

SUMMARY:
Hyperprogression (HP) is a recently defined clinical phenomenon in which patients treated with immunotherapy paradoxically exhibit rapid tumor growth. The mechanisms of HP remain ill-defined though recent studies in this issue point to a possible role for Fc receptors in this process.
In this issue of Clinical Cancer Research, Lo Russo and colleagues describe potential cellular mediators of HP in non-small cell lung cancer (NSCLC)(1). Although the clinical phenomenon of hyperprogression remains ill-defined, this and work of others highlights an important cautionary tale of how little we know about the mechanism of these therapies.
The majority of immune checkpoint inhibitors (ICIs) in the clinic are recombinant IgG antibodies targeting various cell surface proteins (e.g. PD-1, CTLA-4). Antibodies are bifunctional molecules containing two identical antigen binding domains (Fabs) and a single Fc (fragment crystallizable). While nature has endowed antibodies with incredible diversity in their Fab domains through somatic hypermutation, it has also evolved diversity within the Fc fragment(2). This is most evident by the divergent subclass activity of various immunoglobulin G proteins through altered binding to Fc receptors (FcRs). In humans, there are four IgG subclasses (IgG1–4). These unique subclasses exhibit varying affinity for activating or inhibitory FcRs. There are 3 main activating receptors (FcγRI, FcγRIIa, and FcγRIIIa) and a sole inhibitory receptor (FcγRIIb) (Fig. 1A). It is the collective balance of signaling through these receptors expressed on any given cell type that determines its effector function (e.g. antibody dependent cellular cytotoxicity or cellular phagocytosis, ADCC/ADCP). In the context of tumors, it is important to not only consider the cells composing the immune infiltrate, but the FcRs they express and the role that different inflammatory cytokines exert on the FcR expression profile.
Figure 1.
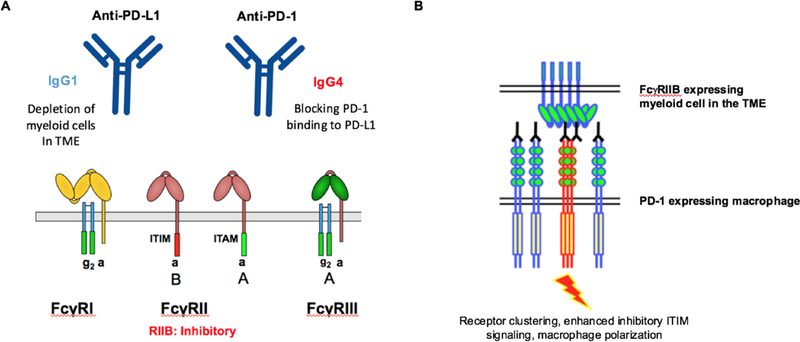
A, Anti-PD-1 antibodies (IgG4) and anti-PD-L1 (IgG1) antibodies have different mechanisms of action through divergent binding to activating or inhibitory Fc receptors (FcRs) B, Proposed mechanism by which FcyRIIB-enhanced clustering of PD-1 on macrophages leads to polarization through enhanced signaling through the ITIM domain.
In general, most cytotoxic antibodies are designed using an IgG1 backbone favoring binding to activating receptors initiating ADCC/ADCP. In contrast, IgG4 antibodies have weak binding to activating receptors and are favored as agents looking to block the in vivo activity of a pathway. Two examples of this are antibodies blocking PD-1 and PD-L1. PD-1 antibodies are usually an IgG4 subclass in which Fc receptor engagement is not required for in vivo anti-tumor activity(3). In contrast, antibodies directed against PD-L1 are of an IgG1 subclass, thus favoring ADCC/ADCP. Pre-clinical studies testing the contributions of the antibody Fc for PD-L1 variants demonstrated they function in part through depletion of intratumoral myeloid cells. Thus, although this has not yet been definitively established in humans there are likely key differences in how ICIs work. The idea that they all fall into the same class is an extreme oversimplification. Much work has been done to define predictors of response to ICI, however, it is equally important to consider which features of a tumor could predispose to immune related adverse events (irAEs) or tumor progression.
Here, Lo Russo et. al. evaluated a cohort of 187 patients with NSCLC being treated with ICIs(1). They defined HP as those having a) treatment failure within 2 months, b) increase in target lesions >50%, c) significant clinical deterioration or d) appearance of 2 or more new lesions or new organ involvement when compared to the previous scan. Using baseline immunohistochemistry (IHC), multiparameter flow cytometry, and immunodeficient mouse models they aimed to define correlates of HP in patients receiving ICIs. They found HP at a rate of 25% in their patient population, which is on the high end of what has been previously reported (between 9 and 29%). Pathologically, they defined a population of tumor associated macrophages (TAMs) that were enriched in patients with HP. These TAMs were polarized to an M2-like CD163+CD33+PD-L1+ phenotype in the 11 HP patients versus 24 patients without HP. Interestingly, this phenotype and clustering of TAMs was recapitulated in their xenografted tumor models. No differences were noted in infiltrating lymphocytes including CD4, CD8, or FOXP3-expressing cells. They didn’t appear to evaluate PD-1 expression in these samples but do note the low level (<1%) PD-1 expression in the tumor cell lines tested.
The authors then went on to see if they could recapitulate HP in murine models as has been previously demonstrated(4). To do this they used two separate immunodeficient models both lacking mouse T cells. Thus, the resultant CD45+ infiltrating cells within the tumor microenvironment (TME) are primarily myeloid cells. They demonstrated that mice bearing H460 NSCLC tumors showed faster tumor growth when treated with the rat IgG2a anti-PD1 antibody and was associated with an increase in tumor infiltrating CD45+ cells. This suggested a possible mechanism for PD-1 expressing myeloid cells within the TME contributing to this process. Although PD-1 expressing myeloid cells have been described, this biology remains in its infancy. Because tumor infiltrating myeloid cells express high levels of activating and inhibitory FcRs(5), the authors went on to determine if the mechanism of HP was Fc-receptor dependent. To do this, they generated F(ab)2 fragments of the anti-mouse PD-1 antibody and saw this abrogated HP. To evaluate the role of PD-1 on infiltrating immune cells they switched to using the human version of this antibody (Nivolumab) which fails to bind to mouse PD-1. In two molecularly distinct NSCLC xenograft models, they found one of them exhibited evidence of HP when treated with Nivolumab and correlated with an increase in CD11b+F4/80high myeloid cells. Collectively, the authors concluded that HP is maintained by myeloid cells within the TME and point to a potential FcR-dependent mechanism.
While these studies are intriguing and provide preliminary evidence for the cellular drivers of HP, many questions remain when determining the contribution of FcRs to this process. First, it is important to consider the clinical question in the correct experimental context. Here, an important lesson remains in that mice are not humans, where murine FcRs do not mirror the structural diversity or unique expression profile observed for human FcRs on human cells. Thus, when considering whether or not the in vivo activity of an antibody is Fc receptor dependent this should be done on an FcR-deficient mouse background or with appropriately modified antibodies (e.g. mouse IgG1-D265A). While treating antibodies with pepsin removes the Fc fragment, it also severely limits any in vivo activity as loss of the Fc fragment alters biodistribution of drug given lack of recycling and trafficking through the neonatal Fc receptor. When F(ab)2 fragments are given systemically in this model only a fraction of the product, if any, is making it to the TME. Finally, testing various ICIs in immunodeficient mice remains a severe limitation to any of these preclinical models, where immunocompetent mice carrying human FcRs in place of the mouse homologues may provide a more relevant in vivo system(3,5).
In the current study, treatment with anti-PD1 antibodies in T cell deficient mice appears to augment tumor growth. Since this is also inhibited through depletion of host macrophages, it is possible that ICIs promote tumor growth through stimulating PD-1 on myeloid cells within the TME. Should this indeed be the case, the most likely Fc-dependent mechanism is through the inhibitory receptor, FcyRIIb. Similar to agonistic antibodies in which FcyRIIb binding is required for optimal activity in vivo, signaling through PD-1 in myeloid cells could be enhanced through FcyRIIb crosslinking (Fig. 1B). Prior data evaluating anti-MARCO antibodies demonstrated repolarization of tumor associated macrophages was also dependent on FcyRIIb. Importantly, it has been demonstrated that Fc receptors, including FcyRIIb are upregulated in the tumor microenvironment compared to other sites (5). This could potentially serve as another biomarker identifying patients at risk for HP. Although intriguing, how do we explain the same rate of HP clinically in NSCLC patients treated with PD-1/PD-L1 antibodies as they have major differences in FcR binding? If the rates of HP are truly the same, this likely supports less of a role for FcR-dependent progression in these patients. It would also be interesting to see the results of PD-L1 treatment in their models or in PD-1 deficient hosts.
As we move forward to more clearly define hyperprogression in lung cancer and other malignancies, the role for FcRs in determining both in vivo anti-tumor activity as well as unwanted effects (e.g. HP and irAEs) should be considered.
Acknowledgments
Funding: Research reported in this publication was supported in part by the National Cancer Institute of the National Institutes of Health (NIH) under Awards R35CA196620 and P01CA190174 (D.A.K. and J.V.R.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. D.A.K. is supported in part by Grant UL1 TR001866 from the National Center for Advancing Translational Sciences, NIH Clinical and Translation Science Award Program.
Conflicts of interest: J.V.R. receives research support from Bristol-Myers-Squibb.
References
- 1.LoRusso G, Moro M, Sommariva M, Cancila V, Boeri M, Centonze G, et al. Antibody-Fc/FcR Interaction on Macrophages as a Mechanism for Hyperprogressive Disease in Non-Small Cell Lung Cancer Subsequent to PD-1/PD-L1 Blockade. Clin Cancer Res 2018. [DOI] [PubMed]
- 2.Bournazos S, Wang TT, Dahan R, Maamary J, Ravetch JV. Signaling by Antibodies: Recent Progress. Annual Review of Immunology 2017;35(1):285–311 10.1146/annurev-immunol-051116-052433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dahan R, Sega E, Engelhardt J, Selby M, Korman AJ, Ravetch JV, et al. FcγRs Modulate the Anti-tumor Activity of Antibodies Targeting the PD-1/PD-L1 Axis. Cancer cell 2015;28(3):285–95 10.1016/j.ccell.2015.08.004. [DOI] [PubMed] [Google Scholar]
- 4.Du S, McCall N, Park K, Guan Q, Fontina P, Ertel A, et al. Blockade of Tumor-Expressed PD-1 promotes lung cancer growth. OncoImmunology 2018;7(4):e1408747 10.1080/2162402X.2017.1408747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Knorr DA, Dahan R, Ravetch JV. Toxicity of an Fc-engineered anti-CD40 antibody is abrogated by intratumoral injection and results in durable antitumor immunity. Proceedings of the National Academy of Sciences 2018. [DOI] [PMC free article] [PubMed]