

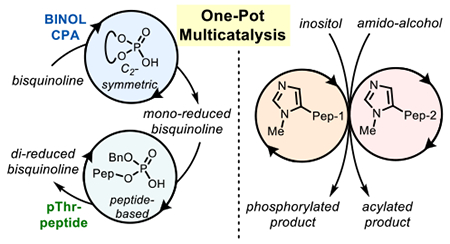
Abstract
We herein report two examples of one pot, simultaneous reactions, mediated by multiple, orthogonal catalysts with the same catalytic motif. First, BINOL-derived chiral phosphoric acids (CPA) and phosphothreonine (pThr)-embedded peptides were found to be matched for two different steps in double reductions of bisquinolines. Next, two π-methylhistidine (Pmh)-containing peptides catalyzed enantio- and chemoselective acylations and phosphorylations of multiple substrates in one pot. The selectivity exhibited by common reactive moieties is adjusted solely by the appended chiral scaffold through outer-sphere interactions.
Graphical Abstract
A remarkable feature of enzymes is their ability to catalyze highly selective transformations on specific substrates, often in tandem and simultaneous pathways and in the presence of competing, reactive analytes.1 The tuning of active sites, which often employ a similar catalytic apparatus for bond-formation, to perform diverse molecular functions is a triumph of evolution.2 However, by comparison, chemists struggle to mimic this level of control over the reactivities and selectivities of multiple catalysts in complex settings,3 as cross-reactivity among diverse chemical species can intervene.4–6 While approaches toward relay catalysis have recently seen a number of advances,7 most of these studies (1) require either sequential addition of catalysts or intermediate purification steps, or (2) are combinations of two catalysts that are chemically distinct (i.e. hetero-combinations of metals, photocatalysts, and organocatalysts), such that both catalysts independently mediate orthogonal transformations.8–10 While simultaneous operations of the same catalytic species is rare, a few reports of one pot multicatalytic reactions, such as with two ruthenium complexes (Figure 1A)11 and a dual imidazolidinone/proline system (Figure 1B),12 create an optimistic prognosis. These studies offer validation that a common, reactive moiety (Ru atom or secondary amine) can be subtly modified to function divergently in situ (i.e. oxidations vs. reductions, and iminium vs. enamine catalysis).
Figure 1.
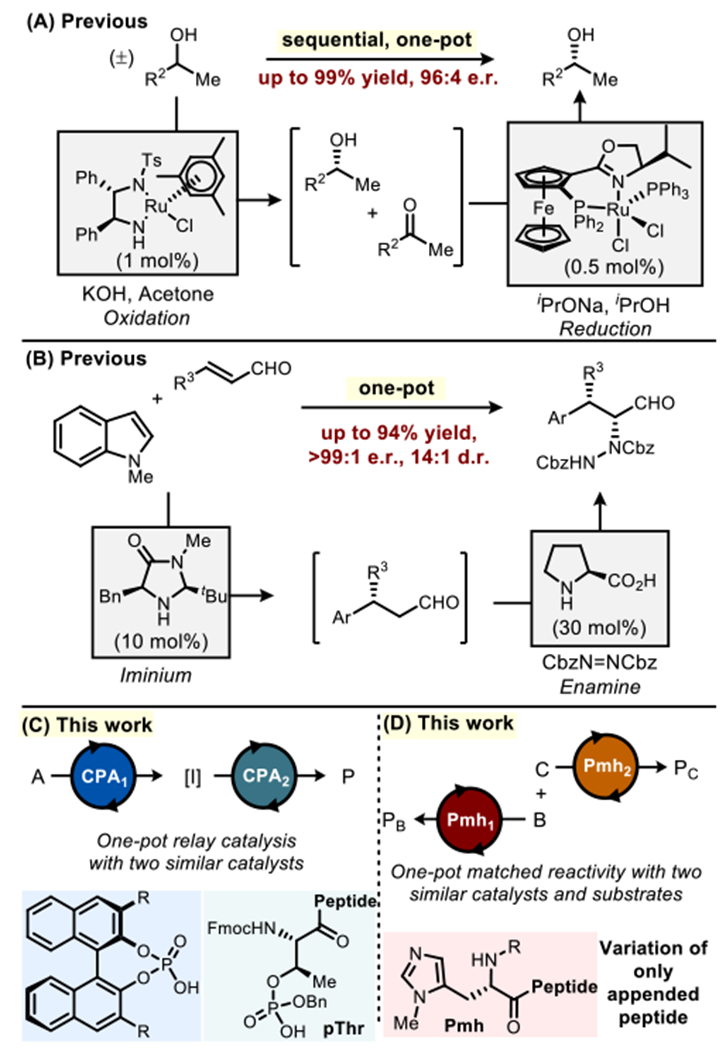
(A,B) Examples of organometallic and organocatalyst multicatalytic systems. (C,D) This work: multicatalysis with similar catalysts.
With these precedents in mind, we sought functional divergence with catalysts of even more closely related intrinsic reactivity. We chose to assess whether (1) two Brønsted acids or (2) two Lewis bases could function in the presence of one another to perform either a tandem reaction, or a parallel processing reaction scheme, simply through modification of the chiral scaffold appended to the catalytically competent group. The crafting of precise outer-sphere interactions between these catalysts and their targeted substrates would be essential in assigning specific catalytic functions, and discriminating between competing, but fully analogous types of bond formations.13 The approach to assigning specific catalyst-substrate interactions described below uses peptide-based catalysts in a biomimetic manner, building on their analogy to enzymes.14 Presented herein are reports involving two different families of catalysts: (1) phosphothreonine (pThr)-embedded peptides,15–18 complementary to BINOL-derived chiral phosphoric acid (CPA) catalysts,19,20 are explored for multicatalytic transfer hydrogenation (Figure 1C); and (2) π-methylhistidine (Pmh)-containing peptides are explored for in situ parallel hydroxyl group transfer reactions (Figure 1D).21
Divergent, One Pot CPA Catalysis.
While BINOL- and peptide-based CPA catalysts (like P1 and 1) maintain a common catalytic phosphoric acid, their overall chiral scaffolds are distinct (Figure 2A). In our previous studies of the transfer hydrogenation of 8-aminoquinolines (2), we reported that both catalyst P1 and 1 were highly selective, affording tetrahydroquinoline 3 in up to 94:6 e.r. and with opposite absolute stereochemical configurations (Figure 2B).15 Given the utility of reduced quinoline scaffolds, we chose to probe the possibility of sequential, high selectivity reductions with these catalysts on biologically relevant bisquinolines,22,23 such as 4, linked through the 8-substituted urea (Figure 2C). Upon initial formation of 5, further reduction can produce a mixture of chiral (6) and meso (7) products. Initial reactions mediated by pThr-derived P1 generated mono-reduced 5 with 67:33 e.r. (Figure 2C–D, entry 1), a lower level of selectivity than previously observed for the reduction of 2 to 3. Yet, further reduction of 5 to 6+7 revealed a substantial amplification of product enantiopurity, producing 6 with 95:5 e.r..24
Figure 2.
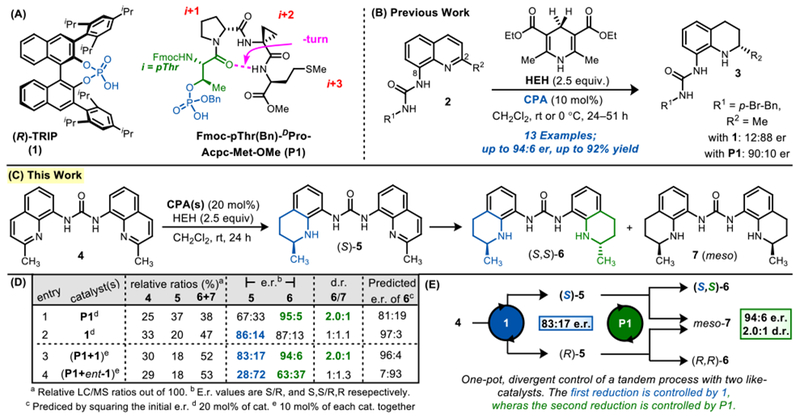
(A) BINOL-derived and pThr-based CPAs. (B) Asymmetric transfer hydrogenation of 8-aminoquinolines with CPAs. (C+D) Scheme and catalyst screening for CPA-mediated reductions of bisquinoline 4. (E) Kinetics diagram for this transformation.
The combination of two subsequent chirality-generating reactions to enhance the e.r. of an asymmetric process is a manifestation of Horeau amplification (Figure 2E).25–29 In this paradigm, e.r. may be enhanced at the expense of d.r., as the disfavored enantiomer in the first reduction is converted to meso-7 through a second reduction. Under ideal conditions, assuming the catalyst reduces 4 and 5 with the same selectivity, the final e.r. of 6 can be predicted by squaring the initial selectivity.30,31 Since catalyst P1 produces mono-reduced 5 in 67:33 e.r. (s1 = 2.0), the theoretical e.r. of 6 would be 81:19 (s12 = 4.1); thus, P1 substantially outperforms ideal Horeau amplification estimates (95:5 e.r., s2 = 19), implying second order outer-sphere effects in the second reduction. In the case of P1 mediated reductions of monoquinolines (2), secondary interactions between the peptide and the 8-aminoquinoline were essential for high selectivities; and these key outer-sphere interactions presumably operate in the reduction of these bisquinolines as well.15
Interestingly, BINOL-derived catalyst 1 produced mono-reduced 5 in higher initial e.r., 86:14 (s1 = 6.1, Figure 2D, entry 2). Yet, upon second reduction, the e.r. of 6 minimally changes to 87:13 (s2 = 6.7), much lower than the Horeau expectation (s12 = 38, 97:3 e.r).32 Thus, catalyst 1 is more selective in the first reduction, while P1 is more selective in the second. Furthermore, while P1 and 1 previously gave the opposite sense of absolute stereochemistry in the reduction of monoquinoline (2 to 3; Figure 2B), both catalysts afford products with the same sense of stereochemistry with 4.31
Given the complementarity of catalysts P1 and 1 for different steps of this bis-reduction, we wondered whether the catalysts could be combined in one pot to mediate their respective, matched reactions with high selectivity (Figure 2E). Indeed, reacting 4 in the presence of a 1:1 mixture of 1 and P1 produced 5 in 83:17 e.r., and 6 in 94:6 e.r. and 2.0:1 d.r., constituting a synergistic effect for the combined catalysts in achieving better e.r. together than in individual screens (Figure 2D, entry 3). It appears that catalyst 1 predominantly controls its matched reduction of 4 to 5 (83:17 e.r. vs. 86:14 e.r. in individual screen). Alternatively, in the second reduction from 5 to 6+7, the dual catalyst system outperforms individual reaction with only catalyst 1 (94:6 e.r. vs. 87:13 e.r.), indicating that P1 affords rate enhancement in the second step (1 also appears to compete to a low, but detectable extent). To further elucidate the level of control exhibited by P1 on this second reduction, reactions were performed in the presence of both P1 and ent-1. The first reduction was still controlled by ent-1, revealing 5 with the reversed (R)-stereochemistry (28:72 e.r., Figure 2D, entry 4). However, even though (R)-5 was favored in the first step, the second reduction results in (S,S)-6 as the major product (63:37 e.r.), revealing that P1 competes effectively. Thus, this reversal of the stereochemical outcome between the first and second reductions elucidates the substantial control that ent-1 and P1 have in conducting their matched reductions in a tandem process. These results imply that, despite the shared identity of the catalytic phosphoric acid of 1 and P1, reactivity can be heavily tuned by appended chiral scaffold and specific, matched outer-sphere interactions (presumably 1 with 4 and P1 with 5).33
Control of Pmh-Catalysts through Peptide Variation.
The divergent selectivity regimes of BINOL- and peptide-based CPAs described above allow for interpretable, catalyst-assigned outcomes in these multicatalytic parallel reactions, specifically due to the different, surrounding molecular architecture.1 Complementary reactivity might not be unexpected for these two families, taking into account the dissimilar nature of the rigid BINOL framework in comparison to the functionalized and flexible nature of peptides. Yet, we are able to document related assignment of catalytic function based on outer-sphere tuning of common catalytic moieties with purely peptide-based systems as well,14,34 specifically relying on Pmh-based catalysts for selective hydroxyl group transfers.
Some time ago, we showed that octameric Pmh catalyst P2 facilitated the selective acylation of racemic trans-aminoalcohol 8 with a krel > 50:1 (Figure 3A).35 With an alternative peptide backbone, Pmh peptide P3 was reported to mediate the phosphorylation of an inositol derivative (10) with high levels of site- and enantioselectivity (>99:1 e.r.; Figure 3B).36 However, catalysts P2 and P3 gave unspectacular, or even stereodivergent selectivity when applied to the reactions for which each was unoptimized. For example, when (±)-8 and 10 were reacted in the presence of only (PhO)2P(O)Cl and P3, phosphorylated (S,S)-12 was obtained with a krel of 1:5 (Figure 3C), the opposite stereochemistry shown with P2 for acylation. These results indicated the crucial nature of the appended peptide sequences in tuning the selectivity of the shared Pmh residue through outer-sphere interactions.35b Furthermore, these divergent reactivities gave us hope that the two catalysts could exhibit specificity for their matched substrates in a one pot, parallel processing experiment wherein catalyst function was assigned by the independent studies of enantioselectivity.
Figure 3.
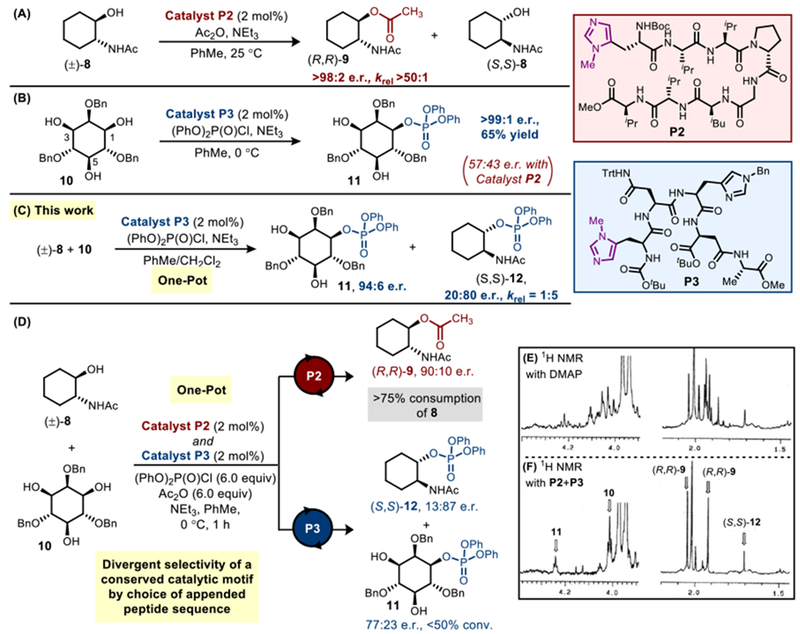
(A) Kinetic resolution of (±)-8 with P2 to yield acylated (R,R)-9. (B) Desymmetrizing phosphorylation of inositol derivative 10 with P3. (C) One pot phosphorylation of (±)-8 and 10 revealed P3 favors reaction with (S,S)-8, opposite to P2. (D–F) Scheme and NMR data for peptide-mediated one-pot reactions.
Competition experiments were performed wherein substrates (±)-8 and 10 were added together in one pot, along with both phosphorylating and acylating reagents(Figure 3D). DMAP provided a complex and intractable mixture of products by 1H NMR (Figure 3E).32 However, when equimolar amounts of the two Pmh-catalysts P2 and P3 were utilized, a vastly simplified product distribution is observed (Figure 3F). Racemic aminoalcohol 8 was mostly consumed (>75% conversion to products), and acylated (R,R)-9 was obtained in 90:10 e.r., presumably under the control of P2. Additionally, P3 was found to phosphorylate the unreacted enantiomer of 8, affording (S,S)-12 with 87:13 e.r.. In parallel and in situ, inositol derivative 10 was consumed more slowly (<50% conversion to products), and phosphate 11 could be isolated with still 77:23 e.r.. Hence, despite the complexity of this one pot reaction, and the conserved nature of the Pmh residue, the peptide sequences of P2 and P3 were indeed tuned to react preferentially with 8 and 10 as assigned. Since mostly acylation of (R,R)-8 and mono-C1-phosphorylation of 10 was observed, with limited crossover reactivity seen, this example represents a striking simplification of the reaction mixture in comparison to the outcome with DMAP, most notably observed in the cleanliness of the crude NMR of the peptide catalytic system compared to that with DMAP (Figure 3E-F). Key outer-sphere interactions between the peptides and specific, assigned substrates, which are absent for DMAP, are key to the observed selectivities.
Multicatalytic systems are a frontier for studies of synthetically-relevant catalysis, as a myriad of selectivity issues emerge, including the efficient performance of different catalysts for different bond-forming operations in one pot. When a common catalytic moiety is assigned aspirationally to perform a different function, the outer-sphere of the catalyst may be called upon to achieve some of the selectivity criteria associated with these complex reaction designs. In the examples explored above, these capabilities have been documented in two distinct reactivity paradigms of considerable generality—chiral phosphoric acid catalysis and chiral Lewis base catalysis. Much study remains to generalize these concepts for efficient process development. Yet, a basis for optimism seems firmly in place as examples of multicatalytic chemistry mount, and as rational control over the outer-sphere of powerful catalytic moieties develops further.
EXPERIMENTAL SECTION
General Information.
Room temperature is defined as 21–25 °C. All reagents were purchased from commercial sources and used as received, unless otherwise noted. Solvents used for reactions, such as methylene chloride (CH2Cl2), tetrahydrofuran (THF), and acetonitrile (MeCN) were either obtained from a Seca Solvent Purification System by Glass Countour, in which the solvents were dried over alumina and dispended under an atmosphere of argon, or distilled from appropriate drying agents prior to use. For all other purposes, solvents were used as received from commercial sources unless otherwise noted.
1H NMR spectra were recorded on 300 MHz, 400 MHz, 500 MHz, or 600 MHz Varian or Agilent spectrometers at ambient temperature. Samples were prepared in chloroform-d (CDCl3) and dimethyl sulfoxided-d6 (d6-DMSO). 1H NMR data are reported as chemical shifts with multiplicity, coupling constants (J) in Hz, and integrations. Proton chemical shifts are reported in ppm (δ) and referenced to tetramethylsilane (TMS δ 0.00 ppm) or residual solvent (CHCl3, δ 7.26 ppm, DMSO δ 2.50 ppm).37 Multiplicity is reported as follows: singlet (s), broad singlet (bs), doublet (d), doublet of doublets (dd), doublet of doublet of doublets (ddd), doublet of doublet of triplets (ddt), doublet of triplets (dt), doublet of triplet of doublets (dtd), doubet of quartets (dq), triplet (t), quartet (q), quartet of doublets (qd), pentet (p), heptet (hept), multiplet (m), and overlapping multiplets (comp). 13C NMR spectra with broadband proton decoupling were recorded on 500 (126) MHz or 600 (151) MHz Agilent spectrometers with complete proton decoupling at ambient temperature, unless otherwise noted. Carbon chemical shifts are reported in ppm (δ) and referenced to tetramethylsilane (TMS) or solvent (CDCl3, δ 77.16 ppm, d6-DMSO δ 39.52 ppm).37 31P NMR spectra were recorded on 500 (202) MHz Agilent spectrometers with complete proton decoupling at ambient temperature.
Low resolution mass spectrometry (MS) was acquired on a Waters SQD2 UPLC/MS equipped with an electrospray ionization (ESI) detector, with a Waters ACQUITY UPLC BEH C18 (1.7 μm, 2.1 × 50 mm) column. High resolution mass spectrometry (HRMS) was conducted by the Mass Spectrometry Laboratory at the University of Illinois at Urbana-Champaign using a Waters Synapt G2-Si instrument equipped with a QToF mass spectrometer and an ESI detector.
Infrared spectra were obtained using a Nicolet ATR/FT-IR spectrometer, and νmax (cm−1) were partially recorded in accordance with convention. Optical rotations were recorded on a Perkin Elmer Polarimeter 341 at the sodium D line (1.0 dm path length) at 20 °C. Analytical thin-layer chromatography (TLC) was performed using EMD Millipore silica gel 60 F254 precoated plates (0.25 mm thickness). The developed plates were visualized by a UV lamp and/or potassium permanganate (KMnO4) stain.
Normal-phase column chromatography was performed with either silica gel 60 Å (32-63 microns) or with a Biotage Isolera One flash purification system equipped with 15 g, 30 g, 60 g, or 120 g SNAP Ultra HP-Sphere 25 μm columns using an appropriate linear gradient of EtOAc/Hexanes. Reversed-phase column hromatography as performed with a Biotage Isolera One flash purification system equipped with 30 g, 60 g, or 120 g SNAP KP-C18-HS or SNAP Ultra-C18 columns with an appropriate gradient of MeCN/H2O. In some cases, 0.1% formic acid or trifluoroacetic acid buffers were utilized.
Enantiomeric ratio (e.r.) values were acquired using both HPLC and GC. Analytical HPLC was performed on both an Agilent 1100 series analytical chiral HPLC equipped with a photodiode array detector (210 nm, 230 nm, 250 nm, and 254 nm) and a Rainin SD-200 chromatograph, equipped with a single wavelength UV detector (214 nm), were used with a Chiralpak IB column (5 μm particle size, 4.5 × 250 mm) and a Chiralcel OD column (Alltech). Analytical GC was performed on a Hewlett-Packard 6890, employing a flame ionization detector and a Chiraldex G-TA column (Alltech).
Double Reductions of Unsymmetrical Bisquinolines with CPAs.
See Supporting Information for Figure S1. Given this significant divergent selectivity displayed by catalysts 1 and P1, we sought to probe the effect further with unsymmetrical bisquinoline 13, which presents the intriguing challenge of both enantio- and site-selectivity with respect to the first reduction (14 vs. 15, Figure S2.A). In this case, amplified enantioselectivities from this tandem process must come from either (1) a secondary kinetic resolution of the two enantiomers of 14 (Figure S2.C, blue),38 or (2) a difference in selectivity for the reduction of 15 to 16 (Figure S2.C, red), as the Horeau-type correction of e.r. is no longer operative. Interestingly, with both catalysts P1 and 1, the initial reduction of 13 leads to mixtures of 14 and 15, despite the quite different level of substitution on each ring (Figure S2.B). Catalyst 1 gives a nearly equivalent ratio of 14 and 15, while P1 favors 14.39 Moreover, we observe that P1 gives similar e.r. for the first reduction of either 4 and 13 (67:33 vs. 65:35), but also produces doubly reduced 15 with lower e.r. (76:24). Catalyst 1 yields substantially lower enantioselectivities than previously observed both at low and high conversion, and with the opposite senses of chirality as P1 for product 15. Taken together, the observations with substrate 13 unveil further the nuanced outer-sphere interactions of catalysts 1 and P1 in the second reduction.
Previously Synthesized Compounds.
Peptide P1 was previously synthesized and characterized in a related publication.15,16 Compound trans-2-acetamidocyclohexanol ((±)-12) was synthesized according to the method of Hawkins.40 For the syntheses and characterizations of catalyst P2 (Boc-Pmh-Val-Val-DPro-Leu-Val-Val-OMe) and acylated 13, please see a related publication.35 Compound 2,4,6-Tribenzyl-myo-inositol (14) was synthesized according to the method of Billington.41 For the syntheses and characterizations of catalyst P3 (Boc-Pmh-Asn(Trt)-His(πBn)-Asp(tBu)-Ala-OMe) and phosphorylated 15, as well as methods for catalyst screening, please see a related publication.36
Compound Synthesis and Characterization.
Reported yields are for the preparation of analytically pure standards. During purifications, mixed fractions were often excluded.
1,3-bis(2-methylquinolin-8-yl)urea (4).
Modified from literature precedent,42 a flame dried 100 mL round bottom flask was charged with 8-aminoquinaldine (2.00 g, 12.6 mmol, 2.10 equiv), 1,1’-Carbonyldiimidazole (CDI, 976 mg, 6.02 mmol, 1.00 equiv), and 32 mL THF. A condenser was added on the flask and was fit with a septum and placed under and Ar balloon. The solution was heated at 76 °C for 24 h, after which the reaction was cooled to room temperature and concentrated. The crude mixture was purified via normal-phase chromatography, with a gradient eluent of 1 to 30% EtOAc/Hex. The isolated product was further crystallized from a solution of CH2Cl2/MeOH, resulting in pure product. Yield: 931 mg beige solid, 45% yield. 1H NMR (400 MHz, CDCl3) δ (s, 1H), 8.64 (dd, J = 7.7, 1.3 Hz, 1H), 8.05 (d, J = 8.4 Hz, 1H), 7.50 (t, J = 7.9 Hz, 1H), 7.42 (dd, J = 8.1, 1.3 Hz, 1H), 7.34 (d, J = 8.4 Hz, 1H), 2.81 (s, 3H). 13C{1H} NMR (151 MHz, CDCl3) δ 157.0, 152.4, 137.8, 136.7, 126.7, 126.4, 122.5, 120.0, 115.3, 25.4. IR (cm−1, neat): 3313, 1691, 1603, 1519, 1487, 1434, 1382, 1333, 1233, 1191, 1140, 1074, 827, 794, 761, 748, 714. HRMS (ESI-QToF) m/z: [M + H]+ calcd for C21H19N4O 343.1559, found 343.1558. Mp: 177–178 °C. TLC: Rf (2:1 Hex/EtOAc) 0.64, visualized with UV light.
1-(2-methyl-1,2,3,4-tetrahydroquinolin-8-yl)-3-(2-methylquinolin-8-yl)urea (5).
A flame dried 100 mL round bottom flask was charged with 4 (200 mg, 0.826 mmol, 1.00 equiv), Hantzsch ester (523 mg, 2.07 mmol, 2.50 equiv), diphenyl phosphate (103 mg, 0.410 mmol, 0.500 equiv), and 17 mL CH2Cl2. The flask was fit with a septum, placed under an N2 atmosphere, and stirred for 24 h. The reaction was washed with NaHCO3 (satd, aq, 20 mL), dried over Na2SO4, filtered, and concentrated. The crude product, which was predominantly 6+7, was purified by automatic normal-phase chromatography, with a gradient eluent of 5% to 75% EtOAc/Hex to yield a mostly pure mixture of 5 with some 6+7. The mixture was suspended in methanol, which dissolves all of 6+7 and some of 5, and the suspended 5 was filtered to yield pure product. Yield: 12.8 mg beige solid, 4%. 1H NMR (600 MHz, d6-DMSO) δ 9.50 (s, 1H), 8.68 (s, 1H), 8.47 (dd, J = 6.1, 2.8 Hz, 1H), 8.23 (d, J = 8.4 Hz, 1H), 7.51–7.40 (comp, 3H), 7.09 (d, J = 7.7 Hz, 1H), 6.78 (d, J = 7.3 Hz, 1H), 6.52 (t, J = 7.6 Hz, 1H), 4.96 (bs, 1H), 2.80 (ddd, J = 16.3, 11.1, 5.4 Hz, 1H), 2.75–2.67 (comp, 3H), 1.88 (dq, J = 11.9, 4.7 Hz, 1H), 1.52 – 1.41 (m, 1H), 1.19 (d, J = 6.3 Hz, 3H). 13C{1H} NMR (151 MHz, d6-DMSO) δ 156.7, 153.4, 137.1, 136.6, 135.5, 126.1, 126.0, 125.7, 122.9, 122.6, 121.4, 119.2, 115.3, 114.2, 114.2, 46.6, 29.3, 26.3, 24.9, 22.3. IR (cm−1, neat):3394, 1665, 1532, 1496, 1433, 1338, 1283, 1226, 1113, 830, 756, 731. HRMS (ESI-QToF) m/z: [M + H]+ calcd for C21H23N4O 347.1872, found 347.1872. Mp: 200–201 °C. TLC: Rf (2:1 Hex/EtOAc) 0.48, visualized with UV light. Assay of enantiomeric purity: Enantiomers of product 5 were separated by a chiral IB column, eluting at a flow rate of 1.0 mL/min with 2.0% EtOH/Hex. Rt[(S)-5] = 27 min; Rt[(R)-5] = 30 min.
Cis- and trans-1,3-bis(2-methyl-1,2,3,4-tetrahydroquinolin-8-yl)urea (6+7).
Two reactions were setup simultaneously: (A) A flame dried 100 mL round bottom flask was charged with 4 (75.0 mg, 0.219 mmol, 1.00 equiv), Hantzsch ester (117 mg, 0.460 mmol, 2.50 equiv), diphenyl phosphate (27.0 mg, 0.110 mmol, 0.500 equiv), and 40 mL CHCl3. The flask was fit with a septum, placed under an Ar balloon, and stirred for 48 h. Upon consumption of all Hantzsch ester, the reaction was washed with NaHCO3 (satd, aq, 40 mL), dried over Na2SO4, filtered, and concentrated. The crude product was purified by automatic normal-phase chromatography, with a gradient eluent of 5% to 75% EtOAc/Hex to yield a mostly pure mixture of 6+7. (B) A flame dried 250 mL round bottom flask was charged with 4 (100 mg, 0.292 mmol, 1.00 equiv), Hantzsch ester (333 mg, 1.31 mmol, 4.50 equiv), diphenyl phosphate (37.0 mg, 0.146 mmol, 0.500 equiv), and 60 mL CH2Cl2. The flask was fit with a septum, placed under an Ar balloon, and stirred for 168 h. Upon consumption of all Hantzsch ester, the reaction was washed with NaHCO3 (satd, aq, 60 mL), dried over Na2SO4, filtered, and concentrated. The crude product was purified by automatic normal-phase chromatography, with a gradient eluent of 5% to 75% EtOAc/Hex to yield a mostly pure mixture of 6+7. (A+B) The crude products from both (A) and (B) were combined and further purified by automatic normal-phase chromatography, with a gradient eluent of 5% to 60% EtOAc/Hex to yield pure product. Yield: 47.7 mg of 6+7 as a beige solid, 26%. 1H NMR (400 MHz, CDCl3) δ 7.04 (d, J = 7.7 Hz, 1H), 6.89 (d, J = 7.5 Hz, 1H), 6.61 (t, J = 7.6 Hz, 1H), 6.07 (s, 1H), 3.44–3.33 (m, 1H), 2.92–2.69 (m, 2H), 1.93 (ddt, J = 12.6, 6.1, 3.5 Hz, 1H), 1.60–1.48 (m, 1H), 1.22 (d, J = 6.3 Hz, 3H). C{1H} NMR (151 MHz, CDCl3) δ 156.1, 140.7, 128.0, 125.1, 123.1, 121.8, 117.0, 47.3, 29.8, 26.8, 22.7. IR (cm−1, neat): 3293, 2956, 2929, 1628, 1560, 1497, 1460, 1336, 1296, 1279, 1247, 1153, 1115, 1064, 961, 742. HRMS (ESI-QToF) m/z: [M + H]+ calcd for C21H27N4O 351.2158, found 351.2188. TLC: Rf (2:1 Hex/EtOAc) 0.29, visualized with UV light. Assay of enantiomeric purity: Stereoisomers of products 6+7 were separated by a chiral IB column, eluting at a flow rate of 1.0 mL/min with 2.0% EtOH/Hex. Rt[(S,S)-6] = 33 min; Rt[meso-7] = 38 min; Rt[(R,R)-6] = 42 min.
(±)-2-acetamidocyclohexyl diphenyl phosphate [(±)-12]:
A 100 mL round bottom flask was charged with (±)-8 (200 mg, 1.27 mmol, 1.00 equiv), DMAP (8.0 mg, 0.070 mmol, 0.050 equiv), PhMe (25 mL), followed by sequential addition of triethylamine (distilled, 173 uL, 1.24 mmol, 1.30 equiv) and diphenylchlorophosphate (251 uL, 1.15 mmol, 1.20 equiv). CH2Cl2 (20 mL) was added to aid solubility. The reaction was stirred at RT for 12 h, followed by quenching by addition of MeOH (2 mL). The solution was concentrated and purified by flash chromatography, eluting with a gradient of 3% to 5% MeOH/CH2Cl2 to yield pure product. Yield: 130 mg light brown oil, 26%. 1H NMR (500 MHz, CDCl3) δ 7.36–7.29 (comp, 4H), 7.22–7.15 (comp, 6H), 6.34 (bd, J = 7.8 Hz, 1H), 4.38–4.28 (m, 1H), 3.93–3.83 (m, 1H), 2.13 (d, J = 12.4 Hz, 1H), 2.04–1.97 (m, 1H), 1.74 (d, J = 10.3 Hz, 1H), 1.69 (s, 3H), 1.62 (d, J = 13.1 Hz, 1H), 1.54 (qd, J = 12.7, 3.9 Hz, 1H), 1.35–1.16 (m, 2H), 1.09 (qd, J = 12.7, 3.6 Hz, 1H). 13C{1H} NMR (151 MHz, CDCl3, for analogous C-P splitting, see a related reference43) δ 170.2, 150.5 (d, JCP = 7.5 Hz), 150.4 (d, JCP = 7.5 Hz), 129.9, 125.7 (d, JCP = 1.1 Hz), 125.6 (d, JCP = 1.0 Hz), 120.3 (d, JCP = 4.6 Hz), 120.2 (d, JCP = 4.8 Hz), 81.0 (d, JCP = 6.4 Hz), 53.6 (d, JCP = 3.3 Hz), 32.61 (d, JCP = 4.1 Hz), 32.1, 24.2, 24.0, 23.2. 31P NMR (202 MHz, CDCl3) δ −10.6. IR (cm−1, thin film from CHCl3): 3300, 2940, 2861, 1657, 1591, 1551, 1488, 1455, 1371, 1272, 1188, 1162, 1021, 952, 907, 726. HRMS (ESI-QToF) m/z: [M + H]+ calcd for C20H24NO5PNa 412.1290, found 412.1292. TLC: Rf (9:1 CH2Cl2/MeOH) 0.33, visualized with UV light. Assay of enantiomeric purity: Enantiomers of product 12 were separated by a chiral OD column (Alltech), eluting at a flow rate of 0.5 mL/min with 1.5% EtOH/Hex. Rt[(S,S)-12] = 38 min; Rt[(R,R)-12] = 41 min.
1-(2-methylquinolin-8-yl)-3-(quinolin-8-yl)urea (13, see Figure S2).
Modified from literature precedent,43 a flame dried 100 mL round bottom flask was charged with 8-aminoquinoline (1.00 g, 6.93 mmol, 1.00 equiv), 1,1’-Carbonyldiimidazole (CDI, 675 mg, 4.16 mmol, 0.600 equiv), and 35 mL CH2Cl2. The solution was fitted with a septum and Ar balloon and stirred for 2 h. Then, 8-aminoquinaldine (1.10 g,6.92 mmol, 1.00 equiv) was added, and the reaction was stirred overnight for 22 h. The solution was concentrated, yielding a mixture of symmetrical urea products (including 4) and the desired unsymmetrical product (13). The mixture was purified by automatic normal-phase chromatography, with a gradient eluent of 1 to 50% EtOAc/Hex. The isolated product was further purified with automatic reversed-phased chromatography, eluting with a gradient of 50% to 100% MeCN/H2O to yield pure product. Yield: 409 mg white solid, 30%. 1H NMR (400 MHz, CDCl3) δ 9.53 (s, 1H), 9.44 (s, 1H), 8.87 (dd, J = 4.2, 1.7 Hz, 1H), 8.70 (dd, J = 7.8, 1.3 Hz, 1H), 8.65 (dd, J = 7.7, 1.3 Hz, 1H), 8.19 (dd, J = 8.3, 1.7 Hz, 1H), 8.06 (d, J = 8.4 Hz, 1H), 7.59 (t, J = 8.0 Hz, 1H), 7.53–7.45 (comp, 3H), 7.42 (dd, J = 8.2, 1.3 Hz, 1H), 7.34 (d, J = 8.4 Hz, 1H), 2.82 (s, 3H). 13C{1H} NMR (151 MHz, CDCl3) δ 157.1, 152.4, 138.5, 137.8, 136.7, 136.7, 135.7, 134.9, 128.3, 127.8, 126.66, 126.4, 122.6, 121.7, 120.3, 120.1, 115.4, 115.4, 25.4. IR (cm−1, neat): 3249, 1667, 1518, 1489, 1434, 1380, 1324, 1254, 1191, 1079, 820, 787, 752, 731, 708. HRMS (ESI-QToF) m/z: [M + H]+ calcd for C20H17N4O 329.1402, found 329.1401. Mp: 149–150 °C. TLC: Rf (2:1 Hex/EtOAc) 0.63, visualized with UV light.
1-(2-methyl-1,2,3,4-tetrahydroquinolin-8-yl)-3-(quinolin-8-yl)urea (14, see Figure S2).
A flame dried 20 mL scintillation vial was charged with 13 (100 mg, 0.305 mol, 1.00 equiv), Hantzsch ester (193 mg, 0.762 mmol,equiv), diphenyl phosphate (38.2 mg, 0.153, 0.500 equiv), and 6 mL CH2Cl2. The vial was flushed with Ar, capped, and the reaction was stirred for 24 h. The solution was washed with NaHCO3 (satd, aq, 6 mL), dried over Na2SO4, filtered, and concentrated. The crude product, which included 13, 14, 15, and 16) was purified by automatic normal-phase chromatography, with a gradient eluent of 5% to 100% EtOAc/Hex. The mostly pure product was further purified with automatic reversed-phase chromatography, eluting with a gradient of 25% to 100% MeCN/H2O to yield pure product. Yield: 8.7 mg pale yellow solid, 9%. 1H NMR (600 MHz, CDCl3) δ 9.48 (s, 1H), 8.66–8.59 (comp, 2H), 8.09 (d, J = 8.3 Hz, 1H), 7.49 (t, J = 8.0 Hz, 1H), 7.40 (d, J = 8.1 Hz, 1H), 7.35 (dd, J = 8.3, 4.2 Hz, 1H), 7.13 (d, J = 7.7 Hz, 1H), 6.98 (d, J = 7.5 Hz, 1H), 6.67 (t, J = 7.6 Hz, 1H), 6.58 (bs, 1H), 4.28 (bs, 1H), 3.44–3.39 (m, 1H), 2.90 (ddd, J = 16.7, 11.4, 5.4 Hz, 1H), 2.81 (dt, J = 16.3, 4.5 Hz, 1H), 1.94 (ddt, J = 6.1, 3.4 Hz, 1H), 1.60 (qd, J = 11.8, 4.9 Hz, 1H), 1.15 (d, J = 6.3 Hz, 3H). 13C(1H} NMR (151 MHz, CDCl2) δ 154.9, 148.0, 141.1, 138.7, 136.3, 135.6, 128.3, 128.1, 127.6, 125.6, 123.0, 121.4, 121.2, 120.2, 116.7, 115.4, 47.3, 29.8, 27.0, 22.6. IR (cm−1, neat): 3270, 2923, 1644, 1542, 1488, 1423, 1382, 1327, 1284, 1252, 1221, 1115, 1102, 822, 787, 746. HRMS (ESI-QToF) m/z: [M + H]+ calcd for C20H21N4O 333.1715, found 333.1715. Mp: 149–150 °C. TLC: Rf (2:1 Hex/EtOAc) 0.40, visualized with UV light. Assay of enantiomeric purity: Enantiomers of product 14 were separated by a chiral IB column, eluting at a flow rate of 1.0 mL/min with 10% EtOH/Hex. Rt[(S)-14] = 15 min; Rt[(R)-14] = 17 min.
1-(2-methylquinolin-8-yl)-3-(1,2,3,4-tetrahydroquinolin-8-yl)urea (15, see Figure S2).
A flame dried 20 mL scintillation vial was charged with 13 (100 mg, 0.305 mol, 1.00 equiv), Hantzsch ester (193 mg, 0.762 mmol,equiv), diphenyl phosphate (38.2 mg, 0.153, 0.500 equiv), and 6 mL CH2Cl2. The vial was flushed with Ar, capped, and the reaction was stirred for 24 h. The solution was washed with NaHCO3 (satd, aq, 6 mL), dried over Na2SO4, filtered, and concentrated. The crude product, which included 13, 14, 15, and 16) was purified by automatic normal-phase chromatography, with a gradient eluent of 5% to 100% EtOAc/Hex. The mostly pure product was further purified with automatic reversed-phase chromatography, eluting with a gradient of 25% to 100% MeCN/H2O, with a 0.1% formic acid buffer (aq), to yield mostly pure 15 as a colorless oil. Yield not reported. Compound 15 was a minor regioisomer of this reaction, and it was difficult to obtain and purify preparative amounts of material for characterization. The product was assigned for conversion measurements by UPLC/MS by a mostly pure NMR. 1H NMR (400 MHz, CDCl3, mostly pure) δ 9.49 (s, 1H), 8.49 (dd, J = 7.7, 1.4 Hz, 1H), 7.94 (d, J = 8.4 Hz, 1H), 7.41 (t, J = 7.9 Hz, 1H), 7.32 (d, J = 6.9 Hz, 1H), 7.20 (d, J = 8.4 Hz, 1H), 7.12 (d, J = 7.7 Hz, 1H), 6.99 (d, J = 7.6 Hz, 1H), 6.67 (t, J = 7.6 Hz, 1H), 5.97 (s, 1H), 4.41 (bs, 1H), 3.39–3.26 (m, 2H), 2.83 (t, J = 6.4 Hz, 2H), 2.49 (s, 3H), 2.01–1.83 (m, 2H). In particular, an observed singlet for the C2 methyl group helps to differentiate this compound from 14. UPLC/MS (ESI) m/z: [M + H]+ calcd for C20H21N4O 333.17, found 333.35.
1-(2-methyl-1,2,3,4-tetrahydroquinolin-8-yl)-3-(1,2,3,4-tetrahydroquinolin-8-yl)urea (16, see Figure S2).
A flame dried 4 mL scintillation vial was charged with 13 (20.0 mg, 0.0600 mol, 1.00 equiv), Hantzsch ester (38.0 mg, 0.150 mmol, 2.50 equiv), diphenyl phosphate (7.50 mg, 0.0300, 0.500 equiv), and 1.25 mL CH2Cl2. The vial was flushed with Ar, capped, and the reaction was stirred for 24 h. The solution was washed with NaHCO3 (satd, aq, 1.5 mL), dried over Na2SO4, filtered, and concentrated. The crude product, which included 13, 14, 15, and 16) was purified by automatic normal-phase chromatography, with a gradient eluent of 5% to 100% EtOAc/Hex to yield pure product. Yield: 5.6 mg beige solid, 28%. 1H NMR (400 MHz, CDCl3) δ 7.05–7.00 (comp, 2H), 6.92–6.84 (comp, 12H), 6.67–6.57 (comp, 2H), 6.23–6.15 (comp, 2H), 3.41–3.33 (m, 1H), 3.33–3.27 (m, 2H), 2.89–2.79 (m, 1H), 2.79–2.71 (comp, 2H), 1.98–1.94 (m, 1H), 1.94–1.86 (comp, 2H), 1.62–1.48 (m, 1H), 1.22 (d, J = 6.3 Hz, 3H). 13C{1H} NMR (151 MHz, CDCl3) δ 155.9, 140.5, 128.2, 125.1, 123.4, 123.4,121.8, 117.4, 117.0, 47.4, 42.1, 29.9, 27.2, 26.8, 22.6, 21.9. IR (cm−1, neat): 3318, 2930, 1630, 1604, 1529, 1472, 1328, 1302, 1232, 1190, 1104, 1026, 744. HRMS (ESI-QToF) m/z: [M + H]+ calcd for C20H25N4O 337.2028, found 337.2022. Mp: >200 °C. TLC: Rf (2:1 Hex/EtOAc) 0.22, visualized with UV light. Assay of enantiomeric purity: Enantiomers of product 16 were separated by a chiral IB column, eluting at a flow rate of 1.0 mL/min with 10% EtOH/Hex. Rt[(S)-16] = 11 min; Rt[(R)-16] = 12 min.
Determination of Absolute Stereochemistry of 5+6.
In our previous study of the reduction of monoquinoline 2 to 3, P1 and 1 gave opposite absolute stereochemical configurations, yet generate the same enantiomers in the reduction of 4. To probe this intriguing reversal, the absolute stereochemistry of enantioenriched 6 was sought. Unfortunately, numerous attempts to grow a single crystal of 5 or 6, in addition to a number of related compounds, proved futile. However, the absolute stereochemistry of 3 had been determined in our previous study,15 and we wondered whether this could be converted into a derivative of 6. As such, (R)-TRIP (1) was utilized to facilitate the HEH-mediated reduction of 17 to (R)-18 (Figure S3A). The secondary amine of (R)-18 was next dimethylated, and the primary amine was deprotected and dimerized with CDI to yield a mixture of (R,R)-19 and meso-20. Intriguingly, the observed e.r. and dr (98:2 e.r., 4.26:1 dr) are similar to the predicted values from squaring the selectivity of the initial reduction (89:11 e.r., s = 8.1), which would give s2 = 65, 98:2 e.r., 4.10 d.r., representing ideal Horeau kinetics for asymmetric amplification. Upon dimethylation of enantioenriched 6, from reduction of 4 with (R)-TRIP (1), chiral-19 proved to have bis-(S) stereochemistry (Figure S2B). Hence, while P1 processes both monoquinoline 2 and bisquinoline 4 with the same absolute stereochemistry of reduction, 1 shows enantiodivergence, representing another intriguing complementary feature of these two catalyst systems.
Benzyl (2-methylquinolin-8-yl)carbamate (17, see Figure S3A).
A flame dried 100 mL round bottom flask was charged with 8-aminoquinaldine (1.00 g, 6.23 mmol, 1.00 equiv), 25 mL THF, and 6.3 mL NaHCO3 (satd, aq). The vessel was cooled to 0 °C, followed by addition of benzyl chloroformate dropwise (948 μL, 6.64 mmol, 1.05 equiv). The reaction was allowed to warm to rt slowly, and stirred overnight for 18 h. Upon completion of the reaction, the reaction was concentrated, redissolved in CH2Cl2 (20 mL), and washed with NaHCO3 (satd, aq, 20 mL). The organics were dried over Na2SO4, filtered, and concentrated. The crude material was purified by automatic normal-phase chromatography, with a gradient eluent of 5% to 100% EtOAc/Hex to yield pure product. Yield: 1.454 g beige solid, 79%. 1H NMR (400 MHz, CDCl3) δ 9.33 (s, 1H), 8.42 (s, 1H), 8.00 (d, J = 8.4 Hz, 1H), 7.54–7.33 (m, 7H), 7.29 (d, J = 8.4 Hz, 1H), 5.31 (s, 2H), 2.71 (s, 3H). 13C{1H} NMR (151 MHz, CDCl3) δ 157.2, 153.5, 137.5, 136.3, 136.3, 134.0, 128.7, 128.5, 128.4, 126.2, 126.1, 122.5, 120.5, 114.6, 67.0, 25.2. IR (cm−1, neat): 3357, 1723, 1604, 1573, 1521, 1492, 1455, 1434, 1383, 1339, 1311, 1237, 1192, 1109, 1023, 978, 862, 797, 748. HRMS (ESI-QToF) m/z: [M + H]+ calcd for C18H17N2O2 293.1290, found 293.1288. Mp: 79–80 °C. TLC: Rf (4:1 Hex/EtOAc) 0.50, visualized with UV light.
(R)-Benzyl (2-methyl-1,2,3,4-tetrahydroquinolin-8-yl)carbamate (18).
The (R)-stereochemistry was assigned in analogy to a similar substrate from a previous study.15 A flame dried 20 mL scintillation vial was charged with 17 (121 mg, 0.413 mmol, 1.00 equiv), Hantzsch ester (261 mg, 1.03 mmol, 2.50 equiv), and catalyst [(A) diphenyl phosphate (52.0 mg, 0.250 mmol, 0.500 equiv); (B) (R)-TRIP (1, 12.0 mg, 0.0159 mmol, 4 mol%)]. The vial was flushed with argon, charged with 8.5 mL CH2Cl2, capped, and stirred for 48 h. Upon completion, the reaction was washed with NaHCO3 (satd, aq, 10 mL), with the aqueous layer being reextracted with CH2Cl2 (10 mL). The combined organics were dried over Na2SO4, filtered, and concentrated. The crude material was purified by automatic normal-phase chromatography, with a gradient eluent of 5% to 100% EtOAc/Hex to yield a mixture of 18 and oxidized HEH byproduct (HEox). For reaction (B), the crude material was used in the next step without further purification assuming full yield. For reaction (A), the crude material was further purified by two automatic reversed-phase chromatography columns, eluting with a gradient of 30% to 100% MeCN/H2O and 50% to 85% MeCN/H2O, both with a 0.1% formic acid buffer (aq). The protonated product was dissolved in 2 mL CH2O2 (2 mL) and washed with 2 mL NaHCO3 (satd, aq), dried over Na2SO4, filtered, and concentrated to yield pure 18 as a colorless oil. Yield not reported. 1H NMR (500 MHz, CDCl3) δ 7.47–7.31 (comp, 5H), 7.16 (bs, 1H), 6.87 (d, J = 7.4 Hz, 1H), 6.67 (t, J = 7.2 Hz, 1H), 6.34 (bs, 1H), 3.41–3.31 (m, 1H), 2.90–2.71 (m, 2H), 1.93 (ddt, J = 11.9, 5.6, 3.1 Hz, 1H), 1.54 (ddt, J = 16.7, 12.0, 5.2 Hz, 1H), 1.23 (d, J = 6.0 Hz, 3H). 13C{1H} NMR (126 MHz, d6-DMSO, 60 °C) δ 154.2, 137.8, 136.7, 128.1, 127.5, 125.4, 122.5, 122.4, 121.3, 115.2, 65.5, 46.4, 28.9, 26.0, 21.7. IR (cm−1, thin film from CHCl3): 3315, 2930, 1697, 1608, 1498, 1453, 1330, 1218, 1065, 1020, 982, 909, 728. HRMS (ESI-QToF) m/z: [M + H]+ calcd for C18H21N2O2 297.1603, found 297.1603. TLC: Rf (4:1 Hex/EtOAc) 0.32, visualized with UV light. Assay of enantiomeric purity: Enantiomers of product 18 were separated by a chiral IB column, eluting at a flow rate of 1.0 mL/min with 15% EtOH/Hex. Rt[(S)-18] = 6.0 min; Rt(R)-18] = 6.5 min.
Cis- and trans-1,3-bis(1,2-dimethyl-1,2,3,4-tetrahydroquinolin-8-yl)urea (19+20). Preparation from 6:
Modified from literature precedent,44 a flame dried 4 mL scintillation vial was charged with 6 (16.7 mg, 0.0476 mmol, 1.00 equiv), 0.75 mL MeCN, and formaldehyde (37% aq soln, 29.0 μL, 0.357 mmol, 7.50 equiv). Sodium cyanoborohydride (22.0 mg, 0.357 mmol, 7.50 equiv), handled entirely in a well ventilated fume hood or capped vials, was added quickly to the reaction. The vessel was fitted with a septum, pierced with a cannula leading to a solution of NaHCO3 (satd, aq) to neutralize any HCN. Glacial acetic acid (10 μL) was added, and the reaction was allowed to stir for 1 h, after which more glacial acetic acid (10 μL) was added. The reaction was stirred overnight for 18 h, after which the mixture was slowly added to a solution of NaHCO3 (satd, aq, 20 mL). This mixture was extracted with CH2Cl2 (20 mL × 2), and the combined organics were dried over Na2SO4, filtered, and concentrated. The crude product was purified by automatic normal-phase chromatography, with a gradient eluent of 5% to 100% EtOAc/Hex, followed by automatic reversed-phase chromatography, with a gradient eluent of 30% to 100% MeCN/H2O, finally followed by normal-phase chromatography with a gradient eluent of 1% to 40% EtOAc/Hex to yield 10.8 mg of 19+20 as a white solid (60% yield). Preparation from 17: Modified from literature precedent,44,45 a 100 mL round bottom flask containing crude 18+HEox (assumed 0.413 mmol of 18, 1.00 equiv) was charged with 3.0 mL MeCN and formaldehyde (37% aq soln, 168 μL, 2.07 mmol, 5.00 equiv). Sodium cyanoborohydride (130 mg, 130 mmol, 5.00 equiv), handled entirely in a well ventilated fume hood or capped vials, was added quickly to the reaction. The vessel was fitted with a septum, pierced with a cannula leading to a solution of NaHCO3 (satd, aq) to neutralize any HCN. Glacial acetic acid (123 μL) was added, and the reaction was allowed to stir for 15 min. A thick sludge formed; as such 2.0 mL of additional MeCN was added. The reaction was stirred overnight for 18 h, after which the mixture was slowly added to a solution of NaHCO3 (satd, aq, 100 mL). This mixture was extracted with CH2Cl2 (100 mL × 2), and the combined organics were dried over Na2SO4, filtered, and concentrated. The crude product was purified by automatic normal-phase chromatography, with a gradient eluent of 5% to 100% EtOAc/Hex, followed by automatic reversed-phase chromatography, with a gradient eluent of 25% to 100% MeCN/H2O, with a 0.1% formic acid buffer (aq). The protonated product was dissolved in 2 mL CH2Cl2 (2 mL) and washed with 2 mL NaHCO3 (satd, aq), dried over Na2SO4, filtered, and concentrated to yield 25.0 mg of mostly pure 21, which was taken forward to the next step without further purification. A 10 mL round bottom containing 21 was charged with 0.75 mL MeOH and 0.75 mL THF. The vessel was fitted with a septum, which was pierced by a balloon of H2, connected to a needle; and H2 was bubbled through the solution. The reaction was stirred at rt overnight for 20 h, after which the crude mixture was filtered through a plug of celite, eluting with MeCN and MeOH. The solution was concentrated to yield 14 mg of mostly pure 22, which was taken forward to the next step without further purification. A 4 mL scintillation vial containing 22 (14.0 mg, 0.0805 mmol, 1.00 equiv) was charged with 0.5 mL THF, followed by 1,1′-Carbonyldiimidazole (CDI, 31.0 mg, 0.191 mmol, 2.4 equiv), which was added portionwise over the course of 1 day. The vial was capped and heated at 70 °C for 96 h. Upon completion, the reaction was concentrated and purified by automatic normal phase chromatography, with a gradient eluent of 1% to 40% EtOAc/Hex, yielding pure product. Yield: 7.0 mg of 19+20 as a white solid, 4% yield from 17, 4 steps. 1H NMR (400 MHz, CDCl3) δ 7.73 (t, J = 6.8 Hz, 1H), 7.29–7.23 (m, 1H), 6.95 (t, J = 7.8 Hz, 1H), 6.77 (d, J = 7.6 Hz, 1H), 3.09 (qd, J = 6.7, 3.5 Hz, 1H), 2.782.72 (m, 2H), 2.54 (d, J = 3.3 Hz, 3H), 2.04–1.88 (m, 1H), 1.69–1.50 (m, 1H), 1.06 (d, J = 6.8 Hz, 3H). 13C{1H} NMR (151 MHz, CDCl3) δ 153.6, 138.9, 138.7, 132.7, 129.9, 124.1, 123.0, 123.0, 118.4, 54.1, 54.0, 41.2, 40.6,24.3, 22.0, 19.2. IR (cm−1, thin film from CH2Cl2): 3272, 2966, 2928, 1654, 1602, 1535, 1468, 1411, 1379, 1315, 1220, 1191, 771. HRMS (ESI-QToF) m/z: [M + H]+ calcd for C23H31N4O 379.2498, found 379.2497. TLC: Rf (4:1 Hex/EtOAc) 0.17, visualized with UV light. Assay of enantiomeric purity: Enantiomers of products 19+20 were separated by a chiral IB column, eluting at a flow rate of 1.0 mL/min with 2.0% EtOH/Hex. Rt[(S,S)-19] = 14 min; Rt[20] = 15 min; Rt[(R,R)-19] = 16 min.
General Procedures for Catalyst Screening.
Bisquinoline reduction.
A flame dried 5 mL scintillation vial was charged with substrate (4 or 13; 0.0600 mmol, 1.00 equiv), Hantzsch ester (38.0 mg, 0.150 mmol, 2.50 equiv), catalyst (DPP, P1, or 1; 0.0120 mmol, 0.200 equiv), and 1.25 mL CH2Cl2. The vial was flushed with Ar, capped, and stirred for 24 h. After conclusion of the reaction, the conversion was measured by LC/MS. The reaction was washed with NaHCO3 (satd, aq, 1.0 mL), the aqueous layer was reextracted with 2 mL CH2Cl2, and the combined organics were dried over Na2SO4, filtered, and concentrated. The crude product, which was purified by automatic normal-phase chromatography, with a gradient eluent of 5% to 75% EtOAc/Hex to yield mono-reduced products (5, 14, or 15), plus di-reduced products (6+7 that were inseparable, or 16). The e.r. and dr of both isolates were measured by HPLC. All reported results are the average of ≥ 2 trials.
Monoquinoline reduction (from literature precedent15):
An oven dried 4 mL vial equipped with a stir bar was charged with catalyst (0.00600 mmol, 0.100 equiv), quinoline 17 (17.6 mg, 0.0600 mmol, 1.00 equiv), and Hantzsch ester (38.0 mg, 0.150 mmol, 2.5 equiv.). The vial was flushed with Ar, charged with 1.25 mL CH2Cl2, capped, and stirred for 24 h. The reaction was quenched by addition of NaHCO3 (1 mL), with the aqueous layer being reextracted with CH2Cl2 (2 mL). The combined organics were dried with Na2SO4, filtered, and concentrated to yield crude product. This was taken up in CDCl3 to determine conversion via 1H NMR and e.r. by HPLC. Reported values are the average of 2 trials. With P1 as catalyst, 18 was produced in 92% conv., 59:41 e.r.; with 1 as catalyst, 18 was produced in 99% conv., 11:89 e.r..
One-pot site- and enantioselective acylations and phosphorylations.
Substrates 10 (14.3 mg, 0.0318 mmol, 1.00 equiv) and (±)-8 (5.0 mg, 0.032 mmol, 1.0 equiv) were dissolved in PhMe (3 mL). Solutions of catalyst P3 (0.7 mg, 6*10−4 mmol, 2 mol%) and P2 (0.6 mg, 6*10−4 mmol, 2 mol%) were added, followed by triethylamine (distilled, 56 μL, 0.40 mmol, 13 equiv). Acetic anhydride (18 μL, 0.19 mmol, 6.0 equiv) and diphenylchlorophosphate (40 μL, 0.19 mmol, 6.0 equiv) were then added in rapid succession. After 1 h, an aliquot (1.5 mL) of the reaction was quenched with methanol (0.75 mL) and the solvent was removed in vacuo. Product distribution was monitored by 1H and 31P NMR spectroscopy (see Figure 3). Products were separated and analyzed by HPLC and GC as detailed below. Separation of reaction products: Reaction products 9, 11, and 12 were separated by normal phase HPLC employing a YMC PVA-Sil column (Waters), eluting at a flow rate of 10 mL/min with the following gradient ramp from Hex to 6.5% iPrOH/Hex over 40 min. Rt(9+12) = 27 min; Rt(11) = 30 min. Assay of enantiomeric purity (11): Enantiomers of product 11 were separated by chiral HPLC employing a Chiralcel OD column (Alltech), eluting at a flow rate of 0.5 mL/min with 30% EtOH/Hex. Rt[11(3-P)] = 11.5 min; Rt[11(1-P)] = 12.5 min.46 Separation of reaction products: Reaction products 9 and 12 were separated by chiral HPLC employing an L-Leucine column (Regis), eluting at a flow rate of 0.5 mL/min with 15% EtOH/Hex. Rt(12) = 16.8 min; Rt(9) = 20.3 min. Assay of enantiomeric purity (9): Enantiomers of product 9 were separated by chiral GC employing a 30m Chiraldex G-TA column (Alltech). Conditions: temperature = 135 °C; flow rate = 60 psi. Rt[(R,R)-9] = 12.0 min; Rt[(S,S)-9] = 12.8 min.35 Assay of enantiomeric purity (12): Enantiomers of product 12 were separated by a chiral OD column (Alltech), eluting at a flow rate of 0.5 mL/min with 1.5% EtOH/Hex. Rt[(S,S)-12] = 38 min; Rt[(R,R)-12] = 41 min.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the National Institutes of Health (NIH R01-GM096403). We also thank Takeda for research support. We graciously thank Angela Lin for assistance in the synthesis of compounds for this study. The earliest work in this project was supported by the National Institutes of Health and the National Science Foundation. C.R.S. thanks the National Science Foundation Graduate Research Fellowship Program for funding (DGE1122492).
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Further commentary on Horeau amplification.Study of CPA-mediated reduction of an unsymmetric bisquinolineSchemes for substrate synthesis and testingTables for conversion dependence of enantioselectivities reduction of bisquinolinesHPLC and UPLC/MS traces for catalytic reductionsX-ray crystallographic data for compounds (±)-5 and (±)-14
Crystallographic data are deposited with the Cambridge Crystallographic Data Centre under the accession numbers CCDC 1882216 and 1882217.
The authors declare no competing financial interests.
REFERENCES
- (1).Anantharaman V; Aravind L; Koonin EV “Emergence of Diverse Biochemical Activities in Evolutionarily Conserved Structural Scaffolds of Proteins.” Curr. Opin. Chem. Biol 2003, 7, 12–20. [DOI] [PubMed] [Google Scholar]
- (2).(a) Denisov IG; Makris TM; Sligar SG; Schlichting I “Structure and Chemistry of Cytochrome P450.” Chem. Rev 2005, 105, 2253–2277. [DOI] [PubMed] [Google Scholar]; (b) Lairson LL; Henrissat B; Davies GJ; Withers SG “Glycosyltransferases: Structures, Functions, and Mechanisms.” Annu. Rev. Biochem 2008, 77, 521–555. [DOI] [PubMed] [Google Scholar]
- (3).Eastgate MD; Schmidt MA; Fandrick KR “On the Design of Complex Drug Candidate Syntheses in the Pharmaceutical Industry.” Nat. Rev. Chem 2017, 1, 1–16. [Google Scholar]
- (4).Lohr TL; Marks TJ “Orthogonal Tandem Catalysis.” Nat. Chem 2015, 7, 477–482. [DOI] [PubMed] [Google Scholar]
- (5).Zhou J Multicatalyst System in Asymmetric Catalysis; Wiley: Hoboken, 2015. [Google Scholar]
- (6).Afewerki S; Córdova A “Combinations of Aminocatalysts and Metal Catalysts: A Powerful Cooperative Approach in Selective Organic Synthesis.” Chem. Rev 2016, 116, 13512–13570. [DOI] [PubMed] [Google Scholar]
- (7).(a) Alachraf MW; Wende RC; Schuler SMM; Schreiner PR; Schrader W “Functionality, Effectiveness, and Mechanistic Evaluation of a Multicatalyst-Promoted Reaction Sequence by Electrospray Ionization Mass Spectrometry.” Chem. Eur. J 2015, 21, 16203–16208. [DOI] [PubMed] [Google Scholar]; (b) Hofmann C; Schuler SMM; Wende RC; Schreiner PR “En Route to Multicatalysis: Kinetic Resolution of Trans-Cycloalkane-1,2-Diols via Oxidative Esterification.” Chem. Commun 2014, 50, 1221–1223. [DOI] [PubMed] [Google Scholar]; (c) Ambrosini LM; Lambert TH “Multicatalysis: Advancing Synthetic Efficiency and Inspiring Discovery.” ChemCatChem 2010, 2, 1373–1380. [Google Scholar]
- (8).Rueping M; Koenigs RM; Atodiresei I “Unifying Metal and Brensted Acid Catalysis-Concepts, Mechanisms, and Classifications.” Chem. Eur. J 2010, 16, 9350–9365. [DOI] [PubMed] [Google Scholar]
- (9).Skubi KL; Blum TR; Yoon TP “Dual Catalysis Strategies in Photochemical Synthesis.” Chem. Rev 2016, 116, 10035–10074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Yang ZP; Zhang W; You SL “Catalytic Asymmetric Reactions by Metal and Chiral Phosphoric Acid Sequential Catalysis.” J. Org. Chem 2014, 79, 7785–7798. [DOI] [PubMed] [Google Scholar]
- (11).Shimada Y; Miyake Y; Matsuzawa H; Nishibayashi Y “Ruthenium-Catalyzed Sequential Reactions: Deracemization of Secondary Benzylic Alcohols.” Chem. Asian J 2007, 2, 393–396. [DOI] [PubMed] [Google Scholar]
- (12).Simmons B; Walji AM; MacMillan DWC “Cycle-Specific Organocascade Catalysis: Application to Olefin Hydroamination, Hydro-Oxidation, and Amino-Oxidation, and to Natural Product Synthesis.” Angew. Chem. Int. Ed 2009, 48, 4349–4353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Davis HJ; Phipps RJ “Harnessing non-covalent interactions to exert control over regioselectivity and site-selectivity in catalytic reactions.” Chem. Sci 2017, 8, 864–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Colby Davie EA; Mennen SM; Xu Y; Miller SJ “Asymmetric Catalysis Mediated by Synthetic Peptides.” Chem. Rev 2007, 107, 5759–5812. [DOI] [PubMed] [Google Scholar]
- (15).Shugrue CR; Miller SJ “Phosphothreonine as a Catalytic Residue in Peptide-Mediated Asymmetric Transfer Hydrogenations of 8-Aminoquinolines.” Angew. Chem. Int. Ed 2015, 54, 11173–11176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Shugrue CR; Featherston AL; Lackner RM; Lin A; Miller SJ “Divergent Stereoselectivity in Phosphothreonine (pThr)-Catalyzed Reductive Aminations of 3-Amidocyclohexanones.” J. Org. Chem 2018, 83, 4491–4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Kwon Y; Chinn AJ; Kim B; Miller SJ “Divergent Control of Point and Axial Stereogenicity: Catalytic Enantioselective C–N Bond-Forming Cross-Coupling and Catalyst-Controlled Atroposelective Cyclodehydration.” Angew. Chem. Int. Ed 2018, 57, 6251–6255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Featherston AL; Shugrue CR; Mercado BQ; Miller SJ “Phosphothreonine (pThr)–Based Multifunctional Peptide Catalysis for Asymmetric Baeyer–Villiger Oxidations of Cyclobutanones.” ACS Catal 2018, Accepted DOI: 10.1021/acscatal.8b04132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Parmar D; Sugiono E; Raja S; Rueping M “Complete Field Guide to Asymmetric BINOL-Phosphate Derived Brensted Acid and Metal Catalysis: History and Classification by Mode of Activation; Brensted Acidity, Hydrogen Bonding, Ion Pairing, and Metal Phosphates.” Chem. Rev 2014, 114, 9047–9153. [DOI] [PubMed] [Google Scholar]
- (20).Reid JP; Goodman JM “Selecting Chiral BINOL-Derived Phosphoric Acid Catalysts: General Model To Identify Steric Features Essential for Enantioselectivity.” Chem. Eur. J 2017, 23, 14248–14260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Copeland GT; Jarvo ER; Miller SJ “Minimal Acylase-like Peptides. Conformational Control of Absolute Stereospecificity.” J. Org. Chem 1998, 63, 6784–6785. [DOI] [PubMed] [Google Scholar]
- (22).(a) Raynes K “Bisquinoline Antimalarials: Their Role in Malaria Chemotherapy.” Int. J. Parasitol 1999, 29, 367–379. [DOI] [PubMed] [Google Scholar]; (b) Kaur K; Jain M; Reddy RP; Jain R“Quinolines and Structurally Related Heterocycles as Antimalarials.” Eur. J. Med. Chem 2010, 45, 3245–3264. [DOI] [PubMed] [Google Scholar]
- (23).(a) Sridharan V; Suryavanshi PA; Menéndez JC “Advances in the Chemistry of Tetrahydroquinolines.” Chem. Rev 2011, 111, 7157–7259. [DOI] [PubMed] [Google Scholar]; (b) Katritzky AR; Rachwal S; Rachwal B “Recent Progress in the Synthesis of 1,2,3,4-Tetrahydroquinolines.” Tetrahedron 1996, 52, 15031–15070. [Google Scholar]
- (24).Compounds 6 and 7 were inseparable in all screened purification conditions.
- (25).Vigneron JP; Dhaenens M; Horeau A “Nouvelle Methode Pour Porter Au Maximum La Purete Optique D’un Produit Partiellement Dedouble sans L’aide D’aucune Substance Chirale.” Tetrahedron 1973, 29, 1055–1059. [Google Scholar]
- (26).Harned AM “From Determination of Enantiopurity to the Construction of Complex Molecules: The Horeau Principle and Its Application in Synthesis.” Tetrahedron 2018, 74, 3797–3841. [Google Scholar]
- (27).Aggarwal VK; Steele RM; Ritmaleni R; Barrell JK; Grayson I “Highly Enantioselective Oxidations of Ketene Dithioacetals Leading to Trans Bis-Sulfoxides.” J. Org. Chem 2003, 68, 4087–4090. [DOI] [PubMed] [Google Scholar]
- (28).Wu MH; Jacobsen EN “Asymmetric Ring Opening of Meso Epoxides with Thiols: Enantiomeric Enrichment Using a Bifunctional Nucleophile An Important Breakthrough in Thiol Addition to Meso.” J. Org. Chem 1998, 63, 5252–5254. [Google Scholar]
- (29).Schreiber SL; Schreiber TS; Smith DB “Reactions That Proceed with a Combination of Enantiotopic Group and Diastereotopic Face Selectivity Can Deliver Products with Very High Enantiomeric Excess: Experimental Support of a Mathematical Model.” J. Am. Chem. Soc 1987, 109, 1525–1529. [Google Scholar]
- (30).See Supporting Information for More Details.
- (31).“Confirmation of the absolute stereochemistry of the product revealed that P1 gives (S) stereochemistry for both 3 and 6, while catalyst 1 produces (R)-3 and (S)-6 with opposite chirality. For assignment of the absolute stereochemistry of the products, see the Supporting Information.
- (32).Utilizing NMI as a catalyst provided no conversion to any products.
- (33).Vedantham R; Vetukuri VPR; Boini A; Khagga M; Bandichhor R “Improved One-Pot Synthesis of Citalopram Diol and Its Conversion to Citalopram.” Org. Process Res. Dev 2013, 17, 798–805. [Google Scholar]
- (34).Alford JS; Abascal NC; Shugrue CR; Colvin SM; Romney DK; Miller SJ “Aspartyl Oxidation Catalysts That Dial In Functional Group Selectivity, along with Regio- and Stereoselectivity.” ACS Cent. Sci 2016, 2, 733–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).(a) Jarvo ER; Copeland GT; Papaioannou N; Bonitatebus PJ; Miller SJ “A Biomimetic Approach to Asymmetric Acyl Transfer Catalysis.” J. Am. Chem. Soc 1999, 121, 11638–11643. [Google Scholar]; (b) Liao R-Z; Santoro S; Gotsev M; Marcelli T; Himo F ACS Catal 2016, 6, 1165–1171. [Google Scholar]
- (36).Sculimbrene BR; Miller SJ “Discovery of a Catalytic Asymmetric Phosphorylation through Selection of a Minimal Kinase Mimic: A Concise Total Synthesis of D-Myo-Inositol-1-Phosphate [14].” J. Am. Chem. Soc 2001, 123, 10125–10126. [DOI] [PubMed] [Google Scholar]
- (37).Fulmer GR; Miller AJM; Sherden NH; Gottlieb HE; Nudelman A; Stoltz BM; Bercaw JE; Goldberg KI “NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist.” Organometallics 2010, 29, 2176–2179. [Google Scholar]
- (38).Merad J; Candy M; Pons JM; Bressy C “Catalytic Enantioselective Desymmetrization of Meso Compounds in Total Synthesis of Natural Products: Towards an Economy of Chiral Reagents.” Synthesis 2017, 49, 1938–1954. [Google Scholar]
- (39).It is difficult to say whether this is the result of site-selective reduction of 8 or chemoselective reduction of 9 to 11 versus 10 to 11.
- (40).Hawkins LR; Bannard RAB “Ammonolysis of 1,2-Epoxycyclohexane and Trans-2-Bromocyclohexanol.” Can. J. Chem 1958, 36, 220–227. [Google Scholar]
- (41).Billington DC; Baker R; Kulagowski JJ; Mawer IM; Vacca JP; Jane S; Huff JR; Sharp M; Point W “The Total Synthesis of Myo-Lnositol Phosphates via Myo-Lnositol Orthoformate.” J. Chem. Soc., Perkin Trans. I 1989, 1423–1429. [Google Scholar]
- (42).Drewe WC; Nanjunda R; Gunaratnam M; Beltran M; Parkinson GN; Reszka AP; Wilson WD; Neidle S “Rational Design of Substituted Diarylureas: A Scaffold for Binding to G-Quadruplex Motifs.” J. Med. Chem 2008, 51, 7751–7767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Liu CY; Pawar VD; Kao JQ; Chen CT “Substitution- and Elimination-Free Phosphorylation of Functionalized Alcohols Catalyzed by Oxidomolybdenum Tetrachloride.” Adv. Synth. Catal 2010, 352, 188–194. [Google Scholar]
- (44).Shaffer CL; Morton MD; Hanzlik RP “N-Dealkylation of an N-Cyclopropylamine by Horseradish Peroxidase. Fate of the Cyclopropyl Group.” J. Am. Chem. Soc 2001, 123, 8502–8508. [DOI] [PubMed] [Google Scholar]
- (45).Lu Q; Yang YT; Chen CS; Davis M; Byrd JC; Etherton MR; Umar A; Chen CS “Zn2+-Chelating Motif-Tethered Short-Chain Fatty Acids as a Novel Class of Histone Deacetylase Inhibitors.” J. Med. Chem 2004, 47, 467–474. [DOI] [PubMed] [Google Scholar]
- (46).Sculimbrene BR; Miller SJ “Discovery of a Catalytic Asymmetric Phosphorylation through Selection of a Minimal Kinase Mimic: A Concise Total Synthesis of D-Myo-Inositol-1-Phosphate.” J. Am. Chem. Soc 2001, 123, 10125–10126. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.