

Abstract
Basal-like breast cancers are an aggressive breast cancer subtype, which often lack estrogen receptor, progesterone receptor and Her2 expression, and are resistant to anti-hormonal and targeted therapy, resulting in few treatment options. Understanding the underlying mechanisms that regulate progression of basal-like breast cancers would lead to new therapeutic targets and improved treatment strategies. Breast cancer progression is characterized by inflammatory responses, regulated in part by chemokines. The CCL2/CCR2 chemokine pathway is best known for regulating breast cancer progression through macrophage dependent mechanisms. Here, we demonstrated important biological roles for CCL2/CCR2 signaling in breast cancer cells. Using the MCF10CA1d xenograft model of basal-like breast cancer, primary tumor growth was significantly increased with co-transplantation of patient derived fibroblasts expressing high levels of CCL2, and was inhibited with CRISP/R gene ablation of stromal CCL2. CRISP/R gene ablation of CCR2 in MCF10CA1d breast cancer cells inhibited breast tumor growth and M2 macrophage recruitment and validated through CCR2 shRNA knockdown in the 4T1 model. Reverse phase protein array analysis revealed that cell cycle protein expression was associated with CCR2 expression in basal-like breast cancer cells. CCL2 treatment of basal-like breast cancer cell lines increased proliferation and cell cycle progression associated with SRC and PKC activation. Through pharmacologic approaches, we demonstrated that SRC and PKC negatively regulated expression of the cell cycle inhibitor protein p27KIP1, and are necessary for CCL2 induced breast cancer cell proliferation.
Keywords: CCL2, CCR2, chemokine, fibroblast, breast cancer, growth, cell cycle, PKC, SRC
Introduction
Breast cancer is the most common form of cancer diagnosed in women in the US, with 250,000 cases diagnosed in the US, and ranks second in the cause of cancer-related deaths (1). Treatment is complicated by the presence of multiple breast cancer subtypes. Luminal A/B breast cancers express Estrogen Receptor, Progesterone Receptor, and/or Her2. Others overexpress Her2+. Basal-like breast cancers (BLBC) often lack ER, PR and Her2 (2,3). BLBC comprise approximately 15% of all of cases diagnosed in North America, but are considered the most aggressive, and are resistant to most treatments other than chemotherapy (4,5). Understanding the mechanisms that regulate BLBC progression would lead to new therapeutic targets and improved treatment strategies.
Cancer progression is associated with recruitment of myeloid immune cells, increased angiogenesis and fibroblast accumulation. These stromal cell responses are regulated in part by chemokines, soluble molecules (8kda), which form molecular gradients to induce cellular chemotaxis during tissue development, inflammation, and cancer, by signaling to seven-transmembrane G protein-coupled receptors (6,7). The Chemokine C-C Ligand 2 (CCL2) regulates recruitment of macrophages and endothelial cells during acute inflammation by primarily signaling to CCR2 receptors (6,8). CCL2 overexpression in breast tumors correlates with macrophage levels (9,10). CCL2 expression in the stroma correlates with poor prognosis for breast cancer patients (10,11). Antibody neutralization of CCL2 inhibits growth, survival and invasion of breast tumor xenografts (10,12,13), correlating with decreased macrophage recruitment. These studies indicate an important role for CCL2 in regulating macrophage recruitment during breast cancer progression.
Studies on CCL2/CCR2 signaling in cancer have focused on its role in regulating immune cell recruitment. Yet, CCR2 expression is over-expressed in epithelial tissues of invasive ductal carcinomas and luminal and BLBC cell lines (14). shRNA knockdown or antibody neutralization of CCL2 inhibits fibroblast-induced survival and invasion of luminal breast cancer cells (14,15). While these studies indicate an important role for epithelial CCR2 signaling in cancer progression, breast tumors exhibit variations in CCL2/CCR2 expression and signaling (14) that are poorly understood.
Compared to other molecular subtypes, BLBC show the highest levels of stromal CCL2 expression (11). Using the MCF10CA1d xenograft model of BLBC, we demonstrate that breast tumor growth was significantly increased with co-transplantation of patient derived fibroblasts, correlating with CCL2 expression. CRISP/R gene ablation of stromal CCL2 or CCR2 in MCF10CA1d breast cancer cells inhibited breast tumor growth and M2 macrophage recruitment. The effects of CCR2 deficiency on tumor growth were validated in the 4T1 model. CCL2 treatment of multiple BLBC cell lines increased proliferation and cell cycle progression associated with SRC and PKC activation, which were decreased in CCR2 knockout cells. Pharmacologic approaches demonstrate that SRC and PKC negatively regulate expression of p27KIP1, and regulates CCL2-induced breast cancer cell proliferation. These studies demonstrate novel mitogenic mechanisms for CCL2/CCR2 chemokine signaling in breast cancer cells, with important implications on therapeutic targeting.
Material and methods
Cell culture
Unless indicated, cell lines were cultured in DMEM/10% fetal bovine serum (FBS)/2mM L-glutamate/1% penicillin-streptomycin. MCF10CA1d cells (16,17) were kindly provided by Fred Miller, Ph.D (University of Michigan). BT-20 and HCC1937 cells were kindly provided by Roy Jensen, M.D (University of Kansas Medical Center). 4T1 cells were purchased from ATCC (cat# 2539). BT-20 cells were cultured in Eagle’s Minimal Essential Medium/10%FBS/2mM L-glutamate/1% penicillin-streptomycin. HCC1937 cells were cultured in RPMI/10% FBS/2mM L-glutamate/1% penicillin-streptomycin. Human cancer associated fibroblasts and normal adjacent fibroblasts were isolated from invasive breast carcinoma tissues obtained from University of Kansas Medical Center Biospecimen Repository, using approaches described (18). For CCL2 knockout, fibroblasts were first immortalized by transfection of hTert, using approaches described (19). Fibroblasts were isolated from MMTV-PyVmT transgenic mice or tumor free mice (C57BL/6xFVB) as described (15,18). Cell lines were cultured less than 3 months at a time and checked for mycoplasma after freeze/thaw, using the Mycoplasma Detection Kit (Lonza, cat# LT07–703).
Recombinant proteins/Inhibitors
PP2 was obtained from Tocris (cat# 1407). Gö 6983 was purchased from Cayman Chemical (cat# 13311). Recombinant human CCL2 was purchased from Peprotech (cat# 300–04).
Orthotopic transplantation
Female athymic nude mice (FoxN1nu/nu) 6–8 weeks old were purchased from Charles River. Breast cancer cells and fibroblasts were transplanted as described (18). Briefly, 100,000 cancer cells and 250,000 fibroblasts were seeded into 50 μl Rat Tail Collagen type I overnight. Mice were anesthetized with 1–3% isoflurane. An incision was made into the skin flap to expose the #4–5 mammary glands. One collagen plug was inserted into a pocket that was made underneath the mammary lymph node using spring scissors. Wounds were closed by gut absorbable suture. Mice were monitored twice a week and measured for tumor size by caliper. Mice bearing MCF10CA1d tumors were sacrificed 6 weeks post-transplantation when control tumors grew 1.5 cm in diameter, the largest allowable size allowed by the Institutional Animal Care and Use committee (IACUC). 4T1 tumor bearing mice were sacrificed 4 weeks post-transplantation. All animal procedures were approved by IACUC under AALAAC guidelines.
Histology/immunohistochemistry
Tissues were fixed in 10% neutral formalin buffer (NBF), paraffin embedded and H&E stained as described (20). For immunostaining, 5-micron sections were dewaxed and heated in 10mM sodium citrate pH6.0 at low pressure for 2 minutes using a pressure cooker. Peroxidases were quenched in PBS/10% methanol/10% H202 for 30 minutes. Slides were blocked in PBS/3% FBS, and incubated overnight with antibodies (1:100) to: arginase-1 (Santa Cruz Biotechnology, cat# 20150), Ly6G (Biolegend, cat# 127602), or Von Willebrand Factor 8 (Millipore, cat#,7356). Arginase I and VWF8 were detected with secondary rabbit biotinylated antibodies (1:1000). Ly6G was detected with secondary rat biotinylated antibodies (1:1000). Slides were immunostained with anti-human Cytokeratin 5 (ThermoFisher, cat #MA5–12596) or anti-GFP (Santa Cruz Biotechnology cat# SC9996), using the MOM kit (Vector Laboratories cat #BMK-2202). Antigens were detected using DAB substrate (Vector Laboratory, cat #SK-4100). Slides were counter-stained with Mayer’s hematoxylin and mounted with Cytoseal (Thermo Fisher, cat #348976). 5 images were captured per sample at 10x magnification using the EVOS FL-Auto Imaging system (Invitrogen). Expression was quantified by Image J using methods described (11).
CRISP/R gene ablation
Exon 1 of human CCL2 was targeted by CRISPR/Cas9 using the gRNA: 5’-GTACCTGGCTGAGCGAGCCCT-3’. The gRNA was cloned into lentivirus vector pLKO5.sgRNA.EFS.GFP (Addgene, cat #57822). hCAF-1 was transfected with lentivirus containing Cas9 and a blasticidin selection marker (Addgene, cat #52962). Cells were selected with 4 μg/ml blasticidin. Cells were transduced with lentivirus expressing CCL2 gRNA or control vector, flow sorted for GFP expression, and seeded into 96-well plates. Single cell colonies were screened by CCL2 ELISA to identify CCL2 deficient clones. CCR2 was targeted by CRISPR/Cas9 with gRNA: 5’TTCACAGGGCTGTATCACAT-3’. The gRNA was cloned into the pL-CRISPR.EFS.GFP vector [86] (Addgene, cat #57818), a lentivirus vector containing Cas9 and a GFP reporter. Transduced cells were flow sorted for GFP expression and seeded into 6-well plates, 1000 cells/well. Individual colonies were manually picked and seeded into 96-well plates. Colonies were screened for CCR2 gene alterations by PCR, using primers flanking the targeting site (primer-F:ACATGCTGGTCGTCCTCATC and primer-R: AAACCAGCCGAGACTTCCTG). Wildtype and CCR2 mutant clones were confirmed by DNA sequencing.
shRNA knockdown
The targeting sequences were 5′-TGGTGAGCCTTGTCATAAA-3′ for CCR2KD#1 and 5′-CCGGTCCATATTTTACTA-3′ for CCR2KD#3. The targeting sequence to silence enhanced GFP as a negative control (CTRL) was 5′-GCTGACCCTGAAGTTCATC-3′. The oligonucleotides were phosphorylated by kinase treatment; complementary oligos were annealed and subcloned into Bgl II and HindIII sites of pRETRO-SUPER vector (21), which was generously provided by Reuven Agami, Ph.D (The Netherlands Cancer Institute). Plasmids were transfected into Phoenix cells by Lipofectamine 2000 (Invitrogen, Carlsbad, CA). 48 hours post-transfection, 4T1 cells were transduced with virus-conditioned medium and selected with 1.5 μg/ml puromycin.
CCL2 ELISA
10,000 fibroblasts/well were seeded in 24-well plates, and incubated in serum free medium for 24 hours. Conditioned medium was assayed for human CCL2 (Peprotech, cat# 900-K31) or murine CCL2 (Peprotech, cat# 900-K126) according to commercial ELISA protocols. Absorbance was read at OD450 nm using a Biotek plate reader. CCL2 levels were normalized to cell density by crystal violet staining as described (22).
Flow cytometry
For antibody staining, 200,000 cells were seeded in 6-cm dishes overnight. Cells were detached from plates with Accutase (EMD Millipore cat #SCR005) at 37oC for 5 minutes, washed in PBS and incubated with anti-CCR2-PE diluted 1:50, (R&D Systems, cat# FAB151P) for 1 hour. For cell cycle studies, 200,000 cells were seeded in 6-cm dishes overnight. Cells were treated with or without 2mM Thymidine (Sigma, cat # T1895–1G) for 16 hours. The cells were washed 3 times with serum free media (BT-20: EMEM, HCC1937: RPMI, and MCF10CA1d: DMEM). Growth media was added for 2 hours for MCF10CA1d cells and 10 hours for BT-20 and HCC1937 cells. Cells were incubated with 2mM thymidine for 16 hours, washed with serum free media and incubated with growth media, with or without 100 ng/ml CCL2. Cells were detached with Accutase and fixed with 70% ethanol at −20ᵒC. Samples were incubated in 500 μL of PBS/0.1% Triton X-100/2mg/ml RNase A (VWR, cat# 97064–064) and 200 μg/ml propidium iodide (Invitrogen, cat# P3566) for 15 minutes at 37ᵒC.
For ALDH activity assay, cells were seeded in 6 well plates (200,000/well), serum starved for 24 hours and incubated in serum free medium with or without 100 ng/ml CCL2 for 24 hours. Cells were subject to AldeRed™ Assay (EMT Millipore, cat #SCR150) according to commercial protocol. All cells were analyzed using a BD LSRII Flow Cytometer.
3D Matrigel:Collagen cultures
1 million cells were cultured in 10-cm dishes in 5 ml DMEM/10% FBS for 24 hours. Medium was collected, centrifuged, and filtered through 0.45 micron pore cellulose acetate membranes. 96-well plates were coated with 40 μl of matrix containing 1:1 ratio of collagen (Corning, cat #354236) and Growth Factor Reduced Matrigel (Corning, cat# 354230). MCF10CA1d cells were seeded 3000 cells/well in 100 μl DMEM/10%FBS containing 2.5% Matrigel, with 100 μl conditioned medium, or 200 μl of DMEM/10% FBS/2% Matrigel with or without CCL2. The media was replaced every 2 days for up to 8 days. Bright field images were captured every two days using an EVOS FL-Auto Imaging system at 10X magnification, 4 fields/well. Sphere size was quantified using Image J.
Mammosphere assay
3000 cells/well were seeded in low attachment 24 well plates (Corning) in 500 μl of DMEM/10% FBS with or without 100 ng/ml CCL2, and incubated for 5 days. Mammospheres were pelleted, and disassociated with 20 mM Trypsin/2 mm EDTA for 7 minutes at 37oC. Cells were quenched in DMEM/10% FBS, pelleted and re-plated. Images were captured at 4x magnification using the EVOS FL-Auto prior to passaging. Mammospheres were counted using Image J, with minimum size of 160 microns2.
Reverse phase protein array (RPPA)
RPPA assays were adapted from previous studies (1–4). Protein lysates were prepared from quadruplicate samples of CCR2 knockout (CCR2KO-G10) or wildtype CCR2 control (WT-A1) using Pierce Tissue Protein Extraction Reagent (VWR cat# 78510) with protease and phosphatase inhibitors. 0.5 mg/ml of protein lysate were denatured in SDS sample buffer. The Aushon 2470 Arrayer (Aushon BioSystems) with a 40 pin (185 μm) configuration was used to spot samples and control lysates onto nitrocellulose-coated slides (Grace Bio-labs) using an array format of 960 lysates/slide (2880 spots/slide). Each sample was probed on triplicate slides. The slides were probed with 345 antibodies against total and phosphoprotein proteins using an automated slide stainer Autolink 48 (Dako). A negative control slide was incubated with antibody diluent only. Primary antibody binding was detected using a biotinylated secondary antibody followed by streptavidin-conjugated IRDye680 fluorophore (LI-COR Biosciences, cat# 926–68079). Total protein content/spot was assessed by Sypro Ruby Protein Blot staining (Invitrogen cat# S11791).
Fluorescence-labeled and negative control slides were scanned on a GenePix 4400 AL scanner at an appropriate PMT. The images were analyzed with GenePix Pro 7.0 (Molecular Devices). Total fluorescence signal intensities/spot were obtained after subtraction of the local background signal for each slide and were normalized for variation in total protein, background and non-specific labeling using a group-based normalization method as described (23). For each spot, the-background-subtracted foreground signal intensity was subtracted by the corresponding signal intensity of the negative control slide (omission of primary antibody) and normalized to the corresponding signal intensity of total protein for that spot. The median of normalized signal intensities/sample was used for statistical analysis.
Immunoblot analysis
Cancer cells were seeded in 6-well plates (200,000 cells/well) in DMEM/10% FBS, serum deprived overnight, and incubated with serum free media with or without recombinant CCL2, DMSO, PP2 or Gö 6983 for 15 minutes. Cells were lysed with RIPA buffer containing protease inhibitors and phosphatase inhibitors. 25 μg proteins were resolved on SDS polyacrylamide gels, and transferred to nitrocellulose membranes. Membranes were blocked in PBS/0.05% Tween-20/5% milk and incubated with primary antibodies (1:1000) to: p-PKC (pan, βII Ser660, CST, cat# 9371), p-SRC (Tyr416, CST, cat# 6943), SRC (CST, cat# 2123), p27KIP1 (Fisher Scientific, cat# BDB610241), p21 (CST, cat# 2947), p-AKT (Ser473, CST, cat# 4060), AKT (R&D,cat# MAB2053), p-ERK (Thr202/Tyr204, CST, cat# 4370), ERK1/2 (CST, cat# 9107), p-SMAD3 (Ser423/425, CST, cat# 9520), SMAD3 (CST, cat# 9523), ACTIN (Sigma, cat# A5441). Proteins were detected with corresponding secondary antibodies conjugated to horse radish peroxidase (1:1000), developed with West Femto chemiluminescence substrate (Thermofisher cat #34094) and imaged using a Biospectrum Imaging System.
Immunocytochemistry/Immunofluorescence
5000 cells/well were seeded in 96 well plates. Cells were fixed in 10% NBF, permeabilized with methanol at −20oC for 10 minutes, and blocked in PBS/3% FBS for 1 hour. For immunocytochemistry studies, cells were incubated for 24 hours with antibodies (1:100) to alpha smooth muscle actin (α-SMA, Abcam, cat # 7187), Fibroblast specific protein 1 (FSP1, Abcam, cat #5550), N cadherin (Santa Cruz Biotechnology cat #sc7939), E-cadherin (BD Biosciences cat # 610181) or Pan-cytokeratin (Santa Cruz Biotechnology, cat #8018). α-SMA, Pan-cytokeratin and N-cadherin were detected using anti-mouse-hrp (1:500). FSP1 and N-cadherin were detected using anti-rabbit-hrp (1:500). Antigens were detected using DAB. For immunofluorescence studies, cells were incubated with antibodies (1:300) to PCNA (Biolegend cat #307901) or p27KIP1 (Fisher Scientific cat #BDB10241) for 24 hours. Cells were incubated with secondary mouse antibodies conjugated to Alexa-Fluor-647 (1:500) for 2 hours. Cells were counterstained with DAPI and mounted in 1:1 PBS:Glycerol. Images were captured at 10X magnification, 3 fields/well using the FL-Auto EVOS Imaging System. Expression was quantified by Image J. PCNA and P27KIP1 expression was normalized to DAPI.
Datamining
METABRIC dataset (24,25) (n=2509) was accessed from http://www.cbioportal.org/ on July 21, 2017. Associations between CCL2 and CCR2 were analyzed using the following parameters: any samples with mutations, putative copy-number alterations from DNAcopy and mRNA expression z-scores from Illumina Human v3 microarray.
Statistical analysis
Statistical analysis was performed using GraphPad software. Two tailed Student’s T- test was used for two groups. One Way ANOVA with Bonferroni post-hoc comparison was used for more than two groups. Associations between continuous variables in TCGA datasets were analyzed by Spearman correlation analysis. RPPA data were analyzed using Two-Tailed Student’s T-test (23), using 1.2 fold change as a cutoff. Heatmapping was performed using https://software.broadinstitute.org/morpheus/. Statistical significance was determined by p<0.05. *p<0.05, **p<0.01, ***p<0.001, n.s=not significant. For cell culture experiments, samples were plated in triplicates/group; experiments were repeated 3 times.
Results
BLBC growth is associated with stromal CCL2 expression
To characterize the significance of CCL2 derived from fibroblasts to progression of BLBC, we utilized the MCF10CA1d model, a breast cancer cell line derived from Ras transformed MCF10A cells (26,27). Fibroblasts were isolated from invasive ductal carcinoma tissues (hCAF) or normal adjacent tissues (hNAF), and characterized for expression of mesenchymal markers and absence of epithelial markers (Supplemental Figure 1). By ELISA, CCL2 expression in fibroblasts were significantly higher than in MCF10CA1d cells, and varied between 0.5 μg/ml and 1 μg/ml, among huNAFs and huCAFs (Figure 1A). To determine associations between stromal CCL2 expression and breast cancer progression, MCF10CA1d breast cancer cells were orthotopically grafted alone, or co-grafted with fibroblasts for 6 weeks. Compared to MC10CA1d cells grafted alone, co-transplantation with hCAF-1 and hNAF-3 significantly enhanced primary tumor mass (Figure 1B). MCF10CA1d breast tumors in all groups were characterized as invasive carcinomas with microvasculature (Figure 1C). Fibroblasts did not significantly affect lung metastasis as determined by analysis of H&E staining (Supplemental Figure 2A-B). These data indicate that MCF10CA1d breast tumor growth is associated with fibroblast expression of CCL2.
Figure 1. CCL2 expressing fibroblasts enhance growth of primary MCF10CA1d breast tumor xenografts.
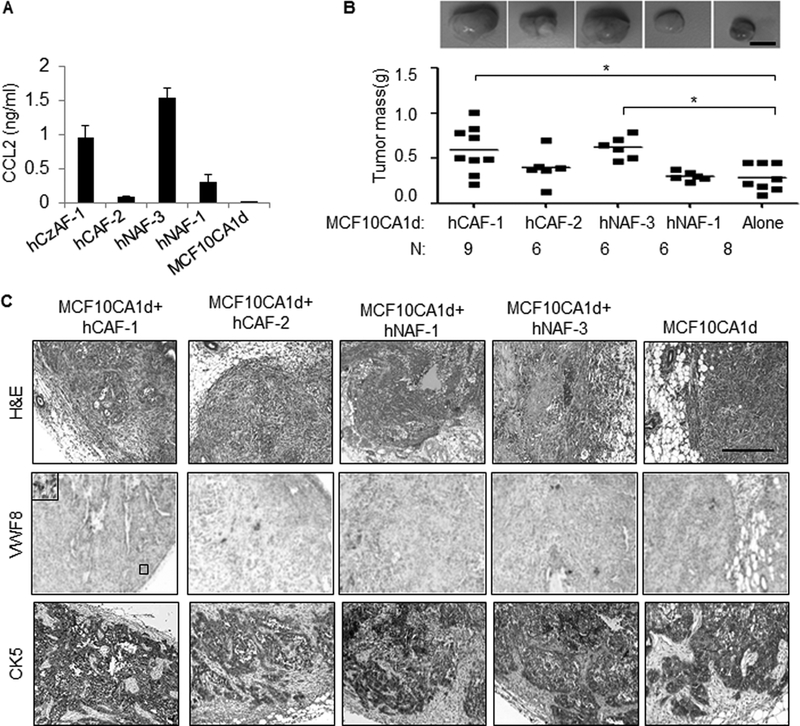
A. CCL2 ELISA of conditioned medium from patient derived carcinoma associated fibroblasts (hCAF-1, hCAF-2), normal adjacent fibroblasts (hNAF-1, hNAF-3) or MCF10CA1d breast cancer cells. B-C. MCF10CA1d breast cancer cells were orthotopically grafted alone or with fibroblasts in nude mice for 6 weeks, and analyzed for: tumor mass (B) by H&E stain and immunostaining for Von Willebrand Factor 8 (VWF8) or Cytokeratin 5 (CK5) expression (C). Scale bar= 200 microns. Statistical analysis was performed using One Way ANOVA with Bonferroni post-hoc comparisons. Statistical significance was determined by p<0.05. *p<0.05. Mean±SEM are shown.
Gene ablation of stromal CCL2 or epithelial CCR2 inhibits breast tumor growth
To determine the contribution of stromal CCL2 derived to MCF10CA1d breast tumor growth, exon 1 of CCL2 was targeted by CRISP/R in hCAF-1. Two clones showed significantly decreased CCL2 expression (CCL2KO#7, CCL2KO#22), compared to wildtype control cells (WT) (Figure 2A). MCF10CA1d cells were co-grafted with WT or CCL2 deficient fibroblast lines, and analyzed for tumor growth over time for up to 42 days (Figure 2B). Up until day 35, CCL2 KO#7 tumors showed decreased tumor volume over time compared to WT tumors, indicating decreased tumor growth rate contributed to early growth delay. From day 35 to 42, CCL2KO#7 appeared to reach a similar growth rate to WT tumors. Throughout the study, the CCL2KO#22 tumor growth rate appeared distinctly lower than WT tumors. At 42 days, CCL2KO#7 and CCL2KO#22 tumors showed significantly decreased mass compared to WT tumors (Figure 2C). CRISP/R resulted in insertion of a GFP reporter, whose expression was detected in stromal tissues, indicating retention of transplanted fibroblasts (Figure 2D). Furthermore, MCF10CA1d breast tumor xenografts with CCL2 deficient fibroblasts showed decreased expression of arginase I, an M2 macrophage marker (28) , but not Ly6G, a neutrophil marker (29) or VWF8, an angiogenesis marker (30) (Figure 2E-G). Overall, these studies indicate that CCL2 derived from fibroblasts regulates MCF10CA1d tumor growth and M2 macrophage levels.
Figure 2. Knockout of stromal CCL2 inhibits growth of primary MCF10CA1d breast tumor xenografts.
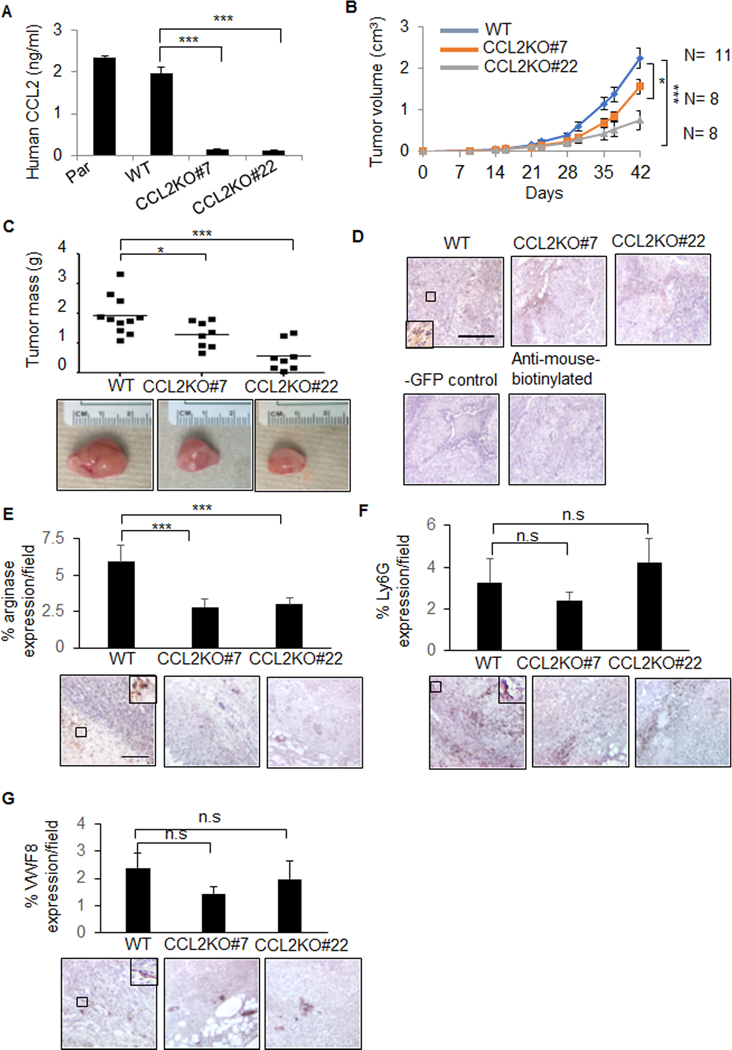
A. CCL2 ELISA of conditioned medium from Parental (Par), control wildtype (WT) or CCL2 knockout (CCL2KO#7, CCL2KO#22) hCAF-1 cell lines. B-C. MCF10CA1d breast cancer cells were co-grafted with WT or CCL2KO fibroblasts for up to 6 weeks and measured for changes in tumor volume over time (B) or endpoint tumor mass (C). D-G. Breast tumor xenografts were immunostained for GFP (D), arginase I (E), Ly6G (F), or Von Willebrand Factor 8 (VWF8) (G). -GFP control is MCF10CA1d co-grafted with hCAF-1 parental fibroblasts.. Expression was quantified by Image J. Statistical analysis was performed using One Way ANOVA with Bonferroni post-hoc comparison. Statistical significance was determined by p<0.05. *p<0.05, ***p<0.001., n.s = not significant. Mean±SEM are shown.
The METABRIC mRNA dataset (24,25) revealed significant correlations between CCL2 and CCR2 expression in invasive breast cancers (Figure 3A). To determine the significance of epithelial CCR2 expression to fibroblast-mediated breast tumor growth, the exon encoding the third transmembrane domain of CCR2 was targeted by CRISP/R. Frameshifts in the coding region generated a premature stop codon, resulting in mutants lacking a C-terminal region. PCR screening identified two mutant clones, CCR2KO-F1 and CCR2KO-G10. DNA sequencing revealed that the CCR2KO-F1 possessed an 81bp deletion with 7bp insertion in the coding region. CCR2KO-G10 appeared to show trans-heterozygous alleles, with one allele containing an 11bp deletion, and the other allele containing a 263bp deletion. To determine the overall levels of CCR2 expression in wildtype and knockout cells, which expressed the C-terminal truncated mutants, we used an antibody that recognized the N-terminus of CCR2. CCR2KO-F1 cells showed a 10% reduction in CCR2 expression, and CCR2KO-G10 cells showed a 75% reduction in CCR2 expression compared to wildtype (WT-A1) control cells (Figure 3B). Despite residual CCR2 expression, we hypothesized that CCR2 truncation would inhibit fibroblast-mediated tumor growth. CCR2KO-G10 and CCR2KO-F10 grafted alone showed decreased tumor growth compared to WT-A1 control cells; only the decreased growth of CCR2KO-G10 tumors was statistically significant. CCR2KO-F1 and CCR2KO-G10 cells co-grafted with fibroblasts showed a significant decrease in tumor growth, compared to WT-A1 cells co-grafted with fibroblasts (Figure 3C). CCR2 deficient tumors showed decreased arginase I expression, indicating decreased M2 macrophage levels (Figure 3D). In summary, CCR2 knockout in MCF10CA1d breast cancer cells inhibits fibroblast-mediated tumor growth.
Figure 3. Knockout of CCR2 inhibits growth of primary MCF10CA1d breast tumor xenografts.
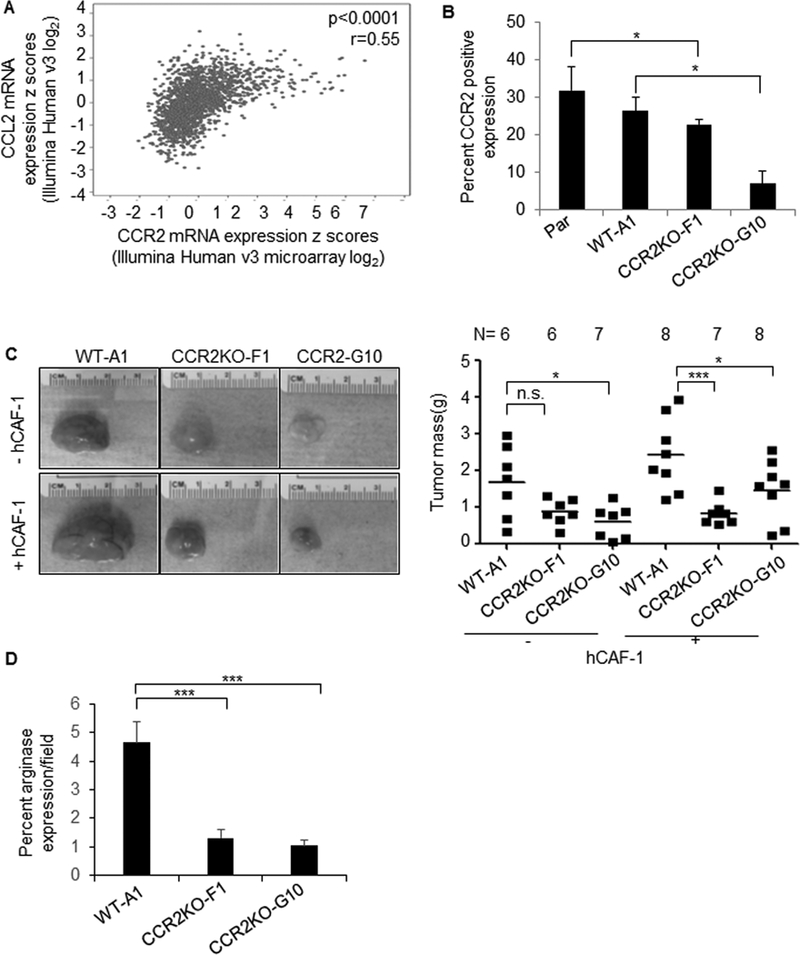
A. Spearman correlation analysis of CCL2 and CCR2 expression in the METABRIC datasets (n=2051). B. Flow cytometry analysis of CCR2 expression in Parental (Par), control wildtype (WT-A1), or CCR2 knockout (CCR2KO-F1, CCR2KO-G10) MCF10CA1d breast cancer cells. C. WT or CCR2KO breast cancer cells were co-grafted with hCAF-1 fibroblasts for up to 6 weeks and analyzed for changes in tumor mass. D. Immunostain for arginase I expression. Expression was quantified by Image J. Expression was normalized to hematoxylin and expressed as percentage per field. Statistical analysis was performed using One Way ANOVA with Bonferroni post-hoc comparison. Statistical significance was determined by p<0.05. *p<0.05, ***p<0.001. n.s = not significant. Mean±SEM are shown.
We further examined the effects of CCR2 knockdown on fibroblast-mediated progression of BLBC using the 4T1 model, which overexpress CCR2 (14,31). Previously, we characterized a tumor promoting role for mammary fibroblasts isolated from MMTV-PyVmT transgenic mice (C57Bl6xFVB) (15,18). Compared to murine normal fibroblasts (mNAF) or 4T1 cells, carcinoma associated fibroblasts (mCAF) expressed the highest levels of CCL2 (Supplemental Figure 3A). Stable CCR2 shRNA in 4T1 cells resulted in approximately 30% knockdown in CCR2KD#1 cells and 20% knockdown in CCR2KD#3 cells, compared to control shRNA expressing cells (Supplemental Figure 3B). CCR2 knockdown did not affect CCL2 expression in 4T1 cells (Supplemental Figure 3C). CCR2KD#1 but not CCR2KD#3 cells co-grafted with mCAFs showed decreased tumor growth and arginase I expression, compared to co-grafting mCAFs with control 4T1 cells (Supplemental Figure 3D-F). CCR2 knockdown did not significantly affect lung metastasis (Supplemental Figure 3G). These data indicate that a 30% CCR2 knockdown but not at 20% is sufficient to inhibit 4T1 tumor growth. Overall, these data support a role for CCR2 expression in regulating growth of BLBC.
Effects of CCL2 and CCR2 expression on MCF10CA1d cell growth were analyzed in 3D Matrigel:Collagen cultures (Figure 4A). MCF10CA1d breast cancer cells treated with huCAF-1 conditioned medium showed increased spheroid growth compared to conditioned medium from MCF10CA1d cells (Figure 4B). CCL2 treatment enhanced spheroid growth (Figure 4C). Conditioned medium from CCL2 deficient fibroblasts inhibited MCF10CA1d spheroid growth, which was rescued with CCL2 treatment (Figure 4D). CCR2 deficiency in MCF10CA1d cells inhibited fibroblast-mediated spheroid growth (Figure 4E). These data indicate that CCL2/CCR2 signaling enhances growth of MCF10CA1d cells.
Figure 4. Stromal CCL2 knockout or CCR2 deficiency in breast cancer cells inhibit the growth and stemness of MCF10CA1d cells.
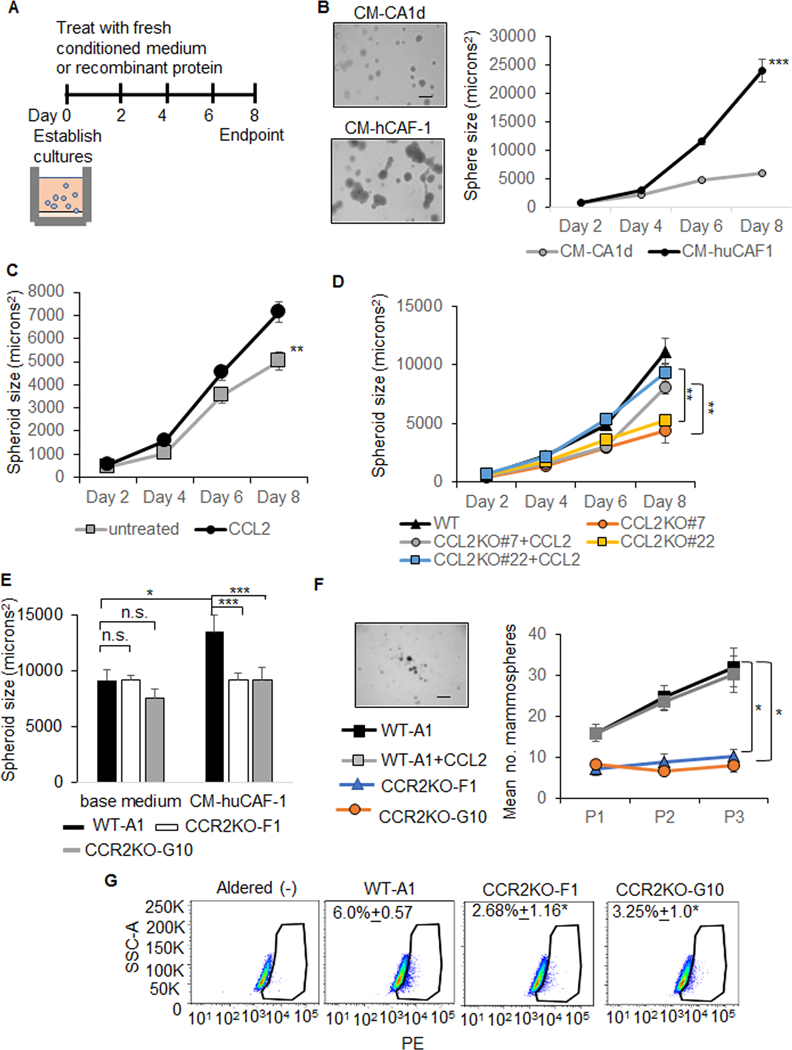
A-E. MCF10CA1d cells were embedded in 3D Matrigel:Collagen. Cultures were treated with conditioned medium or recombinant protein at the establishment of cultures (Day 0). Fresh media was added every 2 days through day 6. Images were captured every 2 days for up to 8 days. Spheroid size was measured using Image J software, normalized to sphere number. Minimum size of spheres analyzed was 80 microns2. Cartoon depicting experiment design is shown (A). The following conditions were used. Treatment with tumor conditioned medium (CM-CA1d) or huCAF-1 conditioned medium (CM-hCAF-1) (B). Mean number spheroids±STDEV: CM-CA1d= 31±9, CM-huCAF-1=23±3. Mean sphere size±STDEV at day 8 (microns2): CM-CA1d= 5891.7±437.6, CM-huCAF-1=23961±3568.3. DMEM/10% FBS with/without 100 ng/ml CCL2 (C). Mean number ±STDEV: Untreated= 65±21, CCL2=50±22. Mean size+STDEV: untreated= 5003.9+2211.8 microns2, CCL2=7132.8±2812.6 microns2. Treatment with conditioned medium from WT control or CCL2KO fibroblasts (CM-CCL2KO#7, CM-CCL2KO#22) with/without 100 ng/ml CCL2 (D). Mean number±STDEV: CM-WT-A1=34±14, CM-CCL2KO#7=29±14, CM-CCL2KO#7+CCL2=34±11, CM-CCL2KO#22=26±14, CM-CCL2KO#22+CCL2=28±15. Mean size ±STDEV (microns2): CM-WT-A1= 14085±8058, CM-CCL2KO#7=5626±2836, CM-CCL2KO#7+CCL2=10296±4540, CM-CCL2KO#22:6653±2340, CM-CCL2KO#22+CCL2=11817±5502 (D). Wildtype (WT-A1) or CCR2KO-F1, CCR2KO-G10 cells incubated in base medium (DMEM/10% FBS) or CM-hCAF-1 (E). Mean number±STDEV: 10% FBS/WT-A1:23±3, 10% FBS/CCR2KO-F1: 31±7, 10%FBS/CCR2KO-G10: 28±8, CM-huCAF-1/WT-A1:22±5, CM-huCAF-1/CCR2KO-F1: 31±3, CM-huCAF-1/CCR2KO-G10: 25±5. Mean size±STDEV (microns2): 10% FBS/WT-A1=13803.4±1398, 10% FBS/CCR2KO-F1=11996.3±3052 10%FBS/CCR2KO-G10=9459±2238, CM-huCAF-1/WT-A=18024.8±1128.8, CM-huCAF-1/CCR2KO-F1=10192±752, CM-huCAF-1/CCR2KO-G10=10714±806. F. Mammosphere assay of WT, CCR2KO-F1 and CCR2KO-G10 MCF10CA1d breast cancer cells treated with or without 100 ng/ml CCL2. Mammospheres were passaged three times, and quantified by Image J. Representative image of WT mammospheres at third passage shown. Scale bar=200 microns. G. MCF0CA1d WT or CCR2KO cells were treated with 100 ng/ml CCL2 for 24 hours and analyzed for AldeRed uptake by flow cytometry. Statistical analysis was performed using One Way ANOVA with Bonferroni post-hoc comparison. Statistical significance was determined by p<0.05. *p<0.05, ***p<0.001. Mean±SEM are shown on graphs.
As stemness is an important factor in tumor growth (32,33), we examined for mammosphere formation and activity of ALDH, a breast cancer stem cell marker {Ginestier C, 2007 #921). CCL2 treatment did not affect mammosphere formation. CCR2 knockout significantly inhibited mammosphere formation and ALDH activity (Figure 4E,F). These data indicate that CCR2 expression regulates stemness of MCF10CA1d cells.
CCL2/CCR2 signaling mediates BLBC cell growth through PKC and SRC pathways
To identify the downstream molecular mechanisms associated with CCL2/CCR2-mediated cell proliferation, we first examined expression of phosphorylated AKT, SMAD3 and ERK1/2 and PKC. These pathways were important in CCL2 signaling in neuronal cells and mediated survival and motility of prostate and mammary carcinoma cells (14,34,35). By immunoblot analysis, CCL2 treatment of MCF10CA1d cells increased phospho-PKC expression (Figure 5A), but did not significantly affect SMAD3, ERK1/2 or AKT phosphorylation (Supplemental Figure 4). For a broader, unbiased analysis of the molecular changes associated with CCR2 expression, CCR2 knockout (CCR2-G10) and wildtype control (WT-A1) cells were subject to RPPA analysis. CCR2 knockout cells showed significant differences in expression of cell cycle and proliferation-related proteins, compared to control cells (Supplemental Figure 5, Supplemental Table 1). Notably, CCR2 deficient cells showed decreased phospho-SRC expression and increased expression of the CDK inhibitor p27KIP1. CCL2 treatment of MCF10CA1d cells increased expression of phospho-SRC, increased expression of proliferating cell nuclear antigen (PCNA) and decreased expression of total p27KIP1. p21 CDK inhibitor expression was weak in BT-20 cells, and not affected with CCL2 treatment in the other cell lines. (Figure 5A). PCNA and p27KIP1 expression in the cytoplasm and nucleus reflect different activity states (36,37). Therefore, changes in expression and cellular localization of PCNA and p27KIP1 were analyzed by immunofluorescence staining. CCL2 significantly decreased nuclear expression of PCNA and p27KIP1 (Figure 5B-C). These data support a mitogenic role for CCL2/CCR2 signaling in MCF10CA1d breast cancer cells. Consistent with RPPA data (Supplemental Table 1), CCL2 treatment of MCF10CA1d cells did not affect cell apoptosis or autophagy, as indicated by immunostaining for cleaved caspase-3 and LC3B (Supplemental Figure 6).
Figure 5. CCL2 enhances SRC and PKC activity associated with proliferation of basal-like breast cancer cells.
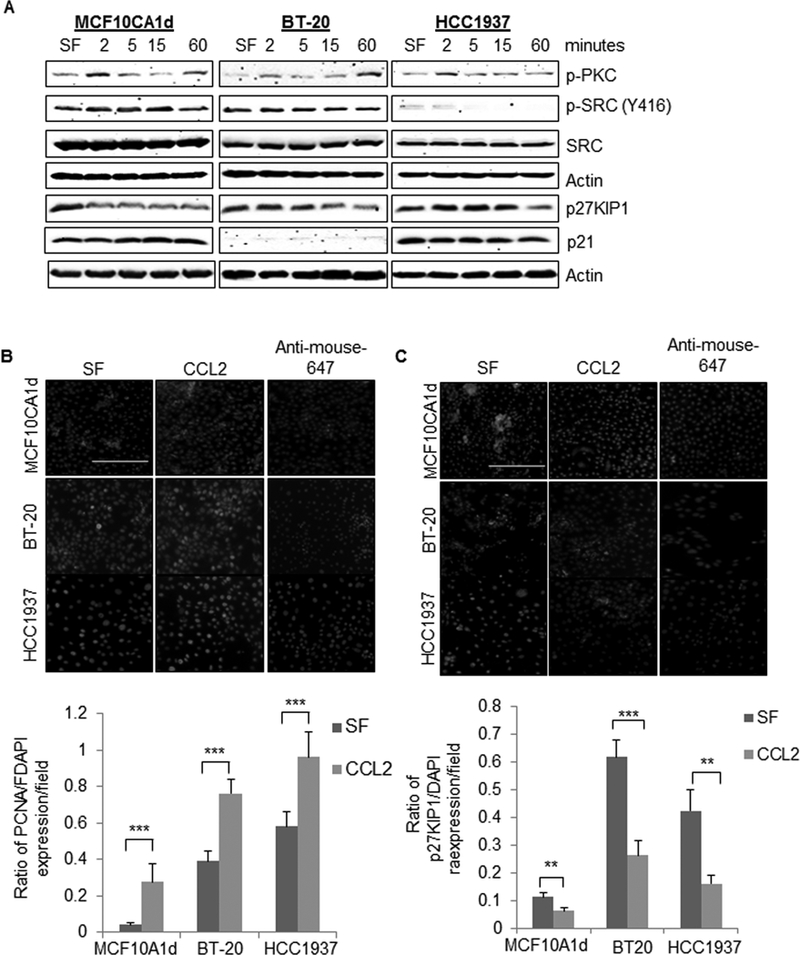
A. MCF10CA1d, BT-20 or HCC1937 cells were treated with or without 100 ng/ml CCL2 for up to 60 minutes and analyzed for expression of the indicated proteins by immunoblot. B-C. MCF10CA1d, BT-20 and HCC1937 breast cancer cells were incubated in serum free (SF) media in the presence or absence of 100 ng/ml CCL2 for 24 hours and analyzed for PCNA (B) or p27KIP1(C) expression by immunofluorescence staining. PCNA and p27 expression was quantified by Image J. Fluorescence intensity was normalized to DAPI. Ratios of PCNA and p27KIP1 staining/DAPI per field are shown. Representative images are shown with secondary antibody only control shown. Scale bar=200 microns. Statistical analysis was performed using Two Tailed T-test, comparing SF vs. CCL2 treatment. Statistical significance was determined by p<0.05. **p<0.01, ***p<0.001. Mean±SEM are shown.
The effects of CCL2 on BLBC cell growth were examined in BT-20 and HCC193 cells, which expressed comparable levels of CCR2 (Supplemental Figure 7). Like MCF10CA1d cells, CCL2 treatment of BT-20 and HCC1937 cells increased expression of phospho-SRC, phospho-PKC and PCNA (Figure 5A-B), and decreased p27KIP1 expression (Figure 5B-C). To examine the effects of CCL2 on cell cycle progression, breast cancer cells were synchronized through thymidine blocking, treated with CCL2 and analyzed by flow cytometry for propidium iodide staining. CCL2 enhanced the percentage of cells in G2/M phase after 2 hours for MCF10CA1d cells and 10 hours for BT-20 and HCC1937 cells (Figure 6A-D). CCR2 deficient MCF10CA1d cells showed a higher percentage of cells in G1/S phase and decreased percentage of cells in G2/M compared to the wildtype CCR2 control (Figure 6E). These data indicate that CCL2/CCR2 signaling promotes proliferation and cell cycle progression in BLBC cells.
Figure 6. CCL2 accelerates cell cycle progression of basal-like breast cancer cells.
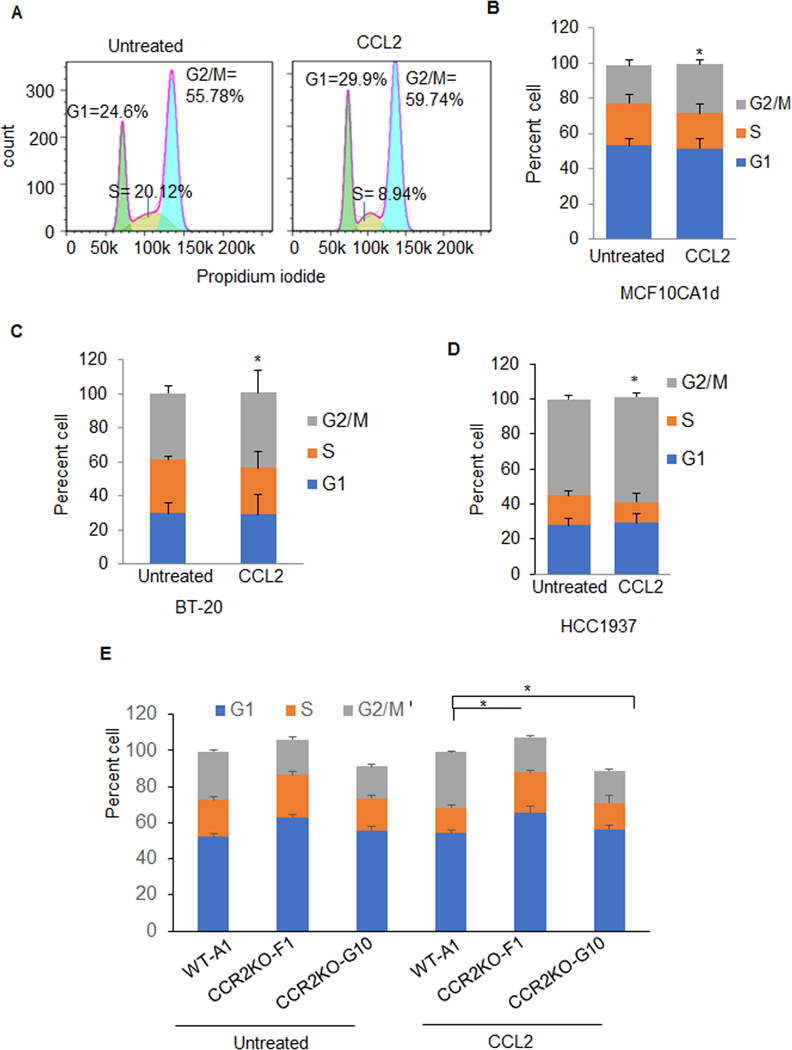
A. Breast cancer cells were synchronized by double thymidine blocking, and then treated with 100 ng/ml CCL2 in growth media for 2 hours (MCF10CA1d) or 10 hours (BT-20 and HCC1937). Cells were stained with propidium iodide and analyzed by flow cytometry. A. Example of histogram analysis showed for CCL2 treatment of HCC1937 cells. Stacked graphs for percentage of cells in G1, S and G2/M phases are shown for B. MCF10CA1d, C. BT-20, D. HCC1937 breast cancer cells or E. MCF10CA1d wildtype control (WT-A1) or F. CCR2 knockout cells (CCR2KO-F1, CCR2KO-G10). Statistical analysis was performed using Two Tailed T-test. Statistical significance was determined by p<0.05. *p<0.05, **p<0.01, ***p<0.001. Statistically significant differences are shown for G2/M in CCL2 vs. untreated for B-E. Mean±SEM are shown.
To determine the relevance of SRC and PKC to CCL2-induced cell growth, we treated breast cancer cells with PP2, a small molecule inhibitor that targets SRC kinases (38), or with the pan-PKC inhibitor Gö 6983 (39). MCF10CA1d cells were tested for responsiveness to PP2 at 10 and 20 μM, and Gö 6983 was tested at 5 and 10 μM, based on previous studies showing 40–50% inhibition of biological activity at these concentrations (40–43). PP2 inhibited CCL2 induced phospho-SRC similarly at both concentrations (Figure 7A). Gö 6983 increased phospho-PKC expression in untreated and CCL2-treated cells similarly at both concentrations (Figure 7A). The increased phospho-PKC expression is consistent with studies showing that Gö 6983 conformational binding to PKC inhibits de-phosphorylation, preventing degradation (44). PP2 or Gö 6983 treatment at lower concentrations inhibited CCL2 mediated expression of PCNA, enhanced expression p27KIP1 (Figure 7B-C). These data indicate that CCL2-mediated breast cancer cell growth through PKC- and SRC-dependent mechanisms.
Figure 7. SRC and PKC inhibition block CCL2 induced growth of basal-like breast cancer cells.
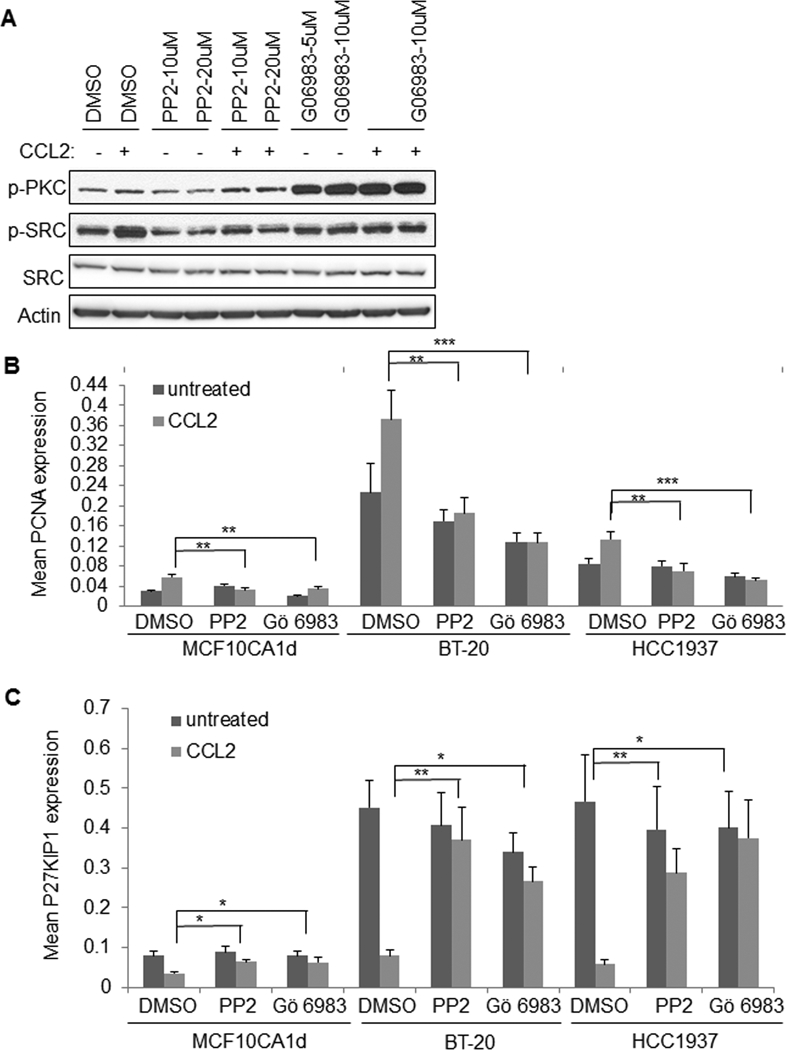
A. MCF10CA1d breast cancer cells were treated with 100 ng/ml CCL2 in the presence or absence of DMSO vehicle control, PP2 or Gö 6983 for 15 minutes and analyzed for expression of the indicated proteins by immunoblot. B-C. Breast cancer cells were treated with CCL2 in the presence or absence of 10 μM PP2 or 5 μM Gö 6983 for 24 hours and analyzed for PCNA (B) or p27KIP1 expression (C) by immunofluorescence staining. Statistical analysis was performed using One Way ANOVA with Bonferroni post-hoc comparison. Statistical significance was determined by p<0.05. *p<0.05, **p<0.01, ***p<0.001. Mean±SEM are shown.
Discussion
We report that stromal derived fibroblasts constitute a major source of CCL2, and regulates growth of BLBC by signaling directly to CCR2 expressing cancer cells. CCL2/CCR2-mediated tumor growth is regulated by PKC and SRC signaling pathways. As CCL2 and CCR2 are most highly expressed in BLBC, the proposed pathways may represent viable therapeutic targets for this subtype.
Breast fibroblasts increased MCF10CA1d breast cancer cell growth through CCL2-dependent mechanisms. CCL2 is not confined to the stroma, but is expressed in the epithelium of some breast tumors (10,11,14). The importance of the stroma may depend on the levels of epithelial CCL2. CCR2 knockout alone in MCF10CA1d cells had minimal effects on tumor growth. These breast cancer cells expressed much lower levels of CCL2, compared to the stroma. As CCR2 knockout inhibited fibroblast-mediated tumor growth, these studies indicate that MCF10CA1d cell growth depend on CCL2 expressing stroma.
CCL2-induction of SMAD3 and ERK1/2 regulates motility and survival of PyVmT and MCF-7 luminal mammary carcinoma cells (14). These cell lines did not show differences in cell proliferation with CCL2 treatment. Here, CCL2-enhanced proliferation but not survival in MCF10CA1d breast cancer cells, and was not associated with invasiveness of MCF10CA1d breast tumors. CCL2-induction of SRC and PKC is important for cell proliferation and cell cycle progression in multiple BLBC cell lines. While CCL2 activated PKC and SRC in MCF10CA1d cells, it did not affect ERK1/2 phosphorylation, a downstream effector of SRC in breast cancer cells mediated by estrogen and progesterone receptors (45–47). These studies suggest that CCL2 mediates PKC and SRC activity in some breast cancer cell lines uncoupled from ERK1/2 signaling. It is not clear why CCL2 induces cell proliferation only in some breast cancer cell lines. Mutations to PKC (48) in breast cancer cell lines could dictate responsiveness to CCL2 treatment. Effects of CCL2 or CCR2 on growth may depend on engagement of co-receptors such as CCR5 (49,50). Mutational and gene expression profiling studies in breast cancer cell lines may reveal further insight into the mechanisms responsible for CCL2 induced-mitogenesis.
CCL2 enhances stemness of some breast cancer cell lines through paracrine mechanisms (51). Here, data indicate that autocrine CCR2 signaling in breast cancer cells regulates stemness of MCF10CA1d cells. While MCF10CA1d express low levels of CCL2, these levels may sufficient for CCR2 mediated stem cell activity. Alternatively, CCR2 mediated stem cell activity could be regulated by CCL7 or CCL8 ligands (52,53), or CCR2 interactions with CCR5 and CXCR4 (49). Within the breast tumor, CCR2 overexpression and activity may sustain a pool of cancer stem cells. Increased bioavailability of CCL2 from the stroma could signal to CCR2+ cancer cells to enhance tumor growth.
CCL2 antibody neutralization inhibits tumor growth and metastasis of 4T1 and MDA-MB-231 breast tumor xenografts associated with decreased macrophage recruitment (12,13,54). Here, knockdown of stromal CCL2 or epithelial CCR2 expression inhibited MCF10CA1d and 4T1 mammary tumor growth and levels of M2 macrophages, but did not affect metastasis. Systemic antibody neutralization may inhibit CCL2 derived from multiple sources in the primary tumor and secondary tissues including lung (55,56), thereby blocking metastatic dissemination. As stromal CCL2 and epithelial CCR2 knockout was confined to the primary tumor, other sources of CCL2 could compensate to sustain metastasis. The importance of macrophages in breast metastasis is well-documented (57,58). As macrophages are heterogenous (59,60), CCL2/CCR2 signaling may regulate different subsets of macrophages important for tumor growth and metastasis.
CCL2/CCR2 signaling in breast cancer cells regulates the levels of M2 macrophages in primary MCF10CA1d and 4T1 tumors, indicating that CCL2 mediated tumor growth could be mediated through immune cells. While the use of immunocompromised mice prevented analysis of T cells in breast tumors, aggressive breast cancers are associated with elevated M2 macrophages and T regulatory cells, and decreased CD8+ cytotoxic T cells (61,62). M2 macrophages inhibit T cell proliferation and activity in tumors, thereby contributing to CD8+ T cell exhaustion in tumors (63,64). Studies from our laboratory support a relationship between CCR2-mediated tumor growth and immune cell activity. Targeting CCR2 expression in MMTV-PyVMT mice (FVB) inhibited tumor growth associated with decreased M2 macrophage polarization and increased cytotoxic CD8+T cell recruitment and activity (unpublished data). Since epithelial CCR2 expression was associated with M2 macrophages in the MCF10CA1d and 4T1 models, future studies could determine the contribution of tumoral CCR2 expression in T cell recruitment activity using syngeneic transgenic or transplantation models of BLBC.
We reveal an important in vivo contribution for CCL2/CCR2 signaling to breast cancer cells. The CCL2/CCR2 pathway is a therapeutic target of interest due to its significant effects on tumor growth and metastasis, but translation to therapy may be complex. One study indicated a rebound in tumor growth with cessation of CCL2 inhibitor treatment, increase macrophage recruitment and angiogenesis (65). However, this rebound may be due to neutralizing antibodies themselves, rather than CCL2 as a target (20). These present studies indicate that targeting CCL2 or CCR2 expression inhibits tumor growth and could provide a therapeutic benefit when combined with other therapies.
Supplementary Material
Implications:
This report sheds novel light on CCL2/CCR2 chemokine signaling as a mitogenic pathway and cell cycle regulator in breast cancer cells.
Acknowledgement
We thank: Gage Brummer, Diana Acevedo, Fang Fan, M.D/Ph.D, Timothy Fields, M.D/Ph.D and Joan Wambi, Ph.D for scientific discussion. We thank Kimal Rajapakshe,Ph.D and Cristian Coarfa,Ph.D for RPPA data processing. We thank Ms. Fuli Jia and Dr. Danli Wu (Antibody-based Proteomics Core/Shared Resource) for their technical assistance in RPPA experiments.
Grant support
This work was supported by the American Cancer Society (RSG-13–182-01-CSM) and National Institutes of Health (R01CA172764) to N Cheng, Cancer Prevention & Research Institute of Texas Proteomics & Metabolomics Core Facility Support Award (RP170005) and NCI Cancer Center Support Grant to Antibody-based Proteomics Core/Shared Resource (P30CA125123) to S Huang.
Footnotes
Competing Interests: The authors declare no competing interests.
References
- 1.DeSantis CE, Ma J, Goding Sauer A, Newman LA, Jemal A . Breast cancer statistics, 2017, racial disparity in mortality by state. CA: a cancer journal for clinicians 2017;67:439–48 [DOI] [PubMed] [Google Scholar]
- 2.Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A 2001;98:10869–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vasconcelos I, Hussainzada A, Berger S, Fietze E, Linke J, Siedentopf F, et al. The St. Gallen surrogate classification for breast cancer subtypes successfully predicts tumor presenting features, nodal involvement, recurrence patterns and disease free survival. Breast 2016;29:181–5 [DOI] [PubMed] [Google Scholar]
- 4.Alabdulkareem H, Pinchinat T, Khan S, Landers A, Christos P, Simmons R, et al. The impact of molecular subtype on breast cancer recurrence in young women treated with contemporary adjuvant therapy. Breast J 2018;24:148–53 [DOI] [PubMed] [Google Scholar]
- 5.Lee A, Djamgoz MBA. Triple negative breast cancer: Emerging therapeutic modalities and novel combination therapies. Cancer treatment reviews 2018;62:110–22 [DOI] [PubMed] [Google Scholar]
- 6.Yao M, Brummer G, Acevedo D, Cheng N. Cytokine Regulation of Metastasis and Tumorigenicity. Adv Cancer Res 2016;132:265–367 [DOI] [PubMed] [Google Scholar]
- 7.Lacalle RA, Blanco R, Carmona-Rodriguez L, Martin-Leal A, Mira E, Manes S. Chemokine Receptor Signaling and the Hallmarks of Cancer. Int Rev Cell Mol Biol 2017;331:181–244 [DOI] [PubMed] [Google Scholar]
- 8.O’Connor T, Borsig L, Heikenwalder M. CCL2-CCR2 Signaling in Disease Pathogenesis. Endocr Metab Immune Disord Drug Targets 2015;15:105–18 [DOI] [PubMed] [Google Scholar]
- 9.Valkovic T, Lucin K, Krstulja M, Dobi-Babic R, Jonjic N. Expression of monocyte chemotactic protein-1 in human invasive ductal breast cancer. Pathology, research and practice 1998;194:335–40 [DOI] [PubMed] [Google Scholar]
- 10.Fujimoto H, Sangai T, Ishii G, Ikehara A, Nagashima T, Miyazaki M, et al. Stromal MCP-1 in mammary tumors induces tumor-associated macrophage infiltration and contributes to tumor progression. Int J Cancer 2009;125:1276–84 [DOI] [PubMed] [Google Scholar]
- 11.Yao M, Yu E, Staggs V, Fan F, Cheng N. Elevated expression of chemokine C-C ligand 2 in stroma is associated with recurrent basal-like breast cancers. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc 2016:810–23 [DOI] [PubMed] [Google Scholar]
- 12.Hembruff SL, Jokar I, Yang L, Cheng N. Loss of transforming growth factor-beta signaling in mammary fibroblasts enhances CCL2 secretion to promote mammary tumor progression through macrophage-dependent and -independent mechanisms. Neoplasia 2010;12:425–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011;475:222–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fang WB, Jokar I, Zou A, Lambert D, Dendukuri P, Cheng N. CCL2/CCR2 chemokine signaling coordinates survival and motility of breast cancer cells through Smad3 protein- and p42/44 mitogen-activated protein kinase (MAPK)-dependent mechanisms. J Biol Chem 2012;287:36593–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fang WB, Jokar I, Chytil A, Moses HL, Abel T, Cheng N. Loss of one Tgfbr2 allele in fibroblasts promotes metastasis in MMTV: polyoma middle T transgenic and transplant mouse models of mammary tumor progression. Clin Exp Metastasis 2011;28:351–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Santner SJ, Dawson PJ, Tait L, Soule HD, Eliason J, Mohamed AN, et al. Malignant MCF10CA1 cell lines derived from premalignant human breast epithelial MCF10AT cells. Breast Cancer Res Treat 2001;65:101–10 [DOI] [PubMed] [Google Scholar]
- 17.Strickland LB, Dawson PJ, Santner SJ, Miller FR. Progression of premalignant MCF10AT generates heterogeneous malignant variants with characteristic histologic types and immunohistochemical markers. Breast Cancer Res Treat 2000;64:235–40 [DOI] [PubMed] [Google Scholar]
- 18.Lambert D, Cheng N. Mammary transplantation of stromal cells and carcinoma cells in C57BL/6 mice. Journal of Visualized Experiments 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O’Hare MJ, Bond J, Clarke C, Takeuchi Y, Atherton AJ, Berry C, et al. Conditional immortalization of freshly isolated human mammary fibroblasts and endothelial cells. Proc Natl Acad Sci U S A 2001;98:646–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yao M, Smart C, Hu Q, Cheng N. Continuous Delivery of Neutralizing Antibodies Elevate CCL2 Levels in Mice Bearing MCF10CA1d Breast Tumor Xenografts. Transl Oncol 2017;10:734–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brummelkamp TR, Bernards R, and Agami R A system for stable expression of short interfering RNAs in mammalian cells. Science 2002;296:550–3 [DOI] [PubMed] [Google Scholar]
- 22.Feoktistova M, Geserick P, Leverkus M. Crystal Violet Assay for Determining Viability of Cultured Cells. Cold Spring Harb Protoc; 2016;2016:pdb prot087379 [DOI] [PubMed] [Google Scholar]
- 23.Chang CH, Zhang M, Rajapakshe K, Coarfa C, Edwards D, Huang S, et al. Mammary Stem Cells and Tumor-Initiating Cells Are More Resistant to Apoptosis and Exhibit Increased DNA Repair Activity in Response to DNA Damage. Stem Cell Reports 2015;5:378–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012;486:346–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pereira B, Chin SF, Rueda OM, Vollan HK, Provenzano E, Bardwell HA, et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nature communications 2016;7:11479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miller FR. Xenograft models of premalignant breast disease. J Mammary Gland Biol Neoplasia 2000;5:379–91 [DOI] [PubMed] [Google Scholar]
- 27.Miller FR, Santner SJ, Tait L, Dawson PJ. MCF10DCIS.com xenograft model of human comedo ductal carcinoma in situ. J Natl Cancer Inst 2000;92:1185–6 [DOI] [PubMed] [Google Scholar]
- 28.Gallina G, Dolcetti L, Serafini P, De Santo C, Marigo I, Colombo MP, et al. Tumors induce a subset of inflammatory monocytes with immunosuppressive activity on CD8+ T cells. J Clin Invest 2006;116:2777–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Daley JM, Thomay AA, Connolly MD, Reichner JS, Albina JE. Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. J Leukoc Biol 2008;83:64–70 [DOI] [PubMed] [Google Scholar]
- 30.Lee AH, Happerfield LC, Bobrow LG, Millis RR. Angiogenesis and inflammation in invasive carcinoma of the breast. Journal of clinical pathology 1997;50:669–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aslakson CJ, Miller FR. Selective events in the metastatic process defined by analysis of the sequential dissemination of subpopulations of a mouse mammary tumor. Cancer Res 1992;52:1399–405 [PubMed] [Google Scholar]
- 32.O’Brien CA, Kreso A, Jamieson CH. Cancer stem cells and self-renewal. Clin Cancer Res 2010;16:3113–20 [DOI] [PubMed] [Google Scholar]
- 33.Yang X, Wang H, Jiao B. Mammary gland stem cells and their application in breast cancer. Oncotarget 2017;8:10675–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roca H, Varsos Z, Pienta KJ. CCL2 protects prostate cancer PC3 cells from autophagic death via phosphatidylinositol 3-kinase/AKT-dependent survivin up-regulation. J Biol Chem 2008;283:25057–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao R, Pei GX, Cong R, Zhang H, Zang CW, Tian T. PKC-NF-kappaB are involved in CCL2-induced Nav1.8 expression and channel function in dorsal root ganglion neurons. Biosci Rep 2014;34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bouayad D, Pederzoli-Ribeil M, Mocek J, Candalh C, Arlet JB, Hermine O, et al. Nuclear-to-cytoplasmic relocalization of the proliferating cell nuclear antigen (PCNA) during differentiation involves a chromosome region maintenance 1 (CRM1)-dependent export and is a prerequisite for PCNA antiapoptotic activity in mature neutrophils. J Biol Chem 2012;287:33812–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao D, Besser AH, Wander SA, Sun J, Zhou W, Wang B, et al. Cytoplasmic p27 promotes epithelial-mesenchymal transition and tumor metastasis via STAT3-mediated Twist1 upregulation. Oncogene 2015;34:5447–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hanke JH, Gardner JP, Dow RL, Changelian PS, Brissette WH, Weringer EJ, et al. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. J Biol Chem 1996;271:695–701 [DOI] [PubMed] [Google Scholar]
- 39.Gschwendt M, Dieterich S, Rennecke J, Kittstein W, Mueller HJ, Johannes FJ. Inhibition of protein kinase C mu by various inhibitors. Differentiation from protein kinase c isoenzymes. FEBS Lett 1996;392:77–80 [DOI] [PubMed] [Google Scholar]
- 40.Liu X, Feng R. Inhibition of epithelial to mesenchymal transition in metastatic breast carcinoma cells by c-Src suppression. Acta Biochim Biophys Sin (Shanghai) 2010;42:496–501 [DOI] [PubMed] [Google Scholar]
- 41.Nam JS, Ino Y, Sakamoto M, Hirohashi S. Src family kinase inhibitor PP2 restores the E-cadherin/catenin cell adhesion system in human cancer cells and reduces cancer metastasis. Clin Cancer Res 2002;8:2430–6 [PubMed] [Google Scholar]
- 42.Kennett SB, Roberts JD, Olden K. Requirement of protein kinase C micro activation and calpain-mediated proteolysis for arachidonic acid-stimulated adhesion of MDA-MB-435 human mammary carcinoma cells to collagen type IV. J Biol Chem 2004;279:3300–7 [DOI] [PubMed] [Google Scholar]
- 43.Holden NS, Squires PE, Kaur M, Bland R, Jones CE, Newton R. Phorbol ester-stimulated NF-kappaB-dependent transcription: roles for isoforms of novel protein kinase C. Cell Signal 2008;20:1338–48 [DOI] [PubMed] [Google Scholar]
- 44.Gould CM, Antal CE, Reyes G, Kunkel MT, Adams RA, Ziyar A, et al. Active site inhibitors protect protein kinase C from dephosphorylation and stabilize its mature form. J Biol Chem 2011;286:28922–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Faivre EJ, Lange CA. Progesterone receptors upregulate Wnt-1 to induce epidermal growth factor receptor transactivation and c-Src-dependent sustained activation of Erk1/2 mitogen-activated protein kinase in breast cancer cells. Mol Cell Biol 2007;27:466–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu X, Du L, Feng R. c-Src regulates cell cycle proteins expression through protein kinase B/glycogen synthase kinase 3 beta and extracellular signal-regulated kinases 1/2 pathways in MCF-7 cells. Acta Biochim Biophys Sin (Shanghai) 2013;45:586–92 [DOI] [PubMed] [Google Scholar]
- 47.Migliaccio A, Di Domenico M, Castoria G, de Falco A, Bontempo P, Nola E, et al. Tyrosine kinase/p21ras/MAP-kinase pathway activation by estradiol-receptor complex in MCF-7 cells. EMBO J 1996;15:1292–300 [PMC free article] [PubMed] [Google Scholar]
- 48.Antal CE, Hudson AM, Kang E, Zanca C, Wirth C, Stephenson NL, et al. Cancer-associated protein kinase C mutations reveal kinase’s role as tumor suppressor. Cell 2015;160:489–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sohy D, Yano H, de Nadai P, Urizar E, Guillabert A, Javitch JA, et al. Hetero-oligomerization of CCR2, CCR5, and CXCR4 and the protean effects of “selective” antagonists. J Biol Chem 2009;284:31270–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Azenshtein E, Luboshits G, Shina S, Neumark E, Shahbazian D, Weil M, et al. The CC chemokine RANTES in breast carcinoma progression: regulation of expression and potential mechanisms of promalignant activity. Cancer Res 2002;62:1093–102 [PubMed] [Google Scholar]
- 51.Tsuyada A, Chow A, Wu J, Somlo G, Chu P, Loera S, et al. CCL2 mediates cross-talk between cancer cells and stromal fibroblasts that regulates breast cancer stem cells. Cancer Res 2012;72:2768–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kurihara T, Bravo R. Cloning and functional expression of mCCR2, a murine receptor for the C-C chemokines JE and FIC. J Biol Chem 1996;271:11603–7 [DOI] [PubMed] [Google Scholar]
- 53.Vande Broek I, Asosingh K, Vanderkerken K, Straetmans N, Van Camp B, Van Riet I. Chemokine receptor CCR2 is expressed by human multiple myeloma cells and mediates migration to bone marrow stromal cell-produced monocyte chemotactic proteins MCP-1, −2 and −3. Br J Cancer 2003;88:855–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lu X, Kang Y. Chemokine (C-C motif) ligand 2 engages CCR2+ stromal cells of monocytic origin to promote breast cancer metastasis to lung and bone. J Biol Chem 2009;284:29087–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nanua S, Zick SM, Andrade JE, Sajjan US, Burgess JR, Lukacs NW, et al. Quercetin blocks airway epithelial cell chemokine expression. Am J Respir Cell Mol Biol 2006;35:602–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kitamura T, Qian BZ, Soong D, Cassetta L, Noy R, Sugano G, et al. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. J Exp Med 2015;212:1043–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Goswami S, Sahai E, Wyckoff JB, Cammer M, Cox D, Pixley FJ, et al. Macrophages promote the invasion of breast carcinoma cells via a colony-stimulating factor-1/epidermal growth factor paracrine loop. Cancer Res 2005;65:5278–83 [DOI] [PubMed] [Google Scholar]
- 58.Lin EY, Nguyen AV, Russell RG, Pollard JW. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J Exp Med 2001;193:727–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol 2011;11:723–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Elliott LA, Doherty GA, Sheahan K, Ryan EJ. Human Tumor-Infiltrating Myeloid Cells: Phenotypic and Functional Diversity. Front Immunol 2017;8:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dongre A, Rashidian M, Reinhardt F, Bagnato A, Keckesova Z, Ploegh HL, et al. Epithelial-to-Mesenchymal Transition Contributes to Immunosuppression in Breast Carcinomas. Cancer Res 2017;77:3982–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Miyan M, Schmidt-Mende J, Kiessling R, Poschke I, de Boniface J. Differential tumor infiltration by T-cells characterizes intrinsic molecular subtypes in breast cancer. Journal of translational medicine 2016;14:227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Huber S, Hoffmann R, Muskens F, Voehringer D. Alternatively activated macrophages inhibit T-cell proliferation by Stat6-dependent expression of PD-L2. Blood 2010;116:3311–20 [DOI] [PubMed] [Google Scholar]
- 64.Peranzoni E, Lemoine J, Vimeux L, Feuillet V, Barrin S, Kantari-Mimoun C, et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc Natl Acad Sci U S A 2018;115:E4041–E50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bonapace L, Coissieux MM, Wyckoff J, Mertz KD, Varga Z, Junt T, et al. Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature 2014;515:130–3 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.