

Abstract
Cells rely on surveillance systems such as autophagy to handle protein alterations and organelle damage. Dysfunctional autophagy, an evolutionarily conserved cellular mechanism for degradation of intracellular components in lysosomes, frequently leads to neurodegeneration. The neuroprotective effect of autophagy stems from its ability to eliminate pathogenic forms of proteins such as α-synuclein or tau. However, the same pathogenic proteins often affect different types and steps of the autophagic process. Furthermore, genetic studies have shown that some proteins related to neurodegeneration, such as huntingtin, participate in autophagy as one of their physiological functions. This complex interplay between autophagy and neurodegeneration suggests that targeting autophagy as a whole might have limited applicability in neurodegenerative diseases, and that future efforts should focus instead on targeting specific types and steps of the autophagic process. This change of strategy in the modulation of autophagy might hold promise for future disease-modifying therapies for patients with neurodegenerative disorders.
Introduction
All cellular proteins and organelles are subjected to quality control. The ubiquitin proteasome system and autophagy contribute to the destruction and recycling of altered cellular components when repair is not possible.1,2 Autophagy is an evolutionarily conserved cellular mechanism for degradation of proteins and organelles in lysosomes. Malfunctioning autophagy has been reported in several diseases, including neurodegenerative disorders such as Parkinson’s disease or Alzheimer’s disease.2–4 Autophagy is an effective neuroprotective mechanism that actively contributes to the removal of pathogenic proteins, but in some instances, it becomes the target of the toxic action of these proteins. This toxicity on the autophagic system can be primary to the main cause of the disease, when the pathogenic proteins directly interfere with components of the autophagic intracellular machinery, or secondary to the main cause of the disease, when the autophagic process is indirectly disturbed. Discriminating between both situations and identifying the specific autophagic defect associated with each neurodegenerative disorder is challenging, because of the relatively small number of studies that have included a detailed molecular dissection of the autophagic defects. However, identifying the autophagic defect becomes essential when autophagy is targeted for therapeutic purposes.
In this Review, we describe the complex interplay between autophagy and neurodegeneration and discuss the possible limitations associated with global enhancement of autophagic activity in the treatment of neurodegenerative diseases. We also discuss how the targeting of autophagy needs to become more selective at two levels: the degraded cargo, and the autophagic step targeted. Lastly, we consider why targeting selective autophagy might hold promise for future therapies for neurodegenerative disorders.
Autophagic pathways
Three types of autophagy have been described in mammals, according to the mechanism of delivery of cargo to the lysosomes (figure 1). In macroautophagy, cytosolic cargo is first trapped inside double membrane vesicles (autophagosomes) that then fuse with lysosomes for complete degradation of cargo. The subset of genes and proteins that participate in this process are called autophagy-related genes and autophagy-related proteins.5,6 In chaperone-mediated autophagy, proteins are identified one by one by a cytosolic chaperone that brings them to the lysosomal surface for translocation across this membrane.3 Finally, in microautophagy and endosomal microautophagy, cargo is internalised through invaginations in the lysosomal and endosomal membrane (panel 1).7 For a more detailed description of the molecular components and regulation of autophagy, and of the dynamic interplay between the different autophagic pathways, readers are directed to extensive reviews on this topic.2–4
Figure 1: Autophagic pathways in mammalian cells.
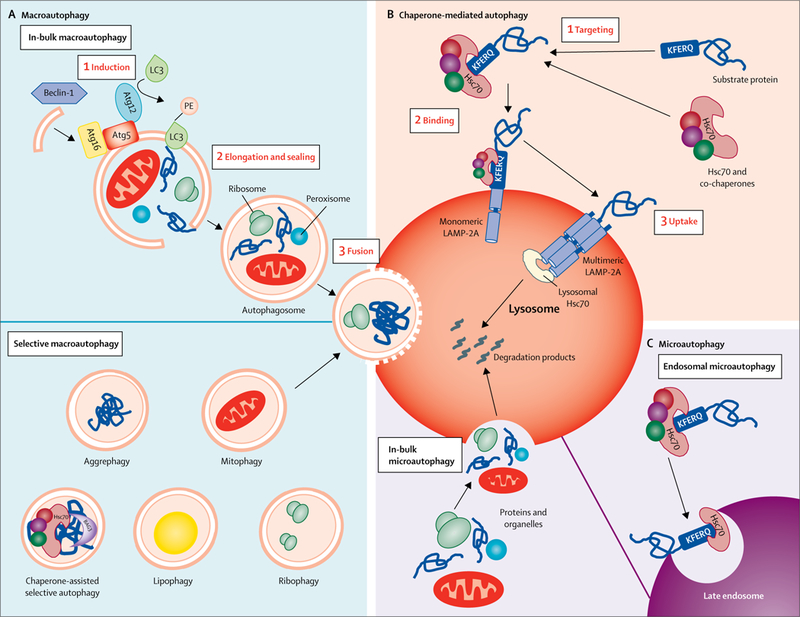
The different autophagic processes that commonly coexist in most mammalian cells can be grouped into three types. (A) Macroautophagy: cytosolic cargo is first trapped inside double-membrane vesicles (autophagosomes) whose membranes form through conjugation of the autophagy-related protein LC3 with the lipid PE and other autophagy-related proteins (ie, Atg5, Atg16, and Atg12, depicted here). Formation of these double-membrane vesicles is initiated by the phosphorylation of lipids in the membrane of organelles such as the endoplasmic reticulum, mitochondria, and Golgi apparatus. This phosphorylation is triggered by a kinase complex regulated by Beclin-1. Cargo can be trapped in bulk (ie, several types of cargo in the same autophagosome) or in a selective manner (ie, only one type of cargo is trapped inside the autophagosome). Examples of selective macroautophagy include aggrephagy (degradation of protein aggregates after their recognition by autophagy cargo receptors such as p62 or neighbour of BCRA1), chaperone-assisted selective autophagy (degradation of protein aggregates targeted to autophagosomes, in this case by chaperones such as Hsc70 and Bag3; in contrast to chaperone-mediated autophagy, chaperone-assisted selective autophagy does not require binding of Hsc70 to a KFERQ-like motif and is dependent on the macroautophagy machinery), mitophagy (selective degradation of mitochondria by macroautophagy, often mediated by autophagy receptors such as PTEN-induced putative kinase 1, Parkin, NIX, and BNIP, which link the mitochondria to be degraded with the autophagy machinery), lipophagy (selective sequestration of lipid droplets by autophagosomes for their degradation in lysosomes), and ribophagy (selective sequestration of lysosomes in autophagosomes for degradation in lysosomes). Autophagosomes are targeted to lysosomes and, after fusion of both vesicles, cargo is delivered to lysosomes for complete degradation. (B) Chaperone-mediated autophagy: single cytosolic proteins bearing a KFERQ-like motif in their sequence are recognised by Hsc70 and brought to the lysosomal membrane for translocation across the LAMP-2A multimeric complex. Lysosomal Hsc70 aids translocation of the substrate protein, which is rapidly degraded once inside the lysosomal lumen. (C) Microautophagy: proteins and organelles can be degraded in bulk through invaginations at the lysosomal membrane. Cytosolic proteins are selectively targeted by Hsc70 to late endosomes, using the same KFERQ-like motif as in chaperone-mediated autophagy, resulting in their internalisation and degradation in a process known as endosomal microautophagy. Hsc70=heat shock cognate 70 kDa protein. LC3=microtubule-associated protein 1 light chain 3. Atg=autophagy-related protein. PE=phosphatidyl ethanolamine. LAMP-2A=lysosome-associated membrane protein-2A. Bag3=Bcl2-associated athanogene 3.
Panel 1: Types of autophagy.
Chaperone-mediated autophagy
Type of autophagy that involves selective delivery of single proteins to lysosomes upon recognition and binding of a specific KFERQ-like pentapeptide motif to cytosolic chaperone heat shock cognate 70 kDa protein (Hsc70). The chaperone-substrate complex binds the cytosolic tail of the lysosome-associated membrane protein type 2A, triggering its assembly into a multimeric complex that mediates substrate translocation. After complete unfolding, and with the assistance of luminal chaperones, the substrate reaches the lysosomal matrix for degradation.
Endosomal microautophagy
Variant of microautophagy that involves trapping of cytosolic proteins and organelles into small vesicles that form on the surface of late endosomes by invagination of their membrane, mediated by the endosomal sorting complexes required for transport. Vesicles pinch off from the membrane into the lumen of the endosome, where they are degraded after lysis of the microvesicle membrane. Some proteins that bear in their sequence a KFERQ-like motif (eg, GAPDH, RNase A, tau) can be selectively targeted for endosomal microautophagy by Hsc70s that recognise the same KFERQ-like pentapeptide motif as in chaperone-mediated autophagy. The factors that determine Hsc70 triage between chaperone-mediated autophagy and endosomal microautophagy remain unknown.
Macroautophagy
Type of autophagy that involves sequestration of cytosolic components by a membrane that elongates through a coordinated assembly of lipids and autophagy-related proteins, and then seals to form a double membrane vesicle called the autophagosome. The sequestered cargo inside the autophagosome is transported towards the lysosomes and, upon fusion of these two vesicles, is rapidly degraded by the resident lysosome hydrolases.
Microautophagy
Type of autophagy where cytosolic cargo, proteins, and organelles are internalised inside lysosomes by invaginations that form at the lysosomal membrane.
Selective macroautophagy
Autophagic processes that use the core machinery of macroautophagy but selectively target specific cytosolic components for degradation. Types of selective macroautophagy include mitophagy (removal of mitochondria), lipophagy (removal of lipids), and aggrephagy (removal of aggregate proteins).
Defective autophagy in the brain
Autophagy is important in maintenance of neurons and glia, because many cells of the CNS are post-mitotic and need to have the most exquisite quality control systems in place to eliminate altered proteins and organelles.5,6 Additionally, maintenance of neuronal and glial activity imposes high energetic demands that are resolved in large part by the recycling of essential components through autophagy.5,6 In cellular and animal models, macroautophagy has been shown to contribute to different aspects of neuronal physiology, including axonal homeostasis, synaptic pruning, and neurogenesis through maintenance of neural progenitors.8–11 Neuronal deletion of key autophagy genes in mice results in neurodegeneration, with age-dependent intracellular accumulation of protein aggregates and dysfunctional organelles,12,13 supporting the role of autophagy in quality control of the CNS. In fact, the process of degeneration observed in mice when autophagy is blocked in neurons is probably a combination of direct proteotoxicity (due to loss of quality control) and of loss of key functions of autophagy in neurons and neuronal progenitors.
Autophagy exerts many of its functions through selective removal of proteins (figure 2), which requires coordinated action of receptors, chaperones, and autophagy proteins (panel 2). Chaperone-mediated autophagy selectively degrades single proteins, but once proteins organise into aberrant oligomeric complexes, degradation through this pathway is no longer possible.1,2 Small aggregates and oligomers can be taken up by endosomal microautophagy,23 but irreversible aggregates can only be eliminated by macroautophagy either in bulk or selectively by aggrephagy.4
Figure 2: Autophagy of pathogenic proteins.
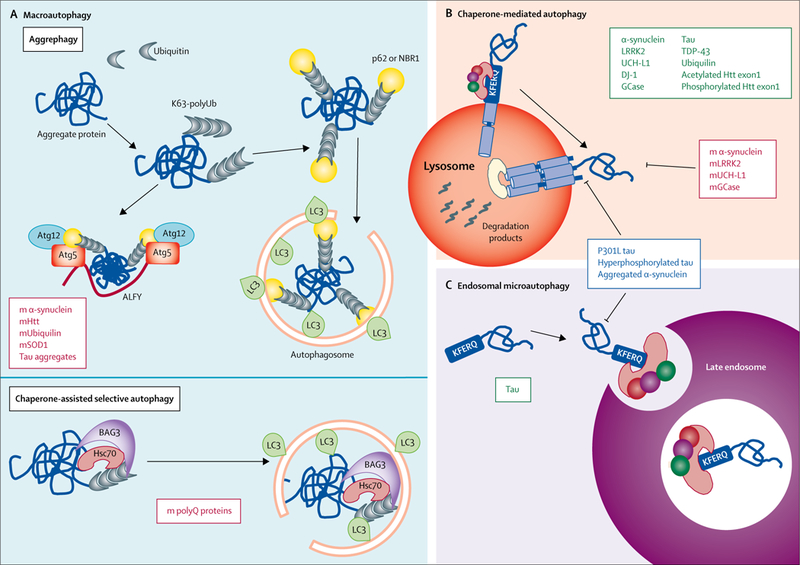
(A) Examples of pathogenic proteins (red boxes) related to neurodegeneration that are cleared up by two types of selective macroautophagy: aggrephagy or chaperone-assisted selective autophagy. In aggrephagy, cytosolic aggregates of these pathogenic proteins labelled with ubiquitin are recognised by p62, which either brings LC3 directly and initiates formation of the autophagosome around the aggregate, or (in a process mediated by ALFY) assembles with Atg12 and Atg5 prior to LC3 conjugation, to initiate formation of the autophagosome. In chaperone-assisted selective autophagy, polyubiquitinated aggregate proteins are brought to the autophagosome by Hsc70 and Bag3. (B) Examples of proteins (green box) related to neurodegeneration that are degraded by chaperone-mediated autophagy, in which they are identified through their KFERQ-like motif by Hsc70, which brings them to the lysosomal surface for translocation across the lysosomal membrane. However, the pathogenic variants of these proteins (red box) are similarly targeted to lysosomes but fail to cross the lysosomal membrane, resulting in blockage of chaperone-mediated autophagy. Some other pathogenic variants of neurodegeneration-related proteins (blue box) are capable of blocking both chaperone-mediated autophagy (B) and endosomal microautophagy (C), wherein substrate proteins bearing KFERQ-like motifs are normally targeted to endosomes (green box) and internalised for degradation through invaginations in their membrane. m=mutant. K63-polyUb=polyubiquitin tagged to proteins through lysine 63. Atg=autophagy-related protein. ALFY=autophagy-linked FYVE protein. Htt=huntingtin. SOD1=superoxide dismutase 1. LC3=microtubule-associated protein 1 light chain 3. Bag3=Bcl2-associated athanogene 3. polyQ=polyglutamine. LRRK2=leucine-rich repeat kinase 2. UCH-L1=ubiquitin carboxy-terminal hydrolase L1. DJ-1=deglycase J-1. GCase=glucocerebrosidase. TDP-43=TAR DNA-binding protein 43.
Panel 2: Cargo recognition machinery.
In normally functioning cells, each autophagic pathway recognises its cargo when it needs to be degraded. In aggrephagy—the selective degradation of aggregates by macroautophagy—the first step is often K63 polyubiquitination. Polyubiquitin chains are recognised by autophagy receptors such as p62, optineurin, neighbour of BCRA1, and nuclear dot protein 52 kDa.14 These receptors simultaneously bind polyubiquitin and microtubule-associated protein 1A/1B-light chain 3B, one of the integral proteins of the autophagosome membrane that forms around the aggregates. Different autophagy receptors display preferences for specific cargos but often also cooperate in cargo recognition.14 Post-translational modifications modulate the cargo recognition abilities of the autophagy receptors. For example, phosphorylation of p62 during proteotoxic insults (ie, overexpression of mutant huntingtin) increases its affinity for ubiquitin residues.15 Adaptors, such as the FYVE domain-containing ALFY protein, are also recruited to ubiquitin-positive inclusions, where they interact with autophagy-related genes and serve as scaffolds in aggregate clearance.16
Ubiquitinated aggregates can also be targeted to macroautophagy by the co-chaperone Bcl2-associated athanogene 3 (Bag3) without involvement of receptors, in a process known as chaperone-assisted selective autophagy. Bag3 overexpression has been proven sufficient to facilitate autophagic removal of polyglutamine-containing proteins, superoxide dismutase 1, and TAR DNA-binding protein 43 (TDP-43).17
Cargo recognition in chaperone-mediated autophagy and endosomal microautophagy occurs by binding of heat shock cognate 70 kDa protein to a KFERQ-like motif in the substrate protein.3 Degradation by chaperone-mediated autophagy has been shown in Parkinson’s disease related proteins α-synuclein, leucine-rich repeat kinase 2, ubiquitin carboxy-terminal hydrolase L1, and deglycase J-1.18–22 Chaperone-mediated autophagy and endosomal microautophagy degradation have also been reported for tau, which accumulates in intracellular neurofibrillary tangles in Alzheimer’s disease and in frontotemporal dementia.21 Other substrates of chaperone-mediated autophagy relevant to neurodegeneration include ubiquilin (associated with amyotrophic lateral sclerosis), TDP-43 (associated with frontotemporal dementia), and huntingtin.3
Reduced ability to eliminate pathogenic proteins via autophagy has been shown in neurodegenerative disorders.4 Failure to remove aggregates of some types of proteins (eg, p38, a protein member of the AMPK family that is prone to form aggregates in Parkinson’s disease) is not due to disruption of the autophagic process, but rather due to primary changes in the protein that prevent its targeting to autophagy (panel 3).24 However, in other cases (ie, huntingtin, α-synuclein, tau), accumulation of pathogenic proteins is often a consequence of defective autophagy.4 Similarly, the functional decline of autophagy in old organisms has been proposed as a primary risk factor for neurodegenerative diseases.1,2 Vulnerability to macroautophagy failure seems to be dependent on neuronal type—eg, Purkinje neurons29 are more vulnerable than midbrain dopaminergic neurons30 to the presence of p62 (also known as sequestosome-1) protein inclusions—and is also affected by age and disease stage.31
Panel 3: Deficient autophagy targeting of neurodegeneration-related proteins.
Deficient aggrephagy in some neurodegenerative disorders is caused by changes in the intrinsic properties of the pathogenic proteins.24 Factors such as the compactness of the aggregate can affect the efficiency of assembly of proteins in autophagosome formation.24 In other cases of neurodegeneration, the primary problem is at the level of the cargo recognition machinery.25 One of the most studied components of this machinery is the cargo receptor p62, whose activity in aggrephagy has been tightly linked to Keap1 and Nrf2, involved in the oxidative stress response.26 Induction of p62 phosphorylation might be beneficial through two separate mechanisms, enhancing p62 function in autophagy and promoting degradation of the Nrf2 inhibitor Keap1, with the subsequent activation of cytoprotective Nrf2 targets.27 Procedures to selectively enhance macroautophagy of pathogenic proteins are still scarce. However, in vitro and in vivo studies have shown that overexpression of specific autophagy receptors (ie, nuclear dot protein 52 kDa) significantly reduces the concentration of toxic proteins (ie, hyperphosphorylated tau).28
Protein targeting to chaperone-mediated autophagy is disrupted once pathogenic proteins organise into higher order molecular complexes and their targeting motif becomes inaccessible to Hsc70. Defective lysosomal targeting contributes to reduced chaperone-mediated autophagy and endosomal microautophagy of the disease-associated P301L tau mutant.21 Although substrate unfolding is not necessary for endosomal microautophagy, recognition by heat shock cognate 70 kDa protein is still a prerequisite and consequently, selective endosomal microautophagy of aggregated pathogenic variants is no longer possible. Post-translational modifications such as hyperphosphorylation, tightly associated with tau pathogenesis, disrupt its targeting to both chaperone-mediated autophagy and endosomal microautophagy in vitro.21 In other instances, these modifications can generate chaperone-mediated autophagy and endosomal microautophagy targeting motifs, thus contributing to toxicity through undesired removal of necessary proteins.28
Changes in autophagic processes in degenerating neurons can be primary or reactive to another underlying cause. For example, mutations of presenilin proteins in Alzheimer’s disease are primary defects, because they directly reduce acidification of lysosomes,32 disturbing their degradative capacity. By contrast, blockage of chaperone-mediated autophagy by mutant α-synuclein3 is a typical example of autophagic malfunction secondary to the main cause of the disease. Mutations in other disease-related proteins can cause autophagy to fail, because their physiological activity is lost. For example, huntingtin contributes to autophagy activation and cargo recognition, and both functions are lost in its mutant counterpart.25,33 In the following sections, we present examples of pathogenic proteins that have a toxic effect on autophagy, the autophagic step that they disrupt, and the genetic mutations that prevent the physiological participation of specific proteins in autophagy.
Pathogenic proteins interfering with autophagy
Defective induction of autophagy
Autophagy effectors and regulators are often found trapped in aggregates in many neurodegenerative disorders. For example, lysosomal proteins or the autophagy cargo receptor protein p62 are common components of Lewy bodies in Parkinson’s disease.34 Although they could aggregate indirectly because of the loss of neuronal proteostasis (a cellular process that ensures proteins maintain the most favourable conformation and do not aggregate), growing evidence supports the direct interaction between pathogenic proteins and components of autophagy as a trigger for aggregation.35,36 This physical sequestration of autophagy regulators by pathogenic proteins has been shown to reduce induction of macroautophagy (figure 3). For example, mutant α-synuclein directly binds transcription factor EB (TFEB), an essential regulator of lysosomal biogenesis and of many of the genes involved in macroautophagy, and prevents its nuclear translocation, thus reducing the transcriptional activation of TFEB-dependent autophagic and lysosomal genes.35 Similarly, mutant huntingtin aberrantly interacts with Ras homologue enriched in striatum (Rhes), a protein that normally activates autophagy by preventing the interaction between Beclin-1 and its endogenous inhibitor Bcl-2.36
Figure 3: Toxic effects of pathogenic proteins on autophagy.
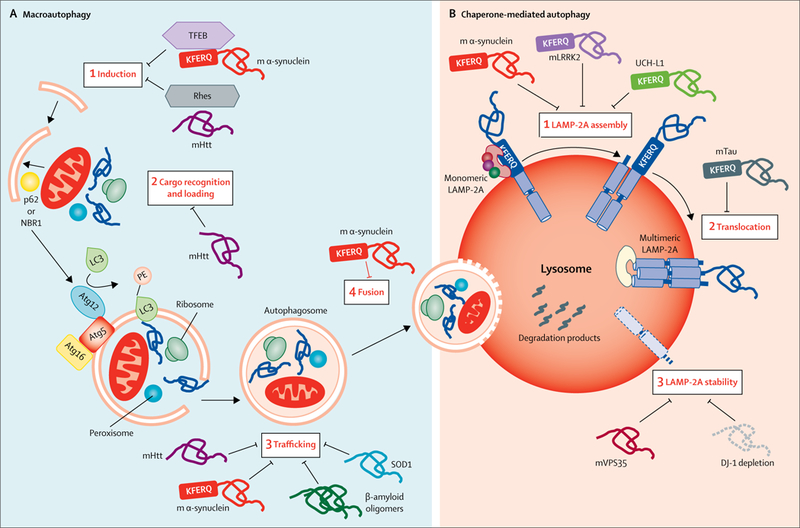
Several proteins related to neurodegeneration have been shown to interfere with macroautophagy (A) and chaperone-mediated autophagy (B). (A) The inhibitory effect of pathogenic proteins on macroautophagy can take place at each of the steps of this process. For instance, binding of mutant α-synuclein to TFEB has been shown to reduce autophagy induction by preventing TFEB nuclear translocation. Initiation of autophagy is also reduced by the interaction of mutant Htt with Rhes, an activator of autophagy through Beclin-1. Mutant Htt also interferes with cargo recognition by autophagy receptors such as p62. Mutant α-synuclein and mutant Htt can also interfere with the trafficking of the autophagosomes to the lysosome, and mutant α-synuclein can also prevent autophagosome fusion with lysosomes. (B) Inhibition of chaperone-mediated autophagy by pathogenic proteins occurs mainly during the steps of this process that take place at the lysosomal membrane. For example, pathogenic variants of α-synuclein, LRKK2, or UCH-L1 can inhibit assembly of monomers of LAMP-2A into the translocation complex, whereas pathogenic forms of tau block translocation of proteins through the translocation complex. Parkinson’s disease-related mutations in VPS35 or DJ-1 seem to promote accelerated degradation of LAMP-2A in lysosomes through still unknown mechanisms, leading to reduced chaperone-mediated autophagy activity. m=mutant. TFEB=transcription factor EB. Rhes=Ras homolog enriched in striatum. Htt=huntingtin. Atg=autophagy-related protein. SOD1=superoxide dismutase 1. LRRK2=leucine-rich repeat kinase 2. UCH-L1=ubiquitin carboxy-terminal hydrolase L1. LAMP-2A=lysosome-associated membrane protein-2A. VPS35=vacuolar protein sorting-associated protein 35. DJ-1=deglycase J-1. LC3=microtubule-associated protein 1 light chain 3.
No direct interaction between pathogenic proteins and signalling molecules involved in induction of chaperone-mediated autophagy (ie, nuclear factor of activated T cells, retinoic acid receptor-α, nuclear factor erythroid 2-related factor 2) have been found yet.3 The relatively poor understanding of the mechanism behind the induction of endosomal microautophagy has limited its possible association with neurodegenerative disease.
Interference with autophagic cargo recognition
Deficient cargo recognition can be due to intrinsic substrate alterations or faulty functioning of the cargo recognition machinery (figure 3). In Huntington’s disease, reduced mitophagy, lipophagy, and aggrephagy have been partly associated with inefficient cargo loading into autophagosomes.37 The formation of similar empty autophagosomes as a result of huntingtin depletion was initially thought of as a gain of toxic function, but instead suggests a physiological role of huntingtin in cargo recognition.25
Mitophagy has been one of the most studied selective types of autophagy in neurodegenerative disorders.38 Inadequate degradation of mitochondria seems to be responsible for the accumulation of defective mitochondria in these conditions.38 Defective mitophagy can occur alongside other defective autophagic pathways (ie, bulk autophagy, lipophagy, aggrephagy), when pathogenic proteins interfere with components of the core machinery involved in autophagy induction, auto-phagosome formation, and clearance (figure 1).35,36 Selective failure of mitophagy has been linked to a primary defect in the components of the mitochondrial cargo recognition machinery. For example, studies have identified a link between TBK1 mutations and defective mitophagy in patients with amyotrophic lateral sclerosis.39,40 Besides its role in immunity, TANK1-binding kinase 1 (TBK1) participates in mitochondria recognition and clearance by mitophagy.41 The best characterised type of mitophagy relies on the cooperation between the E3-ubiquitin ligase Parkin and the mitochondrial resident PTEN-induced putative kinase 1 (PINK1).42 Activation of PINK1 recruits Parkin to depolarised mitochondria, which facilitates their recognition as cargo by the autophagy machinery. Mutations in both PINK1 and Parkin have been described in familiar forms of Parkinson’s disease. Upon mitochondrial depolarisation, the PINK1–Parkin axis mediates ubiquitination of outer membrane mitochondrial proteins and recruitment of cargo receptors such as optineurin or p62. Both receptors are regulated by phosphorylation by TBK1, and optineurin phosphorylation has been shown to increase mitophagy.43 Mutations in TBK1 associated with amyotrophic lateral sclerosis reduce mitophagy, resulting in aggregation of defective mitochondria and neuronal toxicity in cultured cells.43 Whether the described PINK1-independent basal mitophagy44 could also be under TBK1 regulation remains unknown.
A direct toxic effect of pathogenic proteins on heat shock cognate 70 kDa protein (Hsc70), responsible for cargo recognition in chaperone-mediated autophagy and endosomal microautophagy, has not been described yet. However, a model of chaperone depletion has been suggested1 in which aggregate-mediated sequestration of Hsc70 reduces its availability for other cellular functions, such as endocytosis or protein folding (figure 3). Although the consequences for chaperone-mediated autophagy and endosomal microautophagy are still unknown, the negative effect of this Hsc70 sequestration on endocytosis has already been shown in Caenorhabditis elegans.1
Compromised mobilisation of autophagic compartments
Because of their extraordinary polarisation, neurons are highly dependent on organelle trafficking. Autophagosomes generated in the distal part of the axon use retrograde transport to reach the soma, where they fuse with lysosomes.45 Pathogenic proteins have been shown to interfere with autophagosome trafficking and fusion (figure 3). Several studies have shown that aggregated α-synuclein and mutant huntingtin decrease autophagosome motility in neurons and lead to their accumulation.46,47 For example, binding of mutant α-synuclein to the autophagosome surface reduces their retrograde transport and might contribute to accumulation of α-synuclein in amyloid fibrils (Lewy neurites), reported in patients with Parkinson’s disease.4 Similarly, abnormal binding of β-amyloid oligomers to the cytoskeletal motor protein dynein might contribute to the accumulation of autophagosomes in the distal part of the axons in patients with Alzheimer’s disease.48
The fusion of autophagosomes with lysosomes is modulated by membrane lipids and SNARE proteins.5,6 Binding of pathogenic proteins to the autophagosome surface, as reported with mutant α-synuclein in some Parkinson’s disease models, alters the dynamics of this process.4 Lysosomal acidification, defective in neurodegenerative diseases such as Alzheimer’s disease and Parkinson’s disease,32,49 is also essential for compartment motility and fusogenicity.
Alterations of the lysosomal compartment
Primary defects in lysosomal membrane stability, enzymatic content, and activity or acidification of the lysosomal lumen can result in incomplete cargo degradation.32,49,50 The toxic effect of most pathogenic proteins on chaperone-mediated autophagy occurs directly at the lysosomal membrane (figure 3). Mutations and post-translational modifications of α-synuclein impair degradation by chaperone-mediated autophagy, because the modifications interfere with the organisation of the monomeric protein lysosome-associated membrane protein-2A (LAMP-2A) into the multimeric complex required for translocation of substrate proteins from the cytosol into the lysosomal lumen (figure 3B).3,18 Abnormal LAMP-2A binding and disruption of its assembly are also responsible for the toxic effect of other proteins associated with Parkinson’s disease, such as leucine-rich repeat kinase 2 (LRRK2),19 ubiquitin carboxy-terminal hydrolase L1,39 and mutant A152T-tau.21 These proteins often oligomerise at the lysosomal surface further, contributing to the blockage of chaperone-mediated autophagy,3,18,19,21 and possibly acting as a primary site of aggregation in neurons.49 Cells expressing mutations related to Parkinson’s disease in vacuolar protein sorting-associated protein 35 (VPS35),51 or depleted of deglycase J-1 (DJ-1) (encoded by the PARK7 gene),22 show reduced chaperone-mediated autophagy as a consequence of the accelerated degradation of LAMP-2A in lysosomes, although the mechanism behind LAMP-2A destabilisation remains unknown.
Genetic basis for defective autophagy in neurodegenerative disorders
Few neurodegenerative diseases have been directly associated with loss-of-function mutations in autophagy-related genes.52,53 Mutations in autophagic receptors such as p62 or optineurin have been associated with frontotemporal dementia,54 Alzheimer’s disease,55 or amyotrophic lateral sclerosis.56 However, studies investigating the basis for autophagy malfunctioning in other neurodegenerative disorders have identified some non-pathogenic variants of proteins as components of the autophagy machinery. In these instances, when the non-pathogenic form of the protein has a physiological function in autophagy, the autophagic failure originates from loss-of-function, rather than from the toxic effect. In cellular and animal models of Parkinson’s disease, for example, depletion of the non-mutant forms of LRRK219 and DJ-122 reproduces, for the most part, the malfunction of chaperone-mediated autophagy reported in the presence of their mutant counterparts. However, mechanistic information on how these proteins modulate chaperone-mediated autophagy activity is not available yet. In the next sections, we provide some examples of these physiological roles for neurodegeneration-related proteins in autophagy.
Parkin functions in cargo recognition
Parkin (encoded by the PARK2 gene) is the cause of some forms of familial Parkinson’s disease, and plays a major role in mitochondrial quality control through the PINK1–Parkin axis.38,42 Mutations in PINK1 are associated with early-onset Parkinson’s disease, reducing the stability of Parkin and leading to an inefficient ubiquitin ligase activity, faulty mitophagy, and subsequent accumulation of defective mitochondria.57 In fact, the differences in the propensity to mitochondrial defects among different brain regions observed in experimental models of Parkinson’s disease coincides with the spatiotemporal expression of Parkin in the brain.58 Inefficient mitochondrial turnover is associated with defective neuronal energetic balance and can contribute to the deregulated lipid metabolism reported in patients with Parkinson’s disease.59 Conversely, the altered cellular lipid composition reported in these patients could potentially perpetuate the mitochondrial defects by interfering with their mitophagy.59 The absence of aggregates containing α-synuclein (the neuropathological hallmark of Parkinson’s disease) in patients with mutant Parkin suggests that parkin-mediated toxicity might act independently of protein aggregation.
Huntingtin plays a dual role in autophagy induction
Growing evidence highlights a complex relationship between huntingtin and macroautophagy. The presence of autophagic receptors such as optineurin60 or p6261 in huntingtin aggregates suggested that the deficiencies in autophagy reported in patients with Huntington’s disease were due to the direct toxic effect of huntingtin. However, most of these autophagic abnormalities were also reported in models of huntingtin depletion,25,33 highlighting a possible physiological role for huntingtin in macroautophagy. Huntingtin seems to participate in this process, from cargo recognition and initiation of autophagosome formation,25,33 to the later steps of autophagy such as vesicle transport before autophagosome and lysosome fusion.62
Studies in mice and Drosophila melanogaster support a stabilising effect of huntingtin on the interactions of p62 with microtubule-associated protein 1A/1B-light chain 3B (LC3B), an essential protein for autophagosome formation, and with the cargo’s ubiquitin tag.25 Additionally, domain homology of huntingtin with autophagy-related proteins63 allows interaction of huntingtin with other components of autophagy such us ULK1, to activate macroautophagy.25 Thus, huntingtin can act as a scaffolding protein that promotes spatial proximity between essential components of autophagy induction and cargo recognition. Huntingtin is only required for certain types of macroautophagy (eg, aggrephagy, lipophagy, mitophagy), but not for basal autophagy or autophagy induced by nutrient deprivation.25
The presence of abnormally long stretches of polyglutamine, which is likely to change the conformation and dynamic properties of mutant huntingtin, might interfere with the binding of huntingtin to autophagy-related proteins and motor proteins, thereby compromising their role in macroautophagy. For example, mutations that alter the conformation of huntingtin will prevent it from interacting with p62,64 leading to poor cargo recognition. Interestingly, the shorter polyglutamine tract present in non-pathogenic huntingtin might regulate its physiological function in autophagy. A similar regulatory role has also been proposed for the polyglutamine tract in other cytosolic proteins such as ataxin 3, which requires the polyglutamine tract to interact with Beclin-1.33 The polyglutamine tract of mutant huntingtin competes for this interaction with Beclin-1 in a length-dependent manner, preventing induction of autophagy under stress conditions.33 This functional interaction between huntingtin and Beclin-1 provides a better overall understanding of the relation between huntingtin and macroautophagy, and the basis for autophagic impairment in patients with Huntington’s disease.
Role of CHMP2B and VPS35 in lysosomes
Several neurodegenerative disorders have been associated with abnormal endosomal or lysosomal maturation and acidification, which negatively affects all autophagic pathways.50,51,65–67 Endosomal microautophagy occurs in late endosomal multivesicular bodies, but these also contribute to autophagosome clearance. Biogenesis of late endosomal multivesicular bodies requires multiprotein endosomal sorting complexes required for transport (ESCRT) machinery.68 Mutations in one of these ESCRT proteins, the charged multivesicular body protein 2B (CHMP2B), have been found in patients with frontotemporal dementia and in patients with amyotrophic lateral sclerosis.65 Cells bearing these CHMP2B mutations reveal defective macroautophagy and accumulate ubiquitinated proteins:65 TAR DNA-binding protein 43 (TDP-43) in amyotrophic lateral sclerosis models, and polyglutamine aggregates in Huntington’s disease models.4 Interestingly, neurons and microglia of CHMP2B-mutant mice display auto-fluorescent aggregates associated with the endolysosomal system, similar to those found in the brains of patients with frontotemporal dementia, highlighting an important lysosomal storage pathology due to defects in the ESCRT machinery.66
Mutations in the VPS35 gene that codes for a core component of the retromer complex, that mediates sorting of proteins to endosomes, can cause late-onset, autosomal dominant, familial Parkinson’s disease.69 VPS35 mutations lead to defects in autophagosome formation due to abnormal trafficking of autophagy-related protein 9,67 and defects in lysosomal biogenesis because of the inability to recycle the mannose 6-phosphate receptor from the lysosomes back to the Golgi apparatus.50 Reduced concentrations of the mannose 6-phosphate receptor limit the amount of enzymes that reach lysosomes (such as cathepsin D), affecting degradation of substrates, including α-synuclein.50 Studies in dopaminergic neurons expressing mutant VPS35 linked to Parkinson’s disease have also confirmed defective endosome-to-Golgi retrieval of lysosomal membrane proteins, including LAMP-2A.51 Accelerated degradation of LAMPs in lysosomes might negatively affect both macroautophagy and chaperone-mediated autophagy, because of the contribution of LAMP-2A and LAMP-2B to these pathways.
Regulatory role of ubiquilin in autophagy
Ubiquilins are a family of small ubiquitin-like proteins involved in presentation of substrate carried by the chaperones HSP90–HSP110 to the ubiquitin proteasome system.70 Mutations in the UBQLN2 gene (encoding ubiquilin-2) have been reported in patients with amyotrophic lateral sclerosis and in patients with frontotemporal dementia.71 Localisation of ubiquilin-2 in autophagosomes suggests a possible role for this protein in macroautophagy.72 Several different ubiquilin family members might be involved in autophagy, as deletion of ubiquilin-1 reduces the number of autophagosomes and slows down their maturation.73 The role of ubiquilin-2 in the biogenesis of autophagosomes could be in part mediated through its inhibitory effect on mTOR complex 1,74 whereas interaction of ubiquilin-1 and ubiquilin-4 with LC3B at the autophagosome membrane seems to modulate autophagosome maturation.73 Because of its degradation by chaperone-mediated autophagy, ubiquilin-1 might be a potential mediator of the compensatory upregulation of macroautophagy that occurs after chaperone-mediated autophagy has failed.75
Therapeutic targeting of autophagy in neurodegenerative disorders
The fact that defective autophagy is a common feature to many neurodegenerative disorders has provided a rationale for interventions aiming to enhance autophagy (figure 4). The challenge involves determining whether the autophagy needs to be upregulated or repaired. An additional concern is whether activation of autophagy, in some instances, could become detrimental. So far, the described toxicity due to excessive autophagosomes originates from failure to degrade these compartments and consequently should benefit from interventions that increase autophagic flux. However, some studies in cell cultures modelling Parkinson’s disease have shown that abnormally upregulated mitophagy is associated with non-apoptotic cell death,86 neuronal atrophy,87 or altered signalling mechanisms.88 Mitochondrial toxins, such as the short mitochondrial ARF protein known to induce neurodegeneration, might in part act by abnormal upregulation of macroautophagy.89 Unfortunately, in many experimental models, the nonspecific nature of the agents used to stimulate macroautophagy make it difficult to directly link the toxic effect to induction of autophagy. Identifying at which point autophagy fails and what the best approaches are to restore autophagy function and reinstate neuronal proteostasis is essential for therapy development for neurogenerative disorders.
Figure 4: Therapeutic modulation of autophagy in neurodegenerative diseases.
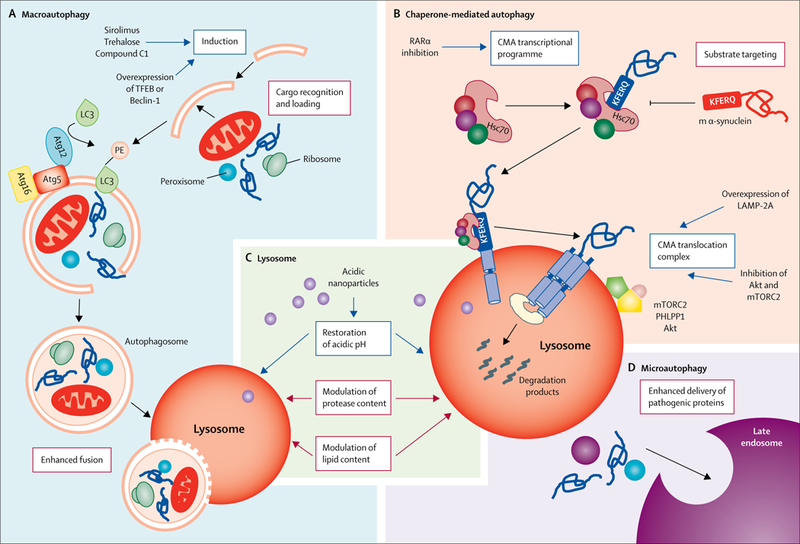
Current strategies (blue boxes) and possible future strategies (red boxes) for enhancing or repairing the autophagic defects described in different neurodegenerative disorders. (A) Macroautophagy: induction of autophagosome biogenesis with chemical compounds (ie, sirolimus or trehalose) or through genetic manipulations (ie, overexpression of TFEB or Beclin-1) has successfully reinstated autophagy function, in cell cultures and in animal models of disease.4,35,76–84 Interventions targeting the other steps in macroautophagy to either enhance their activity or prevent the inhibitory effect of pathogenic proteins have not been attempted yet. (B) Chaperone-mediated autophagy: chemical activation of the transcriptional programme of chaperone-mediated autophagy through inhibition of the retinoic acid receptor-α signaling has been proven effective in reducing α-synuclein toxicity in vitro.85 Drugs such as inhibitors of Akt, inhibitors of mTORC2, or activators of PHLPP1 (proteins that modulate LAMP-2A dynamics of assembly and disassembly at the lysosomal membrane) are also available, but are not selective to this autophagic pathway and their effect on proteotoxicity has not been tested in contexts with defective chaperone-mediated autophagy. Genetic upregulation of LAMP-2A expression to enhance chaperone-mediated autophagy has proven effective in preventing α-synuclein-mediated toxicity. Interventions at the level of substrate targeting that could prevent delivery of pathogenic proteins to lysosomes to avoid their toxic effect on chaperone-mediated autophagy have also not been attempted. (C) Lysosome: restoration of lysosomal acidification with acidic nanoparticles has been successful in restoring autophagy function in animal models.32,49 Interventions to modulate protease or lipid content have not been attempted yet. (D) Endosomal microautophagy: although no interventions to modulate this form of degradation have been attempted, the existence of this degradation pathway at the neuronal synapsis provides a good opportunity to enhance delivery of pathogenic proteins to alleviate their toxicity. m=mutant. TFEB=transcription factor EB. LC3=microtubule-associated protein 1 light chain 3. Atg=autophagy-related protein. RARα=retinoic acid receptor-α. CMA=chaperone-mediated autophagy. Hsc70=heat shock cognate 70 kDa protein. LAMP-2A=lysosome-associated membrane protein-2A. mTORC2=mTOR complex 2. PHLPP1=PH domain leucine-rich repeat-containing protein 1.
Macroautophagy induction
Among the different approaches to induce macro-autophagy in neurodegenerative disorders, sirolimus (also known as rapamycin) has been shown to prevent protein aggregation and neurodegeneration in experimental models of Parkinson’s disease, Alzheimer’s disease, and Huntington’s disease (figure 4A). Sirolimus is the best known mTOR inhibitor, and has been used extensively as an immunosuppressor in patients undergoing organ transplantation.4 However, the neuroprotective efficacy of sirolimus remains to be established in clinically relevant experimental models.76 Although sirolimus and other drugs targeting mTOR (such as torin 1 and WYE-125132)4 effectively upregulate autophagy, they can affect many other autophagy-independent functions of mTOR and also have an immunosuppressive effect, motivating the search for more selective autophagy activators.
Efforts to avoid mTOR inhibition have brought the use of autophagy activators to the forefront, such as trehalose, a disaccharide not synthesised by vertebrates that induces autophagy in an mTOR-independent manner.77 Trehalose has been shown to be beneficial in several animal models of Parkinson’s disease, tauopathies, and amyotrophic lateral sclerosis.4,78 Other strategies to activate autophagy include inhibition of inositol synthesis by carbamazepine90 or lithium,91 inhibition of acetyl transferases by spermidine, and inhibition of the cytosolic protease calpain by calpastatin.92
Pathway-specific activation of autophagy
Disease-specific differences in autophagy dysfunction and in the defective autophagic step require approaches that allow activation of a given autophagic pathway or step (figure 4). For example, in diseases with lysosomal dysfunction, such as some forms of familiar Alzheimer’s disease, a beneficial effect of enhancing lysosomal biogenesis is unlikely, as newly formed lysosomes will probably remain dysfunctional.
Attempts to increase selectivity in activation of autophagy have relied on the overexpression of a specific component of a pathway. Overexpression of Beclin-1 to induce macroautophagy reduces neurodegeneration and aggregation of α-synuclein in a mouse model of Parkinson’s disease.79 Similarly, overexpression of LAMP-2A to boost chaperone-mediated autophagy activity protects from α-synuclein toxicity in a rat model of Parkinson’s disease.93 Efforts are now focused on developing smaller molecules that could mimic the effect of these genetic approaches for use in clinical settings. For instance, a peptide derived from Beclin-1 was able to restore proteostasis in the context of polyglutamine expansion, leading to reduced polyglutamine-repeat aggregates in cultured cells.80 The discovery that chaperone-mediated autophagy is under the negative regulation of retinoic acid receptor-α enabled the development of small molecules that decrease its inhibitory effect, without interfering with other downstream signals of this receptor.85 These novel activators of chaperone-mediated autophagy improved survival after proteotoxic and oxidative insults in vitro.85 Further studies are now needed to show in vivo efficacy of these selective activators in the context of disease.
Targeting lysosomal function
Approaches that only target the lysosomal compartment, desirable in conditions with preferential failure of this organelle, are also gaining momentum (figure 4C). For example, the use of acidic nanoparticles has been shown to restore lysosomal acidification in the context of presenilin-1 mutations that dissipate the lysosomal pH through calcium leakage from this compartment.32 Acidic nanoparticles have also been beneficial in Parkinson’s disease associated with mutations in ATP13A2 or GBA (encoding glucocerebrosidase, also known as GCase), and in models of chemically induced Parkinson’s disease.49 In both experimental models, restoration of lysosomal pH reduced the autophagy blockage, thus supporting the value of directly targeting the lysosome in these conditions.
Modulation of TFEB, known to regulate lysosome biogenesis and macroautophagy,81 has generated considerable interest as a basis for treatment against neurodegeneration.82 Overexpression of TFEB has already been proven beneficial in experimental mouse models of diseases such as the metabolic myopathy Pompe disease94 and alcoholic and non-alcoholic fatty liver disease,95 as well as in neurodegeneration.35,83 The search for more translatable chemical modulators of TFEB has identified a curcumin derivative termed C1 as an mTOR-independent activator of TFEB.96 Confirmation of the efficacy of all these lysosome-targeting compounds in patients is still pending. The combined action of TFEB on lysosomes and macroautophagy has been suggested to drive a general neuroprotective effect on Parkinson’s disease.84 However, TFEB and upregulation of macroautophagy have been shown to cause harm in the context of cellular and Drosophila models of amyotrophic lateral sclerosis, when TDP-43 mutation blocks maturation of autolysosomes.97
The steps of chaperone-mediated autophagy that occur at the lysosomal surface are most vulnerable to neurodegeneration-associated proteotoxicity, compared with the steps taking place in the cytosol.18,19,21 Although chemical inhibition of the signalling through retinoic acid receptor-α increases the concentration of LAMP-2A in lysosomes,85 selective modulation of LAMP-2A dynamics in this compartment might be desirable in specific diseases such as Parkinson’s disease or frontotemporal dementia, wherein pathogenic forms of α-synuclein or tau, respectively, directly interfere with assembly of the LAMP-2A multimeric complex required for chaperone-mediated autophagy.18,19 In this respect, activators of PH domain leucine-rich repeat-containing protein 1 (PHLPP1) or Rac1 (the small GTPase that brings PHLPP1 to the lysosomal membrane to counteract the inhibitory effect of the mTORC2–Akt1 axis on chaperone-mediated autophagy)98 could become attractive targets for selective modulation of chaperone-mediated autophagy in Parkinson’s disease and frontotemporal dementia.
Conclusions
The involvement of defective autophagy in the pathogenesis and progression of neurodegenerative diseases such as Alzheimer’s disease, amyotrophic lateral sclerosis, Huntington’s disease, and Parkinson’s disease is now well established. Evidence supports many mechanisms of autophagy failure in neurodegenerative disorders, which highlight that autophagy can be the primary defect responsible for the loss of proteostasis, or be the target of toxicity caused by pathogenic proteins. Stepwise failures for each of the types of autophagy have been described in neurodegenerative diseases, and more detailed molecular dissection of autophagy has identified common pathogenic proteins as regulators or effectors of autophagy.
However, clinical translation of this wealth of knowledge is still at an early stage. Current limitations range from gaps in methodology to difficulties in selective drug development. A long-standing challenge in the implementation of autophagy-based therapies, from a methodological standpoint, is the difficulty to dynamically evaluate autophagy in vivo. This limitation becomes important both for diagnosis and for monitoring efficiency of any autophagy-based intervention. At the experimental level, progress has been made through expression of the tandem macroautophagy reporter mRFP-GFP-LC3 in mouse models, either as a transgene99 or by the intraventricular delivery of adeno-associated viruses to the brain.100 The mRFP-GFP-LC3 reporter system monitors overall autophagy, whereas for selective types of macroautophagy only a mitophagy reporter model is currently available.101 Autophagy reporters for use in the clinical setting are not available yet. Until this type of dynamic study becomes possible in humans, efforts should be made to identify the molecular signature of defective autophagy in body fluids (ie, CSF or serum). Developing methods to identify this signature will be also important for early diagnosis, because autophagy failure might precede appearance of neurodegenerative symptoms.
The initial methods of pharmacological modulation of autophagy in neurodegenerative disorders were based on overall induction of autophagy, but these are being replaced by more selective approaches that allow only some autophagic pathways or steps to be targeted.85 Although evidence of a beneficial effect of these interventions in clinical settings is still scarce, we believe that the future for therapeutic development lies in the activation of specific types of autophagy, correction of the individual steps, or both.
When designing autophagy-based interventions, more consideration should be given to the regional and cell-specific differences in autophagic requirements, and how each of these are affected during disease. Most studies so far have focused on neuronal autophagy, despite the extensive cellular heterogeneity of the CNS. Autophagy failure in glial cells has been reported in the presence of mutant glucocerebrosidase in experimental models of Parkinson’s disease,102 in cells from patients with Alzheimer’s disease carrying TREM2 risk variants, and in TREM2-deficient mouse models of Alzheimer’s disease.103 Moreover, the close relationship between glia and neurons explains why impairment in glial autophagy can affect the survival of nearby cells,104,105 inducing profound degeneration of neighbouring neurons. In light of these findings, restoring defective glial autophagy should also be beneficial to neurons. In this respect, it is encouraging to see the recent increased emphasis on astrocyte-based therapeutic approaches, including studies using cell-specific microRNAs to selectively target glial cells.106,107
Different cell types also display distinctive autophagy responses to stressors,108 which should be considered when generating drugs that target specific cell types. Furthermore, growing evidence shows that vulnerability to autophagy malfunction changes in different brain regions.13,30 Thus, disease-modifying compounds capable of targeting autophagy in specific brain regions and cell types are required. However, before this type of selective targeting can be done, a more complete picture of the cell-autonomous and non-cell-autonomous regulation of CNS autophagy should be obtained, and more drug screening and testing should be done to identify compounds with good bioavailability in the brain and good capability to modulate different types of selective autophagy.
Closing the gap between bench and bedside for translation of autophagy-based therapeutics will also require development of better methods to monitor autophagy function in patients with neurodegenerative disorders. Few markers of autophagy dysfunction are known in humans (eg, reduced autophagy flux in peripheral blood cells),109 and they often do not provide organ-specific information on the status of autophagy. Identifying surrogate markers of CNS autophagy malfunction and developing non-invasive, image-based methods for dynamic measurement of CNS autophagy (similar to those used to track pathogenic proteins in the brains of patients), are among the most urgent challenges that should be overcome in the next years, so that therapies based on autophagy modulation can be implemented to treat neurodegenerative disorders.
Search strategy and selection criteria.
We searched Entrez (PubMed) and Google Scholar for articles in English that had been published between Jan 1, 2013, to May 25, 2018, using the search terms “autophagy” or “lysosome” in combination with “Alzheimer’s disease”, “Parkinson’s disease”, “Huntington’s disease”, “Amyotrophic lateral sclerosis”, “neurodegeneration”, “protein aggregation”, and “aging”. The final reference list was selected on the basis of originality and topical relevance.
Acknowledgments
We thank the members of our labs and collaborators for their comments and insightful discussions, and especially Susmita Kaushik for critically reading the manuscript.
Footnotes
Declaration of interests
None of the authors declares a competing interest.
Contributor Information
Aurora Scrivo, Department of Developmental and Molecular Biology and Institute for Aging Studies, Albert Einstein College of Medicine, Bronx, NY, USA.
Mathieu Bourdenx, Department of Developmental and Molecular Biology and Institute for Aging Studies, Albert Einstein College of Medicine, Bronx, NY, USA.
Olatz Pampliega, Université de Bordeaux and Centre National de la Recherche Scientifique, Institut des Maladies Neurodégénératives, Bordeaux, France.
Ana Maria Cuervo, Department of Developmental and Molecular Biology and Institute for Aging Studies, Albert Einstein College of Medicine, Bronx, NY, USA.
References
- 1.Labbadia J, Morimoto RI. The biology of proteostasis in aging and disease. Annu Rev Biochem 2015; 84: 435–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaushik S, Cuervo AM. Proteostasis and aging. Nat Med 2015; 21: 1406–15. [DOI] [PubMed] [Google Scholar]
- 3.Kaushik S, Cuervo AM. The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Biol 2018; 19: 365–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Menzies FM, Fleming A, Caricasole A, et al. Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron 2017; 93: 1015–34. [DOI] [PubMed] [Google Scholar]
- 5.Bento CF, Renna M, Ghislat G, et al. Mammalian autophagy: how does it work? Annu Rev Biochem 2016; 85: 685–713. [DOI] [PubMed] [Google Scholar]
- 6.Levine B, Klionsky DJ. Autophagy wins the 2016 Nobel Prize in Physiology or Medicine: breakthroughs in baker’s yeast fuel advances in biomedical research. Proc Natl Acad Sci USA 2017; 114: 201–05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tekirdag K, Cuervo AM. Chaperone-mediated autophagy and endosomal microautophagy: joint by a chaperone. J Biol Chem 2018; 293: 5414–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hegde AN. Proteolysis, synaptic plasticity and memory. Neurobiol Learn Mem 2017; 138: 98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim HJ, Cho MH, Shim WH, et al. Deficient autophagy in microglia impairs synaptic pruning and causes social behavioral defects. Mol Psychiatry 2017; 22: 1576–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xi Y, Dhaliwal JS, Ceizar M, Vaculik M, Kumar KL, Lagace DC. Knockout of Atg5 delays the maturation and reduces the survival of adult-generated neurons in the hippocampus. Cell Death Dis 2016; 7: e2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu X, Fleming A, Ricketts T, et al. Autophagy regulates Notch degradation and modulates stem cell development and neurogenesis. Nat Commun 2016; 7: 10533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hara T, Nakamura K, Matsui M, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006; 441: 885–89. [DOI] [PubMed] [Google Scholar]
- 13.Komatsu M, Waguri S, Chiba T, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006; 441: 880–84. [DOI] [PubMed] [Google Scholar]
- 14.Khaminets A, Behl C, Dikic I. Ubiquitin-dependent and independent signals in selective autophagy. Trends Cell Biol 2016; 26: 6–16. [DOI] [PubMed] [Google Scholar]
- 15.Lim J, Lachenmayer ML, Wu S, et al. Proteotoxic stress induces phosphorylation of p62/SQSTM1 by ULK1 to regulate selective autophagic clearance of protein aggregates. PLoS Genet 2015; 11: e1004987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Isakson P, Holland P, Simonsen A. The role of ALFY in selective autophagy. Cell Death Differ 2013; 20: 12–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crippa V, Boncoraglio A, Galbiati M, et al. Differential autophagy power in the spinal cord and muscle of transgenic ALS mice. Front Cell Neurosci 2013; 7: 234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 2004; 305: 1292–95. [DOI] [PubMed] [Google Scholar]
- 19.Orenstein SJ, Kuo SH, Tasset I, et al. Interplay of LRRK2 with chaperone-mediated autophagy. Nat Neurosci 2013; 16: 394–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kabuta T, Furuta A, Aoki S, Furuta K, Wada K. Aberrant interaction between Parkinson disease-associated mutant UCH-L1 and the lysosomal receptor for chaperone-mediated autophagy. J Biol Chem 2008; 283: 23731–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Caballero B, Wang Y, Diaz A, et al. Interplay of pathogenic forms of human tau with different autophagic pathways. Aging Cell 2018; 17: e12692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu CY, Kang WY, Chen YM, et al. DJ-1 inhibits α-synuclein aggregation by regulating chaperone-mediated autophagy. Front Aging Neurosci 2017; 9: 308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cannizzo ES, Clement CC, Morozova K, et al. Age-related oxidative stress compromises endosomal proteostasis. Cell Reports 2012; 2: 136–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wong E, Bejarano E, Rakshit M, et al. Molecular determinants of selective clearance of protein inclusions by autophagy. Nat Commun 2012; 3: 1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rui YN, Xu Z, Patel B, et al. Huntingtin functions as a scaffold for selective macroautophagy. Nat Cell Biol 2015; 17: 262–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ichimura Y, Waguri S, Sou YS, et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol Cell 2013; 51: 618–31. [DOI] [PubMed] [Google Scholar]
- 27.Jo C, Gundemir S, Pritchard S, Jin YN, Rahman I, Johnson GV. Nrf2 reduces levels of phosphorylated tau protein by inducing autophagy adaptor protein NDP52. Nat Commun 2014; 5: 3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wen Z, Shu Y, Gao C, et al. CDK5-mediated phosphorylation and autophagy of RKIP regulate neuronal death in Parkinson’s disease. Neurobiol Aging 2014; 35: 2870–80. [DOI] [PubMed] [Google Scholar]
- 29.Komatsu M, Wang QJ, Holstein GR, et al. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc Natl Acad Sci USA 2007; 104: 14489–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Friedman LG, Lachenmayer ML, Wang J, et al. Disrupted autophagy leads to dopaminergic axon and dendrite degeneration and promotes presynaptic accumulation of α-synuclein and LRRK2 in the brain. J Neurosci 2012; 32: 7585–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rué L, López-Soop G, Gelpi E, Martínez-Vicente M, Alberch J, Pérez-Navarro E. Brain region-and age-dependent dysregulation of p62 and NBR1 in a mouse model of Huntington’s disease. Neurobiol Dis 2013; 52: 219–28. [DOI] [PubMed] [Google Scholar]
- 32.Lee JH, McBrayer MK, Wolfe DM, et al. Presenilin 1 maintains lysosomal Ca(2+) homeostasis via TRPML1 by regulating vATPase-mediated lysosome acidification. Cell Reports 2015; 12: 1430–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ashkenazi A, Bento CF, Ricketts T, et al. Polyglutamine tracts regulate beclin 1-dependent autophagy. Nature 2017; 545: 108–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bourdenx M, Bezard E, Dehay B. Lysosomes and α-synuclein form a dangerous duet leading to neuronal cell death. Front Neuroanat 2014; 8: 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Decressac M, Mattsson B, Weikop P, Lundblad M, Jakobsson J, Björklund A. TFEB-mediated autophagy rescues midbrain dopamine neurons from α-synuclein toxicity. Proc Natl Acad Sci USA 2013; 110: E1817–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mealer RG, Murray AJ, Shahani N, Subramaniam S, Snyder SH. Rhes, a striatal-selective protein implicated in Huntington disease, binds beclin-1 and activates autophagy. J Biol Chem 2014; 289: 3547–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martinez-Vicente M, Talloczy Z, Wong E, et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat Neurosci 2010; 13: 567–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martinez-Vicente M Neuronal mitophagy in neurodegenerative diseases. Front Mol Neurosci 2017; 10: 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cirulli ET, Lasseigne BN, Petrovski S, et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 2015; 347: 1436–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Freischmidt A, Wieland T, Richter B, et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat Neurosci 2015; 18: 631–36. [DOI] [PubMed] [Google Scholar]
- 41.Heo JM, Ordureau A, Paulo JA, Rinehart J, Harper JW. The PINK1-Parkin mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol Cell 2015; 60: 7–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eiyama A, Okamoto K. PINK1/Parkin-mediated mitophagy in mammalian cells. Curr Opin Cell Biol 2015; 33: 95–101. [DOI] [PubMed] [Google Scholar]
- 43.Richter B, Sliter DA, Herhaus L, et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci USA 2016; 113: 4039–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McWilliams TG, Prescott AR, Montava-Garriga L, et al. Basal mitophagy occurs independently of PINK1 in mouse tissues of high metabolic demand. Cell Metab 2018; 27 (2): 439–449.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maday S, Holzbaur EL. Autophagosome biogenesis in primary neurons follows an ordered and spatially regulated pathway. Dev Cell 2014; 30: 71–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cuervo AM, Wong E. Chaperone-mediated autophagy: roles in disease and aging. Cell Res 2014; 24: 92–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Volpicelli-Daley LA, Gamble KL, Schultheiss CE, Riddle DM, West AB, Lee VM. Formation of α-synuclein Lewy neurite-like aggregates in axons impedes the transport of distinct endosomes. Mol Biol Cell 2014; 25: 4010–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tammineni P, Cai Q. Defective retrograde transport impairs autophagic clearance in Alzheimer disease neurons. Autophagy 2017; 13: 982–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bourdenx M, Daniel J, Genin E, et al. Nanoparticles restore lysosomal acidification defects: implications for Parkinson and other lysosomal-related diseases. Autophagy 2016; 12: 472–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miura E, Hasegawa T, Konno M, et al. VPS35 dysfunction impairs lysosomal degradation of α-synuclein and exacerbates neurotoxicity in a Drosophila model of Parkinson’s disease. Neurobiol Dis 2014; 71: 1–13. [DOI] [PubMed] [Google Scholar]
- 51.Tang FL, Erion JR, Tian Y, et al. VPS35 in dopamine neurons is required for endosome-to-golgi retrieval of LAMP2A, a receptor of chaperone-mediated autophagy that is critical for α-synuclein degradation and prevention of pathogenesis of Parkinson’s disease. J Neurosci 2015; 35: 10613–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Saitsu H, Nishimura T, Muramatsu K, et al. De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat Genet 2013; 45: 445–49. [DOI] [PubMed] [Google Scholar]
- 53.Kim M, Sandford E, Gatica D, et al. Mutation in ATG5 reduces autophagy and leads to ataxia with developmental delay. eLife 2016; 5: e12245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van der Zee J, Van Langenhove T, Kovacs GG, et al. Rare mutations in SQSTM1 modify susceptibility to frontotemporal lobar degeneration. Acta Neuropathol 2014; 128: 397–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cuyvers E, van der Zee J, Bettens K, et al. Genetic variability in SQSTM1 and risk of early-onset Alzheimer dementia: a European early-onset dementia consortium study. Neurobiol Aging 2015; 36: 2005.e15–22 [DOI] [PubMed] [Google Scholar]
- 56.Teyssou E, Takeda T, Lebon V, et al. Mutations in SQSTM1 encoding p62 in amyotrophic lateral sclerosis: genetics and neuropathology. Acta Neuropathol 2013; 125: 511–22. [DOI] [PubMed] [Google Scholar]
- 57.Ando M, Fiesel FC, Hudec R, et al. The PINK1 p.I368N mutation affects protein stability and ubiquitin kinase activity. Mol Neurodegener 2017; 12: 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Damiano M, Gautier CA, Bulteau AL, et al. Tissue-and cell-specific mitochondrial defect in parkin-deficient mice. PLoS One 2014; 9: e99898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lobasso S, Tanzarella P, Vergara D, Maffia M, Cocco T, Corcelli A. Lipid profiling of parkin-mutant human skin fibroblasts. J Cell Physiol 2017; 232: 3540–51. [DOI] [PubMed] [Google Scholar]
- 60.Schwab C, Yu S, McGeer EG, McGeer PL. Optineurin in Huntington’s disease intranuclear inclusions. Neurosci Lett 2012; 506: 149–54. [DOI] [PubMed] [Google Scholar]
- 61.Bjørkøy G, Lamark T, Brech A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 2005; 171: 603–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wong YC, Holzbaur EL. The regulation of autophagosome dynamics by huntingtin and HAP1 is disrupted by expression of mutant huntingtin, leading to defective cargo degradation. J Neurosci 2014; 34: 1293–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ochaba J, Lukacsovich T, Csikos G, et al. Potential function for the Huntingtin protein as a scaffold for selective autophagy. Proc Natl Acad Sci USA 2014; 111: 16889–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sun X, Fu Y, Pan Y, Lu B. Conformation-dependent recognition of mutant HTT (huntingtin) proteins by selective autophagy. Autophagy 2017; 13: 2111–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ghazi-Noori S, Froud KE, Mizielinska S, et al. Progressive neuronal inclusion formation and axonal degeneration in CHMP2B mutant transgenic mice. Brain 2012; 135: 819–32. [DOI] [PubMed] [Google Scholar]
- 66.Clayton EL, Mizielinska S, Edgar JR, et al. Frontotemporal dementia caused by CHMP2B mutation is characterised by neuronal lysosomal storage pathology. Acta Neuropathol 2015; 130: 511–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zavodszky E, Seaman MN, Rubinsztein DC. VPS35 Parkinson mutation impairs autophagy via WASH. Cell Cycle 2014; 13: 2155–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sahu R, Kaushik S, Clement CC, et al. Microautophagy of cytosolic proteins by late endosomes. Dev Cell 2011; 20: 131–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tsika E, Glauser L, Moser R, et al. Parkinson’s disease-linked mutations in VPS35 induce dopaminergic neurodegeneration. Hum Mol Genet 2014; 23: 4621–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hjerpe R, Bett JS, Keuss MJ, et al. UBQLN2 mediates autophagy-independent protein aggregate clearance by the proteasome. Cell 2016; 166: 935–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Radzicki D, Liu E, Deng HX, Siddique T, Martina M. Early impairment of synaptic and intrinsic excitability in mice expressing ALS/dementia-linked mutant UBQLN2. Front Cell Neurosci 2016; 10: 216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Osaka M, Ito D, Yagi T, Nihei Y, Suzuki N. Evidence of a link between ubiquilin 2 and optineurin in amyotrophic lateral sclerosis. Hum Mol Genet 2015; 24: 1617–29. [DOI] [PubMed] [Google Scholar]
- 73.Lee DY, Arnott D, Brown EJ. Ubiquilin4 is an adaptor protein that recruits Ubiquilin1 to the autophagy machinery. EMBO Rep 2013; 14: 373–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Coffey RT, Shi Y, Long MJ, Marr MT 2nd, Hedstrom L Ubiquilin-mediated small molecule inhibition of mammalian target of rapamycin complex 1 (mTORC1) signaling. J Biol Chem 2016; 291: 5221–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rothenberg C, Srinivasan D, Mah L, et al. Ubiquilin functions in autophagy and is degraded by chaperone-mediated autophagy. Hum Mol Genet 2010; 19: 3219–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dehay B, Bourdenx M, Gorry P, et al. Targeting α-synuclein for treatment of Parkinson’s disease: mechanistic and therapeutic considerations. Lancet Neurol 2015; 14: 855–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sarkar S, Davies JE, Huang Z, Tunnacliffe A, Rubinsztein DC. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J Biol Chem 2007; 282: 5641–52. [DOI] [PubMed] [Google Scholar]
- 78.Castillo K, Nassif M, Valenzuela V, et al. Trehalose delays the progression of amyotrophic lateral sclerosis by enhancing autophagy in motoneurons. Autophagy 2013; 9: 1308–20. [DOI] [PubMed] [Google Scholar]
- 79.Spencer B, Potkar R, Trejo M, et al. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson’s and Lewy body diseases. J Neurosci 2009; 29: 13578–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shoji-Kawata S, Sumpter R, Leveno M, et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature 2013; 494: 201–06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Settembre C, Fraldi A, Medina DL, Ballabio A. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol 2013; 14: 283–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Martini-Stoica H, Xu Y, Ballabio A, Zheng H. The autophagy-lysosomal pathway in neurodegeneration: a TFEB perspective. Trends Neurosci 2016; 39: 221–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Polito VA, Li H, Martini-Stoica H, et al. Selective clearance of aberrant tau proteins and rescue of neurotoxicity by transcription factor EB. EMBO Mol Med 2014; 6: 1142–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Torra A, Parent A, Cuadros T, et al. Overexpression of TFEB drives a pleiotropic neurotrophic effect and prevents Parkinson’s disease-related neurodegeneration. Mol Ther 2018; 26: 1552–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Anguiano J, Garner TP, Mahalingam M, Das BC, Gavathiotis E, Cuervo AM. Chemical modulation of chaperone-mediated autophagy by retinoic acid derivatives. Nat Chem Biol 2013; 9: 374–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Carroll RG, Hollville E, Martin SJ. Parkin sensitizes toward apoptosis induced by mitochondrial depolarization through promoting degradation of Mcl-1. Cell Reports 2014; 9: 1538–53. [DOI] [PubMed] [Google Scholar]
- 87.Curry DW, Stutz B, Andrews ZB, Elsworth JD. Targeting AMPK signaling as a neuroprotective strategy in Parkinson’s disease. J Parkinsons Dis 2018; 8: 161–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Li S, Li H, Yang D, et al. Excessive autophagy activation and increased apoptosis are associated with palmitic acid-induced cardiomyocyte insulin resistance. J Diabetes Res 2017; 2017: 2376893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Reef S, Zalckvar E, Shifman O, et al. A short mitochondrial form of p19ARF induces autophagy and caspase-independent cell death. Mol Cell 2006; 22: 463–75. [DOI] [PubMed] [Google Scholar]
- 90.Li L, Zhang S, Zhang X, et al. Autophagy enhancer carbamazepine alleviates memory deficits and cerebral amyloid-β pathology in a mouse model of Alzheimer’s disease. Curr Alzheimer Res 2013; 10: 433–41. [DOI] [PubMed] [Google Scholar]
- 91.Shimada K, Motoi Y, Ishiguro K, et al. Long-term oral lithium treatment attenuates motor disturbance in tauopathy model mice: implications of autophagy promotion. Neurobiol Dis 2012; 46: 101–08. [DOI] [PubMed] [Google Scholar]
- 92.Menzies FM, Garcia-Arencibia M, Imarisio S, et al. Calpain inhibition mediates autophagy-dependent protection against polyglutamine toxicity. Cell Death Differ 2015; 22: 433–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Xilouri M, Brekk OR, Landeck N, et al. Boosting chaperone-mediated autophagy in vivo mitigates α-synuclein-induced neurodegeneration. Brain 2013; 136: 2130–46. [DOI] [PubMed] [Google Scholar]
- 94.Gatto F, Rossi B, Tarallo A, et al. AAV-mediated transcription factor EB (TFEB) gene delivery ameliorates muscle pathology and function in the murine model of Pompe disease. Sci Rep 2017; 7: 15089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang C, Niederstrasser H, Douglas PM, et al. Small-molecule TFEB pathway agonists that ameliorate metabolic syndrome in mice and extend C. elegans lifespan. Nat Commun 2017; 8: 2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Song JX, Sun YR, Peluso I, et al. A novel curcumin analog binds to and activates TFEB in vitro and in vivo independent of MTOR inhibition. Autophagy 2016; 12: 1372–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Xia Q, Wang H, Hao Z, et al. TDP-43 loss of function increases TFEB activity and blocks autophagosome-lysosome fusion. EMBO J 2016; 35: 121–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Arias E, Koga H, Diaz A, Mocholi E, Patel B, Cuervo AM. Lysosomal mTORC2/PHLPP1/Akt regulate chaperone-mediated autophagy. Mol Cell 2015; 59: 270–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 2007; 3: 452–60. [DOI] [PubMed] [Google Scholar]
- 100.Castillo K, Valenzuela V, Matus S, et al. Measurement of autophagy flux in the nervous system in vivo. Cell Death Dis 2013; 4: e917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sun N, Yun J, Liu J, et al. Measuring in vivo mitophagy. Mol Cell 2015; 60: 685–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Osellame LD, Rahim AA, Hargreaves IP, et al. Mitochondria and quality control defects in a mouse model of Gaucher disease—links to Parkinson’s disease. Cell Metab 2013; 17: 941–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ulland TK, Song WM, Huang SC, et al. TREM2 maintains microglial metabolic fitness in Alzheimer’s disease. Cell 2017; 170: 649–663.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Soria FN, Engeln M, Martinez-Vicente M, et al. Glucocerebrosidase deficiency in dopaminergic neurons induces microglial activation without neurodegeneration. Hum Mol Genet 2017; 26: 2603–15. [DOI] [PubMed] [Google Scholar]
- 105.Ray A, Speese SD, Logan MA. Glial draper rescues Aβ toxicity in a Drosophila model of Alzheimer’s disease. J Neurosci 2017; 37: 11881–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Taschenberger G, Tereshchenko J, Kügler S. A microRNA124 Target sequence restores astrocyte specificity of gfaABC1D-driven transgene expression in AAV-mediated gene transfer. Mol Ther Nucleic Acids 2017; 8: 13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Finsterwald C, Magistretti PJ, Lengacher S. Astrocytes: new targets for the treatment of neurodegenerative diseases. Curr Pharm Des 2015; 21: 3570–81. [DOI] [PubMed] [Google Scholar]
- 108.Chapin HC, Okada M, Merz AJ, Miller DL. Tissue-specific autophagy responses to aging and stress in C. elegans. Aging (Albany NY) 2015; 7: 419–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Raz Y, Guerrero-Ros I, Maier A, et al. Activation-induced autophagy is preserved in CD4+ T-Cells in familial longevity. J Gerontol A Biol Sci Med Sci 2017; 72: 1201–06. [DOI] [PMC free article] [PubMed] [Google Scholar]