

Abstract
Familial hypercholesterolemia (FH) is a monogenic dominant inherited disorder of lipid metabolism characterized by elevated low-density lipoprotein levels, and is mainly attributable to mutations in low-density lipoprotein receptor (LDLR), apolipoprotein B (APOB), and proportein convertase subtilisin/kexin type 9 (PCSK9) genes. Next-generation and exome sequencing studies have primarily involved genome-wide association analyses, and meta-analyses and next-generation studies examined a few single-nucleotide polymorphisms (rs151009667 and Val2095Glu) in the ApoB gene. The present study was conducted to investigate the association of APOB and patients with FH in a Saudi population.
We genotyped 100 patients with FH and 100 controls for 2 polymorphisms in APOB using polymerase chain reaction-restriction fragment length polymorphism, followed by 3% agarose gel electrophoresis. The strength of the association between the genotype and allele frequencies with the risk of developing FH was evaluated. Clinical details and genotype analysis results were recorded.
For the rs151009667 polymorphism, 18% of the CT genotypes were observed only in patients with FH. There was a positive association between CT and CC (odds ratio [OR] 45.07 [95% conflict of interest (CI), 2.67–759.1]; P = .0001) and between T and C (OR 87.8 [95% CI, 5.34–144.2]; P < .0001). However, no Val2095Glu mutations were found in patients with FH or controls. There was also no correlation between clinical characteristics and the rs151009667 polymorphism.
In conclusion, we confirmed the association between the rs151009667 polymorphism and FH in a Saudi population. The Val2095Glu novel variant did not appear in either patients with FH or controls. Similar studies should be performed in different ethnic populations to rule out the role of this polymorphism in FH.
Keywords: ApoB gene, familial hypercholesterolemia, rs151009667, Saudi population, Val2095Glu
1. Introduction
Familial hypercholesterolemia (FH; OMIM# 143890) is defined as a common genetic condition categorized by increased plasma levels of low-density lipoprotein-cholesterol (LDL-C) and premature atherosclerotic cardiovascular disease.[1] The disease FH was initially discovered in 1920 by Beeson,[2] and is typically considered to be a single-gene disorder.[3] The FH inheritance pattern was initially described by Khachadurian[4] in Lebanon, before the genes contributing to the disease were identified. FH is defined as an autosomal dominant disease with a clinical presentation based on the phenotype severity of homozygous and heterozygous forms, and with serum LDL-C levels that are 2- to 4-fold the normal level, respectively.[5] In FH, the frequency varies between heterozygous (1/500) and homozygous (1/1,000,000) FH. However, a recent population analysis estimated the prevalence to be as high as 1/250.[6] FH develops between the ages of 30 and 50 years in men and 40 and 60 years in women. Patients with FH are not always diagnosed properly, leading to inappropriate treatment strategies. FH-associated complications are common because of premature diagnosis and therapeutic interventions.[7] FH may be caused by a gain-of-function mutation in the LDL receptor (LDLR), apolipoprotein B (APOB), and pro-protein convertase subtilisin-Kexin type 9 (PCSK9) genes. LDLR may suppress protein synthesis, which is translocated to the cell surface in monogenic mutations and by an additional mechanism that involves mutation affecting APOB, which encodes a key structural component of LDL and very low-density lipoprotein (VLDL). In addition, ApoE and LDLR adaptor protein 1 are required for LDLR and FH.[8,9] FH-affecting mutations were present in 60% to 80% of patients with a clinical diagnosis of pure FH, and 20% to 30% of the affecting mutations may appear in conceivable FH.[10] Genetic variations in LDLR are loss of function mutations, whereas APOB and PCSK9 show similar lipid profile homeostasis functional defects. To date, >1000 genetic variations in LDLR, APOB, and PCSK9 have been reported in the British Heart Foundation and other databases.[11] The FH diagnostic criteria are based on the Simon Broome criteria (UK); (Dutch Lipid Clinic Network Criteria (Netherland); and MedPed criteria (USA).[12] Various proteins, cholesterol internalization, and cellular metabolism have been connected to FH (e.g., ApoB-100, PCSK9, and LDLR). Genetic variations originating in the proteins include large rearrangements of intronic regions, coding, synonymous, nonsynonymous substitutions, and mutations in regulatory regions or splicing sites. Missense mutations were the most frequent mutation type and were identified using second-generation sequencing techniques such as exome and next-generation sequencing (NGS) technologies within the exon coding region.[13] Radovica-Spalvina et al[3] previously performed NGS, and confirmed novel and documented variants in their cohort subjects. Our study was conducted to investigate the novel mutation Val2095Glu and familial variant rs151009667 in APOB in a case–control study of patients with FH in a Saudi population.
2. Materials and methods
2.1. FH participants
The Institutional Review Board of the College of Medicine at the King Saud University (KSU) provided ethics approval (E-12-829) for this study. All subjects who participated in study including patients with FH and control subjects signed an informed consent form. This study was performed according to the principles of the Declaration of Helsinki. As described in our prior publications,[7,14,15] 100 patients with FH were recruited from King Khalid University Hospital (KKUH) at KSU. Inclusion criteria were as follows: FH diagnosis made according to the Dutch group criteria[7]; male or female; subjects who underwent regular checkups; and subjects with no endocrine, metabolic, chronic, and other diseases. Exclusion criteria included the following: subjects with abnormal body mass index (BMI); subjects with diabetes; subjects with liver, renal, or thyroid disease or any other type of diseases; and subjects recruited outside the KKUH. Sex-matched controls (n = 100) were recruited from contract-based KKUH staff who may or may not have been outpatients.
2.2. Blood collection
From each patient, 5 mL of the peripheral blood was collected into 2 tubes (plain and EDTA tubes) by an experienced nurse. A 3-mL sample was used for biochemical analysis of the lipid profile, such as triglycerides (TGs), total cholesterol (TC), high-density lipoprotein-cholesterol (HDL-C), and LDL-C. The remaining 2 mL was used for molecular analysis and was collected into an EDTA tube. Biochemical indications were analyzed using an automated clinical chemistry analyzer (KoneLab, Espoo, Finland).[14]
2.3. Molecular genotyping
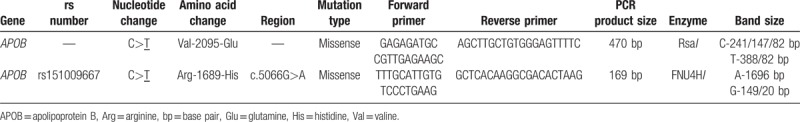
Genomic DNA was extracted from the EDTA blood using a commercial human DNA kit as described by Alharbi et al.[7] To quantify genomic DNA, a NanoDrop spectrophotometer (Thermo Fischer Scientific, Waltham, MA) was used and primers were designed for the selected ApoB variants based on Radovica-Spalvina et al.[3] The complete details of ApoB variants are listed in Table 1. The primers were designed using Primer 3 software. For the Val2095Glu and rs151009667 variants, genotyping was performed by polymerase chain reaction with a 25-μL sample consisting of nuclease-free water, buffer, 2.5 μL MgCl2, 0.5 μL dNTPs, 0.5 μL Taq DNA polymerase, 100 pmol of both sense and antisense primers, and 50 ng quantified genomic DNA. Initial denaturation was carried out at 95°C for 5 min followed by 35 cycles of 30 s at 64°C (for both variants), and 95°C for 45 min for initial elongation. The final elongation step was carried out at 72°C for 5 min. For both variants, restriction fragment length polymorphism analysis was conducted. Both the RsaI (GT↓AC) and FNU4HI (GC↓GGC) restriction enzymes (NEB England BioLabs, Ipswich, MA) were used to digest the samples for 18 h at 37°C. The digestion products were separated on a 3% agarose gel, which was stained with Ethidium bromide and visualized using ultraviolet light.
Table 1.
Genetic details of the APOB gene evaluated in this study.
2.4. Statistical analysis
Clinical data between patients with FH and controls were evaluated using SAS software (version 9.3, SAS Institute, Cary, NC). Hardy-Weinberg equilibrium was tested between patients and controls. Descriptive characteristics of categorical variables are presented as the count (%), and continuous variables are presented as the mean and standard deviation. Between patients with FH and controls, the association was tested using 2 independent sample t tests.[7] Genotype and allele frequencies between patients with FH and controls were assessed using Openepi software (version 2.3.1) to determine the odds ratios, χ2, and P values with and without Yates correction. Analysis of variance was also performed between the FH genotypes for rs151009667 and clinical characteristics.
3. Results
3.1. Clinical analysis
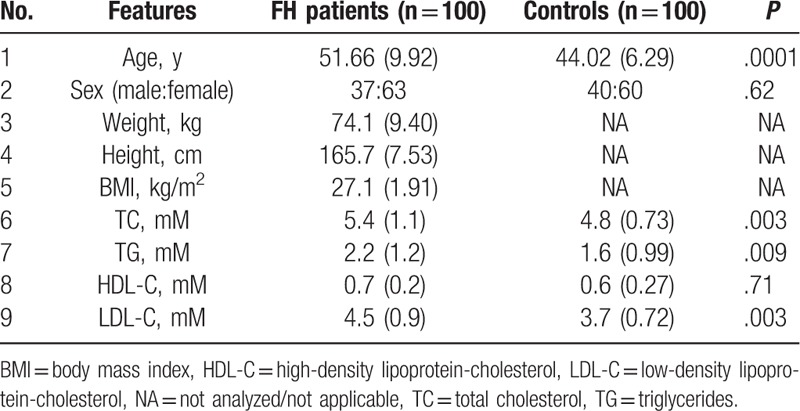
The results of anthropometric and biochemical measurements of patients with FH and controls are presented in Table 2. The mean ages of patients with FH and controls were 51.66 ± 9.92 and 44.02 ± 6.29 years, respectively, which was significantly different (P = .0001). Patients with FH and controls had a nearly equal sex ratio with no significant difference between groups (P > .05). We measured the height (165.7 ± 7.53 cm) and weight (74.1 ± 9.40 kg) only of patients with FH, and the overall BMI was 27.1 ± 1.91 kg/m2 for this group. Lipid profile analysis revealed a positive association with TC, TG, and LDL-C (P < .05), whereas HDL-C showed a negative association when comparing patients with FH and controls (P = .71).
Table 2.
Clinical features of familial hypercholesterolemia (FH) patients and controls.
3.2. Genetic analysis of the rs151009667 polymorphism
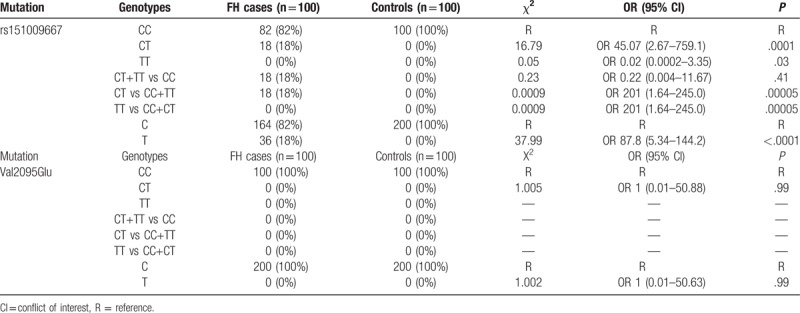
No deviation from Hardy–Weinberg equilibrium was detected in the control group for both variants. Allele and genotype analyses for FH patients and controls are presented in Table 3. All alleles and genotypes in both variants were adjusted by Yates correction. The rs151009667 genotype frequencies for CC and CT were 82% and 18%, respectively, for patients with FH patients, whereas the genotypes in the control group were all CC. However, a strongly significant association was observed only for CT versus CC (odds ratio [OR] 45.07 [95% conflict of interest (CI), 2.67–759.1]; P = .0001) and T vs C: (OR 87.8 [95% CI, 5.34–144.2]; P < .0001). No mutations were detected for CT genotypes within control subjects. No homozygous variants appeared in either patients with FH or control subjects for rs151009667.
Table 3.
Yates correction for allele and genotype frequencies of APOB polymorphisms in patients with familial hypercholesterolemia (FH) and controls.
3.3. Val2095Glu allele and genotype frequencies
A 470 bp polymerase chain reaction product encompassing the novel variant Val2095Glu was digested using the FNU4H1 restriction enzyme, which yielded a 241/147/82 bp fragment, confirming the presence of the C allele. Only the CC genotype was detected in patients with FH and controls. No significant association was observed between the alleles and genotypes for CT vs CC (OR 1.00 [95% CI, 0.01–50.88]; P = .99) and T vs C (OR 1.00 [95% CI, 0.01–50.63]; P = .99). Thus, this locus was not further analyzed. Sanger sequencing was performed on 30 FH samples, which showed the same results.
3.4. Analysis of variance
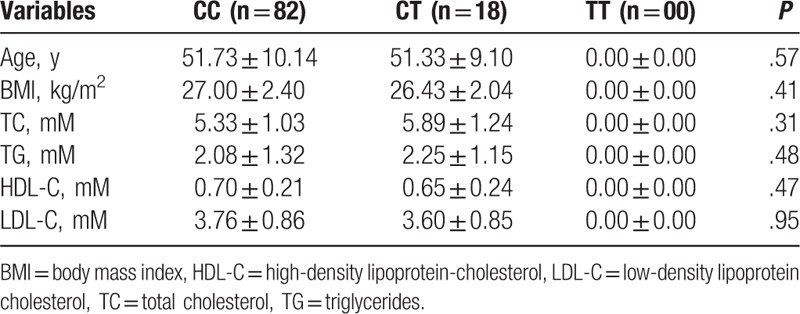
Anthropometric measurements and the lipid profiles of patients with FH and controls with the CC, CT, and TT genotypes of the rs151009667 polymorphism were compared (Table 4). No FH patient characteristics showed a positive association between the rs151009667 polymorphism and age (P = .57), BMI (P = .41), TC (P = .31), TG (P = .48), HDL-C (P = .47), and LDL-C (P = .95).
Table 4.
ANOVA analysis performed between the rs151009667 polymorphism of APOB and nongenetic variables.
4. Discussion
No case–control studies have investigated rs151009667 and the novel Val2095Glu variant in the global population. This is the first study to analyze these factors in a Saudi population and the world. We investigated the association between 2 genetic variants and patients with FH in a Saudi population. To confirm the findings of this study, we screened the Val2095Glu and Arg1689His (rs151009667) variants in patients with FH and controls. The rs151009667 SNP was found to be associated in patients with FH compared to controls (P < .05). However, Val2095Glu showed no heterozygous (CT) and homozygous variant (TT) genotypes in patients with FH or controls (P > .05). These 2 variants were screened by exome sequencing analysis, as used in a previous study of a Latvian population of patients with FH. Generally, heterozygous variants detected in FH disease assists with detection using a sequence base as the prime methodology, and NGS/ES techniques have made these methods easier to use for diagnostic purposes.[3]
Alharbi et al[7] defined FH as a well-known autosomal dominant disorder with an earlier increased risk of coronary heart disease. FH is the only disease diagnosed based on cascade screening of genetic mutations such as LDLR, APOB, and PCSK9 and based on high cholesterol levels and a family history. It is recommended to diagnose FH as soon as possible to prevent morbidity and mortality. However, the latest statins in pharmacotherapy have helped to lower LDL-C levels, decreasing morbidity and mortality related to cardiovascular disease.[16] A recent study by Ghaleb et al[17] confirmed that ApoE is an additional causative gene that contributes to screening for FH. Genetic variants in LDLR, APOB, APOE, and PCSK9 are present in 80% of patients with FH. Genetic risk profiling improved after genome-wide screening was introduced and can simultaneously screen millions of polymorphisms for any complex disease.[18] In France, genetic cascade screening is currently used to help diagnose FH. NGS techniques enable examination of the monogenic and polygenic origins of elevated LDL-C levels observed in patients with FH.[19] There are a few common misconceptions in case control studies, such as matching in these studies does not always eliminate confounding factors; and matching analysis may not always be required. A previous study by Pearce[20] clarifies the misconceptions in case–control studies. In this study, we selected sex-based samples in the recruited subjects. Patients with FH included 37% male and 63% female subjects, whereas controls included 40% male and 60% female subjects. However, age-matched controls were not recruited in this study because FH can develop at any age.
APOB is one of the major genes used for the molecular diagnosis of FH. The chromosomal region is located in the 2p24.1 region.[21]APOB was initially discovered in 1987 in phenotypic patients with FH, which revealed that FH is not related to mutations in LDLR.[22]ApoB has a specific role in FH disease, and variants in APOB typically occur in the exons. Patients with FH with a familial deficiency in APOB may have a milder form of the disease, which can affect LDLR mutations.[23]APOB is known to be associated with FH and it very important in the diagnosis of FH disease. Patients with homozygous FH may be documented in a proband with the appearance of biallelic pathogenic variants.[24] This gene encodes 2 proteins (ApoB-48 and ApoB-100), which play a major role in disease diagnosis. ApoB-48 is present in the intestine and ApoB-100 is in the liver. FH disease with APOB is confirmed by documenting 5 known mutations in ApoB-100. Each mutation blocks cholesterol in the walls of coronary arteries, increasing cholesterol levels and thus the risk of heart attack. Each mutation modifies a single protein building block in a critical region of ApoB-100. LDLs are removed from the blood by the modified proteins, leading to increased cholesterol levels. This is the major relationship between FH and APOB.[25] In any population, patients with mutations in Val2095Glu may have a greater risk of developing FH disease. The 2 SNPs (rs151009667 and Val2095Glu) examined in the present study may promote FH disease development because of the appearance of homozygous and heterozygous variants.
The role of the rs151009667 polymorphism is mostly related to FH and hypertriglyceridemia,[26] whereas for the novel variant Val2095Glu, its genetic role in any disease is unknown. More studies enrolling patients of different ethnicities should be performed to understand the connection with the novel variant in FH. The in vivo relationship between the molecular mechanisms of APOB variation and FH disease is well documented.
Few genetic studies have been performed in Saudi populations with FH and no NGS/ES studies have been performed in the Saudi population.[27]APOB is commonly used to diagnose all patients with FH; however, in this study, we identified novel SNPs and examined their association with FH. Few studies have used meta-analysis to examine FH disease.[28–31] Currently, only one ES study was performed in the global population,[32] whereas a limited number of NGS studies have been performed worldwide.[3,33–37] Radovica-Spalvina et al[3] identified 111 variants as synonymous, nonsynonymous, close proximity to the intron-exon boundary region, and other using NGS techniques in Latvian patients with FH. A meta-analysis of GWAS data can expand the associated data documented in earlier studies using imputed genotype data. However, preferred data selection should consider that the GWAS and meta-analysis results are combined with a unique number of genetic associations that have strong statistical support. GWAS and meta-analysis have shown that convincing size effects are always moderate when using the current platforms and the sample sizes can explain most of the large genetic risks common to most diseases.[38]
The present study had several limitations. We did not collect smoking status, exercise, diet, or clinical and medication details from the enrolled patients with FH. Only 100 FH cases and 100 control subjects were evaluated and thus larger samples sizes are needed to confirm our results. However, we enrolled all subjects from a native Saudi population, including both patients with FH and controls. Some unknown SNPs were screened in this study, and one novel variant was identified as well as another SNP reported in only a few studies. Most sex-matched subjects were recruited for both patients with FH and controls, which was another limitation of our study. In conclusion, we confirmed the association between the rs151009667 polymorphism and FH in a Saudi population. The Val2095Glu novel variant did not appear in either patients with FH or controls. These similar SNPs should be screened in different ethnic populations with FH disease to document their role in FH diagnosis. Accurate meta-analysis results will reveal the genetic role of the diagnostic markers. Future studies should also include NGS/ES studies in the Saudi population. Large-scale studies would be required from different areas and races to screen such a polymorphism.
Author contributions
Conceptualization: Khalid Khalaf Alharbi.
Data curation: Turky H. Almigbal, Imran Ali Khan.
Formal analysis: Mohammed Ali Batais, Fawaziah Khalaf Alharbi, Khalid Khalaf Alharbi, Imran Ali Khan.
Funding acquisition: Mohammed Ali Batais, Turky H. Almigbal.
Investigation: Mohammed Ali Batais, Turky H. Almigbal.
Methodology: Turky H. Almigbal, Noor Ahmad Shaik, Imran Ali Khan.
Project administration: Khalid Khalaf Alharbi.
Resources: Fawaziah Khalaf Alharbi.
Software: Khalid Khalaf Alharbi, Imran Ali Khan.
Supervision: Imran Ali Khan.
Validation: Imran Ali Khan.
Visualization: Imran Ali Khan.
Writing – original draft: Imran Ali Khan.
Writing – review and editing: Fawaziah Khalaf Alharbi, Khalid Khalaf Alharbi, Imran Ali Khan.
Imran Ali Khan orcid: 0000-0002-9746-5300.
Footnotes
Abbreviations: ApoB = apolipoprotein B, BMI = body mass index, CI = conflict of interest, FH = familial hypercholesterolemia, Glu = glutamine, HDLC = high-density lipoprotein receptor, LDLR = low-density lipoprotein receptor, NGS = next-generation sequencing, PCSK9 = pro-protein convertase subtilisin-Kexin type 9, SD= standard deviation, TC = total cholesterol, TG = triglyceride, Val = valine.
This project was supported by the College of Medicine Research Centre, Deanship of Scientific Research, King Saud University, Riyadh, Saudi Arabia.
The authors have no conflicts of interest to disclose.
References
- [1].Paquette M, Genest J, Baass A. Familial hypercholesterolemia: experience from the French-Canadian population. Curr Opin Lipidol 2018;29:59–64. [DOI] [PubMed] [Google Scholar]
- [2].BEESON BB, Albrecht P. A contribution to the study of xanthoma tuberosum, with report of a case. Arch Dermatol Syphilol 1923;8:695–710. [Google Scholar]
- [3].Radovica-Spalvina I, Latkovskis G, Silamikelis I, et al. Next-generation-sequencing-based identification of familial hypercholesterolemia-related mutations in subjects with increased LDL-C levels in a latvian population. BMC Med Genet 2015;16:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Khachadurian AK. The inheritance of essential familial hypercholesterolemia. Am J Med 1964;37:402–7. [DOI] [PubMed] [Google Scholar]
- [5].Fahed AC, Nemer GM. Familial hypercholesterolemia: the lipids or the genes? Nutr Metab 2011;8:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Baila-Rueda L, Lamiquiz-Moneo I, Jarauta E, et al. Association between non-cholesterol sterol concentrations and Achilles tendon thickness in patients with genetic familial hypercholesterolemia. J Transl Med 2018;16:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Alharbi KK, Alnbaheen MS, Alharbi FK, et al. Q192R polymorphism in the PON1 gene and familial hypercholesterolemia in a Saudi population. Ann Saudi Med 2017;37:425–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Durst R, Ibe UK, Shpitzen S, et al. Molecular genetics of familial hypercholesterolemia in Israel-revisited. Atherosclerosis 2017;257:55–63. [DOI] [PubMed] [Google Scholar]
- [9].Mollazadeh H, Carbone F, Montecucco F, et al. Oxidative burden in familial hypercholesterolemia. J Cell Physiol 2018;233:5716–25. [DOI] [PubMed] [Google Scholar]
- [10].Sharifi M, Walus-Miarka M, Idzior-Walus B, et al. The genetic spectrum of familial hypercholesterolemia in south-eastern Poland. Metabolism 2016;65:48–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lye SH, Chahil JK, Bagali P, et al. Genetic polymorphisms in LDLR, APOB, PCSK9 and other lipid related genes associated with familial hypercholesterolemia in Malaysia. PLoS One 2013;8:e60729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Sharifi M, Futema M, Nair D, et al. Genetic architecture of familial hypercholesterolaemia. Curr Cardiol Rep 2017;19:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Angarica VE, Orozco M, Sancho J. Exploring the complete mutational space of the LDL receptor LA5 domain using molecular dynamics: linking SNPs with disease phenotypes in familial hypercholesterolemia. Hum Mol Genet 2016;25:1233–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Alharbi KK, Kashour TS, Al-Hussaini W, et al. Association of angiotensin converting enzyme gene insertion/deletion polymorphism and familial hypercholesterolemia in the Saudi population. Lipids Health Dis 2013;12:177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Alharbi KK, Kashour TS, Al-Hussaini W, et al. Screening for genetic mutations in LDLR gene with familial hypercholesterolemia patients in the Saudi population. Acta Biochim Pol 2015;62:559–62. [DOI] [PubMed] [Google Scholar]
- [16].Hovingh GK, Davidson MH, Kastelein JJ, et al. Diagnosis and treatment of familial hypercholesterolaemia. Eur Heart J 2013;34:962–71. [DOI] [PubMed] [Google Scholar]
- [17].Ghaleb Y, Elbitar S, El Khoury P, et al. Usefulness of the genetic risk score to identify phenocopies in families with familial hypercholesterolemia? Eur J Hum Genet 2018;26:570–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Nurnberg ST, Zhang H, Hand NJ, et al. From loci to biology: functional genomics of genome-wide association for coronary disease. Circ Res 2016;118:586–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Rabes JP, Beliard S, Carrie A. Familial hypercholesterolemia: experience from France. Curr Opin Lipidol 2018;29:65. [DOI] [PubMed] [Google Scholar]
- [20].Pearce N. Analysis of matched case-control studies. BMJ 2016;352:i969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hartgers ML, Defesche JC, Langslet G, et al. Alirocumab efficacy in patients with double heterozygous, compound heterozygous, or homozygous familial hypercholesterolemia. J Clin Lipidol 2018;12:390–6. [DOI] [PubMed] [Google Scholar]
- [22].Innerarity TL, Weisgraber KH, Arnold KS, et al. Familial defective apolipoprotein B-100: low density lipoproteins with abnormal receptor binding. Proc Natl Acad Sci 1987;84:6919–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Henderson R, O’Kane M, McGilligan V, et al. The genetics and screening of familial hypercholesterolaemia. J Biomed Sci 2016;23:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Youngblom E, Pariani M, Knowles JW. Familial hypercholesterolemia; 2016. [Google Scholar]
- [25].Whitfield AJ, Barrett PHR, Van Bockxmeer FM, et al. Lipid disorders and mutations in the APOB gene. Clin Chem 2004;50:1725–32. [DOI] [PubMed] [Google Scholar]
- [26].Johansen CT, Wang J, Lanktree MB, et al. Excess of rare variants in genes identified by genome-wide association study of hypertriglyceridemia. Nat Genet 2010;42:684–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Al-Allaf FA, Athar M, Abduljaleel Z, et al. Next generation sequencing to identify novel genetic variants causative of autosomal dominant familial hypercholesterolemia associated with increased risk of coronary heart disease. Gene 2015;565:76–84. [DOI] [PubMed] [Google Scholar]
- [28].Akioyamen LE, Genest J, Shan SD, et al. Estimating the prevalence of heterozygous familial hypercholesterolaemia: a systematic review and meta-analysis. BMJ Open 2017;7:e016461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Eslami SM, Nikfar S, Ghasemi M, et al. Does Evolocumab, as a PCSK9 Inhibitor, Ameliorate the Lipid Profile in Familial Hypercholesterolemia Patients? A Meta-Analysis of Randomized Controlled Trials. J Pharm Pharm Sci 2017;20:81–96. [DOI] [PubMed] [Google Scholar]
- [30].Peng W, Qiang F, Peng W, et al. Therapeutic efficacy of PCSK9 monoclonal antibodies in statin-nonresponsive patients with hypercholesterolemia and dyslipidemia: A systematic review and meta-analysis. Int J Cardiol 2016;222:119–29. [DOI] [PubMed] [Google Scholar]
- [31].Qian LJ, Gao Y, Zhang YM, et al. Therapeutic efficacy and safety of PCSK9-monoclonal antibodies on familial hypercholesterolemia and statin-intolerant patients: A meta-analysis of 15 randomized controlled trials. Sci Rep 2017;7:238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Tada H, Kawashiri MA, Okada H, et al. A Rare Coincidence of Sitosterolemia and Familial Mediterranean Fever Identified by Whole Exome Sequencing. J Atheroscler Thromb 2016;23:884–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Faiz F, Allcock RJ, Hooper AJ, et al. Detection of variations and identifying genomic breakpoints for large deletions in the LDLR by Ion Torrent semiconductor sequencing. Atherosclerosis 2013;230:249–55. [DOI] [PubMed] [Google Scholar]
- [34].Hinchcliffe M, Le H, Fimmel A, et al. Diagnostic validation of a familial hypercholesterolaemia cohort provides a model for using targeted next generation DNA sequencing in the clinical setting. Pathology 2014;46:60–8. [DOI] [PubMed] [Google Scholar]
- [35].Kim HN, Kweon SS, Shin MH. Detection of familial hypercholesterolemia using next generation sequencing in two population-based cohorts. Chonnam Med J 2018;54:31–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Reiman A, Pandey S, Lloyd KL, et al. Molecular testing for familial hypercholesterolaemia-associated mutations in a UK-based cohort: development of an NGS-based method and comparison with multiplex polymerase chain reaction and oligonucleotide arrays. Ann Clin Biochem 2016;53:654–62. [DOI] [PubMed] [Google Scholar]
- [37].Schaefer EJ, Tsunoda F, Diffenderfer M. De Groot LJ, Chrousos G, Dungan K, Feingold KR, Grossman A, Hershman JM, et al. The measurement of lipids, lipoproteins, apolipoproteins, fatty acids, and sterols, and next generation sequencing for the diagnosis and treatment of lipid disorders. MDText.com, Inc, Endotext. South Dartmouth, MA: 2000. [PubMed] [Google Scholar]
- [38].Zeggini E, Ioannidis JP. Meta-analysis in genome-wide association studies. Pharmacogenomics 2009;10:191–201. [DOI] [PMC free article] [PubMed] [Google Scholar]