

Abstract
PROTOCADHERIN 7 (PCDH7), a transmembrane receptor and member of the Cadherin superfamily, is frequently overexpressed in lung adenocarcinoma and is associated with poor clinical outcome. While PCDH7 was recently shown to promote transformation and facilitate brain metastasis in lung and breast cancers, decreased PCDH7 expression has also been documented in colorectal, gastric, and invasive bladder cancers. These data suggest context-dependent functions for PCDH7 in distinct tumor types. Given that PCDH7 is a potentially targetable molecule on the surface of cancer cells, further investigation of its role in tumorigenesis in vivo is needed to evaluate the therapeutic potential of its inhibition. Here we report the analysis of novel PCDH7 gain- and loss-of-function mouse models and provide compelling evidence that this cell-surface protein acts as a potent lung cancer driver. Employing a Cre-inducible transgenic allele, we demonstrated that enforced PCDH7 expression significantly accelerates KrasG12D -driven lung tumorigenesis and potentiates MAPK pathway activation. Furthermore, we performed in vivo somatic genome editing with CRISPR/Cas9 in KrasLSL-G12D; Tp53fl/fl (KP) mice to assess the consequences of PCDH7 loss of function. Inactivation of PCDH7 in KP mice significantly reduced lung tumor development, prolonged survival, and diminished phospho-activation of ERK1/2. Together, these findings establish a critical oncogenic function for PCDH7 in vivo and highlight the therapeutic potential of PCDH7 inhibition for lung cancer. Moreover, given recent reports of elevated or reduced PCDH7 in distinct tumor types, the new inducible transgenic model described here provides a robust experimental system for broadly elucidating the effects of PCDH7 overexpression in vivo.
Keywords: PROTOCADHERIN 7 (PCDH7), mouse models of lung cancer, NSCLC, lung tumorigenesis, MAPK signaling, KrasG12D driven lung cancer, CRISPR/Cas9, somatic genome editing, PCDH7 transgenic mouse
INTRODUCTION
Protocadherins (PCDHs) are transmembrane proteins and members of the Cadherin superfamily that play well-established roles in cell adhesion and regulation of downstream signaling pathways (1,2). A growing body of evidence has demonstrated that PCDH expression is dysregulated in tumorigenesis (3–7). Both oncogenic and tumor suppressive roles have been assigned to PCDHs. However, the roles of individual PCDHs in cancer and the mechanisms through which their gain-of-function and loss-of-function drive tumorigenesis in vivo remain poorly understood. We previously reported that PROTOCADHERIN 7 (PCDH7) is frequently overexpressed in human non-small cell lung cancer (NSCLC) tumors (8). Moreover, high expression of PCDH7 was associated with poor clinical outcome of lung adenocarcinoma patients. While PCDH7 was recently shown to mediate brain metastasis in breast and lung cancers (9–12), decreased PCDH7 expression has been documented in colorectal, gastric, and invasive bladder cancers (13–15). Collectively, these data suggest context-dependent functions for PCDH7 in distinct tumor types. Because this molecule is a cell surface receptor that is potentially accessible to antibody-based therapies, rigorous in vivo studies are needed to interrogate PCDH7 function in cancer pathogenesis.
Lung cancer is the leading cause of cancer-associated deaths worldwide (16). Given the limited effectiveness of current treatments, there is a critical need to identify new therapeutic targets. PCDH7 transforms human bronchial epithelial cells (HBECs) and synergizes with KRAS to induce MAPK signaling and tumorigenesis in immunocompromised mice (8). One mechanism through which PCDH7 potentiates ERK signaling is by facilitating interaction of Protein Phosphatase 2A (PP2A) with its potent inhibitor, the SET oncoprotein, thereby suppressing PP2A activity (8). Depletion of PCDH7 suppressed ERK activation, sensitized NSCLC cells to MEK inhibitors, and reduced growth of lung cancer cells in xenograft assays. Nevertheless, all prior studies of PCDH7 function in cancer utilized established cell lines. Thus, investigation of the oncogenic activity of PCDH7 in vivo using autochthonous tumor models, which more accurately model multi-stage tumor progression and the role of the tumor microenvironment, is needed to dissect the role of this cell surface receptor in cancer pathogenesis and to evaluate the therapeutic potential of PCDH7 inhibition for non-small cell lung cancer.
In this study, we sought to establish the importance of PCDH7 in KrasG12D-driven lung cancer pathogenesis. To examine the effects of PCDH7 upregulation in lung tumor initiation and progression, we generated an inducible PCDH7 transgenic mouse model, allowing the demonstration that hyperactivity of PCDH7 promoted lung tumorigenesis and induced MAPK pathway activation in KrasLSL-G12D mutant mice (17). Additionally, to validate PCDH7 as a therapeutic target, we performed in vivo somatic gene editing in KrasLSL-G12D; Tp53fl/fl (KP) mice (18). Somatic depletion of PCDH7 in KP mice with CRISPR/Cas9 significantly reduced lung tumor development and prolonged survival, suggesting that targeting PCDH7 may benefit patients with lung adenocarcinoma. Interrogation of downstream signaling pathways revealed diminished phospho-ERK1/2 and phospho-RB in PCDH7 knockout tumors. These findings demonstrate a key oncogenic role for PCDH7 in vivo, supporting the potential therapeutic efficacy of PCDH7 inhibition for patients with lung cancer.
MATERIALS AND METHODS
Constructs
Human PCDH7 isoform-A (NM_002589.2) cDNA was cloned into the CTV vector (Addgene #15912). The resulting CTV-PCDH7 was used to generate transgenic mice. Depletion of PCDH7 in vitro was performed by using Lenti-CRISPR-V2 (Addgene #52961) to introduce Cas9 and sgRNA directed against Pcdh7. Depletion of mouse Pcdh7 in vivo was performed using pSECC (Addgene #60820). The sgRNAs are as follows: human PCDH7, CGACGTCCGCATCGGCAACG; Mouse Pcdh7, GAGGATGCGGACCACGGGAT; human non-specific control, CGCTTCCGCGGCCCGTTCAA; mouse non-specific control, GCGAGGTATTCGGCTCCGCG. Human KrasG12V cDNA was cloned into pLenti CMV Hygro DEST (Addgene #17454) for ectopic expression of KrasG12V in HBECs. The human PCDH7 isoform-A cDNA was cloned into pLX303 lentivirus vector (Addgene #25897) for PCDH7 overexpression in vitro.
Mice
Generation and genotyping of PCDH7 transgenic mice - PCDH7 transgenic mice were generated by UTSW transgenic core facility, through microinjection of the linearized CTV-PCDH7 vectors into the pronuclei of fertilized eggs (C57BL/6J strain). For genotyping of PCDH7 transgenic mice, genomic DNA was isolated from tail clippings using the Gentra Puregene Tissue Kit (Qiagen) according to the manufacturer’s protocol. PCR was performed using 10–100 ng of genomic DNA as template and the following transgene specific primers: forward: 5ʹ-TCCCCAGTCACCAACTGCAGGAAAAAAACACCAG-3ʹ, reverse: 5ʹ-GAATAGGAACTTCGGTACCGAATTGATCGCG-3ʹ (amplicon = 299 bp). Thermal cycling conditions using GoTaq Green Master Mix (Promega) were as follows: initial denaturation at 95 °C for 5 min, followed by 95°C for 30 s, 60°C for 30 s, 72°C for 30 s, in a total of 35 cycles, followed by a final extension at 72°C for 5 min. The samples were stored at 4°C until separated on a 1.2% (wt/vol) agarose gel.
B6.129S4-Krastm4Tyj/J mice, also known as KrasLSL-G12D mice were purchased from The Jackson Laboratory (008179). KrasLSL-G12D; PCDH7LSL/LSL (maintained on a B6 genetic background) were established by breeding KrasLSL-G12D with PCDH7LSL mice. KrasLSL-G12D; Tp53fl/fl mice (maintained on a FVB/B6 mixed genetic background) were provided by James Kim (UTSW). Immunocompromised mice NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG mice, 005557) were purchased from The Jackson Laboratories. CAG-Cre mice (19) were provided by Eric Olson (UTSW).
Ethics Statement.
Mice were monitored closely throughout all experimental protocols to minimize discomfort, distress, or pain. Signs of pain and distress include disheveled fur, decreased feeding, significant weight loss (>20% body mass), limited movement, or abnormal gait. If any of these signs were detected, the animal was removed from the study immediately and euthanized. All sacrificed animals were euthanized with CO2. The animals were placed in a clear chamber and 100% CO2 was introduced. Animals were left in the container until clinical death ensured. To ensure death prior to disposal, cervical dislocation was performed while the animal was still under CO2 narcosis. All methods were performed in accordance with the recommendations of the Panel on Euthanasia of the American Veterinary Medical Association and protocols approved by the UT Southwestern Institutional Animal Care and Use Committee (protocol # 2017–102112).
Tissue collection and immunohistochemistry
Mice were euthanized by intraperitoneal administration of an overdose of Avertin at the time points indicated. Lungs were inflated and perfused through the trachea with 4% paraformaldehyde (PFA), fixed overnight, transferred to 50% ethanol and subsequently embedded in paraffin. Sections were cut and stained with H&E by the UTSW Histology Core, which also provided assistance with the histopathological examination. For immunohistochemistry (IHC) staining, slides were de-paraffinized and treated with Antigen Unmasking Solution (Vector laboratory, H-3300), blocked with BLOXALL™ Blocking Solution (Vector lab, SP-6000), and then incubated with anti-Ki67 (CST #9027, 1:400) or anti-pERK (CST #9101S, 1:200) at 4°C overnight. After washing extensively, slides were incubated with SignalStain® Boost Detection Reagent from Cell Signaling Technology (Rabbit #8114) at room temperature (RT) for 30 minutes. The signal was developed with ImmPACT™ DAB Substrate (Vector lab, SK-4105), and sections were counterstained with hematoxylin (Vector lab, H-3404), and mounted with VectaMount Mounting Reagent (Vector lab, H-5000). All pictures were obtained using a Zeiss microscope (Observer.Z1) with an Axiocam-MRC camera.
Ki67 and pERK1/2 index – Ki67-positive and pERK1/2 staining was distinguished by counting brown nuclei and hematoxylin (blue) counterstain. The Ki67 and pERK1/2 index for each mouse was calculated as follows: Percent positive cells = number of positive nuclei/total cell nuclei x 100, as described previously (20). For Ki67 quantification, n=2–4 animals/group were analyzed, and up to 10 independent fields/animal were quantified. For pERK1/2 quantification, 2–3 animals/group were analyzed, and 6–10 independent fields were quantified/animal.
Tumor burden analysis
For tumor quantification (Figure 1c and2D), the total tumor number for each animal was determined by analyzing H&E-stained sections and categorizing tumors into different stages (including atypical adenomatous hyperplasia (AAH), bronchiolar hyperplasia (BH), adenomas, and adenocarcinomas). Five lobes per animal (one section per lobe) were quantified, and 5–6 animals were analyzed per group.
Figure 1. PCDH7 accelerates lung tumorigenesis in the KrasLSL-G12D model.
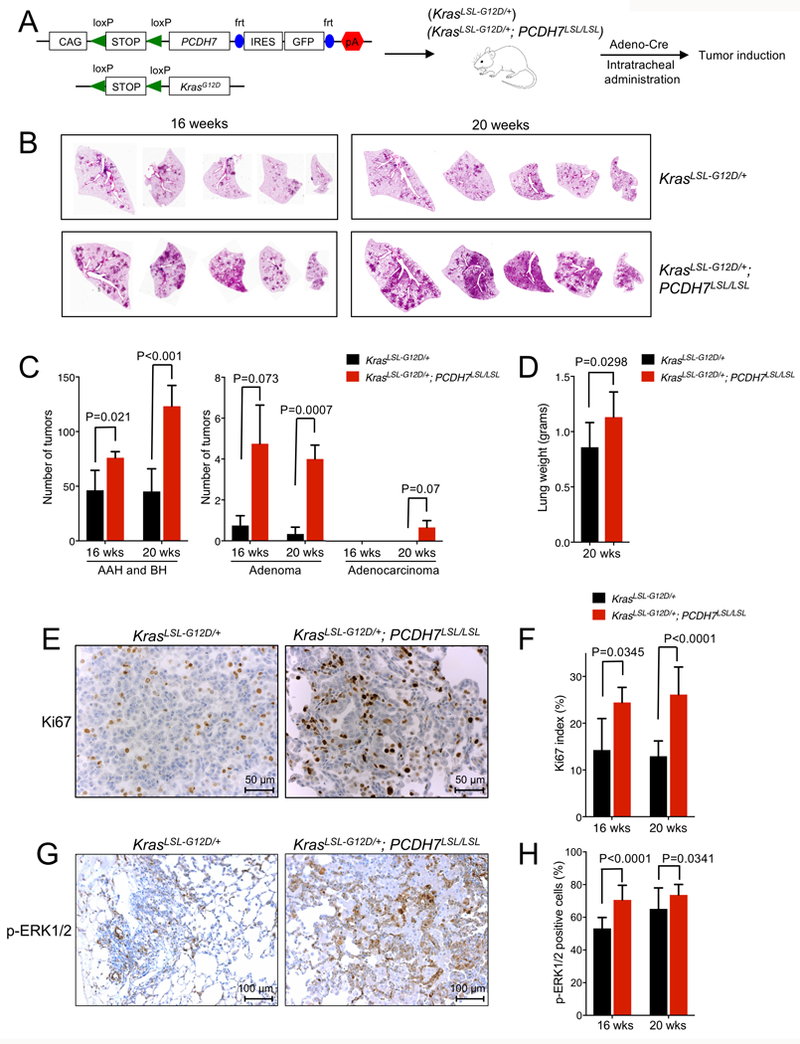
A, Schematic depicting the PCDH7LSL and Kras LSL-G12D transgenes and overview of experimental design. To induce lung tumors, KrasLSL-G12D; PCDH7LSL/LSL or control KrasLSL-G12D mice were infected with Adeno-Cre through intratracheal administration. B, H&E staining of lung lobes harvested at 16 and 20 weeks post-infection. Five independent lobes from one animal of each genotype are shown. C, Tumor burden analysis at 16 and 20 weeks post-infection. n=5–6 mice/group, 5 sections/mouse analyzed. Tumors were counted and classified into atypical adenomatous hyperplasia (AAH)/bronchiolar hyperplasia (BH), adenomas, or adenocarcinomas. D, Weight of lungs collected from KrasLSL-G12D; PCDH7LSL/LSL or KrasLSL-G12D mice at 20 weeks post-infection. E, IHC staining of Ki67 at 20 weeks and F, quantification of Ki67 index for lung sections harvested from KrasLSL-G12D; PCDH7LSL/LSL or control KrasLSL-G12D mice at 16 and 20 weeks post-infection. n=2–4 mice/group, up to 10 fields quantified/animal. G, IHC staining of pERK1/2 for lung sections from KrasLSL-G12D; PCDH7LSL/LSL or control KrasLSL-G12D mice at 16 weeks post-infection. H, quantification of p-ERK1/2 IHC staining at 16 and 20 weeks post-infection. n=2 mice/group, 7 fields quantified/animal.
Figure 2. In vivo inactivation of Pcdh7 reduces lung tumor burden and prolongs survival of KrasLSL-G12D; Tp53fl/fl mice.
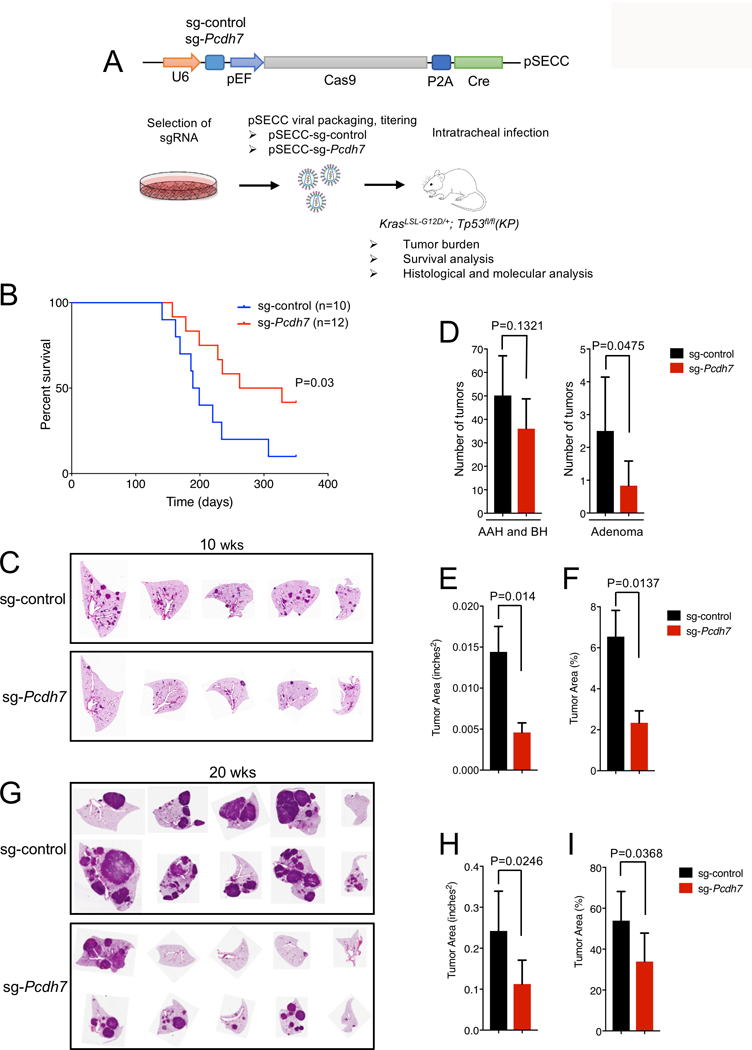
A, Schematic of in vivo CRISPR/Cas9 editing. KP mice were infected with a sg-control or sg-Pcdh7 through intratracheal administration of pSECC lentivirus expressing Cas9 and Cre. B, Survival analysis of KP mice infected with sg-control or sg-Pcdh7 lentivirus. C, H&E staining of lung lobes harvested at 10 weeks post-infection. D, Tumor burden analysis at 10 weeks post-infection. Tumors were counted and classified into AAH, BH, or adenomas based on histopathologic analysis. n=5–7 animals/group, 5 lobes/animal, 1 section/lobe analyzed. E, Total tumor area (inches2) at 10 weeks post-infection based on histopathologic analysis of lung sections using the NIH Image J program. F, Percent tumor area compared to total area of lung lobes at 10 weeks post-infection (determined using NIH Image J). G, H&E staining of lung lobes harvested at 20 weeks post-infection. H, Total tumor area (inches2) at 20 weeks post-infection based on histopathologic analysis of lung sections. I, Percent tumor area compared to total area of lung lobes at 20 weeks post-infection.
To analyze tumor burden of CRISPR/Cas9-targeted mice, tumor number (as described above) and tumor area was quantified. Total tumor area was determined by analyzing H&E-stained sections using the NIH Image J Program (five lobes per animal, one section per lobe, n=5–7 animals per group). The average tumor area (inches2) and percent of tumor vs. total lung area are shown (Figure 2 E-Fand H-I).
TUNEL staining and quantification
TUNEL staining was performed using DeadEND™ Fluorometric TUNEL System (Promega, Part # TB235) following the manufacturer’s guidelines. Paraffin embedded tissue sections were de-paraffinized. After washing in PBS for 5min, tissue sections were fixed in 4% formaldehyde in PBS for 15 minutes. Slides were washed twice in PBS for 5 minutes each and subjected to permeabilization by adding 100μl of a 20μg/ml Proteinase K solution diluted in PBS for 10 min at room temperature. Tissue sections were washed in PBS for 5 minutes, re-fixed by immersing in 4% formaldehyde in PBS for 5 minutes and washed again in PBS for 5 minutes. Tissue sections were equilibrated with100μl of equilibration buffer at room temperature for 10 minutes, labeled by adding 50μl of TdT reaction mix, and incubated for 60 minutes at 37°C in a humidified chamber in a dark container to avoid exposure to light. The reaction was stopped by immersing the slides in 2X SSC for 15 minutes, washed three times in PBS for 5 minutes each. The slides were counterstained and mounted with Vectashield® hard set mounting media with DAPI (H-1500). Localized green fluorescent apoptotic cells in tissue sections were detected and images were captured using a ZEISS confocal fluorescence microscope LSM-800. The TUNEL score was calculated by counting all the positive and negative stained tumor cells within a given field of view. Percent TUNEL positive nuclei was calculated by dividing the number of positive nuclei by the total number of cell nuclei x 100. 15 independent fields were quantified for each animal and 2–4 animals per group were analyzed.
Cell culture
Immortalized normal human bronchial epithelia cells (HBECs) with stable knockdown of TP53 with shRNA (HBEC-shp53) were provided by John Minna at UTSW (21–23). Ectopic expression of KRASG12V in HBEC-shp53 was performed with lentiviral infection followed by selection with hygromycin B for 7 days. HBEC-shp53-PCDH7 or HBEC-shp53-KRASG12V-PCDH7 cells were established with the pLX303-PCDH7 lentivirus followed by selection with blasticidin for 10 days. All HBECs were cultured in keratinocyte serum-free medium (KSFM; Life Technologies Inc.) containing 50 μg/mL of bovine pituitary extract (BPE; Life Technologies, Inc.) and 5 ng/mL of EGF (Life Technologies, Inc.). Human lung adenocarcinoma H1944 cells were cultured in RPMI-1640 media supplemented with 10% fetal bovine serum (Life Technologies, Inc.), 100 units/ml of penicillin and streptomycin (Life Technologies, Inc.). PCDH7 depletion in H1944 cells was achieved using Lenti-CRISPRv2 followed by selection with puromycin for 7 days (1 μg/mL). All cell lines used in this study were cultured in 5% CO2 at 37C, tested negative for mycoplasma contamination, and were authenticated in 2016 with the PowerPlex 1.2 kit (Promega).
Virus preparation, titration and intratracheal administration
Cre-expressing adenovirus (Adeno-Cre) was purchased from Viral Vector Core Facility (University of Iowa). The PCDH7LSL and KrasLSL-G12D transgenes were induced with intratracheal administration of Adenovirus-Cre (Adeno-Cre, 2×107 IFU/mouse) (24).
Pcdh7 sgRNA-1 (sg-Pcdh7) or a non-specific control sgRNA (sg-control) were cloned into pSECC lentiviral vectors, packaged, and titered with Green-Go cells as previously described (18). Lentiviruses were produced by co-transfection of 293T cells with lentiviral backbone constructs and packaging vectors (psPAX2 and pMD2.G) using Lipofectamine 3000 (Invitrogen). Supernatants were collected 72 hours post-transfection, concentrated by ultracentrifugation at 25,000 RPM for 120 minutes and re-suspended in an appropriate volume of Opti-MEM (Gibco). Green-Go cells, which were generated by transducing retrovirus containing an inverted GFP (flanked by two sets of incompatible loxP sites), were provided by Tyler Jacks (Massachusetts Institute of Technology), and maintained in DMEM supplemented with 10% Fetal Bovine Serum and gentamicin (18). Upon infection with pSECC lentivirus, Green-Go cells become GFP+, allowing for titering by fluorescence-activated cell sorting (FACS). Briefly, 2×104 Green-Go cells were seeded into each well of 24-well plates and incubated overnight. The next day, cells were infected with 20ul, 10ul, 5ul or 2ul pSECC lentivirus. At 48 hours post-infection, cells were collected and GFP+ cells were quantified by FACS. The titer of pSECC lentivirus is designated as the average number of GFP+ cells in the four groups. Intratracheal administration of pSECC lentivirus was performed according to established protocols (24). 4×104 virus/mouse was administered for survival analysis and 2×105 virus/mouse was administered for tumor burden analysis at 10 and 20 weeks post-infection (24).
siRNA knockdown
PCDH7 siRNA (Accell™ SMARTpool) or non-specific control siRNA were purchased from Dharmacon. HBECs were incubated in Accell Delivery Media (Dharmacon) in 6 well-plates overnight to reach 50% confluence, and siRNAs diluted in siRNA Buffer (Dharmacon) were added at final concentration of 1μM. 72 hours after transfection, cells were collected, and gene expression was assessed by western blot.
Western blot analysis
Protein lysates were prepared with RIPA Lysis and Extraction Buffer (ThermoFisher, 89900) including Halt™ Protease Inhibitor Cocktail, EDTA-free diluted 1:100. Protein concentrations were determined by BCA assay (Thermo, 23228 and 23224). 25–30μg of each protein lysate was loaded into each well of a Bolt™ 4–12% Bis-Tris Plus Gels (Life Technologies, NW04120BOX), electrophoresed, and transferred to nitrocellulose using iBlot2 Western Blotting System (Life Technologies). The primary antibodies (1:1000 dilution) for western blot are as follows: PCDH7 (Abcam, ab139274), pERK (Cell Signaling Techonolgy, CST # 9101S), total ERK (CST #4695), pRb (Ser807/811, CST #8516), total Rb (CST #9309) and β-ACTIN (CST #4970). Horseradish peroxidase (HRP)–conjugated anti-rabbit or anti-mouse secondary antibodies (1:5,000–20,000 dilution; BioRad) were used and signals developed with the SuperSignal™ West Dura Extended Duration Substrate (Thermo Fisher 34075), according to the manufacturer’s instructions.
Verification of gene editing
To detect CRISPR-induced indels, tumors from a sg-Pcdh7 mouse (20 weeks after infection) were dissected and surrounding lung tissues carefully removed. Tumor tissues were homogenized, and genomic DNA (gDNA) was isolated using the Gentra Puregene Tissue Kit (Qiagen). To amplify the Pcdh7 sgRNA-targeted region, PCR was performed with tumor gDNA using PrimeSTAR HS DNA Polymerase (Clontech). The products were gel purified and cloned into the Zero Blunt TOPO sequencing vector (Invitrogen). At least eight colonies for each tumor were sequenced.
Statistical Analysis
All statistical analyses were performed using GraphPad Prism for Windows. Quantitative variables were analyzed by Student’s t-test, Fisher’s exact test, or Chi-squared test. All statistical analyses were two-sided, and p<0.05 was considered statistically significant.
RESULTS
PCDH7 accelerates lung tumorigenesis in a mouse model of KrasG12D-driven lung adenocarcinoma
To assess the oncogenic activity of PCDH7 in vivo and to investigate the mechanisms underlying PCDH7-mediated transformation, we generated a transgenic mouse model that allows precise control of PCDH7 expression through Cre-mediated recombination. We selected human PCDH7 isoform A, which is the predominant isoform expressed in human lung cancer cell lines from the Cancer Cell Line Encyclopedia and a panel of NSCLC cell lines (8,25). The transgene is depicted in Fig. 1A. A LoxP-Stop-LoxP (LSL) cassette allows control with Cre-recombinase. After removal of the LSL cassette, the CAG promoter drives expression of human PCDH7 and green fluorescent protein (GFP). To validate inducible expression of the transgene, PCDH7LSL mice were crossed to CAG-Cre mice that ubiquitously express Cre recombinase (Supplementary Fig. S1A). Cre-dependent transgene activation was confirmed in lung tissue using quantitative real-time PCR and western blot analysis (Supplementary Fig. S1A-C).
Replication-deficient viruses have been widely used to deliver Cre to the lung, thereby activating a mutant Kras allele to generate sporadic lung tumors in KrasLSL-G12D mice (17,26–28). To determine whether enforced expression of PCDH7 is sufficient to accelerate KrasG12D-mediated lung tumorigenesis in vivo, we bred PCDH7LSL mice with KrasLSL-G12D mice and induced both transgenes via intratracheal administration of Adenovirus-Cre (24) (Fig. 1A). Tumors were detectable in all lobes at 16 and 20 weeks post-infection (Fig. 1B). At 16 weeks post-infection, the majority of tumors were early-stage and categorized as atypical adenomatous hyperplasia (AAH) or bronchiolar hyperplasia (BH) (17,18,24) (Fig. 1C and Supplementary Fig. S1D). KrasLSL-G12D; PCDH7LSL/LSL mice exhibited a significant increase in AAH and BH tumors compared to KrasLSL-G12D mice. KrasLSL-G12D; PCDH7LSL/LSL mice also developed more adenomas than KrasLSL-G12D mice, indicating accelerated disease progression. At 20 weeks post-infection, tumor burden was significantly higher in KrasLSL-G12D; PCDH7LSL/LSL mice than in KrasLSL-G12D mice, as determined by quantifying tumor numbers at different stages (Fig. 1C) and lung weight (Fig. 1D and Supplementary Fig. S1E). No tumors were observed in PCDH7LSL/LSL mice after 16 months post-infection, suggesting that isolated expression of the transgene is not sufficient to initiate tumorigenesis by this time-point.
PCDH7 cooperates with oncogenic KRAS to promote ERK activation and cell proliferation in human bronchial epithelial cells (HBECs) (8). Consistent with these effects, tumors from KrasLSL-G12D; PCDH7LSL/LSL mice exhibited greater numbers of of Ki67 and pERK1/2 positive cells compared to tumors from KrasLSL-G12D mice (Fig. 1E-H). Moreover, the apoptotic index as measured by quantification of TUNEL staining, was significantly lower in tumors from KrasLSL-G12D; PCDH7LSL/LSL mice (Supplementary Fig. S2A-B). Taken together, these data support a pro-tumorigenic role for PCDH7 in KrasG12D-mutant lung cancer and demonstrate that PCDH7 modulates MAPK pathway activity in vivo.
In vivo inactivation of Pcdh7 reduces lung tumor burden and prolongs survival of KrasLSL-G12D; Tp53fl/fl mice
Inhibition of PCDH7 reduced tumorigenesis of KRAS mutant NSCLC cells in xenograft assays (8). However, xenografts do not fully recapitulate all aspects of tumorigenesis, including contributions from the microenvironment and immune system. Therefore, examination of in vivo loss-of-function is critical to establish whether PCDH7 represents a potential therapeutic target in NSCLC. We took advantage of a recently described in vivo somatic genome editing approach to rapidly interrogate PCDH7 function in KrasLSL-G12D;Tp53fl/fl (KP) mice (Fig. 2A) (18). Interestingly, PCDH7 is upregulated in KP tumors relative normal lung, suggesting it may act as an oncogenic driver in this context (Supplementary Fig. S3A). CRISPR/Cas9-based gene editing efficiently produces loss-of-function mutations in this model that result in phenotypes that closely mirror those observed following traditional germline gene-targeting approaches.
To identify an optimal Pcdh7 single-guide (sgRNA) for in vivo studies, immortalized mouse embryonic fibroblasts were infected with a lentivirus expressing the Cas9 nuclease and a control sgRNA or one of five sgRNAs directed against murine Pcdh7 (Fig. 2A) (29). Pcdh7 sgRNA-1 most dramatically diminished PCDH7 expression by western blot analysis (Supplementary Fig. S3B). CRISPR/Cas9-induced double strand DNA breaks were verified with the SURVEYOR assay (30) (Supplementary Fig. S3C), thus Pcdh7 sgRNA-1 was selected for in vivo targeting.
KrasLSL-G12D; Tp53fl/fl (KP) mice were infected with pSECC-sg-control or pSECC-sg-Pcdh7 lentiviruses through intratracheal administration, following established protocols (24). Because the lentivirus expresses Cas9 and Cre, the Pcdh7 sgRNA generates mutations in the same lung cells that undergo activation of the KrasLSL-G12D allele and deletion of theTp53fl/fl allele. Somatic mutation of Pcdh7 prolonged survival of KrasLSL-G12D; Tp53fl/fl mice (Fig. 2B). At an early time-point (10 weeks after infection), we observed fewer adenomas but not AAH or BH lesions in Pcdh7-targeted KP lungs (Fig. 2C-D) and decreased overall tumor area (Fig. 2E-F). Moreover, at 20 weeks post-infection, a time-point associated with robust adenocarcinoma formation in KP mice, we documented significantly reduced tumor burden (Fig. 2G-I).
Given recent studies documenting a role for PCDH7 in metastasis, we assessed invasive properties of tumors in these mice. We observed evidence of micro- and macro-metastases, lymphovascular invasion, and intravascular tumor spreading in control-treated KrasLSL-G12D; Tp53fl/fl mice (Supplementary Fig. S4). sg-Pcdh7-treated mice exhibited a lower incidence of these malignant features and a higher incidence of relatively benign neoplastic lesions, including AAH and adenomas. Thus, Pcdh7 depletion reduced tumor invasiveness in vivo.
To verify gene editing events, genomic DNA was harvested from Pcdh7-targeted tumors at 20 weeks after infection, mutations in the sgRNA-targeted region of Pcdh7 were sequenced, and PCDH7 expression was measured by western blotting. All control tumors from KrasLSL-G12D; Tp53fl/fl mice expressed high levels of PCDH7 relative to normal lung tissue (Fig. 3A). As expected, tumors isolated from sg-Pcdh7-targeted KP mice exhibited diminished PCDH7 expression to varying degrees across analyzed tumors (Fig. 3A and Supplementary Fig. S5A). Accordingly, sequencing of the sg-Pcdh7-targeted region revealed homozygous or heterozygous deletions resulting in a frameshift in 8/9 tumors analyzed, with residual wild-type or in-frame alleles in tumors with higher PCDH7 expression (Supplementary Fig. S5B).
Figure 3. Somatic knockout of Pcdh7 inhibits proliferation and reduces MAPK signaling in KrasLSL-G12D; Tp53fl/fl mice.
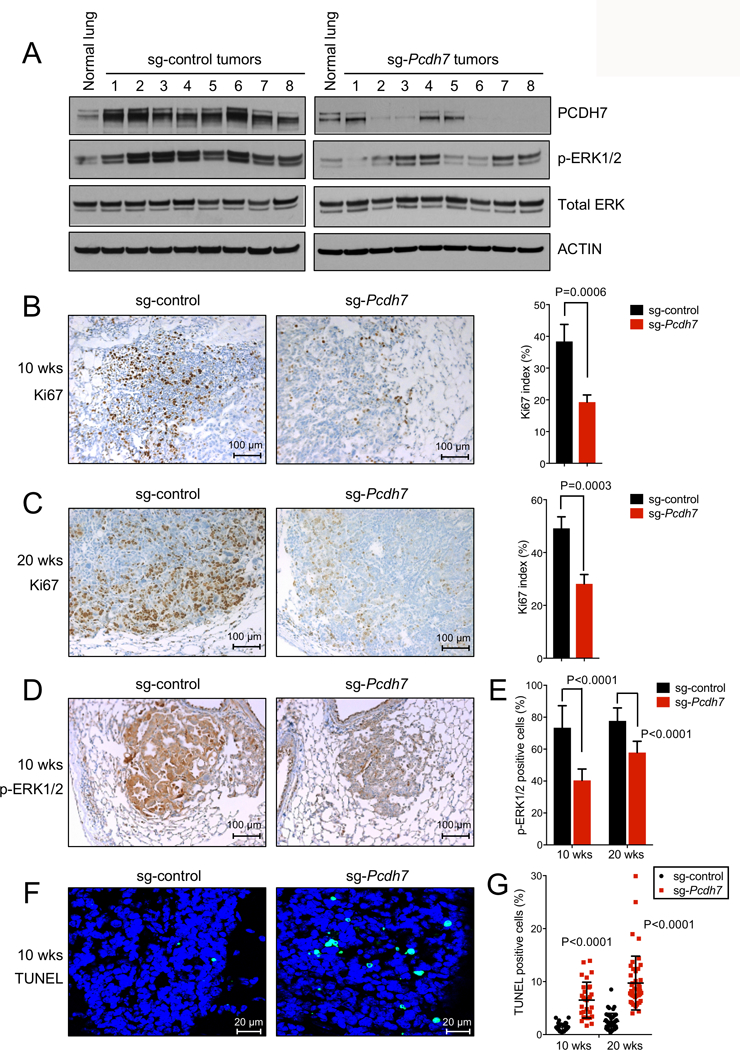
A, Western blot analysis of PCDH7, pERK1/2, and total ERK in normal lung tissues from uninfected KP mice, and lung tumors from KP mice infected with sg-control or sg-Pcdh7 lentivirus (20 weeks post-infection). B-C, Left, Ki67 immunohistochemistry (IHC) staining of lung sections harvested from KP mice infected with sg-control or sg-Pcdh7 lentivirus at 10 weeks (B) or 20 weeks (C) post-infection. Right, Ki67 index = percent of cells with a Ki67 positive signal / total cell number in each field. n=3 animals/group. D, pERK1/2 IHC staining of lung lobes from KrasLSL-G12D; Tp53 fl/fl mice infected with sg-control or sg-Pcdh7 lentivirus at 10 weeks post-infection. E, Quantification of p-ERK1/2 IHC staining (represented by percent of p-ERK1/2 positive cells) at 10 and 20 weeks post-infection. n=2–3 animals/group, 6–10 fields quantified/animal. F, TUNEL staining of lung lobes from KrasLSL-G12D; Tp53 fl/fl mice infected with sg-control or sg-Pcdh7 lentivirus at 10 weeks post-infection. G, Quantification of TUNEL index (represented by percent of TUNEL positive cells) at 10 and 20 weeks post-infection. n=2–3 animals/group, 15 independent fields quantified/animal.
Somatic knockout of Pcdh7 inhibits proliferation and reduces MAPK signaling in KrasLSL-G12D; Tp53fl/fl mice
To examine the effects of PCDH7 depletion on downstream signaling in vivo, we harvested tumors from KP mice infected with either sg-control or sg-Pcdh7 lentivirus and analyzed phospho-ERK1/2 and total ERK expression by western blotting (Fig. 3A). Although phospho-ERK1/2 was uniformly lower in sg-Pcdh7-targeted tumors compared to control tumors, some heterogeneity existed, consistent with additional genetic events affecting the MAPK pathway. Immunohistochemistry demonstrated that pERK1/2 and Ki67 staining were reduced in tumors from sg-Pcdh7-targeted mice compared to control KP tumors (Fig. 3B-E), further supporting a role for PCDH7 in stimulating MAPK pathway activity. TUNEL staining was significantly increased in sg-Pcdh7-targeted mice, demonstrating that inhibition of Pcdh7 also promotes apoptosis (Fig. 3F-G).
PCDH7 modulates expression of PP2A targets
PCDH7 overexpression in HBECs impact several important cancer-relevant pathways. One reported target of PP2A is pRB (31). Accordingly, the pRB pathway was previously identified as a significantly upregulated gene set in RNAseq analysis of HBEC-shp53-PCDH7 cells (p=0; FDR q value = 2.35 × 10−4) (8). To determine whether PCDH7 regulates the pRB pathway in the context of mutant KRASG12V, we generated HBEC-shp53 cells with enforced expression of mutant KRASG12V, PCDH7, or both in combination. Western blot analysis revealed that while KRASG12V increased pERK and pRB signaling, PCDH7 cooperated with KRASG12V to further enhance both pERK and pRB signaling (Fig. 4A). Furthermore, both pERK and pRB signaling were significantly reduced upon PCDH7 inhibition with siRNAs in shp53-KRASG12V-PCDH7 cells (Fig. 4B).
Figure 4. PCDH7 modulates expression of PP2A targets pERK1/2 and pRB.
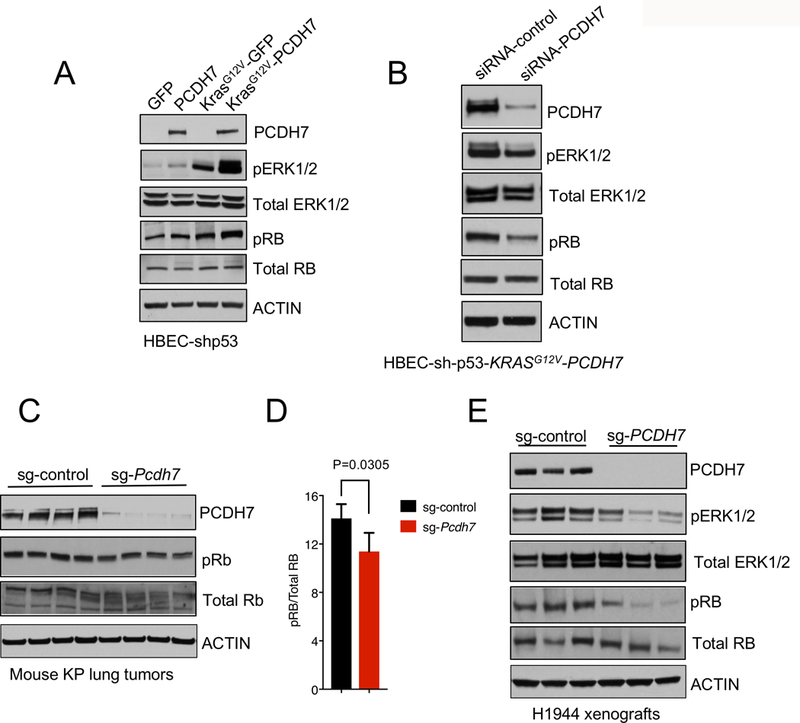
A, Western blot analysis of PCDH7, pERK, total ERK, pRb and total RB levels in HBECs expressing KRASG12V, PCDH7, or both. B, PCDH7 inhibition with siRNAs in HBEC-shp53-KRASG12V-PCDH7 cells and its effects on pERK and pRb signaling, as indicated by western blot analysis. C, Western blot analysis of PCDH7, pRB, and total RB expression in lung tumors harvested from KP mice with sg-control or sg-Pcdh7 lentivirus at 20 weeks post-infection. D, Quantification of pRB protein levels shown in (C). E, Western blot analysis showing diminished pERK1/2 and pRB protein in human NSCLC H1944 xenografts with PCDH7 sgRNA.
To determine the extent to which PCDH7 inhibition impacts the RB pathway in vivo, sg-control or sg-Pcdh7-treated tumors from KP mice were collected at 20 weeks post-infection, and pRB and total RB were examined by western blotting. A minor, but significant, decrease in pRB signaling was observed in PCDH7-depleted tumors compared to control tumors (Fig. 4C-D). Finally, we extended these studies to human NSCLC xenografts. In KRAS mutant H1944 lung adenocarcinoma cells, CRISPR/Cas9-mediated depletion of PCDH7 suppressed the growth of xenografts in immunocompromised NOD/SCID IL2Rγnull NSG mice (8). Western blotting demonstrated that PCDH7 depletion diminished both pRB and pERK signaling in vivo (Fig. 4E). Collectively, these data show that PCDH7 enhances phosphoactivation of multiple PP2A targets including pERK1/2, and to a lesser extent pRB, in KRAS-mutant lung cancer cells.
DISCUSSION
PCDH7 is an actionable therapeutic target in lung adenocarcinoma
The inducible PCDH7 transgenic mouse described here provides a valuable model for the evaluation of PCDH7 function in various tumorigenic contexts. Given the prior reports of elevated or reduced PCDH7 in distinct tumor types (8,10,13–15), the PCDH7LSL model, when combined with various tissue-specific Cre driver lines, provides a robust experimental system for elucidating the effects of PCDH7 overexpression in different in vivo settings. Furthermore, this animal model represents an ideal system for future testing of therapeutics directed at PCDH7, including monoclonal antibodies.
We previously demonstrated that PCDH7 interacts with the PP2A phosphatase, and SET (a potent PP2A inhibitor), thereby inhibiting PP2A activity in HBECs and human NSCLC cells (8). PP2A loss of function results in aberrant phosphorylation of substrates in a variety of pathways linked to cancer, including the MAPK, RB, AKT, and JAK/STAT pathways (32–35). Our in vivo gain- and loss-of-function studies demonstrate that PCDH7 promotes ERK phosphoactivation, an event known to initiate lung tumorigenesis and promote the rapid progression of adenomas to more invasive adenocarcinomas (36). Importantly, the magnitude and duration of ERK signaling are tightly controlled by regulators that provide negative feedback, including PP2A (37,38). Moreover, the consequences of PCDH7-mediated PP2A inhibition likely extend beyond MAPK signaling. Indeed, we show here that PCDH7 modulates pRB levels in human NSCLC xenografts and, to a lesser extent, in KP mouse tumors. PCDH7 likely modulates other PP2A targets in addition to pERK and pRB. Taken together, our data establish PCDH7 as a cooperative oncogenic driver in KRAS-mutant lung cancer that functions to enhance pro-tumorigenic signaling in cancer cells.
PCDH7 was recently shown to promote metastasis of lung and breast cancer cells (10,39), further supporting the importance of this protein in multiple aspects of tumor biology. Consistent with these observations, our studies revealed that Pcdh7 depletion in KP mice reduced tumor invasiveness in vivo. Overall, the results reported here establish PCDH7 as an oncogenic driver of lung tumor initiation and progression in vivo, setting the stage for future efforts to target this cell-surface protein with novel therapeutic strategies in lung adenocarcinoma.
Supplementary Material
Implications:
In this study, we establish a critical oncogenic function for PCDH7 in vivo using novel mouse models and CRISPR/Cas9 genome editing, and we validate the therapeutic potential of PCDH7 inhibition for lung cancer.
ACKNOWLEDGEMENTS
We thank John Shelton in the University of Texas Southwestern Histology Core for assistance with histology, John Minna and Michael Peyton for sharing cell lines, and Tyler Jacks, David McFadden, James Kim, and Eric Olson for sharing reagents and mice. We also thank Joshua Mendell and members of the O’Donnell laboratory for critical reading of the manuscript. This work was supported by the NCI (R01 CA207763 to K.A.O.), the Sidney Kimmel Foundation (SKF-15–067 to K.A.O), the Cancer Prevention Research Institute of Texas (CPRIT, R1101 and RP150676 to K.A.O., RP140110 and RP160157 to S.S.), The Welch Foundation (I-1881 to K.A.O.), the LUNGevity Foundation (2015–03 to K.A.O.), and a SPORE in Lung Cancer CDA (P50CA70907–17). K.A.O. is a CPRIT Scholar in Cancer Research and a Kimmel Scholar. X. Zhou was supported by the Lung Cancer Research Foundation (LCRF 2015) and the National Natural Science Foundation of China (NSFC 81571527, 81771681).
REFERENCES
- 1.Kahr I, Vandepoele K, van Roy F. Delta-protocadherins in health and disease. Progress in molecular biology and translational science 2013;116:169–92. [DOI] [PubMed] [Google Scholar]
- 2.Pan D The hippo signaling pathway in development and cancer. Developmental cell 2010;19:491–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van Roy F Beyond E-cadherin: roles of other cadherin superfamily members in cancer. Nature reviews Cancer 2014;14:121–34. [DOI] [PubMed] [Google Scholar]
- 4.Vincent A, Omura N, Hong SM, Jaffe A, Eshleman J, Goggins M. Genome-wide analysis of promoter methylation associated with gene expression profile in pancreatic adenocarcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research 2011;17:4341–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang X, Chen MW, Terry S, Vacherot F, Chopin DK, Bemis DL, et al. A human- and male-specific protocadherin that acts through the wnt signaling pathway to induce neuroendocrine transdifferentiation of prostate cancer cells. Cancer research 2005;65:5263–71. [DOI] [PubMed] [Google Scholar]
- 6.Yu B, Yang H, Zhang C, Wu Q, Shao Y, Zhang J, et al. High-resolution melting analysis of PCDH10 methylation levels in gastric, colorectal and pancreatic cancers. Neoplasma 2010;57:247–52. [DOI] [PubMed] [Google Scholar]
- 7.Yu JS, Koujak S, Nagase S, Li CM, Su T, Wang X, et al. PCDH8, the human homolog of PAPC, is a candidate tumor suppressor of breast cancer. Oncogene 2008;27:4657–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou X, Updegraff BL, Guo Y, Peyton M, Girard L, Larsen JE, et al. PROTOCADHERIN 7 Acts through SET and PP2A to Potentiate MAPK Signaling by EGFR and KRAS during Lung Tumorigenesis. Cancer research 2017;77:187–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bos PD, Zhang XH, Nadal C, Shu W, Gomis RR, Nguyen DX, et al. Genes that mediate breast cancer metastasis to the brain. Nature 2009;459:1005–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen Q, Boire A, Jin X, Valiente M, Er EE, Lopez-Soto A, et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 2016;533:493–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li AM, Tian AX, Zhang RX, Ge J, Sun X, Cao XC. Protocadherin-7 induces bone metastasis of breast cancer. Biochemical and biophysical research communications 2013;436:486–90. [DOI] [PubMed] [Google Scholar]
- 12.Valiente M, Obenauf AC, Jin X, Chen Q, Zhang XH, Lee DJ, et al. Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell 2014;156:1002–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bujko M, Kober P, Mikula M, Ligaj M, Ostrowski J, Siedlecki JA. Expression changes of cell-cell adhesion-related genes in colorectal tumors. Oncol Lett 2015;9:2463–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen HF, Ma RR, He JY, Zhang H, Liu XL, Guo XY, et al. Protocadherin 7 inhibits cell migration and invasion through E-cadherin in gastric cancer. Tumour Biol 2017;39:1010428317697551. [DOI] [PubMed] [Google Scholar]
- 15.Lin YL, Wang YL, Fu XL, Li WP, Wang YH, Ma JG. Low expression of protocadherin7 (PCDH7) is a potential prognostic biomarker for primary non-muscle invasive bladder cancer. Oncotarget 2016;7:28384–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Swanton C, Govindan R. Clinical Implications of Genomic Discoveries in Lung Cancer. N Engl J Med 2016;374:1864–73. [DOI] [PubMed] [Google Scholar]
- 17.Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev 2001;15:3243–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sanchez-Rivera FJ, Papagiannakopoulos T, Romero R, Tammela T, Bauer MR, Bhutkar A, et al. Rapid modelling of cooperating genetic events in cancer through somatic genome editing. Nature 2014;516:428–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sakai K, Miyazaki J. A transgenic mouse line that retains Cre recombinase activity in mature oocytes irrespective of the cre transgene transmission. Biochemical and biophysical research communications 1997;237:318–24. [DOI] [PubMed] [Google Scholar]
- 20.Bologna-Molina R, Damian-Matsumura P, Molina-Frechero N. An easy cell counting method for immunohistochemistry that does not use an image analysis program. Histopathology 2011;59:801–3. [DOI] [PubMed] [Google Scholar]
- 21.Ramirez RD, Sheridan S, Girard L, Sato M, Kim Y, Pollack J, et al. Immortalization of human bronchial epithelial cells in the absence of viral oncoproteins. Cancer research 2004;64:9027–34. [DOI] [PubMed] [Google Scholar]
- 22.Sato M, Larsen JE, Lee W, Sun H, Shames DS, Dalvi MP, et al. Human lung epithelial cells progressed to malignancy through specific oncogenic manipulations. Mol Cancer Res 2013;11:638–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sato M, Vaughan MB, Girard L, Peyton M, Lee W, Shames DS, et al. Multiple oncogenic changes (K-RAS(V12), p53 knockdown, mutant EGFRs, p16 bypass, telomerase) are not sufficient to confer a full malignant phenotype on human bronchial epithelial cells. Cancer research 2006;66:2116–28. [DOI] [PubMed] [Google Scholar]
- 24.DuPage M, Dooley AL, Jacks T. Conditional mouse lung cancer models using adenoviral or lentiviral delivery of Cre recombinase. Nat Protoc 2009;4:1064–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012;483:603–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jackson EL, Olive KP, Tuveson DA, Bronson R, Crowley D, Brown M, et al. The differential effects of mutant p53 alleles on advanced murine lung cancer. Cancer research 2005;65:10280–8. [DOI] [PubMed] [Google Scholar]
- 27.Kumar MS, Erkeland SJ, Pester RE, Chen CY, Ebert MS, Sharp PA, et al. Suppression of non-small cell lung tumor development by the let-7 microRNA family. Proc Natl Acad Sci U S A 2008;105:3903–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sutherland KD, Song JY, Kwon MC, Proost N, Zevenhoven J, Berns A. Multiple cells-of-origin of mutant K-Ras-induced mouse lung adenocarcinoma. Proc Natl Acad Sci U S A 2014;111:4952–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 2014;11:783–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 2013;8:2281–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Magenta A, Fasanaro P, Romani S, Di Stefano V, Capogrossi MC, Martelli F. Protein phosphatase 2A subunit PR70 interacts with pRb and mediates its dephosphorylation. Mol Cell Biol 2008;28:873–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kurimchak A, Grana X. PP2A: more than a reset switch to activate pRB proteins during the cell cycle and in response to signaling cues. Cell cycle 2015;14:18–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perrotti D, Neviani P. Protein phosphatase 2A: a target for anticancer therapy. Lancet Oncol 2013;14:e229–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ross JA, Cheng H, Nagy ZS, Frost JA, Kirken RA. Protein phosphatase 2A regulates interleukin-2 receptor complex formation and JAK3/STAT5 activation. J Biol Chem 2010;285:3582–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ruvolo PP. The broken “Off” switch in cancer signaling: PP2A as a regulator of tumorigenesis, drug resistance, and immune surveillance. BBA Clin 2016;6:87–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cicchini M, Buza EL, Sagal KM, Gudiel AA, Durham AC, Feldser DM. Context-Dependent Effects of Amplified MAPK Signaling during Lung Adenocarcinoma Initiation and Progression. Cell Rep 2017;18:1958–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kiely M, Kiely PA. PP2A: The Wolf in Sheep’s Clothing? Cancers (Basel) 2015;7:648–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lake D, Correa SA, Muller J. Negative feedback regulation of the ERK1/2 MAPK pathway. Cell Mol Life Sci 2016;73:4397–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ren D, Zhu X, Kong R, Zhao Z, Sheng J, Wang J, et al. Targeting Brain-Adaptive Cancer Stem Cells Prohibits Brain Metastatic Colonization of Triple-Negative Breast Cancer. Cancer research 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.