

Abstract
The Mizoroki–Heck reaction is one of the most efficient methods for alkenylation of aryl, vinyl, and alkyl halides. Given its innate nature, this protocol requires the employment of compounds possessing a halogen atom at the site of functionalization. However, the accessibility of organic molecules possessing a halogen atom at a particular site in aliphatic systems is extremely limited. Thus, a protocol that allows a Heck reaction to occur at a specific nonfunctionalized C(sp3)–H site is desirable. Reported here is a radical relay Heck reaction which allows selective remote alkenylation of aliphatic alcohols at unactivated β-, γ-, and δ-C(sp3)–H sites. The use of an easily installed/removed Si-based auxiliary enables selective I-atom/radical translocation events at remote C–H sites followed by the Heck reaction. Notably, the reaction proceeds smoothly under mild visible-light-mediated conditions at room temperature, producing highly modifiable and valuable alkenol products from readily available alcohols feedstocks.
Keywords: C–H activation, Heck reaction, palladium, photochemistry, radicals
Graphical Abstract

A selective Heck reaction at β-, γ-, and δ-C(sp3)–H sites of aliphatic alcohols has been developed. The radical hydrogen-atom transfer/I-atom translocation process, combined with the palladium-catalyzed Heck reaction, allows selective remote alkenylation at unactivated C-(sp3)–H sites under mild visible-light-induced conditions at room temperature. Neither exogenous oxidants nor photosensitizers are necessary.
The ability to convert ubiquitous C–H bonds of aliphatic molecules into useful functionalities is an ongoing endeavor in organic synthesis. Among the established C–H functionalization methods, remote C–H alkenylation is arguably the most attractive approach because of its capability to associate important olefin functionality with organic molecules by the C–C bond-forming process.[1] In the last decades, considerable progress has been made for alkenylation of C(sp2)–H sites through directed transition metal catalyzed reactions.[2] In contrast, alkenylation of remote C(sp3)–H sites still remains one of the most challenging C–H functionalization processes. For instance, employment of the most efficient heteroatom-containing directing groups, frequently resulted in the cyclized products rather than in the desired alkenylated products.[3] Moreover, remote C(sp3)–H alkenylation often requires difficult to install/modify directing groups, as well as forcing reaction conditions for the activation of strong C(sp3)–H bonds.[4] To solve this challenging problem, we thought of utilizing one of the most powerful tools for C–C bond alkenylation, the Noble-Prize-winning Mizoroki–Heck reaction (Scheme 1a).[5] While the Heck reaction is well known for C(sp2)–X sites, the alkyl Heck reaction is less developed because of the slower oxidative addition at C(sp3)–X sites, and an undesired premature β-hydride elimination reaction.[6] Recently, the development of hybrid Pd/radical species has enabled Heck reactions to occur at unactivated C(sp3)–X sites with high efficiency.[7] Nevertheless, the Heck reaction at remote C(sp3)–H sites has not been reported to date. Thus, if a protocol could be developed that would guide a halogen atom (X) or a hybrid Pd/radical species to a targeted C(sp3)–H site, followed by a Heck reaction, it will create a novel avenue toward remote alkenylation at aliphatic C(sp3)–H sites. Herein, we report a site-selective radical relay Heck reaction of aliphatic alcohols (Scheme 1b). The reaction proceeds under mild visible-light-induced conditions at room temperature, producing β-, γ-, and δ-alkenylation products at remote unactivated sites selectively without the use of exogenous photosensitizers[8] or external oxidants.[9] The control over the targeted C(sp3)–H sites is enabled by an employment of easily installed/removable Si auxiliary through an I-atom/radical translocation event (1→2/3) followed by a Heck reaction at the remote sites (2/3→5). Overall, this method expeditiously converts aliphatic alcohols, which are featured in many complex natural products and are abundant, inexpensive, and sustainable feedstocks, into more complex and valuable organic synthons.[10]
Scheme 1.

Development of radical relay Heck reaction.
In our previously developed method for selective desaturation of aliphatic alcohols,[11] we discovered a remarkable capability of the iodide-containing auxiliary (Scheme 1b, T) to undergo a 1,n-hydrogen atom-transfer (1,n-HAT) event, producing a hybrid Pd/radical species at a targeted C(sp3)–H site (Scheme 1b, 1→3), which upon a facile hydrogen loss was converted into the dehydrogenated product (6). Accordingly, we hypothesized that if the β-hydride elimination step (3→6) could be interrupted, there would be a possibility to engage the translocated species 3 either in a direct coupling with an alkene[12] (3→5) or in the I-atom translocation process,[13] leading to the alkyl halide 2, a capable substrate for the alkyl Heck reaction.[7] If either of these processes are achieved, it would constitute the desired remote Heck reaction at an unactivated C(sp3)–H site (1→5). The success of this reaction hinges on overcoming several challenges (Scheme 1c), such as competitive desaturation (6), premature Heck coupling at the Si-auxiliary site (7), and the hydrodehalogenation side-reaction (8). To test this hypothesis, alkyl radical relay Heck reaction of the tethered-aliphatic alcohol 1a with acrylonitrile (4) was tested under our previously reported desaturation conditions using the ferrocene ligand L1 (Scheme 1d, preliminary results). However, these reported conditions produced only trace amounts of the desired remote Heck product 5, whereas the desaturation product 6 was the major outcome of the reaction. Evidently, under these reaction conditions, the β-hydride elimination of the translocated hybrid Pd/radical species is faster than the desired coupling reaction with 4. Therefore, screening of better chelating ligands with larger bite angle compared to that in L1 was performed to circumvent the undesired β-hydride elimination process.[14] Gratifyingly, xantphos was identified as the best ligand for the radical relay Heck reaction (Scheme 1d). Upon exposure of 1a to the fully optimized reaction conditions,[15] followed by a one-pot deprotection of the Si auxiliary, the γ-Heck product 5a was isolated in 71% yield. Moreover, the reaction proceeded efficiently under mild visible-light-induced conditions at room temperature! This result showcases the first radical relay Heck reaction that produces a remote alkene at a sterically demanding site, resulting in the formation of a quaternary carbon center. We find this result quite remarkable, as the remote C–H alkenylation at a tertiary C–H site is unprecedented.
The generality of this γ-radical relay Heck reaction was found to be relatively broad (Scheme 2a). Thus, alkenes containing electron-withdrawing groups were found to be suitable substrates, as products the 5a–d were isolated in good yields, favoring the Z isomers (for 5a, 5d).[16] It was found that styrene derivatives with different electronic properties all reacted smoothly to selectively generate substituted bis-homoallylic alcohols (5e–m). Notably, this class of alkenes is an uncommon coupling partner for remote alkenylation at C(sp3)–H sites.[3,4,12] Next, substrates containing competitive tertiary C–H sites (β- vs. γ- for 1p,q, and γ- vs. δ- for 1r,s) were tested. Noticeably, because of the higher preference of the Si auxiliary for 1,6-HAT,[11a] γ-functionalized alkenols were obtained as the sole regioisomers (5p–s). Cyclic substrates were also applicable for the reaction, furnishing the products 5t–w in moderate yields. Interestingly, the internal olefin existing in substrate 1x did not hamper the reaction. Next, bulkier tertiary aliphatic alcohols were tested. In these cases, for the ease of installation of the reacting Si tether, the less sterically congested dimethyl Si auxiliary was used (1y–ac). Gratifyingly, acyclic, monocyclic, bicyclic, and tricyclic tertiary alcohols all reacted well, efficiently producing the corresponding Heck reaction products with perfect regioselectivities (5y–ac). The primary alcohol 1ad containing kinetically more accessible tertiary C–H site was coupled at the γ-C–H site selectively. Also, we examined the feasibility of this transformation in a more-complex setting. Substrates derived from natural lauric acid (1ae), oleic acid (1af), and stearic acid (1ag) all furnished the radical relay Heck products in an effective manner. The sclareolide derivative 1ah underwent selective γ-functionalization, where the free hydroxy group did not compromise the reaction. A lithocholic-acid-derived substrate also reacted under these reaction conditions at the γ-site to produce the alkene 5ai in moderate yield. A brief additive-based robustness screening[17] was also performed to evaluate the functional-group tolerance of this reaction.[18] After establishing the scope for γ-Heck reaction, we targeted the more challenging β-alkenylation reaction (Scheme 2b). Since for Si-tethered alcohols 1,5-HAT is kinetically less favorable than 1,6-HAT,[11a] a direct application of our optimized reaction conditions was not efficient for β-relay Heck reaction of 1aj, resulting in the formation of the premature Heck product 7 (Scheme 1c) as a major product. However, employing more diluted conditions[15] provided the desired β-Heck products 5aj and 5ak in good yields (Scheme 2b). The study of the selectivity preference between β- and δ-C–H sites on substrate 1al indicated the preferential alkenylation at the β- over the δ-position (5al). Likewise, the secondary alcohol 1am underwent selective β-alkenylation at the tertiary site, whereas γ-alkenylation at the secondary site was observed as a minor process. Next, the possibility of achieving a δ-Heck reaction was examined (Scheme 2c). Remarkably, styrene, chlorostyrene, and acrylonitrile all smoothly underwent selective δ-alkenylation of alcohols 1an–ap. Then, we turned our attention to another challenging aspect of this reaction, the abstraction of a hydrogen atom at less reactive secondary C(sp3)–H sites[19] (Scheme 2d). Gratifyingly, under slightly modified reaction conditions, tertiary alcohols (1aq–at), as well as a secondary alcohol (1au), all underwent remote Heck reaction at unactivated secondary C–H sites in good yields. Nevertheless, several substrates were found to be incapable reaction partners for this Heck relay. Because of the significantly higher BDE of the C–H bond in the cyclo-propane[20] of 1 av, a HAT event was outcompeted by the premature coupling process. In contrast, the γ-benzylic C–H site of law underwent 1,6-HAT to produce stable benzylic radical, which was inefficient in addition to the alkene,[21] thus, producing the desaturation byproduct exclusively.
Scheme 2.

Scope of radical relay Heck reaction. Experimental details are provided In the supplementary material, a) γ-Heck reactions, b) β-Heck reactions. c) δ-Heck reactions, d) Heck reaction at secondary C–H sites, d.r. = diastereomeric ratio, e) Inefficient substrates, R = iPr for 1a–z, 1ad–am, 1 ao, 1 ap, 1 au, R = Me for 1 aa–ac, 1 an, 1 aq–at, [a] Different deprotection procedures were applied depending on the products: TBAF in THF; CSA in MeOH; or AcCI and Montmorillonite K10 in CH2Cl2, See the Supporting Information for details, [b] Methyl vinyl ketone was used in the radical relay Heck reaction followed by reduction with NaBH4 [c] Premature coupling product was observed as a major product (66%), [d] Desaturation product was observed as a major product (57%), CSA = camphorsulfonic acid, TBAF = tetra-n-butylammonium fluoride, THF = tetrahydrofuran.
The synthetic utility of this reaction was demonstrated by applying synthetically useful alkene transformations to the remote Heck products 5e and 5a (Scheme 3). Formation of a 1,2-diol (9a) and epoxide (9b) at the remote sites to the original hydroxy group proceeded smoothly by dihydroxylation and epoxidation, respectively, of the alkene moiety. Dibromination of an olefin provided two new functionalizable reaction sites at the δ- and ε- positions in a reasonable yield (9c). Formal remote carbonylation (9d) and methylhydroxylation reactions (9e) were also achieved after ozonolysis of the alkene. Interestingly, bromooxygenation of 5e provided the densely substituted tetrahydropyran 9f in good yield. Finally, partial 1,4- and exhaustive reductions of the alkenyl nitrile 5a produced ε-cyano (9g) and ζ-amino (9h) products efficiently.
Scheme 3.
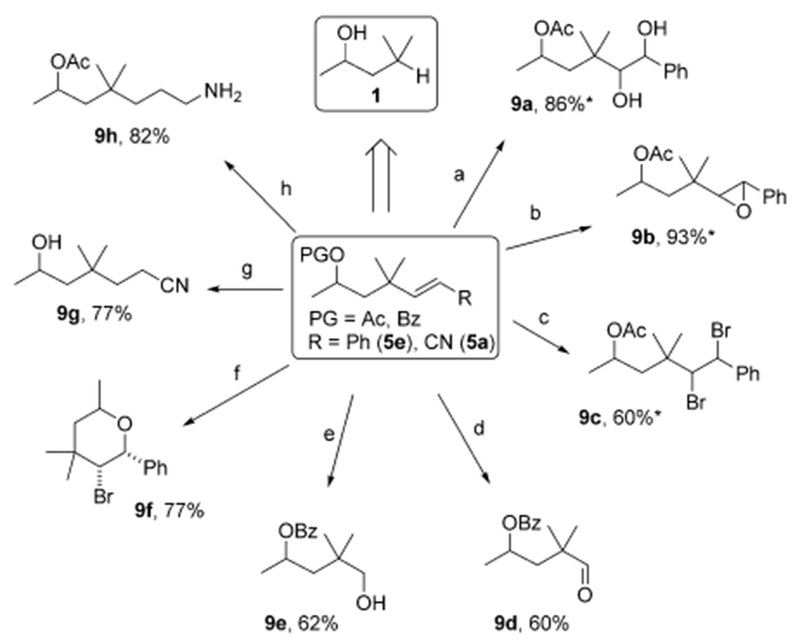
Transformations of alkenol products. Experimental details are provided in the Supporting Information. a) 10 mol% OsO4, 1.2 equiv NMO, acetone/water. b) 1.5 equiv mCPBA, CH2Cl2. c) 2 equiv LiBr, 0.5 equiv NaIO4, CH3CN. d) O3, 2 equiv Me2S, acetone. e) O3, 2 equiv Me2S, acetone, then NaBH4 in MeOH. f) 5 equiv HBr, CHCI3/DMSO. g) 1.2 equiv LiAlH4, Et2O. h) 10 mol% NiCl2, 7 equiv NaBH4, MeOH. *≈1:1 d.r. DMSO = dimethylsulfoxide, mCPBA=m-chloroperbenzoic acid, NMO = N-methylmorpholine N-oxide.
Based on the literature precedent for visible-light-induced palladium-catalyzed alkyl Heck reactions, the mechanism of the transformation is expected to occur by a hybrid Pd/radical pathway.[7f–h] To further investigate the reaction mechanism, we conducted a series of mechanistic studies, including radical scavenger tests and radical-trapping experiments, analysis of the reaction profile, UV-vis analysis, Stern–Volmer studies,[15] and studies on the formation of the proposed I-atom translocation intermediate (2) [Eqs. (1) and (2)]. Indeed, radical scavengers and radical-trapping studies provided support for the radical nature of this transformation, as the employment of radical scavengers greatly suppressed the reaction.[15] Moreover, the radical-trapping product was observed when TEMPO was used.[15] Photophysical studies revealed that the Pd0 complex is the single-photo-absorbing species engaged in an SET event with 1, as demonstrated by Stern–Volmer studies.[15] Analysis of the reaction profile also supports the radical relay Heck hypothesis.[15] Thus, formation of the I-atom translocation intermediate 2 was observed at the beginning of the reaction at a comparable rate with that of the Heck product 5. Then, the translocated alkyl iodide 2 was consumed gradually together with an increasing concentration of the product 5. Consequently, an experiment was tested without base and alkene to prevent the elimination step and/or remote coupling with alkene. Expectedly, formation of 2 was observed under these reaction conditions [Eq. (1)]. Furthermore, when the independently synthesized 2 was subjected to the Heck reaction conditions with both 4-chlorostyrene and acrylonitrile, it was smoothly converted into the γ-Heck products [Eq. (2)]. All together, these results provided evidence for the I-atom/radical translocation process in this radical relay Heck protocol, and strongly supports our initial hypothesis. Based on these studies, the reaction mechanism is proposed (Scheme 4). First, the Pd0 complex undergoes excitation with visible light to form its an excited state species which promotes an SET event with 1 to produce the hybrid Pd-radical species 11. Subsequently, the latter undergoes a 1,n-HAT process, generating the translocated radical species 3. The radical species 3 could reversibly form the observed I-atom transfer intermediate 2 either by a direct atom transfer from the PdI species or by recombination with the Pd complex followed by reductive elimination. Coupling of alkene 4 with 2/3 results in the Pd/radical species 12, which upon β-hydride elimination produces the radical relay Heck product 5 and regenerates the Pd catalyst.
Scheme 4.

Proposed mechanism.
 |
(1) |
 |
(2) |
Through the engagement of selective HAT strategy, the radical relay Heck reaction was achieved. This method expands the boundaries for C–H functionalizations, allowing remote alkenylation reactions to proceed at unactivated β-, γ- and δ-C(sp3)–H sites. It was demonstrated that remote Heck products can easily be obtained from inexpensive, naturally abundant aliphatic alcohols at room temperature, providing access to a diverse classes of more complex molecules. It is anticipated that this approach would provide a more general method for the installation of olefins at remote C–H sites and will find broad applications in synthesis.
Supplementary Material
Acknowledgements
We thank the National Institutes of Health (GM120281) and National Science Foundation (CHE-1663779) for the financial support of this work.
Footnotes
A previous version of this manuscript has been deposited on a preprint server (https://doi.org/10.26434/chemrxiv.7108304).
Supporting information and the ORCID identification number(s) for the author(s) of this article can be found under: https://doi.org/10.1002/anie.201812398.
Conflict of interest
The authors declare no conflict of interest.
References
- [1].a) Daugulis O, Roane J, Tran LD, Acc. Chem. Res 2015, 48, 1053–1064; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Larock RC, Comprehensive Organic Transformations, 2nd ed., Wiley, New York, 1999. [Google Scholar]
- [2].Ma W, Gandeepan P, Li J, Ackermann L, Org. Chem. Front 2017, 4, 1435–1467. [Google Scholar]
- [3].a) He J, Li S, Deng Y, Fu H, Laforteza BN, Spangler JE, Homs A, Yu J-Q, Science 2014, 343, 1216–1220; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Jiang H, He J, Liu T, Yu J-Q, J. Am. Chem. Soc 2016, 138, 2055–2059; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Stowers KJ, Fortner KC, Sanford MS, J. Am. Chem. Soc 2011, 133, 6541–6544; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) He C, Gaunt MJ, Chem. Sci 2017, 8, 3586–3592; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Zhuang Z, Yu C-B, Chen G, Wu Q-F, Hsiao Y, Joe CL, Qiao JX, Poss MA, Yu J-Q, J. Am. Chem. Soc 2018, 140, 10363–10367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].See Ref. [3b] and:; Thrimurtulu N, Khan S, Maity S, Volla CMR, Maiti D, Chem. Commun 2017, 53, 12457–12460. [DOI] [PubMed] [Google Scholar]
- [5].a) Oestreich M, The Mizoroki – Heck Reaction, Wiley, Chichester, 2009; [Google Scholar]; b) Beletskaya IP, Cheprakov AV, Chem. Rev 2000, 100, 3009–3066. [DOI] [PubMed] [Google Scholar]
- [6].Brase S, de Meijere A in Metal-Catalyzed Cross-Coupling Reactions (Eds.: Diederich F, Stang PJ), Wiley-VCH, Weinheim, 1998. [Google Scholar]
- [7].a) Firmansjah L, Fu GC, J. Am. Chem. Soc 2007, 129, 11340–11341; [DOI] [PubMed] [Google Scholar]; b) Bloome KS, McMahen RL, Alexanian EJ, J. Am. Chem. Soc 2011, 133, 20146–20148; [DOI] [PubMed] [Google Scholar]; c) McMahon CM, Alexanian EJ, Angew. Chem. Int. Ed 2014, 53, 5974–5977; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2014, 126, 6084–6087; [Google Scholar]; d) Venning ARO, Kwiatkowski MR, Roque PeÇa JE, Lainhart BC, Guruparan AA, Alexanian EJ, J. Am. Chem. Soc 2017, 139, 11595–11600; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Parasram M, Iaroshenko VO, Gevorgyan V, J. Am. Chem. Soc 2014, 136, 17926–17929; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Kurandina D, Parasram M, Gevorgyan V, Angew. Chem. Int. Ed 2017, 56, 14212–14216; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2017, 129, 14400–14404; [Google Scholar]; g) Kurandina D, Rivas M, Radzhabov M, Gevorgyan V, Org. Lett 2018, 20, 357–360; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Wang G-Z, Shang R, Cheng W-M, Fu Y, J. Am. Chem. Soc 2017, 139, 18307–18312; [DOI] [PubMed] [Google Scholar]; i) Wang G-Z, Shang R, Fu Y, Org. Lett 2018, 20, 888–891; [DOI] [PubMed] [Google Scholar]; j) Koy M, Sandfort F, Tlahuext-Aca A, Quach L, Daniliuc CG, Glorius F, Chem. Eur. J 2018, 24, 4552–4555. [DOI] [PubMed] [Google Scholar]
- [8].a) Prier CK, Rankic DA, MacMillan DWC, Chem. Rev 2013, 113, 5322–5363; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Romero NA, Nicewicz DA, Chem. Rev 2016, 116, 10075–10166; [DOI] [PubMed] [Google Scholar]; c) Skubi KL, Blum TR, Yoon TP, Chem. Rev 2016, 116, 10035–10074; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Studer A, Curran DP, Nat. Chem 2014, 6, 765–773; [DOI] [PubMed] [Google Scholar]; e) Parasram M, Gevorgyan V, Chem. Soc. Rev 2017, 46, 6227–6240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Voica A-F, Mendoza A, Gutekunst WR, Fraga JO, Baran PS, Nat. Chem 2012, 4, 629–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Warren S, Designing Organic Syntheses: A Programmed Introduction to the Synthon Approach, Wiley, Chichester, 1978. [Google Scholar]
- [11].a) Parasram M, Chuentragool P, Wang Y, Shi Y, Gevorgyan V, J. Am. Chem. Soc 2017, 139, 14857–14860; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Parasram M, Chuentragool P, Sarkar D, Gevorgyan V, J. Am. Chem. Soc 2016, 138, 6340–6343; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Chuentragool P, Parasram M, Shi Y, Gevorgyan V, J. Am. Chem. Soc 2018, 140, 2465–2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].a) Tang S, Liu K, Liu C, Lei A, Chem. Soc. Rev 2015, 44, 1070–1082; [DOI] [PubMed] [Google Scholar]; b) Chu JCK, Rovis T, Nature 2016, 539, 272; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Choi GJ, Zhu Q, Miller DC, Gu CJ, Knowles RR, Nature 2016, 539, 268–271; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Jiang H, Studer A, Angew. Chem. Int. Ed 2018, 57, 1692–1696; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2018, 130, 1708–1712; [Google Scholar]; e) Zhang J, Li Y, Zhang F, Hu C, Chen Y, Angew. Chem. Int. Ed 2016, 55, 1872–1875; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2016, 128, 1904–1907; [Google Scholar]; f) Shen X, Zhao J-J, Yu S, Org. Lett 2018, 20, 5523–5527; [DOI] [PubMed] [Google Scholar]; g) Wang C, Harms K, Meggers E, Angew. Chem. Int. Ed 2016, 55, 13495–13498; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2016, 128, 13693–13696; [Google Scholar]; h) Shu W, Genoux A, Li Z, Nevado C, Angew. Chem. Int. Ed 2017, 56, 10521–10524; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2017, 129, 10657–10660. [Google Scholar]
- [13].a) “Iodine atom transfer reactions in organic synthesis”: Curran DP in Free Radicals in Synthesis and Biology. NATO ASI Series Series C, Vol. 260 (Ed.: Minisci F), Springer, Dordrecht, 1989, chap. 3; [Google Scholar]; b) Stateman LM, Nakafuku KM, Nagib DA, Synthesis 2018, 50, 1569–1586; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Ratushnyy M, Parasram M, Wang Y, Gevorgyan V, Angew. Chem. Int. Ed 2018, 57, 2712–2715; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2018, 130, 2742–2745. [Google Scholar]
- [14].It is known that ligands with larger bite angles, such as xantphos, accelerate reductive elimination step and do not favor β-Helimination process. See:; a) van Leeuwen PWNM, Kamer PCJ, Reek JNH, Dierkes P, Chem. Rev 2000, 100, 2741–2770. [DOI] [PubMed] [Google Scholar]; For ligand bite angle values of ferrocene ligands, see:; b) Trivedi M, Ujjain SK, Singh G, Kumar A, Dubey SK, Rath NP, J. Organomet. Chem 2014, 772–773, 202–209. [Google Scholar]
- [15].See the Supporting Information for details.
- [16].It was reported that radical Heck reaction employing small alkenes favors Z isomers. See: Ref. [7c]. For radical recombination process favoring the Z isomer, see:; a) Singh K, Staig SJ, Weaver JD, J. Am. Chem. Soc 2014, 136, 5275–5278; [DOI] [PubMed] [Google Scholar]; b) Zheng C, Cheng W-M, Li H-L, Na R-S, Shang R, Org. Lett 2018, 20, 2559–2563. [DOI] [PubMed] [Google Scholar]
- [17].a) Collins KD, Rühling A, Glorius F, Nat. Protoc 2014, 9, 1348–1353; [DOI] [PubMed] [Google Scholar]; b) Collins KD, Glorius F, Nat. Chem 2013, 5, 597–601. [DOI] [PubMed] [Google Scholar]
- [18].It was found that internal alkynes and alkenes were perfectly tolerated, while other functional groups such as heterocycles, nitrile, ester, and alcohol were reasonably tolerated. Expectedly, additives containing acidic protons hampered the reaction significantly. See the Supporting Information for details.
- [19].Blanksby SJ, Ellison GB, Acc. Chem. Res 2003, 36, 255–263. [DOI] [PubMed] [Google Scholar]
- [20].Tian Z, Fattahi A, Lis L, Kass SR, J. Am. Chem. Soc 2006, 128, 17087–17092. [DOI] [PubMed] [Google Scholar]
- [21].Franz JA, Suleman NK, Alnajjar MS, J. Org. Chem 1986, 51, 19–25. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.