

Abstract
Isocitrate dehydrogenase 1 (IDH1) is the most commonly mutated metabolic enzyme in human malignancy. A heterozygous genetic alteration, arginine 132, promotes the conversion of α-ketoglutarate to D-2-hydroxyglutarate (2-HG). While pharmacologic inhibitors of mutant IDH1 are promising, resistance mechanisms to targeted therapy are not understood. Additionally, the role of wild-type IDH1 (WT.IDH1) in cancer requires further study. Recently it was observed that the regulatory RNA-binding protein, HuR (ELAVL1), protects nutrient deprived cancer cells without IDH1 mutations, by stabilizing WT.IDH1 transcripts. In the present study, a similar regulatory effect on both mutant (Mut.IDH1) and WT.IDH1 transcripts in heterozygous IDH1 mutant tumors is observed. In ribonucleoprotein immunoprecipitation assays of IDH1 mutant cell lines, wild-type and mutant IDH1 mRNAs each bound to HuR. Both isoforms were profoundly down-regulated at the mRNA and protein levels after genetic suppression of HuR (siRNAs or CRISPR deletion) in HT1080 (R132C IDH1 mutation) and BT054 cells (R132H). Proliferation and invasion were adversely affected after HuR suppression and metabolomic studies revealed a reduction in Pentose Phosphate Pathway metabolites, nucleotide precursors, and 2-HG levels. HuR-deficient cells were especially sensitive to stress, including low glucose conditions or a mutant IDH1 inhibitor (AGI-5198). IDH1 mutant cancer cells were rescued by WT.IDH1 overexpression to a greater extent than Mut.IDH1 overexpression under these conditions. This study reveals the importance of HuR’s regulation of both mutant and wild-type IDH1 in tumors harboring a heterozygous IDH1 mutation with implications for therapy.
Keywords: HuR; IDH1; CRISPR/Cas9; 2-hydroxyglutarate; AGI-5198, IDH1-mutated cancer
Introduction
Isocitrate dehydrogenase 1 (IDH1) is the most commonly mutated metabolic enzyme in human cancer. Gain-of-function mutations occur in the majority of secondary glioblastomas and low grade gliomas (>80%), sarcomas (e.g., chondrosarcomas and fibrosarcomas, 55%), intrahepatic cholangiocarcinomas (20%), acute myelogenous leukemia (20%), angioimmunoblastic T-cell lymphoma (20%), melanoma (10%), and anaplastic thyroid cancer (10%) (1, 2). Together, more than 25,000 cancers are diagnosed each year with IDH1 mutations, indicating that there is an urgent need to develop effective therapies (1).
In virtually all cases, the IDH1 mutation is a heterozygous missense substitution at arginine 132, creating an altered catalytic pocket. Most commonly a histidine is substituted at this position (R132H; CGT -> CAT), but other changes have been reported (R132C, R132G, R132S, and R132L)(3). The abnormal enzyme diverts α-ketoglutarate into a non-canonical reductive pathway requiring NADPH. The reaction produces the oncometabolite, D-2-hydroxyglutarate (2-HG) (Figure. 1A)(4). Increased 2-HG produced by mutant IDH1 impairs cellular differentiation by inhibiting the TET family demethylating enzymes leading to increased DNA methylation (5). Histone methylation is also increased due to a reduction in Jumonji C domain histone demethylase activity. In contrast, the wild type isoenzyme catalyzes the interconversion of isocitrate and α-ketoglutarate. NADPH is either produced or consumed depending on the direction of the wild type IDH1 reaction. Under metabolic stress (e.g., hypoxia, nutrient deprivation), oxidative decarboxylation is likely favored to generate more NADPH for antioxidant defense (Figure. 1A)(6–8).
Figure 1: HuR is required for cell proliferation and invasion in IDH1 mutated cancer cells.
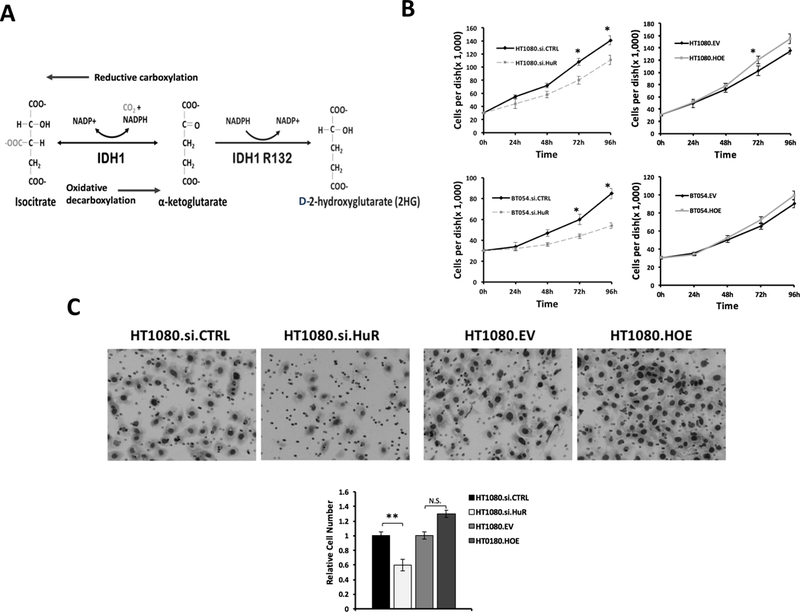
A, Schematic of the WT.IDH1 and Mut.IDH1 catalytic reaction. B, Cell growth (PicoGreen, dsDNA content) of HT1080 and BT054 after HuR knockdown (si.HuR) or overexpression (HOE) compared with control (si.CTRL or EV) for indicated time points. Each data point represents the mean of 5 independent experiments ± standard error of the mean (SEM). *, p < 0.05. C, Representative images of Matrigel invasion assays performed in HT1080 cells after HuR silencing (si.HuR) or overexpression (HOE). Cells that invaded through the Matrigel and onto the basal surface of transwell inserts were stained and photographed at 20X magnification. Quantification of cell number is represented by the bar graph. Data are presented as mean -fold change in invaded cells relative to si.CTRL or EV. Each bar represents the mean of 3 independent experiments ± SEM. N.S. non-significant; **, p < 0.005.
While the biologic impact of mutant IDH1 is well described, the importance of wild type IDH1 (WT.IDH1) in cancer biology is also recognized, particularly in the context of severe metabolic stress (8–14). These observations may have important implications for IDH1 mutant (Mut.IDH1) tumors since tumors with this aberrant genotype only express half the usual transcript dose of WT.IDH1 (with only a single copy of wild type IDH1 per cancer cell). It makes sense that this deficiency in WT.IDH1 expression sensitizes Mut.IDH1 tumors to metabolic stress and chemotherapy (13, 15). In fact, IDH1 mutant tumors are less necrotic, less hypoxic, and less aggressive than their wild type IDH1 counterparts, providing further indirect evidence that WT.IDH1 contributes to cancer virulence. Not surprisingly, Mut.IDH1 tumors are associated with improved long-term outcomes (2, 3). These observations do not discount that Mut.IDH1 has oncogenic properties, but rather suggests that the tumorigenic changes may come with at a cost of impaired adaptative capabilities under metabolic stress. We speculate that in certain scenarios (e.g., nutrient withdrawal or chemotherapy), Mut.IDH1 tumors may suffer from haploinsufficiency of the wild type allele. If true, then both IDH1 alleles would be important therapeutic targets in tumors harboring a heterozygous gain-of-function IDH1 mutation.
We recently reported that the RNA stability factor, HuR (ELAVL1), regulates WT.IDH1 transcript expression in pancreatic cancer (which do not typically acquire IDH1 mutations)(16). Mechanistically, the RNA binding protein impacts mRNA expression by first recognizing and binding AU-rich RNA elements (AREs) within specific mRNA 3’ untranslated regions (3’ UTRs)(17). HuR is ubiquitously expressed in cancer cell nuclei. Under stress, the pro-survival regulatory protein translocates to the cytoplasm with bound mRNAs, stabilizes transcripts, and facilitates protein translation(18). While 5% of transcripts have HuR binding sites (19), transcripts that are especially important for cancer cell survival and adaptation to stress are enriched with HuR binding sequences (20). Our prior work identified three separate and biologically relevant HuR binding sequences within a 153-base pair region in the WT.IDH1 3’-untranslated region (UTR) (11). We showed that HuR-deficient pancreatic cancer cells almost completely lost WT.IDH1 expression, and this loss impaired cell viability under stress. WT.IDH1 overexpression completely restored pancreatic cancer viability in both in vitro and in vivo models of HuR-deficient cancer.
Since mutant and wild type IDH1 transcripts have identical 3’UTR sequences, we hypothesize here that HuR regulates both of these enzymes in heterozygous IDH1 mutant tumors. Further, this regulatory interaction likely plays an important role in mutant IDH1 cancer cell survival under relevant stressors, such as glucose withdrawal or mutant IDH1 inhibition. This work, if validated, would prioritize HuR, as well as the wild type IDH1 isoenzyme, as additional therapeutic targets in IDH1-mutant tumors.
Materials and Methods
Cell lines and cell culture
HT1080, a fibrosarcoma cell line with a heterozygous IDH1 R132C mutation, was obtained from the American Type Culture Collection (Manassas, VA, 2014) and authenticated in our laboratory. Mutational analysis for the IDH1 gene was performed by PCR amplification and Sanger sequencing of DNA. Briefly, DNA was isolated from 3 million cells using the DNeasy blood and tissue kit (Qiagen) according to the manufacturer’s protocol. A portion of the IDH1 gene exon 4 containing the Arg132 was amplified using two pairs of primers: IDH1F1: 5’CGGTCTTCAGAGAAGCCATT; IDH1R1: 5’GCAAAATCACATTATTGCCAAC IDH1F2: 5’ACCAAATGGCACCATACGA; IDH1R2: 5’TTCATACCTTGCTTAATGGGTGT. The PCR products were gel-purified using QIAquick PCR purification Kit (Qiagen) and sequenced using one of the amplification primers. Under standard culture conditions, DMEM (containing 25 mM glucose) was used. Media was supplemented with 10% fetal bovine serum, 1% L-glutamine (200 mM), and 1% penicillin/streptomycin (Invitrogen). An oligodendroglioma cell line, BT054, was kindly provided by Dr. Samuel Weiss (University of Calgary)(21), and authenticated to reveal a heterozygous IDH1 R132H mutation. IDH1 mutational status was further verified by Western blotting with an antibody against IDH1-R132H (MilliporeSigma; MABC 171; 1:500). The cells are grown in media permissive of neural stem cell growth as previously described (22–24). Briefly, for BT054 maintenance, cells at passage 12 were seeded in 6-well plates at 200,000 cells per well. The next day, proliferation medium (DMEM plus 10% FCS) was replaced with neural stem cell medium made from serum-free DMEM supplemented with B27 and N2 supplements (Invitrogen), bFGF(20 ng/mL, R&D Systems), EGF(20 ng/mL, PeproTech) and PDGFAA (20 ng/mL, PeproTech). Medium was replaced every 2–3 days.
In the cell line model, 5 mM glucose was typically used to simulate nutrient withdrawal, since cells were adapted to supraphysiologic glucose levels (25 mM) at baseline. Notably, when cells were cultured at 5 mM glucose over a multi-day experiment without media changes, glucose levels declined into the sub-physiologic range (25). All cell lines were tested routinely and prior to all metabolomic analyses, for mycoplasma contamination and grown at 37 °C and 5% CO2. For in vitro assays, experiments were performed in triplicate. Specific parental lines used, descendant clones harboring genetic modifications, and relevant cell line nomenclature for this manuscript are summarized in Table S1.
CRISPR/Cas9-mediated knockout of HuR in HT1080 cells was accomplished using a guide RNA targeting HuR, fused with CRISPR/Cas9 and GFP protein as previously described (26). cDNA plasmids and the CRISPR Universal Negative Control plasmid (CRISPR06–1EA) were purchased from Sigma-Aldrich. HuR knockout cells are designated herein as HuR −/− (Table S1). Stable cell line cultures with IDH1 overexpression were generated in HT1080 −/− cells by transducing IDH1 cDNA plasmids. Retroviral vectors were used to generate stable WT.IDH1 and Mut.IDH1 overexpression. Plasmids (pBABE-puro-WT.IDH1 and pBABE-puro-.Mut.IDH1.R132C) were generously provided by Kun-Liang Guan (Moores Cancer Center, University of California, San Diego, CA)(27). Scrambled pBABE-puro was used as a negative control plasmid for stable transfections (Addgene; 1764).
Cell proliferation and Drug sensitivity assays
AGI-5198 is a mutant IDH1 inhibitor with activity against IDH1 harboring R132H and R132C mutations (28, 29). The inhibitor was purchased from Selleckchem and dissolved in DMSO. Cells were plated in 96-well plates at 103 cells per well, and treated after 24 hours with AGI-5198 at various concentrations. Cells were assayed for DNA quantitation as a marker of cell viability using the Quant-it PicoGreen® dsDNA assay kit (Invitrogen) at 5 days as previously described(30). Fluorescence intensity was measured by a microplate reader (Tecan) using an excitation wavelength of 485 nm and emission wavelength of 535 nm. Data were plotted relative to day 0 to provide estimates of cell proliferation based on dsDNA content. To estimate cell death, cells were trypsinized every 24 hours and counted after Trypan blue staining (Invitrogen) with a Hausser bright-line hemocytometer (Fisher Scientific).
Soft agar colony formation assay
The experiment was performed as previously described (31). Briefly, the base agarose layer was prepared in 6-well culture plates by pouring 2 ml of base agarose mixture comprised of 1X DMEM, 10% FBS, and 0.5% agarose (Affymetrix). Cells were then prepared in the top agarose mixture comprised of 1X DMEM, 10% FBS, and 0.35% agarose, and poured over the solidified base agarose layer at a final seeding density of 5,000 cells/well. After solidification of the top layer, 2 ml of 25mM or 5mM glucose media (with or without AGI-5198(0.3 μM)) were added to each well, and samples were placed in a 37°C incubator. Cells incubated for 4 weeks, with the overlaid media and AGI-5198 exchanged thrice weekly. At the termination of the experiment, samples were rinsed twice with DPBS and fixed with 3.7% formaldehyde (Sigma-Aldrich) for 10 minutes. After two more washes with PBS, cells were stained with 0.01% crystal violet (Santa Cruz Biotechnology) for 1 hour. Pictures were taken using Floid Cell Imaging Station (Life Technologies) and colonies were counted using ImageJ (http://imagej.nih.gov/ij/)).
Matrigel invasion assay
Permeable supports (Corning, 353097) were coated with 200 μg/ml Matrigel basement membrane matrix (Corning, 354234), and inserted in 24-well companion plates (Corning, 353504). Cell suspensions were prepared in serum-free DMEM, and seeded in invasion chambers at 25,000 cells/chamber. DMEM with 20% FBS was added to each well as a chemoattractant. Samples were incubated at 37°C for 24 hours to allow for cell migration through the Matrigel. Non-invading cells on the apical surface of the Matrigel-coated supports were removed with cotton swabs, and cells that had migrated to the lower surface of the supports were stained using the Differential Quik Stain Kit (Polysciences). Photographs were taken, and cells were counted using ImageJ.
Small RNA interference, cDNA transfections
Cells were plated to 60% confluency in 6-well plates, and transient HuR overexpression(1μg) and (1 μM) siRNA transfections were performed using Lipofectamine 2000 (Invitrogen) and Optimen (Invitrogen) according to manufacturer’s protocol as previously described (31–33). Cells were treated and analyzed, as described, 48 hours after transfections. Overexpression (OE) and empty vector (EV) plasmids were purchased from OriGene Technologies (pCMV6-XL5; SC116430). Small interfering RNA (siRNA) oligos were purchased from Life Technologies (Grand Island, NY; siCTRL (control siRNA, AM4635).
Whole cell extracts and SDS-PAGE/Western blotting
Cytoplasmic and nuclear extracts were isolated using the NE-PER Nuclear and Cytoplasmic Extraction Kit (Thermo-Scientific) as per manufacturer’s instructions. Whole cell lysates were isolated using RIPA lysis buffer (Invitrogen) by incubating the cell pellets on ice for 10 minutes followed by centrifugation at 13,000*g for 15 minutes at 4°C as previously described(31) . Samples were mixed 4:1 with 5X Laemmli buffer and boiled for 5 minutes. Proteins were measured with Pierce BCA kit (ThermoFisher) and approximately 50 μg of protein was then separated using a 12% Bis-Tris polyacrylamide gel and transferred to a PVDF membrane (Invitrogen). Membranes were probed with antibodies against HuR (Santa Cruz Biotechnologies; 5261 clone 3A2), IDH1 (Abcam; ab 184615), IDH1-R132H (MilliporeSigma; MABC 171), Myc-Tag (Cell Signaling Technology; 2272), GAPDH (Cell Signaling Technology; 5014), Lamin A/C (Cell Signaling Technology; 2032) and α-Tubulin (Invitrogen; 32–2500).
Immunofluorescence
Approximately 5,000 cells per well were plated on coverslips in 24 well plates. After appropriate treatments, cells were fixed with 3.7% paraformaldehyde for 10 min, permeabilized with 0.1% Triton-X 100 for 30 min, blocked with 5% goat serum for 1 hour at room temperature and incubated with primary antibody (HuR; Santa Cruz Biotechnologies; 5261 clone 3A2; 1:200) overnight at 4°C. Alexa Fluor 488 F anti-mouse secondary antibody was applied to coverslips for 1 hour the following day, nuclei were stained with 4’,6’-diamidino-2-phenylindole (DAPI) and mounted (ProLong Gold, Life Technologies) for analysis with a Zeiss LSM-510 Confocal Laser Microscope. All images were taken at 40X magnification.
RNP-IP and qPCR
Cells were plated at 50% confluency in 100 mm dishes. The following day, immunoprecipitation was performed using either anti-HuR or IgG control antibodies as previously described (25). RNA quantitation (RT-qPCR) was performed using standard methodology. Total RNA was extracted using RNAeasy kit (Qiagen) according to manufacturer’s instructions. cDNA was synthesized from 1 μg of total RNA using TagMan probes (HuR, 00171309; IDH1, 00271858; 18s, 99999901, Life Technologies) and MultiScribe Reverse Transcriptase (Life Technologies). RT-qPCR analysis was performed using the Applied Biosystems 7500 Fast Real-Time PCR System (Life Technologies) and TagMan RT-PCR Master Mix (Life Technologies). Fold changes in mRNA expression were normalized according to the ΔΔCt method, as previously described(34).
Metabolite extraction and MS sample preparation
Metabolite extraction was performed as described earlier (35), with some modifications. After confirming 80% confluence of the cells, we replaced the media with fresh media for 2 h before metabolite extraction. Media was aspirated and the cells were washed twice with PBS to remove residual media before lysing the cells. The polar metabolites were then extracted with cryogenically cold 80% methanol/water mixture. LC/MS grade water (Fisher Scientific, Pittsburgh, PA, USA) and LC/MS grade methanol (Fisher Scientific, Pittsburgh, PA, USA) were utilized. The cells from the cold plates were scraped with a cell scraper, pipetted into an Eppendorf tube, and centrifuged at 13,000 rpm for 5 min to collect the supernatant. Samples were dried using a speed vacuum evaporator (Savant Speed Vac® Plus, Thermo Electron Corporation, USA) to evaporate the methanol and lyophilized using freeze dry system (Labconco, Kansas City, USA) to remove water. The dried sample was then prepared for mass spectrometry by dissolving in LC/MS grade water. Liquid chromatography/tandem mass spectrometry (LC-MS/MS) was performed as described below.
As a separate measure of D-2-hydroxyglutarate (D2-HG) levels, the metabolite was independently measured using colorimetric method (BioVision; K213–100), as per the manufacturer’s instructions.
LC-MS/MS experiment and analysis
Lyophilized concentrates were resuspended in equal volumes of LC/MS grade water and subjected to LC-MS/MS analysis, using a single reaction monitoring (SRM) method by utilizing XevoTQ-S Triple quadrupole mass spectrometer (Waters Inc.), as described previously (36). Data acquisition was carried out using MassLynx software (Waters Inc.), and peaks were integrated with TargetLynx (Waters Inc.). Peak areas were normalized with the respective protein concentrations and the resultant peak areas were subjected to relative quantification analyses with MetaboAnalyst 2.0 (37).
Xenograft studies
All experiments involving mice were approved by the Thomas Jefferson University Institutional Animal Care Regulations and Use Committee. Six-week-old, male, athymic nude mice (Nude-Foxn1nu) were purchased from Harlan Laboratories (6903M). HT1080 cells, or genetically modified variants of this cell line are summarized in Table S1, were prepared in 100 μl solution comprised of 70% DPBS and 30% Matrigel. Suspensions of 5 × 106 cells were then injected subcutaneously into the left and right flanks of mice. Tumor volumes were measured three times per week using a caliper (Volume = Length × Width2/2), along with body weights.
AGI-5198 was suspended in 0.5% methylcellulose and 0.2% Tween 80 at 30 mg/mL, and delivered orally (450 mg/kg). Treatment was initiated once the tumor diameter reached 50 mm3, for 28 days. Upon termination of the experiment, mice were euthanized using carbon dioxide inhalation followed by cervical dislocation, and tumors were harvested.
Statistical analysis
Statistical analyses were performed using a two-tailed unpaired Student’s t-test. P values <0.05 were considered statistically significant. The IC50 values were calculated by GraphPad software and a non-linear fit curve model.
Results
HuR promotes cell proliferation and invasion in Mut.IDH1 cancer cells
Previous studies showed that HuR promotes proliferation in cancer cells(20, 38), including wild type gliomas(39, 40). Here, we investigated the effect of HuR expression on cell proliferation in cancer cells that harbor a natural heterozygous IDH1 mutation. In two different IDH1 mutant cell lines (HT1080 cells have an IDH1 R132C mutation; BT054 cells have an IDH1 R132H), quantitative PCR (qPCR) and immunoblot experiments revealed expected HuR expression changes after genetic modulation with HuR-specific siRNAs (HuR silencing) or cDNA plasmids (HuR overexpression) (Figures.S1A and B). HuR transcript silencing resulted in a significant reduction of cell growth and proliferation over 4 days, as assessed by PicoGreen staining of double-stranded DNA (dsDNA) content (Figure. 1B). Interestingly, HuR overexpression did not have a significant effect on cell proliferation in either HT1080.HOE or BT054.HOE cells (Figure. 1B).
Next, we assessed the importance of HuR expression on cell migration using Matrigel invasion assays. HuR silencing reduced invasion by 60% (p < 0.001), while HuR overexpression increased invasion by 1.4-fold (N.S) (Figure. 1C). Collectively, these data suggest that reduced HuR expression may impact mutant IDH1 cancer cell viability and aggressiveness. Further, they raise the possibility that HuR represents a novel therapeutic target in Mut.IDH1 cancer cells. Although HuR has been shown to impact tumor aggressiveness through many different biologic targets(31, 41, 42), we were particularly interested in its role as a regulator of wild type and mutant IDH1, particularly in light of our recent work demonstrating HuR’s consequential interaction with WT.IDH1 in other cancer types.
HuR inhibition down-regulates both wild type and mutant IDH1 expression
HuR regulates wild type IDH1 mRNA at AU-rich elements in the 3’-untranslated region (11). Since this non-coding sequence is identical for WT.IDH1 and Mut.IDH1 transcripts, HuR should regulate both IDH1 isoforms (mutant and wild type) in tumors harboring the heterozygous mutation. Regulation of each transcript should therefore have measurable biologic consequences. To test this hypothesis, we first confirmed the expectation that wild type and mutant IDH1 transcripts bound to HuR with high affinity through HuR protein enrichment assays in HT1080 cells. Indeed, IDH1 mRNA was elevated in the HuR ribonucleoprotein immunoprecipitation (RNP-IP), compared to the IgG control (Figure. 2A). Direct Sanger sequencing of bound IDH1 mRNAs, with primers anchored across the known IDH1 mutation site (exon 4, C394T), confirmed that both wild and mutant isoforms bind to HuR (Figure. 2B). The similarly sized peaks at the altered base pair in the chromatogram provides a semi-quantitative assessment of template copy number, and suggests similar enrichment of the wild type and mutant isoforms. Equal expression at baseline is reflected by similar peak heights in the background IgG control. Next, we silenced HuR in HT1080 cells (HT1080.siHuR) and observed a corresponding decrease in total IDH1 mRNA expression (> 84% reduction) compared with HT1080.siCTRL cells. Total IDH1 protein expression was correspondingly reduced (Figure. 2C). Again, direct Sanger sequencing revealed both alleles to be present in equal ratios at baseline and after HuR silencing (as well as the control), suggesting that HuR regulates both wild type and mutant transcripts (Figure. 2D).
Figure 2: HuR inhibition down-regulates both wild type and mutant IDH1 expression.
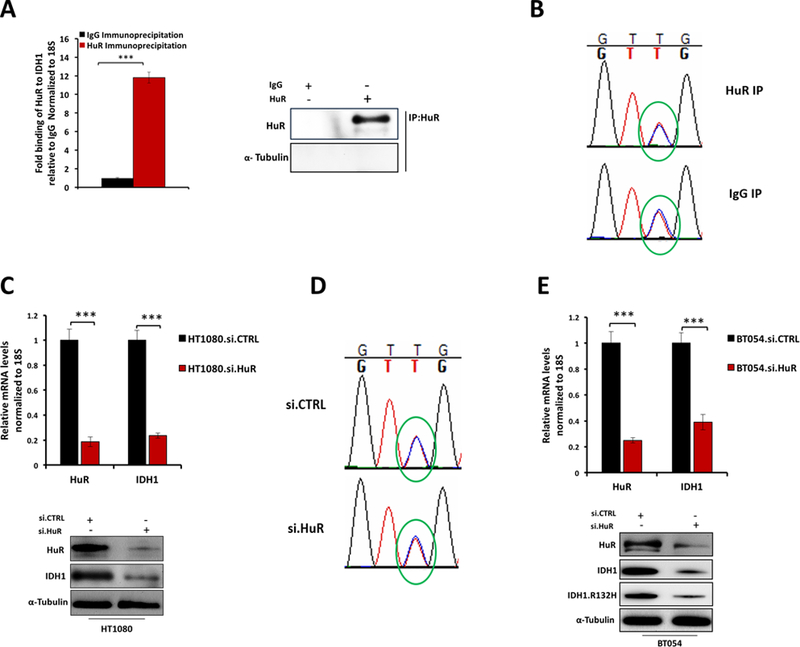
A, mRNP-IP assay and qPCR for IDH1 of mRNAs bound to HuR protein, relative to IgG in HT1080 cells. Results were represented as means of three independent experiments ± SEM. B, Chromatograms show allelic ratios of wild type and mutant IDH1 by semi-quantitiative Sanger sequencing (overlapping red and blue peaks are circled) for HuR RNP-IP relative to IgG control. C, HT1080 cells were transfected with siRNAs against HuR (or control siRNA) and IDH1 mRNA expression was measured by qPCR (Top panel), Representative immunoblots are provided for HuR and IDH1 from HT1080 cell lysates 72 h following transfection with siRNA oligos. ***, p < 0.001. D, Chromatograms show allelic ratios of wild type and mutant IDH1 by semi-quantitiative Sanger sequencing (overlapping red and blue peaks are circled) after HuR silencing relative to si.CTRL. E, Representative qPCR analysis of HuR and IDH1 mRNA expression 48 h after transfection with si.CTRL or si.HuR in BT054 cells. (Top panel), Representative immunoblots for HuR, IDH1 and IDH1.R132H of BT054 cell lysates 72 h following transfection with siRNA oligos. ***, p < 0.001.
We next evaluated the effect of HuR silencing in BT054 cells, which contain the R132H mutation. Targeted HuR mRNA suppression resulted in a 61% reduction in IDH1 mRNA expression. Additionally, we observed a marked decrease in protein expression of the mutant isoform, detectable with a R132H-specific IDH1 antibody (Figure. 2E). These data offer the first direct evidence, to our knowledge, that HuR regulates the expression of the neomorphic, oncogenic mutant IDH1 enzyme. More broadly, we showed that HuR post-transcriptionally regulates both IDH1 isoforms in heterozygous IDH1 mutant tumors.
HuR inhibition sensitizes IDH1 mutant cells to a mutant IDH1 inhibitor
Previously, we showed that low nutrient levels induce adaptive resistance to cytotoxic chemotherapy in pancreatic cancer cells (11). At a molecular level, nutrient withdrawal activated HuR biology to orchestrate a pro-survival network that included enhanced wild type IDH1 expression and activity. This action led to a rise in intracellular reductive power (NADPH synthesis) to promote antioxidant defense under oxidative stress (11). We used Mut.IDH1 cells to determine if a similar adaptive response was present. In mutant IDH1 cancer cells, nutrient withdrawal increased resistance to pharmacologic inhibition with a mutant IDH1 inhibitor (AGI-5198). Under low levels of glucose, both HT1080 and BT054 cells acquired a relative resistance to the drug exceeding 10- fold, as compared to the same cells cultured under glucose abundance (Figure.3A).
Figure 3: HuR inhibition sensitizes IDH1 mutant cells to a mutant IDH1 inhibitor, AGI-5198.
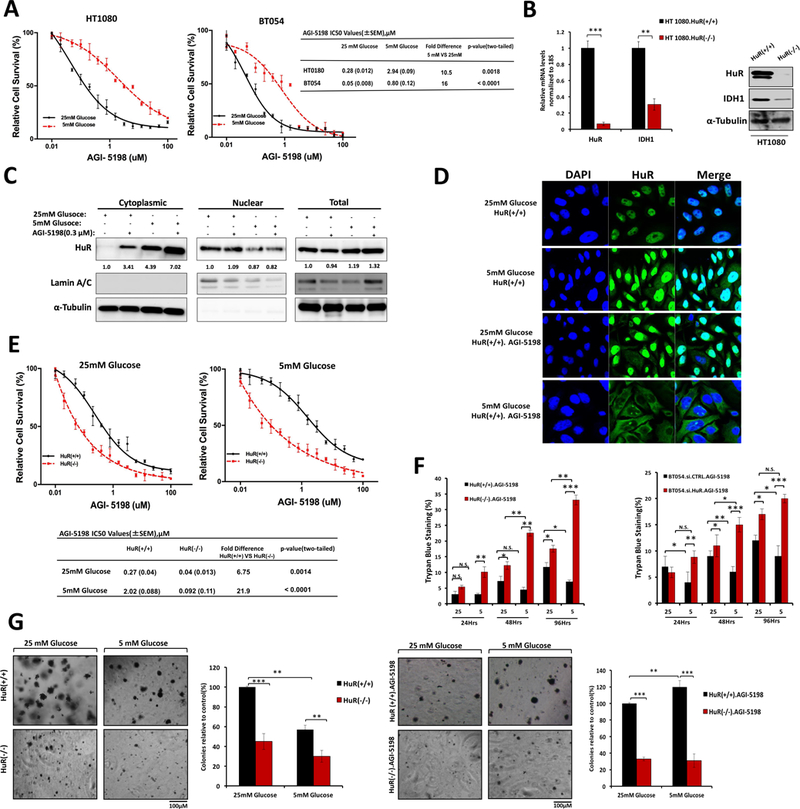
A, Cell viability (PicoGreen DNA quantitation) of Mut.IDH1 cell lines treated at the indicated doses of AGI-5198. IC50 values are provided. B, Representative qPCR analysis of HuR and IDH1 mRNA expression in HT1080.HuR(+/+) and HT1080.HuR(−/−) cells (HuR-knockout by CRISPR gene editing); representative immunoblots for HuR and IDH1 of HT1080.HuR(+/+) and HT1080.HuR(−/−) whole cell lysates, **P ≤ 0.01 and ***P ≤ 0.001. C, Immunoblot analysis of fractionated lysates from HT1080.HuR(+/+) cells upon glucose withdrawal, AGI-5198 treatment (0.3 μM), and a combination of both conditions for 24 hours. Lamin A/C and α-Tubulin were used as controls to determine the integrity of nuclear and cytosolic lysates respectively. D, Immunofluorescence demonstrates HuR subcellular localization to the cytoplasm (green cytoplasmic signal) in HT1080 cells upon glucose withdrawal, AGI-5198 treatment, and a combination of both conditions for 24 hours. Magnification 40×. E, Drug sensitivity measured by PicoGreen DNA quantitation, in HT1080 cells under the indicated culture conditions, and with varying doses of AGI-5198. IC50 values are provided. F, Trypan blue staining in HT1080 and BT054 cells after HuR silencing or CRISPR gene editing, cultured under high or low glucose conditions, with or without AGI-5198 treatment (0.3 μM). G, Long-term cell survival assessed by colony formation in soft agar. HT1080 cells were cultured under the indicated conditions. AGI-5198 was dosed at 0.3 μM for 4 weeks, as indicated. Each data point represents the mean ± SEM of three independent experiments. N.S. non-significant; * p < 0.05; ** p < 0.01; *** p < 0.001.
To determine if HuR drives AGI-5198 resistance, HuR was genetically deleted using CRISPR-Cas9 editing in HT1080 cells. Gene editing was validated at genetic (211delG, Figure.S2), mRNA, and protein levels (Figure.3B). Near total loss of IDH1 expression was observed in HT1080.HuR(−/−) cells, highlighting HuR’s potent regulatory effect on this enzyme. HuR normally resides in the nucleus at baseline, but translocates to the cytoplasm with bound mRNA cargo in response to cancer-associated stressors, such as nutrient withdrawal or chemotherapy (11, 25). This biology was demonstrable in Mut.IDH1 cells, as shown by immunofluorescence (Figure.3C and D). Glucose withdrawal and the Mut.IDH1 inhibitor (AGI-5198) each induced HuR to move to the cytoplasm in HuR-proficient HT1080.HuR(+/+) cells. Maximal cytoplasmic HuR protein expression was observed when both stressors were present simultaneously.
HT1080.HuR(−/−) cells were more sensitive to AGI-5198 than HT1080.HuR(+/+) cells, under both high glucose and low glucose conditions (Figure. 3E). As the drug sensitivity curves illustrate, HuR deletion prevented the adaptive resistance response seen in HuR-proficient cells under glucose withdrawal. While HT1080 parental and HT1080.HuR(+/+) cells were each 10-fold more resistant to AGI-5198 under glucose withdrawal (Figure. 3A), HT1080.HuR(−/−) cells were 10-fold more sensitive to the drug under these conditions, reflecting a 100-fold swing in AGI-5198 sensitivity attributable to HuR genetic deletion. These findings were further validated using a Trypan Blue assay. In the HuR-proficient cancer cells, reduced cell death was observed after AGI-5198 treatment in glucose-depleted media, again pointing to an HuR-dependent drug resistance mechanism against the Mut.IDH1 inhibitor (Figure. 3F). In contrast, an increase in cell death was observed by 96 hours in HT1080.HuR(−/−) and BT054.si.HuR (20% and 5% more death, respectively) relative to isogenic controls (Figure. 3F). While the magnitude of greater cell death is small in BT054 cells after HuR silencing, the magnitude is nevertheless noteworthy since cell killing is reduced in HuR-proficient BT-054 cells after glucose withdrawal.
Anchorage-independent growth was also tested over a 3-week time period to further measure in vitro proliferation. HuR(−/−) colonies were strikingly sparse in number and small in size compared to HuR(+/+) colonies, in both 25mM glucose and 5 mM glucose (Figure. 3G, left panel). Amazingly, we once again observed resistance to AGI-5198 under glucose withdrawal in HT1080.HuR(+/+) cells (Figure. 3G, right panel). In contrast, HT1080.HuR(−/−) were especially sensitive to AGI-5198 under both nutrient abundant and deprived conditions.
HuR inhibition induces global metabolic changes in mutant IDH1 cancer cells and downregulates mutant IDH1 production of 2-HG
Since HuR regulates both WT.IDH1 and Mut.IDH1 expression, we hypothesized that HuR deletion drives metabolic perturbations in IDH1 mutant cancer cells. Additionally, downregulation of Mut.IDH1 should specifically diminish 2-HG and glutamate levels(43, 44). Endogenous metabolites were extracted from HT1080.HuR(+/+) and HT1080.HuR(−/−) cells cultured under complete media and analyzed by LC-MS/MS. A two dimensional-partial least squares discriminant analysis (2D-PLS-DA) plot shows clustering of triplicate experiments, with especially tight reproducibility in the HT1080.HuR(−/−) cells (Figure. 4A). Pathway enrichment analysis of metabolites demonstrated that glutamine/glutamate and glutathione pathway metabolites, as well as some other metabolic pathways, were highly dysregulated after HuR inhibition (Figure. 4B, Figure. S3A and Table S2). More granular pathway analyses revealed a reduction in levels of several Pentose Phosphate Pathway (PPP) metabolites, particularly those involved in the non-oxidative arm (Figure.4C). PPP feeds metabolites into the nucleotide biosynthesis pathway; reduced PPP metabolite levels may negatively impact the biosynthesis of nucleotides, which are the building blocks for nucleic acid synthesis (45). Along these lines, we observed a significant reduction in several nucleotides, including UMP (−71%) and dTDP (−84%), as well as nucleotide precursors in HuR(−/−) cells (Figure. 4D). In the tricarboxylic acid cycle, fumarate and malate were diminished in HT1080.HuR(−/−) cells. It is unknown if these global changes were related to HuR’s regulation of WT.IDH1 or other key transcripts. More specifically, the end products of both WT.IDH1 and MutIDH1 catalysis (α-ketoglutarate and 2-HG, respectively) were both reduced in the analysis (Figure. 4E). The impact of HuR deletion on 2-HG in IDH1 mutant cancer cells was independently confirmed by a separate colorimetric assay indirectly measuring 2-HG levels (Figure. 4F and Figure. S3B). Overall, our LC-MS/MS metabolomic-based studies demonstrate that HuR inhibition significantly reduced PPP activity and nucleoside synthesis, supporting earlier findings that HuR inhibition decreases cancer cell proliferation and viability in IDH1 mutant cells. The effect on 2-HG is most likely a direct effect of HuR’s regulation of Mut.IDH1 expression.
Figure 4: HuR inhibition induces metabolic alterations in mutant IDH1 cancer cells.
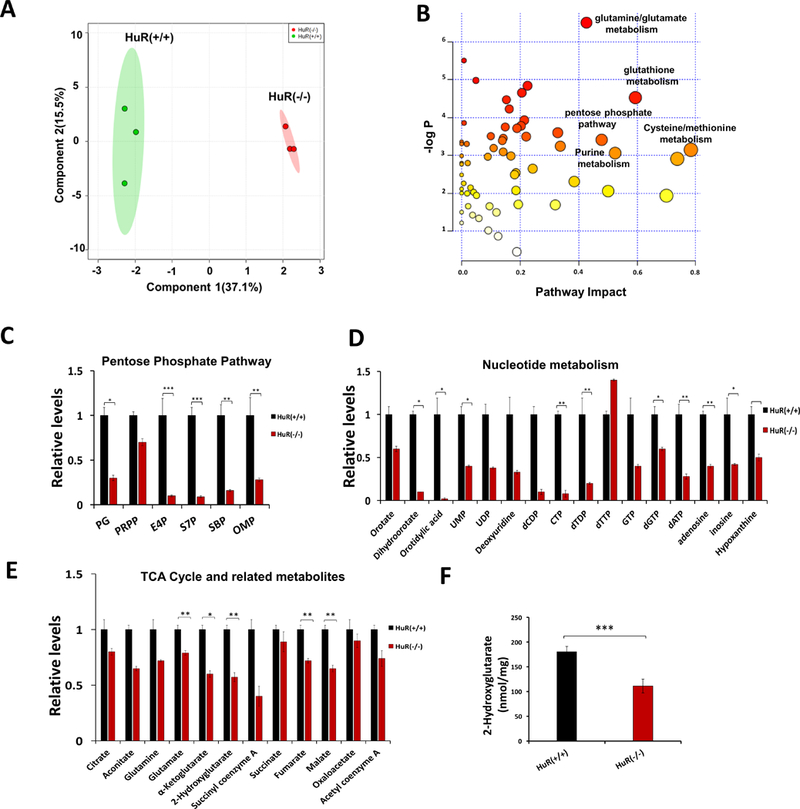
A, PLS-DA (partial least squares discriminant analysis) plot of metabolites generated from LC-MS/MS performed on HT1080.HuR(+/+) and HT1080.HuR(−/−) cells. B, Pathway enrichment analysis of metabolites from HT1080.HuR(+/+) and HT1080.HuR(−/−) cells. C, Relative levels of Pentose Phosphate Pathway metabolites from HT1080.HuR(+/+) and HT1080.HuR(−/−) cells. D, Relative levels of nucleotide metabolites from HT1080.HuR(+/+) and HT1080.HuR(−/−) cells. E, Relative levels of the Tricarboxylic acid cycle metabolites, including 2-HG (Hydroxyglutarate), from HT1080.HuR(+/+) and HT1080.HuR(−/−) cells, as measured by LC-MS/MS. F, 2-HG levels in HT1080.HuR(+/+) or HT1080.HuR(−/−) cells, as measured by a colorimetry-based method. Values represented are mean ± SEM. * p ≤ 0.05, ** p ≤ 0.01 and *** p ≤ 0.001.
HuR inhibition increases tumor response to mutant IDH1 inhibition in vivo
We subcutaneously injected nude mice (n=8) with an equal number of HT1080.HuR(+/+) or HT1080.HuR(−/−) cells in their hind flanks. Tumor volumes were measured on alternate days. Consistent with previous reports, AGI-5198 [450 (mg/kg)] resulted in 59% growth inhibition in genetically unmodified HT1080 cells (28). (Figures. 5A and 5B). HuR deletion markedly diminished tumor growth on its own, similar to our observation in pancreatic cancer(11, 26). However, AGI-5198 treatment combined with HuR deletion had the greatest impact on tumor growth over 3 weeks of treatment. Reduced HuR mRNA in HT1080.HuR(−/−) xenografts was confirmed by immunoblotting at 54 days (Figure. 5C).
Figure 5: HuR inhibition combined with mutant IDH1 inhibition by AGI-5198 suppresses tumor growth in vivo.
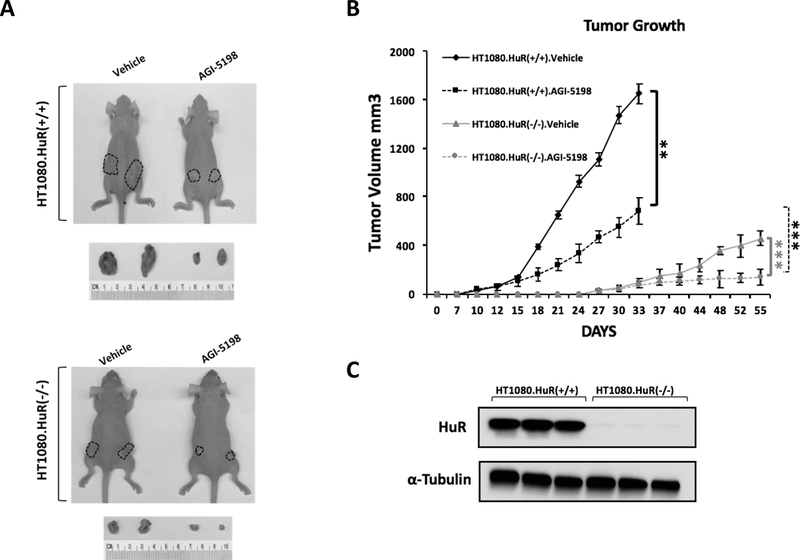
A, Mice were treated with vehicle or AGI-5198. Representative images of excised tumors of HT1080.HuR(+/+) and HT1080.HuR(−/−)(HuR-knockout by CRISPR gene editing) at the termination of the experiment (top panel, day 33; bottom panel, day 55). B, Tumor growth curves of HT1080.HuR(+/+) and HT1080.HuR(−/−) xenografts. Mice were treated with vehicle or AGI-5198. Each data point represents the mean ± SEM (n=8 per group). C, Representative immunoblots depict validation of HuR inhibition in HT1080.HuR(−/−) xenografts compared to HT1080.HuR(+/+) xenografts. Each data point represents the mean ± SEM of three independent experiments. ** p < 0.01; *** p < 0.001.
HuR’s impact on IDH1 mutant cancer growth and AGI-5198 response is largely mediated through HuR’s regulation of the wild type IDH1 enzyme
In order to determine the relative importance of WT.IDH1 and Mut.IDH1 alleles in the HuR-IDH1 regulatory axis for IDH1 mutant tumors, we performed a series of in vitro rescue experiments with WT.IDH1 and Mut.IDH1 overexpression in an HuR (−/−) background (Figure. 6A). IDH1 overexpression at the protein level was validated in HuR(−/−) cells. Functional validation (Figure. S4A) reveals a decrement in 2-HG levels quantified colorimetrically in HuR (−/−) cells. 2-HG levels were rescued (i.e., restored) to baseline in isogenic HuR(−/−).Mut.IDH1OE cells. In vitro drug sensitivity studies revealed that overexpression of WT.IDH1 rescued HuR (−/−) cells treated with AGI-5198. However, overexpression of the Mut.IDH1 enzyme did not have the same impact (Figure. 6B).
Figure 6: The ability of wild type and mutant IDH1 overexpression to rescue HT1080 cells treated with AGI-5198, in vitro and in vivo.
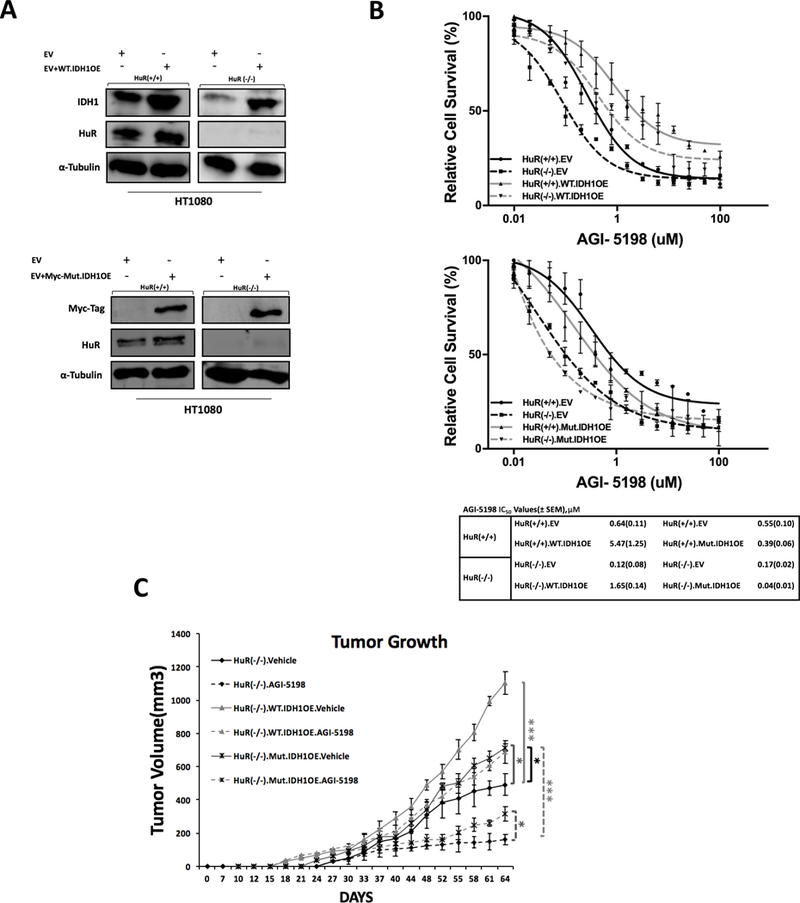
A, Immunoblots depict WT.IDH1 or Mut.IDH1 overexpression in HT1080.HuR(+/+) or HuR(−/−) knockout cell lines. B, PicoGreen drug sensitivity assays in HT1080 cells treated with AGI-5198. WT.IDH1 (top panel) or Mut.IDH1 (bottom panel) were stably overexpressed (or empty vector) in HT1080.HuR(−/−) and HT1080.HuR(+/+) cells. IC50 values are provided in table. C, Xenograft growth of HT1080.HuR(−/−) cells with stable overexpression of WT.IDH1 or MT.IDH1, treated with vehicle or AGI-5198. Each data point represents the mean ± SEM (n=8 per group). * p ≤ 0.05, ** p < 0.01; *** p < 0.001.
We further explored this line of investigation in vivo. Nude mice (n=8) were subcutaneously injected with three groups of genetically modified HT1080-derived cells: 1) Negative-control HT1080.HuR(−/−) cells, 2) HT1080.HuR(−/−).Mut.IDH1 (stable overexpression of Mut.IDH1) cells and 3) HT1080.HuR(−/−).WT.IDH1 cells (stable overexpression of WT.IDH1). Mice bearing isogenic xenografts were treated with either vehicle alone or AGI-5198, yielding six treatment groups in total. Representative tumors from each experimental group are provided in Figure. S4B. Overexpression of WT.IDH1 and Mut.IDH1 in HT1080.HuR(−/−) xenografts was confirmed by immunoblotting at 64 days (Figure. S4C). Similar to the in vitro experiments, restoration of WT.IDH1 expression in the HuR-null cells resulted in marked drug resistance to AGI-5198. Mut.IDH1 overexpression was also protective in HT1080.HuR(−/−) cells, but to a lesser extent (Figure. 6C).
Discussion
Multiple clinical trials are ongoing that test the utility of mutant IDH1 inhibitors in cancers with gain-of-function IDH1 alterations. An oral drug from Bayer, BAY1436032, is in phase I studies in patients with acute myeloid leukemia (NCT03127735) and solid tumors (NCT02746081). Oral drugs produced by Agios Pharmaceuticals, AG-120 and AG-881, will be tested in low grade gliomas (NCT03343197) and acute myeloid leukemia (NCT02677922). Nearly a dozen other drugs targeting mutant IDH1 are under pre-clinical development (29). Interest in these targeted therapies has grown due to the efficacy of lead compounds in pre-clinical studies, along with a relatively high prevalence of IDH1 mutations in diverse tumor types(28, 29, 46).
Biologic studies uncovered tumor-promoting properties of the oncometabolite (2-HG) produced by neomorphic Mut.IDH1 (2). Mutant IDH1 activity promotes hyper-methylation of DNA and histones, which suppresses cell differentiation(5). These findings led to the discovery of AGI-5198 and other drugs which potently block mutant IDH1 (R132H or R132C) with IC50 ranges around 100 nM(28). Treatment of Mut.IDH1 tumors with these and related compounds reverse epigenetic marks. More importantly, Mut.IDH1 inhibitors slow tumor growth(47, 48).
Until now, the field has specifically focused on targeting the mutant IDH1 allele in these tumors. For instance, AGI-5198 has limited activity against WT.IDH1 and IDH2 isoforms (IC50>100 μM), and the same is true for most of the other mutant IDH1 inhibitors under clinical development (29). Indeed, IDH1 inhibitors are generally prioritized based on their specificity for the mutant allele over wild type IDH1 (29). However, there are emerging data that highlight the importance of WT.IDH1 in cancers. For instance, we recently demonstrated that pancreatic cancers, which do not have IDH1 mutations, rely on WT.IDH1 expression to generate reductive power needed to combat severe oxidative stress ubiquitously present in the tumor microenvironment (11, 25). For this reason, our group has postulated that IDH1 mutations may even be deleterious in pancreatic cancer due to the characteristically harsh microenvironment (49), and could account for the rarity of this genetic alteration in that lethal cancer(3). Others have shown the importance of WT.IDH1 for reductive carboxylation and lipogenesis under hypoxic conditions in other IDH1 wild type cancers (e.g., lung) (8, 10).
Beyond the world of wild type IDH1 cancers though, we speculate that the wild type isoform also plays a key role in tumors harboring heterozygous IDH1 mutations (a single copy of the wild type and mutant alleles) (50). In support of this idea, others have demonstrated that the wild type isoform is required in IDH1 mutant tumors for reductive carboxylation of glutamine-derivatives (e.g., α-ketoglutarate) during lipogenesis (14). In a separate study, mutant IDH1 tumors were especially dependent on oxidative metabolism and susceptible to mitochondrial inhibitors, compared to wild type IDH1 tumors (51). Similarly, IDH1-mutant tumors are more susceptible to chemotherapy (15). Thus, haploinsufficiency of wild type IDH1 could expose metabolic vulnerabilities in IDH1 mutant tumors, attributable to reduced metabolic flexibility under stress. If true, therapeutic strategies that target both IDH1 alleles (mutant and wild type) in heterozygous tumors, or a common regulator of these isoenzymes, are particularly compelling.
Based on prior work elucidating HuR’s regulation of WT.IDH1 through interactions in the IDH1 3’UTR (11), we reasoned that this RNA binding protein likely regulates both isoforms in tumors with a heterozygous IDH1 R132 mutation. In the present study, we show that HuR binds both isoforms in ribonucleoprotein immunoprecipitation assays. Through this binding interaction, HuR potently regulates Mut.IDH1 and WT.IDH1 transcripts, as well as their associated protein expression. Transient HuR silencing impaired cell growth, and this finding was validated in isogenic cells with genetically (CRISPR) deleted HuR. Metabolomic profiling revealed significant perturbations in key metabolic pathways in HuR-deficient cells, including the PPP, nucleotide synthesis, and glutathione production. Moreover, reduced 2-HG levels functionally in HuR-deficient cells link HuR to Mut.IDH1 biology.
Low glucose conditions, a hallmark of the tumor microenvironment, induced cancer resistance to mutant IDH1 inhibition by AGI-5198. Mechanistically, a role for HuR was evident. Low glucose levels and AGI-5198 both engaged HuR to translocate to the cytoplasm, and HuR-deletion abrogated resistance to AGI-5198 under glucose withdrawal. Furthermore, HuR-null cells failed to grow significantly in nude mice treated with AGI-5198, in contrast to HuR-proficient cells treated with the drug. Finally, we demonstrated that HuR impacts cell growth and viability in IDH1-mutant tumors through regulatory effects on both WT.IDH1 and Mut.IDH1 alleles. In vitro, WT.IDH1 overexpression completely rescued HuR (−/−) from AGI-5198 treatment. Interestingly, Mut.IDH1 overexpression had an observable, albeit more modest effect. Indeed, the growth of AGI-5198 treated HuR(−/−).WT.IDH1OE cells even surpassed the growth of untreated HT1080.HuR(−/−) cells.
These data provide novel and clinically relevant insights. First, HuR is a compelling therapeutic target in IDH1-mutant tumors. A proposed role for the HuR-IDH1 axis in the survival of mutant IDH1 cancer cells under stress is summarized in Figure 7. Existing drugs with anti-HuR properties (e.g., pyrvinium pamoate)(52) or even novel HuR inhibitors (53) should be explored in conjunction with Mut.IDH1 inhibitors. Second, WT.IDH1 likely plays an important role in these tumors, providing a rationale for pan-IDH1 inhibition.
Figure 7:
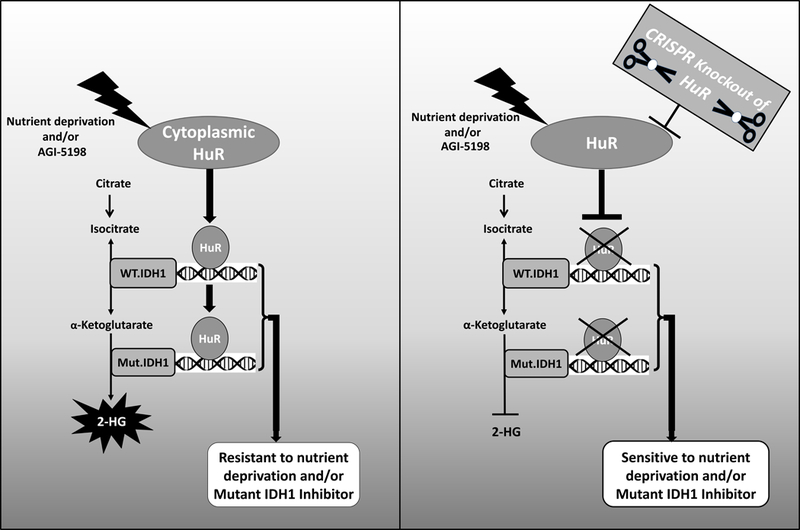
Schematic showing how HuR regulates resistance to glucose withdrawal and pharmacologic IDH1 inhibition through post-transcriptional regulation of both wild type and mutant IDH1 transcripts. The impact of HuR expression and targeting on resistance and susceptibility to these stressors is illustrated.
Supplementary Material
Implications:
This study highlights the HuR-IDH1 (mutant and wild-type IDH1) regulatory axis as a critical, actionable therapeutic target in IDH1-mutated cancer, and incomplete blockade of the entire HuR-IDH1 survival axis would likely diminish the efficacy of drugs that selectively target only the mutant isoenzyme.
Acknowledgments
Funding: This work was supported by American Cancer Society Mentored Research Scholar Grant-14–019-01-CDD (J.M.W), 1R37CA227865 (J.M.W.), R01 CA212600 (J.R.B. and J.M.W.), R01CA227865 (J.M.W and J.R.B), R01 CA210439, R01 CA216853 and SPORE 2P50 CA127297 (P.K.S). The Gail V. Coleman-Kenneth M. Bruntel Charitable Grant Fund provided additional support. Mark Levine’s contributions to this work were made in memory of Ethel Levine. K.O. was supported by NIH institutional award T32 GM008562 for Postdoctoral Training in Clinical Pharmacology. We would also like to acknowledge the Fred & Pamela Buffett Cancer Center Support Grant (P30CA036727, NCI) for supporting shared resources.
Abbreviations:
- IDH1
Isocitrate dehydrogenase 1
- RNP-IP
ribonucleoprotein immunoprecipitation
- qRT-PCR
quantitative reverse transcription polymerase chain reaction
- α-KG
α-ketoglutarate
- 2-HG
2-hydroxyglutarate
- LC-MS/MS
Liquid chromatography/tandem mass spectrometry
- PPP
Pentose Phosphate Pathway
- UMP
uridine 5’-monophosphate
- dTDP
Thymidine 5’-pyrophosphate
Footnotes
Disclosure of Potential Conflicts of Interest: The authors have no conflicts of interest to disclose.
Author Contributions:
Study concept and design: M.Z., J.R.B. and J.M.W., Acquisition of data: M.Z., S.L., K.O., P.K.S., J.R.B., J.M.W., Analysis and interpretation of data: M.Z., S.L., V.G, P.K.S., J.R.B., J.M.W., Drafting of the manuscript: M.Z., J.R.B., J.M.W., Critical revision of the manuscript for important intellectual content: M.Z., A.V., P.K.S., J.R.B., J.M.W.
References:
- 1.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9–29. [DOI] [PubMed] [Google Scholar]
- 2.Ohgaki H, Kleihues P. The definition of primary and secondary glioblastoma. Clin Cancer Res. 2013;19(4):764–72. [DOI] [PubMed] [Google Scholar]
- 3.Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321(5897):1807–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462(7274):739–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. 2012;21(3):297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Metallo CM. Expanding the reach of cancer metabolomics. Cancer Prev Res (Phila). 2012;5(12):1337–40. [DOI] [PubMed] [Google Scholar]
- 8.Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, et al. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature. 2011;481(7381):385–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Filipp FV, Scott DA, Ronai ZA, Osterman AL, Smith JW. Reverse TCA cycle flux through isocitrate dehydrogenases 1 and 2 is required for lipogenesis in hypoxic melanoma cells. Pigment Cell Melanoma Res. 2012;25(3):375–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. 2011;481(7381):380–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zarei M, Lal S, Parker SJ, Nevler A, Vaziri-Gohar A, Dukleska K, et al. Posttranscriptional Upregulation of IDH1 by HuR Establishes a Powerful Survival Phenotype in Pancreatic Cancer Cells. Cancer Res. 2017;77(16):4460–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grassian AR, Parker SJ, Davidson SM, Divakaruni AS, Green CR, Zhang X, et al. IDH1 mutations alter citric acid cycle metabolism and increase dependence on oxidative mitochondrial metabolism. Cancer Res. 2014;74(12):3317–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leonardi R, Subramanian C, Jackowski S, Rock CO. Cancer-associated isocitrate dehydrogenase mutations inactivate NADPH-dependent reductive carboxylation. J Biol Chem. 2012;287(18):14615–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reitman ZJ, Duncan CG, Poteet E, Winters A, Yan LJ, Gooden DM, et al. Cancer-associated isocitrate dehydrogenase 1 (IDH1) R132H mutation and d-2-hydroxyglutarate stimulate glutamine metabolism under hypoxia. J Biol Chem. 2014;289(34):23318–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang JB, Dong DF, Wang MD, Gao K. IDH1 overexpression induced chemotherapy resistance and IDH1 mutation enhanced chemotherapy sensitivity in Glioma cells in vitro and in vivo. Asian Pac J Cancer Prev. 2014;15(1):427–32. [DOI] [PubMed] [Google Scholar]
- 16.Brody JR, Yabar CS, Zarei M, Bender J, Matrisian LM, Rahib L, et al. Identification of a novel metabolic-related mutation (IDH1) in metastatic pancreatic cancer. Cancer Biol Ther. 2018;19(4):249–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hinman MN, Lou H. Diverse molecular functions of Hu proteins. Cell Mol Life Sci. 2008;65(20):3168–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang J, Guo Y, Chu H, Guan Y, Bi J, Wang B. Multiple functions of the RNA-binding protein HuR in cancer progression, treatment responses and prognosis. Int J Mol Sci. 2013;14(5):10015–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mukherjee N, Corcoran DL, Nusbaum JD, Reid DW, Georgiev S, Hafner M, et al. Integrative regulatory mapping indicates that the RNA-binding protein HuR couples pre-mRNA processing and mRNA stability. Mol Cell. 2011;43(3):327–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jimbo M, Blanco FF, Huang YH, Telonis AG, Screnci BA, Cosma GL, et al. Targeting the mRNA-binding protein HuR impairs malignant characteristics of pancreatic ductal adenocarcinoma cells. Oncotarget. 2015;6(29):27312–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luchman HA, Stechishin OD, Dang NH, Blough MD, Chesnelong C, Kelly JJ, et al. An in vivo patient-derived model of endogenous IDH1-mutant glioma. Neuro Oncol. 2012;14(2):184–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S, et al. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004;64(19):7011–21. [DOI] [PubMed] [Google Scholar]
- 23.De Filippis L, Lamorte G, Snyder EY, Malgaroli A, Vescovi AL. A novel, immortal, and multipotent human neural stem cell line generating functional neurons and oligodendrocytes. Stem Cells. 2007;25(9):2312–21. [DOI] [PubMed] [Google Scholar]
- 24.Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012;483(7390):479–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burkhart RA, Pineda DM, Chand SN, Romeo C, Londin ER, Karoly ED, et al. HuR is a post-transcriptional regulator of core metabolic enzymes in pancreatic cancer. RNA Biol. 2013;10(8):1312–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lal S, Cheung EC, Zarei M, Preet R, Chand SN, Mambelli-Lisboa NC, et al. CRISPR Knockout of the HuR Gene Causes a Xenograft Lethal Phenotype. Mol Cancer Res. 2017;15(6):696–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Y, Xiao M, Chen X, Chen L, Xu Y, Lv L, et al. WT1 recruits TET2 to regulate its target gene expression and suppress leukemia cell proliferation. Mol Cell. 2015;57(4):662–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science. 2013;340(6132):626–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Urban DJ, Martinez NJ, Davis MI, Brimacombe KR, Cheff DM, Lee TD, et al. Assessing inhibitors of mutant isocitrate dehydrogenase using a suite of pre-clinical discovery assays. Sci Rep. 2017;7(1):12758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Costantino CL, Witkiewicz AK, Kuwano Y, Cozzitorto JA, Kennedy EP, Dasgupta A, et al. The role of HuR in gemcitabine efficacy in pancreatic cancer: HuR Up-regulates the expression of the gemcitabine metabolizing enzyme deoxycytidine kinase. Cancer Res. 2009;69(11):4567–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lal S, Burkhart RA, Beeharry N, Bhattacharjee V, Londin ER, Cozzitorto JA, et al. HuR posttranscriptionally regulates WEE1: implications for the DNA damage response in pancreatic cancer cells. Cancer Res. 2014;74(4):1128–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McAllister F, Pineda DM, Jimbo M, Lal S, Burkhart RA, Moughan J, et al. dCK expression correlates with 5-fluorouracil efficacy and HuR cytoplasmic expression in pancreatic cancer: a dual-institutional follow-up with the RTOG 9704 trial. Cancer Biol Ther. 2014;15(6):688–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pineda DM, Rittenhouse DW, Valley CC, Cozzitorto JA, Burkhart RA, Leiby B, et al. HuR’s post-transcriptional regulation of Death Receptor 5 in pancreatic cancer cells. Cancer Biol Ther. 2012;13(10):946–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–8. [DOI] [PubMed] [Google Scholar]
- 35.Shukla SK, Gebregiworgis T, Purohit V, Chaika NV, Gunda V, Radhakrishnan P, et al. Metabolic reprogramming induced by ketone bodies diminishes pancreatic cancer cachexia. Cancer Metab. 2014;2:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shukla SK, Purohit V, Mehla K, Gunda V, Chaika NV, Vernucci E, et al. MUC1 and HIF-1alpha Signaling Crosstalk Induces Anabolic Glucose Metabolism to Impart Gemcitabine Resistance to Pancreatic Cancer. Cancer Cell. 2017;32(1):71–87 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xia J, Mandal R, Sinelnikov IV, Broadhurst D, Wishart DS. MetaboAnalyst 2.0--a comprehensive server for metabolomic data analysis. Nucleic Acids Res. 2012;40(Web Server issue):W127-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brody JR, Dixon DA. Complex HuR function in pancreatic cancer cells. Wiley Interdiscip Rev RNA. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Filippova N, Yang X, Ananthan S, Sorochinsky A, Hackney JR, Gentry Z, et al. Hu antigen R (HuR) multimerization contributes to glioma disease progression. J Biol Chem. 2017;292(41):16999–7010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Holmes B, Benavides-Serrato A, Freeman RS, Landon KA, Bashir T, Nishimura RN, et al. mTORC2/AKT/HSF1/HuR constitute a feed-forward loop regulating Rictor expression and tumor growth in glioblastoma. Oncogene. 2018;37(6):732–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blanco FF, Jimbo M, Wulfkuhle J, Gallagher I, Deng J, Enyenihi L, et al. The mRNA-binding protein HuR promotes hypoxia-induced chemoresistance through posttranscriptional regulation of the proto-oncogene PIM1 in pancreatic cancer cells. Oncogene. 2016;35(19):2529–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chand SN, Zarei M, Schiewer MJ, Kamath AR, Romeo C, Lal S, et al. Posttranscriptional Regulation of PARG mRNA by HuR Facilitates DNA Repair and Resistance to PARP Inhibitors. Cancer Res. 2017;77(18):5011–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Izquierdo-Garcia JL, Viswanath P, Eriksson P, Chaumeil MM, Pieper RO, Phillips JJ, et al. Metabolic reprogramming in mutant IDH1 glioma cells. PLoS One. 2015;10(2):e0118781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reitman ZJ, Jin G, Karoly ED, Spasojevic I, Yang J, Kinzler KW, et al. Profiling the effects of isocitrate dehydrogenase 1 and 2 mutations on the cellular metabolome. Proc Natl Acad Sci U S A. 2011;108(8):3270–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Patra KC, Hay N. The pentose phosphate pathway and cancer. Trends Biochem Sci. 2014;39(8):347–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chaturvedi A, Herbst L, Pusch S, Klett L, Goparaju R, Stichel D, et al. Pan-mutant-IDH1 inhibitor BAY1436032 is highly effective against human IDH1 mutant acute myeloid leukemia in vivo. Leukemia. 2017;31(10):2020–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li L, Paz AC, Wilky BA, Johnson B, Galoian K, Rosenberg A, et al. Treatment with a Small Molecule Mutant IDH1 Inhibitor Suppresses Tumorigenic Activity and Decreases Production of the Oncometabolite 2-Hydroxyglutarate in Human Chondrosarcoma Cells. PLoS One. 2015;10(9):e0133813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dang L, Su SM. Isocitrate Dehydrogenase Mutation and (R)-2-Hydroxyglutarate: From Basic Discovery to Therapeutics Development. Annu Rev Biochem. 2017;86:305–31. [DOI] [PubMed] [Google Scholar]
- 49.Brody JR, Yabar CS, Zarei M, Bender J, Matrisian LM, Rahib L, et al. Identification of a novel metabolic-related mutation (IDH1) in metastatic pancreatic cancer. Cancer Biol Ther. 2016:0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jin G, Reitman ZJ, Duncan CG, Spasojevic I, Gooden DM, Rasheed BA, et al. Disruption of wild-type IDH1 suppresses D-2-hydroxyglutarate production in IDH1-mutated gliomas. Cancer Res. 2013;73(2):496–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Grassian AR, Pagliarini R, Chiang DY. Mutations of isocitrate dehydrogenase 1 and 2 in intrahepatic cholangiocarcinoma. Curr Opin Gastroenterol. 2014;30(3):295–302. [DOI] [PubMed] [Google Scholar]
- 52.Guo J, Lv J, Chang S, Chen Z, Lu W, Xu C, et al. Inhibiting cytoplasmic accumulation of HuR synergizes genotoxic agents in urothelial carcinoma of the bladder. Oncotarget. 2016;7(29):45249–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meisner NC, Hintersteiner M, Mueller K, Bauer R, Seifert JM, Naegeli HU, et al. Identification and mechanistic characterization of low-molecular-weight inhibitors for HuR. Nat Chem Biol. 2007;3(8):508–15. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.