

Abstract
Rationale:
Pantothenate kinase-associated neurodegeneration (PKAN), also called Hallervorden–Spatz Syndrome (HSS), is a rare neurodegeneration with brain iron accumulation from pantothenate kinase 2 gene (PANK2) mutation characterized as extrapyramidal symptoms. However, few studies involving PKAN patients were reported in China. This study was conducted to identify the genetic mutations in a Chinese boy with PKAN, and to review all PANK2 mutations reported in Chinese cases with PKAN.
Patient concern:
We reported a 23-year-old Chinese boy with PKAN, exhibiting difficulty in writing and manipulation using right hand with slow progression for 12 years. He spoke with a severe stutter when he was 15 years old.
Diagnosis:
Considering results of magnetic resonance images, brain computed tomography and medical history, the case was suspected to be related to genetic factors.
Interventions:
Whole exome sequencing was arranged, and the mutations were identified in his parents’ genome.
Outcomes:
In the present study, whole exome sequencing revealed 2 novel mutations (c.1696C > G in exon 7 and c.1160_c.1161insG in exon3) of the PANK2 gene in the proband. c.1696C > G and c.1160_c.1161insG, respectively, were confirmed in his father and mother. We also reviewed 14 different PANK2 mutations, most of which were missense type in Chinese cases. Those mutations did not show apparent hotspots, but exon 3 and 4 were frequently involved.
Lessons:
Two novel compound heterozygous mutations were identified and considered to be pathogenic in PKAN patients. This review of the reports indicated that atypical PKAN is the more common phenotype in China and no apparent genotype-phenotype correlation was found.
Keywords: novel heterozygous mutation, PANK2 gene mutation, pantothenate kinase-associated neurodegeneration, review
1. Introduction
Pantothenate kinase associated neurodegeneration (PKAN) which is an autosomal recessive disease mutating from pantothenate kinase 2 gene (PANK2) manifests as progressive extrapyramidal dysfunction.[1] The feature of neuroimaging is “eye of the tiger sign” which is connected with brain iron accumulation in the globus pallidus.[2] PKAN is classified into 2 phenotypes: a “typical PKAN” with early onset (the first decade), speedy development and homogenous form in majority of the patients. An “atypical PKAN” with late onset, slow progression and variable clinical features. Compared to typical PKAN, atypical PKAN is characterized by more prominent speech problems, psychiatric symptom, much milder cognitive impairment and gait abnormalities. In addition, patients with atypical PKAN develop rarely retinopathy.
The PANK2 gene which is considered as main causative gene of PKAN is located on chromosome 20p13 and encodes pantothenate kinase-2 which plays an important role in coenzyme A (CoA) biosynthesis. It has been hypothesized that CoA deficiency due to PANK2 mutations might cause secondary accumulation of iron.[3] But molecular mechanism of PKAN still remained unclear.
In this article, we reported the novel compound heterozygous PANK2 mutations of a Chinese boy with an atypical phenotype PKAN. In addition, we summarized clinical and genetic features of PKAN patients which were reported in Chinese population.
2. Methods
2.1. Ethical approval and consent for publication
PANK2 gene mutation analysis was approved by the ethical committee of Sichuan University. Peripheral blood samples for total DNA extraction from the proband, his parents and100 healthy controls were obtained after conformed consent.
2.2. Detection of mutation
To determine all possible mutations, whole-exome sequencing (WES) in the proband was performed using IDT xGenExome Research Panel 1.0. If possible pathogenicity mutations were found in the proband, the mutations would be identified in his parents.
2.3. Review of the literature
We searched for literatures about PKAN on the Chinese Biological Medicine (CBM) Database (http://www.sinomed.ac.cn/zh/) and PubMed. We excluded the literatures lack of genetic analysis or complete data by reading the full text and finally 9 studies[4–12] were included in the review. The genetic and clinical features of PKAN in China were summarized.
3. Case report
This proband, a 23-year-old male, was the only child of a non-consanguineous family. No history of genetic diseases was found in his family. He was born at full-term without birth injury and was in good health till the age of 11 years when he had difficulty in writing and manipulation using right hand with slow progression. Now he had to write with his left hand. By the time he was 15, he spoke with a severe stutter accentuated by tension and had bilateral hands’ mild static tremor.
Examination of the nervous system revealed mild tremor with both hands and severe stutter. Both lower extremities were revealed no lesions. No gait and feeding difficulties or balance impairment or cranial nerve deficits were found. Fundus examination discloses nothing of note and the rest of the eye examinations including extraocular movements, visual acuity, visual field and pupillary evaluation were normal. He did not have cognitive problems despite poor academic performance. The score of self-rating anxiety scale was 32 which revealed mild anxiety state. In addition, his parents complained that he was irritable in life but lack of related psychological examination.
Laboratory tests including blood biochemistry, ceruloplasmin, thyroid function, parathormone, calcitonin, serum HIV antibody, syphilis antibody and autoimmune antibody were normal. Neuroimaging findings: the whole-spine magnetic resonance images (MRI) was normal. Brain computed tomography scan revealed symmetric calcifications of the bilateral globus pallidus (Fig. 1A). Brain T2-weighted (Fig. 1B) and fluid-attenuated inversion recovery (FLAIR) (Fig. 1C) images showed bilateral symmetrical low signal intensity in the globus pallidus with central hyperintense foci termed as “eye-of-the-tiger” sign. Brain susceptibility weighted imaging (SWI) showed bilateral symmetrical low signal in the globus pallidus which demonstrated brain iron accumulation (Fig. 1D).
Figure 1.
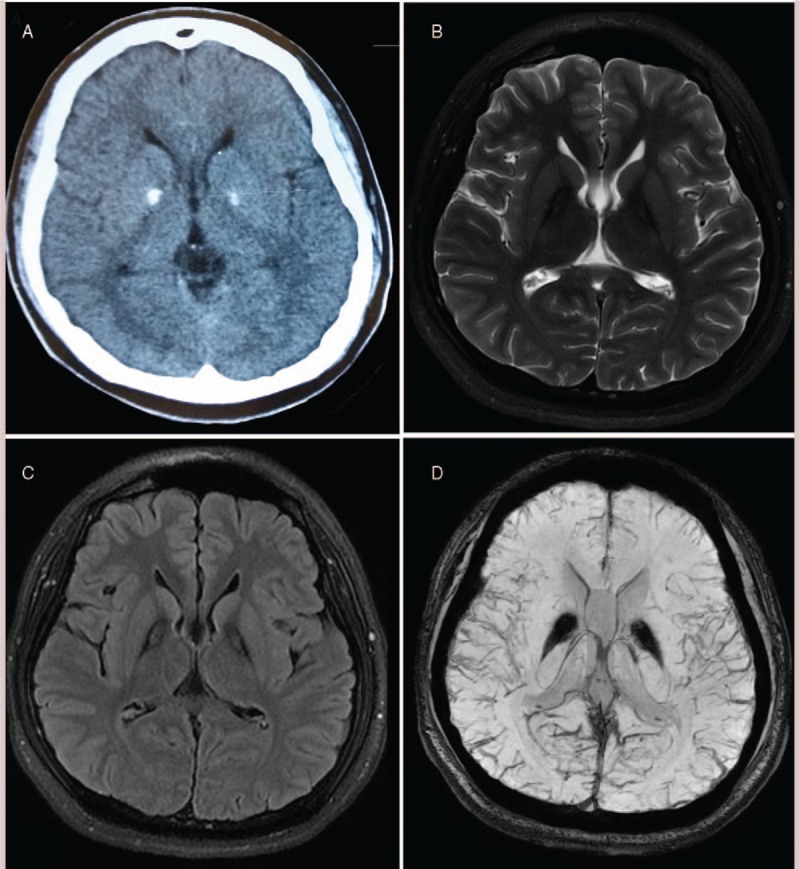
T2-weighted brain MRI showing the “eye-of-the-tiger” sign. (A) Brain CT scan showed a bilateral calcification of the globus pallidus; (B and C) T2 weighted and FLAIR brain MRI displayed typical “eye of the tiger”; (D) brain SWI showed bilateral symmetrical low signal in the globus pallidus. CT = computed tomography, FLAIR = fluid-attenuated inversion recovery, MRI = magnetic resonance imaging, SWI = susceptibility weighted imaging.
Two compound heterozygous mutations c.1696C > G and c.1160_c.1161insG respectively located on exon 7 and 3 of PANK2 were identified in the proband (Fig. 2). The mutation, c.1696C > G, changed the amino acid at position 566 from leucine to valine (p.L566 V). And the mutation, c.1160_c.1161insG, denotes a frame shifting change with leucine97 as the first influenced amino acid and the new reading frame being open for 9 amino acids (p L387Lfs∗9).
Figure 2.
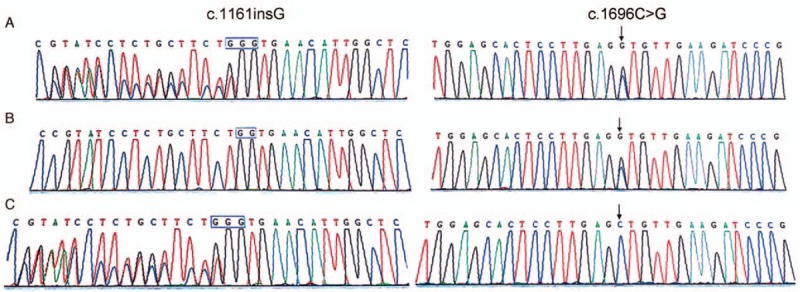
The family with compound heterozygous PANK2 gene mutations. (A) Proband with 2 compound heterozygous mutations (c.1696C > G and c.1160_c.1161insG); (B) father with mutation (c.1696C > G); (C) mother with mutation (c.1160_c.1161insG).
His father and mother were respective heterozygous carriers of c.1696C > G and c.1160_c.1161insG. In addition, we did not detect single nucleotide polymorphism in the healthy controls. Two compound heterozygous mutations in our study were absent in the 1000 Genomes Project database (http://www.1000genomes.org/).
4. Discussion
The PANK2 gene was firstly identified as being responsible for PKAN. And for now, more than 100 mutations of this gene including aberrant splicing, missense and nonsense mutations have been reported in PKAN patients (http://www.hgmd.org). In this study, a Chinese 23-year-old male with a 12-year history of mainly difficulty in writing was described. Typical eye-of-the-tiger sign was presented in neuroimaging. Novel compound heterozygous mutations in exon 7 (c.1696C > G) and 3 (c.1160_c.1161insG) of PANK2 gene were detected and were found for the first time in this study, as far as we know.
Our patient with the primary symptom of difficulty in writing and manipulation using right hand were observed when he was about 11. Then the patient developed dysarthria and tremor symptoms. Difficulty in writing with right hand has been described in a Caucasian female with atypical PKAN, which should be considered at clinical practice.[13]
Human PANK2 mainly gathered in mitochondria of neurons.[3] Deficiency of PANK2 would cause the accumulation of cysteine-containing substrates, which lead to the generation of free radicals and trigger cell death.[14] Most nonsense PANK2 mutations with typical form caused premature protein termination, whereas a larger proportion of missense PANK2 mutations with atypical form resulted in amino acid substitutions.[15] In the present study, 2 novel mutations (c.1696C > Gandc.1160_c.1161insG)of the PANK2 gene were identified. In addition, c.1160_c.1161insG and c.1696C > G mutations were respectively considered as pathogenic and likely pathogenic according to AGMG guide. To sum up, the clinical manifestations, neuroimaging finding showing typical eye-of-the-tiger sign and the finding of 2 novel mutations on the PANK2 gene described in our patient support the clinical diagnostic criteria of atypical PKAN.
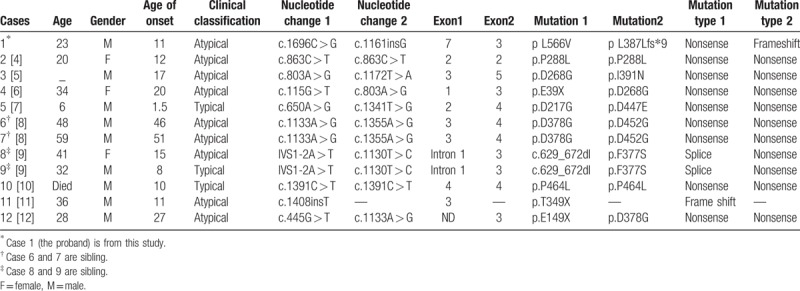
Our literature review of 12 Chinese cases suggests that PKAN is very rare in China. To date, 14 different PANK2 sequence variants have been reported in Chinese PKAN patients, the most common being missense mutations (Table 1). These mutations of PANK2 are located in exons1–5 and 7 and intron 1. Exons 3 and 4 are frequently involved, which are the site of relatively more mutations (5 in exon 3, 3 in exon 4). Only 2 recurrent mutations (c.803A > G occurring twice and c.1133A > G occurring twice) were found in unrelated cases. We did not find mutation hotspots in Chinese cases, which might be associated with low PANK2 detective rate. It is reported that the missense mutation c.1583C > T (p.T528M) in exon 6 is relatively PANK2 gene mutation hotspots observed in other countries and an apparent phenotype-genotype correlation in a cohort of homozygous c.1583C > T Gypsy patients with a milder phenotype was identified.[1,15–18] But in Chinese cases, we did not find an evident relationship between genotype and phenotype.
Table 1.
Genetic features of Chinese with PANK.
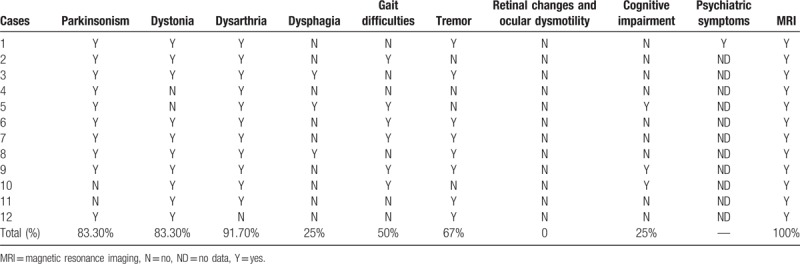
Our study also indicated that atypical PKAN is the more common phenotype among Chinese patients with PKAN (9/12,75%) (Table 2). Dystonia (91.7%,) and parkinsonism (83.3%) are the core symptoms of PKAN and more common manifestation observed in Chinese series. Pyramidal signs, mental impairment, and parkinsonism had a higher prevalence among Caucasian patients. In addition, tremor and dysarthria also are frequent symptoms in our study. However, half of our PKAN patients have gait difficulty which will cause them unable to walk or need assistance, having a major effect on patients’ quality of life. Although psychiatric symptoms were considered as common features of PKAN,[19] psychiatric symptoms were not involved in other Chinese studies except our patient, which hinted that Chinese clinician should pay attention to PKAN patients’ psychiatric symptoms. Cognitive impairment is more common in typical PKAN, which in line with Chinese series that cognitive impairment occurred in all 3 typical PKAN patients. Retinal changes and ocular dysmotility were not found in our series, which indicated that these symptoms were not easily involved in Chinese cases with PKAN. In short, genetic and clinical features in Chinese with PKAN still remain to be summarized due to a limited number of cases reported at present.
Table 2.
Clinical characteristics of Chinese with PANK.
We identified 2 novel mutations in the PANK2 gene in PKAN patient, which expands the inventory of PANK2 mutations involved in PKAN. This review of the reports indicated that atypical PKAN is the more common phenotype in China and also provide a more complete gene mapping in the Chinese population with PKAN, although PKAN is uncommon in China. In addition, no apparent genotype–phenotype correlation was found.
Acknowledgments
We sincerely thank this patient and his parents.
Author contributions
Conceptualization: Dong Zhou, Tianhua Yang.
Data curation: Yingying Zhang, Dong Zhou, Tianhua Yang.
Formal analysis: Yingying Zhang, Dong Zhou, Tianhua Yang.
Funding acquisition: Tianhua Yang.
Investigation: Yingying Zhang.
Methodology: Yingying Zhang, Dong Zhou, Tianhua Yang.
Project administration: Yingying Zhang, Tianhua Yang.
Resources: Yingying Zhang, Tianhua Yang.
Software: Yingying Zhang, Dong Zhou.
Supervision: Dong Zhou, Tianhua Yang.
Validation: Dong Zhou, Tianhua Yang.
Visualization: Dong Zhou, Tianhua Yang.
Writing – original draft: Yingying Zhang.
Writing – review & editing: Yingying Zhang, Dong Zhou, Tianhua Yang.
Footnotes
Abbreviations: CBM = Chinese Biological Medicine, CoA = coenzyme A, CT = computed tomography, FLAIR = fluid-attenuated inversion recovery, HSS = Hallervorden–Spatz Syndrome, MRI = magnetic resonance images, PANK2 = pantothenate kinase2 gene, PKAN = Pantothenate kinase–associated neurodegeneration, SWI = susceptibility weighted imaging, WES = whole-exome sequencing.
Funding: This study was supported by the National High Technology Research and Development Program of China (863 Program), (Item Number: 0040205403097).
Statement: The patient has provided informed consent for publication of the case.
The authors have no conflicts of interest to disclose.
References
- [1].Zhou B, Westaway SK, Levinson B, et al. A novel pantothenate kinase gene (PANK2) is defective in Hallervorden–Spatz syndrome. Nat Genet 2001;28:345–9. [DOI] [PubMed] [Google Scholar]
- [2].Sethi KD, Adams RJ, Loring DW, et al. Hallervorden–Spatz syndrome: clinical and magnetic resonance imaging correlations. Ann Neurol 1988;24:692–4. [DOI] [PubMed] [Google Scholar]
- [3].Johnson MA, Kuo YM, Westaway SK, et al. Mitochondrial localization of human PANK2 and hypotheses of secondary iron accumulation in pantothenate kinase-associated neurodegeneration. Ann N Y Acad Sci 2004;1012:282–98. [DOI] [PubMed] [Google Scholar]
- [4].Zhang Y, Tang B, Guo J, et al. Studies on PANK2 gene mutations in Chinese patients with Hallervorden-Spatz syndrome. Chin J Med Genet 2005;22:189–91. [PubMed] [Google Scholar]
- [5].Chan KY, Lam CW, Lee LP, et al. Pantothenate kinase-associated neurodegeneration in two Chinese children: identification of a novel PANK2 gene mutation. Hong Kong Med J 2008;14:70–3. [PubMed] [Google Scholar]
- [6].Song XW, Wang YL, Shi YW, et al. Clinical manifestations and detection of pantothenate kinase 2 gene mutation in a patient with Hallervorden–Spatz syndrome. Zhonghua Yi Xue Za Zhi 2009;89:3320–3. [PubMed] [Google Scholar]
- [7].Wu YR, Chen CM, Chao CY, et al. Pantothenate kinase-associated neurodegeneration in two Taiwanese siblings: identification of a novel PANK2 gene mutation. Mov Disord 2009;24:940–1. [DOI] [PubMed] [Google Scholar]
- [8].Mak CM, Sheng B, Lee HH, et al. Young-onset parkinsonism in a Hong Kong Chinese man with adult-onset Hallervorden-Spatz syndrome. Int J Neurosci 2011;121:224–7. [DOI] [PubMed] [Google Scholar]
- [9].Lee CH, Lu CS, Chuang WL, et al. Phenotypes and genotypes of patients with pantothenate kinase-associated neurodegeneration in Asian and Caucasian populations: 2 cases and literature review. ScientificWorldJournal 2013;2013:860539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Shan J, Wen B, Zhu J, et al. Novel PANK2 gene mutations in two Chinese siblings with atypical pantothenate kinase-associated neurodegeneration. Neurol Sci 2013;34:561–3. [DOI] [PubMed] [Google Scholar]
- [11].Li YF, Li HF, Zhang YB, et al. Novel homozygous PANK2 mutation identified in a consanguineous Chinese pedigree with pantothenate kinase-associated neurodegeneration. EMBO Mol Med 2016;5:217–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Shi X, Zheng F, Ye X, et al. Basal ganglia calcification and novel compound heterozygous mutations in the PANK2 gene in a Chinese boy with classic pantothenate kinase-associated neurodegeneration: a case report. Medicine 2018;97:e0316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ibrahimagic OC, Smajlovic D, Dostovic Z, et al. Change of the writing-hand: unusual manifestation of Hallervorden–Spatz disease. Psychiatria Danubina 2017;29:507–9. [DOI] [PubMed] [Google Scholar]
- [14].Kotzbauer PT, Truax AC, Trojanowski JQ, et al. Altered neuronal mitochondrial coenzyme A synthesis in neurodegeneration with brain iron accumulation caused by abnormal processing, stability, and catalytic activity of mutant pantothenate kinase 2. J Neurosci 2005;25:689–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hayflick SJ, Westaway SK, Levinson B, et al. Genetic, clinical, and radiographic delineation of Hallervorden–Spatz syndrome. N Engl J Med 2003;348:33–40. [DOI] [PubMed] [Google Scholar]
- [16].Hartig MB, Hortnagel K, Garavaglia B, et al. Genotypic and phenotypic spectrum of PANK2 mutations in patients with neurodegeneration with brain iron accumulation. Ann Neurol 2006;59:248–56. [DOI] [PubMed] [Google Scholar]
- [17].Zhang YM, Rock CO, Jackowski S. Biochemical properties of human pantothenate kinase 2 isoforms and mutations linked to pantothenate kinase-associated neurodegeneration. J Biol Chem 2006;281:107–14. [DOI] [PubMed] [Google Scholar]
- [18].Wu YW, Hess CP, Singhal NS, et al. Idiopathic basal ganglia calcifications: an atypical presentation of PKAN. Pediatr Neurol 2013;49:351–4. [DOI] [PubMed] [Google Scholar]
- [19].Darling A, Tello C, Martí M, et al. Clinical rating scale for pantothenate kinase-associated neurodegeneration: a pilot study. Mov Disord 2017;32:1620–30. [DOI] [PubMed] [Google Scholar]