pinacolborane. Dalton Trans 2004, 1556–1562. [DOI] [PubMed] [Google Scholar]
- (47).Vanchura BA; Preshlock SM; Roosen PC; Kallepalli VA; Staples RJ; Maleczka RE; Singleton DA; Smith MR Electronic Effects in Iridium C-H Borylations: Insights from Unencumbered Substrates and Variation of Boryl Ligand Substituents. Chem. Commun 2010, 46, 7724–7726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Wei CS; Jimenez-Hoyos CA; Videa MF; Hartwig JF; Hall MB Origins of the Selectivity for Borylation of Primary over Secondary C-H Bonds Catalyzed by Cp*-Rhodium Complexes. J. Am. Chem. Soc 2010, 132, 3078–3091. [DOI] [PubMed] [Google Scholar]
- (49).Roosen PC; Kallepalli VA; Chattopadhyay B; Singleton DA; Maleczka RE; Smith MR Outer-Sphere Direction in Iridium C-H Borylation. J. Am. Chem. Soc 2012, 134, 11350–11353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Green AG; Liu P; Merlic CA; Houk KN Distortion/Interaction Analysis Reveals the Origins of Selectivities in Iridium-Catalyzed C-H Borylation of Substituted Arenes and 5-Membered Heterocycles. J. Am. Chem. Soc 2014, 136, 4575–4583. [DOI] [PubMed] [Google Scholar]
- (51).Larsen MA; Hartwig JF Iridium-Catalyzed C-H Borylation of Heteroarenes: Scope, Regioselectivity, Application to Late-Stage Functionalization, and Mechanism. J. Am. Chem. Soc 2014, 136, 4287–4299. [DOI] [PubMed] [Google Scholar]
- (52).Larsen MA; Wilson CV; Hartwig JF Iridium-Catalyzed Borylation of Primary Benzylic C-H Bonds without a Directing Group: Scope, Mechanism, and Origins of Selectivity. J. Am. Chem. Soc 2015, 137, 8633–8643. [DOI] [PubMed] [Google Scholar]
- (53).Haines BE; Saito Y; Segawa Y; Itami K; Musaev DG Flexible Reaction Pocket on Bulky Diphosphine-Ir Complex Controls Regioselectivity in para-Selective C-H Borylation of Arenes. ACS Catal 2016, 6, 7536–7546. [Google Scholar]
- (54).Zhu L; Qi XT; Li YZ; Duan M; Zou LF; Bai RP; Lan Y Ir(III)/Ir(V) or Ir(I)/Ir(III) Catalytic Cycle? Steric-Effect-Controlled Mechanism for the para-C-H Borylation of Arenes. Organometallics 2017, 36, 2107–2115. [Google Scholar]
- (55).Semproni SP; Milsmann C; Chirik PJ Four-Coordinate Cobalt Pincer Complexes: Electronic Structure Studies and Ligand Modification by Homolytic and Heterolytic Pathways. J. Am. Chem. Soc 2014, 136, 9211–9224. [DOI] [PubMed] [Google Scholar]
- (56).Gunanathan C; Milstein D Bond Activation and Catalysis by Ruthenium Pincer Complexes. Chem. Rev 2014, 114, 12024–12087. [DOI] [PubMed] [Google Scholar]
- (57).Zell T; Milstein D Hydrogenation and Dehydrogenation Iron Pincer Catalysts Capable of Metal Ligand Cooperation by Aromatization/Dearomatization. Acc. Chem. Res 2015, 48, 1979–1994. [DOI] [PubMed] [Google Scholar]
- (58).Li H; Hall MB Computational Mechanistic Studies on Reactions of Transition Metal Complexes with Noninnocent Pincer Ligands: Aromatization Dearomatization or Not. ACS Catal 2015, 5, 1895–1913. [Google Scholar]
- (59).Li H; Hall MB Mechanism of the Formation of Carboxylate from Alcohols and Water Catalyzed by a Bipyridine-Based Ruthenium Complex: A Computational Study. J. Am. Chem. Soc 2014, 136, 383–395. [DOI] [PubMed] [Google Scholar]
- (60).Chai J-D; Head-Gordon M Long-range Corrected Hybrid Density Functionals with Damped Atom-atom Dispersion Corrections. Phys. Chem. Chem. Phys 2008, 10, 6615–6620. [DOI] [PubMed] [Google Scholar]
- (61).Dunning TH Gaussian-basis Sets for Use in Correlated Molecular Calculations .1. the Atoms Boron Through Neon and Hydrogen. J. Chem. Phys 1989, 90, 1007–1023. [Google Scholar]
- (62).Weigend F; Ahlrichs R Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys 2005, 7, 3297–3305. [DOI] [PubMed] [Google Scholar]
- (63).Marenich AV; Cramer CJ; Truhlar DG Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [DOI] [PubMed] [Google Scholar]
- (64).Hay PJ; Wadt WR Ab Initio Effective Core Potentials for Molecular Calculations – Potentials for the Transition-Metal Atoms Sc to Hg. J. Chem. Phys 1985, 82, 270–283. [Google Scholar]
- (65).Andrae D; Häussermann U; Dolg M; Stoll H; Preuss H Energy-Adjusted Ab Initio Pseudopotentials for the 2nd and 3rd Row Transition-Elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar]
- (66).Zhao Y; Truhlar DG The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals. Theor. Chem. Acc 2008, 120, 215–241. [Google Scholar]
- (67).Tao JM; Perdew JP; Staroverov VN; Scuseria GE Climbing the Density Functional Ladder: Nonempirical Meta-Generalized Gradient Approximation Designed for Molecules and Solids. Phys. Rev. Lett 2003, 91, 146401. [DOI] [PubMed] [Google Scholar]
- (68).Grimme S; Ehrlich S; Goerigk L Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem 2011, 32, 1456–1465. [DOI] [PubMed] [Google Scholar]
- (69).Zhao Y; Truhlar DG A New Local Density Functional for Main-Group Thermochemistry, Transition Metal Bonding, Thermochemical Kinetics, and Noncovalent Interactions. J. Chem. Phys 2006, 125, 194101. [DOI] [PubMed] [Google Scholar]
- (70).Perdew JP; Burke K; Ernzerhof M Generalized Gradient Approximation Made Simple. Phys. Rev. Lett 1996, 77, 3865–3868. [DOI] [PubMed] [Google Scholar]
- (71).Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Mennucci B; Petersson GA; Nakatsuji H; Caricato M; Li X; Hratchian HP; Izmaylov AF; Bloino J; Zheng G; Sonnenberg JL; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Montgomery JA Jr.; Peralta JE; Ogliaro F; Bearpark M; Heyd JJ; Brothers E; Kudin KN; Staroverov VN; Keith T; Kobayashi R; Normand J; Raghavachari K; Rendell A; Burant JC; Iyengar SS; Tomasi J; Cossi M; Rega N; Millam JM; Klene M; Knox JE; Cross JB; Bakken V; Adamo C; Jaramillo J; Gomperts R; Stratmann RE; Yazyev O; Austin AJ; Cammi R; Pomelli C; Ochterski JW; Martin RL; Morokuma K; Zakrzewski VG; Voth GA; Salvador P; Dannenberg JJ; Dapprich S; Daniels AD; Farkas O; Foresman JB; Ortiz JV; Cioslowski J; Fox DJ Gaussian 09, Revision D01, Gaussian Inc., Wallingford CT, 2013. [Google Scholar]
- (72).Manson J; Webster CE; Pérez LM; Hall MB; Jimp2 version 091, released on May 19, 2006 http://www.chem.tamu.edu/jimp2/index.html.
- (73).Hall MB; Fenske RF Electronic Structure and Bonding in Methyl- and Perfluoromethyl(pentacarbonyl)manganese. Inorg. Chem 1972, 11, 768–775. [Google Scholar]
- (74).Bursten BE; Jensen JR; Fenske RF An Xα Optimized Atomic Orbital Basis. J. Chem. Phys 1978, 68, 3320–3321. [Google Scholar]
- (75).Berry RS Correlation of Rates of Intramolecular Tunneling Process, With Application to Some Group-V Compounds. J. Chem. Phys 1960, 32, 933–938. [Google Scholar]
- (76).Xu H; Bernskoetter WH Mechanistic Considerations for C–C Bond Reductive Coupling at a Cobalt(III) Center. J. Am. Chem. Soc 2011, 133, 14956–14959. [DOI] [PubMed] [Google Scholar]
- (77).Ben-Ari E; Leitus G; Shimon LJW; Milstein D Metal-ligand Cooperation in C-H and H-2 Activation by an Electron-rich PNPIr(I) System: Facile Ligand Dearomatization-Aromatization as Key Steps. J. Am. Chem. Soc 2006, 128, 15390–15391. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.