

Abstract
Rationale:
This article describes a child with a life-threatening multiorgan failure with disseminated intravascular coagulation (DIC) and massive complement consumption. To our knowledge this therapeutic approach was for the first time effectively applied in a pediatric patient.
Patient concerns:
A 14-month-old boy was presented with a severe, rapidly progressing, life-threatening disease because of sudden onset of fever, hemathemesis, hematuria, and bloody diarrhoea alongside fast spreading hematomas and general corporeal edema.
Diagnosis:
The most plausible diagnosis in our patient is Clostridium difficile sepsis-induced thrombotic microangiopathy alongside with DIC and consumption coagulopathy. The diagnosis was confirmed by positive C difficile bacteria strain in coproculture, clinical, and laboratory tests affirming DIC and global complement activation and consumption.
Interventions:
The patient was treated with antibiotics (Metronidazole, Vancomycin), plasmapheresis, dialysis, methylprednisolone, mycophenolate mofetil, and Eculizumab.
Outcomes:
The child is in fair overall condition in a 2 year follow-up with no complications save chronic renal failure.
Lessons:
In rare cases of sepsis with massive complement consumption, a case-sensitive Eculizumab therapy may be at least considered after the resolution of life-threatening multiorgan failure. The application of this drug can be performed only after sepsis induced disease is put under control. A fast withdrawal of Eculizumab after control of massive complement consumption is recommended to prevent triggering of second sepsis reactivation.
Keywords: child, disseminated intravascular coagulation, Eculizumab, multiorgan failure, sepsis
1. Introduction
Clostridium difficile (C difficile) colitis is constantly an increasing cause of severe infection disease in children.[1,2] A child with life-threatening multiorgan failure (MOF) caused by C difficile is still considered as a challenge for the most plausible treatments.[1,3] Eculizumab, a humanized anti-C5 monoclonal antibody that inhibits terminal pathway activation by blocking the generation of C5b-9 (membrane attack complex) is currently considered as an effective and safe choice for the treatment of patients with uncontrolled complement consumption. Herein, we present a child with MOF and massive complement consumption caused by acute C difficile colitis who among other acknowledged treatment also received a short course of Eculizumab.
2. Case presentation
A 14-month-old boy with severe, rapidly progressing, life-threatening disease was admitted in intensive care unit because of sudden onset of fever, hemathemesis, hematuria, and bloody diarrhoea alongside fast spreading hematomas and general corporeal edema. A day before onset of the disease the child consumed a small portion of plant soil. Sedation was performed with the goal of life-saving interventions tolerance, blood pressure was maintained via medications and as oliguria/anuria soon progressed a continuous veno-venous hemodialysis was set forth. Anuria was maintained throughout the disease. Previously he was a healthy child with no relevant medical history of similar disease or in family history. No prodromal symptoms were noticed.
Relevant clinical, laboratory, diagnostic, and medication follow-up is shown in Figure 1. The initial presentation of MOF begun with sepsis including gastrointestinal, renal, cardiac, and liver impairment. Life-threatening anemia and thrombocytopenia required frequent packed RBC and platelet transfusions, avoided as much as possible. Massive bloody diarrhoea was lasting during the first 10 days following by hose outflow. Bleeding edematous gut mucosa was visualized by colonoscopy without signs of intestinal perforation. The child received initially multiple fresh frozen plasma infusions and a short course of plasma exchange with plasma replacement during 4 consecutive days, until stabile vital functions were achieved. Kidney ultrasound showed hyperechogenic kidneys without corticomedullar differentiation. As positive C difficile bacteria was isolated in 2 separate coprocultures by 2 independent laboratories, Metronidazole and Vancomycin were administered. Unfortunately, due to technical difficulties we did not achieve positive C difficile toxin identification. Shiga toxin was proved negative as well as E. coli, Shigella sp., Streptococcus pneumoniae, Campylobacter sp., and other bacteria causing HUS. Extensive search for poisonous substances were proved negative alongside with PNH, methylmalonic aciduria and homocystinuria. No fragmentocytes were found in periferal blood. Direct Coombs test was negative.
Figure 1.
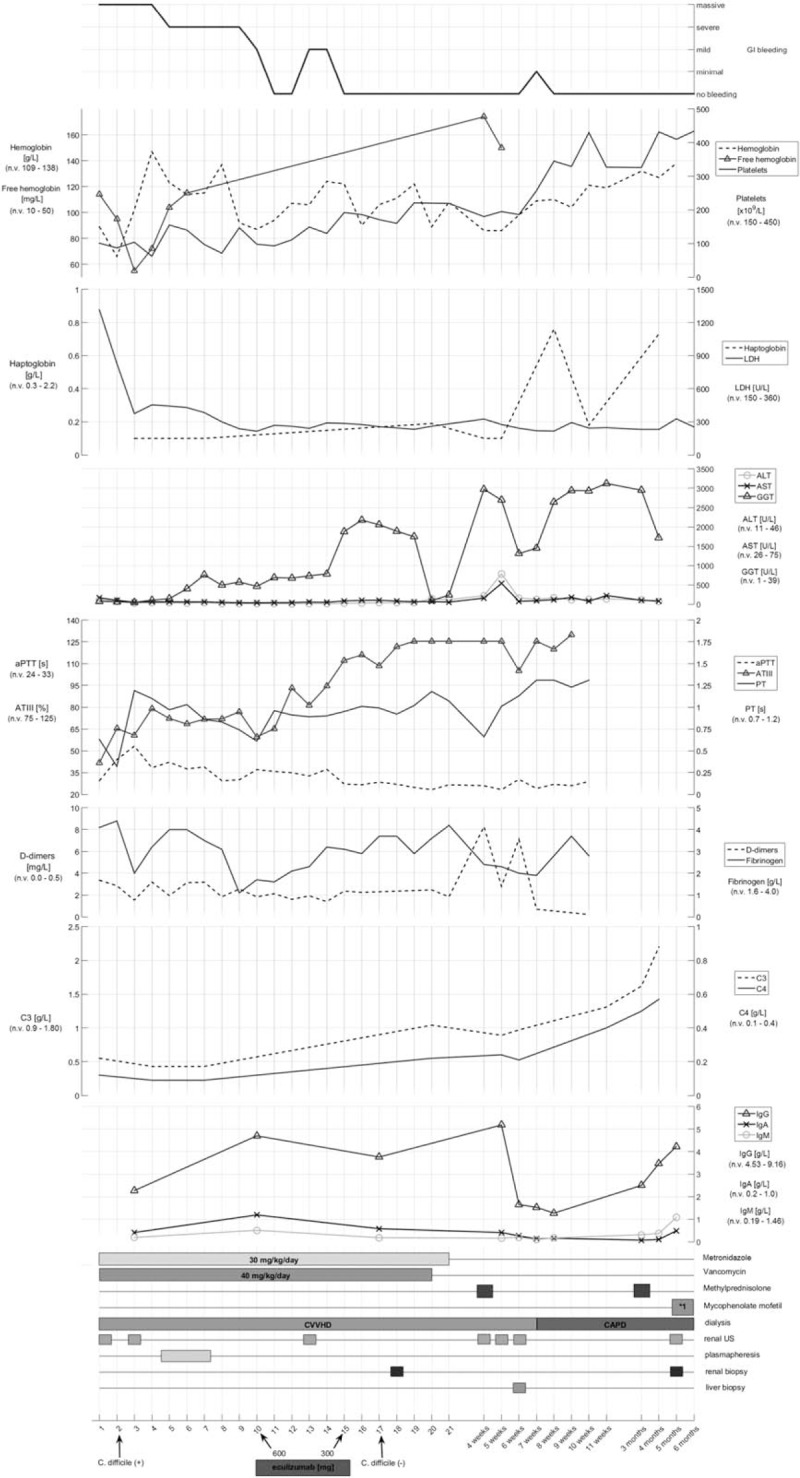
Clinical, laboratory, and treatment time-line of our case.
Coagulation tests revealed normal prothrombin time (PT), elevated activated partial thromboplastin time (aPTT), and increased D-dimers. Fibrinogen was found within normal range. LDH and free hemoglobin were elevated from the beginning of the disease. Antithrombin III values were low and slowly improved during time. All values were tested according to ISTH diagnostic scoring system for DIC guidelines and found negative <5.[4–6] Haptoglobin values were low from the beginning of the disease. C3, factor H levels, and alternative pathway (AP) activity were decreased, without elevation of complement activation markers sC5b9 or C3a. This is probably due to a protein-loss related activity rather than activation and consumption. C1q was severely decreased and with negative anti-C1q, supporting the presence of global complement activation and consumption. Decreased but not deficient ADAMTS13 metalloprotease activity (46%) excluded thrombotic thrombcytopenic purpura (TTP). Liver enzymes (AST, ALT, GGT) were elevated from the beginning of the disease. Vitamin K was supplemented. Immunoglobulins levels (IgG, IgA, and IgM) were found to be lower for the age and in need of occasional substitution even after active disease ceased. Abdominal and brain CT scans were proven negative for gut perforation or brain damage.
From clinical judgment and laboratory findings (normal plasma fibrinogen, elevated D-dimers and free hemoglobin, low haptoglobin and decreased platelets), as well as alternative pathway activation indicating the presence of severe, global complement activation and consumption, we suspected of uncontrollable complement regulator loss and activation process underway the infective C difficile disease trigger. Therefore, a secondary thrombotic microangiopathy (TMA) triggered by the infection and by the coagulopathy was assumed. Negative complement activation marker sC5b9 may be explained by plasma sample collection which was taken before ongoing TMA activation.
After administration of Eculizumab in dosage on 10th day of the disease (600 mg) the patient's physical condition soon improved with ceasing of bloody diarrhoea. Second onset of bloody diarrhoea after 5 days also ceased with the second dose of Eculizumab (300 mg). Small amount of bloody diarrhoea was again noticed after 1 month of last Eculizumab administration but treated via symptomatic medications. After administration of Eculizumab, a rapid improvement and normalization of platelets and other hematologic data were noticed alongside with further significant clinical improvement. Haptoglobin level was normalized 6 weeks after the administration of Eculizumab. As patient's clinical state and laboratory values were normalized and with negative support from kidney biopsy, we decided to cease further use of Eculizumab.
As elevation of liver enzymes continued throughout the course of the disease, a liver biopsy was performed showing no tissue pathology. Kidney biopsy was performed 1 week after the start of Eculizumab treatment and showed interstitial nephritis. Therefore, a pulse methylprednisolone therapy (5 days of duration) was administered followed by oral methylprednisolone therapy during following month. As kidney function was not improved, a second kidney biopsy was performed with the same histological features. A second pulse of methylprednisolone was thus performed 2 months after the first one, followed by oral mycofenolate mofetil with no recovery of kidney function afterward. Soon after methylprednisolone/mycofenolate use all liver enzymes fall to normal values after 6 months of its continuous elevation. Other supportive therapy (i.v. immunoglobulins) were also periodically administered. During the genetic workup, the whole coding regions of the genes encoding complement factor H (CFH), complement factor H-related protein 5 (CFHR5), factor I (CFI), membrane cofactor protein (CD46), thrombomodulin (THBD), factor B (CFB), C3 (C3), and diacylglycerol kinase-epsilon (DGKE) were analyzed by direct DNA sequencing following PCR amplification. The patient was found to be heterozygous for a substitution in exon 2 of the CFHR5 gene (c.136C>T) causing a proline to serine change at codon 46 of the complement factor H-related protein 5 (p. P46S), interpreted as variant with unknown functional significance. Furthermore, the CFH H3 haplotype was identified in the patient, reported as a risk factor of aHUS.
3. Discussion
Since verotoxin test was negative, and the disease showed unusually severe course with MOF, typical HUS could be excluded safely despite obvious triggering of the disease by C difficile.[7–10] It is possible that the development of secondary HUS/TTP was related to severe protein loss, but predisposition by the presence of CFH H3 aHUS risk haplotype and/or by the p.P46S CFHR5 mutation should also be considered. Pathogenic factors identified for this case may represent rather a continuum: based on the current pathogenesis model, weak predispositing factors with strong trigger may jointly lead to the development of thrombotic microangiopathy. We consider protein loss (loss of plasma factors and regulators in the GI tract) as the major obstacle in pathogenesis, which may also explain the lack of activation product (sC5b9) elevation. Elevated LDH at the beginning of the disease and free hemoglobin lasting 5 weeks alongside with elevated D-dimers, low platelets, decreased ADAMTS13 activity, consistent and long lasting low level of haptoglobin supports the presence of secondary thrombotic microangiopathy, manifested as MOF and HUS. Genetic analysis does not fully support aHUS either. The boy's mutation was not described previously in patients with aHUS but was observed in 3 patients with MPGN II/DDD (PMID: 16299065) and also in healthy subjects with a relatively low frequency (0.8–1.8%). Based on these data this mutation may or may not have a pathogenic role in the development of aHUS, and was considered as variant with unknown functional significance. Importantly, the possibility of thrombotic microangiopathy triggered by disseminated intravascular coagulation (DIC)—specific pathology seems plausible explanation.[11]
Therefore, one of the most presumable solutions for the diagnosis in our patient is DIC or consumption coagulopathy which can arise in sepsis-induced thrombotic microangiopathy (TMA), especially in severe cases with reduced ADAMTS13 activity.[12–17] The presence of DIC is supported by aPTT moderate elevation (normalized after 8 days) and reduced antitrombin III (probably due to protein loss) which lasts 12 days to full recovery alongside low platelets count.[4,6] A 7 weeks of elevated plasma D-dimers as well as decreased ADAMTS13 activity are nevertheless consistent with DIC but also with concomittant TMA.[4] Both, ADAMTS13 and aPTT have prognostic significance for DIC evolution indicating increased possibility of renal failure.[4,18,19] Normal level of fibrinogen as well as ISTH score repeatedly negative results are not compatible with DIC (low plasma fibrinogen is often associated with severe forms of DIC.[4–6] However, normal level of fibrinogen observed in severe septic DIC may be due to its substantial overproduction which compensates the rate of actual consumption.[6] We should expect prolonged PT (usually >50%) associated with liver disease, vitamin K deficiency and loss of the coagulation proteins due to massive bleeding.[4] The possible explanation may be the presence of circulating activated clotting factors, such as thrombin and Xa.[6]
After successful treatment of sepsis, symptoms of DIC soon decreased. Eculizumab treatment in sepsis-induced DIC with decreased ADAMTS13 activity was already successfully administered in adult patients.[12,13] The same treatment applies for the secondary TMA.[20] Judging from clinical feature and Eculizumab response, a secondary TMA seems to be probable. It is also supported by long lasting low level of haptoglobin and platelets from the beginning of the disease as well as by minor but steady hemolysis. Fast normalization of LDH is probably partly explained by plasma exchange. Low C3 supports potential concurrent TMA as well. Decreased C3, HF, and AP activity may support complement loss or activation and ongoing TMA; however, these parameters may also be related to infection related consumption. Since, due to a technical difficulties kidney biopsy was performed only 1 week after the start of Eculizumab treatment, it is not possible to draw firm conclusion about the presence or absence of TMA before treatment.
A potential crosstalk between complement and coagulation systems may be involved in our patient as complement is activated in most patients with DIC and active coagulation cascade.[11,12,21–23] Some authors suggested that patients with amplification of the complement and coagulation cascades with clinical progression of the disease may benefit from Eculizumab, preventing the generation of C5a and membrane attack complex impairment.[11,23] Rapid clinical and laboratory response (rise of platelets and C3) after Eculizumab administration best fits such possibility. Infection with C difficile supports all aforementioned diagnostic possibilities, (HUS, aHUS, secondary TMA DIC). Unrecognized immune deficiency afore present illness probably contributed to onset of C difficile infection-mediated disease since decreased levels of immunoglobulins (IgG, IgA, IgM) IgG) still exist after the cease of active disease.
To our best judgement Eculizumab administration in such clinical cases remains controversial. We are fully aware that if administered sooner than time-optimal without sufficient infection control of triggering MOF, Eculizumab might increase the risk of infection by meningococcus and encapsulated bacteria. By reducing massive complement activation, Eculizumab may temporarily help to prevent tissue damage involved in MOF. We do not recommend Eculizumab administration in all of MOF patients. Instead, a case-sensitive approach in patients with massive complement consumption with short course of Eculizumab may be considered as a temporary solution for complement inhibition. It may look like driving between Scylla and Charybdis but carefully timing Eculizumab administration will temporary reduce devastating complement activation and consumption, minimazing the risk of infection activation. Plausible withdrawal of the drug by its short course of administration may be the proper answer.
To our knowledge, this is the first report of a child treated with Eculizumab in such conditions. Carefully monitoring symptoms and signs of infection with real-time laboratory data and daily evaluation of patient's overall condition may lead physicians to optimal therapy approach. We believe that in similar clinical circumstances apart from C difficile triggering infection, the administration of Eculizumab should be at least considered.
4. Declarations
This publication has been approved for publishing in the journal BMC Pediatrics by the ethical committee of the University Hospital Centre Zagreb. Informed written consent was obtained from the patient for publication of this case report and accompanying images.
Author contributions
Conceptualization: Danko Milosevic.
Data curation: Slobodan Galic, Dorottya Csuka, Zoltán Prohászka, Daniel Turudic, Petra Dzepina.
Formal analysis: Dorottya Csuka, Zoltán Prohászka, Daniel Turudic.
Investigation: Slobodan Galic, Daniel Turudic, Danko Milosevic.
Methodology: Danko Milosevic.
Project administration: Danko Milosevic.
Software: Daniel Turudic, Petra Dzepina.
Supervision: Danko Milosevic.
Validation: Danko Milosevic.
Writing – original draft: Danko Milosevic.
Writing – review & editing: Daniel Turudic.
Footnotes
Abbreviations: AP = alternative pathway, aPTT = activated partial thromboplastin time, CD46 = membrane cofactor protein 46, CFB = complement factor B, CFH = complement factor H, CFHR5 = complement factor H-related protein 5, CFI = complement factor I, DGKE = diacylglycerol kinase-epsilon, HUS = hemolytic-uremic syndrome, MOF = multiorgan failure, PNH = paroxysmal nocturnal hemoglobinuria, PT = prothrombin time, RBC = red blood cell, THBD = thrombomodulin, TMA = thrombotic microangiopathy, TTP = thrombotic thrombcytopenic purpura.
The authors of this publication have no competing interests or financial disclosures.
Data and material are available for review on demand.
The authors have no conflicts of interest to disclose.
References
- [1].Tamma PD, Sandora TJ. Clostridium difficile infection in children: current state and unanswered questions. J Pediatric Infect Dis Soc 2012;1:230–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Clayton JA, Toltzis P. Recent issues in pediatric Clostridium difficile infection. Curr Infect Dis Rep 2017;19:49. [DOI] [PubMed] [Google Scholar]
- [3].Shen EP, Surawicz CM. Current treatment options for severe Clostridium difficile-associated disease. Gastroenterol Hepatol (NY) 2008;4:134–9. [PMC free article] [PubMed] [Google Scholar]
- [4].Wada H, Matsumoto T, Yamashita Y. Diagnosis and treatment of disseminated intravascular coagulation (DIC) according to four DIC guidelines. J Intensive Care 2014;2:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Gando S, Saitoh D, Ogura H, et al. A multicenter, prospective validation study of the disseminated intravascular coagulation scoring system in patients with severe sepsis. Crit Care 2013;17:R11120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Levi M, Toh CH, Thacil J, et al. BJH guideline. Guidelines for the diagnosis and management of disseminated intravascular coagulation 2009;145:24–33. [DOI] [PubMed] [Google Scholar]
- [7].Alvarado AS, Brodsky SV, Nadasdy T, et al. Hemolytic uremic syndrome associated with Clostridium difficile infection. Clin Nephrol 2014;81:302–6. [DOI] [PubMed] [Google Scholar]
- [8].Mbonu CC, Davison DL, El-Jazzar KM, et al. Clostridium difficile colitis associated with hemolytic-uremic syndrome. Am J Kidney Dis 2003;41:E14. [DOI] [PubMed] [Google Scholar]
- [9].Kalmanovich E, Kriger-Sharabi O, Shiloah E, et al. Atypical hemolytic uremic syndrome associated with Clostridium difficile infection and partial membrane cofactor protein (CD46) deficiency. IMAJ 2012;14:586–7. [PubMed] [Google Scholar]
- [10].Rafiq A, Hassan Tariq H, Naeem Abbas N, et al. Atypical hemolytic-uremic syndrome: a case report and literature review. Am J Case Rep 2015;16:109–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kurosawa S, Stearns-Kurosawa DJ. Complement, thrombotic microangiopathy and disseminated intravascular coagulation. J Intensive Care 2014;2:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Abe T, Sasaki A, Ueda T, et al. Complement-mediated thrombotic microangiopathy secondary to sepsis-induced disseminated intravascular coagulation successfully treated with eculizumab. Medicine 2017;96:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Omura T, Watanabe E, Otsuka Y, et al. Complete remission of thrombotic microangiopathy after treatment with eculizumab in a patient with non-Shiga toxin associated bacterial enteritis. Medicine 2016;95:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Sakamaki Y, Konishi K, Hayashi K, et al. Renal thrombotic microangiopathy in a patient with septic disseminated intravascular coagulation. BMC Nephrol 2013;14:260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Booth KK, Terrell DR, Vesely SK, et al. Systemic infections mimicking thrombotic thrombocytopenic purpura. Am J Hematol 2011;86:743–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Coppo P, Adrie C, Azoulay E, et al. Infectious diseases as a trigger in thrombotic microangiopathies in intensive care unit (ICU) patients? Intensive Care Med 2003;29:564–9. [DOI] [PubMed] [Google Scholar]
- [17].Douglas KW, Pollock KG, Young D, et al. Infection frequently triggers thrombotic microangiopathy in patients with preexisting risk factors: a single-institution experience. J Clin Apher 2010;25:47–53. [DOI] [PubMed] [Google Scholar]
- [18].Habe K, Wada H, Ito-Habe N, et al. Plasma ADAMTS 13, von Willebrand factor (VWF) and VWF propeptide profiles in patients with DIC and related diseases. Thromb Res 2012;129:598–602. [DOI] [PubMed] [Google Scholar]
- [19].Ono T, Mimuro J, Madoiwa S, et al. Severe secondary deficiency of von Willebrand factor-cleaving protease (ADAMTS 13) in patients with sepsis-induced disseminated intravascular coagulation: its correlation with development of renal failure. Blood 2006;107:528–34. 15. [DOI] [PubMed] [Google Scholar]
- [20].Román E, Mendizábal S, Jarque I, et al. Secondary thrombotic microangiopathy and eculizumab: a reasonable therapeutic option. Nefrologia 2017;37:478–91. [DOI] [PubMed] [Google Scholar]
- [21].Lupu F, Keshari RS, Lambris JD, et al. Crosstalk between the coagulation and complement systems in sepsis. Thromb Res 2014;133(0 1):S28–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kenawy HI, Boral I, Alan Bevington A. Complement-coagulation cross-talk: a potential mediator of the physiological activation of complement by low pH. Front Immunol 2015;6:215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Oikonomopoulou K, Ricklin D, Ward PA, et al. Interactions between coagulation and complement—their role in inflammation. Semin Immunopathol 2012;34:151–65. [DOI] [PMC free article] [PubMed] [Google Scholar]