

Abstract
Resistance to standard therapy remains a major challenge in the treatment of pancreatic ductal adenocarcinoma (PDA). Although anti-VEGF therapy delays PDA progression, therapy-induced hypoxia results in a less differentiated mesenchymal-like tumor cell phenotype, which reinforces the need for effective companion therapies. COX-2 inhibition has been shown to promote tumor cell differentiation and improve standard therapy response in PDA. Here, we evaluate the efficacy of COX-2 inhibition and VEGF blockade in preclinical models of PDA. In vivo, the combination therapy was more effective in limiting tumor growth and metastasis than single-agent therapy. Combination therapy also reversed anti-VEGF–induced epithelial-mesenchymal transition, collagen deposition and altered the immune landscape by increasing tumor-associated CD8+ T cells while reducing FoxP3+ T cells and FasL expression on tumor endothelium.
Keywords: COX-2, VEGF, tumor microenvironment, pancreatic cancer
Graphical Abstract
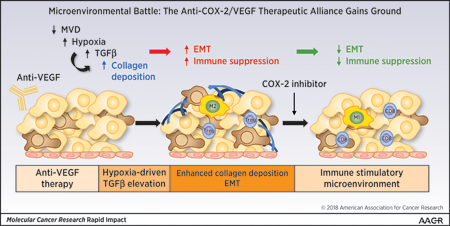
Introduction
Primary tumors and metastases require nutrients and oxygen delivered by blood vessels1. Although angiogenesis is complex, it is widely recognized that vascular endothelial growth factor-A (VEGF-A) is the predominant angiogenic factor that promotes tumor neovascularization2,3. Inhibitors of angiogenesis have become a central part of systemic therapy for a variety of malignancies4,5. However, angiogenesis inhibition has in general resulted in only modest gains in clinical outcomes in cancer patients as many patients treated with anti-angiogenic/anti-VEGF therapy either fail to respond or relapse on therapy6,7. Additionally, anti-angiogenic therapy has been implicated in promoting tumor progression and accelerating metastasis in preclinical models8,9.
Pancreatic cancer, the third-leading cause of cancer-related death10, is highly metastatic and poorly responsive to standard therapy11,12. It is also an immunologically “cold” tumor that has remained largely refractory to immune checkpoint blockade12,13. Anti-VEGF therapy has been studied in pancreatic cancer patients4; however, it has not provided significant clinical benefit in combination with gemcitabine, the standard chemotherapy for pancreatic ductal adenocarcinoma (PDA)14–16. Previously, we investigated the efficacy and biology of anti-VEGF therapy in preclinical models of PDA using the antibody mcr849,17. We found that mcr84 alone or in combination with gemcitabine slowed the growth of PDA but induced hypoxia-induced epithelial plasticity that resulted in a less differentiated tumor cell phenotype and continued metastatic burden9. These observations reinforce the need to develop companion therapies that combat therapy-induced epithelial plasticity.
Inflammation is a pathological phenotype that facilitates the “hallmarks” of cancer18. Further, the incidence of several cancers is associated with inflammation, which contributes to tumor initiation and cancer cell survival by producing reactive oxygen species, cytokines, and proinflammatory mediators19. Among mediators of inflammation that are associated with tumor progression is cyclooxygenase-2 (COX-2), an inducible enzyme that catalyzes the rate-limiting step in the synthesis of the prostaglandin E2 (PGE2). COX-2 is induced at sites of inflammation and during the process of tumor progression20. Multiple studies have demonstrated that elevated COX-2 expression is prevalent in human malignancies, including PDA21,22. In addition, elevated expression of COX-2 in tumors correlates with advanced stage and worse outcome by promoting chemoresistance, metastasis, and angiogenesis23,24. COX-2 has also been identified as a potential mediator of VEGF-independent tumor angiogenesis25. Thus, targeting COX-2 inhibition has been explored as a potential anticancer therapy26. Additionally, COX-2 blockade can enhance the efficacy of anti-angiogenic treatments in breast cancer, which supports the investigation of COX-2 inhibitors with VEGF blockade in other tumors27.
Apricoxib is a selective COX-2 inhibitor that has shown significant anti-tumor activity in various xenograft models28 and has been under clinical investigation. Previously, we demonstrated that apricoxib improved the efficacy of standard therapy in preclinical models of PDA29. Further, we found that inhibition of COX-2 reversed epithelial-mesenchymal transition (EMT), leading to a shift toward a more epithelial phenotype in xenograft models of PDA29. In the present study, we investigated the combination of anti-VEGF therapy and COX-2 inhibition as a therapeutic strategy in robust preclinical models of PDA with the hypothesis that apricoxib would prevent or reduce therapy-induced epithelial plasticity. We also investigated the effect of anti-VEGF and COX-2 inhibition on the immune landscape of PDA given that prior reports have demonstrated VEGF and PGE2 can limit T cell infiltration into tumor cell nests30 and reports that EMT can be a significant driver of immune suppression in tumors31–34.
Materials and Methods
Cell culture
Human pancreatic cancer cell lines AsPC-1, HPAF-II were obtained from ATCC. Colo357 was obtained from MD Anderson Cancer Center. AsPC-1 was grown in DMEM, Colo357 and HPAF-II in MEM. All cell lines were grown in a humidified atmosphere with 5% CO2, at 37˚C, and were DNA fingerprinted for provenance using the Power-Plex 1.2 kit (Promega) and confirmed to be the same as the DNA fingerprint library maintained by ATCC. Cell lines were confirmed to be free of mycoplasma (e-Myco kit, Boca Scientific) before use. In vitro PGE2 and VEGF response to apricoxib treatment was evaluated by enzyme-linked immunosorbent assay (ELISA; R&D Systems) of conditioned media over different time points. To induce EMT, cells were grown on collagen I-coated plates and treated with 50 ng/ml transforming growth factor-β (TGFβ) for 24 hours35. EMT changes were confirmed by probing cell lysates for E-Cadherin, N-Cadherin and Vimentin (Cell Signaling) (see Supplementary Table 1).
Animal studies
All animals were housed in a pathogen-free facility with 24-hour access to food and water. Experiments were approved by and conducted in accordance with the Institutional Animal Care and Use Committee at UT Southwestern (Dallas, TX). KrasLSL-G12D; Cdkn2afl/fl; Ptf1aCre/+ (KIC) mice were generated as previously described36. At 3 weeks of age, mice were randomized to receive saline, mcr84 500 µg/dose i.p. once weekly, Apricoxib 10 mg/kg by oral gavage daily or mcr84 plus Apricoxib. All mice were sacrificed when they were 7 weeks old. Four-to-6-week-old female NOD/SCID mice were obtained from a campus supplier. A total of 1×106 Colo357 cells were injected orthotopically and tumor growth was monitored by ultrasound. Mice with established tumors were randomized to receive therapy. Treatment groups were the same as described above. Mice bearing Colo357 tumors received 4 weeks of therapy prior to sacrifice. Tissues from all animal experiments were fixed in 10% formalin or snap-frozen in liquid nitrogen for further studies.
Histology and tissue analysis
Formalin-fixed tissues were embedded in paraffin and cut in 5 µm sections. Sections were evaluated by Masson’s Trichrome staining and immunohistochemical analysis using antibodies specific for VEGF, COX-2 (Abcam), E-cadherin, N-cadherin, Slug, Snail (Cell Signaling), CD3 (Bio-Rad), CD8 (Bioss), FoxP3 (eBioscience), CD31 (dianova), FasL (Santa Cruz), CD11b (Abcam), iNOS, Arginase 1 (Thermo Fisher, Arg-1), endomucin (Santa Cruz), NG2 (Millipore) and F4/80 (Novus Biologicals). Fluorescent images were captured with Zeiss Aixoscan Z1 using ZenLite software. Color images were obtained with Hamamatsu Nanozoomer 2.0HT using NDPview2 software. Pictures were analyzed using NIS Elements (Nikon) and Fiji software.
Statistical analysis
Data were analyzed using GraphPad software. Results are expressed as mean ± SEM. Data were analyzed by ANOVA with the Dunn’s test for multiple comparisons and results are considered significant at p< 0.05.
Results
Pharmacologic blockade of COX-2 and VEGF inhibits tumor growth and limits metastatic burden in pancreatic cancer models
To investigate the efficacy of COX-2 inhibition with apricoxib and VEGF blockade with mcr8417 in preclinical models of PDA, we used a genetically engineered mouse model of PDA and SCID mice bearing established orthotopic pancreatic xenografts. Therapy was initiated in 3-week-old KIC mice. Mice were randomized to receive saline, mcr84, apricoxib, or mcr84 + apricoxib and were sacrificed after 4 weeks (7 weeks old). Therapy with mcr84 or apricoxib reduced primary tumor weight by ~30% whereas mcr84 + apricoxib reduced primary tumor weight by 62% compared to the control group (P < 0.0001; Fig. 1a). At the time of sacrifice, the extent of liver metastasis was determined based on gross metastasis. Seven out of 10 evaluable mice in the control group had at least 1 macroscopic metastasis; this number was reduced to 1/5, 2/7, 1/6 for the mcr84, apricoxib, and combination therapy groups, respectively (Fig. 1a). To further define the effect of COX-2 inhibition and anti-VEGF therapy on tumor burden and liver metastases, we established human PDA xenografts in mice by orthotopically injecting Colo357, a human pancreatic cancer cell line, into the pancreas of SCID mice. Similar to in vitro data published previously29, Colo357 cells showed high COX-2 expression and were responsive to apricoxib (data not shown). Mice with established tumors, which was confirmed by ultrasound, were randomized to receive treatment as described above. After 4 weeks of therapy, we found that single-agent therapy had a minimal effect on primary tumor growth (Fig. 1b) and metastatic incidence, although the mean metastatic events per treatment group was reduced by mcr84 or apricoxib (Fig. 1b). In contrast, combination therapy significantly reduced primary tumor weight (P < 0.05 vs. control) and substantially limited metastases (P < 0.01 vs. control; P < 0.05 vs. single-agent therapy; Fig. 1b). H&E analysis of livers confirmed metastatic lesions in Colo357 tumor bearing mice (Supplementary Fig. S1a). The effect of mcr84 + apricoxib on primary tumor growth compared favorably to the effect of gemcitabine + erlotinib in the same model reported in our prior study37 (Supplementary Fig. S1b).
Figure 1. Combination therapy with apricoxib and mcr84 reduced tumor growth and metastasis in murine models of pancreatic cancer.

(a) At 3 weeks of age, KrasLSL-G12D; Cdkn2afl/fl; Ptf1aCre/+ (KIC) mice were randomized to receive saline (n = 11), mcr84 (n = 10), apricoxib (n = 13), or mcr84 plus apricoxib (n = 13). All mice were sacrificed when they were 7 weeks old. Mean tumor weight and metastasis burden were compared. (b) A total of 1106 Colo357 cells were injected orthotopically into NOD/SCID mice. Treatment began when established tumors were visible by ultrasound and consisted of control (n = 8), mcr84 (n = 10), apricoxib (n = 10) or mcr84 plus apricoxib (n = 10) and continued for 4 weeks, after which mean tumor weight and metastasis burden were shown. Data are displayed in a scatter plot with mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.005 vs. control; #P < 0.05 vs. single-agent mcr84 or apricoxib by ANOVA with Dunn’s MCT. (c) Human pancreatic cancer cell lines, HPAF-II, Colo357, and AsPC-1 were treated with 500 nM apricoxib and evaluated by ELISA for the production of VEGF. Colo357 and AsPC-1 were plated under normal conditions or conditions of forced EMT (50 ng/ml TGFβ on collagen I -coated plates for 24 hours). VEGF levels were evaluated by ELISA after 500 nM apricoxib treatment. Biological repeats have been performed (n=3) and data are displayed as mean ± SEM. Paraffin-embedded tumor sections from Colo357 tumor-bearing mice were analyzed for (d) VEGF and COX-2 expression by immunofluorescence. Quantification of percentage area fraction is shown. Data are displayed as mean ± SEM and represent 5 images per tumor with 3 animals per group. Representative images (COX-2, red; DAPI, blue) are shown for Colo357 tumors. Total magnification is 400X. Scale bars are presented as indicated.
COX-2 activity has been implicated in promoting angiogenesis25,38,39. Previously, prostaglandins, products of COX-2 activity were shown to elevate VEGF expression and inhibition of COX-2 was shown to contribute to anti-angiogenic effects40,41. Furthermore, fibroblasts from Cox-2-deficient mice were reported to produce significantly less VEGF than fibroblasts from wild-type or Cox-1-deficient animals42. Additionally, treatment of wild-type fibroblasts with a selective COX-2 inhibitor resulted in a 90% reduction in VEGF production42. However, recently Xu et al25 determined that PGE2 can contribute to angiogenesis in a VEGF-independent manner in colon cancer models. Given these data, we sought to investigate the relationship between COX-2 activity and VEGF production in PDA cell lines, we selected a COX-2 negative cell line, AsPC-1, and 2 COX-2 positive cell lines, one with a high expression of COX-2, Colo357, the other with moderate COX-2 expression, HPAF-II29. Cells were treated with 500 nM apricoxib and the level of VEGF produced was determined by ELISA. Only in high COX-2 cell line, Colo357, COX-2 inhibition reduces VEGF production transiently. In HPAF-II and AsPC-1 cells, VEGF production was unaffected by apricoxib, with VEGF production in HPAF-II cells elevated over time (Fig. 1c). To determine if EMT induction altered VEGF production and/or the effect of apricoxib, we plated Colo357 and AsPC-1 cells under conditions that stimulate EMT. Under normal culture conditions we observed similar trends as shown before. However, under EMT-inducing conditions VEGF production was elevated significantly and was largely independent of COX-2 inhibition in Colo357 cells. In AsPC-1 cells, VEGF production increased faster over time under induced EMT conditions compared to normal conditions (Fig. 1c). We also investigated the effect of apricoxib on PGE2 production by Colo357 and Aspc-1 cells under normal and EMT-inducing culture conditions (Supplementary Fig. S2a, b). The induction of EMT was confirmed by evaluating the expression level of E-Cadherin, N-Cadherin and Vimentin (Supplementary Fig. S2c). The induction of EMT reduced the effect of apricoxib on PGE2 production in Colo357 cells. In contrast, AsPC-1 cells produced minimal PGE2 under either culture condition (Supplementary Fig. S2a, b). We corroborated these findings by examining the level of VEGF expression in Colo357 pancreatic xenografts by immunofluorescence staining and found that VEGF expression was not affected by apricoxib (Fig. 1d). Importantly, apricoxib did reduce COX-2 expression in Colo357 tumors supporting the pharmacodynamic activity of the drug. The induction of hypoxia by mcr84 is consistent with prior studies9 and the reduction of microvessel density by mcr84 in Colo357 tumors (Supplementary Fig. S3a, b). We found that apricoxib alone did not reduce microvessel density in Colo357 tumors (Supplementary Fig. S3a, b), which further supports that apricoxib anti-tumor activity is not mediated by inhibition of angiogenesis. However, we did observe that apricoxib alone or in combination with mcr84 increased the percentage of pericyte-associated blood vessels in Colo357 tumors (Supplementary Fig. S3c). These data suggest that COX-2 functions in a VEGF-independent manner in PDA to promote tumor progression.
Apricoxib in combination with mcr84 reverses anti-VEGF–induced EMT and collagen deposition
Although anti-VEGF therapy with mcr84 restricts tumor growth and improves the survival of KIC mice9, therapy-induced hypoxia results in a less differentiated tumor cell phenotype9. We previously found that COX-2 inhibition with apricoxib reverses EMT in HT29 xenografts28 and Colo357 tumor-bearing mice29. To determine whether apricoxib can prevent or reduce hypoxia-induced epithelial plasticity as a result of mcr84 treatment, we analyzed tumor tissue from KIC mice in each treatment group. Treatment with mcr84 alone increased the expression of N-cadherin, a common marker of mesenchymal cells and Slug, an EMT-inducing transcription factor (EMT-TF). Apricoxib alone or in combination with mcr84 significantly reduced N-cadherin expression and downregulated Slug expression to the same level of control group. Although the expression of Snail, another EMT-TF43 was not affected by mcr84, treatment with apricoxib or apricoxib combined with mcr84 decreased Snail expression significantly (Fig. 2a). We also observed that collagen deposition was increased in KIC and Colo357 tumors after treatment with mcr84, a feature we identified previously that is associated with epithelial plasticity9. This effect was attenuated by apricoxib alone or in combination with mcr84 (Fig. 2b).
Figure 2. Apricoxib in combination with mcr84 reverses anti-VEGF–induced EMT and collagen deposition.

(a) KIC pancreatic tissues from the treated mice underwent immunohistochemistry for E-cadherin, N-cadherin, Slug or Snail. Images are shown at 400X. Scale bars are presented as indicated. (b) Pancreatic tissues from Colo357 tumor-bearing mice and KIC mice were stained with Masson’s trichrome. Total magnification is 200X. Scale bars are presented as indicated. The whole tumor areas were scanned with Hamamatsu Nanozoomer 2.0HT. Images of whole tumor areas were analyzed using ImageJ software. Quantification of percentage area fraction is shown. Data are displayed as mean ± SEM with 3 animals per group analyzed. *P < 0.05, **P < 0.01, ***P < 0.005 vs. control; by ANOVA with Dunn’s MCT.
Blockade of VEGF and COX-2 pathways promotes an immune stimulatory microenvironment
Eicosanoids, including PGE2, contribute to the immune microenvironment of solid tumors20. For example, PGE2 can induce a shift in cytokine expression in myeloid-derived suppressor cells (MDSCs) and macrophages towards an immune suppressive profile (e.g., IL-4, IL-10, IL-6) and PGE2 can directly reduce T effector cell activity44. Furthermore, EMT is also associated with an immunosuppressive tumor microenvironment31,32,34. Thus, given our observations that COX-2 inhibition with apricoxib reduces PGE2 production and decreases therapy-induced EMT, we investigated the immune landscape in KIC tumors from the different treatment groups shown in Figure 1. Tumors harvested from mice that received mcr84 or apricoxib alone had an increase in the number of tumor-associated CD3+ and CD8+ T cells. Combination therapy further elevated CD3+ and CD8+ T cell levels (Fig. 3a). Additionally, apricoxib alone and in combination with mcr84 significantly decreased FoxP3+ regulatory T (Treg) cells (Fig. 3a). Motz et al30 previously reported that selective expression of the death mediator Fas ligand (FasL) on endothelial cells in human and mouse solid tumors was associated with scarce T-cell infiltration. They also identified that FasL was induced on endothelium by VEGF, IL-10, and PGE2. Thus, we evaluated FasL expression in the vasculature of KIC tumors by dual staining of the endothelium for CD31 and FasL. We found that FasL was indeed present on CD31+ endothelial cells in control-treated KIC tumors and that treatment with mcr84, apricoxib, or the combination significantly reduced endothelial FasL expression (Fig. 3b). To determine the effect of VEGF blockade and COX-2 inhibition on macrophages in the tumor microenvironment, we stained for CD11b, iNOS, and Arginase 1. We found that mcr84 alone and mcr84 in combination with apricoxib reduced CD11b+iNOS+ macrophages but apricoxib alone did not. In contrast, mcr84 or apricoxib alone decreased CD11b+Arg1+ macrophages, while the effect was more significant with combination therapy (Fig. 3c). Although the number of total myeloid cells that were marked by CD11b was elevated in the combination treatment group, the total macrophage number (F4/80) was reduced with anti-VEGF and COX-2 inhibition (Supplementary Fig. S4a, b).
Figure 3. Combination blockade of VEGF and COX-2 pathway restores antitumor immunity.

KIC pancreatic tissue was subjected to immunohistochemistry for (a) CD3, CD8, FoxP3, (b) CD31 and FasL, (c) CD11b and iNOS, CD11b and Arginase 1. The whole tumor areas were scanned with Hamamatsu Nanozoomer 2.0HT and Zeiss Aixoscan Z1. Images of whole tumor areas were analyzed using NIS Elements (Nikon) and Fiji software. Representative images are shown at 400X in (a) and (b). Scale bars are presented as indicated. Schematic Quantification of percentage area fraction for each target in each treatment group is shown. Data are displayed as mean ± SEM with 3 animals per group analyzed. *P < 0.05, **P < 0.01, ***P < 0.005, ****P < 0.0001 vs. control, by ANOVA with Dunn’s MCT.
Discussion
Our data support that VEGF production by tumor cells is independent of COX-2, especially following COX-2 inhibition, and the data also strongly support that COX-2 activity on tumor cells is linked closely to the induction and/or maintenance of a less differentiated tumor cell phenotype. Epithelial plasticity is a common pathway exploited by tumors to resist therapeutic interventions, including chemotherapy and targeted therapy. Our data demonstrate that reducing hypoxia-induced epithelial plasticity by blocking COX-2 enhances the therapeutic activity of anti-VEGF in PDA. We have shown previously that anti-VEGF therapy (mcr84) of PDA induces hypoxia, which drives an increase of TGFβ and subsequent increase in collagen deposition. Furthermore we found that collagen and TGFβ in the tumor microenvironment stimulated tumor cell EMT9. Additionally, we previously reported that COX-2 inhibition (apricoxib) reduces EMT in models of GI cancer in vivo and TGFβ-induced EMT in vitro28,37. Therefore, we further investigated the effect of COX-2 inhibition on the level of active TGFβ in orthotopic Colo357 pancreatic tumors. We found that anti-VEGF (mcr84) increased active TGFβ levels, as anticipated but that this increase was blunted by COX-2 inhibition (data not shown), which suggests that COX-2 inhibition reduces EMT and immune suppression in part by reducing hypoxia-induced TGFβ expression (Supplementary Fig. S5). TGFβ, a multifunctional cytokine can drive tumor cell EMT and is also a potent immunosuppressive factor produced by tumor cells, fibroblasts and tumor-infiltrating lymphocytes45. TGFβ can inhibit innate and adaptive immune responses in the tumor microenvironment. For example, TGFβ can polarize macrophages towards an immunosuppressive phenotype, support regulatory T cell differentiation and directly inhibit effector T cell activity46. In addition, our results are consistent with reports that celecoxib, another selective COX-2 inhibitor, reduces hepatic expression of TGFβ thereby attenuating EMT of hepatocytes47. Furthermore, COX-2 has been shown to participate in TGFβ-driven EMT in human hepatocellular carcinoma48. Thus there are multiple examples of a connection between COX-2 activity and TGFβ-driven tumor progression.
We also found that COX-2 inhibition might reduce immune suppression in PDA. The immunosuppressive microenvironment is a major limitation for the efficacy of cancer immune therapy49. Our data are consistent with other studies that have shown that anti-angiogenic agents and COX-2 inhibitors have the potential to reduce the immunosuppressive tumor microenvironment and enhance immunotherapy50,51,52. Our results support the findings of Motz et al30, who found that pharmacologic blockade of VEGF and COX-2 resulted in a significant increase in infiltrating CD8+ T cells and a reduction in FoxP3+ Treg cells by downregulating FasL expression on tumor endothelial cells in multiple murine cancer models. Our data indicate that in KIC tumors, VEGF blockade or COX-2 inhibition alone could reduce FasL expression on the tumor endothelium but combination therapy resulted in higher T effector cell recruitment and lower Treg infiltration than single-agent therapy.
In summary, our data support the rationale of a combination of anti-VEGF and COX-2 inhibition in PDA patients and also provide evidence that this combination might prime PDA or other tumors for increased efficacy with immune therapy.
Supplementary Material
Implications:
Together, these findings demonstrate that COX-2 inhibition enhances the efficacy of anti-VEGF therapy by reducing hypoxia-induced epithelial plasticity and promoting an immune landscape that might facilitate immune activation.
Acknowledgements
We acknowledge helpful discussions from members of the Brekken laboratory and editorial assistance by Dave Primm in the Department of Surgery at UT Southwestern.
Financial support: The work was supported by NIH grants R01 CA192381 and U54 CA210181 (PI: M. Ferrari) to R.A. Brekken, T32 CA136515 (PI: J. Schiller) to A. Kirane, F31 CA19603301 to N.B. Sorrelle, a sponsored research agreement from Tragara Pharmaceuticals, Inc., to R.A. Brekken, and the Effie Marie Cain Scholarship in Angiogenesis Research to R.A. Brekken. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Disclosure of potential conflicts of interest: R.A. Brekken declares research funding from Tragara Pharmaceuticals, Inc. Additionally, F.J. Burrows was an employee of Tragara Pharmaceuticals at the time of the majority of the studies. The remaining authors have declared that no conflict of interest exists.
References
- 1.Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307(5706):58–62. doi: 10.1126/science.1104819 [DOI] [PubMed] [Google Scholar]
- 2.Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473(7347):298–307. doi: 10.1038/nature10144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nagy JA, Dvorak AM, Dvorak HF. VEGF-A and the Induction of Pathological Angiogenesis. Annu Rev Pathol Mech Dis. 2007;2(1):251–275. doi: 10.1146/annurev.pathol.2.010506.134925 [DOI] [PubMed] [Google Scholar]
- 4.Kerbel RS. Tumor Angiogenesis. N Engl J Med. 2008;358(19):2039–2049. doi: 10.1056/NEJMra0706596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goel HL, Mercurio AM. VEGF targets the tumour cell. Nat Rev Cancer. 2013;13(12):871–882. doi: 10.1038/nrc3627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Welti J, Loges S, Dimmeler S, Carmeliet P. Recent molecular discoveries in angiogenesis and antiangiogenic therapies in cancer. J Clin Invest. 2013;123(8):3190–3200. doi: 10.1172/JCI70212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lambrechts D, Lenz H-J, de Haas S, Carmeliet P, Scherer SJ. Markers of Response for the Antiangiogenic Agent Bevacizumab. J Clin Oncol. 2013;31(9):1219–1230. doi: 10.1200/JCO.2012.46.2762 [DOI] [PubMed] [Google Scholar]
- 8.Pàez-Ribes M, Allen E, Hudock J, et al. Antiangiogenic Therapy Elicits Malignant Progression of Tumors to Increased Local Invasion and Distant Metastasis. Cancer Cell. 2009;15(3):220–231. doi: 10.1016/j.ccr.2009.01.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aguilera KY, Rivera LB, Hur H, et al. Collagen signaling enhances tumor progression after anti-VEGF therapy in a murine model of pancreatic ductal adenocarcinoma. Cancer Res. 2014;74(4):1032–1044. doi: 10.1158/0008-5472.CAN-13-2800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30. doi: 10.3322/caac.21387 [DOI] [PubMed] [Google Scholar]
- 11.Steeg PS. Targeting metastasis. Nat Publ Gr. 2016;16. doi: 10.1038/nrc.2016.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feng M, Xiong G, Cao Z, et al. PD-1/PD-L1 and immunotherapy for pancreatic cancer. Cancer Lett. August 2017. doi: 10.1016/j.canlet.2017.08.006 [DOI] [PubMed] [Google Scholar]
- 13.Foley K, Kim V, Jaffee E, Zheng L. Current progress in immunotherapy for pancreatic cancer. Cancer Lett. 2016;381(1):244–251. doi: 10.1016/j.canlet.2015.12.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Van Cutsem E, Vervenne WL, Bennouna J, et al. Phase III Trial of Bevacizumab in Combination With Gemcitabine and Erlotinib in Patients With Metastatic Pancreatic Cancer. J Clin Oncol. 2009;27(13):2231–2237. doi: 10.1200/JCO.2008.20.0238 [DOI] [PubMed] [Google Scholar]
- 15.Kindler HL, Niedzwiecki D, Hollis D, et al. Gemcitabine plus bevacizumab compared with gemcitabine plus placebo in patients with advanced pancreatic cancer: phase III trial of the Cancer and Leukemia Group B (CALGB 80303). J Clin Oncol. 2010;28(22):3617–3622. doi: 10.1200/JCO.2010.28.1386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wolfgang CL, Herman JM, Laheru DA, et al. Recent progress in pancreatic cancer. CA Cancer J Clin. 2013;63(5):318–348. doi: 10.3322/caac.21190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sullivan LA, Carbon JG, Roland CL, et al. r84, a novel therapeutic antibody against mouse and human VEGF with potent anti-tumor activity and limited toxicity induction. Gartel AL, ed. PLoS One. 2010;5(8):e12031. doi: 10.1371/journal.pone.0012031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 19.Sobolewski C, Cerella C, Dicato M, Ghibelli L, Diederich M. The role of cyclooxygenase-2 in cell proliferation and cell death in human malignancies. Int J Cell Biol. 2010;2010:215158. doi: 10.1155/2010/215158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang D, Dubois RN. Eicosanoids and cancer. Nat Rev Cancer. 2010;10(3):181–193. doi: 10.1038/nrc2809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eberhart CE, Coffey RJ, Radhika A, Giardiello FM, Ferrenbach S, DuBois RN. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology. 1994;107(4):1183–1188. http://www.ncbi.nlm.nih.gov/pubmed/7926468. Accessed April 20, 2017. [DOI] [PubMed] [Google Scholar]
- 22.Lunardi S, Muschel RJ, Brunner TB. The stromal compartments in pancreatic cancer: are there any therapeutic targets? Cancer Lett. 2014;343(2):147–155. doi: 10.1016/j.canlet.2013.09.039 [DOI] [PubMed] [Google Scholar]
- 23.de Groot DJA, de Vries EGE, Groen HJM, de Jong S. Non-steroidal anti-inflammatory drugs to potentiate chemotherapy effects: from lab to clinic. Crit Rev Oncol Hematol. 2007;61(1):52–69. doi: 10.1016/j.critrevonc.2006.07.001 [DOI] [PubMed] [Google Scholar]
- 24.Ghosh N, Chaki R, Mandal V, Mandal SC. COX-2 as a target for cancer chemotherapy. Pharmacol Rep. 62(2):233–244. http://www.ncbi.nlm.nih.gov/pubmed/20508278. Accessed April 20, 2017. [DOI] [PubMed] [Google Scholar]
- 25.Xu L, Stevens J, Hilton MB, et al. COX-2 inhibition potentiates antiangiogenic cancer therapy and prevents metastasis in preclinical models. Sci Transl Med. 2014;6(242):242ra84. doi: 10.1126/scitranslmed.3008455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khan Z, Khan N, Tiwari RP, Sah NK, Prasad GBKS, Bisen PS. Biology of Cox-2: an application in cancer therapeutics. Curr Drug Targets. 2011;12(7):1082–1093. http://www.ncbi.nlm.nih.gov/pubmed/21443470. Accessed April 21, 2017. [DOI] [PubMed] [Google Scholar]
- 27.Ben-Batalla I, Cubas-Cordova M, Udonta F, et al. Cyclooxygenase-2 blockade can improve efficacy of VEGF-targeting drugs. Oncotarget. 2015;6(8):6341–6358. doi: 10.18632/oncotarget.3437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kirane A, Toombs JE, Larsen JE, et al. Epithelial-mesenchymal transition increases tumor sensitivity to COX-2 inhibition by apricoxib. Carcinogenesis. 2012;33(9):1639–1646. doi: 10.1093/carcin/bgs195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kirane A, Toombs JE, Ostapoff K, et al. Apricoxib, a novel inhibitor of COX-2, markedly improves standard therapy response in molecularly defined models of pancreatic cancer. Clin Cancer Res. 2012;18(18):5031–5042. doi: 10.1158/1078-0432.CCR-12-0453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Motz GT, Santoro SP, Wang L-P, et al. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med. 2014;20(6):607–615. doi: 10.1038/nm.3541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dongre A, Rashidian M, Reinhardt F, et al. Epithelial-to-Mesenchymal Transition Contributes to Immunosuppression in Breast Carcinomas. Cancer Res. 2017;77(15):3982–3989. doi: 10.1158/0008-5472.CAN-16-3292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kudo-Saito C, Shirako H, Takeuchi T, Kawakami Y. Cancer Metastasis Is Accelerated through Immunosuppression during Snail-Induced EMT of Cancer Cells. Cancer Cell. 2009;15(3):195–206. doi: 10.1016/j.ccr.2009.01.023 [DOI] [PubMed] [Google Scholar]
- 33.Sangaletti S, Tripodo C, Santangelo A, et al. Mesenchymal Transition of High-Grade Breast Carcinomas Depends on Extracellular Matrix Control of Myeloid Suppressor Cell Activity. Cell Rep. 2016;17(1):233–248. doi: 10.1016/j.celrep.2016.08.075 [DOI] [PubMed] [Google Scholar]
- 34.Thiery JP, Acloque H, Huang RYJ, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871–890. doi: 10.1016/j.cell.2009.11.007 [DOI] [PubMed] [Google Scholar]
- 35.Aguilera KY, Rivera LB, Hur H, et al. Collagen signaling enhances tumor progression after anti-VEGF therapy in a murine model of pancreatic ductal adenocarcinoma. Cancer Res. 2014;74(4):1032–1044. doi: 10.1158/0008-5472.CAN-13-2800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aguirre AJ, Bardeesy N, Sinha M, et al. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003;17(24):3112–3126. doi: 10.1101/gad.1158703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kirane A, Toombs JE, Ostapoff K, et al. Apricoxib, a novel inhibitor of COX-2, markedly improves standard therapy response in molecularly defined models of pancreatic cancer. Clin Cancer Res. 2012;18(18):5031–5042. doi: 10.1158/1078-0432.CCR-12-0453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gately S The contributions of cyclooxygenase-2 to tumor angiogenesis. Cancer Metastasis Rev. 2000;19(1–2):19–27. http://www.ncbi.nlm.nih.gov/pubmed/11191059. Accessed August 14, 2017. [DOI] [PubMed] [Google Scholar]
- 39.Gately S, Li WW. Multiple roles of COX-2 in tumor angiogenesis: a target for antiangiogenic therapy. Semin Oncol. 2004;31(2 Suppl 7):2–11. http://www.ncbi.nlm.nih.gov/pubmed/15179620. Accessed August 14, 2017. [DOI] [PubMed] [Google Scholar]
- 40.Cheng T, Cao W, Wen R, Steinberg RH, LaVail MM. Prostaglandin E2 induces vascular endothelial growth factor and basic fibroblast growth factor mRNA expression in cultured rat Müller cells. Invest Ophthalmol Vis Sci. 1998;39(3):581–591. http://www.ncbi.nlm.nih.gov/pubmed/9501870. Accessed July 21, 2018. [PubMed] [Google Scholar]
- 41.Jones MK, Wang H, Peskar BM, et al. Inhibition of angiogenesis by nonsteroidal anti-inflammatory drugs: insight into mechanisms and implications for cancer growth and ulcer healing. Nat Med. 1999;5(12):1418–1423. doi: 10.1038/70995 [DOI] [PubMed] [Google Scholar]
- 42.Williams CS, Tsujii M, Reese J, Dey SK, DuBois RN. Host cyclooxygenase-2 modulates carcinoma growth. J Clin Invest. 2000;105(11):1589–1594. doi: 10.1172/JCI9621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ye X, Tam WL, Shibue T, et al. Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nature. 2015;525(7568):256–260. doi: 10.1038/nature14897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang D, Dubois RN. Eicosanoids and cancer. Nat Rev Cancer. 2010;10(3):181–193. doi: 10.1038/nrc2809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limón P. The polarization of immune cells in the tumour environment by TGFbeta. Nat Rev Immunol. 2010;10(8):554–567. doi: 10.1038/nri2808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li MO, Wan YY, Sanjabi S, Robertson A-KL, Flavell RA. TRANSFORMING GROWTH FACTOR-β REGULATION OF IMMUNE RESPONSES. Annu Rev Immunol. 2006;24(1):99–146. doi: 10.1146/annurev.immunol.24.021605.090737 [DOI] [PubMed] [Google Scholar]
- 47.Wen SL, Gao JH, Yang WJ, et al. Celecoxib attenuates hepatic cirrhosis through inhibition of epithelial-to-mesenchymal transition of hepatocytes. J Gastroenterol Hepatol. 2014;29(11):1932–1942. doi: 10.1111/jgh.12641 [DOI] [PubMed] [Google Scholar]
- 48.Sambasivarao SV Cox-2 and Akt mediate multiple growth factor-induced epithelial-mesenchhymal transition in human hepatocellular carcinoma. J Gastroenterol Hepatol. 2013;18(9):1199–1216. doi: 10.1111/j.1440-1746.2011.06980.x.COX-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kerkar SP, Restifo NP. Cellular constituents of immune escape within the tumor microenvironment. Cancer Res. 2012;72(13):3125–3130. doi: 10.1158/0008-5472.CAN-11-4094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huang Y, Yuan J, Righi E, et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc Natl Acad Sci U S A. 2012;109(43):17561–17566. doi: 10.1073/pnas.1215397109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sharma S, Stolina M, Yang S-C, et al. Tumor Cyclooxygenase 2-dependent Suppression of Dendritic Cell Function. Clin Cancer Res. 2003;9(3). http://clincancerres.aacrjournals.org/content/9/3/961.long. Accessed August 27, 2017. [PubMed] [Google Scholar]
- 52.Chen JH, Perry CJ, Tsui Y-C, et al. Prostaglandin E2 and programmed cell death 1 signaling coordinately impair CTL function and survival during chronic viral infection. Nat Med. 2015;21(4):327–334. 10.1038/nm.3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.