

Abstract
Objective:
Hcy is an independent risk factor for cerebrovascular disease and cognitive impairment. The purpose of this study was to elucidate the role of mGluR5 in Hcy-mediated impairment of cerebral endothelial wound repair.
Methods:
Mouse CMVECs (bEnd.3) were used in conjunction with directed pharmacology and shRNA. AutoDock was used to simulate the docking of ligand-receptor interactions.
Results:
Hcy (20 μM) significantly increased Cx43-pS368 by mGluR5- and PKC-dependent mechanisms. Hcy attenuated wound repair by an mGluR5-dependent mechanism over the six-day study period but did not alter cell proliferation in a proliferation assay, suggesting that the attenuation of wound repair may be due to dysfunctional migration in HHcy. Hcy increased the expression of Cx43 and Cx43-pS368 at the wound edge by activating mGluR5. Direct activation of mGluR5, using the specific agonist CHPG, was sufficient to reproduce the results whereas KO of mGluR5 with shRNA, or inhibition with MPEP, blocked the response to Hcy.
Conclusions:
Inhibition of mGluR5 activation could be a novel strategy for promoting endothelial wound repair in patients with HHcy. Activation of mGluR5 may be a viable strategy for disrupting angiogenesis.
Keywords: connexin43, Cx43-pS368, bEnd.3, wound healing, angiogenesis, protein kinase C
INTRODUCTION
An elevated plasma concentration of Hcy, HHcy, is an independent risk factor for cardiovascular and neurocognitive diseases [9,17,40], including stroke and dementia [13,22]. Clinically, HHcy is classified as mild (15–30 μM), moderate (31–100 μM), or severe (>100 μM). In 1976, Harker et al. [12] showed striking images of the loss of endothelium from the vascular lumen in baboons with HHcy. Over subsequent years, several laboratories reported that Hcy dose-dependently decreases EC wound repair and angiogenesis [11,23,25,33,34]. After stroke or brain injury, EC wound repair and angiogenesis are essential for optimal neurological and functional recovery [4]. Patients with greater vascular remodeling and endothelial growth survive the longest following stroke [19]. A fundamental gap in our knowledge is the identity of the receptor on vascular endothelium that is activated by Hcy to inhibit wound repair.
mGluRs are members of the group C family of G-protein-coupled receptors [2]. Hcy activates mGluR5 in neurons [41]. Although Hcy activation of mGluR5 has not been tested in endothelium, vascular ECs do express mGluR5 in vitro [6] and in vivo [10]. mGluR5 is coupled to the αq/11 G-protein subunit, which activates phospholipase C with subsequent production of inositol trisphosphate and DAG from phosphatidylinositol bisphosphate. DAG remains in the cell membrane and activates PKC. We recently demonstrated that Hcy induces endothelial nitrative stress by activating mGluR5 (in press, Journal of Cerebral Blood Flow & Metabolism). Hence, mGluR5 is a rational candidate for Hcy-mediated inhibition of wound repair. The overall goals of our studies have been to determine whether there is a cell surface receptor for extracellular Hcy and to elucidate the intracellular players in Hcy-dependent impairment of EC wound repair.
Wound repair requires cell proliferation and migration. Cx43 is a ubiquitous protein that plays a key role in both of these processes. Cxs are a family of proteins that form selective membrane channels, often creating gap junctions that couple the cytoplasm of adjacent cells. Cx43 can regulate cell cycle traverse independent of gap junctional communication by slowing cell proliferation [16]. Cx43 also regulates cell migration [28,29], possibly through signaling to the cytoskeleton through proteins such as zonula occludens-1. This regulation may involve phosphorylation of serine residues [1,7,37,38], especially S368 (Cx43-pS368), which is involved in the regulation of skin wound repair [32]. The dynamics of Cx43 expression and phosphorylation at S368 during wound repair of cerebral ECs are not defined and the effect of Hcy on these dynamics is unknown. In this study, we tested the specific hypothesis that mGluR5 activation is responsible for impaired wound healing of cerebral ECs caused by Hcy and that this is associated with sustained expression of Cx43 and phosphorylation at Cx43-S368 at the wound edge.
MATERIALS AND METHODS
All procedures and safety measures were approved by the Idaho State University IACUC. All chemicals were purchased from Fisher Scientific (Waltham, MA, USA) or Sigma-Aldrich (St. Louis, MO, USA) unless indicated otherwise. Hcy was D/L-Hcy purchased from Sigma-Aldrich.
Cell Culture and Chemicals
Mouse brain microvascular ECs (bEnd.3; American Type Culture Collection, Manassas, VA, USA, catalog no. CRL-2299) were grown to confluence in T25 flasks, 12-, 24-, or 96-well plates in the DMEM-H (Hyclone, Logan, UT, USA) supplemented with 10% bovine calf serum. Cells were maintained in a humidified incubator at 37°C with 5% CO2/95% room air. Experiments began at three days postconfluence. The treatment reagents included PBS and DMSO as vehicle controls; Hcy, PMA, and MPEP, dissolved in sterile PBS; and BIM and CHPG, dissolved in DMSO.
Stable KO of mGluR5
shRNA-mGluR5 KO was achieved using the lentiviral vector GIPZ containing the shRNAmir sequences V3LMM_455250 (shRNA-mGluR5) from Open Biosystems (Huntsville, AL, USA), and the NS shRNAmir (catalog no. RHS4348) was used as a negative control. Optimal multiplicity of infection was determined following the manufacturer’s instructions. A stable culture was generated by growing these cells in the presence of 10 μg/mL puromycin for seven days. Percentage of mGluR5 KO was quantified with cell-surface ELISA and Western blot by immunolabeling for the extracellular domain of mGluR5 (Santa Cruz Biotechnology, Santa Cruz, CA, USA, catalog no. sc-47147).
In-Cell ELISA
Cells were fixed in 4% paraformaldehyde (in PBS) at room temperature for 15 minutes. The cells were washed twice with TBS for five minutes each and permeabilized in TBS with 1% Triton X-100 at room temperature for 15 minutes, blocked with 3% bovine serum albumin (w/v) in TBS with 0.05% Tween-20 at room temperature for 30 minutes, and probed using the polyclonal rabbit anti-Cx43 (Santa Cruz Biotechnology, catalog no. sc-9059) or goat anti-Cx43-pS368 primary antibody (Santa Cruz Biotechnology, catalog no. sc-25165) at a 1:500 dilution at 4°C overnight. The sample proteins were washed three times with wash buffer (TBS with 1% Tween-20) for five minutes each and incubated in the alkaline phosphatase-conjugated goat antirabbit secondary antibody (Vector Laboratories, Burlingame, CA, USA) at a 1:5000 dilution at room temperature for 30 minutes followed by three washes in wash buffer and TBS. The absorbance of blue phosphatase substrates (KPL, Gaithersburg, MO, USA) was quantified at a wavelength of 620 nm using a plate reader (Synergy HT, BioTek Instruments, Winooski, VT, USA).
Immunohistochemistry
Primary antibodies were rabbit anti-Cx43 (Santa Cruz Biotechnology, catalog no. sc-9059) and goat anti-Cx43-pS368 (Santa Cruz Biotechnology, catalog no. sc-25165). Secondary antibodies were conjugated to AlexaFluor-564 (for Cx43; Invitrogen, Eugene, OR, USA) and AlexaFluor-488 (for Cx43-pS368; Invitrogen, Eugene, OR, USA). Images were acquired by a fluorescence microscopy (Leica DMLFS, Deerfield, IL, USA) with a digital camera (Micropublisher 3.3 RTV, QImaging Corporate Headquarters, Surrey, BC, Canada) using QCapture 5.1 Software (QImaging Corporate Headquarters, Surrey, BC, Canada).
AutoDock Simulation
AutoDock applies protein-protein interaction computational models to calculate the binding energies of molecules to molecular receptors. PDB was used to input the three dimensional crystal structures of three ligands (glutamate, CHPG, and Hcy) and the molecular receptor mGluR5 (PDB code is 3LMK). All ligands were given hydrogen atoms, defined ROOT and rotatable bonds, and assigned charges by default settings. Then the macromolecule mGluR5 was given hydrogen atoms, checked for missing atoms, and assigned default charges and solvation parameters. Map types and grid dimension were chosen as each ligand and 62, respectively. After running AutoGrid, docking parameters including setting protein and ligand, choosing search method (genetic algorithm), and setting docking parameters were chosen in order to run the AutoDock. Then the best conformers and docked energies were computed.
Cell Proliferation Assays
Cell proliferation was assessed by plating cells at 20–30% confluence in 96-well plates in the late afternoon. Experiment timing began the following morning, designated as time zero, after a media change to remove any cells that did not adhere. At the times indicated in respective experiments, proliferation was stopped by rinsing a plate with PBS, removing all fluid, and storing at −20°C beginning with the time zero. Cell number was quantified by counting the number of cells in one microscope field of view (10× objective) at the center of each well. The perimeter of each well was inspected; there were no differences in cell density between center and periphery for any well (i.e., cell density was homogeneous throughout each well). In an identical set of experiments, total cell protein in each well was quantified using a BCA colorimetric assay (Thermo Scientific, Rockford, IL, USA, catalog no. 23227). Proliferation was defined as the number of new cells or protein: count/protein for that day minus count/protein at day zero. Cells for all time points of a given experiment were seeded at the same time to ensure that the starting population and density were essentially identical.
Wound Repair Assays
Wound repair assays were performed using an in vitro scrape model. bEnd.3 cells were seeded in 12-well plates and studied three days postconfluence. Cells were removed (“scraped”) using a 5-mm wide plastic cell scraper by drawing the scraper across the middle of each well (top to bottom, through the center). Cells were rinsed three times with warm Minimal Essential Medium (Hyclone, Logan, UT, USA, catalog no. SH30235.02) to remove scraped and loosened cells followed by incubation in DMEM-H with respective drug or control treatments. The DMEM-H with treatments were replaced every 24 hours. At the time of media change, culture plates were rapidly moved across the microscope stage, pausing momentarily at each measurement site while the images were captured on a digital video device recorder for later playback and measurement. Cell wound repair distance was quantified as the distance between initial wound edges minus the distance between wound edges each day divided by 2 (to determine average wound repair of each edge). Three measurements were made and averaged as a single measurement for each well on each day.
Statistical Analyses
Alpha was set at 0.05 for statistical significance. One-way ANOVA (Tukey post hoc test) was used to compare groups using computer software programs (SPSS, IBM Corporation, Armonk, NY, USA; Minitab, State College, PA, USA; SAS, Cary, NC, USA). Data are presented as mean ± standard error mean.
RESULTS
Hcy Increases Cx43-pS368 But Not Total Cx43 in the Endothelial Monolayer
As shown in Figure 1A, Cx43 was generally not observed at cell membranes in either control or Hcy-treated conditions, consistent with other studies of this cell line [21]. These results provide the opportunity to investigate the roles of Cx43 in the intracellular domain. Therefore, we explored the expression and phosphorylation of Cx43 using an in-cell ELISA format. Acute treatment with Hcy (two hours) did not change the total Cx43 expression (Figure 1B), but significantly increased Cx43-pS368 (Figure 1C).
Figure 1.
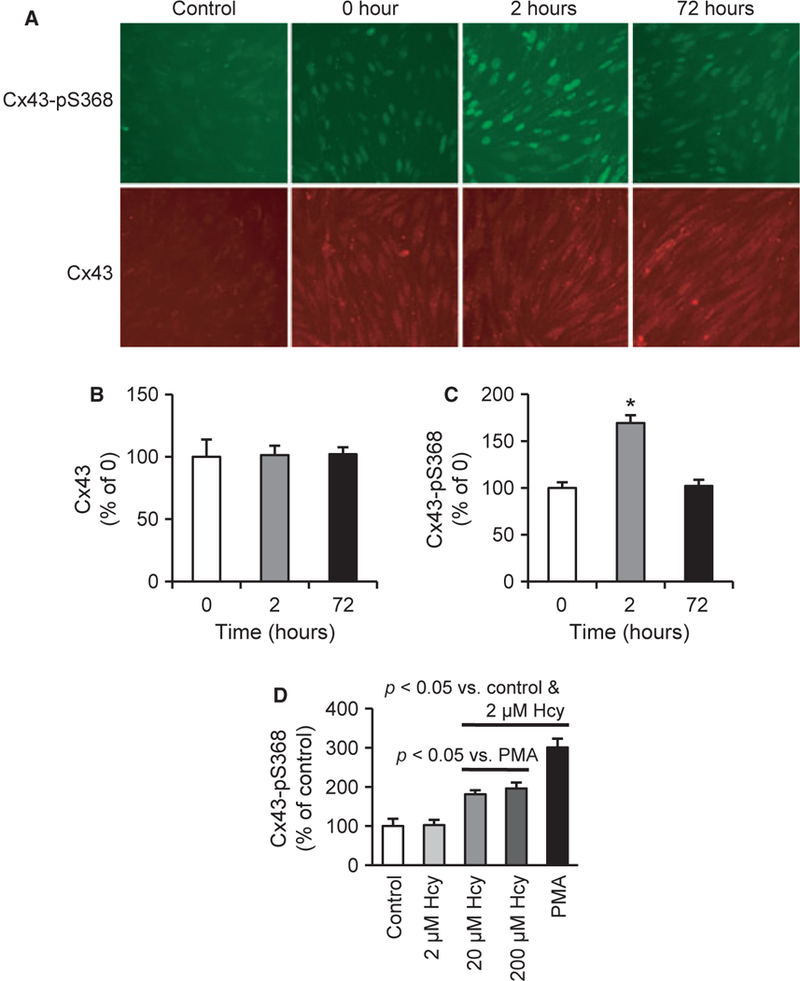
Hcy increases Cx43 phosphorylation on S368 (Cx43-pS368). (A) Cx43-pS368 (green) and total Cx43 (red) expression after treating with Hcy for the indicated times; control is secondary antibody only (omission of the primary antibody). Scale bar is 80 μm and applies to all images. (B-C) ELISA for the expression of Cx43 (B) and Cx43-pS368 (C) (n= 10). (D) Two hour treatments with Hcy and PMA, an activator of PKC, induce significantly greater expression of Cx43-pS368 (n = 10).
PMA, a direct activator of PKC [36], increased Cx43-pS368 suggesting that PKC activation produces the phosphorylation of Cx43 at S368 in this cell line. Figure 1D shows that 20 and 200 μM Hcy significantly increase Cx43-pS368 following two hours treatment.
Hcy Induces Cx43-pS368 Via PKC and mGluR5
Figure 2 shows that both Hcy and PMA induced the phosphorylation of Cx43 at S368 expression, without a change in total Cx43 expression. This finding is consistent with data presented in Figure 1D and suggests that Hcy may activate PKC in cerebral ECs. Treatment with the PKC inhibitor, BIM (1 μM), blocked the Hcy-mediated increase in Cx43-pS368 (Figure 2C). Figure 2D confirms that the Hcy-mediated increase in Cx43-pS368 was also blocked by specific mGluR5 inhibition with MPEP, which did not influence the total Cx43 expression (Figure 2B). As internal controls, neither BIM-1 nor MPEP alone altered the expression of Cx43-pS368. These data suggest that Hcy induces Cx43-pS368 via PKC and mGluR5.
Figure 2.
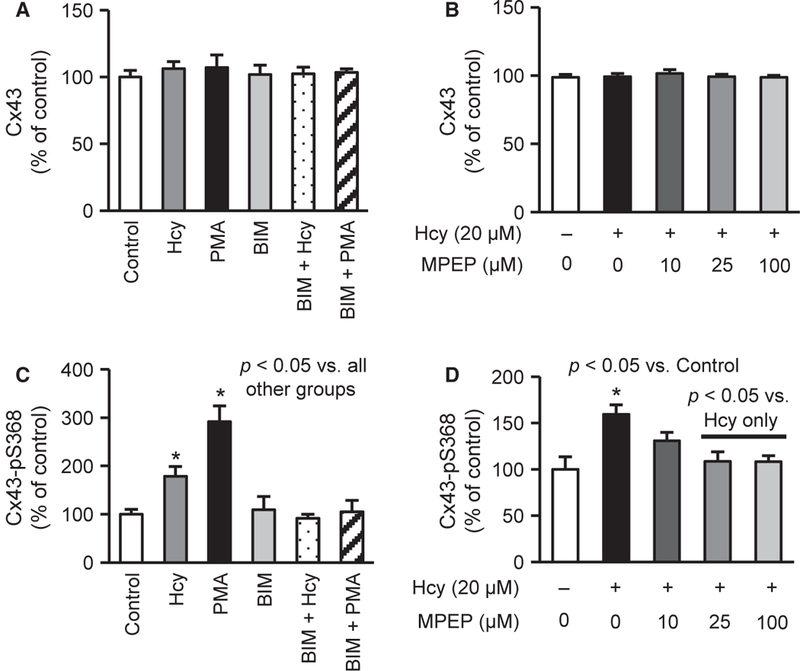
Hcy elevates Cx43-pS368 via PKC and metabotropic glutamate receptor 5 (mGluR5). (A-B) Two-hour treatments with Hcy alone or in combinations with PKC or mGluR5 inhibition do not alter Cx43 expression (n = 10). (C) Two-hour treatments with 20 μM Hcy and 250 nM PMA induce Cx43-pS368, which is blocked by PKC inhibition (1 μM BIM) (n = 8–14). BIM alone does not affect Cx43-pS368. (D) Twenty-micromolar Hcy treatment significantly elevates Cx43-pS368, which is rescued by treating with 25 or 100 μM MPEP, an antagonist of mGluR5 (n = 5–10). MPEP alone does not alter the Cx43-pS368 response (data not shown).
mGluR5 Activation Increases Cx43-pS368 Via PKC
If Hcy leads to Cx43 phosphorylation at S368 by activating mGluR5, as shown in Figure 2D, then activating mGluR5 should directly produce a similar result. Indeed, the activation of mGluR5 with the specific agonist CHPG did not affect Cx43 expression (Figure 3A,B) but produced a dose-dependent increase in Cx43-pS368 expression (Figure 3C), which was inhibited by PKC inhibition (Figure 3D). Collectively, these data demonstrate that Hcy elevates Cx43-pS368 by activating the mGluR5-PKC pathway in cerebral ECs.
Figure 3.
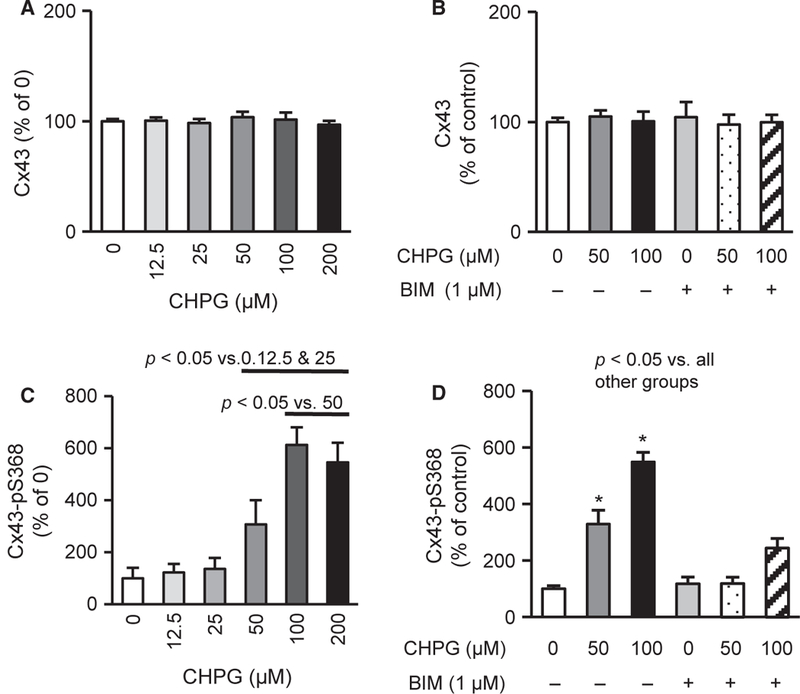
mGluR5 activation induces Cx43-pS368 via PKC. (A-B) Two-hour treatment with CHPG, mGluR5 agonist, and BIM, PKC inhibitor, do not alter Cx43 expression (n = 10). (C) Two-hour treatments with CHPG dose-dependently raises Cx43-pS368 expression (n = 9–12). (D) About 50 and 100 μM CHPG increases Cx43-pS368 expression, which is blocked by PKC inhibition (1 μM BIM) (n = 8–10), suggesting that mGluR5 activation raises Cx43-pS368 via PKC
Predicted Binding of Hcy to mGluR5
Given the results of pharmacologic data, we questioned whether it is likely that Hcy binds mGluR5 directly. Figure 4 shows that binding sites bound by glutamate (Figure 4A) and CHPG (Figure 4B) are those already reported [8,26,27], demonstrating the accuracy of the model prediction in our hands as an internal control. Hcy (Figure 4C) was predicted to bind the same region as glutamate and CHPG, which is consistent with activation of the orthosteric region of the receptor. The docked energy for Hcy is not as strong (negative) as for glutamate but stronger than for CHPG, which provides further evidence that Hcy may activate the receptor directly through the orthosteric region.
Figure 4.
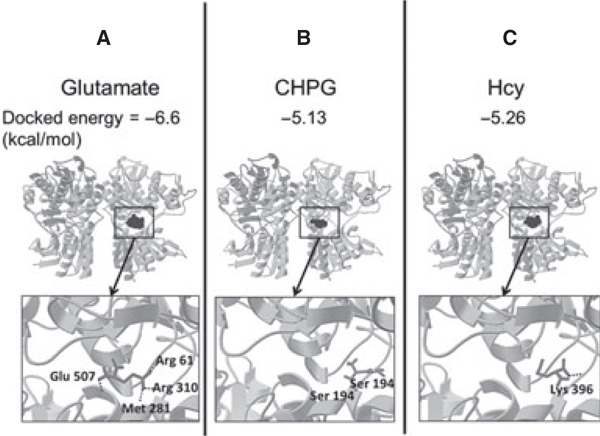
AutoDock predicts docked energies and ligand binding sites on mGluR5. AutoDock, a computer-based modeling software program, was used to predict mGluR5-binding sites and calculate docked energies for (A) glutamate, (B) CHPG (a selective agonist), and (C) Hcy. Glutamate and CHPG are known to bind mGluR5, demonstrating the accuracy of the modeling program in our hands. The binding sites of mGluR5 for Hcy are in the same orthosteric pocket as the two known agonists, glutamate and CHPG. The docked energy for Hcy is intermediate between mGluR5 and CHPG, which suggests that Hcy is likely to bind mGluR5.
Hcy Slows Endothelial Wound Repair by Activating mGluR5
The effect of Hcy on proliferation of brain microvascular ECs has not previously been explored. Hcy treatments slowed EC wound repair (Figure 5A,B) and wound repair rate for the duration of experiments (six days) in a dose-dependent manner (2, 20, and 200 μM).
Figure 5.
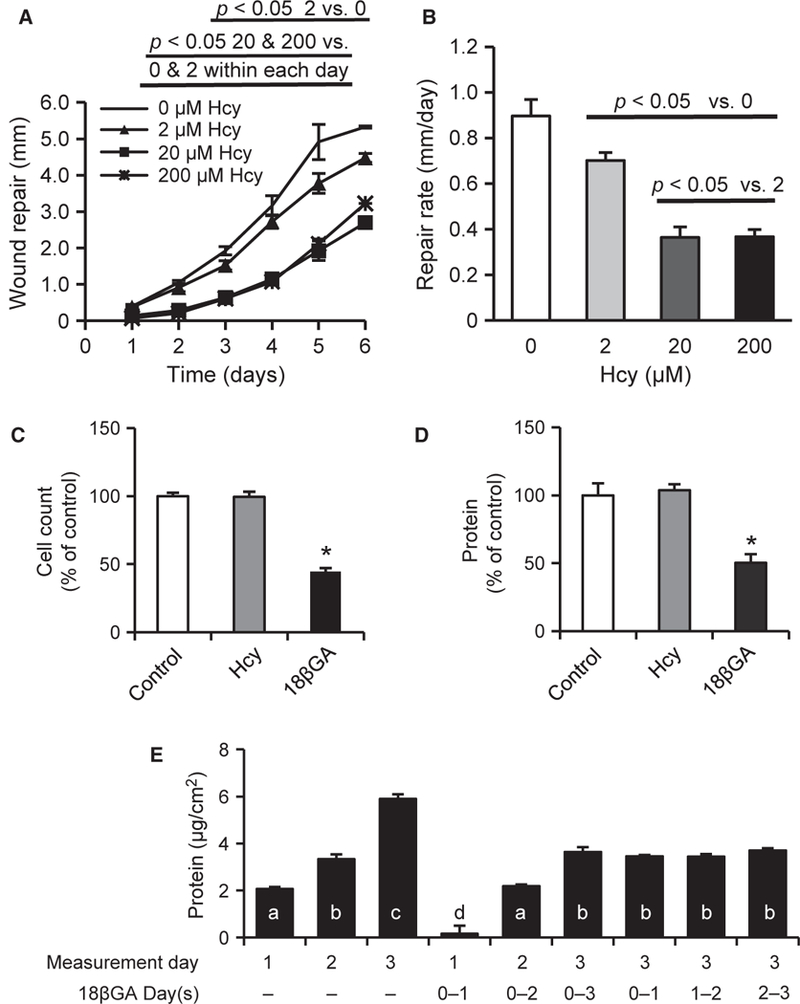
Hcy impairs EC wound repair but not proliferation. (A) About 20 and 200 μM Hcy significantly attenuates daily wound repair distance in in vitro scrape model (n = 6). (B) Hcy dose-dependently impairs the wound repair rate for the duration of one to six days (n = 6). (C-D) Single-cell suspension was plated at 20–30% confluence. The following morning, cells were treated with Hcy, 18βGA, or vehicle control for three days (n = 10). Data are the increase from values determined in parallel cultures harvested at the time of treatment (morning after plating). Cell number (C) and total protein content (D) were not affected by Hcy but were significantly decreased by 18βGA treatment. (E) To better understand the effect of 18βGA, proliferation experiments were repeated with daily measurements in the context of continuous treatment (first six columns) and with treatment for 24 hours only beginning at each of the three days of study (last three columns). Bars with different letters (a-d) are significantly different from one another (p <0.05). Although 18βGA reduced proliferation, the effect appears to be blocking of one day’s worth of proliferation regardless of the timing or duration of treatment (n = 10).
Hcy Does Not Alter Proliferation of bEnd.3 Cells
Hcy treatment (20 μM) did not affect the rate of proliferation of bEnd.3 ECs when plated at 20–30% confluence and quantified daily for three days (Figure 5C,D). The gap junction blocker 18βGA (60 μM) significantly impaired cell proliferation (Figure 5C and5D). However, follow-up studies provided evidence that this treatment may be transient because a series of experiments with treatment for 24 hours or multiple days, and beginning on different days all produced a consistent effect that can be summarized as the loss of one day’s worth of proliferation (Figure 5E). These results demonstrate that Hcy does not alter the proliferation of bEnd.3 cells under these conditions.
To further examine whether mGluR5 is responsible for Hcy-mediated slowing of wound repair, the mGluR5-specific antagonist MPEP was used. Figure 6A,B shows that MPEP blocked the effects of Hcy. These findings are consistent with the idea that Hcy suppresses wound repair by activating mGluR5 signal transduction pathways. To determine whether the activation of mGluR5 is sufficient to slow wound repair, cells were treated with 0, 25, 50, and 100 μM CHPG. Figure 6C,D shows that mGluR5 activation dose-dependently attenuated the repair rate of CMVECs. To approach the question from an alternative direction, we created a stable KO of mGluR5 using shRNA methodology. Figure 7A,B illustrates the results of these experiments and demonstrates that mGluR5 is necessary for the slow wound repair in response to Hcy treatment.
Figure 6.
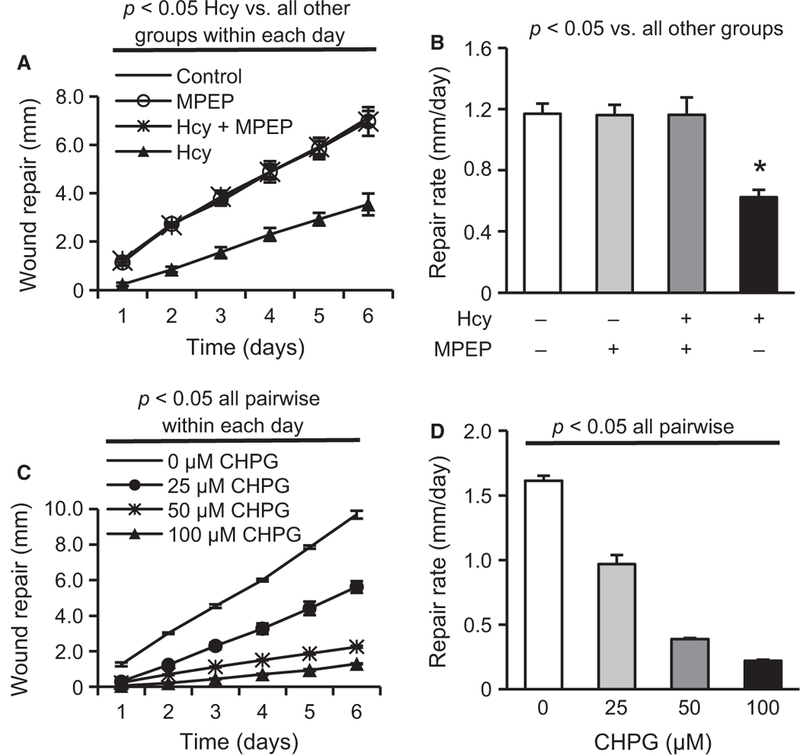
Hcy impairs EC wound repair via mGluR5. Hcy (20 μM) significantly reduces EC (A) daily repair distance and (B) repair rate for one to six days, which are rescued by treatment with the mGluR5-selective antagonist MPEP (25 μM) (n = 3). CHPG, a mGluR5-specific agonist, dose-dependently impairs (C) EC repair distance within each day (n = 3) and (D) EC repair rate for one- to six- day observations (n = 3).
Figure 7.
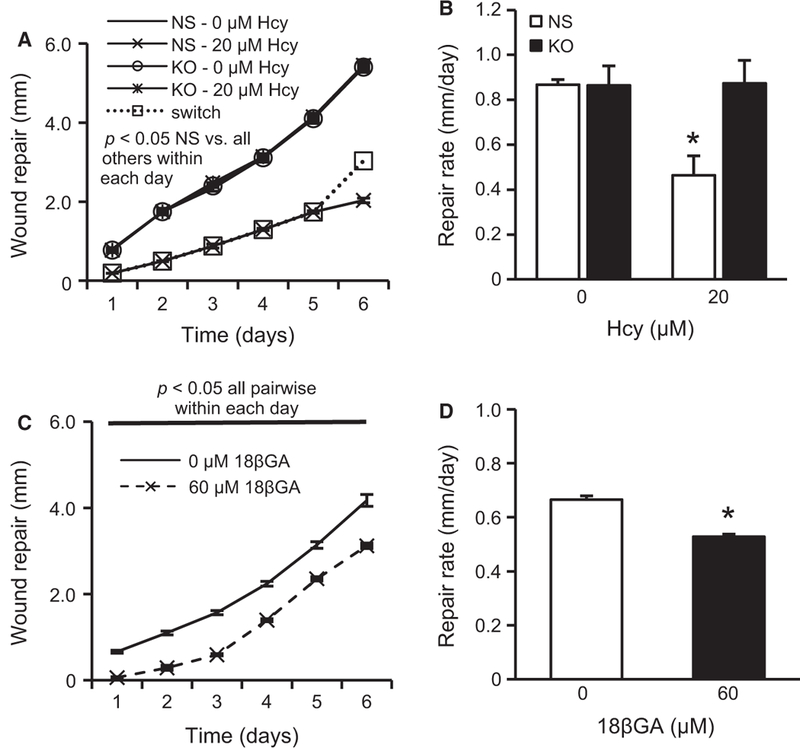
Stable KO of mGluR5 using shRNA rescues the wound repair inhibiting effects of Hcy. Chronic treatment with Hcy (20 μM) impaired the daily wound repair distance (A) and six-day repair rate (B) in cells with an NS control shRNA, which was rescued by shRNA KO of mGluR5 to levels observed in both cell types without Hcy treatment (n = 3). A subset of NS cells (“switch”) was treated with Hcy for five days followed by change to normal medium (no Hcy) for the final day, resulting in a rescued rate (slope of line) to levels observed in the other cell lines. (C-D) Treatment with 18βGA reduced the wound repair distance at each day (C) and the overall six-day repair rate (D) but these effects were entirely due to the nearly complete arrest of repair during the first 24 hours following wounding/treatment because the slope of the daily repair was identical beyond the first day (n = 6).
The gap junction blocker 18βGA also reduced the overall rate of wound repair (Figure 7D) but the daily record (Figure 7C) demonstrates that all of these effects were due to the complete arrest of growth into the wound region during the first 24 hours (i.e., no repair). Beyond the first 24 hours, cells treated with 18βGA filled in the wound area at the same rate as untreated cells (same slope of the daily wound repair line). This pattern was very different from that of cells treated with Hcy where the repair rate was significantly lower throughout the six days of wound repair, which provides evidence that Hcy does not slow wound repair by blocking gap junction or hemichannel communication.
Hcy Increases Cx43 Expression and Phosphorylation of Cells at the Wound Edge by Activating mGluR5
KO and NS control cells were grown to confluence in 96-well plates. Three days postconfluence, cells were scraped with a sterile pipette tip to create wounds in the shape of a plus (+) symbol. This created four edges of EC wounds in a reproducible manner. In-cell ELISAs were used to quantify the expression of Cx43 and Cx43-pS368 at 0, 24, and 72 hours. Hcy significantly increased Cx43 (Figure 8A) and Cx43-pS368 (Figure 8B) expression at 24 and 72 hours compared with zero hour in control cells but not in mGluR5 KO cells. Immunohistochemistry demonstrated that the Hcy-induced expression of Cx43 and Cx43-pS368 (Figure 9) occurs in a band of cells, approximately seven cells deep along the wound edge and not in the confluent monolayer behind. These results are consistent with those in the earlier experiments (Figure 1), which found no change in the expression of Cx43 or Cx43-pS368 in the cell monolayer at 72 hours. We hypothesize that Hcy-mediated activation of mGluR5 may impair EC wound repair by regulating the expression and phosphorylation of Cx43 at the wound edge.
Figure 8.
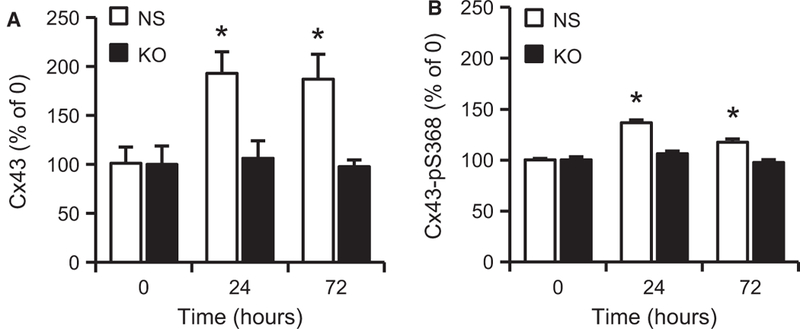
Hcy increases the expression of Cx43 and its phosphorylation at S368 expression via mGluR5 during wound repair. (A) ELISA results demonstrate that Hcy (20 μM) significantly increases Cx43 expression, which is blocked by mGluR5 KO with shRNA but not NS control shRNA (n = 7; *p < 0.05 vs. KO) at 24 and 72 hours after wounding. (B) ELISA results further demonstrate that Hcy significantly induces the phosphorylation of Cx43 at S368, which is blocked by mGluR5 KO with shRNA but not NS control shRNA (n = 7; *p < 0.05 vs. KO) at 24 and 72 hours after wounding.
Figure 9.
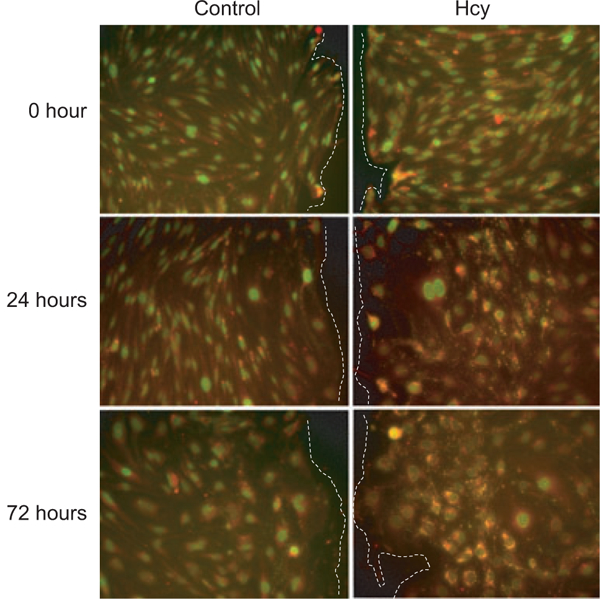
Hcy induces increased expression of Cx43 (red) and Cx43-pS368 (green; yellow is overlap of red and green) along the edge of wounded ECs at 24 and 72 hours after scrape wounding. Wound edges are toward the center line of the paired images and are indicated by dashed lines (i.e., to the right of control images and to the left of Hcy images). Scale bar is 100 μm and applies to all images.
DISCUSSION
This study provides the first evidence for a receptor-dependent pathway by which Hcy impairs cerebral EC wound repair. First, we showed that Hcy induced the intracellular phosphorylation of Cx43 on S368 (Figure 1), which was PKC-and mGluR5-dependent (Figure 2) and that the activation of mGluR5 drove PKC-dependent increases in Cx43-pS368 (Figure 3). Second, Hcy dose-dependently decreased the rate of wound repair by an mGluR5-dependent process (Figures 5 and 6). Third, the activation of mGluR5 was sufficient to slow wound repair (Figure 6). mGluR5 is necessary for the slow wound repair in response to Hcy treatment, but Hcy is unlikely to slow wound repair by blocking the gap junction or hemichannel communication (Figure 7). Fourth, we demonstrated that Hcy raises the expression of both Cx43 and Cx43-pS368 in cells at the wound edge by activating mGluR5 (Figures 8 and 9).
Wound repair requires both cell proliferation and migration. Our results do not support the idea that Hcy impairs cell proliferation in ECs, at least in this model. Cx proteins are best known for their role in creating hemichannels and gap junctions where they couple cells with the extracellular space or the cytosol of adjacent cells, respectively. The gap junction blocker 18βGA impaired cell proliferation and wound repair with a profile completely different from that of Hcy. Specifically, 18βGA seemed to arrest cells for the 24-hour period following the treatment. This could be explained by a resetting of the cell cycle when treated with 18βGA (the cell doubling rate for bEnd.3 cells is approximately 24 hours), although further mechanistic studies are needed. Beyond the initial 24 hours following wounding, 18βGA had no effect on the rate of wound repair. We conclude that the effect of Hcy on slowing wound repair is not through 18βGA-sensitive mechanisms and unlikely to be mediated by gap junctions or hemichannels. Because cell proliferation was not affected by Hcy, we hypothesize that Hcy most likely slowed wound repair by disrupting some aspect of cell migration. Migration of the leading cells requires a complex regulation of orientation, adhesion, microtubule alignment, detachment, and motility. Wound repair requires the proliferation of cells behind the leading edge, which was not specifically tested in the present study. More extensive and focused studies will be required to dissect the mechanisms by which Hcy regulates one or more of these steps in wound repair.
Previous studies concluded that Hcy inhibits cell proliferation by inhibiting DNA methylation through the suppression of DNA methyltransferase 1 in HUVECs [15,38]. These results seem to differ from the results of our study because we did not find an effect of Hcy on cell proliferation perse. The reason for the discrepancy is unclear but may be due to the cell type used (large vein vs. microvascular) or may have to do with methodology. Our studies of proliferation were based on an assay of cell proliferation from single cells at moderate plating density. In the studies using HUVECs, cells were grown to near confluence (~80–90%) before the treatment and study. Cells cultures that are 80% confluent have many patches that are confluent with some bare patches that cells are growing to fill. It is possible that Hcy impairs the proliferation of cells near the wound edge by a mechanism that is not activated in our proliferation assay (i.e., proliferation behind a leading edge is mechanistically different from proliferation of dispersed cells). It is also possible that impaired migration at the leading edge of a wound causes impaired proliferation of cells behind the leading edge. Either of these possibilities would explain the apparent differences between our study and those using HUVECs. Dissecting these mechanisms will require further studies.
Kwak et al. observed that scrape-wounding of bEnd.3 cells enhances Cx43 expression at the leading edge of the wound 24 hours after injury [21]. Results from immunohistochemistry support this observation (Figure 9, control cells); however, the increase did not reach statistical significance for the control group in the complementary ELISA experiments (Figure 8). For these ELISA experiments, we created a wound in the shape of a “+” in 96-well plates. This created a substantial mass of cells at a wound edge but not all cells were at a wound edge. Therefore, significance will only be found if the changes at the wound edge are large enough to numerically overwhelm the cells that do not change expression throughout the confluent regions behind the leading edges. Results from the ELISA experiments in Figure 8 are best viewed as a relative measure and, in these experiments, they are viewed relative to the results with Hcy treatment. Hcy treatment doubled Cx43 expression, relative to control, along with a smaller but significant increase in pS368. The increased expression was not confined to the leading cells but extended approximately seven cells deep.
The study by Kwak et al. also found no effect of inhibiting gap junction communication on bEnd.3 cell proliferation but did find a delay in the overall time to wound closure [21]. Our results, which quantify the daily wound repair progression, extend these results by suggesting that the majority of this effect may be the result of inhibiting gap junction communication during the first 24 hours of treatment with 18βGA. This idea is supported by results from the Lampe laboratory, which reported an important regulatory role of downregulation of Cx43 and upregulation of Cx43-pS368 expression during the first 24 hours of skin wound repair [32]. The rate of skin wound repair is significantly increased by antisense KO of Cx43 expression [30] and by KO of the Cx43 gene [18], suggesting that an increase in Cx43 expression is associated with delayed wound repair.
It is not clear whether slowed wound repair in our studies was a result of the large increase in Cx43 expression, the relatively smaller increase in Cx43-pS368, or a ratio of Cx43 to Cx43-pS368. However, the carboxyl tail of Cx43 (which includes S368) is critical for the regulation of cell migration in several types of cells. For example, the carboxyl tail of Cx43 is required for the regulation of cell polarity and motility through interaction with tubulin in developing epicardial tissue [31]. During brain development, deleting the terminal 125 residues of the carboxyl tail of Cx43 arrests neuronal migration in the neocortex [5]. In breast cancer cells, the carboxyl tail of Cx43 associates with CYR61/Connective Tissue Growth Factor/Nephroblastoma Overexpressed (CCN3/NOV), which regulates actin cyto-skeleton reorganization. In glioma cells, high levels of Cx43 upregulate CCN3 expression, which results in reduced cell motility [35]. These and other studies implicate Cx proteins and the carboxyl tail of Cx43 in particular in the regulation of cell motility, although the mechanisms remain largely unknown.
We used computer-based protein-ligand docking software to predict the binding sites of mGluR5 for glutamate, CHPG, and Hcy. Results of these experiments suggested that Hcy may directly bind to mGluR5. The flexibility of all ligands was modeled, but the mGluR5 was simulated as a rigid macromolecule, which means that the spatial shape of the receptor was not changed during the docking process [14]. The flexibility limitation of the receptor should be improved in future work. However, the predicted binding domain is the same as for glutamate [8,20] and CHPG, lending confidence to the hypothesis that Hcy is an agonist of mGluR5 in vascular endothelium.
EC wound repair plays an important role in various disorders and conditions, including atherosclerosis, carcinogenesis, vascular repair following injury such as wounding and stroke, and in normal responses to stresses such as exercise [3]. Our data suggest that Hcy concentrations in the low micromolar range create a dose-dependent attenuation of microvascular EC wound repair. Inhibition of endothelial mGluR5 activation may be a novel strategy for promoting endothelial repair in patients with HHcy. These studies also suggest that the activation of mGluR5 may be an anti-angiogenic strategy in conditions such as cancer.
PERSPECTIVE
Elevated blood levels of Hcy, a condition known as HHcy, impair EC wound repair and angiogenesis, although the mechanism remains unknown. We have identified a cell surface receptor, mGluR5, which is activated by Hcy and is both necessary and sufficient for the inhibition of wound repair. These results are the first identification of a receptor-mediated effect of Hcy on microvascular endothelial wound repair and offer new opportunities for exploring potential therapeutic strategies to modulate EC growth in patients.
ACKNOWLEDGMENTS
We thank Dr. Vernon Winston for assistance in the use of AutoDock and also thank Dr. Teri Peterson, ISU Faculty Statistical Consultant, for consultation on statistical analyses. This work was supported by NIH Grant P20RR016454 and ISU Grants FRC 1019, GSRSC S09–4, and S11–28U.
Abbreviations used:
- 18βGA
18-beta-glyccyrhetinic acid
- BIM
bisindolylmaleimide
- CHPG
2-amino-2-(2-chloro-5-hydroxyphenyl) acetic acid
- CMVECs
cerebral microvascular endothelial cells
- Cx
connexin
- Cx43
connexin43
- Cx43-pS368
phosphorylation of Cx43 at S368
- DAG
diacyl glycerol
- DMEM-H
Dulbecco’s Modified Eagle’s Medium high glucose
- DMSO
dimethyl sulfoxide
- EC
endothelial cell
- ELISA
enzyme-linked immunosorbent assay
- Hcy
homocysteine
- HHcy
hyperhomocysteinemia
- HUVECs
human umbilical vein endothelial cells
- KO
knockout
- mGluR
metabotropic glutamate receptor
- mGluR5
metabotropic glutamate receptor 5
- MPEP
2-methyl-6-(phenylethynyl)pyridine
- NS
nonsilencing
- PBS
phosphate buffered saline
- PDB
protein data bank
- PKC
protein kinase C
- PMA
phorbol 12-myristate 13-acetate
- TBS
Tris-buffered saline
REFERENCES
- 1.Boassa D, Solan JL, Papas A, Thornton P, Lampe PD, Sosinsky GE. Trafficking and recycling of the connexin43 gap junction protein during mitosis. Traffic 11: 1471–1486, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brauner-Osborne H, Wellendorph P, Jensen AA. Structure, pharmacology and therapeutic prospects of family C G-protein coupled receptors. Curr Drug Targets 8: 169–184, 2007. [DOI] [PubMed] [Google Scholar]
- 3.Carmeliet P Angiogenesis in life, disease and medicine. Nature 438: 932–936, 2005. [DOI] [PubMed] [Google Scholar]
- 4.Chopp M, Li Y. Treatment of neural injury with marrow stromal cells. Lancet Neurol 1: 92–100, 2002. [DOI] [PubMed] [Google Scholar]
- 5.Cina C, Maass K, Theis M, Willecke K, Bechberger JF, Naus CC. Involvement of the cytoplasmic C-terminal domain of connexin43 in neuronal migration. J Neurosci 29: 2009–2021, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Collard CD, Park KA, Montalto MC, Ala-pati S, Buras JA, Stahl GL, Colgan SP. Neutrophil-derived glutamate regulates vascular endothelial barrier function. J Biol Chem 277: 14801–14811, 2002. [DOI] [PubMed] [Google Scholar]
- 7.Cooper CD, Solan JL, Dolejsi MK, Lampe PD. Analysis of connexin phosphorylation sites. Methods 20: 196–204, 2000. [DOI] [PubMed] [Google Scholar]
- 8.De Blasi A, Conn PJ, Pin J, Nicoletti F. Molecular determinants of metabotropic glutamate receptor signaling. Trends Pharmacol Sci 22: 114–120, 2001. [DOI] [PubMed] [Google Scholar]
- 9.Debreceni L Homocysteine: a risk factor for atherosclerosis. Orv Hetil 142: 1439–1444, 2001. [PubMed] [Google Scholar]
- 10.Gillard SE, Tzaferis J, Tsui HC, Kingston AE. Expression of metabotropic glutamate receptors in rat meningeal and brain microvasculature and choroid plexus. J Comp Neurol 461: 317–332, 2003. [DOI] [PubMed] [Google Scholar]
- 11.Girelli D, Martinelli N, Olivieri O, Pizzolo F, Friso S, Faccini G, Bozzini C, Tenuti I, Lotto V, Villa G, Guarini P, Trabetti E, Pig-natti PF, Mazzucco A, Corrocher R. Hype-rhomocysteinemia and mortality after coronary artery bypass grafting. PLoS ONE 1: e83, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harker LA, Ross R, Slichter SJ, Scott CR. Homocystine-induced arteriosclerosis. The role of endothelial cell injury and platelet response in its genesis. J Clin Invest 58: 731–741, 1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hogervorst E, Ribeiro HM, Molyneux A, Budge M, Smith AD. Plasma homocysteine levels, cerebrovascular risk factors, and cerebral white matter changes (leu-koaraiosis) in patients with Alzheimer disease. Arch Neurol 59: 787–793, 2002. [DOI] [PubMed] [Google Scholar]
- 14.Huang SY, Zou X. Advances and challenges in protein-ligand docking. Int J Mol Sci 11: 3016–3034, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jamaluddin MD, Chen I, Yang F, Jiang X, Jan M, Liu X, Schafer AI, Durante W, Yang X, Wang H. Homocysteine inhibits endothelial cell growth via DNA hypome-thylation of the cyclin A gene. Blood 110: 3648–3655, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johnstone SR, Best AK, Wright CS, Isak-son BE, Errington RJ, Martin PE. Enhanced connexin 43 expression delays intra-mito-tic duration and cell cycle traverse independently of gap junction channel function. J Cell Biochem 110: 772–782, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kalita J, Kumar G, Bansal V, Misra UK. Relationship of homocysteine with other risk factors and outcome of ischemic stroke. Clin Neurol Neurosurg 111: 364–367, 2009. [DOI] [PubMed] [Google Scholar]
- 18.Kretz M, Euwens C, Hombach S, Eckardt D, Teubner B, Traub O, Willecke K, Ott T. Altered connexin expression and wound healing in the epidermis of connexin-deficient mice. J Cell Sci 116: 3443–34B2, 2003. [DOI] [PubMed] [Google Scholar]
- 19.Krupinski J, Kaluza J, Kumar P, Kumar S, Wang JM. Role of angiogenesis in patients with cerebral ischemic stroke. Stroke 2B: 1794–1798, 1994. [DOI] [PubMed] [Google Scholar]
- 20.Kunishima N, Shimada Y, Tsuji Y, Sato T, Yamamoto M, Kumasaka T, Nakanishi S, Jingami H, Morikawa K. Structural basis of glutamate recognition by a dimeric metabotropic glutamate receptor. Nature 4G7: 971–977, 2000. [DOI] [PubMed] [Google Scholar]
- 21.Kwak BR, Pepper MS, Gros DB, Meda P. Inhibition of endothelial wound repair by dominant negative connexin inhibitors. Mol Biol Cell 12: 831–84B, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luchsinger JA, Tang MX, Shea S, Miller J, Green R, Mayeux R. Plasma homocysteine levels and risk of Alzheimer disease. Neurology 62: 1972–1976, 2004. [DOI] [PubMed] [Google Scholar]
- 23.Martinez-Poveda B, Chavarria T, Sanchez-Jimenez F, Quesada AR, Medina MA. An in vitro evaluation of the effects of homocysteine thiolactone on key steps of angiogenesis and tumor invasion. Bio-chem Biophys Res Commun 311: 649–6B3, 2003. [DOI] [PubMed] [Google Scholar]
- 24. Mayo JN, Beard RS Jr, Price TO, Chen CH, Erickson MA, Ercal N, Banks WA, Bearden SE. Nitrative stress in cerebral endothelium is mediated by mGluRB in hyperhomocysteinemia. J Cereb Blood Flow Metab. 2011. December 21. doi: 1G.1G38/ jcbfm.2G11.18B. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nagai Y, Tasaki H, Takatsu H, Nihei S, Yamashita K, Toyokawa T, Nakashima Y. Homocysteine inhibits angiogenesis in vitro and in vivo. Biochem Biophys Res Commun 281: 726–731, 2GG1. [DOI] [PubMed] [Google Scholar]
- 26.Nicoletti F, Bockaert J, Collingridge GL, Conn PJ, Ferraguti F, Schoepp DD, Wrob-lewski JT, Pin JP. Metabotropic glutamate receptors: from the workbench to the bedside. Neuropharmacology 60: 1017–1041, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Niswender CM, Conn PJ. Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol 50: 295–322, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Olk S, Turchinovich A, Grzendowski M, Stuhler K, Meyer HE, Zoidl G, Dermietzel R. Proteomic analysis of astroglial connexin43 silencing uncovers a cytoskeletal platform involved in process formation and migration. Glia 58: 494–505, 2010. [DOI] [PubMed] [Google Scholar]
- 29.Olk S, Zoidl G, Dermietzel R. Connexins, cell motility, and the cytoskeleton. Cell Motil Cytoskeleton 66: 1000–1016, 2009. [DOI] [PubMed] [Google Scholar]
- 30.Qiu C, Coutinho P, Frank S, Franke S, Law LY, Martin P, Green CR, Becker DL. Targeting connexin43 expression accelerates the rate of wound repair. Curr Biol 13: 1697–1703, 2003. [DOI] [PubMed] [Google Scholar]
- 31.Rhee DY, Zhao XQ, Francis RJ, Huang GY, Mably JD, Lo CW. Connexin 43 regulates epicardial cell polarity and migration in coronary vascular development. Development 136: 3185–3193, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Richards TS, Dunn CA, Carter WG, Usui ML, Olerud JE, Lampe PD. Protein kinase C spatially and temporally regulates gap junctional communication during human wound repair via phosphorylation of connexin43 on serine368. J Cell Biol 167: 555–562, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shastry S, Tyagi N, Hayden MR, Tyagi SC. Proteomic analysis of homocysteine inhibition of microvascular endothelial cell angiogenesis. Cell Mol Biol (Noisy-le-grand) 50: 931–937, 2004. [PubMed] [Google Scholar]
- 34.Shukla N, Angelini GD, Jeremy JY. Interactive effects of homocysteine and copper on angiogenesis in porcine isolated saphenous vein. Ann Thorac Surg 84: 43–49, 2007. [DOI] [PubMed] [Google Scholar]
- 35.Sin WC, Bechberger JF, Rushlow WJ, Naus CC. Dose-dependent differential upregulation of CCN1/Cyr61 and CCN3/NOV by the gap junction protein Connexin43 in glioma cells. J Cell Biochem 103: 1772–1782, 2008. [DOI] [PubMed] [Google Scholar]
- 36.Solan JL, Fry MD, TenBroek EM, Lampe PD. Connexin43 phosphorylation at S368 is acute during S and G2/M and in response to protein kinase C activation. J Cell Sci 116: 2203–2211, 2003. [DOI] [PubMed] [Google Scholar]
- 37.Solan JL, Lampe PD. Connexin 43 in LA-25 cells with active v-src is phosphorylated on Y247, Y265, S262, S279/282, and S368 via multiple signaling pathways. Cell Commun Adhes 15: 75–84, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Solan JL, Lampe PD. Connexin43 phosphorylation: structural changes and biological effects. Biochem J 419: 261–272, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang H, Jiang X, Yang F, Chapman GB, Durante W, Sibinga NE, Schafer AI. Cyclin A transcriptional suppression is the major mechanism mediating homocysteine-induced endothelial cell growth inhibition. Blood 99: 939–945, 2002. [PMC free article] [PubMed] [Google Scholar]
- 40.Wierzbicki AS. Homocysteine and cardiovascular disease: a review of the evidence. Diab Vasc Dis Res 4: 143–149, 2007. [DOI] [PubMed] [Google Scholar]
- 41.Zieminska E, Lazarewicz JW. Excitotoxic neuronal injury in chronic homocysteine neurotoxicity studied in vitro: the role of NMDA and group I metabotropic glutamate receptors. Acta Neurobiol Exp 66: 301–309, 2006. [DOI] [PubMed] [Google Scholar]