

Abstract
Background
DNA methylation is involved in the regulation of gene expression. Although bisulfite-sequencing based methods profile DNA methylation at a single CpG resolution, methylation levels are usually averaged over genomic regions in the downstream bioinformatic analysis.
Results
We demonstrate that on the genome level a single CpG methylation can serve as a more accurate predictor of gene expression than an average promoter / gene body methylation. We define CpG traffic lights (CpG TL) as CpG dinucleotides with a significant correlation between methylation and expression of a gene nearby. CpG TL are enriched in all regulatory regions. Among all promoters, CpG TL are especially enriched in poised ones, suggesting involvement of DNA methylation in their regulation. Yet, binding of only a handful of transcription factors, such as NRF1, ETS, STAT and IRF-family members, could be regulated by direct methylation of transcription factor binding sites (TFBS) or its close proximity. For the majority of TF, an alternative scenario is more likely: methylation and inactivation of the whole regulatory element indirectly represses functional TF binding with a CpG TL being a reliable marker of such inactivation.
Conclusions
CpG TL provide a promising insight into mechanisms of enhancer activity and gene regulation linking methylation of single CpG to gene expression. CpG TL methylation can be used as reliable markers of enhancer activity and gene expression in applications, e.g. in clinic where measuring DNA methylation is easier compared to directly measuring gene expression due to more stable nature of DNA.
Electronic supplementary material
The online version of this article (10.1186/s12864-018-5387-1) contains supplementary material, which is available to authorized users.
Keywords: CpG traffic lights, DNA methylation, Transcription regulation, Enhancers, CAGE, Chromatin states, NRF1, ETS, STAT, IRF
Background
Epigenetic regulation of gene expression has been thoroughly investigated over last decades. DNA methylation, usually in CpG context, is probably the most well-studied mechanism of epigenetic regulation. DNA methylation is linked to many normal and pathological biological processes: organism development, cell differentiation, cell identity and pluripotency maintenance (reviewed in [1–3]), aging [4], memory formation [5, 6], responses to environmental exposures, stress and diet [7–9]. Abnormalities in DNA methylation play an important role in various diseases, including metabolic [10], cardiovascular [11], neurodegenerative [12, 13] diseases and cancers (reviewed in [14]). For about a decade, DNA demethylating drugs (Decitabine, Azacytidine) are used in clinic for the treatment of acute myeloid leukemia and myelodysplastic syndrome [15]. Recent advances in site-specific editing of DNA methylation [16] suggest DNA methylation as a promising target for non-invasive therapies against diseases linked to aberrant methylation.
Functionally, DNA methylation of promoter regions is tightly associated with repression of transcription initiation, while high levels of gene body methylation, on the contrary, are linked to the increased gene expression (reviewed in [17]). Enhancers, distant regulatory regions, that contribute to the establishment of the correct temporal and cell-type-specific gene expression pattern, have been shown to initiate transcription of short RNAs [18]. Therefore, it is no surprise that DNA methylation also regulates the enhancer functioning [19–22].
Methods based on bisulfite sequencing allow detection of single cytosine methylation. Yet, in downstream bioinformatic analysis, methylation levels of several dozens of cytosines are often averaged to increase statistical power [23, 24]. At the same time, multiple examples show that changes in methylation of a single CpG can affect transcription [25–39]. Recently, we have shown that methylation of particular single CpG dinucleotides are tightly linked to gene expression [40]. We have called such positions CpG traffic lights (CpG TL) and have demonstrated a strong negative selection against them in computationally predicted transcription factor binding sites. In the current study we show enrichment of CpG TL in regulatory elements of different types: in transcription start sites (TSS), in particular, in poised promoters, as well as in enhancers and regions with active chromatin marks. Although CpG TL may regulate transcription factors, co-factors and epigenetic regulators, binding of only a handful of transcription factors could be regulated by direct methylation of a CpG TL within a transcription factor binding site (TFBS). For the majority of TF, an alternative scenario is more likely: inactivation of the whole regulatory element via DNA methylation repress TF binding indirectly; and CpG TL are reliable markers of inactivation. We believe that CpG TL provide a promising insight into mechanisms of enhancer activity and gene regulation linking methylation of single CpG to gene expression.
Results
CpG traffic lights detection
DNA methylation in promoter regions often repress gene expression. Nevertheless, the link between expression and promoter or gene body methylation is not straightforward, suggesting the need to deconvolute DNA methylation profiles into regulatory regions of a smaller size. To thoroughly investigate the connection between methylation and expression, we focus on methylation levels of single CpG dinucleotides. Following the logic previously reported in our works [40, 41], we expand our approach and use whole-genome DNA methylation (genome wide bisulfite sequencing, WGBS) and expression (RNA-seq) data for 48 normal human primary cells and tissues from the Roadmap Epigenomics Project. We selected non-related cell types to capture CpG position which are most variable in methylation between cell types. We define CpG traffic lights (CpG TL) as CpG dinucleotides with significant Spearman correlation coefficient (SCC) between DNA methylation and expression levels of a neighboring gene (FDR<0.01, Fig. 1).
Fig. 1.

Schematic representation of a CpG traffic light detection. Left panel. Suppose we analyze a particular genomic region (chr1:123..11654), which contains for simplicity one gene. For each CpG in this region and the gene we have methylation and expression vectors in 6 cell lines, respectively. CpG dinucleotides are represented by dark blue lollipops (filled: methylated CpG, empty: unmethylated CpG). First three CpGs are located within the promoter region, while the last three are located in the gene body. Gene expression or lack of it is represented by green arrows. Right panel. The yellow column shows methylation of a random CpG (used as a background), the methylation vector of this CpG demonstrates low correlation with the gene expression (the green box on the right, in RPKM). Correlation between the average promoter/gene body methylation (shown in the light blue and light purple columns, respectively) and the corresponding gene expression is also low. However, for the CpG TL (shown in the red box), the methylation significantly correlates with the gene expression
Here we show that the average methylation of promoter/gene body less frequently correlates significantly with the gene expression compared to the methylation of CpG TL, even applying a proper multiple testing correction. In particular, at FDR<0.01 we find only 764/762 genes for which average promoter/gene body methylation correlates with expression, while at the same level of significance we observe 7997 genes correlating significantly with CpG TL methylation levels (Table 1, Additional file 1: Table S1, Table S2). Similar tendencies are observed for different promoter/gene body boundaries (Additional file 1: Table S3).
Table 1.
The number of genes with significant correlation between expression and methylation
| FDR-corrected p-value (significance level) | Total number of genes, which have significant correlations between gene expression and methylation | |||
|---|---|---|---|---|
| Average methylation of promoter regions (-1000..500) (1) | Average methylation of gene bodies (+500..TTS) (2) | Methylation of CpG TL (3) | Permutation test (4) | |
| 0.001 | 263 | 186 | 1463 | 14.5 |
| 0.005 | 537 | 505 | 4905 | 15.4 |
| 0.01 | 764 | 762 | 7997 | 16.2 |
| 0.05 | 2038 | 2125 | 22957 | 21.8 |
| 0.1 | 3251 | 3401 | 34095 | 27.5 |
Note: for multiple testing correction the number of genes was used in (1) and (2), while the number of all CpG - gene pairs was used for the same purpose in (3) and (4). (4) Permutation test (RPKM) results: the number of genes with significant correlation between expression and methylation obtained by chance (averaged over 10 random permutations). (TTS) refers to a Transcription Termination Site
The majority of promoter CpG TL demonstrate negative SCC, while the majority of those located in intronic regions demonstrate positive SCC, which is in line with the previous findings. Exonic CpG TL demonstrate comparable number of both positive and negative SCC with an increase in positive SCC towards gene 3’ end (Additional file 1: Figure S1). CpG TL are uniformly distributed along the genome (Manhattan plot, Additional file 1: Figure S2).
CpG traffic lights are conserved across mammals and primates
To address functionality of CpG TL, we first investigate their evolutionary conservation and find that CpG TL are preserved in mammals and in primates according to GERP RS [42] and PhyloP [43] scores respectively (Fig. 2a, b). Also, CpG TL are depleted in repetitive sequences determined by repeatMasker (Fig. 2c) (see “Methods”) and chromatin states (chromHMM [44], Fig. 3g). Eigen non-coding scores [45] that reflects non-coding functionality are significantly higher for CpG TL (Fig. 2d). Taken together, these results suggest the regulatory role of CpG TL in the genome.
Fig. 2.
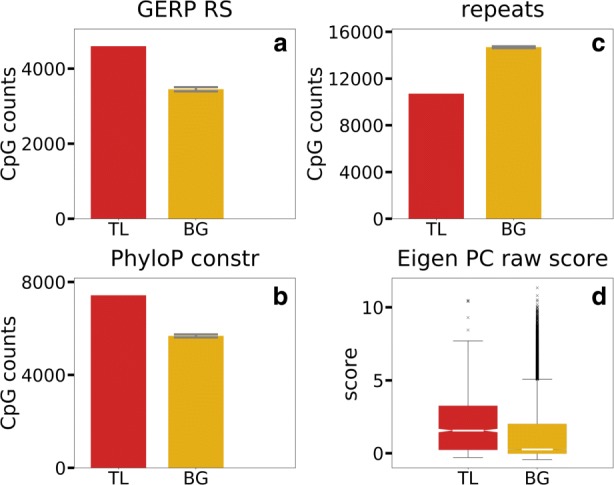
Evolutionary conservation of the CpG TL compared to the background CpG sites (BG). a Conservation in mammals and b in primates, c repeats determined by RepeatMasker, d Eigen non-coding functionality score. Whiskers (abc) represent standard deviation out of the 50 random background samples. Fisher exact test, p-value <5E−4 (a - c), Kolmogorov-Smirnov statistic for 2 samples p-value <5E−4 (d)
Fig. 3.

CpG TL in regulatory regions. Over-representation of CpG TL in a open chromatin regions (DNaseI), b transcription start sites determined by CAGE, c enhancers determined by histone modifications, d enhancers determined by FANTOM5. No difference between CpG TL and CpG BG counts in e CpG islands while f CpG TL are over-represented in CpG islands shores. Panel g represents averaged across 127 cell types ratio of TL / BG in chromatin states determined by chromHMM. The color g reflects absolute number of the CpG TL located in a given chromatin state. Whiskers (a-f) represent standard deviation of the 50 random background samples. Fisher exact test, p-value <5E−4
CpG traffic lights are enriched in regulatory elements
To narrow down the regulatory role of CpG TL we tested for the overlap between CpG TL and various functional genomic elements. CpG TL are enriched in the open chromatin regions (Fig. 3a) supporting the claim of their regulatory potential. In particular, they are 2-fold enriched at exact transcription start sites (Fig. 3b) determined by CAGE (Cap Analysis of Gene Expression) [46], as well as in all promoter types determined by chromHMM [44], including active, bivalent, and poised promoters but not in the regions of transcription elongation (Fig. 3g). Interestingly, the strongest enrichment was observed in poised promoters (>3.5 fold). Since the poised or bivalent chromatin is thought to be able to easily switch between active and repressed states [47], such enrichment may suggest a contribution of CpG TL to the maintenance of the bivalent state of the chromatin.
CpG TL are also highly enriched in chromatin states corresponding to regulatory elements (Fig. 3c-g), in particular in enhancers, determined by a combination of histone marks (Fig. 3c), by CAGE bidirectional transcription (Fig. 3d), and by chromatin states (Fig. 3g). Among all enhancer types the most enriched are various stem cell and hematopoietic cell enhancers suggesting potential role of CpG TL methylation in regulation of pluripotency and hematopoesis (Fig. 4, Additional file 1: Table S4). Surprisingly, CpG TL are enriched in CpG island shores but not in CpG islands (Fig. 3e, f).
Fig. 4.

Functional categories of human enhancers enriched with CpG TL (negative SCC). Fisher’s exact test and FDR (Benjamini-Hochberg) correction for multiple testing (implemented in python scipy.stats.fisher_exact and p_adjust (method=’fdr’) from R) were used to calculate the p-values
CpG traffic lights are enriched in regulatory genes but avoid the majority of transcription factor binding sites
By analyzing the functionality of genes harboring CpG TLs we found a strong enrichment of such genes with known transcription regulators — transcription factors, co-factors and epigenetic regulators (Table 2, see “Methods”). Previously we reported that CpG TL avoided computationally predicted TFBS suggesting that direct methylation of TFBS may not be the main mechanism of TF binding regulation [40]. In this work, using aggregated data on in vivo binding (ChIP-Seq) we support this claim showing that for the majority of TFBS (with those of NRF1 being a notable exception) there is no enrichment for the CpG TL (Fig. 5a). Yet, surprisingly for some TF, CpG TL were enriched in the close proximity of their TFBS (Fig. 5b, c).
Table 2.
Enrichment of CpG TL in regulatory genes
| Gene type | # genes of in the annotation | # genes with CpG TL | # genes expected | fold enrichment | over-repre-senta-tion | p-value |
|---|---|---|---|---|---|---|
| Epigenetic regulators | 719 | 279 | 98.56 | 2.83 | + | 1.4E-63 |
| Histones | 94 | 17 | 12.89 | 1.32 | + | 0.23 |
| Transcription factors | 1751 | 599 | 240.02 | 2.50 | + | 1.06E-108 |
| Transcription co-factors | 951 | 356 | 130.36 | 2.73 | + | 4.69E-76 |
Fig. 5.
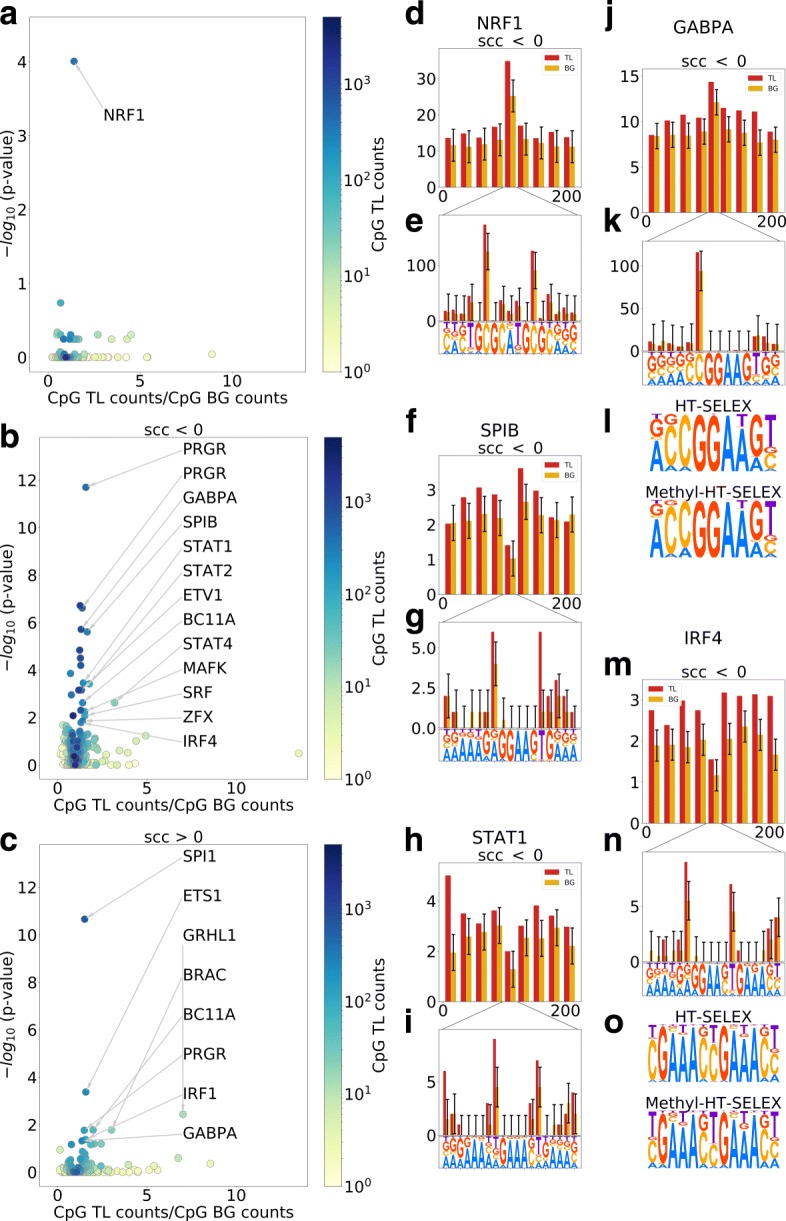
CpG TL within transcription factor binding sites. CpG TL to CpG BG ratio vs Fisher’s exact test p-value within a all predicted TFBS; b TFBS and 50 nt shores for CpG TL with a negative SCC and c the same for CpG TL with a positive SCC. Length-normalized distribution of CpG TL / CpG BG counts (negative SCC) within TFBS and 100 nt shores: d NRF1; f SPIB; h STAT1; j GABPA; m IRF4. Per position distribution of CpG TL / CpG BG counts (negative SCC) within TFBS with logo: e NRF1; g SPIB; i STAT1; k GABPA; n IRF4. In vitro binding preferences of unmethylated and methylated oligos: l GABPA; o IRF4
NRF1 binding sites
Despite the observation that overall TFBS do not co-locate with CpG TL, binding sites of NRF1 (Fig. 5a, d) — a transcription factor involved into activation of key metabolic genes — are enriched in CpG TL even when overall enrichment for regulatory regions is taken into account (see “Methods”). Interestingly, core CpG positions of NRF1 binding sites are the most enriched with CpG TL supporting their functional importance for NRF1 binding (Fig. 5e). Being in line with the previous findings [48], these observations imply that NRF1 may be one of the very few TF whose binding may be directly regulated by DNA methylation.
ETS-family binding sites
Exact binding sites of GABPA (ETS-motif binding TF) and their close proximity (50bp) are 1.3-folds enriched in CpG TL (Fig. 5j, k). The strongest enrichment is observed in C neighboring the core GGAA box. In vitro binding data (HT-SELEX and Methyl-SELEX) [49] show that methylated C is less frequent in this position (Fig. 5l). Similar CpG TL enrichment was observed for binding sites of another members of ETS-familty: SPIB (Fig. 5f, g) and ETV1 (Additional file 1: Figure S3a-c). Binding of ETS-family members might be directly affected by DNA methylation, yet enrichment of the CpG TL in the closest proximity also supports the hypothesis of the indirect effect of regional methylation.
STAT-family and IRF-family binding sites
Surprisingly, such GA-rich motifs as those bound by STAT1,2,4 and IRF1,4 are also enriched in CpG TL but in their weak positions and in close proximity to the TFBS (Fig. 5h, i, m, n, Additional file 1: Figure S3d-k). In vitro binding data for IRF4 (HT-SELEX and Methyl-SELEX) shows an avoidance of methylated C in this motif position (Fig. 5o). Since the enrichment in CpG TL is observed only in weak motif positions we speculate that binding of the TF from STAT- and IRF-families is indirectly affected by methylation of the whole regulatory region.
Discussion
In this work we demonstrate that methylation profiles of single CpG dinucleotides (CpG TL) more often significantly correlate with gene expression as compared to average promoter / gene body methylation. It is a surprising observation, since it is widely accepted that DNA methyltransferases once bound to DNA move along it [50] or multimerize [51] methylating all neighboring CpG dinucleotides unless a boundary protein, such as Sp1, is reached (reviewed in [52]). Yet, only a small fraction of CpG TL are co-located within the promoter (or body) of the same gene. We speculate that local change in DNA methylation could be achieved through active DNA demethylation probably with the help of TET proteins. A direct experiment with the use of CRISPR/TALEN-based technology is required to test this hypothesis.
It should be noted that our procedure of CpG TL detection based on correlation (SCC) cannot be applied to CpG dinucleotides that are fully methylated or methylated in all studied cell types. Our dataset consists of 48 cell types and does not cover the whole spectrum of human cell types. Due to this limitation, a significant fraction of regulatory CpG might be missing from our analysis. Novel data on DNA methylation and expression in various cell types will improve our understanding of CpG TL functions.
The enrichment of CpG TL in enhancers, in particular in hematopoietic enhancers, is in line with the recent reports that DNA methylatransferases DNMT3a/b can bind enhancers and regulate the enhancer RNA production in hematopoietic cells [22]. Also, distal regulatory regions can initiate transcription themselves, being in turn regulated by DNA methylation [53], contributing to the similarity of TSS and enhancers in terms of CpG TL enrichment.
Previously, it has been reported that NRF1 binding is directly regulated by DNA methylation [48]. In our work we demonstrate that such regulated binding is functional and regulate corresponding gene expression at least in some cases when NRF1 TFBS harbor a CpG TL. We also observed the enrichment of CpG TL in the close proximity to the ETS-, STAT- and IRF-family motifs hits. Interestingly, the majority of TF from these families are involved in hemapoietic regulation being in line with the strong enrichment of CpG TL in hematopoietic enhancers. These observations support the importance of the enhancer methylation in the regulation of the hematopoetic cells.
In the light of over-representation in regulatory regions, lack of enrichment of CpG TL within the majority of TFBS is puzzling. We can see several possible explanations. CpG TL may target unknown TFBS, although we believe that this scenario is unlikely. It was previously shown that almost all novel motifs obtained from regulatory regions correspond to known families of TFBS [46, 54, 55]. Furthermore, the HOCOMOCO v11 collection covers almost all structural families of transcription factors, except for the zinc finger family. Among those, there might be some important isolated cases enriched with CpG TL but their contribution to the overall picture is expected to be negligible. Alternatively, cytosine methylation could accumulate as a consequence of the absence of TF binding, which makes methylation of CpG TL not a primary cause, but just a “passive” marker of absent gene expression resulting from inactivation of its regulatory element. The last alternative is supported by previous works [56, 57]. More studies are needed to confirm which alternative is the most accurate. Yet, even if the “passive” marker explanation is true, CpG TL methylation could be a reliable marker of enhancer activity and gene expression, and can be used in practical applications, for example, in clinic where testing for DNA methylation is easier than testing directly for gene expression due to more stable nature of DNA.
Conclusions
In this work we demonstrate that CpG TL are enriched in regulatory regions, including poised/bivalent promoters and enhancers, in particular in hematopoietic enhancers. Only a handful of TFBS, such as those bound by NRF1, could be directly regulated by DNA methylation, while binding of several TF families (ETS-, STAT-, IRF-) could be affected indirectly through methylation and repression of the entire regulatory region. CpG traffic lights provide a promising insight into gene regulation linking single CpG methylation to gene expression.
Methods
DNA methylation and expression data processing
We selected 48 tissues and cell types (see Additional file 1: Table S5) for which both WGBS and RNA-seq data were available in Roadmap Epigenomics Project. For all samples sequenced with the Illumina platform read trimming and adapter removal were performed by Trimmomatic [58] (up to 2 mismatches between an adapter and a read sequence; 5bp sliding window; quality threshold of 20; removing sequences shorter than 20 bp after trimming). For the samples sequenced with the SOLiD platform we used Cutadapt [59] (up to 10% error rate relative to the length of the matching region; quality threshold of 20; removing sequences shorter than 20 bp after trimming).
We mapped WGBS data to the genome (assembly GRCh38-Ensembl 78) with Bismark [60] (zero mismatches in the seed, 20bp seed length, 0/500bp the min/max insert size for valid paired-end alignments). Further we consider only methylated cytosines in the CpG context, covered with not less than 4 reads on both strands. For each CpG position in every of the 48 samples, the methylation values were averaged between replicates. We removed all CpG positions if methylation values were available for less than 20 samples.
We mapped RNA-Seq data with Tophat v2.0.13 [61] (up to 2 mismatches and 2 gaps per read, paired-end reads are reported only if both reads are mapped). We generated an expression matrix using FeatureCount [62], the expression profiles were normalized to RPKM values.
CpG traffic lights detection
To determine CpG TL we considered all pairs of genes and CpG located within 10000 bp upstream of TSS to 3’ gene ends. One CpG might be associated with multiple genes, similarly, one gene might be associated with multiple CpG. For each CpG-gene pair we created two k-dimensional vectors (where k =20..48) of methylation levels (beta-values, [0,1]) and gene expression (RPKM). The length of the vectors (k) varies due to the fact that WGBS does not provide uniform coverage for all genomic CpG leading to missing values in the methylation profile of many CpGs. To avoid vague correlations we did not consider the CpG positions having less than 20 defined values in the respective methylation profiles. We further refer to each of the two vectors as a methylation and expression profiles. In total we had 18,830,232 CpGs associated with 59,396 genes (in total, 25,813,295 pairs).
For each CpG position, we calculated SCC between the methylation and expression profiles for all available samples. We refereed to a CpG position as a CpG traffic light (CpG TL) if it had a significant Spearman correlation coefficient (SCC) between methylation and expression profiles at the level of FDR<0.01 (Benjamini-Hochberg correction for the total number of pairs). We found 33,276 such CpG TL (0.18% of the original number of CpGs) that corresponded to 7997 genes.
Construction of background datasets
To explore enrichment of CpG TL within various genomic regions we constructed background sets (CpG BG) of the same size. We required CpG BG to be similar to CpG TL based on the following criteria:
GC content (the total number of C and G nucleotides) of the surrounding region of CpG BG must be similar to that of CpG TL. We calculated GC content in 200 bp windows centered on each CpG TL. For each such TL-centered window, we searched for another genomic CpG with the surrounding window having no more than 5% difference in GC content. For example, if there are 80 cytosines and guanines in a 200 bp window around CpG TL, we were looking for a CpG BG having from 76 to 84 cytosines and guanines in a 200 bp window.
CpG content (the total number of CG pairs) of the surrounding region of CpG BG should be similar to that of CpG TL. Again, for each CpG TL we allowed no more than 5% difference in CpG content in a 200 bp window.
CpG BG should have a similar distance to the TSS of the associated gene (while accounting for upstream or downstream location). For this purpose, we separately considered CpG TLs in [−100;TSS] and [TSS;100] distance bins by collecting CpG BG from the respective regions. For CpG TLs located farther than 100 bp from TSS, we considered log10(distance) and allowed up to 5% difference between CpG TL and its respective CpG BG. E.g. if a CpG TL is located + 1000 from a TSS, we are looking for a CpG BG located [708;1413].
A background CpG for a CpG TL with a SCC<0 (SCC>0) should also have a negative (positive) SCC with at least one of the associated genes). We repeated the selection process 50 times.
It is important to note that we did not control for the presence of a CpG island (CGI). Recently it has been shown that even methylated CpG dinucleotides within CpG islands were more conserved in primate evolution compared to methylated CpG outside the CGI [63]. Yet algorithms for CGI search use arbitrary parameters and may not be accurate in determination of CGI boundaries [64]. Therefore, controlling for a presence of a CGI would not necessarily reduce this bias.
Genomic annotations
We annotated all CpG positions with overlapping genomic features. For each feature we calculated the over-representation of CpG TL over CpG BG within each annotation using the exact Fisher’s test (in the total number of CpG TL and for CpG TL with positive/negative SCC separately). The following genomic annotations were tested: repeats (RepeatMasker http://hgdownload.soe.ucsc.edu/goldenPath/hg38/database/rmsk.txt.gz); the robust CAGE clusters [46]; the robust enhancers [65] (mapped to hg38 with the liftOver); the DNaseI hypersensitivity clusters (http://hgdownload.soe.ucsc.edu/goldenPath/hg38/database/wgEncodeRegDnaseClustered.txt.gz). Functional annotation of the enhancers was obtained from [46, 54, 55].
Evolutionary conservation and Eigen scores
Conservation of CpG TL and background sites in mammals and primates was assessed with UCSC Genome Browser GERP RS [42] and PhyloP [43] hg19 tracks, respectively. We calculated how many sites in each dataset had GERP RS score greater than 2, which we considered as conserved in mammals and PhyloP score greater than 0.5, which we considered as conserved in primates. Overall functional scores for each site were calculated with Eigen [45]. Higher Eigen scores imply more likely functionality of respective genome sites.
Histone modifications and chromatin states
The Roadmap Epigenomics Consortium 25-state segmentation of 127 epigenomes predicted with ChromHMM [44, 66] was used to assess chromatin states co-located with CpG TL. The annotation based on the imputed data for 12 chromatin marks (H3K4me1, H3K4me2, H3K4me3, H3K9ac, H3K27ac, H4K20me1, H3K79me2, H3K36me3, H3K9me3, H3K27me3, H2A.Z, and DNaseI) was downloaded from http://egg2.wustl.edu/roadmap/web_portal/imputed.html#chr_imp. We calculated a CpG TL/CpG BG ratio for each of the 25 chromatin states in each of the 127 epigenomes and then averaged the ratios for a representation on a figure.
Additionally, to verify CpG TL enrichment in the enhancers we selected regions having H3K27ac and H3K4me1 but lacking H3K4me3 (ENCODE, averaged among all samples mapped to hg38 with pre-calculated narrowPeak available, files with major errors and warnings excluded) (Additional file 1: Table S6).
TFBS prediction
For transcription factor binding sites prediction, we used position weight matrices (PWM) of human TFs provided in full HOCOMOCO v11 [67] collection and its default PWM thresholds according to the pre-calculated motif P-value of 0.0005 as in [68]. In HOCOMOCO v11, the thresholds and P-value were estimated against whole-genome dinucleotide composition. However, prediction of TFBS using PWMs alone can result in a notable number of false positives. Having this in mind, out of all predicted TFBS, we considered only those located in the reproducible and control data-supported cistrome [69] (only A, B, and C cistrome categories) for each TF. The cistrome was constructed from the ChIP-Seq data on transcription factors provided in the GTRD database [70] and processed by a common pipeline involving several computational ChIP-Seq peak callers, allowing to capture binding events routinely detected in different experiments. Thus, the TFBS considered in our study, were supported both by computational sequence analysis and by experimental ChIP-Seq data.
Gene enrichment analysis
We tested if genes that harbor CpG TL were enriched in transcription factors, co-factors and epigenetic regulators using Fisher’s exact test (implemented in python library scipy.stats) with Bonferroni correction. A list of TF and co-TF was obtained from Tcof DB [71] and the list of epigenetic regulators was obtained from EpiFactors [72].
Additional file
Supplementary materials. Figure S1: SCC of the CpG TL located in various gene regions; Figure S2: Distribution of CpG TLs along the genome; Figure S3: TFBS; Table S1: Number of significant SCC between average methylation of genomic region and gene expression; Table S2: Number of significant SCC between CpG methylation and gene expression; Table S3: Number of significant SCC between gene expression and average methylation of the genome region; Table S4: Most enriched with CpG TLs categories of enhancers; Table S5: Names of the cell samples in the study; Table S6: Enhancers = H3K27ac+H3K4me1-H3K4me3; Table S7: Expression data source; Table S8: Methylation data source. (PDF 2562 kb)
Acknowledgements
The authors are very grateful to Marina Lizio and Hideya Kawaji for their help with FANTOM5 datasets.
Funding
CpG TL detection was supported by RFBR grant 14-04-00180 to YAM. Functional analysis of CpG TL was supported by RFBR grant 17-54-80033 to YAM. AK and VBB were supported by the base research fund of the King Abdullah University of Science and Technology (KAUST). ChIP-Seq data analysis was supported by Russian Science Foundation [17-74-10188 to I.V.K.]. SELEX data analysis was supported the Program of fundamental research for state academies for 2013-2020 years (No 01201363825). These funding bodies had no role in the design of the study, collection, analysis, and interpretation of data, or in writing the manuscript.
Availability of data and materials
The datasets analysed during the current study are available in the Roadmap Epigenomics Project and FANTOM5, the links to the main datasets are in Additional file 1: Table S7 (expression) and Additional file 1: Table S8 (methylation). The cistrome data are available at Figshare [73].
Abbreviations
- CAGE
Cap analysis of gene expression
- CGI
CpG island
- ChIP-Seq
Chromatin immunoprecipitation (ChIP) sequencing
- CpG
CpG dinucleotide, 5’—C—phosphate—G—3’
- CpG BG
background CpG dinucleotide
- CpG TL
CpG traffic light
- ENCODE
Encyclopedia of DNA elements
- FANTOM5
Functional annotation of mammalian genome 5
- FDR
False discovery rate
- PWM
Position weight matrice
- RNA-seq
RNA sequencing
- RPKM
Reads per kilobase per million
- SCC
Spearman correlation coefficient
- TF
Transcription factor
- TFBS
Transcription factor binding sites
- TSS
Transcription start sites
- WGBS
genome wide bisulfite sequencing
Authors’ contributions
AL contributed to data processing and performed the over-representation analysis; AK processed the raw data and contributed to data analysis; AA contributed to statistical analysis; EB performed SELEX data analysis; VR performed data analysis; VBB contributed to study design; IVK contributed to study design and TFBS analysis; YAM designed the study, contributed to statistical analysis and drafted the MS. All authors contributed to MS preparation. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Only publicly available data were used, ethics approval could be found in the cited papers.
Consent for publication
Not applicable.
Competing interests
The authors claim no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Anna V. Lioznova, Email: anna.lioznova@gmail.com
Abdullah M. Khamis, Email: abdullahkhamis@gmail.com
Artem V. Artemov, Email: artem.v.artemov@gmail.com
Elizaveta Besedina, Email: bio_besedina@mail.ru.
Vasily Ramensky, Email: ramensky@gmail.com.
Vladimir B. Bajic, Email: vladimir.bajic@kaust.edu.sa
Ivan V. Kulakovskiy, Email: ivan.kulakovskiy@gmail.com
Yulia A. Medvedeva, Email: ju.medvedeva@gmail.com
References
- 1.Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33 Suppl:245–54. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 2.Messerschmidt DM, Knowles BB, Solter D. DNA methylation dynamics during epigenetic reprogramming in the germline and preimplantation embryos. Genes Dev. 2014;28(8):812–28. doi: 10.1101/gad.234294.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tomazou EM, Meissner A. Advances in Experimental Medicine and Biology. Vol 695. Boston: Springer; 2010. Epigenetic regulation of pluripotency. [DOI] [PubMed] [Google Scholar]
- 4.Christensen BC, Houseman EA, Marsit CJ, Zheng S, Wrensch MR, Wiemels JL, Nelson HH, Karagas MR, Padbury JF, Bueno R, Sugarbaker DJ, Yeh R-F, Wiencke JK, Kelsey KT. Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet. 2009;5(8):1000602. doi: 10.1371/journal.pgen.1000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miller CA, Sweatt JD. Covalent modification of DNA regulates memory formation. Neuron. 2007;53(6):857–69. doi: 10.1016/j.neuron.2007.02.022. [DOI] [PubMed] [Google Scholar]
- 6.Feng J, Zhou Y, Campbell SL, Le T, Li E, Sweatt JD, Silva AJ, Fan G. Dnmt1 and dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat Neurosci. 2010;13(4):423–30. doi: 10.1038/nn.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jirtle RL, Skinner MK. Environmental epigenomics and disease susceptibility. Nat Rev Genet. 2007;8(4):253–62. doi: 10.1038/nrg2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ladd-Acosta C, Fallin MD. The role of epigenetics in genetic and environmental epidemiology. Epigenomics. 2016;8(2):271–83. doi: 10.2217/epi.15.102. [DOI] [PubMed] [Google Scholar]
- 9.Pacchierotti F, Spanò M. Environmental impact on DNA methylation in the germline: State of the art and gaps of knowledge. Biomed Res Int. 2015;2015:123484. doi: 10.1155/2015/123484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Desai M, Jellyman JK, Ross MG. Epigenomics, gestational programming and risk of metabolic syndrome. Int J Obes. 2015;39(4):633–41. doi: 10.1038/ijo.2015.13. [DOI] [PubMed] [Google Scholar]
- 11.Zhong J, Agha G, Baccarelli AA. The role of DNA methylation in cardiovascular risk and disease: Methodological aspects, study design, and data analysis for epidemiological studies. Circ Res. 2016;118(1):119–31. doi: 10.1161/CIRCRESAHA.115.305206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wüllner U, Kaut O, deBoni L, Piston D, Schmitt I. DNA methylation in parkinson’s disease. J Neurochem. 2016;139 Suppl 1:108–20. doi: 10.1111/jnc.13646. [DOI] [PubMed] [Google Scholar]
- 13.Sanchez-Mut JV, Gräff J. Epigenetic alterations in alzheimer’s disease. Front Behav Neurosci. 2015;9:347. doi: 10.3389/fnbeh.2015.00347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baylin SB, Jones PA. Epigenetic determinants of cancer. Cold Spring Harb Perspect Biol. 2016; 8(9). [DOI] [PMC free article] [PubMed]
- 15.Derissen EJB, Beijnen JH, Schellens JHM. Concise drug review: azacitidine and decitabine. Oncologist. 2013;18(5):619–24. doi: 10.1634/theoncologist.2012-0465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shawn Liu X, Wu H, Ji X, Stelzer Y, Wu X, Czauderna S, Shu J, Dadon D, Young RA, Jaenisch R. Editing DNA methylation in the mammalian genome. Cell. 2016;167(1):233–24717. doi: 10.1016/j.cell.2016.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13(7):484–92. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 18.Kim T-K, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, Harmin DA, Laptewicz M, Barbara-Haley K, Kuersten S, Markenscoff-Papadimitriou E, Kuhl D, Bito H, Worley PF, Kreiman G, Greenberg ME. Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010;465(7295):182–7. doi: 10.1038/nature09033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Petell CJ, Alabdi L, He M, Miguel PS, Rose R, Gowher H. An epigenetic switch regulates de novo DNA methylation at a subset of pluripotency gene enhancers during embryonic stem cell differentiation. Nucleic Acids Res. 2016;44(16):7605. doi: 10.1093/nar/gkw426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heyn H, Vidal E, Ferreira HJ, Vizoso M, Sayols S, Gomez A, Moran S, Boque-Sastre R, Guil S, Martinez-Cardus A, Lin CY, Royo R, Sanchez-Mut JV, Martinez R, Gut M, Torrents D, Orozco M, Gut I, Young RA, Esteller M. Epigenomic analysis detects aberrant super-enhancer DNA methylation in human cancer. Genome Biol. 2016;17. [DOI] [PMC free article] [PubMed]
- 21.Kozlenkov A, Roussos P, Timashpolsky A, Barbu M, Rudchenko S, Bibikova M, Klotzle B, Byne W, Lyddon R, Di Narzo AF, Hurd YL, Koonin EV, Dracheva S. Differences in DNA methylation between human neuronal and glial cells are concentrated in enhancers and non-CpG sites. Nucleic Acids Res. 2014;42(1):109. doi: 10.1093/nar/gkt838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rinaldi L, Datta D, Serrat J, Morey L, Solanas G, Avgustinova A, Blanco E, Pons JI, Matallanas D, Von Kriegsheim A, Di Croce L, Benitah SA. Dnmt3a and dnmt3b associate with enhancers to regulate human epidermal stem cell homeostasis. Cell Stem Cell. 2016;19(4):491–501. doi: 10.1016/j.stem.2016.06.020. [DOI] [PubMed] [Google Scholar]
- 23.Bock C. Analysing and interpreting DNA methylation data. Nat Rev Genet. 2012;13(10):705–19. doi: 10.1038/nrg3273. [DOI] [PubMed] [Google Scholar]
- 24.Klein H-U, Hebestreit K. An evaluation of methods to test predefined genomic regions for differential methylation in bisulfite sequencing data. Brief Bioinform. 2016;17(5):796–807. doi: 10.1093/bib/bbv095. [DOI] [PubMed] [Google Scholar]
- 25.Mamrut S, Harony H, Sood R, Shahar-Gold H, Gainer H, Shi Y-J, Barki-Harrington L, Wagner S. DNA methylation of specific CpG sites in the promoter region regulates the transcription of the mouse oxytocin receptor. PLoS ONE. 2013;8(2):56869. doi: 10.1371/journal.pone.0056869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang X, Wu M, Xiao H, Lee MT, Levin L, Leung YK, Ho SM. Methylation of a single intronic CpG mediates expression silencing of the PMP24 gene in prostate cancer. Prostate. 2010;70(7):765–76. doi: 10.1002/pros.21109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kitazawa R, Kitazawa S. Methylation status of a single CpG locus 3 bases upstream of TATA-box of receptor activator of nuclear factor-kappaB ligand (RANKL) gene promoter modulates cell- and tissue-specific RANKL expression and osteoclastogenesis. Mol Endocrinol. 2007;21(1):148–58. doi: 10.1210/me.2006-0205. [DOI] [PubMed] [Google Scholar]
- 28.Wang T, Li J, Ding K, Zhang L, Che Q, Sun X, Dai Y, Sun W, Bao M, Wang X, Yang L, Li Z. The CpG Dinucleotide Adjacent to a kB Site Affects NF-kB Function through Its Methylation. Int J Mol Sci. 2017;18(3). [DOI] [PMC free article] [PubMed]
- 29.Lim KH, Park ES, Kim DH, Cho KC, Kim KP, Park YK, Ahn SH, Park SH, Kim KH, Kim CW, Kang HS, Lee AR, Park S, Sim H, Won J, Seok K, You JS, Lee JH, Yi NJ, Lee KW, Suh KS, Seong BL, Kim KH. Suppression of interferon-mediated anti-HBV response by single CpG methylation in the 5’-UTR of TRIM22. Gut. 2018;67(1):166–78. doi: 10.1136/gutjnl-2016-312742. [DOI] [PubMed] [Google Scholar]
- 30.Claus R, Lucas DM, Stilgenbauer S, Ruppert AS, Yu L, Zucknick M, Mertens D, Buhler A, Oakes CC, Larson RA, Kay NE, Jelinek DF, Kipps TJ, Rassenti LZ, Gribben JG, Dohner H, Heerema NA, Marcucci G, Plass C, Byrd JC. Quantitative DNA methylation analysis identifies a single CpG dinucleotide important for ZAP-70 expression and predictive of prognosis in chronic lymphocytic leukemia. J Clin Oncol. 2012;30(20):2483–91. doi: 10.1200/JCO.2011.39.3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pogribny IP, Pogribna M, Christman JK, James SJ. Single-site methylation within the p53 promoter region reduces gene expression in a reporter gene construct: possible in vivo relevance during tumorigenesis. Cancer Res. 2000;60(3):588–94. [PubMed] [Google Scholar]
- 32.Qiang M, Denny A, Chen J, Ticku MK, Yan B, Henderson G. The site specific demethylation in the 5’-regulatory area of NMDA receptor 2B subunit gene associated with CIE-induced up-regulation of transcription. PLoS ONE. 2010;5(1):8798. doi: 10.1371/journal.pone.0008798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mamrut S, Harony H, Sood R, Shahar-Gold H, Gainer H, Shi YJ, Barki-Harrington L, Wagner S. DNA methylation of specific CpG sites in the promoter region regulates the transcription of the mouse oxytocin receptor. PLoS ONE. 2013;8(2):56869. doi: 10.1371/journal.pone.0056869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang T, Chen M, Liu L, Cheng H, Yan YE, Feng YH, Wang H. Nicotine induced CpG methylation of Pax6 binding motif in StAR promoter reduces the gene expression and cortisol production. Toxicol Appl Pharmacol. 2011;257(3):328–37. doi: 10.1016/j.taap.2011.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ceccarelli V, Racanicchi S, Martelli MP, Nocentini G, Fettucciari K, Riccardi C, Marconi P, Di Nardo P, Grignani F, Binaglia L, Vecchini A. Eicosapentaenoic acid demethylates a single CpG that mediates expression of tumor suppressor CCAAT/enhancer-binding protein delta in U937 leukemia cells. J Biol Chem. 2011;286(31):27092–102. doi: 10.1074/jbc.M111.253609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Snow JW, Trowbridge JJ, Fujiwara T, Emambokus NE, Grass JA, Orkin SH, Bresnick EH. A single cis element maintains repression of the key developmental regulator Gata2. PLoS Genet. 2010;6(9):1001103. doi: 10.1371/journal.pgen.1001103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nile CJ, Read RC, Akil M, Duff GW, Wilson AG. Methylation status of a single CpG site in the IL6 promoter is related to IL6 messenger RNA levels and rheumatoid arthritis. Arthritis Rheum. 2008;58(9):2686–93. doi: 10.1002/art.23758. [DOI] [PubMed] [Google Scholar]
- 38.Pant V, Kurukuti S, Pugacheva E, Shamsuddin S, Mariano P, Renkawitz R, Klenova E, Lobanenkov V, Ohlsson R. Mutation of a single CTCF target site within the H19 imprinting control region leads to loss of Igf2 imprinting and complex patterns of de novo methylation upon maternal inheritance. Mol Cell Biol. 2004;24(8):3497–504. doi: 10.1128/MCB.24.8.3497-3504.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mesquita P, Peixoto AJ, Seruca R, Hanski C, Almeida R, Silva F, Reis C, David L. Role of site-specific promoter hypomethylation in aberrant MUC2 mucin expression in mucinous gastric carcinomas. Cancer Lett. 2003;189(2):129–36. doi: 10.1016/S0304-3835(02)00549-9. [DOI] [PubMed] [Google Scholar]
- 40.Medvedeva YA, Khamis AM, Kulakovskiy IV, Ba-Alawi W, Bhuyan MSI, Kawaji H, Lassmann T, Harbers M, Forrest ARR, Bajic VB. Effects of cytosine methylation on transcription factor binding sites. BMC Genomics. 2014;15:119. doi: 10.1186/1471-2164-15-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pardo LM, Rizzu P, Francescatto M, Vitezic M, Leday GGR, Sanchez JS, Khamis A, Takahashi H, van de Berg WDJ, Medvedeva YA, van de Wiel MA, Daub CO, Carninci P, Heutink P. Regional differences in gene expression and promoter usage in aged human brains. Neurobiol Aging. 2013;34(7):1825–36. doi: 10.1016/j.neurobiolaging.2013.01.005. [DOI] [PubMed] [Google Scholar]
- 42.Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, Batzoglou S. Identifying a high fraction of the human genome to be under selective constraint using GERP++ PLoS Comput Biol. 2010;6(12):1001025. doi: 10.1371/journal.pcbi.1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010;20(1):110–21. doi: 10.1101/gr.097857.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ernst J, Kellis M. ChromHMM: automating chromatin-state discovery and characterization. Nat Methods. 2012;9(3):215–6. doi: 10.1038/nmeth.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ionita-Laza I, McCallum K, Xu B, Buxbaum JD. A spectral approach integrating functional genomic annotations for coding and noncoding variants. Nat Genet. 2016;48(2):214–20. doi: 10.1038/ng.3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.FANTOM Consortium and the RIKEN PMI and CLST (DGT) Forrest ARR, Kawaji H, Rehli M, Baillie JK, de Hoon MJL, Haberle V, Lassmann T, Kulakovskiy IV, Lizio M, Itoh M, Andersson R, Mungall CJ, Meehan TF, Schmeier S, Bertin N, Jørgensen M, Dimont E, Arner E, Schmidl C, Schaefer U, Medvedeva YA, Plessy C, Vitezic M, Severin J, Semple CA, Ishizu Y, Young RS, Francescatto M, Alam I, Albanese D, Altschuler GM, Arakawa T, Archer JAC, Arner P, Babina M, Rennie S, Balwierz PJ, Beckhouse AG, Pradhan-Bhatt S, Blake JA, Blumenthal A, Bodega B, Bonetti A, Briggs J, Brombacher F, Burroughs AM, Califano A, Cannistraci CV, Carbajo D, Chen Y, Chierici M, Ciani Y, Clevers HC, Dalla E, Davis CA, Detmar M, Diehl AD, Dohi T, Drabløs F, Edge ASB, Edinger M, Ekwall K, Endoh M, Enomoto H, Fagiolini M, Fairbairn L, Fang H, Farach-Carson MC, Faulkner GJ, Favorov AV, Fisher ME, Frith MC, Fujita R, Fukuda S, Furlanello C, Furino M, Furusawa J-I, Geijtenbeek TB, Gibson AP, Gingeras T, Goldowitz D, Gough J, Guhl S, Guler R, Gustincich S, Ha TJ, Hamaguchi M, Hara M, Harbers M, Harshbarger J, Hasegawa A, Hasegawa Y, Hashimoto T, Herlyn M, Hitchens KJ, Ho Sui SJ, Hofmann OM, Hoof I, Hori F, Huminiecki L, Iida K, Ikawa T, Jankovic BR, Jia H, Joshi A, Jurman G, Kaczkowski B, Kai C, Kaida K, Kaiho A, Kajiyama K, Kanamori-Katayama M, Kasianov AS, Kasukawa T, Katayama S, Kato S, Kawaguchi S, Kawamoto H, Kawamura YI, Kawashima T, Kempfle JS, Kenna TJ, Kere J, Khachigian LM, Kitamura T, Klinken SP, Knox AJ, Kojima M, Kojima S, Kondo N, Koseki H, Koyasu S, Krampitz S, Kubosaki A, Kwon AT, Laros JFJ, Lee W, Lennartsson A, Li K, Lilje B, Lipovich L, Mackay-Sim A, Manabe R-I, Mar JC, Marchand B, Mathelier A, Mejhert N, Meynert A, Mizuno Y, de Lima Morais DA, Morikawa H, Morimoto M, Moro K, Motakis E, Motohashi H, Mummery CL, Murata M, Nagao-Sato S, Nakachi Y, Nakahara F, Nakamura T, Nakamura Y, Nakazato K, van Nimwegen E, Ninomiya N, Nishiyori H, Noma S, Noma S, Noazaki T, Ogishima S, Ohkura N, Ohimiya H, Ohno H, Ohshima M, Okada-Hatakeyama M, Okazaki Y, Orlando V, Ovchinnikov DA, Pain A, Passier R, Patrikakis M, Persson H, Piazza S, Prendergast JGD, Rackham OJL, Ramilowski JA, Rashid M, Ravasi T, Rizzu P, Roncador M, Roy S, Rye MB, Saijyo E, Sajantila A, Saka A, Sakaguchi S, Sakai M, Sato H, Savvi S, Saxena A, Schneider C, Schultes EA, Schulze-Tanzil GG, Schwegmann A, Sengstag T, Sheng G, Shimoji H, Shimoni Y, Shin JW, Simon C, Sugiyama D, Sugiyama T, Suzuki M, Suzuki N, Swoboda RK, ’t Hoen PAC, Tagami M, Takahashi N, Takai J, Tanaka H, Tatsukawa H, Tatum Z, Thompson M, Toyodo H, Toyoda T, Valen E, van de Wetering M, van den Berg LM, Verado R, Vijayan D, Vorontsov IE, Wasserman WW, Watanabe S, Wells CA, Winteringham LN, Wolvetang E, Wood EJ, Yamaguchi Y. A promoter-level mammalian expression atlas. Nature. 2014;507(7493):462–70. doi: 10.1038/nature13182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lesch BJ, Page DC. Poised chromatin in the mammalian germ line. Development. 2014;141(19):3619. doi: 10.1242/dev.113027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Domcke S, Bardet AF, Adrian Ginno P, Hartl D, Burger L, Schubeler D. Competition between DNA methylation and transcription factors determines binding of NRF1. Nature. 2015;528(7583):575–9. doi: 10.1038/nature16462. [DOI] [PubMed] [Google Scholar]
- 49.Yin Y, Morgunova E, Jolma A, Kaasinen E, Sahu B, Khund-Sayeed S, Das PK, Kivioja T, Dave K, Zhong F, Nitta KR, Taipale M, Popov A, Ginno PA, Domcke S, Yan J, Schubeler D, Vinson C, Taipale J. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science. 2017;356(6337). [DOI] [PMC free article] [PubMed]
- 50.Hermann A, Goyal R, Jeltsch A. The dnmt1 DNA-(cytosine-C5)-methyltransferase methylates DNA processively with high preference for hemimethylated target sites. J Biol Chem. 2004;279(46):48350–9. doi: 10.1074/jbc.M403427200. [DOI] [PubMed] [Google Scholar]
- 51.Stepper P, Kungulovski G, Jurkowska RZ, Chandra T, Krueger F, Reinhardt R, Reik W, Jeltsch A, Jurkowski TP. Efficient targeted DNA methylation with chimeric dCas9-Dnmt3a-Dnmt3L methyltransferase. Nucleic Acids Res. 2016; 45(4). [DOI] [PMC free article] [PubMed]
- 52.Turker MS. Gene silencing in mammalian cells and the spread of DNA methylation. Oncogene. 2002;21(35):5388–93. doi: 10.1038/sj.onc.1205599. [DOI] [PubMed] [Google Scholar]
- 53.Sarda S, Das A, Vinson C, Hannenhalli S. Distal CpG islands can serve as alternative promoters to transcribe genes with silenced proximal-promoters. Genome Res. 2017;27(4). [DOI] [PMC free article] [PubMed]
- 54.Lizio M, Harshbarger J, Shimoji H, Severin J, Kasukawa T, Sahin S, Abugessaisa I, Fukuda S, Hori F, Ishikawa-Kato S, Mungall CJ, Arner E, Baillie JK, Bertin N, Bono H, de Hoon M, Diehl AD, Dimont E, Freeman TC, Fujieda K, Hide W, Kaliyaperumal R, Katayama T, Lassmann T, Meehan TF, Nishikata K, Ono H, Rehli M, Sandelin A, Schultes EA, ‘t Hoen PA, Tatum Z, Thompson M, Toyoda T, Wright DW, Daub CO, Itoh M, Carninci P, Hayashizaki Y, Forrest AR, Kawaji H, the FANTOM consortium Gateways to the fantom5 promoter level mammalian expression atlas. Genome Biol. 2015;16(1):22. doi: 10.1186/s13059-014-0560-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lizio M, Harshbarger J, Abugessaisa I, Noguchi S, Kondo A, Severin J, Mungall C, Arenillas D, Mathelier A, Medvedeva YA, Lennartsson A, Drabl?s F, Ramilowski JA, Rackham O, Gough J, Andersson R, Sandelin A, Ienasescu H, Ono H, Bono H, Hayashizaki Y, Carninci P, Forrest AR, Kasukawa T, Kawaji H. Update of the FANTOM web resource: high resolution transcriptome of diverse cell types in mammals. Nucleic Acids Res. 2017; 45(D1):737–43. [DOI] [PMC free article] [PubMed]
- 56.Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, Garg K, John S, Sandstrom R, Bates D, Boatman L, Canfield TK, Diegel M, Dunn D, Ebersol AK, Frum T, Giste E, Johnson AK, Johnson EM, Kutyavin T, Lajoie B, Lee B-K, Lee K, London D, Lotakis D, Neph S, Neri F, Nguyen ED, Qu H, Reynolds AP, Roach V, Safi A, Sanchez ME, Sanyal A, Shafer A, Simon JM, Song L, Vong S, Weaver M, Yan Y, Zhang Z, Zhang Z, Lenhard B, Tewari M, Dorschner MO, Hansen RS, Navas PA, Stamatoyannopoulos G, Iyer VR, Lieb JD, Sunyaev SR, Akey JM, Sabo PJ, Kaul R, Furey TS, Dekker J, Crawford GE, Stamatoyannopoulos JA. The accessible chromatin landscape of the human genome. Nature. 2012;489(7414):75–82. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang H, Maurano MT, Qu H, Varley KE, Gertz J, Pauli F, Lee K, Canfield T, Weaver M, Sandstrom R, Thurman RE, Kaul R, Myers RM, Stamatoyannopoulos JA. Widespread plasticity in CTCF occupancy linked to DNA methylation. Genome Res. 2012;22(9):1680–8. doi: 10.1101/gr.136101.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics. 2014;30(15):2114–20. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17(1):10–12. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 60.Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011;27(11):1571–2. doi: 10.1093/bioinformatics/btr167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25(9):1105–11. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liao Y, Smyth GK, Shi W. featurecounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923–30. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 63.Panchin AY, Makeev VJ, Medvedeva YA. Preservation of methylated CpG dinucleotides in human CpG islands. Biol Direct. 2016;11(1):11. doi: 10.1186/s13062-016-0113-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Medvedeva YA. Bioinformatics - Trends and Methodologies. London: IntechOpen Limited; 2011. Algorithms for CpG islands search: New advantages and old problems. [Google Scholar]
- 65.Andersson R, Gebhard C, Miguel-Escalada I, Hoof I, Bornholdt J, Boyd M, Chen Y, Zhao X, Schmidl C, Suzuki T, Ntini E, Arner E, Valen E, Li K, Schwarzfischer L, Glatz D, Raithel J, Lilje B, Rapin N, Bagger FO, Jørgensen M, Andersen PR, Bertin N, Rackham O, Burroughs AM, Baillie JK, Ishizu Y, Shimizu Y, Furuhata E, Maeda S, Negishi Y, Mungall CJ, Meehan TF, Lassmann T, Itoh M, Kawaji H, Kondo N, Kawai J, Lennartsson A, Daub CO, Heutink P, Hume DA, Jensen TH, Suzuki H, Hayashizaki Y, Müller F, FANTOM Consortium. Forrest ARR, Carninci P, Rehli M, Sandelin A. An atlas of active enhancers across human cell types and tissues. Nature. 2014;507(7493):455–61. doi: 10.1038/nature12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Roadmap Epigenomics Consortium. Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, Ziller MJ, Amin V, Whitaker JW, Schultz MD, Ward LD, Sarkar A, Quon G, Sandstrom RS, Eaton ML, Wu Y-C, Pfenning AR, Wang X, Claussnitzer M, Liu Y, Coarfa C, Harris RA, Shoresh N, Epstein CB, Gjoneska E, Leung D, Xie W, Hawkins RD, Lister R, Hong C, Gascard P, Mungall AJ, Moore R, Chuah E, Tam A, Canfield TK, Hansen RS, Kaul R, Sabo PJ, Bansal MS, Carles A, Dixon JR, Farh K-H, Feizi S, Karlic R, Kim A-R, Kulkarni A, Li D, Lowdon R, Elliott G, Mercer TR, Neph SJ, Onuchic V, Polak P, Rajagopal N, Ray P, Sallari RC, Siebenthall KT, Sinnott-Armstrong NA, Stevens M, Thurman RE, Wu J, Zhang B, Zhou X, Beaudet AE, Boyer LA, De Jager PL, Farnham PJ, Fisher SJ, Haussler D, Jones SJM, Li W, Marra MA, McManus MT, Sunyaev S, Thomson JA, Tlsty TD, Tsai L-H, Wang W, Waterland RA, Zhang MQ, Chadwick LH, Bernstein BE, Costello JF, Ecker JR, Hirst M, Meissner A, Milosavljevic A, Ren B, Stamatoyannopoulos JA, Wang T, Kellis M. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518(7539):317–30. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kulakovskiy IV, Vorontsov IE, Yevshin IS, Soboleva AV, Kasianov AS, Ashoor H, Ba-Alawi W, Bajic VB, Medvedeva YA, Kolpakov FA, Makeev VJ. HOCOMOCO: expansion and enhancement of the collection of transcription factor binding sites models. Nucleic Acids Res. 2016;44(D1):116–25. doi: 10.1093/nar/gkv1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vorontsov IE, Khimulya G, Lukianova EN, Nikolaeva DD, Eliseeva IA, Kulakovskiy IV, Makeev VJ. Negative selection maintains transcription factor binding motifs in human cancer. BMC Genomics. 2016;17 Suppl 2:395. doi: 10.1186/s12864-016-2728-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vorontsov IE, Fedorova AD, Yevshin IS, Sharipov RN, Kolpakov FA, Makeev VJ, Kulakovskiy IV. Genome-wide map of human and mouse transcription factor binding sites aggregated from chip-seq data. BMC Res Notes. 2018;11(1):756. doi: 10.1186/s13104-018-3856-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yevshin I, Sharipov R, Valeev T, Kel A, Kolpakov F. GTRD: a database of transcription factor binding sites identified by ChIP-seq experiments. Nucleic Acids Res. 2017;45(D1):61–7. doi: 10.1093/nar/gkw951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schmeier S, Alam T, Essack M, Bajic VB. TcoF-DB v2: update of the database of human and mouse transcription co-factors and transcription factor interactions. Nucleic Acids Res. 2017;45(D1):145–50. doi: 10.1093/nar/gkw1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Medvedeva YA, Lennartsson A, Ehsani R, Kulakovskiy IV, Vorontsov IE, Panahandeh P, Khimulya G, Kasukawa T, FANTOM Consortium. Drabløs F. EpiFactors: a comprehensive database of human epigenetic factors and complexes. Database. 2015;2015:067. doi: 10.1093/database/bav067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vorontsov IE, Fedorova AD, Yevshin IS, Sharipov RN, Kolpakov FA, Makeev VJ, Kulakovskiy IV. Human and mouse cistromes: genomic maps of putative cis-regulatory regions bound by transcription factors. 2018. 10.6084/m9.figshare.7087697.v1.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary materials. Figure S1: SCC of the CpG TL located in various gene regions; Figure S2: Distribution of CpG TLs along the genome; Figure S3: TFBS; Table S1: Number of significant SCC between average methylation of genomic region and gene expression; Table S2: Number of significant SCC between CpG methylation and gene expression; Table S3: Number of significant SCC between gene expression and average methylation of the genome region; Table S4: Most enriched with CpG TLs categories of enhancers; Table S5: Names of the cell samples in the study; Table S6: Enhancers = H3K27ac+H3K4me1-H3K4me3; Table S7: Expression data source; Table S8: Methylation data source. (PDF 2562 kb)
Data Availability Statement
The datasets analysed during the current study are available in the Roadmap Epigenomics Project and FANTOM5, the links to the main datasets are in Additional file 1: Table S7 (expression) and Additional file 1: Table S8 (methylation). The cistrome data are available at Figshare [73].