

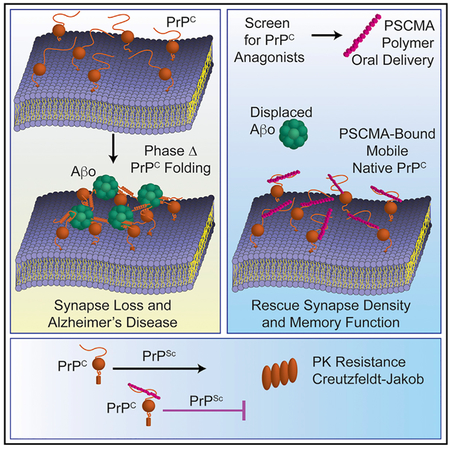
SUMMARY
Cellular prion protein (PrPC) binds the scrapie conformation of PrP (PrPSc) and oligomeric β-amyloid peptide (Aβo) to mediate transmissible spongiform encephalopathy (TSE) and Alzheimer’s disease (AD), respectively. We conducted cellular and biochemical screens for compounds blocking PrPC interaction with Aβo. A polymeric degradant of an antibiotic targets Aβo binding sites on PrPC with low nanomolar affinity and prevents Aβo-induced pathophysiology. We then identified a range of negatively charged polymers with specific PrPC affinity in the low to sub-nanomolar range, from both biological (melanin) and synthetic (poly [4-styrenesulfonic acid-co-maleic acid], PSCMA) origin. Association of PSCMA with PrPC prevents Aβo/PrPC-hydrogel formation, blocks Aβo binding to neurons, and abrogates PrPSc production by ScN2a cells. We show that oral PSCMA yields effective brain concentrations and rescues APPswe/PS1ΔE9 transgenic mice from AD-related synapse loss and memory deficits. Thus, an orally active PrPC-directed polymeric agent provides a potential therapeutic approach to address neurodegeneration in AD and TSE.
In Brief
Gunther et al. search for antagonists for Aβ oligomer binding to PrPC and identify a class of potent polymeric compounds. These molecules bind PrPC and competitively antagonize Aβo action at synapses, while also clearing PrPSc replication from neuroblastoma cells. An orally available PrPC antagonist rescues transgenic mouse Alzheimer phenotypes.
Graphical Abstract
INTRODUCTION
Extensive evidence points to the oligomeric form of β-amyloid peptide (Aβo) as the trigger to initiate Alzheimer’s pathology (Citron et al., 1997; Cleary et al., 2005; Hardy and Selkoe, 2002; Kostylev et al., 2015), but clinical measures to reduce brain Aβ burden have been therapeutically ineffective (Schneider et al., 2014), inspiring exploration for alternate strategies. Discovery that cellular prion protein (PrPC) acts as a high-affinity neuronal receptor required for toxic Aβo signaling (Gimbel et al., 2010; Laurén et al., 2009; Purro et al., 2018; Salazar et al., 2017) has led to the identification of several effectors downstream of Aβo/PrPC interaction, such as mGluR5 (Haas et al., 2014, 2017; Haas and Strittmatter, 2016; Um et al., 2013), Fyn kinase (Kaufman et al., 2015; Smith et al., 2018; Um et al., 2012), and Pyk2 kinase (Haas and Strittmatter, 2016; Kaufman et al., 2015), that can be targeted pharmacologically to rescue the murine brain from AD model pathology. Abrogation of Aβo/PrPC interaction itself in vivo, genetically (Gimbel et al., 2010; Laurén et al., 2009) or with antibodies directed against the Aβo-binding domains on PrPC (Chung et al., 2010; Freir et al., 2011; Klyubin et al., 2014), also reverses synaptic degeneration and restores behavioral performance to impaired AD model mice, even as Aβ load is unaltered. These data indicate the possibility of disease intervention independently of Aβo clearance and identify Aβo/PrPC interaction as an opportune nexus for pharmacological intervention after Aβ accumulation occurs.
Cell-surface PrP is a conformationally diverse protein, originally identified as effecting transmissible spongiform encephalopathy (TSE) via a template-induced proteinase K-resistant form (Colby and Prusiner, 2011). Conversion of PrPC to infectious scrapie (PrPSc) refolds the C-terminal segment, while the toxic action of PrPSc also requires natively unfolded N terminus (Sonati et al., 2013). Recently, we showed that cell surface PrPC engages in phase state changes between soluble, liquid, and hydrogel (Kostylev et al., 2018). The mobile liquid state exists at endogenous PrPC levels present in lipid rafts. The relatively immobile hydrogel phase is induced upon association with multivalent Aβo. Lateral mobility of PrPC in the membrane is restricted upon Aβo association, and mGluR5 is trapped in the hydrogel. Upon Aβo association, the unstructured N-terminal PrP domain adopts an α-helical structure, which coincides with the engagement of mGluR5 and consequent synaptotoxic signaling though Fyn and Pyk2 kinases.
Transgenic APPswe/PS1DE9 mice (APP/PS1) expressing the human mutant amyloid precursor protein (APP) and presenilin 1 (PS1) proteins that cause early onset AD exhibit certain pathological characteristics (Jankowsky et al., 2004). Age-dependent accumulation of Aβo and abundant amyloid plaques, synaptic degeneration, dendritic spine loss, Fyn dysregulation, microglial and astrocytic activation, and multiple memory deficits are among APP/PS1 histopathologies and functional deficits (Brody and Strittmatter, 2018). Because later-stage AD symptoms such as tau tangle accumulation and cell loss are not evident in APP/PS1 and similar strains (Drummond and Wisniewski, 2017), these models may reflect an early AD stage, at which Aβo-directed intervention might have the greatest impact.
Here, we describe competitive antagonists of Aβo/PrPC interaction. These compounds target PrPC Aβo-binding domains, thereby preventing Aβo association with PrPC, Aβo action in vitro, and APP/PS1 phenotypes in vivo. Additionally, these N terminus-directed ligands potently inhibit PrPSc propagation in culture, suggesting efficacy across PrPC-mediated neurodegenerative diseases.
RESULTS
Ceftazidime Degradation Yields a Potent Polymeric Aβo/PrPC Inhibitor Termed Compound “Z”
To search for inhibitors of Aβo/PrPC interaction we engaged in a high throughput cell-based screen using stably PrPC-transfected CV-1 cells. Aβo prepared from biotinylated synthetic Aβ42 peptide associates with these cells in a PrPC-dependent fashion that can be blocked by an antibody (6D11) directed against the Aβo-binding domain at PrPC 90–111 (Figures 1A and 1B). From a screen of 2,560 known drug and 10,130 diverse small molecules, the cephalosporin antibiotic cefixime sample was found to be highly inhibitory. Upon attempted validation, neither fresh cefixime nor a range of cephalosporins was found to possess inhibitory activity, suggesting an impurity or degradation product of cefixime was responsible (compound “X”). To investigate this possibility, five different cephalosporins were allowed to stand in DMSO at 23C for 6 days before re-testing. In addition to cefixime, ceftazidime exhibited activity resulting from prolonged incubation (compound “Z”), while other cephalosporins (cefdinir, cefotaxime, and ceftriaxone) exhibited zero activity either freshly diluted or after 6 days in DMSO (Figure 1C). Inhibitory activity developed progressively from ceftazidime incubated in sodium carbonate at 23°C over 9 days (Figure S1).
Figure 1. Cefixime or Ceftazidime Degradation Produces HMW Inhibitor of Aβo/PrPC Interaction.

(A) Human stably PrPC-transfected CV1 cells treated with biotinylated Aβo (~20 nM Aβo, 1 μM Aβ monomer equivalent) followed by 555-streptavidin exhibit signal that is inhibited by the 6D11 antibody (1 μg/mL) directed against the PrPC 90–111 Aβo-binding domain. Scale bar, 5 μm.
(B) Quantitation of Aβo-binding in (A), background subtracted. Data are mean ± SEM, n = 3 wells. *p < 0.05, Student’s t test.
(C) Aβo/PrPC interaction-inhibiting activity of five cephalosporin antibiotics applied to stably PrPC-transfected CV1 cells followed by treatment with biotinylated Aβo (500 nM monomer equivalent). “Fresh” column: 10 μM drug applied immediately upon dissolution. “Aged” column: 10 μM drug applied 6 days post dissolution. Red circles: acid groups common to cephalosporins that acquire activity post-aging. Scale bar, 5 μm.
(D) Absorbance trace from size exclusion chromatography (SEC) fractionation of aged ceftazidime activity (compound “Z”). MW standards at top of graph.
(E) Aβo/PrPC interaction-inhibitory activity of SEC fractions from (D) as measured by PLISA biochemical assay. Maximal activity resides in the HMW fractions of aged ceftazidime.
Compound Z Binds PrPC with Nanomolar Affinity
Potency and rate of generation from ceftazidime led to a focus on compound Z. Fractionation by size exclusion chromatography (SEC) demonstrated broad high molecular weight of the activity (Figures 1D and 1E), consistent with the reported polymerization of a negatively charged R group degradant of ceftazidime (Baertschi et al., 1997; Ercanli and Boyd, 2006). Both cefixime and ceftazidime possess a negatively charged R group, while other cephalosporins do not, consistent with this group being required (Figure 1C). Elution of Z by high NaCl (100–130 millisiemens) in anion exchange chromatography confirmed that Z is highly negatively charged. Measurement of Aβo-binding inhibitory activity of the 20 kDa Z SEC fraction by PrPC-linked immunosorbent assay (PLISA) (Figure 2B) or PrPC binding affinity by biolayer interferometry (BLI) (Figure 2A), indicated an EC50 of 900 pM and KD of 1.7 nM, respectively. Conversely, Z showed no affinity for Aβo by BLI (Figure 2A), indicating specificity of affinity for PrPC.
Figure 2. Purified Compound Z Binds PrPC Reversibly, Inhibiting Aβo/PrPC Interaction via Competition for PrPC Aβo-Binding Domains.

(A) Biolayer interferometric association (60–180 s) and dissociation (180–300 s) traces of 10–20 kDa Z with PrPC-coated sensor in 4-fold dilution steps from 1 μM top concentration, indicating a dissociation constant of 1.7 nM (top graph). Aβo-coated sensor detects soluble full-length PrPC interaction but not compound Z (middle and bottom graphs, respectively).
(B) PLISA measurement of 10–20 kDa Z Aβo/PrPC inhibitory activity, indicating an IC50 of 910 pM. Data are mean ± SEM, n = 3 replicates per sample.
(C) PrPC immunoblot of non-denaturing gel-shift assay of full-length PrPC incubated with 10–40 kDa Z. Laddering indicates multiple PrPC molecules bound per Z molecule. Incubation at 65°C after co-incubation shows reversible association of Z and PrPC.
(D) Biotinylated 10–20 kDa Z binds full-length PrPC-coated plate concentration-dependently. Data are mean ± SEM, n = 3 replicates per sample.
(E) Binding of biotinylated Z to PrPC-coated plate is inhibited by antibodies directed against either of the two Aβo-binding domains on PrPC and not by antibodies against other regions of PrP, indicating direct and selective Z interaction with PrPC Aβo-binding domains. Data are mean ± SEM, n = 3 replicates per sample. *p <0.05, Student’s t test.
Aβo associates with two lysine-rich domains near the PrPC N terminus: 23–31 and 90–111. To determine whether Z directly associates with these epitopes, we tested whether antibodies against specific PrPC epitopes could inhibit Z binding to PrPC. Biotinylated Z exhibited unaltered PrPC affinity in a plate-based Z-linked immunosorbent assay (ZLISA) (Figure 2D), enabling the evaluation of PrPC-directed agents to compete with Z. Antibodies directed against PrPC in the 23–31 or 90–111 regions were able to block soluble biotin-Z binding to plate-bound PrPC, while antibodies directed against other PrPC domains did not (Figure 2E), consistent with occupation of these sites by Z. PrPC gel shift assay showed laddering of PrPC in the presence of Z that could be reversed by heating to 65°C, indicating non-covalent reversible binding between multivalent Z and PrPC (Figure 2C). Taken together, these data indicate Z is a PrPC-binding reversible competitive antagonist of Aβo/PrPC interaction.
Compound Z Blocks Aβo Action and PrPSc Propagation In Vitro
Functionally, Z blocks numerous metrics of Aβo action (Figure 3). Aβo association with days in vitro (DIV) 19 mouse cortical neuron cultures is reduced by 80% in the presence of Z (Figure 3A). In addition, co-incubation with Z fully blocks Aβo-induced Fyn activation in cortical neuron cultures detected with a phosphospecific anti-Fyn pY416 antibody (Figure 3B). The PrPC-mediated synaptotoxic action of Aβo is evidenced in hippocampal neuronal culture by an 8-fold increase in dendritic spine loss (Figure 3D). Co-administration of 100 nM Z with 1 μM Aβo prevented 92% of Aβo-induced spine loss in hippocampal cultures (Figure 3D). Treatment of DIV 21 hippocampal neurons for 6 hr with a higher concentration of Aβo (3 μM) exerted a neurotoxic action evidenced by LDH release. Aβo-induced LDH release was blocked dose-dependently by Z, with an IC50 of 2 nM (Figure 3C). Given the efficacy of Z with regard to PrPC-mediated AD phenotypes, we sought to determine whether Z could also affect prion propagation underlying TSE. In an scN2A cell culture PrPSc propagation assay, treatment with Z (1 μM) cleared PrPSc infection, as detected by the elimination of proteinase K-resistant PrP (Figure 3E).
Figure 3. Z Blocks Neuronal Action of Aβo and Propagation of Proteinase K-Resistant PrPSc in Cell Culture.

(A) Aβo (1 μM monomer equivalent) binding to DIV 19 mouse hippocampal neurons is blocked by 50 nM 10–20 kDa Z. 80% and 87% of neuronal Aβo binding is inhibited relative to the neuronal markers SV2a and actin, respectively. Scale bar, 10 μM. Data are mean ± SEM, n = 3 wells. **p < 0.01; ***p <0.001 Student’s t test.
(B) Induction of phospho-SFK (Src Family Kinase) in DIV 21 mouse cortical neurons by 30-min application of Aβo (1 μM) is blocked by 10–20 kDa Z (50 nM). Phospho-SFK is normalized to total Fyn that is the predominant neuronal SFK family member activated by Aβo (Um et al., 2012). Data are mean ± SEM, n = 3 wells. *p < 0.05 by one-way ANOVA with Tukey’s multiple comparisons test.
(C) Neurotoxic action of 6 hr Aβo (3 μM) treatment of DIV 21 hippocampal neurons is blocked dose-dependently by 10–20 kDa Z, as indicated by LDH release, with maximal effect reached at 5 nM Z. *p < 0.05 by one-way ANOVA with Tukey’s multiple comparisons test.
(D) Induction of DIV 20 hippocampal neuronal dendritic spine loss by 6 hr application of Aβo (500 nM) is blocked by co-incubation with 10–20 kDa Z (100 nM). *p < 0.05; **p < 0.01 by one-way ANOVA with Tukey’s multiple comparisons test.
(E) Propagation of proteinase K-resistant PrPSc prion in scN2a cell culture is blocked by 6 day application of 10–20 kDa Z (1 μM) as revealed by anti-PrP immunoblot.
Compound Z Rescues Transgenic APP/PS1 Mouse Memory Deficits
Next, we considered whether the in vitro efficacy of Z might be translated in vivo. Spatial memory performance by Morris water maze (MWM) of aged APP/PS1 mice was assessed after 1 month of treatment (Figure 4). The large size and negative charge of Z predicted inefficient transit across the blood-brain barrier such that polymerized compound might need to be administered centrally. Therefore, we delivered Z chronically by intracerebroventricular (ICV) minipump infusion beginning at 12–14 months of age (Figures 4A–4C), when Aβ accumulation and learning deficits are well established (Gimbel et al., 2010; Haas et al., 2016; Kaufman et al., 2015; Kostylev et al., 2015; Salazar et al., 2017; Um et al., 2013). CNS administration rescued mice from phenotypic learning impairment across six twice-daily blocks of four learning trials to a hidden platform (Figure 4A) and during reversal trials to a new location (Figure 4B). Memory performance during a 60-s probe trial performed 24 hr after the learning trials was impaired in vehicle-treated APP/PS1 mice compared to wild-type (WT) and restored by ICV Z treatment (Figure 4C).
Figure 4. Intracerebroventricular Z, but Not Peripherally Administered Fresh Ceftazidime, Rescues APP/PS1 Transgenic AD Model Mice from Memory Pathology.

(A and B) Ceftazidime (as Fortaz) was dissolved at 333 mg/mL in sodium carbonate per manufacturer’s instructions and allowed to stand 14 days at 23°C. Vehicle (veh) or aged Fortaz (Z) containing PrP antagonist activity equal to 2 μM purified 10–20 kDa compound Z was administered intracerebroventricularly (ICV) by minipump to 12- to 14-month-old wild-type (WT) or APP/PS1 (TG) mice at a constant rate of 0.11 μL/hr for 4 weeks before memory assessment by MWM. Assuming the 24 hr dose distributes equally through the brain and is cleared within 24 hr, this yields a 10 nM predicted steady-state Z level. The same dose was continued throughout the behavior testing period via pump exchange. The time to reach the hidden platform during learning of an initial platform location (A) or a subsequent reversed platform location (B) is plotted as a function of swim block. The vehicle-treated APP/PS1 group differed significantly from all other groups by one-way RM-ANOVA over the last eight trials (two blocks) with Tukey’s multiple comparisons test (*p < 0.05), whereas other comparisons were not different (p > 0.05).
(C) A 60-s probe trial test was performed 24 hr after completion of platform reversal training in the MWM. Plotted is the time spent in the area where the platform was previously located (target area). Vehicle-treated APP/PS1 transgenic Alzheimer’s model (TG) mice spent significantly less time in the target area than WT mice, reflecting AD memory deficit. TG mice treated with Z underwent normalization of time spent in the target area relative to WT mice treated with Z, reflecting restored memory function. Data are mean ± SEM of 9–12 mice/group. One-way ANOVA with Tukey’s comparisons as indicated.
(D and E) 100 mg/kg ceftazidime (as Fortaz freshly solubilized at 333 mg/mL in sodium carbonate) or vehicle (veh) was administered by intraperitoneal (IP) injection twice daily for 6 weeks to 12- to 14-month-old wild-type (WT) or APP/PS1 transgenic Alzheimer’s model (TG) mice. Four weeks post-treatment initiation, spatial memory was assayed by Morris water maze (MWM) and plotted as the latency to locate a hidden platform during either the initial (forward) set of training blocks (D) or the set of training blocks post-platform relocation (reversal) (E). Data are mean ± SEM of 10–12 mice/group. Both APP/PS1 groups differed significantly from WT groups by one-way RM-ANOVA over the last eight trials (two blocks) with Tukey’s multiple comparisons test (*p < 0.05), whereas there were no differences within genotype groups (p > 0.05).
We considered the possibility that fresh ceftazidime (as Fortaz) might polymerize in vivo and be distributed across the blood-brain barrier. Intraperitoneal (i.p.) twice-daily administration of fresh non-polymerized ceftazidime (as Fortaz) to APP/PS1 mice had no detectable effect on learning trials of spatial memory testing (Figures 4D and 4E). Similarly, i.p. twice-daily administration of aged 100 mg/kg Fortaz containing prepolymerized active Z did not rescue APP/PS1 memory deficits (not shown). Thus, antagonism of PrPC and effective rescue of APP/PS1 behavioral deficits by Z requires central administration.
Class of Acidic Polymers as Competitive Inhibitors of Aβo/PrPC Interaction
The activities of Z provide proof-of-principle that Aβo/PrPC interaction can be pharmaceutically targeted with non-biologic agents. Because inability to cross the blood-brain barrier constrains translational utility, we undertook an expanded PLISA screen of 56,610 small molecules for Aβo/PrPC inhibitory activity in an effort to identify molecules with greater potential to transit the blood-brain barrier, followed by extensive medicinal chemical optimization of 121 candidates. Although numerous activities were developed, none achieved an IC50 below 1 μM, and thus were deemed insufficiently potent for development.
Consequently, we sought insight from the chemical nature of Z for a directed approach to inhibitor development. A polymeric ceftazidime degradant has been reported to possess anti-HIV activity (Hobi et al., 2001) with a hypothetical structure containing repeating acidic subunits (Baertschi et al., 1997; Ercanli and Boyd, 2006). To explore the composition of Z, we purified active Z from aged ceftazidime by either size exclusion or anion exchange chromatography, followed by replicated elemental analysis. Elemental analysis of Z did not conform precisely to the published predicted structure but does agree with an alternate structure with water and sodium adducts (Figure S2). Both potential structures allow for repeating acidic polar subunits. Based on these features, we tested structurally related polymers to derive a structure-activity relationship (SAR) for activity. Simple polyanionic structures, such as poly-acrylic acid co-maleic acid, were minimally inhibitory, while the inclusion of a hydrophobic moiety, such as in poly (styrene co-maleic acid) partial isobutyl ester, dramatically increased Aβo/PrPC inhibition (Figure 5A). A range of polymers featuring acidic groups proximal to cyclic hydrophobic groups exhibit low nM activity, comparable or superior to Z (Figures 5B–5E), indicating that acidic-hydrophobic polymers are selectively active against PrPC.
Figure 5. Specific Anionic Polymers Are Potent PrPC-Directed Inhibitors of Aβo/PrPC Interaction.

(A) Dilution series of anionic polymer activities were assayed by PLISA. Presence of the hydrophobic phenyl group in polar polystyrene co-maleic acid correlates with the high activity of the polymer. Data are mean ± SEM, n = 3 replicates per sample.
(B) Size-dependence of Aβo/PrPC inhibitory activity was assayed by PLISA, using polystyrene sulfonate polymers of specific average lengths and repeat number. Activity positively correlated with size up to 3.4 kDa, when an IC50 plateau of ~5 nM was reached. Data are mean ± SEM, n = 3 replicates per sample.
(C) Aβo/PrPC inhibitory activity of selected anionic polymers exceed the activity of Z as assayed by PLISA. IC50s = 900 pM, 700 pM and 300 pM for Z, polystyrene co-maleic acid, and poly (4-styrenesulfonic acid-co-maleic acid) (PSCMA), respectively. Data are mean ± SEM, n = 3 replicates per sample.
(D) Known PrPSc inhibitors, pentosan polysulfate and dextran sulfate, exhibit much weaker PLISA activity than identified polar anionic polymers. Data are mean ± SEM, n = 3 replicates per sample.
(E) Table of IC50 and molecular weight (MW) of selected polymers assayed for Aβo/PrPC inhibitory activity by PLISA. MWs represent the weight average MW of a bell-shaped range of polymer sizes. IC50 represents activity based on concentrations calculated using that weight average MW of each polymer sample.
(F) Biolayer interferometric measurement of PSCMA binding to PrPC-coated sensor tip. Association (100–360 s) and dissociation (360–600 s) traces of 3.4 kDa PSCMA in 4-fold dilution steps from 1 μM top concentration, indicated a dissociation constant of 540 pM.
(G) Biolayer interferometric measurement of soluble full-length PrPC binding to Aβo-coated sensor tip in assay buffer alone (left sensorgram) or in the presence of PSCMA (P) (right sensorgram). Association (200–400 s) and dissociation (400–600 s) traces of PrPC in 4-fold dilution steps from 500 nM top concentration, are completely inhibited by PSCMA (1 μM). PSCMA exhibits no affinity for Aβo (100–200 s, right sensorgram) indicating specificity for PrPC.
Given the high affinity of PrPC for these polymers, we considered whether similar biological polymers might exhibit such activity. We evaluated melanin and related molecules for inhibitory activity by PLISA. Neuromelanin produced from 72 hr aqueous degradation of norepinephrine, but not freshly solubilized norepinephrine, exhibited a PLISA IC50 of 19 nM monomer equivalents (not shown). Because the neuromelanin is comprised of numerous subunits originating from monomeric norepinephrine, the activity relative to polymeric neuromelanin MW is likely much more potent than 19 nM. Commercial adrenochrome free base exhibited an IC50 of 700 pM, suggesting pre-existing polymerization (not shown).
The efficacy of specific polymers contrasts with our inability to derive high-affinity small molecule inhibitors and suggests a minimum size or valency. PLISA evaluation of specific molecular weights of polystyrene sulfonate (PSS) indicates 1–10 nM IC50 for species of 17 kDa, 6.8 kDa, and 4.3 kDa, with potency decreasing dramatically at 1.7 kDa or smaller (Figure 5B). We speculate this inhibitor size threshold may relate to the minimum dimension required for simultaneous contact with both Aβo-binding domains at PrP 23–31 and 90–111, either intramolecularly or intermolecularly.
Our hypothesis is that polymers blocking Aβo binding to PrPC do so by the same mechanism as does Z, namely by competitive inhibition upon PrPC binding. We examined this for poly (4-styrenesulfonic acid-co-maleic acid) (PSCMA). Like Z, PSCMA directly binds PrPC, with an observed KD of 540 pM by BLI (Figure 5F). Concordantly, PSCMA affinity for Aβo is undetectable by BLI and, when co-applied with full-length PrPC in solution, completely blocks PrPC binding to an Aβo-coated BLI sensor (Figure 5G).
Next, we evaluated 17 kDa PSCMA or 3.4 kDa polystyrene sulfonate (PSS) activity in cellular assays. Soluble Aβo interaction with cell membrane-associated PrPC is blocked by PSCMA with an IC50 of 3.4 nM in PrPC-transfected COS cells (Figures 6A and 6B) and 32 nM in primary neurons with endogenous PrPC (Figures 6E and 6F). To assess Aβo-induced neuronal phenotypes, we determined dendritic spine density of mouse 14 DIV hippocampal neurons after 4-day exposure to 1 μM Aβo in the presence of PSS. In close agreement with PLISA activity (Figure 5B), PSS completely blocked Aβo-induced spine toxicity at 10 nM or greater concentration, with an EC50 of 1–10 nM (Figures 6G and 6H). Thus, the PrPC-directed polymeric inhibitors effectively inhibit a direct functional consequence of Aβo interaction with neurons.
Figure 6. Active Polymers Block Aβo Neuronal Action and Terminate Pathological PrPSc Propagation at Low nM Concentrations.

(A) PrPC-transfected COS7 cells were treated with 0 or 200 nM PSCMA for 30 min at RT, followed by addition of 1 μM (monomer equivalent) biotinylated Aβo for 1 hr, fixation and staining of PrPC (green), Aβo (red), DAPI (blue) as indicated. Scale bar, 20 μm.
(B) Quantification of Aβo signal for individual PrPC-transfected cells as in (A), expressed as the ratio of Aβo signal to PrPC immunoreactivity. The PSCMA IC50 of 3.4 nM is indicated. n = 3 wells per condition, 10 randomly selected cells per well. Data are mean ± SEM.
(C) SNAP-tagged PrPC-transfected COS7 cells were treated with SNAP-Surface Alexa Fluor647 to fluorescently label cell-surface PrP. Cells were treated with vehicle 1 hr,1 μM Aβo for 1 hr, 1 μM PSCMA for 1 hr, or PSCMA for 15 min followed by Aβo for 1 hr. A 50 μm2 square of fluorescent PrP was bleached with a burst of laser light, and recovery of PrP into the bleached area though lateral PrP diffusion in the plasma membrane monitored over 250 s. Scale bar, 1 μm.
(D) Quantification of recovery in (C). Vehicle and PSCMA treatment fluorescence recovery curves were indistinguishable. Aβo treatment strongly inhibited PrPC recovery kinetics, indicating arrested lateral mobility of PrPC in the plasma membrane as a result of Aβo/PrPC complexation. PSCMA pre-treatment completely prevented Aβo-induced inhibition PrPC recovery kinetics.
(E) 18 DIV mouse hippocampal neurons were treated with 0 or 200 nM polystyrene sulfonate (PSS) for 30 min at RT, followed by addition of 0.5 μM (monomer equivalent) biotinylated Aβo for 1 hr, fixation and staining for the dendritic marker MAP2 (green) and for Aβo (red). Scale bar, 15 μm.
(F) Quantification of total Aβo signal per well for cultures similar to those in (E), measuring PSCMA inhibition of binding 21 DIV rat cortical neurons. The PSCMA IC50 of 32 nM is indicated. n = 3 wells per condition. Data are mean ± SEM.
(G) 14 DIV mouse hippocampal neurons were treated 4 days with vehicle or Aβo (1 μM) +/[C0] the designated concentrations of polystyrene sulfonate (PSS). Scale bar, 15 μm (top), 5 μm (bottom). Aβo induced a consistent reduction in spine density per mm dendrite length.
(H) Quantitation of dendritic spine density from experiments as in (G.) Co-administered PSS blocked Aβo action dose-dependently with an IC50 between 1–10 nM. n = 3 dendrites per neuron, 5–7 neurons per coverslip, 4–8 coverslips per condition. Data are mean ± SEM (***p < 0.001), one-way ANOVA with Dunnett’s multiple comparisons test.
(I) Propagation of proteinase K-resistant PrPSc prion in scN2a cell culture is blocked by 6 day application of PSCMA, with an EC50 between 10–40 nM, as revealed by PrPC immunoblot. Data are mean band densitometry ± SEM, n = 3 replicates per condition. **p < 0.01; ***p < 0.001 relative to veh control, one-way ANOVA with Tukey’s corrected pairwise comparisons.
To investigate the specificity of PSCMA for PrPC as opposed to other Aβo binding sites, we considered that microglial scavenger receptors such as SCARA1 have been reported to act as Aβo receptors. In transfected HEK cell Aβo binding, we tested scavenger receptors related to SCARA1. We found SCARF1 to be a high-affinity receptor with selectivity for Aβo over Aβm, similar to PrPC. However, in contrast to PrPC, SCARF1 binding of Aβo is not PSCMA-sensitive even at 1 mM, 1,000-fold above the IC50 for PrPC (Figure S3). Thus, PSCMA exhibits selectivity for blockade of Aβo binding PrPC.
We recently observed that upon Aβo binding, the lateral movement of PrPC within the plasma membrane is greatly reduced as monitored by fluorescence recovery after photobleaching (FRAP), with a shift from rapid to slow recovery from photobleaching (Kostylev et al., 2018). To investigate the ability of PSCMA to block Aβo-triggered restriction of PrPC in a hydrogel with Aβo, we performed a set of FRAP experiments in COS7 cells transiently transfected with a human PrP/N terminus SNAP tag fusion construct, enabling specific fluorescent labeling of PrPC on the cell surface. In these cells, cell-surface SNAP-Alexa Fluor647-PrPC exhibits rapid lateral translation within the plasma membrane, as indicated by recovery of fluorescent signal within the laser-bleached zone over 250 s. Treatment with 1 μM PSCMA had no measurable effect upon FRAP kinetics compared with vehicle control (Figures 6C and 6D). Critically, PSCMA pre-treatment completely blocked the PrPC immobilizing action of Aβo (Figures 6C and 6D).
Both PSS and PSCMA function like Z with regard to potent inhibition of Aβo binding and to protection of dendritic spines from Aβo-induced loss. We sought to test if PSCMA would also clear PrPSc replication as does Z. PSCMA exhibits pronounced activity in an scN2A cell PrPSc propagation assay, with an IC50 between 10–40 nM (Figure 6I). PSCMA induced no observable toxicity in N2A cells even after 5 days at 5 μM, as indicated by unchanged cell morphology and density in culture. Thus, a common characteristic among pharmacologically active acidic polymers appears to be efficacy against both AD and TSE cellular pathophysiologies.
Peripherally Administered PSCMA Enters the Brain and Rescues APP/PS1 AD Model Mice
The ideal PrPC antagonist would be orally bioavailable and cross the blood-brain barrier. It is not clear that relatively large acidic polymers would have such characteristics. Nevertheless, high-affinity macromolecules (antibodies) targeting PrPC have been peripherally administered and shown to penetrate the brain at sufficient concentration to rescue model mice from AD phenotypes (Chung et al., 2010; Freir et al., 2011; Klyubin et al., 2014). Because the polymers have nM affinities, comparable to anti-PrPC antibodies, we explored the ability of orally administered PSCMA to reach the brain at PrPC-inhibitory concentrations. PSCMA-spiked brain lysate defied mass spectrometry-based efforts of detection, likely due to charge and mass heterogeneity of the polymer. However, phenol-chloroform extraction retrieved activity in a “nucleic acid” fraction due to general chemical similarities between PSCMA and RNA, enabling us to characterize PSCMA-dependent Aβo/PrPC inhibitory activity from brain. Adult mice were treated by oral gavage with 20 kDa PSCMA, followed by assessment of brain lysate for Aβo/PrPC inhibitory activity. Twice daily administration of 40 mg/kg for 10 days yielded brain bioactivity equivalent to 40 nM (Figure 7A). Although this represents only ~2% of the 2 μM concentration expected from a single dose of a perfectly bioavailable compound, it is nearly 100-fold above the PSCMA/PrPC KD of 540 pM (Figure 5F) and sufficiently high to evaluate in a disease model. A 3 mg/kg PSCMA twice daily dose is calculated to achieve brain concentrations 10-fold greater than the PSCMA/PrPC KD. Studies of systemic toxicity by survival, body weight, organ weight, blood chemistry, and hematology revealed no concerns at this dose (Figures S4 and S5). Survival was reduced at 40-fold higher doses.
Figure 7. Oral Administration Yields Active PSCMA in Mouse Brain and Rescues APP/PS1 Memory Deficits and Synaptopathology.

(A) WT mice were administered 40 mg/kg (mpk) PCMA by oral gavage twice daily for 10 days, followed by perfusion, brain lysis, extraction, and PLISA assessment of Aβo/PrPC inhibitory activity. Data are mean ± SEM, n = 6 mice. Standard curve was made by spiking brain lysate from untreated mice with the designated concentrations of PSCMA, prior to identical processing as brains from treated mice. Orally dosed mouse brain contains an average 40 nM PSCMA. Data are mean ± SEM, n = 3 replicates per sample.
(B) APP/PS1 transgenic and wild-type mice were orally gavaged with vehicle (n = 13 transgenic, 15 WT) or 3 mpk PSCMA twice daily (n = 14 transgenic, 14 WT) for a total of 30 days prior to behavioral analysis. After behavioral testing on day 42, PFA-perfused brains were sectioned and immunostained for the pre-synaptic marker SV2A. Representative dentate gyrus confocal images are shown. Scale bar, 10 μm.
(C) Synaptotoxicity-induced 42% reduction in SV2a immunoreactivity in the hippocampi of AD model mice was significantly improved by PSCMA treatment, which restored the hippocampal area occupied by SV2a, as determined by one-way ANOVA with Tukey’s multiple comparisons test (*p < 0.05).
(D) Dentate gyrus sections prepared as in (B) were immunostained for the post-synaptic marker PSD95. Scale bar, 10 μm.
(E) A trend toward PSD95 area reduction was observed in the hippocampus of vehicle-treated transgenic mice compared to WT, but it did not reach statistical significance, as determined by one-way ANOVA and Tukey’s multiple comparisons test. PSCMA reversed this trend.
(F) PSCMA-treated APP/PS1 mice are rescued from learning and memory deficits. After treating for 4 weeks twice daily by oral gavage with PSCMA, mice were evaluated for memory performance by Morris water maze. Latency to find the hidden platform is plotted as a function of trial block number. Statistical differences between groups are indicated in the figure. Data are mean ± SEM (*p < 0.05; ***p < 0.001). One-way repeated-measures ANOVA comparing each group to the transgenic group treated with PSCMA, with Dunnett’s multiple comparisons test.
(G) For the probe trial, percent time spent in the target quadrant after platform removal is indicated. Vehicle-treated transgenic animals spent significantly less time in the target quadrant compared to either drug-treated WT animals or drug-treated transgenic animals. Data are mean ± SEM (*p < 0.05; **p < 0.01). One-way ANOVA with Dunnett’s test comparing to APP/PS1, veh.
The PrPC antagonist, PSCMA, was administered orally to APP/PS1 mice for 30 days at the 3 mg/kg twice daily dose, followed by MWM spatial memory testing. Importantly, treatment began at 12 months of age, after Aβ accumulation, synapse loss, and learning and memory deficits are well established in this strain (Gimbel et al., 2010; Haas et al., 2016; Kaufman et al., 2015; Kostylev et al., 2015; Salazar et al., 2017; Um et al., 2013). This provides a therapeutic disease-modifying regime rather than a prophylactic design. Mice remained on treatment throughout testing and were examined histologically after behavioral analysis (Figures 7B–7G). Transgene-dependent behavioral impairment was evidenced by increased swim latency to the hidden platform by APP/PS1 mice compared to age- and sex-matched WT mice (Figure 7F). Oral PSCMA rescued mice from pre-existing phenotypic learning and memory impairment, as evidenced by a return of APP/PS1 swim latency to a duration indistinguishable from WT, in both forward trials (Figure 7F) and training trials performed after reversal of the submerged platform location (Figure 7F). One day after the final learning swim, a probe trial in the absence of the platform was conducted to test memory for the learned location. The APP/PS1 mice treated with vehicle spent significantly less time in the target quadrant than did APP/PS1 or WT mice treated with PSCMA (Figure 7G), indicating that PSCMA treatment corrected a pre-existing learning and memory deficit in APP/PS1 mice.
To determine whether behavioral recovery reflected repair of transgene-dependent synaptic damage, we examined hippocampal synaptic architecture. The presynaptic marker SV2a exhibited a 42% reduction in area occupied by immunoreactive puncta within the molecular layer of the dentate gyrus of APP/PS1 compared to WT mice (Figures 7B and 7C). PSCMA treatment rescued APP/PS1 mice from synapse loss with restoration of presynaptic SV2a immunoreactivity to WT levels (Figures 7B and 7C). Post-synaptic degeneration detected by PSD-95 staining showed a similar but non-significant statistical trend between transgenic and WT mice, with PSCMA correcting the transgene-dependent deleterious trend (Figures 7D and 7E).
Previous investigations of the role of the Aβo/PrPC axis in AD have shown restoration of learning and memory performance as well as synapse density by PrPC pathway blockade, while Aβ plaque load and neuroinflammatory hallmarks of astrogliosis and microgliosis persisted (Chung et al., 2010; Gimbel et al., 2010; Salazar et al., 2017; Um et al., 2013). Similar to previous PrPC-pathway-directed experiments, the treatment with PSCMA did not alter Aβ plaque area in APP/PS1 mice (Figure S6). In addition, there was no change in the elevated astrocytic GFAP and microglial Iba1 in APP/PS1 animals treated with PSCMA as compared to vehicle (Figure S6). This is consistent with PSCMA selectively blocking PrPC-mediated synaptic effects in APP/PS1 mice, without modulating Aβ metabolism or glial reaction to protein deposition.
DISCUSSION
The PrPC-dependence of the AD phenotype in APP/PS1 transgenic model mice has enabled elucidation of AD signaling pathway components (Brody and Strittmatter, 2018). Antibodies directed against the Aβo-binding domains of PrPC have proven effective preclinically, but no chemical antagonist has yet been developed to target this proximal site of AD induction by Aβo in vivo (Chung et al., 2010; Freir et al., 2011; Klyubin et al., 2014; Purro et al., 2018). Although the large surface area contact of protein-protein interactions are challenging targets for pharmacological intervention with small molecules, the potential experimental and therapeutic utility of a chemical tool for Aβo/PrPC blockade compelled us to search for such agents. High-affinity small molecule inhibitors were not identified during extensive screening and medicinal chemistry efforts. However, higher MW polymers with repeating negatively charged and hydrophobic moieties possess anti-PrPC activity. Our findings show that such polymers can be optimized to bind PrPC with robust affinity, effectively block Aβo binding, and intervene in the AD pathway in vitro and in vivo.
The discovery of compound Z, a polymer that spontaneously forms from degradation products of the cephalosporin antibiotic ceftazidime, revealed these active polymers bind the PrPC N terminus specifically in the two domains mediating high-affinity Aβo interaction. Individual positively charged lysine residues within a given Aβo-binding region of PrPC contribute in an additive fashion to Aβo affinity, potentially via electrostatic interactions with negatively charged domains of Aβo (Kostylev et al., 2018). Avidity effects between bivalent PrPC and multivalent Aβo yield low nM affinity. We posit the charge distribution structure of Z and analogs mimics Aβo, interacting electrostatically with positively charged residues in the Aβo binding domains of PrPC. As a result, Z and its analogs act as potent reversible competitive antagonists of Aβo/PrPC interaction.
We independently identified numerous compounds as active Aβo/PrPC inhibitors that had been previously reported as PrPSc inhibitors, including epigallocatechin gallate, tannic acid, tricyclic antidepressants and antipsychotics, porphyrins, and dyes (unpublished observations). In our hands, the PLISA activity of these molecules was variable and reduced by charcoal filtration or size filtration. All submicromolar-potent hits from a 56,610-compound screen were not reproduced upon resynthesis or charcoal filtration (unpublished observations). This contrasts with the stability and reproducibility of polymeric inhibitors. We hypothesize that these observations derive from a necessity for inhibitor multivalency in order to interact with the two distinct Aβo-binding domains of PrPC. The covalent linkage among charged polymer regions maintains capacity for multivalent association with bivalent PrPC and consequent high avidity. We speculate that nominally “small” molecules acquire the multivalency though non-covalent aggregation, accounting for ephemeral activity. This requirement for multiple charged regions within an inhibitor is evidenced by the three order-of-magnitude difference between the IC50s of 3.4 kDa and 1.7 kDa polystyrene sulfonate. Given the mapping of Z interaction to two Aβo sites on PrPC, as well as the polymer activity size-dependence, it is likely that high avidity requires interaction between two basic Aβo-binding sites on PrPC with two acidic polymer regions, intensified by hydrophobic polymer constituents locally driving polymer to protein affinity.
While active polymers appear to mimic Aβo in binding site and mechanism, they lack agonist activity. Downstream functional consequences of Aβo/PrPC interaction, such as PrPC immobilization at the cell surface, Fyn kinase activation, dendritic spine loss, neurotoxicity, and memory impairment are not induced by the polymers. We tested Z, PSCMA, and PSS for antagonist activity toward multiple consequences of Aβo/PrPC interaction. Upon Aβo binding at the cell surface, PrPC exhibits dramatic lateral mobility inhibition, as evidenced by FRAP. Subsequent dendritic spine retraction and cytotoxicity results from toxic PrPC signaling mediated in part by Fyn activation. Each of these Aβo-induced neuropathologies were completely blocked by Z,PSCMA, or PSS, indicating effective abrogation of the Aβo/PrPC axis by competitive inhibition.
Numerous neurodegenerative diseases have recently been found to involve proteins that undergo liquid-liquid phase transitions (LLPT), including FUS, TIA1, TDP-43, hnRNPA1, and tau (Banani et al., 2017; Hyman et al., 2014; Zhang et al., 2017). The role of the phase states in the pathophysiology of the associated diseases remains an open area of investigation. Previously, no drug-based method of phase change intervention has been developed and shown to influence disease mechanism. Recently, we described PrPC as the first example of cell surface membrane-bound protein to undergo LLPT (Kostylev et al., 2018) Additionally, we found that PSCMA prevents the Aβo-induced conversion of PrPC to hydrogel state and reverses Aβo/PrPC hydrogel once formed, establishing that it is possible to pharmacologically target phase transition of disease-related proteins. Our findings here, that PSCMA blocks Aβo-induced PrPC immobility at the cell surface and rescues APP/PS1 mice from AD model pathophysiology, demonstrates that it is possible to develop phase change-based intervention to address neurodegeneration.
Previous studies have identified polymeric inhibitors of the conversion of PrPC to PrPSc in TSE. Specifically, dextran sulfate sodium (DSS) and pentosan polysulfate (PPS) exhibit anti-prion activity in vitro (Caughey and Raymond, 1993). As with the polymers here, DSS and PPS possess charged and hydrophobic groups, suggesting they may also possess Aβo/PrPC inhibitory activity. Indeed, DSS has been reported to partially inhibit Aβo/PrPC interaction (Aimi et al., 2015). Our evaluation confirmed Aβo/PrPC inhibition by DSS and PPS, but with a potency orders of magnitude weaker than the single nM activity observed for the active polymers reported here. Given the evident similarities but superior potency of the polymers described here, we considered whether they were capable of inhibiting PrPSc propagation in vitro. Both Z and PSCMA blocked PrPSc propagation in scN2A culture, with PSCMA exhibiting an IC50 between 10 and 40 nM to clear PrPSc from neuroblastoma cells. To the extent that the PrPC N terminus Aβo-binding domains mediates interaction with these compounds, inhibition of PrPSc propagation implicates the N terminus domains prion formation. Given our finding that twice-daily oral administration of 40 mg/kg PSCMA yields ~40 nM PSCMA in brain, it will be of interest to investigate PSCMA inhibition of TSE. Because our biochemical assays utilized human PrPC and our functional AD assays were in murine systems, it is clear these polymers act across species. It remains to be tested whether these polymers might have broader strain and species specificity than previously reported anti-PrPSc compounds (Lu et al., 2013).
The non-pathological function of PrPC is an open matter of study. Our finding that neuromelanin potently interferes with PrPC binding to Aβo raises the possibility that a physiological role for PrPC may involve high-affinity interaction with melanin produced endogenously. Given the synaptic presence of PrPC, where high catecholamine concentrations may lead to extracellular melanin formation, this interaction may bear significance in processes yet to be investigated, such as neuromelanin clearance or toxicity mediation.
Blockade of PrPC with PSCMA reversed synaptic loss and memory impairment in APP/PS1 mice even though microgliosis and astrocytosis persisted. While a range of human genetic data implicate microgliosis and microglia proteins such as TREM2 in AD (Guerreiro et al., 2013; Jonsson et al., 2013; Ulrich et al., 2017), this is not inconsistent with the lack of microglial change during rescue with PrPC antagonist. On the one hand, the observations may be explained by an upstream microglial action related to Aβ accumulation or plaque compaction (Condello et al., 2018), which is unaffected by downstream blockade of Aβo/PrPC at postsynaptic sites. Other data suggest that microglia participate in synaptic damage triggered by Aβo (Hong et al., 2016). In this case, the action of Aβo/PrPC complexes may be essential to “tag” damaged synapses for downstream engulfment by microglia. When PrPC/mGluR5 signaling is blocked by PSCMA, subsets of synapses may no longer be marked for destruction by microglia. Confirmation of current results and investigation of potential synaptic mechanisms in additional AD models will aid in establishing the relevance to clinical translation.
It has been shown that PrPC accounts for 50% of punctate neuronal Aβo binding (Laurén et al., 2009). While evidence points to Aβo/PrPC interaction mediating AD synaptopathology, other pathological aspects of AD are not explained by Aβo action though PrPC (Purro et al., 2018; Salazar et al., 2017; Smith and Strittmatter, 2017). As mentioned above, neuroinflammation is unaffected in APP/PS1 transgenic mice by Prnp deletion or by drugs targeting PrP-dependent downstream mechanisms (Haas et al., 2016, 2017; Salazar et al., 2017; Um et al., 2013). The finding that PSCMA and other polymers block 90% of Aβo binding to neurons, but at concentrations greater than the IC50 for Aβo/PrPC inhibition (Figures 3A, 6E, and 6F), suggests that other non-PrPC lower-polymer-affinity neuronal Aβo receptors may also be inhibited by PSCMA. These receptors remain to be identified, but apparently share a common Aβo binding mechanism with PrPC, which is similarly inhibited by these polymers. At the low PSCMA dose we achieved in vivo during treatment of APP/PS1 mice, it is unlikely that alternate low-affinity receptors were inhibited. Clearly, not all Aβo receptors are sensitive to PSCMA, because we found that the scavenger receptor SCARF1 binds Aβo with high affinity but, in contrast to PrPC, is not PSCMA-sensitive. The essentially complete extinction of Aβo binding to neurons, while binding to microglial SCARF1 is unaffected, demonstrates a cell-type specificity for PSCMA action and suggests neuronally mediated APP/PS1 phenotype rescue. Further experimentation will be necessary to determine whether PSCMA action is limited to neurons or also has a beneficial role in blocking Aβo disruption of vascular function or blood-brain barrier integrity via endothelial PrPC.
The development of acidic polymeric inhibitors of Aβo/PrPC interaction provides a tool for investigation of AD and demonstrates the feasibility of pharmaceutically intervening with a compound directed at this mechanism of AD etiology. Although these high-affinity PrP-interactors share characteristics with Aβo, such as multivalency, rapid on- and slow off-rate kinetics, and dual PrPC binding-domain specificity, they do not share the toxic pathway-inducing activity of Aβo, and thus act as potent competitive inhibitors of Aβo function. Rescue of AD model mice with oral administration suggests that these compounds, or compounds similar to them, could provide a therapeutically tractable strategy at early stages of AD, when the role of Aβ accumulation in disease progression is most clear. The inhibition of PrPSc propagation as well suggests that a single PrPC-targeted drug might address both AD and TSE pathologies, and that targeting phase transitions will be therapeutically effective in neurodegenerative conditions.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Stephen Strittmatter (stephen.strittmatter@yale.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Transgenic and control mouse strains
Mice were cared for by the Yale Animal Resource Center and all experiments were approved by Yale’s Institutional Animal Care and Use Committee and performed in accordance with the American Association for Accreditation of Laboratory Animal Care (AAALAC). Wild-type and APPswe/PS1DE9 mice (APP/PS1) (Jankowsky et al., 2003, 2004) were purchased from Jackson Laboratory and maintained on a C57/Bl6J background as described previously (Gimbel et al., 2010; Um et al., 2012, 2013). All experiments were conducted in a blinded fashion with respect to genotype and treatment, and groups were matched for age and sex, and groups contained 45%–55% of each sex.
METHOD DETAILS
Aßo preparation
Biotinylated and unlabeled synthetic Aβ1–42 peptide are obtained as lyophilized powder from The ERI Amyloid Laboratory, LLC (Oxford, CT). Preparation and characterization of Aβ1–42 oligomers (Aβo) have been described previously (Um et al., 2012). Aβ monomer is dissolved at 10 mg/ml in HFIP and boiled in water bath 1 h at 70C, then cooled on ice, transferred to 2 mL microfuge and tubes spun 7 min at 12,000 × g. Avoiding pellet, 50 μl (0.5 mg) is aliquoted in a 1.6 mL microfuge tube and allowed to evaporate completely in chemical hood (24 h), followed by a minimum of 1 h in a speed vac. An observable clear film surrounds the inside tip of the tube. Closed tubes are stored at RT for later oligomer preparation. To prepare oligomers, add 40 μl DMSO to tube, allow to stand 20 min with occasional flicking/trituration to suspend peptide, wiping down tube sides to insure no un-dissolved peptide remains. Aliquot 20 μl/1.6 mL microfuge tube. Add 1 mL phenol red-free F12 (Atlanta Biologicals cat # M15350) to tubes for 0.25 mg/ml final conc. Let stand O/N at RT. Spin 15 min at 14000 rpm - there is usually no pellet; a large pellet indicates an unsuccessful prep. Aliquots can be frozen for later use. Binding assays show consistent results over at least 72 h post F12 addition. Concentrations of Aβo are expressed in monomer equivalents, with 1 μM total Aβ1–42 peptide corresponding to approximately 10 nM oligomeric species (Laurén et al., 2009).
Cell-based Screen for small molecule inhibitors of Aßo/PrPC interaction
CV1 cells stably transfected with rat PrP were plated in 96 well tissue culture plates (Corning, 354461) 24 h prior to application of small molecule library components dissolved at 10 mM in DMSO (10 μM final concentration) for 1 h prior to addition of biotinylated Aßo (1 μM final concentration). Wells were fixed 25 min in 4% paraformaldehyde, washed twice with PBS, blocked 1 h in PBS containing 5% goat serum (GIBCO, 16210–064). 50 μl 1:1000 Eu-labeled streptavidin (PerkinElmer, 1244–360) in DELFIA assay buffer (PerkinElmer Life Sciences) was added per well for 30 min. After washing five times in PBST, 50 μL of DELFIA Enhancement Solution (PerkinElmer Life Sciences) was applied to each well, and time-resolved europium fluorescence was measured using a Victor 3 plate reader (PerkinElmer Life Sciences). Compounds were assayed in singlet, with hits defined as inhibition exceeding 50% of Aßo-dependent signal, with assay plates exhibiting a Z’ over 0.5 relative to anti-PrP positive control antibody 6D11 (Covance). Hits were validated at 10 μM in triplicate in a similar assay, with visual inspection of each treatment for cell toxicity and detachment. Compounds inducing obvious cellular toxicity were eliminated from further study. Non-toxic active compounds were subjected to dose-response evaluation in triplicate in the same cell-based format. Libraries screened were Enzo FDA Approved Drugs Library (Enzo), Microsource Pharm 1600 (Microsource) and Yale Small Molecule Discovery Center compound collection.
Immunocytochemistry
COS-7 cells were maintained in DMEM supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. COS-7 cells transfected 2 days earlier with PrPC expression vector were pre-incubated with inhibitor in F12 media at 22°C for 30 min. Biotin–Aβ42 oligomers were added to a concentration of 500 nM monomer equivalent for 2 h. Cells were washed twice with PBS, fixed 25 min with 4% paraformaldehyde in PBS, washed twice with PBS, blocked 1 h with 5% goat serum in PBS, incubated in 0.1% 488-streptavidin (Life technologies, S11223) in PBS 1 h, washed twice and visualized on an ImageExpress Micro (Molecular Devices). Images were analyzed using ImageJ. Alternatively, higher resolution images were capture from cells cultured on poly-lysine coated glass with a Zeiss confocal microscope.
Size-exclusion Chomatography (SEC)
Aged ceftazidime (as 14-day reconstituted Fortaz) was separated on a Superdex 75 10/300 GL gel filtration column (GE Healthcare Bio-Sciences) using AKTA purifier FPLC system (GE Healthcare). 200 μl of sample was injected at a flow rate of 0.75 ml/min. PBS, pH 7.4, was used as a mobile phase. Fractions of 0.5 mL were continuously collected thoughout the run and analyzed for PLISA activity or selected for Z quantitation.
Z concentration measurement
The SEC fraction of aged Fortaz corresponding to 15 kDa was collected, exchanged into water though extensive washes with a 3 kDa filter (Amicon, UFC500396), and desiccated. Weighed material (a light brown powder) was resolubilized in PBS and absorbance determined at 280 nm to determine concentration.
Anion-exchange Chomatography
Aged ceftazidime (as 14-day 440 mM reconstituted Fortaz in sodium carbonate) was separated on an XK 50/20 column (GE Healthcare Life Sciences) packed with Q Sepharose Fast Flow (GE Healthcare, 17-0510-01) with a 0.5–2.0 M NaCl gradient. Fractions eluting at 100–120 millisiemens were used as Z.
Biolayer Interferometry
Biotinylated Aßo or full-length human PrPC biotinylated by incubation in 2-fold molar excess biotin-NHS (Thermo Scientific, 21329) 2 h in PBS at RT were coated onto streptavidin Biosensors (ForteBio, 18–5019) and exposed to dilution series of solutes as described in Figures 2A, 5F, and 5G in PBS with 0.05% Tween 20 (Sigma, P7949) and 0.1% BSA. Association curves were detected and analyzed using an Octet biolayer interferometer (ForteBio).
Blue Native PAGE shift assay
Recombinant human PrPC (1 μM) was incubated in the presence or absence of 2 μM Z for 10 min in 0.25X PBS at room temperature. Following the incubation, the samples were loaded on 4%–16% NOVEX Bis-Tris gel (ThermoFisher) and separated according to manufacturer’s recommendations. To demonstrate the requirement of native PrPC structure for complexation with compound Z, some samples were heated in 65C heatblock for 10 minutes prior to BN-PAGE separation to denature the protein. Following the BN-PAGE, the gels were transferred onto PVDF membranes using iBlot semi-dry transfer (ThermoFisher), the membranes were dried and the excess of Coomassie dye was removed by washing the membrane in methanol thee times for 5 min to avoid interference with subsequent immunoblotting. The membranes were then washed 3X with water, blocked using fluorescent western blot blocking buffer (Rockland) and immunoblotted with 1:500 SAF-32 mouse anti-PrPC antibody (Cayman Chemical) in TBS-T followed by 800CW-conjugated donkey anti-mouse secondary antibody (Li-Cor). Immunoblots were imaged using Li-Cor Odyssey near-infrared scanner.
Assay for Aßo binding to immobilized PrPC (PLISA)
MaxiSorp 384 well white microplates (ThermoFisher Scientific, 460372) were coated overnight with 20 μl/well of 250 nM human full length PrPC in 30 mM Na2CO3, 80 mM NaHCO3, pH 9.6, at 4°C. After washing two times with PBST (PBS, 0.05% Tween 20), the plates were blocked with 100 μl/well protein-free T20 PBS blocking buffer (Pierce, 37573) for 1 h at 23C. After thee PBST washes, 20 μl samples diluted in PBST (PBS, 0.05% Tween 20) were applied to microplates in triplicate and incubated 1 h at RT. 20 μl of synthetic biotinylated Aβo in PBSTB (5 nM monomer equivalent) was added for 2 h. Plates were then washed four times with PBST and incubated 1 h with 20 μl 1:1000 Eu-labeled streptavidin (PerkinElmer, 1244–360) in DELFIA assay buffer (PerkinElmer Life Sciences). Finally, after washing five times in PBST, 20 μL of DELFIA Enhancement Solution (PerkinElmer Life Sciences) was applied, and time-resolved europium fluorescence measured using a Victor 3 plate reader (PerkinElmer Life Sciences).
Biochemical small molecule screen
PLISA was used to screen 56,610 unique compounds not included in the initial cell-based screen. Libraries screened were MicroSource GenPlus (Microsource), Yale compound collection, and ChemDiv Diversity Library (ChemDiv). Small molecule library components are stored dissolved at 10 mM in DMSO (10 μM final concentration) and added in singlet to PrP-coated wells of a 384-well plate containing PBST for a final concentration of 10 μM. After 30 min, PBST containing biotinylated Aβo was added for a final Aβo concentration of 5 nM, incubated @ RT 2 h and developed per PLISA protocol. Hits exceeding 50% signal inhibition were evaluated further.
Z-binding PrP-epitope mapping
A MaxiSorp 384 well white microplate (ThermoFisher Scientific, 460372) was coated overnight with 20 μl/well of 250 nM human full length PrPC in 30 mM Na2CO3, 80 mM NaHCO3, pH 9.6, at 4°C. After two PBST washes, the plates were blocked with 100 μl/well protein-free T20 PBS blocking buffer (Pierce, 37573) for 1 h at 23°C. After 3 PBST washes, 20 μL of anti-PrP antibodies (8B4, Santa Cruz Biotech, sc-47729; 5058, Millipore Sigma, AB5058; 8G8, Cayman chemical,189760; 6D11, Covance, SIG-399810; Pri 308, Cayman Chemical, 189750; 8H4, abcam, ab61409; SAF70, Cayman Chemical, 189770) diluted 1:50 in PBST were applied to wells in triplicate and incubated 1 h at RT. Twenty μl of 50 nM Z biotinylated by incubation in 10-fold molar excess biotin-NHS (Thermo Scientific, 21329) 2 h in PBS at RT was added per well for 2 h. Plates were washed four times with PBST and incubated 1 h with 20 μl 1:1000 Eu-labeled streptavidin (PerkinElmer, 1244–360) in DELFIA assay buffer (PerkinElmer Life Sciences). Finally, after 3 PBST washes, 20 μL of DELFIA Enhancement Solution (PerkinElmer Life Sciences) was applied, and time-resolved europium fluorescence \ measured (Victor 3V PerkinElmer Life Sciences).
Melanin Synthesis
Norepinephine bitartrate salt (Sigma Aldrich A0937) was dissolved at 50 mg/ml in H2O, brought to pH 8.5 with ammonium hydroxide and allowed to stand at RT at least 72 h, forming a black solution.
PrPSc Propagation Assay
Chonically PrPSc-infected scN2a cells, RML strain, were cultured in Delbucco’s Modified Eagle Medium (DMEM) with L-glutamine and 4.5 g/L glucose plus 10% fetal bovine serum (FBS) and 50 U/ml penicillin, 50 μg/ml streptomycin. 10 mM and 5 mM stock solutions of compounds Z and PSCMA, respectively, were prepared in PBS and stored at 4°C for no longer than 1 week prior to use. Compound stock solutions or PBS alone (vehicle) were added to culture medium and working concentrations obtained via serial dilution. Trypsinized ScN2a cells were split 1:10 and allowed to adhere to plates in compound-free media for 12 h, at which point medium containing the treatment compound was added. Cells were grown for 3 days to confluence with a media exchange at 36 h. Cells were trypsinized, split 1:10 and again allowed to adhere in compound-free media for 12 h, and then returned to compound-containing medium for an additional 3 days prior to processing. Cells were lysed in ice-cold lysis buffer (10 mM Tris pH 7.5, 150 mM NaCl, 0.5% w/v Na deoxycholate, 0.5% v/v NP-40). Lysate was centrifuged at 2,100 × g for 30 s to pellet DNA. 10% of the resulting supernatant was added to an equal volume of 2x SDS-PAGE loading buffer, boiled at 95°C for 10 min and saved as the minus proteinase K (–PK) sample. To the remaining supernatant, PK was added to a final concentration of 20 μg/ml and samples were digested shaking at 37°C for 30 min prior to quenching with the addition of phenylmethylsulfonyl fluoride (PMSF) to a final concentration of 5 mM. PK-digested samples were centrifuged at 100,000 × g for 1 hour at 4°C and the pellet was resuspended in equal volumes of lysis buffer and 2x SDS-PAGE loading buffer prior to boiling and analysis via SDS-PAGE and western blot. Western blotting was carried out with GE8 primary antibody at 1:2000 and HP-conjugated sheep anti-mouse secondary antibody at 1:5000.
Immunoblots
Proteins were electrophoresed though precast 4%–20% tris-glycine gels (Bio-Rad) and transferred with an iBlotTM Gel Transfer Device (Novex-Life Technologies) onto nitrocellulose membranes (Invitrogen). Membranes were blocked in blocking buffer for fluorescent western blotting (Rockland MB-070–010) for 1 hour at room temperature and incubated overnight in primary antibodies at 4°C. The following primary antibodies were used: anti-Fyn (Cell Signaling Technology 4023; 1:1,000), anti-phospho-Src (Cell Signaling Technology 2101; 1:1,000). appropriate secondary antibodies were applied for 1 h at room temperature (Odyssey donkey anti-mouse or donkey anti-rabbit conjugated to IRDye 680 or IRDye 800, LI-COR Biosciences) and proteins were visualized with a LI-COR Odyssey infrared imaging system. Quantification of band intensities was performed within a linear range of exposure.
Neuronal culture and Aßo binding
Brain cortices and hippocampi were dissected from embryonic day 13 pups removed from CO2-euthanized pregnant C57/Bl6 mice, dissociated by incubating in 0.25% trypsin 10 min at 37C, followed by gentle trituration in Neurobasal A medium supplemented with 2% B27, 1% Glutamax, 1% sodium pyruvate, 1% pen/strep and 0.2% FBS, filtration though a 40 um filter and plated at 30,000 cells/well in a polylysine-coated 96 well plate. After DIV 14, wells were treated with Z or PSCMA at the specified concentrations 30 min by adding to the conditioned media, followed by addition of biotinylated Aßo for 2 h, after which cells were washed once with PBS, fixed 25 min in 4% formaldehyde in PBS, washed 3 times with PBS, blocked 1 h in PBST with 5% goat serum, incubated overnight at 4°C with designated antibody in PBST (SV2a, Abcam 32942, 1:250; NeuN, Millipore, mab377, 1:500 or actin, Cell Signaling Technology, 4967S, 1:500), followed by washing twice with PBS and incubating 2 h in cognate secondary antibodies, DAPI and 0.1% 555-streptavidin (Invitrogen, S32355) in PBST, followed by washing twice in PBS and imaging with an ImagExpress (Molecular Devices). Signal was quantitated with ImageJ. Alternatively, stained neurons cultured on poly-D-lysine-coated glass were imaged with a Nikon Eclipse Ti Spinning Disk Confocal Microscope.
Cell Transfection and Aßo Staining
Human embryonic kidney cells (HEK293) were plated in 24-well plates and transfected with full length human PrPC or human SCARF1 (Transomic, clone ID pCS6(BC039735)) using lipofectamine 3000 transfection reagent. Thee days post transfection, cells in conditioned media were treated with 1 μM PSCMA or PBS vehicle for 30 min prior to addition of biotinylated Aßo at final 1 μM monomer equivalent concentration for an additional 2 h. Cells were washed 1 x with PBS, fixed 25 min in 4% formaldehyde, washed 3x in PBS, blocked 1 h in 5% normal goat serum, incubated 1 h in 488-streptavidin (Molecular Probes), washed 3x in PBS and imaged with an ImageXpress Micro XLS Widefield High-Content Analysis System (Molecular Devices).
Imaging of Dendritic Spine Stability (Z)
Hippocampal neurons of various genotype were obtained from E17–19 mouse embryos (Um et al., 2012). After hippocampal digestion with papain (37°C; 5% CO2 for 30 min), the neurons were transfected with myristoyl-GFP expression vector by Amaxa Nucleofector. Cells were plated at 100,000 cells per well on poly-D-lysine-coated glass 8 well plates (Lab-Tek Chambered Coverslip 155411). The culture medium was Neurobasal A supplemented with 1X penicillin/streptomycin, 1 mM Na-pyruvate, 2 mM GlutaMax, and B27 supplement with weekly replenishment. After 19–23 DIV, neurons were imaged with a 100X objective on a Nikon Eclipse Ti Spinning Disk Confocal Microscope using a 488 laser. A 10 μm Z stack at 0.1 μm intervals was obtained every 15 min over 6 hours from multiple fixed locations per 8-well dish with an automated stage. 500 nM Aß oligomer or F12 vehicle control were added after one hour of imaging and additional images collected over 5 hours. In some conditions, 50 nM Z or drug vehicle control were added immediately before Aß or vehicle. Spine number in consecutive images for specific dendritic segments was measured using ImageJ software without knowledge of drug or genotype. For each condition, at least 4 segments with 30 spines at time zero were assessed.
LDH release
Cell toxicity was quantitatively assessed by the measurement of Lactose dehydrogenase (LDH) activity in the medium. LDH activity in the culture medium was measured by Cytotoxicity Detection Kit (Roche) according to manufacturer’s procedure. In brief, 60 μL of supernatant from each well was transferred to a 96 well plate and 60 μL of reconstituted substrate solution was added to each well and then the plates were incubated for 30 min. Total LDH release was achieved by adding 2% Triton X-100 solution to untreated control cells. The absorbance of the samples was measured at 490 nm using a VictorX3 Multilabel Plate Reader (PerkinElmer). The values were expressed as a percent of the total LDH release.
Animal Treatment
For Morris water maze, mice were randomly assigned to treatment groups and the experimenter was unaware of both genotype and treatment group. Groups were balanced for age, sex, and weight. Mice used were 12–14 months of age at experiment initiation. During treatment, the experimenter was blinded to genotype. For drug treated mice, Poly(4-styrenesulfonic acid co-maleic acid) was administered by twice daily oral gavage of 5.0 mg and 15.0 mg Poly(4-styrenesulfonic acid co-maleic acid) per kg body weight in a vehicle of 0.5% w/v hydroxypropyl methylcellulose and 0.1% w/v polysorbate 80 to APP/PS1 and WT mice, respectively. Vehicle treated animals were gavaged twice daily with vehicle. 500 mg ceftazidime as Fortaz was dissolved per manufacturer’s instructions in 1.5 mL sterile milliQ H2O to obtain 333 mg/ml in sodium carbonate solution. Animals treated peripherally with fresh ceftazidime were injected directly after dissolution intraperitoneally with 100 mg ceftazidime per kg body weight in a vehicle of PBS. Animals treated centrally with aged ceftazidime were fitted with an intracerebroventricular cannula (Alzet brain infusion kit 0008663) and subcutaneous osmotic minipump (Alzet model 1004) loaded with aged ceftazidime diluted in PBS. All animals were treated for 4 weeks prior to the Morris water maze and thoughout assessment.
Behavioral Testing
Morris water maze was performed as previously described (Morris, 1984; Smith et al., 2018). Thoughout experimentation, the experimenter was blinded to treatment group, and genotype. Each animal was handled by the experimenter for five minutes each day for thee consecutive days preceding the initiation of behavioral experiments to minimize animal stress on testing days. The testing pool was ~1 m in diameter with four unique spatial cues placed evenly around the perimeter. For learning swims, a clear plastic platform was submerged 1 cm below the surface of the water and fixed to the bottom of the pool in the target quadrant. For each swim a mouse was placed in the water facing the pool wall opposite the target quadrant in one of four positions. The sequence of the entry positions was changed for each of the six trial blocks.
For a single trial block, each animal was swum four times. For each swim mice were given 60 s to locate the hidden platform. Mice were given a 60 s rest interval with access to a heating lamp between swims. Trial blocks were initiated every 12 hours over thee consecutive days for a total of 6 trial blocks. If a mouse failed to locate the hidden platform in the allotted time during trial blocks one or two, the mouse was gently guided to the platform and placed there for 15 s. The reverse swim began the day after completion of the forward swims and followed the same protocol with the hidden platform placed in the quadrant opposite that of the forward swims.
Twenty-four hours after the last learning trial block of the reverse swim, the platform was removed from the pool for the probe trial. During the probe trial, each animal was placed in the pool once and allowed to freely swim for 60 s. For all swims animals were tracked using SMART 3.0 software (Panlab, S.L. - Harvard Apparatus, Inc, Holliston, MA) with a JVC Everio G-series camcorder (Yokohama, Japan).
To account for differences in visual acuity, a marker that extended above the surface of the water was placed on the platform and mice we placed in the water facing the wall opposite the platform. Latency to reach the visible platform was recorded and any animals that did not reach the platform within two standard deviations of the mean were excluded from analysis. For analysis of the learning swims, a single mouse’s latency to find the platform was averaged across four swims to generate a trial average.
Fluorescence Recovery After Photobleaching (FRAP)
COS-7 green monkey kidney cells (ATCC® CRL-1651) were passaged in high-glucose DMEM (ThermoFisher, 11965092) supplemented with sodium pyruvate, 10% Fetal Bovine Serum and Pen/Strep antibiotic mix. Trypsinized cells were seeded in 8-well chambered sterile coverglass slides (ThermoFisher, 155411) at 10000 cells/well in 250 μl of complete growth medium and cultured overnight. The following day, the cells were transfected with a total of 200 ng DNA/well using Lipofectamine 3000 lipid transfection reagent (ThermoFisher, L3000015) according to manufacturer’s protocol.
For fluorescent labeling, cells expressing SNAP-PrP were incubated with 500 nM SNAP-Surface Alexa Fluor647 in complete medium for 30 min at 37°C. Cells were washed twice with PBS supplemented with calcium and magnesium (Sigma, D8662) to remove the excess labeling fluorophores and then incubated for 15 min in PBSCa,Mg with 1 μM PSCMA or PBSCa,Mg alone as a control. Aβo or PBS (vehicle) was subsequently applied to 1 μM final concentration for 1 h at 37°C and cells were then imaged at room temperature.
All FRAP experiments were performed on UltraVIEW VoX (Perkin Elmer) SDC microscope equipped with PhotoKinesis FRAP unit using 60x oil immersion objective. Images were collected every second for 7 s. before bleaching to measure the baseline fluorescence. Following the bleaching cycle, the imaging was performed with 1 s intervals for the first 30 s and with 4 s intervals for an additional 220 s. 640 nm laser was used for selective photobleaching of SNAP-PrP conjugated with Alexa 647. All the imaging was performed in the apical membrane of the cells and at least thee 2×2 μm areas per cell were bleached to average the intrinsic variability in the protein mobility between the regions of the plasma membrane. Quantitation of fluorescence recovery was performed in Volocity software (PerkinElmer).
Immunohistology
Mice were euthanized by CO2 asphyxiation, perfused with cold PBS and brains were dissected and post-fixed in 4% paraformaldehyde for 72 h at 4°C. Brains were sliced into 40 μm coronal brain sections using a Leica WT1000S vibratome. Sections were permeabilized in PBS + 0.1% Triton X-100 for 15 min. All slices underwent an antigen retrieval step prior to exposure to primary antibody by incubating slices in 1x Reveal Decloaker buffer (RV1000M, Biocare Medical) for 15 min at 90C in an oven. After antigen retrieval, sections were blocked in 10% normal horse serum (Jackson ImmunoResarch Laboratories) in PBS for 1 hour at room temperature and then incubated with primary antibodies for 24 hours at 4C. The following primary antibodies were used: anti-GFAP (glial fibrillary acidic protein; Abcam ab4674; 1:500), anti-Iba1 (ionized calcium-binding adaptor molecule 1; Wako 019–19741; 1:250), anti-PSD95 (postsynaptic density protein 95; Invitrogen 51–6900; 1:250), and anti-SV2a (synaptic vesicle glycoprotein 2A; Abcam 32942; 1:250). Sections were washed 3 times in PBS and incubated with secondary antibodies (donkey anti-rabbit or donkey anti-chicken fluorescent antibodies; Invitrogen Alexa Fluor; 1:500) for 1 hour at room temperature. After 3 washes in PBS, the sections were mounted onto glass slides (Superfrost Plus, Fisher Scientific) and coverslipped with Vectashield (Vector Laboratories H-1200) antifade aqueous mounting medium.
Imaging and analysis of immunohistochemistry
For imaging of synapse density stained by anti-SV2a and anti-PSD95 antibodies, a Zeiss 800 confocal microscope with a 63X 1.4 NA oil-immersion lens was used. The area occupied by immunoreactive synaptic puncta from the molecular layer of the dentate gyrus was measured as described previously (Gimbel et al., 2010). For imaging and analysis of the tissue stained for Iba1 and GFAP, a Zeiss 800 confocal microscope with a 20X 0.3 NA air-objective lens was used and a full tiled z stack of the hippocampus was taken. β-amyloid plaque load was imaged on a Zeiss AxioImager Z1 fluorescent microscope with a 4X air-objective lens. ImageJ software was used for quantification of image area containing tissue only, excluding image area not containing tissue. Statistical analysis was based on separate mice.
Thioflavin S Aß plaque staining
60 μm sections were incubated in pre-heated 10 mM sodium citrate with 0.05% Tween 20, pH 6 at 95°C for 1 hour, rinsed twice with PBST and blocked with 10% donkey serum (Jackson ImmunoResearch 017-000-121) for 1 hour. Next, sections were incubated in 0.1% Thioflavin S (Sigma T1892) in 70% ethanol at room temperature for 15 min, washed twice with 70% ethanol, then twice with distilled water. Images of cortical Thioflavin S staining were collected from thee slices for each animal and quantified using ImageJ. Thee values for a single animal were averaged and graphed as a single data point per animal.
Mouse pharmacokinetic (PK) study
Brain penetration of 20 kDa PSCMA (Sigma, 434566) was characterized in six male C57BL/6 mice. Mice received drug at 40 mg/kg as a solution in 95% PEG400/5% Solutol (dose volume = 5 mL/kg) by oral gavage. After 10 days of treatment, mice were euthanized by CO2 asphyxiation, perfused with ice-cold PBS for 60 s, and brains were rapidly dissected. Whole brains were Dounce homogenized in 1:10 w:v brain:PBS, followed by polyanion extraction using TRIzol reagent (Invitrogen, 15596026). 1 mL trizol per 100 mg tissue was added, vortexed, 0.2 mL chloroform per ml TRIzol added, vortexed, centrifuged 15 min at 12,000 × g, aqueous phase transferred to a new tube, 0.5 mL isopropanol added per ml TRIzol used for lysis, incubated 15 min, centrifuged at 12,000 × g, supernatant retrieved leaving RNA pellet, supernatant speed vacuumed overnight, resuspended in H2O and purified using Oasis® WAX cartridges (Waters, 186002489). Eluted extracted PSCMA was assayed for Aßo/PrPC inhibitory activity by PrP-ELISA or PLISA (Kostylev et al., 2015) using similarly Dounce homogenized brain, spiked with varying concentrations of PSCMA followed by extraction, to establish a standard curve.
Toxicological Assessment of PSCMA
For the survival study, WT mice were orally gavaged with 120 mg/kg, 40 mg/kg, 2.4 mg/kg, or vehicle twice daily for 10 days. For the tissue sampling study, groups of 12-month-old WT mice balanced for sex and weight were orally gavaged with 15 mg/kg, 3 mg/kg, or vehicle twice daily for 1 week. All group received the same volume of vehicle per gram of body weight. After treatment, animals were sacrificed and whole blood was collected via cardiac puncture using a 21 gauge needle, and aliquoted into a plasma separator tube (BD Microtainer 365985) and a K2 EDTA tube (BD Microtainer 365974). Plasma separator tubes were centrifuged at 6,000 × g for 90 s. Following blood collection, heart, liver, spleen, and left kidney were removed and weighed. Blood samples were sent to ANTECH Diagnostics for complete blood counts and chemistry panels.
Analysis of Dendritic Spine Density (PSS)
For Aßo-induced spine loss, Aßo (1 μM monomer, 10 nM oligomer), vehicle (veh), Aßo + PSS, or PSS alone were applied at the designated dose to GFP transfected neurons at DIV 17, replacing 50% culture medium with fresh Aßo + veh, Aßo + PSS, or Veh-containing conditioned culture medium every 24 hours for 4 days thereafter. Neurons were fixed and imaged with a 40X objective oil lens on a Nikon Eclipse Ti Spinning Disk Confocal Microscope driven by Volocity software (PerkinElmer). Images were obtained as a 1 μm Z stack with 0.5 μm spacing using a 488 laser. All imaging and analyses were completed by an observer unaware of genotype or treatment group. Analysis and quantification of data were performed with Volocity software after max intensity projection. The number of dendritic spines were counted manually to estimate the density of primary or secondary dendritic branch by observer unaware of treatment. For each condition, at least 3 dendrites were measured from each neuron and 5–7 neurons were imaged per coverslip.
QUANTIFICATION AND STATISTICAL ANALYSIS
All results are presented as mean ± SEM. Microsoft Excel, Prism 6 software and IBM SPSS Statistics were used for statistical analysis. Data were analyzed using one-way ANOVA, or repeated-measures one-way ANOVA, followed by Tukey’s or Dunnett’s multiple comparisons test, as specified in the Figure legends. Only two-sided tests were used, and all data analyzed met the assumption for the specific statistical test that was performed. Probability levels of p ≤ 0.05 were considered statistically significant.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Eu-labeled streptavidin | PerkinElmer | Cat# 1244–360 |
| 488-streptavidin | Life technologies | Cat# S11223; RRID:AB_2336881 |
| SAF32 mouse anti-PrP | Cayman Chemical | Cat# 189720-; RRID:AB_327961 |
| IRDye 800CW Donkey anti-Mouse IgG (H + L) | Li-Cor | Cat# 925–32212; RRID:AB_2716622 |
| 8B4 mouse anti-PrP | Santa Cruz Biotech | Cat# Sc-4772; AB_628171 |
| 5058 mouse anti-PrP | Millipore Sigma | Cat# AB5058; RRID:AB_2172149 |
| 8G8 mouse anti-PrP | Cayman Chemical | Cat# 189760; RRID:AB_10078469 |
| 6D11 mouse anti-PrP | Covance | Cat#SIG-399810; RRID:AB_2564735 |
| Pri 308 mouse anti-PrP | Cayman Chemical | Cat# 189750; RRID:AB_327964 |
| 8H4 mouse anti-PrP | abeam | Cat# Ab61409; RRID:AB_944979 |
| SAF70 mouse anti-PrP | Cayman Chemical | Cat# 189770; AB_10104643 |
| Rabbit anti-SV2A | Abeam | Cat# 32942; RRID:AB_778192 |
| Mouse anti-NeuN | Millipore | Cat# Mab37; RRID:AB_2298772 |
| Rabbit anti-beta-actin | Cell Signaling Technology | Cat# 4967; RRID:AB_330288 |
| 555-streptavidin | Invitrogen | Cat# S3235; RRID:AB_2571525 |
| Rabbit anti-Fyn | Cell Signaling Technology | Cat# 4023; AB_10698604 |
| Rabbit anti-phospho-Src | Cell Signaling Technology | Cat# 210; RRID:AB_331697 |
| IRDye 680 CW Donkey anti-rabbit IgG (H + L) | Li-Cor | Cat# 925–6807; RRID:AB_2716687 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Thioflavin S | Sigma-Aldrich | Cat# 230456 |
| Aβ1–42: DAEFRHDSGYEVHHQKLVFFAEDVG SNKGAIIGLMVGGWIA | The ERI Amyloid Laboratory | amyloid.peptides@att.net |
| Z | This paper | Z |
| Proteinase | Roche Applied Science | Cat# 1373–196 |
| Sepharose Q fast-flow medium | GE Healthcare | Cat# 17051001 |
| DELFIA assay buffer | Perkin Elmer | Cat# 4002–0010 |
| phenol red-free F12 | Atlanta Biologicals | Cat#: M15350 |
| Goat serum | Life technologies | Cat# 16210–064 |
| DELFIA Enhancement Solution | Perkin Elmer | Cat# 4001–0010 |
| 1,1,1,3,3,3-Hexafluoro-2-propanol | Sigma-Aldrich | Cat#: 105228 |
| DMSO | Americanbio | Cat#: AB00435–00500 |
| Enzo FDA Approved Drugs Library | Enzo Life Sciences | SCREEN-WELL FDA approved drug library V2 |
| Microsource Pharm 1600 library | Microsource | Pharm 1600 |
| ChemDiv Targeted Diversity library | ChemDiv | Targeted Diversity library |
| Yale Small Molecule Discovery Center compound collection | This paper | Yale Small Molecule Discovery Center compound collection |
| Formaldehyde, 37% | J.T. Baker | Cat# 2106–01 |
| Ceftazidime (Fortaz) | GlaxoSmithKline | Cat# NDC 0173-0377-10 |
| biotin-NHS | Thermo Scientific | Cat# 21329 |
| Tween 20 | Sigma-Aldrich | Cat# P7949 |
| T20 PBS blocking buffer | Pierce | Cat# 37573 |
| ChemDiv Diversity Library | ChemDiv | Targeted Diversity Library |
| Trypsin 0.25% | GIBCO | Cat# 25200–056 |
| GlutaMAX | GIBCO | Cat# 35050–061 |
| Sodium pyruvate | GIBCO | Cat# 11360–070 |
| Pen strep | GIBCO | Cat# 15140–122 |
| Neurobasal media | GIBCO | Cat# 10888–022 |
| Triton X-100 | American bioanalytical | Cat# AB02025–00500 |
| Lipofectamine 3000 | ThermoFisher | Cat# L3000015 |
| pBSCa,Mg | Sigma Aldrich | Cat# D8662 |
| Polysorbate 80 | Sigma Aldrich | Cat# W291706 |
| SNAP-Surface Alexa Fluor 647 | New England Biolabs | Cat# S9136 |
| PBS, pH 7.4 1 OX | Americanbio | Cat# AB11072 |
| Papain | Sigma-Aldrich | Cat# P5306 |
| Polystyrene sulfonate (PSS) | PSS Polymer Standards Service GmbH | Cat# PSS-pss3.4K or PSS-pss17K |
| Polyethylene glycol) | Sigma-Aldrich | Cat# 94646 |
| Poly(4-styrenesulfonic acid-co-maleic acid) sodium salt (PSCMA) | Sigma-Aldrich | Cat# 434566 |
| Poly(acrylic acid) sodium salt | Sigma-Aldrich | Cat# 447013 |
| Poly(methacrylic acid) sodium salt | Sigma-Aldrich | Cat# 434507 |
| Poly(acrylic acid co-maleic acid) solution | Sigma-Aldrich | Cat# 416053 |
| Pentosan polysulfate sodium | BOC Sciences | Cat# 116001-96-8 |
| Poly-d-glutamic acid sodium salt | Sigma-Aldrich | Cat# P4033 |
| Dextran sulfate sodium salt | Sigma-Aldrich | Cat# D4911 |
| Poly (2-acrylamido-2-methyl-1-propanesulfonic acid) | Sigma-Aldrich | Cat# 191973 |
| Poly (styrene-co-maleic acid) partial isobutyl ester | Sigma-Aldrich | Cat# 435287 |
| Poly (styrene-alt-maleic acid) sodium salt | Sigma-Aldrich | Cat# 662631 |
| Adrenochome free base | Sigma-Aldrich | Cat# A5762 |
| Norepinephine bitartrate salt | Sigma-Aldrich | Cat# A0937 |
| Critical Commercial Assays | ||
| Cytotoxicity Detection Kit | Roche | Cat# 11644793001 |
| Experimental Models: Cell Lines | ||
| CV-1 cells | ATCC | Cat# CCL-70 |
| Green monkey: COS-7 kidney cells | ATCC | Cat# CCL-1651 |
| Experimental Models: Organisms/Strains | ||
| APPswe/PS1 AE9 (APP/PS1), age 12–14 months at the time of testing, male percentage 45%−55% | Jackson Laboratory | RRID: MMRRC: 34832-JAX; Jankowsky et al., 2003 |
| Recombinant DNA | ||
| SNAP-PrP in pcDNA3.1 vector | This paper | SNAP-PrP |
| pSNAP-tag vector | New England Biolabs | Cat#: N9183 |
| Full-length PrPc (AA23–231) in pRSET A vector | Zahnetal., 1997 | PrP, PrP-FL |
| Full-length PrPc in PCDNA3 | This paper | N/A |
| Software and Algorithms | ||
| GraphPad Prism 7 | graphpad | Graphpad.com |
| Volocity | PerkinElmer | Volocity 6.3 |
| ImageJ | lmagej.nih.gov | ImageJ |
| Image Studio | Li-Cor | https://www.licor.com/bi |
| Other | ||
| 3 kDa filter | Amicon | Cat# UFC500396 |
| XK 50/20 column | GE Healthcare Life Sciences | Cat# 9621706 |
| Q Sepaharose Fast Flow | GE Healthcare Life Sciences | Cat# 17-0510-01 |
| BLI streptavidin Biosensors | Forte Bio | Cat# 18–5019 |
| 4–16% NOVEX Bis-Tris gel | ThermoFisher | Cat# BN1002BOX |
| western blot blocking buffer | Rockland | Cat# MB-070–010TF |
| MaxiSorp 384 microplates | ThermoFisher | Cat# 460372 |
| D-lysine-coated glass 8 well coverslip | Lab-Tek | Cat# 155411 |
| intracerebroventricular cannula | Alzet | Cat# 0008663 |
| AKTApurifier 10 FPLC System | GE Healthcare | Cat# 28406264 |
| iBIot dry transfer system | Thermo Fisher Scientific | Cat# IB1001 |
Highlights.
Screen for antagonist of PrPC binding to Aβo identifies polymeric antibiotic degradant
Class of polymeric nM-potent PrPC antagonists rescue Aβo-induced phenotypes in vitro
PrPC antagonists also clear neuroblastoma cells of PrPSc replication
Oral PrPC antagonist rescues transgenic AD mouse synapse loss and memory deficits
ACKNOWLEDGMENTS
We thank Stefano Sodi for expert technical assistance. This work was supported by grants to S.M.S. from the Falk Medical Research Trust and the NIH (R01AG034924, P50AG047270, RF1AG053000, R35NS097283, and P30DA018343).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes six figures and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.12.021.
DECLARATION OF INTERESTS
S.M.S. and E.C.G. are inventors on patents disclosing PrP-C antagonism for treatment of Alzheimer’s disease. G.M. is a full-time employee of ReNetX Bio, Inc.
REFERENCES
- Aimi T, Suzuki K, Hoshino T, and Mizushima T (2015). Dextran sulfate sodium inhibits amyloid-β oligomer binding to cellular prion protein. J. Neurochem 134, 611–617. [DOI] [PubMed] [Google Scholar]
- Baertschi SW, Dorman DE, Occolowitz JL, Collins MW, Spangle LA, Stephenson GA, and Lorenz LJ (1997). Isolation and structure elucidation of the major degradation products of cefaclor formed under aqueous acidic conditions. J. Pharm. Sci 86, 526–539. [DOI] [PubMed] [Google Scholar]
- Banani SF, Lee HO, Hyman AA, and Rosen MK (2017). Biomolecular condensates: organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol 18, 285–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brody AH, and Strittmatter SM (2018). Synaptotoxic signaling by amyloid beta oligomers in Alzheimer’s disease through prion protein and mGluR5. Adv. Pharmacol 82, 293–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caughey B, and Raymond GJ (1993). Sulfated polyanion inhibition of scrapie-associated PrP accumulation in cultured cells. J. Virol 67, 643–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung E, Ji Y, Sun Y, Kascsak RJ, Kascsak RB, Mehta PD, Strittmatter SM, and Wisniewski T (2010). Anti-PrPC monoclonal antibody infusion as a novel treatment for cognitive deficits in an Alzheimer’s disease model mouse. BMC Neurosci. 11, 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citron M, Westaway D, Xia W, Carlson G, Diehl T, Levesque G, Johnson-Wood K, Lee M, Seubert P, Davis A, et al. (1997). Mutant presenilins of Alzheimer’s disease increase production of 42-residue amyloid b-protein in both transfected cells and transgenic mice. Nat. Med 3, 67–72. [DOI] [PubMed] [Google Scholar]
- Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, and Ashe KH (2005). Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat. Neurosci 8, 79–84. [DOI] [PubMed] [Google Scholar]
- Colby DW, and Prusiner SB (2011). Prions. Cold Spring Harb. Perspect. Biol 3, a006833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condello C, Yuan P, and Grutzendler J (2018). Microglia-mediated neuroprotection, TREM2, and Alzheimer’s disease: evidence from optical imaging. Biol. Psychiatry 83, 377–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond E, and Wisniewski T (2017). Alzheimer’s disease: experimental models and reality. Acta Neuropathol. 133, 155–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ercanli T, and Boyd DB (2006). Exploration of the conformational space of a polymeric material that inhibits human immunodeficiency virus. J. Chem. Inf. Model 46, 1321–1333. [DOI] [PubMed] [Google Scholar]
- Freir DB, Nicoll AJ, Klyubin I, Panico S, Mc Donald JM, Risse E, Asante EA, Farrow MA, Sessions RB, Saibil HR, et al. (2011). Interaction between prion protein and toxic amyloid b assemblies can be therapeutically targeted at multiple sites. Nat. Commun 2, 336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimbel DA, Nygaard HB, Coffey EE, Gunther EC, Laurén J, Gimbel ZA, and Strittmatter SM (2010). Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J. Neurosci 30, 6367–6374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JS, Younkin S, et al. ; Alzheimer Genetic Analysis Group (2013). TREM2 variants in Alzheimer’s disease. N. Engl. J. Med 368, 117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas LT, and Strittmatter SM (2016). Oligomers of amyloid b prevent physiological activation of the cellular prion protein-metabotropic glutamate receptor 5 complex by glutamate in Alzheimer disease. J. Biol. Chem 291, 17112–17121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas LT, Kostylev MA, and Strittmatter SM (2014). Therapeutic molecules and endogenous ligands regulate the interaction between brain cellular prion protein (PrPC) and metabotropic glutamate receptor 5 (mGluR5). J. Biol. Chem 289, 28460–28477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas LT, Salazar SV, Kostylev MA, Um JW, Kaufman AC, and Strittmatter SM (2016). Metabotropic glutamate receptor 5 couples cellular prion protein to intracellular signalling in Alzheimer’s disease. Brain 139, 526–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas LT, Salazar SV, Smith LM, Zhao HR, Cox TO, Herber CS, Degnan AP, Balakrishnan A, Macor JE, Albright CF, and Strittmatter SM (2017). Silent allosteric modulation of mGluR5 maintains glutamate signaling while rescuing Alzheimer’s mouse phenotypes. Cell Rep. 20, 76–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, and Selkoe DJ (2002). The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356. [DOI] [PubMed] [Google Scholar]
- Hobi R, Hübscher U, Neftel K, Alteri E, Poncioni B, Walker MR, Woods-Cook K, Schneider P, and Lazdins JK (2001). Anti-HIV-1 activity in vitro of ceftazidime degradation products. Antivir. Chem. Chemother 12, 109–118. [DOI] [PubMed] [Google Scholar]
- Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres BA, et al. (2016). Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman AA, Weber CA, and Jülicher F (2014). Liquid-liquid phase separation in biology. Annu. Rev. Cell Dev. Biol 30, 39–58. [DOI] [PubMed] [Google Scholar]
- Jankowsky JL, Xu G, Fromholt D, Gonzales V, and Borchelt DR (2003). Environmental enrichment exacerbates amyloid plaque formation in a transgenic mouse model of Alzheimer disease. J. Neuropathol. Exp. Neurol 62, 1220–1227. [DOI] [PubMed] [Google Scholar]
- Jankowsky JL, Fadale DJ, Anderson J, Xu GM, Gonzales V, Jenkins NA, Copeland NG, Lee MK, Younkin LH, Wagner SL, et al. (2004). Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum. Mol. Genet 13, 159–170. [DOI] [PubMed] [Google Scholar]
- Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, Bjornsson S, Huttenlocher J, Levey AI, Lah JJ, et al. (2013). Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med 368, 107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman AC, Salazar SV, Haas LT, Yang J, Kostylev MA, Jeng AT, Robinson SA, Gunther EC, van Dyck CH, Nygaard HB, and Strittmatter SM (2015). Fyn inhibition rescues established memory and synapse loss in Alzheimer mice. Ann. Neurol 77, 953–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klyubin I, Nicoll AJ, Khalili-Shirazi A, Farmer M, Canning S, Mably A, Linehan J, Brown A, Wakeling M, Brandner S, et al. (2014). Peripheral administration of a humanized anti-PrP antibody blocks Alzheimer’s disease Ab synaptotoxicity. J. Neurosci 34, 6140–6145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostylev MA, Kaufman AC, Nygaard HB, Patel P, Haas LT, Gunther EC, Vortmeyer A, and Strittmatter SM (2015). Prion-protein-interacting amyloid-b oligomers of high molecular weight are tightly correlated with memory impairment in multiple Alzheimer mouse models. J. Biol. Chem 290, 17415–17438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostylev MA, Tuttle MD, Lee S, Klein LE, Takahashi H, Cox TO, Gunther EC, Zilm KW, and Strittmatter SM (2018). Liquid and hydrogel phases of PrP(C) linked to conformation shifts and triggered by Alzheimer’s amyloid-beta oligomers. Mol. Cell 72, 426–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurén J, Gimbel DA, Nygaard HB, Gilbert JW, and Strittmatter SM (2009). Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 457, 1128–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu D, Giles K, Li Z, Rao S, Dolghih E, Gever JR, Geva M, Elepano ML, Oehler A, Bryant C, et al. (2013). Biaryl amides and hydrazones as therapeutics for prion disease in transgenic mice. J. Pharmacol. Exp. Ther 347, 325–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris R (1984). Developments of a water-maze procedure for studying spatial learning in the rat. J. Neurosci. Methods 11, 47–60. [DOI] [PubMed] [Google Scholar]
- Purro SA, Nicoll AJ, and Collinge J (2018). Prion protein as a toxic acceptor of amyloid-b oligomers. Biol. Psychiatry 83, 358–368. [DOI] [PubMed] [Google Scholar]
- Salazar SV, Gallardo C, Kaufman AC, Herber CS, Haas LT, Robinson S, Manson JC, Lee MK, and Strittmatter SM (2017). Conditional deletion of Prnp rescues behavioral and synaptic deficits after disease onset in transgenic Alzheimer’s disease. J. Neurosci 37, 9207–9221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider LS, Mangialasche F, Andreasen N, Feldman H, Giacobini E, Jones R, Mantua V, Mecocci P, Pani L, Winblad B, and Kivipelto M (2014). Clinical trials and late-stage drug development for Alzheimer’s disease: an appraisal from 1984 to 2014. J. Intern. Med 275, 251–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith LM, and Strittmatter SM (2017). Binding sites for amyloid-β oligomers and synaptic toxicity. Cold Spring Harb. Perspect. Med 7, a024075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith LM, Zhu R, and Strittmatter SM (2018). Disease-modifying benefit of Fyn blockade persists after washout in mouse Alzheimer’s model. Neuropharmacology 130, 54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonati T, Reimann RR, Falsig J, Baral PK, O’Connor T, Hornemann S, Yaganoglu S, Li B, Herrmann US, Wieland B, et al. (2013). The toxicity of antiprion antibodies is mediated by the flexible tail of the prion protein. Nature 501, 102–106. [DOI] [PubMed] [Google Scholar]
- Ulrich JD, Ulland TK, Colonna M, and Holtzman DM (2017). Elucidating the role of TREM2 in Alzheimer’s disease. Neuron 94, 237–248. [DOI] [PubMed] [Google Scholar]
- Um JW, Nygaard HB, Heiss JK, Kostylev MA, Stagi M, Vortmeyer A, Wisniewski T, Gunther EC, and Strittmatter SM (2012). Alzheimer amyloid-β oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat. Neurosci 15, 1227–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Um JW, Kaufman AC, Kostylev M, Heiss JK, Stagi M, Takahashi H, Kerrisk ME, Vortmeyer A, Wisniewski T, Koleske AJ, et al. (2013). Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer ab oligomer bound to cellular prion protein. Neuron 79, 887–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahn R, von Schroetter C, and Wüthrich K (1997). Human prion proteins expressed in Escherichia coli and purified by high-affinity column refolding. FEBS Lett. 417, 400–404. [DOI] [PubMed] [Google Scholar]
- Zhang X, Lin Y, Eschmann NA, Zhou H, Rauch JN, Hernandez I, Guzman E, Kosik KS, and Han S (2017). RNA stores tau reversibly in complex coacervates. PLoS Biol. 15, e2002183. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.