

Abstract
Downregulation of the urea cycle enzyme argininosuccinate synthase (ASS1) by either promoter methylation or by HIF1α is associated with increased metastasis and poor prognosis in multiple cancers. We have previously shown that in normoxic conditions, ASS1 downregulation facilitates cancer cell proliferation by increasing aspartate availability for pyrimidine synthesis by the enzyme complex CAD. Here we report that in hypoxia, ASS1 expression in cancerous cells is downregulated further by Hif1α-mediated induction of miR224-5p, making the cells more invasive and dependent on upstream substrates of ASS1 for survival. ASS1 was downregulated under acidic conditions, and ASS1-depleted cancer cells maintained a higher intracellular pH (pHi), depended less on extracellular glutamine, and displayed higher glutathione levels. Depletion of substrates of urea cycle enzymes in ASS1-deficient cancers decreased cancer cell survival. Thus, ASS1 levels in cancer are differentially regulated in various environmental conditions to metabolically benefit cancer progression. Understanding these alterations may help uncover specific context-dependent cancer vulnerabilities that may be targeted for therapeutic purposes.
Introduction
The urea cycle (UC) is the main metabolic pathway in mammals, responsible for detoxifying excess nitrogen, carried in the forms of glutamine and ammonia, by converting it to urea. The UC enzyme argininosuccinate synthase (ASS1) catalyzes an important step in this cycle, which conjugates nitrogen from two sources – aspartate, derived from glutamine, and citrulline, containing nitrogen from ammonia – to form argininosuccinate (1, 2) (Figure 1A). The importance of ASS1 for normal nitrogen metabolism is underscored by manifestations of citrullinemia type I, an inborn error of metabolism (IEM) caused by biallelic germline mutations in ASS1. Individuals with citrullinemia type I present biochemically with elevated plasma levels of ammonia and citrulline, and clinically with neurological impairment and even death, when the enzymatic deficiency is severe. However, counter-intuitively to its critical role in normal homeostasis, ASS1 is downregulated in multiple diverse cancers, making tumors auxotrophic for arginine (3, 4). This feature has been exploited for therapy in the treatment of individuals with ASS1-deficient tumors, by employing arginine-depleting agents (5).
Fig. 1: ASS1 expression is downregulated during hypoxia and acidic states.
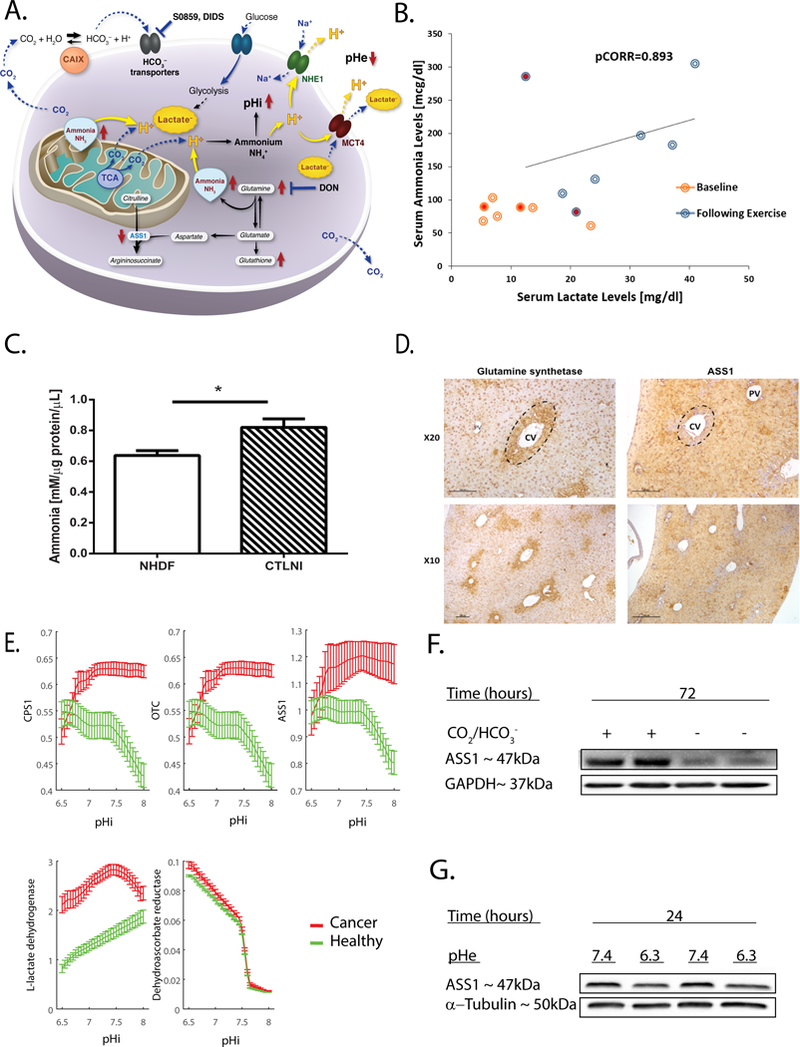
(A) Illustration of the hypothesized metabolic effects of ASS1 downregulation on cancer pH gradient. During aerobic metabolism, hydrated CO2 is a main cellular source of acidity. Inactivation of ASS1 in cancer would be expected to lead to accumulation of upstream metabolites such as glutamine and ammonia (derived from glutamine). Increase in these metabolites could be vital for the pH gradient in the maintenance of an alkalized pHi that is crucial for cancer cell survival, invasion and migration. In contrast, depletion of ASS1’s upstream substrates in ASS1 deficient cancer cells would be expected to confer vulnerability. Abbreviations: ASS1 (Argininosuccinate Synthase 1), pHi (pH intracellular), pHe (pH extracellular), TCA (tricarboxylic acid), NHE-1 (Na(+)/H(+) Exchanger 1), MCT4 (monocarboxylate transporter 4), CAIX (Carbonic anhydrase 9), DIDS (4,4′-Diisothiocyanatostilbene-2,2′-disulfonic acid), DON (6-Diazo-5-oxo-L-norleucine). (B) The FIT was performed for individuals who presented at the Metabolic Disease Unit of the Sheba Medical Center for evaluation of elevated serum Creatine Phosphokinase levels. The graph demonstrates the results of seven male subjects tested in the last five years in the age range of 8–28 years. Five out of the tested subjects were considered to have normal results since they had a coupled elevation of both lactate and ammonia in their serum after three minutes of exercise (blue circles) as compared to baseline levels (orange circles), while two subjects depicted by the filled red circles showed elevation of lactate without the expected accompanying elevation of ammonia levels and was thus suspected to have inborn error of metabolism. The normal ranges for serum lactate and ammonia levels are 6.0–18.0 mg/dl and 31.0–123.0 mcg/dl, respectively. pCORR=0.893 was calculated for healthy subjects following after three minutes of exercise. pCORR=0.311 was calculated for all tested subjects including those suspected to have IEM. (C) Extracellular ammonia measurements in media of fibroblasts generated from a patient with CTLN I as compared to normal human dermal fibroblasts (NHDF). The experiment was performed in triplicates and repeated twice. (D) A representative immunohistochemistry staining (DAB) of a wild type mouse liver for ASS1 and glutamine synthetase (GS). Magnification X10, X20. Portal Vein (PV); Central Vein (CV); glutamine synthetase is used as a CV biomarker. (E) The predicted metabolic activity of the three proximal urea cycle enzymes CPS1, OTC, and ASS1 following pHi changes. The activity is predicted to decrease in cancer cells with acidic pHi, in comparison to normal healthy cells (green), while that of Glutathione dehydrogenase (Dehydroascorbate reductase), is predicted to increase in acidic pH in both cancer and healthy cells. The activity of lactate dehydrogenase (LDH), which has been reported to decrease in cancer cells with acidity(31), serves as an internal control for the other predictions. Fluxes are given in mmol/mgDW/h. Mean values and their corresponding standard deviations are estimated across a dozens of genome-scale metabolic models, representing cell lines from each of the NCI-60 and HapMap panels. (F) A representative western blot image showing ASS1 protein levels extracted from osteosarcoma cells following 72 hours of incubation with or without bicarbonate in the media and CO2 in the incubator (−CO2/−HCO3− or +CO2/+HCO3− conditions). ASS1 protein levels are normalized to the expression levels of GAPDH (G) A representative western blot image showing ASS1 protein levels extracted from osteosarcoma cells following 24 hours of incubation in either pHe 7.4 or pHe 6.3 media using 25mM PIPES. ASS1 protein levels are normalized to the expression levels of α−tubulin. We used student’s t-test (2-tail) for statistical analysis in (C). The graph is presented as an average ± S.D. *, P ≤ 0.05.
We recently determined that ASS1 deficiency under normoxic conditions promotes proliferation of cancer cells by increasing the availability of the ASS1 substrate- aspartate, for pyrimidine synthesis via the carbamoyl-phosphate synthase 2-aspartate transcarbamylase-dihydroorotase (CAD) complex (6). Interestingly, under hypoxic conditions, when decreased proliferation is seen, hypoxia inducible factor alpha (HIF1α) has been shown to bind and downregulate the expression levels of both ASS1 and CAD (7–9). Thus, we set out to investigate the metabolic advantages that ASS1 downregulation confers on cancer cells, under acidic and hypoxic conditions.
Tumor hypoxia has been shown to promote increased acidosis by inducing a shift in cellular metabolism that generates a high acid load (10). Recent findings proposed that glutamine plays an important role in increasing cellular compatibility with an acidic environment, by releasing ammonia to neutralize protons derived from acids (11, 12). In addition, the recycling of glutamine-derived ammonia has been shown to support cancer biomass through its incorporation into amino acids, rather than its disposal as a waste product (13). Based on these findings, and the biochemical abnormalities found in ASS1-deficient patients, we hypothesized that under hypoxia-related acidosis, the metabolic benefits of ASS1 downregulation involve the build-up of ASS1 upstream metabolites – glutamine and glutamine derived ammonia– thus enabling cancer cell survival (Figure 1A).
Materials and Methods
Metabolic measurements in human subjects and cells-
The Forearm Ischemic Test (FIT) was performed as part of the clinical evaluation of patients who presented due to unexplained consistent or recurrent elevations of serum CPK levels, at the Metabolic Disease Unit of the Sheba Medical Center, Israel.
Ammonia measurements from fibroblasts were performed on anonymized cells devoid of all identifiers. Cells were cultured in a 10 cm plate to a confluence of 80%. At time 0, the cells were rinsed with PBS, fed with 5 mL of the test medium (20% FBS contained DMEM) and cultured for 24h. Ammonia was measured in the test medium with a colorimetric assay (Abcam). Ammonia concentration in the cells’ free medium was subtracted.
Metabolic modelling-
pH profiles of metabolic enzymes of all organisms were fetched from BRENDA database. These experimental in vitro measurements included at most 6 discrete, 3 acidic and 3 basic pH points corresponding to 0%, 50% and 100% of enzymatic activity. When no experimental value for human was available for one or more of these points, we used the available data of the closest homolog. pH activity profiles of enzymes at any given pH were then linearly interpolated (14).
We applied pH-activity profiles into a panel of genome-scale metabolic models (GSMM), developed recently based on the human GSMM, representing the NCI-60 cell-lines panel and the normal healthy lymphoblastic cell-lines from the HapMap project. In a given intracellular pH (pHi), the pH activity profiles were used to adjust the permissible flux bounds of each reaction in the GSMM, allowing us to estimate the metabolic state of the network at each pHi. Reactions’ fluxes were measured using flux-variability analysis (FVA) under standard constraint of cell viability, of cellular proliferation (Biomass yield) > 80% of its maximum (14).
TCGA tumor data analysis-
We downloaded TCGA gene expression profiles of 8,224 patients encompassing 27 cancer types from the Broad Firehose resources (analyzed on Jan 28, 2016, doi:10.7908/C11G0KM9). Gene expression values are normalized in each cancer type to account for the distinct basal expression levels in different cancer types. A gene is determined to be downregulated/ upregulated in a sample if its gene expression is below bottom tertile/ above top tertile across samples in each cancer type. For metastasis analysis, we used gene expression profiles of 648 patients across 14 cancer types. A sample is termed metastatic if the tumor is diagnosed stage IV, and we only considered those cancer types for which greater than or equal to 10 such samples are available. To overcome the differences in tumor heterogeneity, we used the tumor samples with high cancer cell fraction (15).
Hypoxia score-
We used gene expression signature of hypoxia to calculate the hypoxia score (16). We took the mean expression level of the signature genes and rank-normalized the values in each cancer type to control for cancer-type specific effect. To make sure our findings are not limited to HIF1α target genes, we repeated the same analysis with the signature genes excluding validated HIF1α target genes.
Patient survival analysis-
We performed two different analyses to study the association between ASS1 together with CA9 and MCT4 and patient survival: Kaplan Meier analysis and Cox proportional hazard model. We compared the survival of patients with high CA9 (top tertile, n=932 patients) and low-CA9 (bottom tertile, n=905 patients) among the ASS1 downregulated samples (bottom tertile) using logrank test (17), and the effect size was quantified by the difference in the area under the curves (ΔAUC). To control for potential confounders, we performed a Cox regression analysis, while controlling for patients’ age, sex, race, and tumor’s genomic instability and the tissue of origin.
| (1) |
where is an indicator variable over all possible combinations of patients’ stratifications based on race, sex and cancer type. is the hazard function (defined as the risk of death of patients per time unit), and is the baseline-hazard function at time t of the sth stratification. The model contains three covariates: (i) CA9: CA9 expression, (ii) age: age of the patient, (iii) GII: genomic instability index. GII measures the relative amplification or deletion of genes in a tumor based on the SCNA. Given be the absolute of log ratio of SCNA of gene i in a sample relative to normal control, GII of the sample is given as in Bilal et al.(18):
| (2) |
The s are the regression coefficients of the covariates, which quantify the effect of covariates on patient survival, determined by standard likelihood maximization (19). We performed a similar analysis to identify the association with survival of MCT4 (low MCT4: n=905 patients; High MCT4: n=931 patients) and CA9+MCT4 combined expression (low CA9+MCT4: n=905 patients; High CA9+MCT4: n=932 patients) with patient survival. We performed a control analysis by repeating the same analysis with the samples that overexpress ASS1 (top tertile).
Metabolomics analysis-
Osteosarcoma cells were seeded at 8*105 cells per 10 cm plate and incubated with 6.5 mL fresh HCO3− free RPMI (Biological Industries 01–104-5A) under a CO2 free atmosphere for 72 h. Subsequently, medium aliquots were collected for glutamine uptake quantification by amino acid analyzer, (Biochrome 30+, Biochrome, UK). For intracellular glutamine levels, cells were than washed with ice cold saline, lysed with a solution of 50% methanol in water with ribitol as internal standard (2 μg/mL) and quickly scraped followed by three freeze thaw cycles in liquid nitrogen. The insoluble material was pelleted in a cooled centrifuge (4 °C) and the supernatant was collected for consequent GC-MS analysis. Samples were dried under airflow at 42°C using Techne Dry-Block Heater with sample concentrator (Bibby Scientific Limited, UK).
Gas chromatography/mass spectrometry; Cellular glutamine measurements-
Dried samples were treated with 40 μl of a methoxyamine hydrochloride solution (20 mg/ml in pyridine) at 37 °C for 90 min while shaking followed by incubation with 70 μl N, O-Bis (trimethylsilyl) trifluoroacetamide (Sigma) at 37 °C for additional 30 min. Samples were then centrifuged and allowed to stand at room temperature for 2 hours before injection. The results were normalized to ribitol as an internal standard and to the protein levels of each sample. GC/MS analysis was performed using a gas chromatograph (7820AN, Agilent Technologies, USA) interfaced with a mass spectrometer (5975 Agilent Technologies, USA). A HP-5ms capillary column 30 m × 250 µm × 0.25 µm (19091S-433, Agilent Technologies, USA) was used. Helium carrier gas was maintained at a constant flow rate of 1.0 mL min−1. The GC column temperature was programmed from 70 to 150 °C via a ramp of 4 °C min−1, 150–215 °C via a ramp of 9 °C min−1, 215–310 °C via a ramp of 25 °C min−1 and maintained at 310 °C for additional 5 min. The electron impact ionization MS operated in full scan mode from m/z, 30–500. The inlet and MS transfer line temperatures were maintained at 250 °C and 310 °C, respectively. The ion source temperature was 280 °C. Sample injection (1–2 μL) was in splitless mode.
Glutamine uptake measurements-
100 μL medium from cell culture was mixed 1:1 with 5% sulfur salicylic acid (Sigma) containing 250 μM of Norleucine (Sigma) as internal standard. The mixture was allowed to stand at 40C for 30 min. The precipitate was centrifuged for 10 min at 10000g at 40C. The supernatant was taken and filtered through 0.2 μm membrane filter and injected into amino acid analyzer (Biochrom 30+). Results were normalized to Norleucine and to the protein levels of each sample. Background glutamine measurements from cell free culture were subtracted.
Glutamine measurements in tumors-
Following euthanization, tumors were removed and incubated for six hours with freshly prepared RPMI supplemented with 4mM L-GLUTAMINE, (ALPHA-15N, 98%, Cambridge Isotope Laboratories, Inc) and dialyzed FBS. Next, tumors were grinded using a stainless steel mesh grid and ice cold saline to produce single cell suspensions. Cells were further washed twice with saline and pelleted. The pellet was lysed with a solution of 50% methanol in water with ribitol as internal standard (2 μg/mL), followed by three freeze thaw cycles in liquid nitrogen. The insoluble material was pelleted in a cooled centrifuge (4 °C) and the supernatant was collected, dried under air flow at 42°C and subjected to GC-MS analysis. Total glutamine masses were normalized to ribitol and to the protein levels of each tumor.
Cell cultures-
All cell lines were authenticated and cultured using standard procedures: 370C humidified incubator with 5% CO2, RPMI (Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum, 1% pen-strep, 10mM glucose and 4 mM glutamine. B16-F10 cells were cultured using DMEM (Gibco) supplemented with the same ingredients. MNNG/HOS, LOX-IMVI, Malme-3M, OVCAR-3, MCF-7, U2OS and B16-F10 cell lines were purchased from the American Type Culture Collection (ATCC). Murine colon MC-38 and Lewis Lung Carcinoma (LLC) cell lines were a kind gift from the laboratories of Profs. Eran Elinav and Lea Eisenbach (Weizmann Institute), respectively. All cells were routinely tested for mycoplasma using a Mycoplasma EZ-PCR test kit (20–700-20, Biological Industries).
Viability assays-
HCO3− free-buffered solution contained RPMI solution w/o glutamine and bicarbonate (Biological Industries 01–104-5A) supplemented with 4 mM glutamine, 1% pen-strep, 10% heat-inactivated fetal bovine serum and titrated to pH 7.4 using 5M NaOH. For control conditions, we used 22 mM HCO3− contained RPMI solution supplemented with the same ingredients. CO2/HCO3− buffers contained RPMI w/o glutamine and bicarbonate (Sigma R7755) supplemented with 4 mM glutamine, 1% pen-strep, 10% heat-inactivated fetal bovine serum and either 22mM HCO3− (pH 7.4) or 1.75 mM HCO3− (pH 6.3) in accordance with the Henderson-Hasselbalch equation . Buffers were left under 5% CO2/95% atmosphere and the pH was adjusted using 1M KOH and 6M HCl. For cell viability assays done under hypoxia, we used RPMI solution w/o glutamine and glucose (Sigma), supplemented with 11 mM glucose, 1% pen-strep, 10% FBS and with or without 4 mM glutamine.
For experiments done under −CO2/−HCO3− vs. +CO2/+HCO3−: Cells were seeded in 6 well plates at 8*104 cells /well in triplicates. After 24 h for adherence of the cells (time 0), cells were washed and the medium was replaced to 1.5 mL of either fresh HCO3− free RPMI or HCO3− contained RPMI (as described before), respectively, and the cells were incubated under the indicated atmosphere, for 72 h. For experiments done under pHe 7.4 (22mM HCO3−; 5% CO2) vs. pHe 6.3 (1.75 mM HCO3−; 5% CO2): 200K cells per well were plated in six well plates in triplicates. After 24 h for adherence of the cells (time 0), cells were washed once with saline solution (0.9%) and incubated under the bicarbonate buffers at the specified pHe for 24 h. At the end of the incubation timings and following several washings, cells were fixated with 4 % Paraformaldehyde and further stained with crystal violet (0.2 %). Color de-staining was performed using 10% acetic acid in H2O. Absorbance was measured at 590 nm. For experiments done under hypoxic conditions with the five different human cell lines: cells were seeded in six well plates at 8*104 cells per well in triplicates. After 24 h (time 0), cells were washed with PBS and the medium was replaced to 1.5 mL of freshly prepared RPMI with or without glutamine, for 48 hours under hypoxic conditions (1% O2) followed by crystal violet staining and de-staining. In the case of control vs. shASS1 MCF-7 cells with either glutamine depletion, DON, S0859 or DIDS under hypoxic conditions: 1.8*105 cells per well in triplicates were seeded. Time 0 was considered after 24 h for adherence of the cells and the cells were incubated with freshly prepared RPMI as described before, using the indicated different inhibitors.
Invasion assays-
U2OS pLKO empty vector (EV) and shASS1 osteosarcoma cells were seeded at 100,000 cells/well in twelve-well plates. At the following day, cells were incubated for 24 hours under either normoxic or hypoxic conditions (1% O2). Then, cells were trypsinized and seeded in duplicates or triplicates in 24-well transwell® (Corning) coated with 50% Matrigel (BD) in the absence of serum in the upper chamber and with 20% serum in the lower chamber as an attractant, followed by another 24 hours incubation under the same conditions. For the miR experiments, U2OS osteosarcoma cells were seeded at 50,000 cells/well in 12-well plates. At the following day, cells were transfected with either 300pmol (diluted in Dharmacon Si buffer X1) non-targeting control (Dharmacon miRIDIAN miRNA Mimic Negative Control) or miR224-5p (Dharmacon miRIDIAN miRNA Mimic). 48 hours post transfection, cells were trypsinized and seeded in duplicates or triplicates in 24-well transwells® (Corning) as described before, followed by another 24 hours incubation under normoxic conditions. For all experiments, we additionally seeded cells in regular 24-well plates in parallel to seeding the cells in 24-well transwells. At the end of incubation, transwells were washed with PBS, fixated in 4% Paraformaldehyde and then stained with 0.2% Crystal violet. Cells that were stained on the lower chamber outer part were captured with an inverse light microscope nicon™ camera and quantified by ImageJ. The percentage of invading cells was calculated as .
Migration assays-
U2OS osteosarcoma cells were seeded at 100,000 cells per well in six well plates. At the following day, cells were transfected with either 300pmol miR224-5p or non-targeting (NT) control and incubated for 24 hours. Then, cells were trypsinized and equal amount of cells was seeded in the upper chamber in a technical triplicate using a 24-transwell plate (Corning) and in the absence of serum. As an attractant we used 10% serum in the lower chamber and incubated the cells for another 24-hours under normoxic conditions. At the end of incubation, transwells were washed with PBS, fixated in 4% Paraformaldehyde and then stained with 0.2% Crystal violet. The migrated cells were stained on the lower chamber outer part and were captured with an inverse light microscope Nikon™ camera. Quantification was done using ImageJ.
pHi measurements-
pH 7.4 HEPES buffer contained 25 mM HEPES, 140 mM NaCl, 5 mM KCl, 1mM MgSO4, 10 mM Glucose and last to add 2mM CaCl2. CO2/HCO3− buffers (bicarbonate buffers) were prepared by bubbling 5% CO2/95% air and contained the amount of NaHCO3− that yielded the desired pH according to the Henderson-Hasselbalch equation: pH = 10−6.15 + log ( [HCO3−]/ [CO2]). Specifically, pH 7.4 CO2/HCO3− buffer contained: 22 mM HCO3−, 115 mM NaCl, 4.5 mM KCl, 11 mM Glucose, 1 mM MgCl2, 1 mM CaCl2; pH 6.6 CO2/HCO3− buffer contained: 3.5 mM HCO3−, 133.5 mM NaCl, 4.5 mM KCl, 11 mM Glucose, 1 mM MgCl2, 1 mM CaCl2. NaCl final concentration was changed in order to allow final osmolarity of 300 mOsm/kg. Nigericin buffer contained 10 μM nigericin in: 25 mM HEPES, 105 mM KCl and 1mM MgCl2. Nigericin buffers were titrated to the required pH using 1M KOH and 1M HCl.
PIPES buffers contained RPMI solution w/o glutamine and bicarbonate (Biological Industries 01–104-5A) and supplemented with 25 mM PIPES, 22 mM HCO3−, 4 mM glutamine, 1% pen-strep, 10% heat-inactivated fetal bovine serum. PIPES buffers were titrated to pH 7.4 or pH 6.3 using 1M KOH or 6M HCl, respectively. In the case of U2OS pLKO empty vector (EV) and shASS1 osteosarcoma under −CO2/−HCO3− conditions, cells were washed twice with HEPES buffer and then Carboxy SNARF-1 AM (5 μM in HEPES buffer) was loaded on the cells as the membrane permeate ester for 5–10 min in 37 0C. Next, cells were washed twice with HEPES buffer before incubation with a fresh HEPES buffer in 37 0C. Cell imaging was performed using 24-well glass bottom black plates (Cellvis) in an Operetta automated imaging system (PerkinElmer) at 40x magnification equipped with a temperature control chamber and with the appropriate excitation and emission filters (Ex 520–550 nm; Em 585–605 nm and 650–700 nm). Each field was defined as a region of interest (ROI). At least five different ROIs with 50 % confluence were chosen per cell type. Fluorescence measurements were taken immediately.Mean fluorescence intensity at the two emission wavelengths was measured per ROI and converted to pHi values using nigericin (10 μM) (20) following a ratio calculation (585–605 nm /650–700 nm). At the end of each measurement, control cells were loaded with EIPA (25 μM) supplemented directly into the well, serving as a positive control to reduce the pHi. All imaging analysis was done by Harmony-image analysis tool of the Operetta system.
In the case of the five different cell lines expressing different levels of ASS1 or in the case of U2OS pLKO empty vector (EV) and shASS1 osteosarcoma incubated under alternating pHe solutions, we used confocal spinning disc (Zeiss) equipped with a temperature control, a CO2 chamber and the appropriate excitation and emission filters (Ex 512 nm; Em 580 and 640 nm). Imaging acquisition was performed using a x40 magnification (oil). In the case of the five different cell lines – following the incubation period, cells were washed twice with a HEPES buffer and loaded with Carboxy SNARF-1 AM (5 μM), as described before. Imaging control cells was performed while using pHe 7.4 bicarbonate buffer (22mM HCO3− ; 5% CO2). Imaging cells that were incubated under −CO2/−HCO3− conditions was performed while using a pHe 7.4 HEPES buffer. For each of the cell lines the appropriate calibration curve was generated using nigericin (10 μM). For imaging U2OS pLKO empty vector (EV) and shASS1 osteosarcoma incubated under alternating pHe solutions we used pHe 7.4 bicarbonate buffer (22mM HCO3−; 5% CO2) and pHe 6.6 bicarbonate buffer (3.5 mM HCO3−; 5% CO2). Each measurement consisted of a few ROIs with n ≥ 12 cells per ROI. Mean fluorescence intensity at the two emission wavelengths was measured per ROI and converted to pHi values using nigericin (10 μM) following a ratio calculation (580 nm/640 nm). Each measurement represents the average of a few ROIs. All measurements were done in biological duplicates or triplicates and were repeated at least three times. Imaging analysis was performed using ImageJ.
Protein analyses; Western blotting-
Cells were lysed in RIPA buffer (Sigma-Aldrich) with 0.5% protease inhibitor cocktail (Calbiochem). Following centrifugation, the supernatant was collected and protein content was evaluated by the Bradford assay. Seventy to one hundred micrograms from each sample under reducing conditions were loaded into each lane and separated by electrophoresis on a 10% SDS polyacrylamide gel. Following electrophoresis, proteins were transferred to Immobilon transfer membranes (Tamar, Israel). Nonspecific binding was blocked by incubation with TBST (10 mM Tris–HCl (pH 8.0), 150 mM NaCl, 0.1 % Tween 20) containing 3% Albumin from Bovine Serum for 1h at room temperature. Membranes were subsequently incubated with antibodies against ASS1 (1:500, 124465, abcam), GAPDH (1:5000, EPR-16891, abcam), α -Tubulin (1:3000, T5168, Sigma), Hif1α (1:500, ab2185, abcam), MCT4 H-90 (1:500, sc-50329, Santa Cruz). Antibody was detected using peroxidase-conjugated AffiniPure goat anti-rabbit IgG or goat anti-mouse IgG (Jackson ImmunoResearch, West Grove, PA) and enhanced chemiluminescence western blotting detection reagents (EZ-Gel, Biological Industries).
Gels were quantified by Gel Doc™ XR+ (BioRad) and analyzed by ImageLab 4.1 software (BioRad). The band area was calculated by the intensity of the band. The obtained value was then divided by the value obtained from the loading control.
Immunofluorescence; Tumor staining.
Hypoxia and ASS1 levels.
Pimonidazole (hypoxyprobe-1 kit, hpi) was IP injected, according to the manufacturer instructions, two hours prior euthanization. Upon euthanization, the mice were rapidly perfused with cold 2.5% PFA in PBS. The tumor was then excised and transferred to 4% PFA in PBS. Next, four-micrometer paraffin embedded tissue sections were deparaffinized and rehydrated. Sections undergoing for ASS1 staining were incubated in cold acetone at −20ºc for 7 minutes and then boiled in 10 mM citric acid pH 6, for 10 min using a low boiling program in the microwave to break protein cross-links and unmask antigens. For hypoxic areas staining, adjacent four-micrometer section was taken and underwent antigen retrieval in 10 mM citric acid pH 6 for 10 min using a low boiling program in the microwave. After pre-incubation with 20% normal horse serum and 0.2% Triton X-100 for 1 hour at RT, sections were incubated with the primary antibodies as follow; ASS1 (1:100, abcam, ab124465, CA, USA); hypoxyprobe-1 (1:35, hpi, USA). All antibodies were diluted in PBS containing 2% normal horse serum and 0.2% Triton. Sections were incubated overnight at RT followed by 48h at 4°C. Sections were washed three times in PBS and incubated with secondary biotinylated IgG at RT for 1.5 hour, washed three times in PBS and incubated with avidin-biotin Complex (Elite-ABC kit, Vector Lab, CA, USA) at RT for additional 90 min.
ASS1 and MCT4.
Staining was performed on four-micrometer paraffin embedded tissue sections as described above or on seven-micrometer OCT embedded LLC sections. Sections were incubated in cold acetone at −20ºc for 7 minutes and then boiled in 10 mM citric acid pH 6, for 10 min using a low boiling program in the microwave to break protein cross-links and unmask antigens. After pre-incubation with 20% normal horse serum and 0.2% Triton X-100 for 1 hour at RT, sections were incubated with both primary antibodies as follow; MCT4 (1:100, Santa Cruz Biotechnology, sc-50329); ASS1 (1:100, abcam, ab124465). All antibodies were diluted in PBS containing 2% normal horse serum and 0.2% Triton. Sections were further treated as described above.
Liver immunohistochemistry staining-
Following euthanization, liver from wild type mice was harvested and transferred to 4% PFA in PBS. Next, two-micrometer paraffin embedded tissue sections were deparaffinized and rehydrated. Endogenous peroxidase was blocked with three percent H2O2 in methanol. Sections undergoing for GS staining were incubated in cold acetone at −20ºc for 7 minutes and then boiled in TRIS-EDTA for 10 min using a low boiling program in the microwave to break protein cross-links and unmask antigens. Sections undergoing for ASS1 staining were incubated as described above. After pre-incubation with 20% normal horse serum and 0.2% Triton X-100 for 1 hour at RT, sections were incubated with the primary antibodies as follow: GS (1:100, abcam, ab73593, CA, USA); ASS1 (1:100, abcam, ab124465). Sections were washed three times in PBS and incubated with secondary biotinylated IgG at RT for 1.5 hour, washed three times in PBS. Following incubation with avidin-biotin complex, a DAB (Sigma) reaction was performed.
RNA extraction and complementary DNA (cDNA) synthesis-
U2OS osteosarcoma or B16 melanoma cells were seeded at 100,000 cells/well in a 6-well plate and at the following day were either incubated for 24 – 48 hours under hypoxic conditions (1% O2) or under normoxic conditions. At the end of incubation, RNA was extracted from the cells by using PerfectPure RNA Cultured Cell Kit (5′-PRIME). cDNA was synthesized from 1μg RNA by using qScript cDNA Synthesis Kit (Quanta).
Quantitative PCR-
Detection of ASS1 and CAD cDNAs was performed using SYBR green PCR master mix (Tamar, Israel) and the required primers. The detection of hsa- or mmu-miR224-5p was performed using PerfeCTa SYBR Green SuperMix (Quanta) and Primers for hsa- or mmu-miR224-5p were supplied by Quanta. Primer sequences were as follows: Human ASS1: forward, 5’- TTGAAATTTGCTGAGCTGGTGTA −3’; reverse, −5’- AGCCTGAGGGAATTGATGTTGAT-3’. Human HPRT: forward, 5′-ATTGACACTGGCAAAACAATGC-3′; reverse: 5′-TCCAACACTTCGTGGGGTCC-3′. Human GAPDH: forward, 5’- AAATTGAGCCCGCAGCCTCCC-3’; reverse: 5′- AGCGATGTGGCTCGGCTGG. Mouse ASS1: forward, 5’- TGTACCTGAACGAAGTTGCGG-3′; reverse, 5′-ACCTCGGGACTTCATTCCAAT-3’. Mouse HPRT: forward, 5’-GCAGTACAGCCCCAAAATGG-3’; reverse, 5’-GGTTCCTTTTCACCAGCAAGCT-3’. Human CAD: forward, 5′-TTTGCAGTCCTTCCCGCTTC-3′; reverse, 5′-GCCGGTTTGAAACACCACTT-3′. For analysis, we used StepOne real-time PCR technology (Applied Biosystems).
Luciferase Assay-
Primers were designed by Primer BLAST software. The primers had forward Xhol (5′-TAATCTCGAGACCCGTGTACAATGAGGAGCT-3′) and reverse Notl (5′-TTATGCGGCCGCTAGAAATAAAAGACTAGTGTC-3′), restriction sites. PCR reaction was performed in a final reaction of 50 µl in 0.2 mL PCR-tubes containing 250 ng of template DNA, 5 µl of each primer at 10 µM, 25 C GoTaq Green Supper Mix x2 (Promega). Sterile ultrapure deionized water was used as a negative control. PCR reaction was carried out with initial denaturation at 95 °C for 5 min followed by 35 thermal cycles of denaturation at 95 °C for 1 min, annealing at 57 °C for 30 sec and extension at 72 °C for 30 sec. Final extension was accomplished at 72 °C for 5 min. The amplified 3’ UTR fragment was analyzed by electrophoresis in 1% agarose gels.
ASS1 3’ UTR DNA fragment was purified (using Qiagen gel extraction kit) and ligated in a 20 µL volume containing 2 µL ligation buffer, 1 µL ligase (New England Biolabs), 8.5 µL fragmented ASS1 3’ UTR and 1.5 µL psiCHECK2 plasmid (Addgene). The ligated plasmid was transformed into dh5α competent bacteria by heat shock transformation. The transformed bacteria were seeded on LB-ampicillin agar plate. The recombinant plasmid was purified (using Wizard plus SV Minipreps DNA Purification System) and the correct sequence and orientation were verified by sequencing with forward (5’-TTCCAACGCTATTGTCGAGG-3’) and reversed (5’- AAACACTCTCATTCGGAGGC-3’) cloning validation primers.
U2OS cell-line was seeded at 100,000 cells per well in a 6-well plate. At the following day, cells were transfected with 300pmol miR224-5p or non-targeting (NT) control (using lipofectamine 2000 transfection reagent). Following incubation of 24 hours cells were transfected again with 1 µg psiCHECK2-ASS1 3’ UTR plasmid and incubated for additional 24 hours. Cells were collected and measured (Cytation 5 Cell Imaging Multi-Mode Reader) according to the dual-luciferase reporter assay system manufacture protocol (Promega).
ASS1 silencing-
In the case of U2OS: Cells were infected with pLKO-based lentiviral vector with or without the human ASS1 short hairpin RNA (shRNA) encoding two separate sequences (RHS4533-EG445, GE Healthcare, Dharmacon). In the case of MCF-7 cells: GFP short hairpin RNA (shGFP) was used to infect control cells. Transduced cells were selected with 2 μg ml−1 puromycin.
miRNA transfection, extraction and complementary DNA (cDNA) synthesis-
U2OS osteosarcoma or B16 melanoma cells were seeded at 100,000 cells/well in a 6-well plate. At the following day, cells were either transfected with 300pmol non-targeting control or with miR224-5p. Following 24 hours, the cells were transfected again with a complementary 600pmol specific miR inhibitor (Dharmacon miRIDIAN miRNA Hairpin Inhibitor) plus a negative nonspecific sequence (Dharmacon miRIDIAN miRNA Inhibitor Negative Control). Cells were incubated for 48h hours under normoxic conditions. In the case of hypoxic conditions (1% O2) vs. normoxic conditions - miR224-5p was extracted after 24 hours (B16) or 48 hours (U2OS). In all cases, at the end of incubation microRNA was extracted from the cells by using miRNeasy mini kit (Quiagen). cDNA for miRNA was synthesized from 0.7–1μg RNA by using qScript MicroRNA cDNA Synthesis kit (Quanta).
Animal studies-
All experiments with mice were approved by the Institutional Animal Care and Use Committee (Weizmann Institute of Science). Tumors did not exceed the limits of more than 10% of the animal weight and were no longer than 1.5 cm in length in any dimension. Induction of tumors: Ten million MALME-3m melanoma cells suspended in 400 μl PBS with 5% Matrigel (4132053 Corning) were subcutaneously injected to 8- to 12-week-old male SCID mice that were purchased from Harlan. MC38 tumors were induced by an orthotopic implantation of 1×105 MC38 CRC into 8–10 week-old male C57BL/6 mice. Tumor sections were isolated from the colonic injection site at day 14. LLC tumors were induced by subcutaneously injecting 2*106 LLC-D122 cells in PBS into the left hind limb of 8–10 week-old male C57BL/6 mice. Tumor sections were isolated at day 21. Mice were monitored for survival and tumor burden twice a week by a veterinarian investigator who was blinded to the expected outcome.
Results
Since cancer cells hijack physiological processes that benefit normal cells’ survival and proliferation, we began our investigations into the relationship between lactate and ammonia levels by analyzing biochemical data of subjects without cancer who underwent a forearm ischemic test (FIT). This test is used to evaluate muscular energy production following complaints of either exercise-induced muscle pain, or unexplained elevations in serum creatine phosphokinase levels (21, 22). As expected, in comparison to lactate and ammonia levels at baseline, we found that after 3 minutes of FIT, a significant correlation exists between the elevations of ammonia and lactate in healthy subjects, while patients with IEM demonstrate an abnormal response (Figure 1B). In addition, we noted higher levels of ammonia, with no significant rise in lactate levels, in primary citrullinemia type I fibroblasts, compared to our findings in the media of fibroblasts from healthy controls (Figure 1C and Supplementary Figure 1A). These results suggest that ammonia and lactate levels function in tandem in normal physiological states, whereas disruption of this association might signify metabolic dysregulation.
Ex vivo, at the tissue level, we studied livers from wild-type mice in which a gradient from high oxygen in the portal vein, to low oxygen levels in the central vein, had been established (23). Here, we found that ASS1 expression is confined to the portal vein, and is diminished in the hypoxic regions surrounding the central vein, where glutamine synthase is expressed (Figure 1D). Since lactate levels near the central vein are relatively high (24), the downregulation of ASS1 expression, together with the biochemical changes associated with its depletion in normal states, suggest that in hypoxic regions, low ASS1 levels might provide a metabolic benefit resulting from increased ammonia/glutamine levels.
To determine the relevance of these findings to cancer, we performed an unbiased exploration of the predicted activity of the proximal UC enzymes at different intracellular pH (pHi) values in normal and in cancerous cells, using genome-scale metabolic models (Methods)(14) . Compared to healthy cells, we found that in cancer cells under acidic pHi conditions, the activity, or “flux,” of ASS1, and of the more upstream enzymes in the UC, is predicted to decrease, while the activity of glutathione dehydrogenase (dehydroascorbate reductase) is predicted to increase (Figure 1E).
To validate the potential metabolic association between the downregulation of ASS1 and pHi, we induced either gradual or acute alterations in extracellular pH (pHe), which trigger intracellular pH acidic stress. First, we grew osteosarcoma cells without bicarbonate in the culture medium, in a CO2-free atmosphere. In this buffer-free system, intracellular acidic stress is increased, due to a gradual decrease of the pHe, and the lack of extrinsic buffering (Supplementary Figure 1B). Consistent with our hypothesis, and in silico predictions, ASS1 expression in osteosarcoma cells decreased concomitant with prolonged acidity (Figure 1F and Supplementary Figure 1C). We further validated our results by inducing acute pHe changes, independent of bicarbonate limitation (25). In line with our previous findings, ASS1 levels were found to be lower under acidic conditions (pHe 6.3, as compared to measurements taken under baseline conditions of pHe 7.4), (Figure 1G and Supplementary Figure 1D). Thus, inducing acidic stress using two different systems, reduces ASS1 levels in osteosarcoma cells, and corroborates our findings in non-transformed cells.
Next, we applied our results in osteosarcoma cells, to evaluate whether ASS1 levels facilitate cancer cells’ survival under acidic conditions in other malignancies. To do so, we grew five different human cancer cell lines, each of which expressed different levels of ASS1, in the buffer free system described above. Two of these cancer lines, MNNG/HOS (osteosarcoma) and Lox IMVI (melanoma), display low ASS1 levels, whereas the other three cancer cell lines, U2OS (osteosarcoma), OVCAR-3 (ovarian carcinoma) and MCF-7 (breast adenocarcinoma) express varying levels of ASS1 (Figure 2A and Supplementary Figure 2A). As expected, all cancer cells, irrespective of ASS1 levels, were less viable and demonstrated a lower pHi when grown under acidic conditions induced by the lack of extrinsic buffering (Supplementary Figures 2B–C). Interestingly, the stress induced by the buffer-free conditions highlighted the differences in cell viability, favoring survival of cancer cells with low ASS1 and a higher pHi, as compared to cancers expressing higher ASS1 levels (Figure 2B).
Figure 2: ASS1 deficient cancer cells have survival advantage in acidic environment.
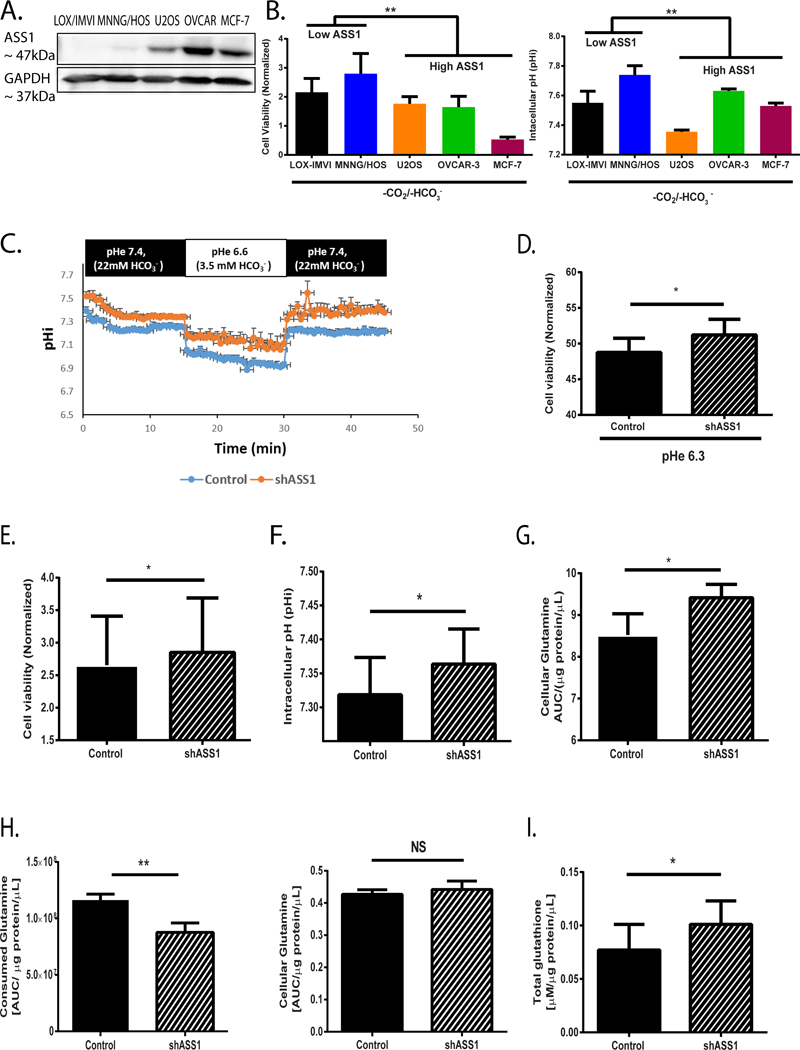
(A) A representative western blot showing ASS1 expression levles in five different cancer cell lines (B) Left panel: viability assays of five different cancer cells divided into two groups based on ASS1 expression as either “High ASS1” or “Low ASS1”, following 72 hours incubation under −CO2/−HCO3− conditions (viability is quantified relative to day 0). Right panel: pHi measurements of five different cancer cells divided into two groups based on ASS1 expression as either “High ASS1” or “Low ASS1”, following 72 hours incubation under −CO2/−HCO3− conditions (C) A Representative pHi curve based on quantifications of live confocal microscope images of U2OS osteosarcoma cells infected with either empty vector (control) or shASS1 and incubated in alternating pH solutions. Using the HH equation, solutions were supplemented with bicarbonate to reach the specified pH. For pH 7.4 buffer we used 22mM HCO3−; 5% CO2 and for pH 6.6 buffer we used 3.5 mM HCO3−; 5% CO2. Each point of a pHi value is an average of n ≥ 10 cells (ROIs). (D) Viability experiments showing increased survival for shASS1 U2OS cells as compared to control cells following 24 hours incubation under pHe 6.3 (1.75 mM HCO3−; 5% CO2). Viability at pHe 6.3 (1.75 mM HCO3−; 5% CO2) of each clone was normalized to its viability at pHe 7.4 (22 mM HCO3− ; 5% CO2). We did not detect significant differences in pHi between control and shASS1 U2OS cells under pHe 6.3. (E) Viability measurements of U2OS osteosarcoma cells infected with either empty vector or shASS1 and incubated under −CO2/−HCO3− conditions (viability is quantified relative to day 0). (F) Averaged pHi measurements of control U2OS osteosarcoma cells infected with either empty vector or shASS1 following incubation under −CO2/−HCO3− conditions for 72 hours. All measurements were done using pH 7.4 HEPES buffer medium in an incubator without CO2. Results are presented as an average of n≥5 independent experiments. (G) Cellular glutamine measurements using GC/MS of control and shASS1 osteosarcoma cells grown under +CO2/+HCO3− conditions at time point 24 hours (baseline). (H) Left panel: Glutamine uptake measurements using amino acid analyzer from the media of either control or shASS1 osteosarcoma cells grown under −CO2/−HCO3− conditions for 72 hours.
Right panel: Cellular glutamine measurements of control and of shASS1 osteosarcoma cells grown under −CO2/−HCO3− conditions at time point 72 hours.
(I) Total glutathione levels in control and shASS1 osteosarcoma cells grown under −CO2/−HCO3− conditions at time point 72 hours. For the multiple cell line comparisons in 2B, we used Two-way ANNOVA (with A-priori contrast: MNNG/HOS, LOX-IMVI vs. U2OS, MCF-7 and OVCAR-3). For all other comparisons, student’s t-test was used (2-tail) for statistical analysis. Unless otherwise specified, each graph represents the average of n≥3 experiments ± S.D. *, P ≤ 0.05; **, P≤ 0.01.
To decipher the biochemical and cellular changes caused by specific alterations in ASS1 expression levels, we next focused on U2OS osteosarcoma cells in which we knocked down ASS1 using shASS1 (6) (Supplementary Figure 2D). To dissect the specific effect of ASS1 downregulation on pHi, and to avoid any contamination of our results due to cell death, we first performed short-term live-cell imaging experiments under both baseline (pHe 7.4; 22mM HCO3−) and acidic conditions (pHe 6.6; 3.5mM HCO3−), by using the Henderson-Hasselbalch equation and limiting the amount of bicarbonate in the media (26). Encouragingly, we found that shASS1 U2OS cells display a higher pHi in both instances (Figure 2C), coupled with a significant survival advantage following their prolonged incubation at pHe 6.3 (Figure 2D). By completely depleting bicarbonate from the media and incubating the cells in the buffer-free system, we further confirmed the survival advantage and associated higher pHi of shASS1 U2OS grown under acidic conditions (Figure 2E–F and Supplementary Figure 2E),
Since glutamine has been associated with cellular defense against acidic stress (25), and since glutamine levels are elevated in instances of inherited ASS1 deficiency (27), we further assessed whether ASS1 downregulation affects both intra- and extracellular glutamine levels. We first determined that ASS1-depleted U2OS cells accumulate higher levels of glutamine when grown under baseline normoxic conditions (Figure 2G). Importantly, we found that when grown in acidic conditions, cancer cells characterized by ASS1 downregulation consume less glutamine from the medium (Figure 2H, left panel), yet retain similar intracellular glutamine levels (Figure 2H, right panel). Since glutathione is synthesized from glutamine, and plays an essential role in anti-oxidant metabolism (25, 28) we measured its levels under these acidic conditions. As expected from our enzymatic activity prediction (Figure 1E), we found higher glutathione levels in ASS1-depleted cells, compared to control cells (Figure 2I). Hence, cancer cells in which ASS1 is downregulated display higher intracellular glutamine levels, enabling them, in acidic states, to depend less on external sources of glutamine, to preserve alkalinized pHi and to synthesize glutathione.
Mechanisms to preserve pHi in cancerous cells are necessary to enable the cells to survive, and maintain their carcinogenic properties (29–31). In recent years, it has been observed that cancer cells generate a pHe that is lower than their pHi. This unique, ‘reversed’ pH gradient across cancer cell membranes has emerged as a new hallmark of cancer, and is thought to empower cancer cells’ metastatic potential, while preserving its normal cellular functions (29–31). One of the main transcription factors that regulates the pH gradient in cancer is HIF1α (10). Under hypoxic conditions, HIF1α is upregulated, to promote pHi preservation by inducing, for example, the expression of the acid extruder monocarboxylate transporter 4 (MCT4), as well as the expression of the membrane CO2 hydrating enzymes carbonic anhydrase 9 and 12 (CA9, CA12)(10, 32). Interestingly, HIF1α has also been reported to downregulate ASS1 levels (33).
To study the association between ASS1 and the hypoxic − gene expression in cancer, we first analyzed the Cancer Genome Atlas (TCGA) database for tumors expressing hypoxia-related genes (16), in which the cancer cell purity was estimated to be >75% (15). Using this dataset, we found that ASS1 expression levels are significantly lower in tumors with a high hypoxic gene signature (Figure 3A). Since cancer cells might express HIF1α irrespective of hypoxia (34), we repeated the analysis, while excluding HIF1α target genes. Again, we found a comparable, significant trend associating low ASS1 levels and hypoxia, independent of HIF1α expression (Supplementary Figure 3A). Our analysis of the TCGA data also yielded a significant inverse correlation between the expression levels of ASS1 and MCT4 in different tumor types (Figure 3B). The inverse correlation between ASS1 and MCT4 was also observed at the protein level in human osteosarcoma cells, following hypoxia induction (Figure 3C and Supplementary Figure 3B). As previously reported (8), we confirmed that HIF1α upregulation associates with the downregulation of CAD expression levels (Supplementary Figure 3C). To test whether the downregulation of ASS1 induced by HIF1α under hypoxia can be further augmented under acidosis, we incubated osteosarcoma cells under acute acidic conditions in the presence or absence of hypoxia. Indeed, ASS1 levels were downregulated more under hypoxic pHe 6.3 as compared to normoxic pHe 6.3 (Supplementary Figure 3D). Taken together, these results suggest that under hypoxic and acidic microenvironment, ASS1 downregulation supports survival mechanisms in cancerous cells that differ from those we previously identified for promoting proliferation under normoxic conditions (6).
Figure 3: ASS1 levels are regulated together with HIF1α− gene network for pH regulation.
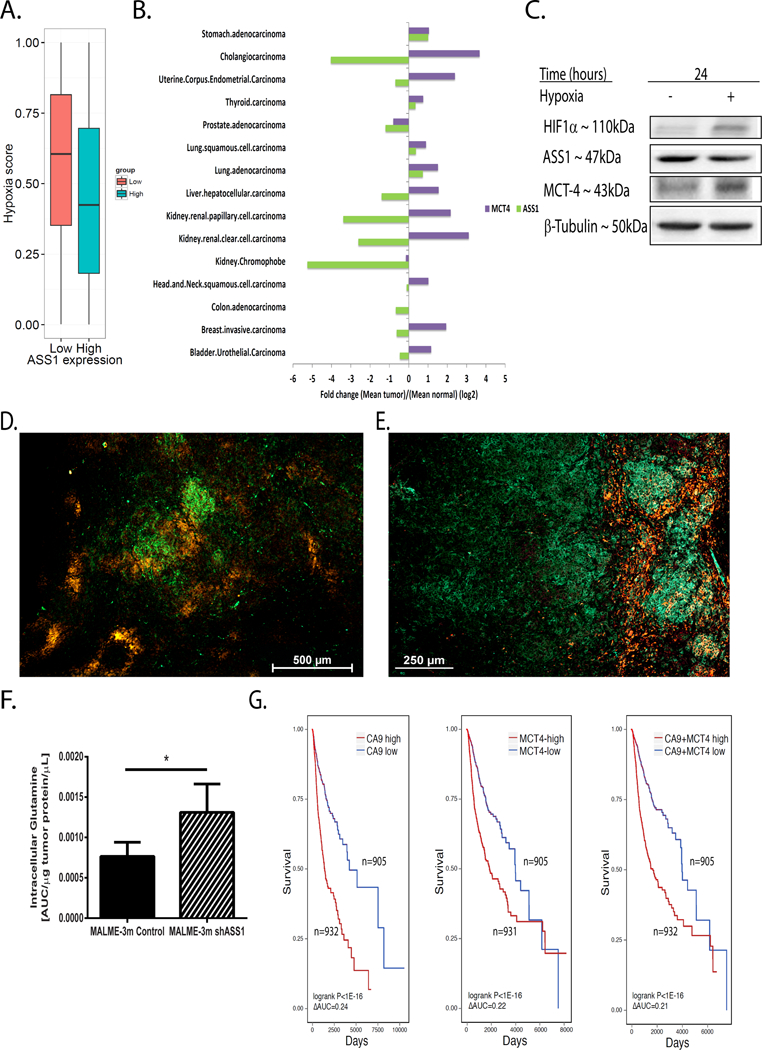
(A) Pan cancer analysis of the TCGA database demonstrating that tumors with low ASS1 expression levels (red) have a higher hypoxic score, based on measurement that includes HIF1α target genes’ expression (effect size: 0.180; Wilcoxon rank sum P<0.05, see Methods). (B) Pan cancer analysis of the TCGA database for ASS1 and MCT4, as compared to the expression of these genes in the respective normal tissues. Monocarboxylate transporter 4 (MCT4; SLC16A3); Argininosuccinate synthase 1(ASS1). (C) A representative western blot for the protein expression of HIF1α target genes in human osteosarcoma (U2OS) cells exposed to either normoxia or hypoxia for 24 hours. (D) A representative immunofluorescence image of a melanoma (Malme-3M) paraffin embedded tumor section following the administration and staining of the hypoxia sensitive pimonidazole probe (red) and of ASS1 (green). Magnification X4. (E) A representative immunofluorescence image of a Malme-3M melanoma paraffin tumor section stained for MCT4 (green) and ASS1 (red). Magnification X6. (F) Ex-vivo measurement of tumor glutamine levels using GC/MS in control Malme-3M vs shASS1 Malme-3M tumor cells. (G) Kaplan-Meier plots showing decreased survival in patients with tumors having either over expression of CA9 (left panel), MCT4 (middle panel) or of both (right panel) when ASS1 expression is downregulated. CA9 (low CA9: n=905; High CA9: n=932 patients) - Cox hazard ratio=1.14 (P<0.06), logrank P<1E-16 (ΔAUC=0.24); MCT4 (low MCT4: n=905 patients; High MCT4: n=931 patients) - Cox hazard ratio=1.19 (P<0.01), logrank P<1E-16, (ΔAUC=0.22). CA9 and MCT4 (low CA9+MCT4: n=905 patients; High CA9+MCT4: n=932 patients) - Cox hazard ratio=1.23 (P<0.004), logrank P<1E-16 (ΔAUC=0.21). We used student’s t-test (2-tail) for statistical analysis in (F) that represents the average of five mice per group ± S.D. *, P ≤ 0.05.
To test these findings in vivo, we studied human melanoma tumors expressing ASS1 implanted in mice, following injection with a hypoxia-dependent reactive probe (35). In these tumors, we found a clear demarcation between the hypoxic regions, and regions expressing ASS1 (Figure 3D). Furthermore, MCT4 expression in melanoma tumors, in murine Lewis Lung Carcinoma (LLC), and in colon MC38 tumors, was limited to regions that did not express ASS1 (Figure 3E and Supplementary Figures 3E–F).
Next, we measured glutamine levels in melanoma tumors, and found that shASS1 tumors grown in vivo, contained higher levels of intracellular glutamine, compared to tumors expressing ASS1 (Figure 3F). To evaluate the clinical relevance of these findings for humans with cancer, we performed a pan-cancer Kaplan Meir survival analysis using the TCGA database. We found that individuals with cancer whose levels of the HIF1α target genes CA9 and MCT4 are elevated, alongside decreased ASS1 expression, have poorer chances of survival, compared to patients whose cancers are characterized by similar elevations in CA9 and MCT4, but have increased ASS1 expression (Figure 3G and Supplementary Figure 3G). Collectively, these results suggest that in vivo, ASS1 expression is downregulated in hypoxic regions, whereas expression of HIF1α−pHi related target genes is upregulated. It would appear that this co-regulation increases glutamine levels, promotes tumor fitness, and worsens patients’ survival.
In accordance with the cellular context, HIF1α can downregulate different target genes either directly, or via microRNA (miR)(36). Interestingly, we found that miR-224–5p, which has been found to be regulated by HIF1α in invasive melanoma and gastric cancers (37, 38) displays a conserved binding site in mammals on the 3’UTR site of ASS1 (Supplementary Figure 4A). Our analysis of the TCGA data, suggested an inverse correlation between ASS1 downregulation and miR-224–5p upregulation, in multiple cancers (Supplementary Figure 4B). We confirmed the specific association of miR-224–5p upregulation and ASS1 downregulation in hypoxia in both osteosarcoma and in murine B16 cancer cells (Supplementary Figure 3C and Supplementary Figure 4C–D). Furthermore, we determined that overexpression of miR-224–5p can downregulate ASS1 levels at the RNA level in both cancers, independent of hypoxia (Figure 4A and Supplementary Figure 4E), and that this downregulation also manifests at the protein level (Figure 4B). Using a luciferase assay, we specifically demonstrated the direct binding of miR-224–5p to ASS1 3’UTR, independent of hypoxic conditions (Figure 4C). Importantly, the downregulation of ASS1 solely by miR-224–5p was sufficient to increase the extent of invasion and migration in osteosarcoma cells (Figure 4D–E). The co-regulation of ASS1 levels by HIF1α and miR-224–5p strongly support the notion that low ASS1 levels in hypoxia provide cancer with a metabolic benefit that has identifiable phenotypic consequences.
Figure 4: miR224-5p downregulates ASS1.
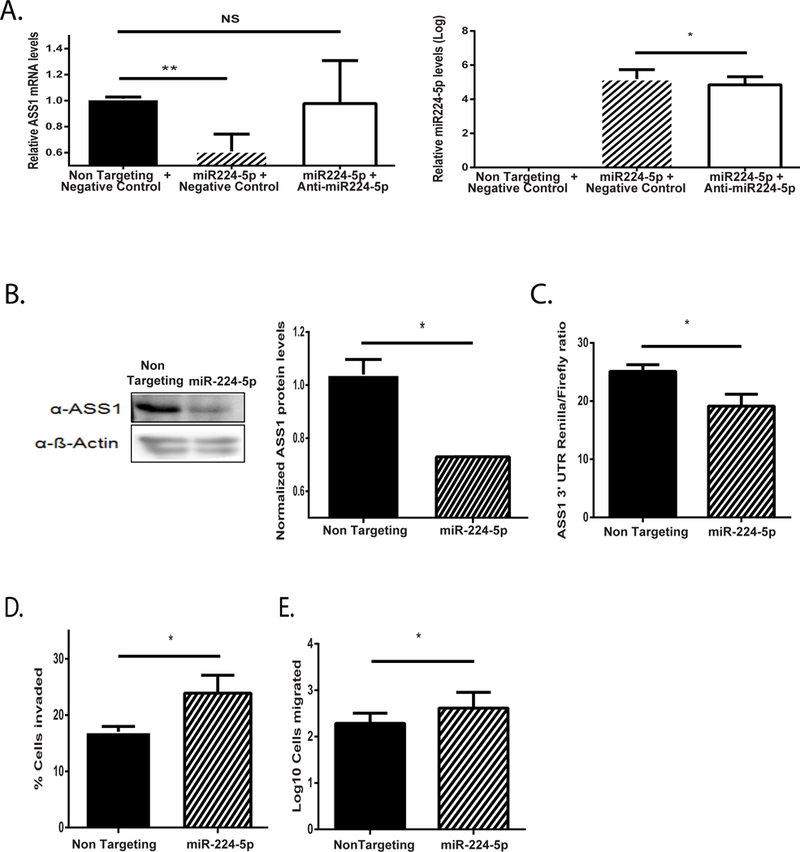
(A) ASS1 mRNA (left panel) levels are downregulated in osteosarcoma cells following transfection with miR-224–5p (right panel) under normoxia. Addition of anti miR224-5p rescued ASS1 levels. (B) Left panel: A representative western blot image showing reduction in ASS1 expression levels in osteosarcoma cells following transfection with miR224-5p (48 hours). Right panel: Averaged gel quantification (C) Luciferase assay showing diminished ratio of ASS1 3’ UTR Renilla to Firefly luciferases following transfection with miR224-5p. (D) Osteosarcoma cells invade significantly more following transfection with miR224-5p under normoxia. (E) Osteosarcoma cells migrate significantly more following transfection with miR-224–5p under normoxia. We used student’s t-test (2-tail) for statistical analysis. Unless otherwise specified, each graph represents the average of n≥3 experiments ± S.D. *, P ≤ 0.05; **, P≤ 0.01.
To assess whether the decreased survival of patients with ASS1 downregulation in cancers expressing hypoxia genes could be associated with pH gradient-related clinical phenotypes such as metastasis formation (31), we utilized the TCGA database to compare gene expression in metastatic tumor samples expressing hypoxia genes, to those that do not. Similar to our findings in solid tumors (Figure 3A), we found that ASS1 expression is significantly lower in metastases expressing hypoxia gene-related signatures (Figure 5A). Even when excluding HIF1α target genes, we observed comparable, significant trend correlating low ASS1 expression levels and hypoxic metastases (Supplementary Figure 5A).
Figure 5: Cancer cells expressing low levels of ASS1 have increased sensitivity to glutamine and bicarbonate depletion.
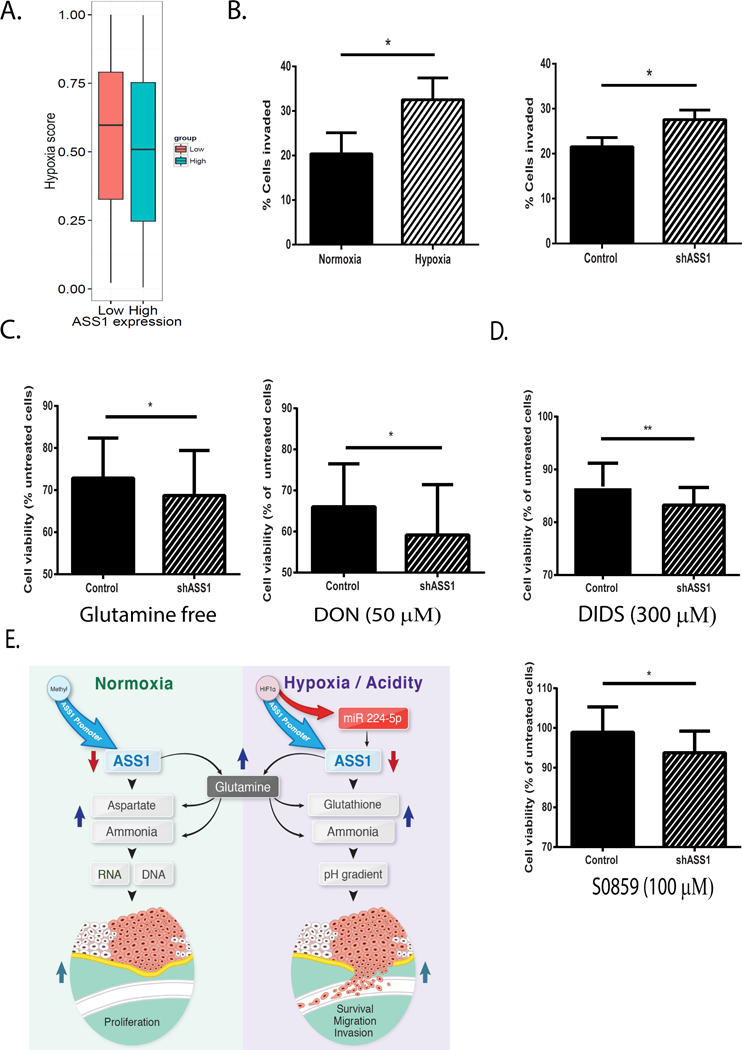
(A) Pan cancer analysis based on TCGA metastatic samples showing that tumors with low ASS1 levels have a higher hypoxia-score (effect size: 0.088; Wilcoxon rank sum P<3.35E-3, see Methods). (B) Left panel: U2OS osteosarcoma cancer cells invade significantly more upon incubation of 48 hours under hypoxic as compared to normoxic conditions. Right panel: U2OS osteosarcoma cells with ASS1 depletion invade significantly more than control cells following incubation of 48 hours under normoxia. (C) MCF-7 breast cancer cells with ASS1 downregulation are less viable following incubation under hypoxia for 48 hours with either: glutamine free conditions (left panel) or 6-Diazo-5-oxo-L-norleucine (DON ; 50 μM; right panel). (D) MCF-7 breast cancer cells with ASS1 downregulation are less viable following incubation under hypoxia with either: DIDS (300 μM; 24 hours incubation) (upper panel) or S0859 (100 μM; 48 hours incubation) (lower panel). Viability is normalized to non-treated cells (cells grown in the absence of inhibitors). The experiments described in C and D have been performed under pHe 7.4. (E) Illustration of the different metabolic consequences following ASS1 downregulation in normoxia vs. hypoxia or acidity.
Unless otherwise specified, we used student’s t-test (2-tail) for statistical analysis. Each graph represents the average of n≥3 experiments ± S.D. *, P ≤ 0.05.
To explore these results in greater depth, we compared the invasive ability of osteosarcoma cells under normoxic and hypoxic conditions, and found higher rates of invasion in the latter (Figure 5B, left panel). To determine the contribution of ASS1 downregulation to hypoxia-independent invasion, we evaluated the invasion of osteosarcoma cells following ASS1 downregulation in normoxia. Similar to our findings following miR-224–5p transfection, we determined that even under normoxic conditions, shASS1-osteosarcoma cells are more invasive than controls (Figure 5B, right panel).
To test whether the survival and invasive advantages we found in cancers with low ASS1 expression in hypoxia, relate to the increase in ASS1 upstream metabolites, we grew the five different human cancer cell lines expressing different levels of ASS1 under hypoxic conditions (Figure 2A), with or without glutamine in the culture media. Indeed, we found that cancer cells with low levels of ASS1 demonstrate increased sensitivity to glutamine depletion, compared to cells with high ASS1 levels (Supplementary Figure 5B). To confirm the specific susceptibility of cancers with low ASS1 levels to ASS1 upstream metabolites, we knocked down ASS1 in MCF-7 breast adenocarcinoma cells (Supplementary Figures 5C–D). We next grew these cells in glutamine- free conditions, with pharmacological inhibitors that decrease the levels of glutamine and bicarbonate: 6-Diazo-5-oxo-L-norleucine (DON), (a glutamine antagonist)(39), and S0859, which inhibits the NBC sodium-bicarbonate co-transporter (40). Since S0859 has been found to have non-specific effects, we repeated our experiments using the HCO3− transport inhibitor, 4,4′-Diisothiocyanatostilbene-2,2′-disulfonic acid (DIDS). Using all agents, we find that MCF-7 breast cancer cells subjected to ASS1 knockdown are more sensitive to the depletion of both glutamine and the alkali bicarbonate (Figures 5C–D and Supplementary Figure 5E).
Discussion
Cancer cells display a distinct ‘reversed’ pH gradient, with a constitutively higher pHi, compared to their pHe. This gradient enables disease progression by promoting malignant cell proliferation, in most instances enabling these cells to evade apoptosis, metabolically adapt, migrate, and invade neighboring tissues, irrespective of tumor pathology, genetics and origins. Not surprisingly, pHi is emerging as a new hallmark in cancer studies, and a potential target for therapy (31, 41). One of the outcomes of this reversed pH gradient is that it can be thermodynamically costly for cells, necessitating that they extrude more acids (42). Hence, cancer cells must promote alternative pathways for venting and buffering acids, to cope with the additional burden of intracellular acidity. Herein, we show that cancer cells with ASS1 downregulation display higher pHi levels as compared to control cells, and that this metabolic benefit is associated with higher glutamine levels. Under hypoxic conditions, ASS1 is directly downregulated in cancer cells by HIF1α and miR-224–5p, with phenotypic consequences on cells’ survival and invasive capacity. Clinically, we find that the downregulation of ASS1, together with the induction of HIF1α -pHi related target genes- MCT4 and CA9 are significantly associated with decreased patient survival. Collectively, our results suggest that ASS1 downregulation in cancerous processes may play an important role in the HIF1α−pHi network.
To clarify our findings concerning the impact of acidity on cancer cells, we used two acidification systems that are independent of hypoxia. In so doing, we determined that ASS1 is downregulated under acidic conditions; notably, under such conditions cancer cells with low ASS1 levels have a higher pHi, and demonstrate increased survival. While ASS1 downregulation by itself reduces the formation of argininosuccinic acid (43), we show here that the survival benefit of ASS1-depleted cancer cells results from the increase in ASS1’s upstream substrate- glutamine, which contributes to the maintenance of pHi and glutathione synthesis. Thus, ASS1 downregulation contributes directly to cellular defenses against both hypoxia-dependent and -independent acidic stresses.
The metabolic rewiring that occurs during carcinogenesis in general, and the decreased vascularization that takes place as the tumor grows, generate acidic levels that requires intracellular pH alkalization for the maintenance of protein activities (44). Although protons generated during hypoxia are not the only source of acidity in tumors, many of the changes related to hypoxia are induced by acidosis. This is best exemplified by HIF1α, which, on one hand increases acid extrusion, while on the other hand, is stabilized and protected from degradation by acidity (45). Whereas reduced pHe in the tumor microenvironment has been linked with metastases formation, alkalinized pHi has been shown to support cell adhesion processes necessary for invasion and migration. Here we find that ASS1 depleted cancer cells maintain a higher pHi at both baseline and acidic conditions, which may explain, at least in part, our findings of augmented invasion for ASS1 depleted cancer cells. We further demonstrate that ASS1 downregulation is regulated by HIF1α and, independently, by acidity, to promote these cancerous phenotypes.
Our findings have translational relevance as they imply that cancers expressing ASS1 may benefit too from treatment with arginine-depleting agents since they may have hypoxic/ acidic regions in which ASS1 is downregulated. In addition, we show that ASS1 downregulation confers on cancerous cells a sensitivity to drugs targeting ASS1 upstream substrates. As such, our results expose cancer-specific vulnerabilities with a therapeutic potential.
Supplementary Material
Statement of Significance:
Cancer cells in an acidic or hypoxic environment downregulate the expression of the urea cycle enzyme ASS1, which provides them with a redox and pH advantage resulting in better survival.
Acknowledgments:
We acknowledge and thank the Weizmann Institute for providing financial and infrastructural support. We greatly appreciate the technical support provided by Yogev Sela and the statistical contributions of Ron Rotkopf. A.Erez is incumbent of the Leah Omenn Career Development Chair and is supported by research grants from the European research program (CIG618113, ERC614204), the Israel Science Foundation (ISF- 1343/13; 1952/13) and from the Minerva grant award (711730). A.Erez received additional support from the Adelis Foundation, the Henry S. and Anne S. Reich Research Fund, the Dukler Fund for Cancer Research, the Paul Sparr Foundation, the Saul and Theresa Esman Foundation, from Joseph Piko Baruch, and from the estate of Fannie Sherr. E.Ruppin is supported by grants from the ISF (658/13) and the NIH (R33 CA225291-01).
Footnotes
The authors declare no potential conflicts of interest
References and Notes:
- 1.Quinonez SC, Thoene JG. Citrullinemia Type I 2014.
- 2.Thoene JG. Citrullinemia Type I In: Pagon RA, Adam MP, Bird TD, Dolan CR, Fong CT, Stephens K, editors. GeneReviews Seattle (WA: )2004. [Google Scholar]
- 3.Long Y, Center TUoTMAC, Tsai W-B, wbtsai@mdanderson.org, Pathology TM, Wangpaichitr M, et al. Arginine deiminase resistance in melanoma cells is associated with metabolic reprogramming, glucose dependence, and glutamine addiction. Molecular Cancer Therapeutics 2015;12(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kobayashi E, Masuda M, Nakayama R, Ichikawa H, Satow R, Shitashige M, et al. Reduced argininosuccinate synthetase is a predictive biomarker for the development of pulmonary metastasis in patients with osteosarcoma. Mol Cancer Ther 2010;9(3):535–44. [DOI] [PubMed] [Google Scholar]
- 5.Delage B, Luong P, Maharaj L, O’Riain C, Syed N, Crook T, et al. Promoter methylation of argininosuccinate synthetase-1 sensitises lymphomas to arginine deiminase treatment, autophagy and caspase-dependent apoptosis. Cell Death Dis 2012;3:e342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rabinovich S, Adler L, Yizhak K, Sarver A, Silberman A, Agron S, et al. Diversion of aspartate in ASS1-deficient tumours fosters de novo pyrimidine synthesis. Nature 2015. [DOI] [PMC free article] [PubMed]
- 7.Tsai WB, Aiba I, Lee SY, Feun L, Savaraj N, Kuo MT. Resistance to arginine deiminase treatment in melanoma cells is associated with induced argininosuccinate synthetase expression involving c-Myc/HIF-1alpha/Sp4. Molecular cancer therapeutics 2009;8(12):3223–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen KF, Lai YY, Sun HS, Tsai SJ. Transcriptional repression of human cad gene by hypoxia inducible factor-1alpha. Nucleic acids research 2005;33(16):5190–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Long Y, Tsai WB, Chang JT, Estecio M, Wangpaichitr M, Savaraj N, et al. Cisplatin-induced synthetic lethality to arginine-starvation therapy by transcriptional suppression of ASS1 is regulated by DEC1, HIF-1alpha, and c-Myc transcription network and is independent of ASS1 promoter DNA methylation. Oncotarget 2016;7(50):82658–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chiche J, Brahimi-Horn MC, Pouyssegur J. Tumour hypoxia induces a metabolic shift causing acidosis: a common feature in cancer. J Cell Mol Med 2010;14(4):771–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nissim I Newer aspects of glutamine/glutamate metabolism: the role of acute pH changes. Am J Physiol 1999;277(4 Pt 2):F493–7. [DOI] [PubMed] [Google Scholar]
- 12.Huang W, Choi W, Chen Y, Zhang Q, Deng H, He W, et al. A proposed role for glutamine in cancer cell growth through acid resistance. Cell research 2013;23(5):724–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spinelli JB, Yoon H, Ringel AE, Jeanfavre S, Clish CB, Haigis MC. Metabolic recycling of ammonia via glutamate dehydrogenase supports breast cancer biomass. Science 2017;358(6365):941–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Persi E, Duran-Frigola M, Damaghi M, Roush WR, Aloy P, Cleveland JL, et al. Systems analysis of intracellular pH vulnerabilities for cancer therapy. Nat Commun 2018;9(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yoshihara K, Shahmoradgoli M, Martinez E, Vegesna R, Kim H, Torres-Garcia W, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun 2013;4:2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Benita Y, Kikuchi H, Smith AD, Zhang MQ, Chung DC, Xavier RJ. An integrative genomics approach identifies Hypoxia Inducible Factor-1 (HIF-1)-target genes that form the core response to hypoxia. Nucleic acids research 2009;37(14):4587–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mantel N Evaluation of survival data and two new rank order statistics arising in its consideration. Cancer Chemother Rep 1966;50(3):163–70. [PubMed] [Google Scholar]
- 18.Bilal E, Dutkowski J, Guinney J, Jang IS, Logsdon BA, Pandey G, et al. Improving breast cancer survival analysis through competition-based multidimensional modeling. PLoS Comput Biol 2013;9(5):e1003047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Therneau TM, Grambsch PM. Modeling survival data : extending the Cox model New York: Springer; 2000. xiii, 350 p. p. [Google Scholar]
- 20.Grillo-Hill BK, Webb BA, Barber DL. Ratiometric Imaging of pH Probes. Method Cell Biol 2014;123:429–48. [DOI] [PubMed] [Google Scholar]
- 21.Hogrel JY, Laforet P, Ben Yaou R, Chevrot M, Eymard B, Lombes A. A non-ischemic forearm exercise test for the screening of patients with exercise intolerance. Neurology 2001;56(12):1733–8. [DOI] [PubMed] [Google Scholar]
- 22.Tarnopolsky M, Stevens L, MacDonald JR, Rodriguez C, Mahoney D, Rush J, et al. Diagnostic utility of a modified forearm ischemic exercise test and technical issues relevant to exercise testing. Muscle Nerve 2003;27(3):359–66. [DOI] [PubMed] [Google Scholar]
- 23.LeCluyse EL, Witek RP, Andersen ME, Powers MJ. Organotypic liver culture models: meeting current challenges in toxicity testing. Crit Rev Toxicol 2012;42(6):501–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kietzmann T Metabolic zonation of the liver: The oxygen gradient revisited. Redox biology 2017;11:622–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang WJ, Choi W, Chen YL, Zhang Q, Deng HT, He W, et al. A proposed role for glutamine in cancer cell growth through acid resistance. Cell Res 2013;23(5):724–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hulikova A, Vaughan-Jones RD, Swietach P. Dual role of CO2/HCO3(−) buffer in the regulation of intracellular pH of three-dimensional tumor growths. J Biol Chem 2011;286(16):13815–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wilson CJ, Lee PJ, Leonard JV. Plasma glutamine and ammonia concentrations in ornithine carbamoyltransferase deficiency and citrullinaemia. Journal of inherited metabolic disease 2001;24(7):691–5. [DOI] [PubMed] [Google Scholar]
- 28.Lamonte G, Tang X, Chen JL, Wu J, Ding CK, Keenan MM, et al. Acidosis induces reprogramming of cellular metabolism to mitigate oxidative stress. Cancer & metabolism 2013;1(1):23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reshkin SJ, Greco MR, Cardone RA. Role of pHi, and proton transporters in oncogene-driven neoplastic transformation. Philosophical transactions of the Royal Society of London Series B, Biological sciences 2014;369(1638):20130100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Parks SK, Pouyssegur J. Targeting pH regulating proteins for cancer therapy-Progress and limitations. Semin Cancer Biol 2017;43:66–73. [DOI] [PubMed] [Google Scholar]
- 31.Webb BA, Chimenti M, Jacobson MP, Barber DL. Dysregulated pH: a perfect storm for cancer progression. Nat Rev Cancer 2011;11(9):671–7. [DOI] [PubMed] [Google Scholar]
- 32.Swietach P, Patiar S, Supuran CT, Harris AL, Vaughan-Jones RD. The role of carbonic anhydrase 9 in regulating extracellular and intracellular ph in three-dimensional tumor cell growths. J Biol Chem 2009;284(30):20299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qing G, Skuli N, Mayes PA, Pawel B, Martinez D, Maris JM, et al. Combinatorial regulation of neuroblastoma tumor progression by N-Myc and hypoxia inducible factor HIF-1alpha. Cancer research 2010;70(24):10351–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krieg M, Haas R, Brauch H, Acker T, Flamme I, Plate KH. Up-regulation of hypoxia-inducible factors HIF-1alpha and HIF-2alpha under normoxic conditions in renal carcinoma cells by von Hippel-Lindau tumor suppressor gene loss of function. Oncogene 2000;19(48):5435–43. [DOI] [PubMed] [Google Scholar]
- 35.Bayin NS, Placantonakis DG. Evaluation of vascularity, blood perfusion, and oxygen tension in tumor xenografts with fluorescent microscopy. Methods in molecular biology 2018;1741:199–207. [DOI] [PubMed] [Google Scholar]
- 36.Liu W, Shen SM, Zhao XY, Chen GQ. Targeted genes and interacting proteins of hypoxia inducible factor-1. Int J Biochem Mol Biol 2012;3(2):165–78. [PMC free article] [PubMed] [Google Scholar]
- 37.Hwang HW, Baxter LL, Loftus SK, Cronin JC, Trivedi NS, Borate B, et al. Distinct microRNA expression signatures are associated with melanoma subtypes and are regulated by HIF1A. Pigment Cell Melanoma Res 2014;27(5):777–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.He C, Wang L, Zhang J, Xu H. Hypoxia-inducible microRNA-224 promotes the cell growth, migration and invasion by directly targeting RASSF8 in gastric cancer. Molecular cancer 2017;16(1):35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hernandez-Davies JE, Tran TQ, Reid MA, Rosales KR, Lowman XH, Pan M, et al. Vemurafenib resistance reprograms melanoma cells towards glutamine dependence. Journal of translational medicine 2015;13:210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ch’en FF, Villafuerte FC, Swietach P, Cobden PM, Vaughan-Jones RD. S0859, an N-cyanosulphonamide inhibitor of sodium-bicarbonate cotransport in the heart. British journal of pharmacology 2008;153(5):972–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sharma M, Astekar M, Soi S, Manjunatha BS, Shetty DC, Radhakrishnan R. pH Gradient Reversal: An Emerging Hallmark of Cancers. Recent Pat Anticancer Drug Discov 2015;10(3):244–58. [DOI] [PubMed] [Google Scholar]
- 42.Hulikova A, Black N, Hsia LT, Wilding J, Bodmer WF, Swietach P. Stromal uptake and transmission of acid is a pathway for venting cancer cell-generated acid. P Natl Acad Sci USA 2016;113(36):E5344–E53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen BC, Ngu LH, Zabedah MY. Argininosuccinic aciduria: clinical and biochemical phenotype findings in Malaysian children. The Malaysian journal of pathology 2010;32(2):87–95. [PubMed] [Google Scholar]
- 44.White KA, Grillo-Hill BK, Barber DL. Cancer cell behaviors mediated by dysregulated pH dynamics at a glance. J Cell Sci 2017;130(4):663–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mekhail K, Gunaratnam L, Bonicalzi ME, Lee S. HIF activation by pH-dependent nucleolar sequestration of VHL. Nature cell biology 2004;6(7):642–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.