

Abstract
Bone growth is dependent upon the presence of self-renewing progenitor cell populations. While the contribution of Tissue Nonspecific Alkaline Phosphatase (TNAP) enzyme activity in promoting bone mineralization when expressed in differentiated bone forming cells is well understood, little is known regarding the role of TNAP in bone progenitor cells. We previously found diminished proliferation in the calvarial MC3T3E1 cell line upon suppression of TNAP by shRNA, and in calvarial cells and tissues of TNAP−/− mice. These findings indicate that TNAP promotes cell proliferation. Here we investigate how TNAP mediates this effect. Results show that TNAP is essential for calvarial progenitor cell cycle progression and cytokinesis, and that these effects are mediated by inorganic phosphate and Erk1/2. Levels of active Erk1/2 are significantly diminished in TNAP deficient cranial cells and tissues even in the presence of inorganic phosphate. Moreover, in the absence of TNAP, FGFR2 expression levels are high and FGF2 rescues phospho-Erk1/2 levels and cell cycle abnormalities to a significantly greater extent than inorganic phosphate. Based upon the data we propose a model in which TNAP stimulates Erk1/2 activity via both phosphate dependent and independent mechanisms to promote cell cycle progression and cytokinesis in calvarial bone progenitor cells. Concomitantly, TNAP feeds back to inhibit FGFR2 expression. These results identify a novel mechanism by which TNAP promotes calvarial progenitor cell renewal and indicate that converging pathways exist downstream of FGF signaling and TNAP activity to control craniofacial skeletal development.
Keywords: TNAP, Erk, FGFR2, Phosphate, Cell Cycle, Bone Progenitor Cell
1.1. Introduction
TNAP is highly expressed in and commonly used as a marker of differentiated bone forming cells. TNAP deficiency causes poor bone mineralization because an essential function of TNAP in osteoblasts is to hydrolyze pyrophosphate (PPi, an inhibitor of tissue mineralization) to phosphate (Pi, a substrate for tissue mineralization) [1–4]. While the contribution of TNAP in promoting bone mineralization when expressed in differentiated bone forming cells is well understood, little is known regarding the role of TNAP in bone progenitor cells. We previously observed decreased proliferation in the MC3T3E1 calvarial progenitor cell line upon suppression of TNAP by shRNA, and in calvarial cells and tissues of TNAP−/− mice [5,6]. Therefore, we hypothesized that TNAP must have an additional essential function in promoting bone progenitor cell proliferation that is also critical to skeletal development. Additional evidence for a role of TNAP in progenitor cells includes the fact that TNAP is expressed in pluripotent migratory primordial germ cells during early embryonic development [7–9]. in neural progenitor cell populations before and after birth [10], and in calvarial rudiments several days prior to the onset of matrix mineralization [11,12]. How TNAP influences calvarial progenitor cell proliferation is unknown. It is tempting to speculate that TNAP functions in progenitor cells to increase cellular Pi levels because TNAP enzyme activity produces Pi, and because Pi is known to be essential for numerous metabolic and biochemical pathways [13]. The goal of this study was to establish mechanisms by which TNAP promotes the proliferation of calvarial progenitor cells.
1.2. Materials and Methods
1.2.1. Mouse Strain and Genotyping
TNAP+/− mice on a 12.5% C57Bl/6 – 87.5% 129SF2/J background were bred and utilized for cell isolation and in vivo analyses. Genotypes of the Alpl allele were established by PCR analysis of DNA isolated from tail biopsies utilizing TNAP+/+ primers TGCTGCTCCACTCACGTCGAT and ATCTACCAGGGGTGCTAACC, and TNAP−/− primers GAGCTCTTCCAGGTGTGTGG and CAAGACCGACCTGTCCGGTG, as previously described [3,5]. TNAP is essential for vitamin B6 metabolism [14]. Therefore, all mice were provided with a modified rodent diet containing pyridoxine at 325 ppm to suppress seizures and extend life span in TNAP−/− mice. Genders were combined for analyses because previous results showed no differences between genders [5,6]. Animal use followed federal guidelines and were performed in accordance with the University of Michigan’s Institutional Animal Care and Use Committee.
1.2.2. Cell Lines and Primary Cells
The MC3T3E1 calvarial pre-osteoblast cell line was generously provided by Dr. Renny Franceschi (University of Michigan, Ann Arbor, MI) and is available through the American Type Culture Collection (ATCC; Gaithersbug, MD). MC3T3E1 cells were transduced with lentiviral particles expressing a puromycin resistance gene and TNAP specific shRNA (Sigma Mission, SHCLNV_NM_007431) or non-target shRNA (Sigma Mission, SHC002V) in the presence of 8 ug/ml hexadimethrine bromide. Puromycin resistant colonies were expanded, tested for expression of TNAP then utilized for experiments [5]. Primary calvarial cells were isolated from dissected calvaria by collagenase digestion, as previously described [6]. Briefly, bones were rinsed with media then serially digested in a solution containing 2 mg/ml collagenase P and 2.5 mg/ml trypsin. Cells from the third digestion were utilized for experiments.
1.2.3. Cell Culture
Cells were cultured in custom formulated alpha MEM media containing no ascorbate, supplemented with 10% fetal bovine serum (FBS) and penicillin/streptomycin (10,000ug/ml). For experiments in which cells were treated with inorganic phosphate, cells were cultured in DMEM containing no phosphate (Thermo Fisher, 11971025). Where indicated, cells were treated with 5 mM (immunoblots) or 2.5 mM (cell counts and Fluorescent Cell Cycle Analyses) sodium phosphate, 50 ng/ml (cell counting and Fluorescent Cell Cycle Analyses) or 10 ng/ml (immunoblots) FGF2 (PeproTech), 10 uM U0126 MEK inhibitor (Cell Signaling), 50uM levamisole (Sigma-Aldrich) and/or 50uM MLS0038949 (EMD Millipore) TNAP inhibitor.
1.2.4. Micro Array
RNA expression of cell cycle proteins in MC3T3E1 cells stably expressing TNAP or non-target shRNA was quantified using the RT² Profiler PCR Array Mouse Cell Cycle (Qiagen, # PAMM-020Z). After establishing control genes as invariant, RNA was normalized using an average of five housekeeping genes Actb (actin beta), B2m (beta-2 microglobulin), Gapd (GAPDH), Gusb (glucuronidase, beta), Hsp90ab1 (heat shock protein 90 alpha cytosolic, class B member 1), Ct data was analyzed using linear models and p values were adjusted for multiple comparisons using the false discovery rate [15,16].
1.2.5. Real Time PCR
Undifferentiated MC3T3E1 cells were seeded at 2×105 cells/well in 6 well culture plates and RNA was isolated upon cell confluence (three days after seeding) using Trizol reagent (Invitrogen) following manufacturer protocols. mRNA levels were assayed by reverse transcription and real time PCR. Real time PCR was performed for murine Gapd, Hus1, E2F2, Aurora B, Cyclin D1 and Fgfr2IIIc using Taman primer sets and Taqman Universal PCR Master Mix (Applied Biosystems). Real-time PCR was performed on a GeneAmp 7700 thermocyler (Applied Biosystems) and quantified by comparison to a standard curve. mRNA levels are reported after normalization to Gapd mRNA levels.
1.2.6. Protein Immunoblotting
Preparation of cell lysate was achieved by solubilization in RIPA buffer (50 mM Tris-Cl pH 7.4, 150 mM NaCl, 1% NaDeoxycholate, 1% Triton-X 100, 0.1% SDS) containing protease inhibitor cocktail (Cell Signaling), followed by removal of insoluble material by centrifugation. Samples were separated by SDS polyacrylamide gel electrophoresis and transferred onto Immobilon. Immunoreactive protein bands were visualized by incubation with primary and HRP conjugated secondary antibodies (Promega, W402B; Sigma-Aldrich, A0545), and enhanced chemiluminescence (Thermo Fisher). The primary antibodies utilized include total and/or phospho-specific antibodies for Cyclin E1 (Cell Signaling, 20808), Cyclin A2 (Abcam, ab38), Cyclin B1 (Cell Signaling, 4138), Cyclin D1 (Cell Signaling, 2978), CDK1 (Cell Signaling, 77055; Cell Signaling, 4539), CDK2 (Cell Signaling, 2546), Aurora B (Cell Signaling, 3094; Cell Signaling, 2914), Histone H3 (Cell Signaling, 9715; Cell Signaling, 3377), Erk1/2 (Cell Signaling, 4695; Cell Signaling, 9101), FGFR2 (Santa Cruz, sc-122) and βtubulin (Cell Signaling, 5346).
1.2.7. Fluorescent Cell Cycle Analyses
The distribution of cells at specific cell cycle stages was evaluated by fluorescence activated flow cytometry (FACS). Cells were seeded 1×105 per well in 6 well plates and grown for two days in media containing 10% fetal bovine serum. 48 hours after seeding, cells were serum starved for 24 hours then serum stimulated for 24 hours. Cells were then collected, counted, fixed and stained for FACS. Briefly, trypsin-EDTA treated cells were neutralized with 10% fetal bovine serum containing media and then centrifuged. After centrifugation, cell pellets were rinsed with PBS and fixed in 70% cold ethanol at 4°C overnight. Subsequently, cells were stained with a propidium iodide-RNAse solution (50ug/ml propidium iodide + 100ug/ml RNAse A in phosphate buffered saline) and subjected to FACS analysis based on DNA content. The samples (1×104 cells) were analyzed by cell cycle distribution using LSR Fortessa (BD Bioscience) and ModFit LT software (version 4.1.7, Verity Software House).
1.2.8. Cell Morphology, Nucleation and Cytokinesis Assays
MC3T3E1 cells stably expressing TNAP or non-target shRNA, and calvarial cells isolated from TNAP−/− or wild type littermates were seeded at 5×104 cells/well onto 4-well chamber slides (Lab-Tek). 24 hours later, the cells were stained with hematoxylin and eosin. Stained slides were imaged using an Olympus BX 51 microscope. Total and bi/multi-nuclear cell counts were quantified (n = 35 cells per genotype or shRNA type). Cytoplasmic area and perimeter were also measured using Image J software (NIH) in triplicate (n=75 cells per genotype or shRNA type). For in vivo analysis, the left liver lobe of 20-day-old mice were fixed and paraffin embedded. Fresh 4 um sections were stained with hematoxylin and eosin. Total and bi/multi-nuclear cells were counted and averaged from several sections from each animal (n > 250 cells per section, n = 3 sections per animal, n=3 animals per genotype). For enhanced visualization of binucleation (Fig. 2H,I), cells cultured on chamber slides were stained by immunofluorescence using Alexa Fluor 488 conjugated anti-phalloidin antibody for F-Actin (Invitrogen, A12379), a primary anti-GM130 golgi marker (Abcam ab52649) with Alexa Fluor 555 secondary antibody and dapi (Sigma-Aldrich, D8417). For enhanced visualization of cytokinesis, cells cultured on chamber slides were stained by immunofluorescence using Alexa Fluor 555 conjugated anti-beta tubulin antibody and dapi. Cells undergoing normal and abnormal cytokinesis were also quantified (n = 130 cells per shRNA type).
Fig. 2: TNAP Deficient Cells are Defective in Cytokinesis.
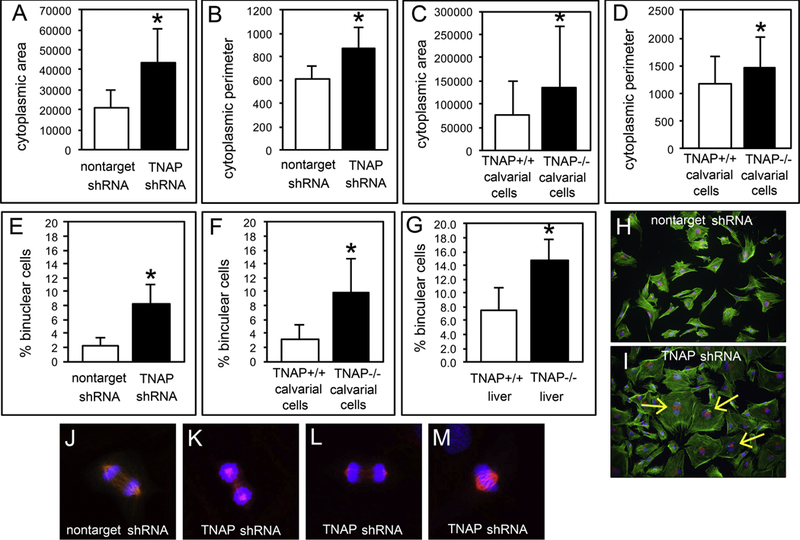
(A,B) MC3T3E1 TNAP shRNA expressing cells are increased in cell size and perimeter (p<005). (C,D) Primary calvarial cells isolated from TNAP−/− mice are increased in cell size and perimeter (p<005). (E,F) MC3T3E1 TNAP shRNA expressing cells and primary calvarial cells isolated from TNAP−/− mice exhibit a greater incidence of binucleation (p<005). (G) Liver tissue of TNAP−/− mice has a greater incidence of binucleated cells as compared to liver tissue of TNAP+/+ mice (p<005). Quantified cell data are represented as mean ± SD. (H,I) MC3T3E1 cells expressing control or TNAP shRNA were stained for F-actin (green), golgi (red) and nuclei (blue) for better visualization of increased cell size and binucleation. Yellow arrows point to binucleation. (J) To visualize the mitotic spindle and chromosomes during cytokinesis, the cells were stained for beta tubulin (red) and dapi (blue). Immunofluorescent staining of control cells shows normal compaction, polarization and migration of chromosomes during cytokinesis. (K-M) TNAP shRNA expressing cells exhibit abnormal compaction, polarization and migration of chromosomes during cytokinesis. The incidence of abnormal cytokinesis in MC3T3E1 TNAP shRNA vs. control cells was also quantified. Control cells exhibited 19 +/− 6 percent of cells with abnormal cytokinesis while TNAP shRNA cells exhibited 42 +/− 3 percent of cells with abnormal cytokinesis (p<.009).
1.2.9. Cell Number Assays
To assay for increases in cell number, cells were seeded and grown in media containing 10% fetal bovine serum for indicated number of days. Cells were stained with trypan blue and counted in sixtuplet at each time point.
1.2.10. Immunohistochemistry
Skulls of five-day-old mice were fixed and paraffin embedded. Fresh 8 µm sagittal sections of the coronal suture and surrounding bones were immunostained using Erk1/2, and phospho-Erk1/2 primary antibodies (Cell Signaling, 4695; Cell Signaling, 9101), HRP-conjugated anti-rabbit secondary antibody (Sigma-Aldrich, A0545), an antigen unmasking solution (Vector Labs) and 3,3’-diaminobenzidine (DAB) colorimetric detection (Abcam) plus counter staining with hematoxylin. The number of brown stained cells and the number of total cells present were counted. Three sections per mouse were calculated and an average value per mouse was utilized to calculate means and standard deviations per genotype (n=4 mice per genotype).
1.2.11. Immunofluorescence
Skulls of five-day-old mice were fixed and paraffin embedded. Fresh 8 µm sagittal sections of the coronal suture and surrounding bones were immunostained using FGFR2 primary antibody (Abcam, ab10648) and fluorescence conjugated secondary antibody (Invitrogen A21428). Sections were mounted with ProLong Gold antifade reagent with DAPI (Invitrogen, P36931) and images were taken on Nikon C2 confocal microscope with 40x objective. Staining was quantified using Image J software. Two sections per mouse were calculated and an average value per mouse was utilized to calculate means and standard deviations per genotype (n=3 mice per genotype).
1.2.12. Statistics
Statistical differences were established using the two-tailed Student’s test. All data are reported as mean +/− standard deviation. T test values of p<.05 were considered significantly different.
1.3. Results
1.3.1. TNAP deficiency inhibits cell cycle progression
We previously showed that TNAP deficiency diminishes calvarial cell proliferation [5,6], yet a functional role for TNAP in osteoblast progenitor cells has yet to be fully established. As a first step towards understanding how TNAP stimulates progenitor cell proliferation, we performed a micro array RNA analysis of cell cycle proteins (Supplemental Fig.1A) in TNAP deficient vs. control cells under standard cell culture conditions (media containing 10% fetal bovine serum). Results indicated significant differences for two cell cycle checkpoint proteins (Hus1 and Sfn), plus diminished RNA levels for the E2F2 transcription factor (promotes transcription of genes that are essential for cell cycle progression), Cyclin D1 (promotes transition from G0 to G1 and G1 to S phase transitions) and Aurora kinase B (essential for mitotic chromosome condensation and for cytokinesis). We confirmed these results by real time PCR, which showed increased expression of Hus1 and decreased expression of E2F2, Aurora B and Cyclin D1 in both MC3T3E1 TNAP shRNA cells and in cells isolated from calvarial tissues of TNAP−/− mice (Supplemental Fig.1B-E).
We next performed immunoblotting for cell cycle proteins in MC3T3E1 cells expressing TNAP or control shRNA after synchronization by serum starvation followed by serum stimulation. Results (Fig. 1A) showed decreased expression of cyclin B1, which normally increases during G2 and functions to promote transition from G2 phase to mitosis [17] and decreased expression of Cyclin D1 which, as stated above, functions to promotes transition from G0 to G1 and G1 to S phase transitions. In addition, Cyclin E1 expression appeared to be poorly regulated in the TNAP shRNA MC3T3E1 cells, in that Cyclin E1 was expressed at all time points in these cells. Cyclin E1 expression normally peaks during G1 to facilitate the transition from G1 to S phase, along with Cyclin D1 [18–20].
Fig. 1: TNAP Deficient Cells Exhibit Aberrant Cyclin and Cytokinesis Protein Expression plus G2/M Arrest.
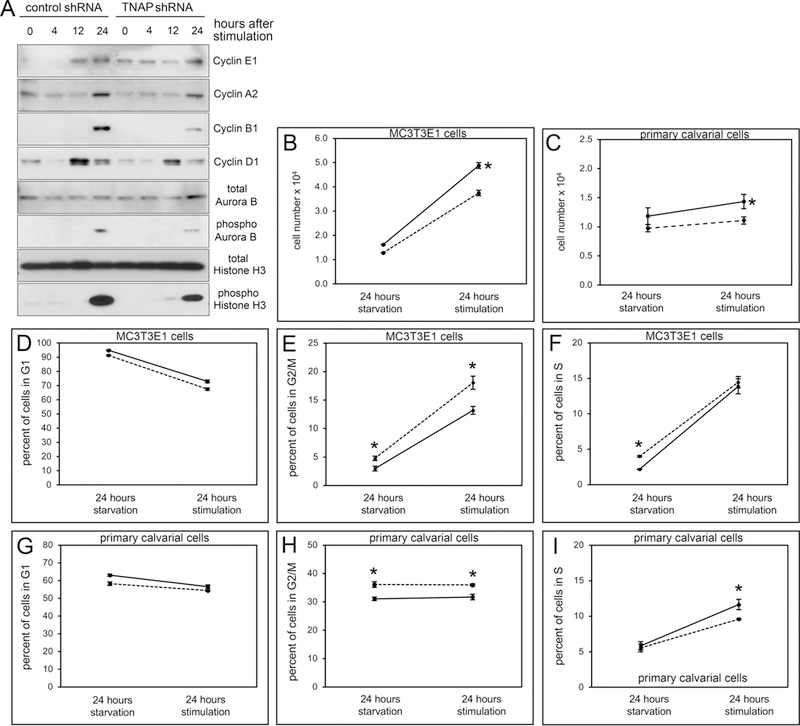
(A) Cell cycle proteins cyclin B1 and cyclin D1 are reduced and cyclin E1 expression appears at all time points in synchronized MC3T3E1 TNAP shRNA expressing cells, as compared to control cells. Phosphorylation of cytokinesis related proteins AuroraB and Histone3 are also reduced in synchronized TNAP shRNA expressing cells. (B) MC3T3E1 TNAP shRNA expressing cells and (D) TNAP−/− primary cells show diminished proliferation compared to control cells after synchronization by serum starvation followed by serum stimulation. (D-F) MC3T3E1 TNAP shRNA expressing cells show more cells entering G2/M phase and fewer cells entering S phase after serum stimulation. (G-I) TNAP−/− primary cells show more cells in the G2/M phase and fewer cells entering S phase after serum stimulation. Solid black lines = controls cells. Dashed black lines = TNAP shRNA expressing or TNAP−/− cells. Data are represented as mean ± SD.
In addition to abnormal cyclin expression, phosphorylated forms of both Aurora B and Histone 3 were diminished in TNAP deficient cells. Activated forms of these proteins are important for chromosome remodeling and condensation during mitosis [21,22], as well as cytokinesis [23,24]. Subsequent analysis of synchronized propidium iodine stained cells by fluorescence activated cell sorting showed significantly more cells in the G2/M phase of the cell cycle before and after serum stimulation in both TNAP shRNA expressing MC3T3E1 cells (Fig. 1E) and in TNAP−/− primary cells (Fig. 1H). Results also showed that fewer TNAP deficient cells entered S phase than control cells, based upon the slope of percentage change after serum stimulation (Fig. 1F,I). Together, these results indicate that TNAP deficient cells appear to be primarily arrested during the G2 and/or M phase. We also confirmed that the TNAP deficient cells showed diminished proliferation compared to control cells during these experiments (Fig. 1B,C).
1.3.2. TNAP deficiency leads to cytokinesis defect
During experimental culture, we noted that TNAP deficient cells were increased in cell size and exhibited binucleation much more often compared to control cells. To establish consistency of these findings, we quantified cell size by perimeter and area, and quantified the percentage of binuclear cells after 24 hours of culture in standard growth media. Results demonstrated that MC3T3E1 TNAP shRNA expressing cells exhibited significantly increased cell perimeter and cell area as compared to control cells (Fig. 2A,B). Concordantly, primary calvarial cells isolated from TNAP−/− mice also exhibited significantly increased cell perimeter and cell area, as compared to cells isolated from wild type littermates (Fig 2C,D). In addition, TNAP deficient cells exhibited significantly higher percentages of binucleation when compared to control cells (Fig 2E,F). To better visualize the increase in cell size and binucleation in TNAP shRNA cells, we also stained cultured cells by immunofluorescence (Fig 2H,I).
To confirm that the increased incidence of binucleation was not an artifact of cell culture, we also looked for evidence of increased binucleation in the TNAP−/− mice. Under normal physiologic conditions, it is expected that polyploid cells will be cleared by apoptosis in most tissues [25]. Polyploidy in the liver, in contrast, is found during normal homeostasis [26]. TNAP is highly expressed in liver tissue [27]. Therefore, to demonstrate increased binucleation in TNAP deficient cells in vivo, we quantified the percentage of binuclear cells in hepatic tissue isolated from twenty-day-old mice. We found a significant increase in binuclear cells in livers of TNAP−/− mice, as compared to TNAP+/+ mice (Fig. 2G). Because cell mass must increase during the cell cycle so that the daughter cells contain the same cell mass as the mother cell [28], the combination of increased cell size and polyploidy suggests that TNAP deficiency leads to a defect in cytokinesis. Subsequent nuclear dapi combined with βtubulin immunofluorescent staining of the mitotic spindle revealed significantly more instances of abnormal chromosome polarization and separation during cytokinesis as compared to control cells (Fig. 2J-M). Control cells exhibited 19 +/− 6 percent of cells with abnormal cytokinesis while TNAP shRNA cells exhibited 42 +/− 3 percent of cells with abnormal cytokinesis (p<.009). This data is consistent with our results showing that levels of the active forms of Aurora B kinase and Histone H3 are diminished in TNAP deficient cells (Fig. 1A), as these protein forms are essential for chromosome condensation and cytokinesis [22,29].
1.3.3. TNAP increases Erk1/2 activity
Erk1/2 stimulates downstream pathways to elicit changes including cell proliferation in response to extracellular mitogens [30,31]. Previous studies showed that inorganic phosphate (Pi) stimulates gene expression by inducing Erk1/2 activity [32–34]. Because TNAP catalytic activity generates Pi which could promote cell proliferation via increased Erk1/2 activity, we next sought to determine if Erk1/2 activity was reduced in TNAP deficient cells and tissues. Consistent with previously published results [32], our data shows that treatment with Pi induces phosphorylation of Erk1/2 in a bi-phasic pattern. Erk1/2 activity increased within 3 minutes of Pi stimulation and was lost by 10 minutes after stimulation. A second and more sustained wave of Erk1/2 activity then occurred starting at approximately 30 minutes after Pi stimulation. In addition, when cells were cultured in media that did not contain Pi, MC3T3E1 TNAP shRNA cell (Fig. 3A) and calvarial cells isolated from TNAP−/− mice (Fig. 3B) exhibited lower Erk1/2 phosphorylation levels when compared to control cells, indicating that TNAP stimulates Erk1/2 activity. Surprisingly, when cells cultured in medium without Pi were treated with Pi, TNAP deficient cells continued to exhibit diminished levels of phosphorylated Erk1/2 as compared to control cells, indicating that TNAP stimulates Erk1/2 activity via both Pi dependent and independent pathways. Total Erk1/2 protein levels were not different between experimental and controls cells, with or without Pi stimulation. Next, we utilized chemical inhibitors of TNAP to determine if TNAP activity promotes Erk1/2 activity (Fig. 3C). Two inhibitors of TNAP enzyme activity diminished Erk1/2 phosphorylation. Additionally, we stained for total Erk1/2 and phosphorylated Erk1/2 in mouse cranial tissues. Results showed diminished levels of phosphorylated Erk1/2 in periosteum and cranial suture cells of TNAP−/− mice (Fig. 4). Together, these results demonstrate that TNAP promotes Erk1/2 activity in calvarial progenitor cells.
Fig. 3: TNAP Mediates Erk1/2 Activity in Vitro.
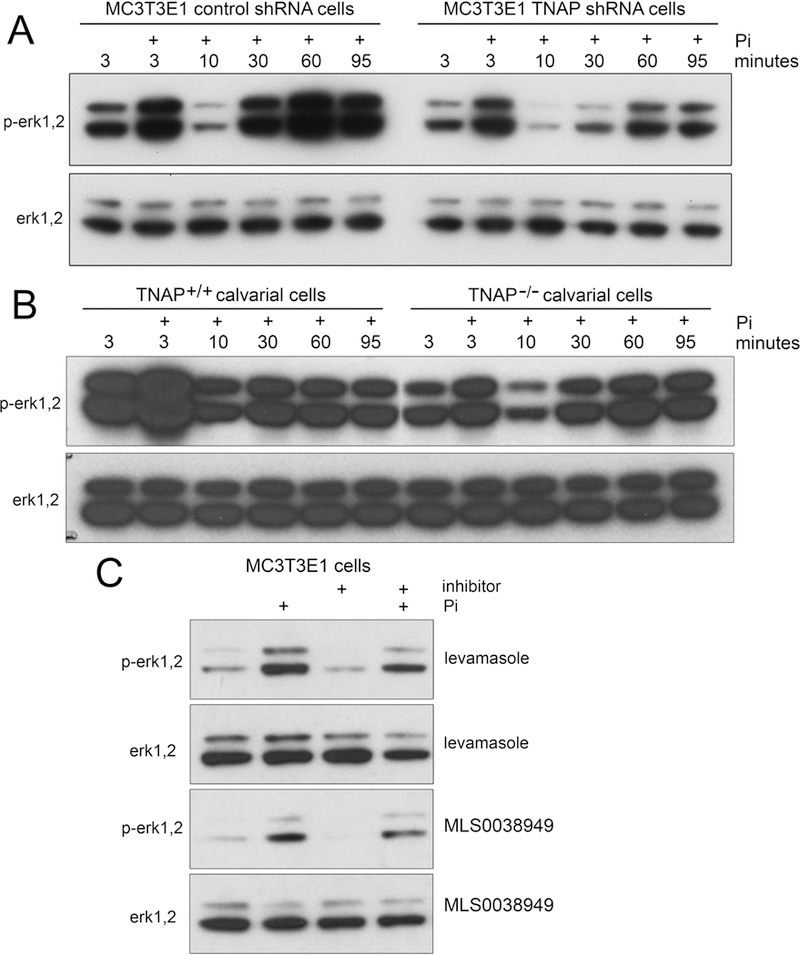
(A) MC3T3E1 TNAP shRNA cells exhibit diminished phosphorylation of Erk1/2 as compared to control shRNA cells. (B) TNAP−/− primary calvarial cells exhibit diminished phosphorylation of Erk1/2 as compared to TNAP+/+ cells. (C) MC3T3E1 cells treated with chemical inhibitors of TNAP (levamasole and MLS0038949) exhibit diminished phosphorylation of Erk1/2 with and without Pi stimulation. Quantified data are represented as mean ± SD.
Fig. 4: TNAP Mediates Erk1/2 Activity in Vivo.
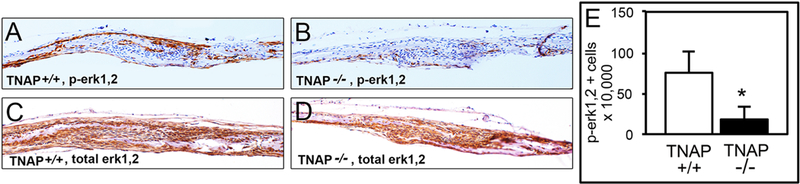
Immunohistochemical stains of frontal and parietal calvarial bones surrounding the coronal suture demonstrate similar total Erk1/2 but diminished phosphorylated Erk1/2 levels in periosteum, calvarial osteogenic fronts and suture tissues of TNAP−/− as compared to TNAP+/+ mice. (A) Representative phospho-Erk1/2 stain of TNAP+/+cranial tissues. (B) Representative phospho-Erk1/2 stain of TNAP−/− cranial tissues. (C) Representative total Erk1/2 stain of TNAP+/+ cranial tissues. (D) Representative total Erk1/2 stain of TNAP−/− cranial tissues. (E) Quantification of phospho-Erk1/2 positive cells (n=4 per genotype). *p<.05 between genotypes. Quantified cell data are represented as mean ± SD.
1.3.4. Diminished proliferation of TNAP deficient cells is mediated by Erk1/2 activity.
Our results show that TNAP promotes cell cycle progression, and that the active forms of Erk1/2 are expressed at lower levels in TNAP deficient calvarial cells with/without exogenous Pi stimulation. Therefore, we next sought to determine if other known inducers of Erk1/2 activity were limited in their ability to stimulate phosphorylation of Erk1/2 in TNAP deficient cells. Results showed that, in contrast to the lower induction of Erk1/2 phosphorylation seen upon stimulation with Pi, FGF2 stimulates Erk1/2 phosphorylation in a more sustained manner in MC3T3E1C4 TNAP shRNA cells when compared to control cells (Fig. 5A). This data shows that pathways other than those induced by Pi are able to fully stimulate Erk1/2 activity and perhaps to an even greater extent in TNAP deficient cells.
Fig. 5: Erk1/2 Mediates the Diminished Proliferation and Cell Cycle Lag of TNAP Deficient Cells.
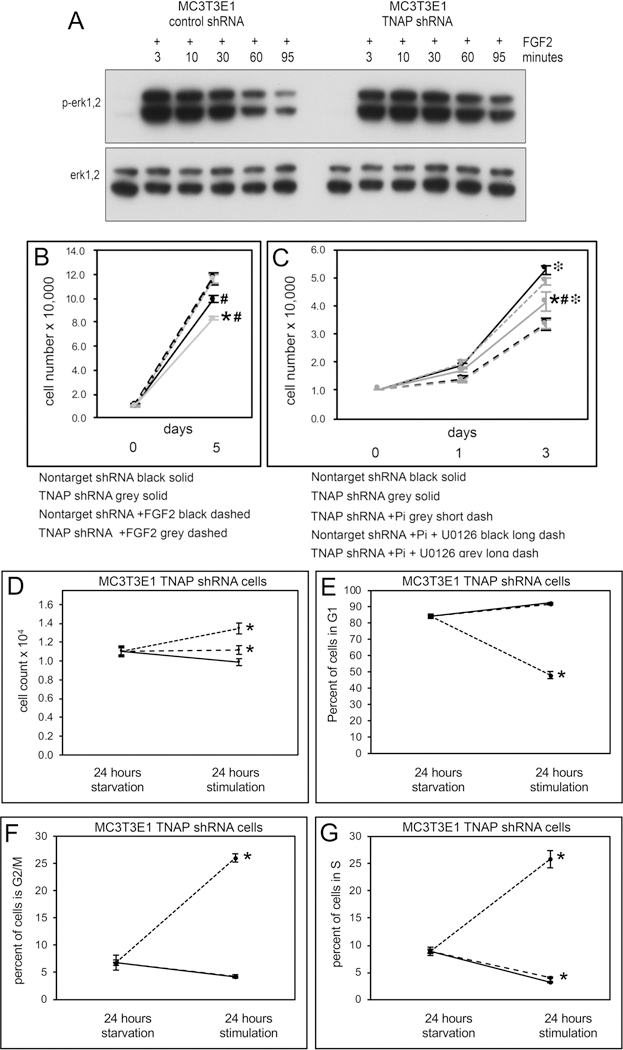
(A) MC3T3E1 TNAP shRNA expressing cells exhibit more sustained levels of phosphorylated Erk1/2 as compared to control cells upon stimulation with FGF2. (B) FGF2 treatment stimulates TNAP deficient cells to proliferate at levels similar to that seen in control cells. (C) Treatment with the MEK inhibitor U0126 prevents the increased proliferation seen in TNAP deficient cells upon treatment with Pi. (D–G) MC3T3E1 TNAP shRNA cells were serum starved to synchronize and then stimulated with FGF2 or Pi. (D) TNAP shRNA cells are stimulated to proliferate upon treatment with FGF2 to a greater degree than with Pi. (E) FGF2 but not Pi promotes G1 phase exit of TNAP shRNA cells. (F) FGF2 but not Pi promotes G2/M phase entrance of TNAP shRNA cells. (G) TNAP shRNA cells are stimulated to enter S phase to a significantly greater extent than Pi. Quantified cell data are represented as mean ± SD.
We next performed experiments to determine if Erk1/2 activity mediates the decreased proliferation of TNAP deficient cells. Assays revealed that treatment of MC3T3E1 TNAP shRNA cells with FGF2 increased rates of proliferation to that of control cells (Fig. 5B). In addition, while treatment of TNAP deficient cells with Pi increased the proliferation of these cells, co-treatment with MEK inhibitor U0126 prevented this increase in proliferation (Fig. 5C). Together, these results indicate that Erk1/2 activity is an essential downstream mechanism by which TNAP stimulates cell proliferation. The results also show that Pi-induced proliferation of TNAP deficient progenitor cells is dependent upon Erk1/2 activity.
1.3.5. FGF2, but not Pi, rescues cell cycle arrest in TNAP deficient cells
Because FGF2 and Pi are able to stimulate Erk1/2 phosphorylation and increase proliferation of TNAP deficient cells, we next sought to determine if FGF2 and/or Pi could rescue the cell cycle progression abnormalities seen in TNAP deficient cells. Results from cells grown in serum and Pi free media show that both Pi or FGF2 can stimulate proliferation of TNAP shRNA expressing cells, although FGF2 does this to a significantly stronger extent than Pi (Fig. 5D). Only FGF2 strongly promoted cell cycle progression (Fig. 5E–F). FGF2 but not Pi stimulated cells to exit G1 and enter G2/M. Pi and FGF2 both stimulated entry of the cells into S phase, though FGF2 did this to a significantly greater extent than Pi. These results indicate that FGF2 is more efficacious than Pi for rescuing cell proliferation and cycle progression defects of TNAP deficient cells.
1.3.6. TNAP regulates FGFR2 expression via Erk1/2 activity
Both FGFR2 [35,36] and TNAP are known to regulate skeletal development. Additionally, mutations in FGFR2 and in TNAP enzyme are both associated with craniosynostosis (the premature fusion of cranial bones that limits skull growth and causes high intracranial pressure) [4,37–40]. Because both FGF signaling [41] and TNAP expression influence Erk1/2 activity and because abnormalities in either FGFR signaling or TNAP levels result in similar craniofacial skeletal phenotypes, we next sought to determine if and how pathways downstream of FGF signaling and TNAP activity might interact. Thus, we next tested for protein expression of the mesenchyme-specific isoforms FGFR1IIIc and FGFR2IIIc in MC3T3E1 TNAP shRNA cells and in calvarial cells isolated from TNAP−/− mice, both before and after osteoblast differentiation. Expression of FGFR1 and FGFR2 was not different in TNAP deficient vs. control cells after osteoblast differentiation, and FGFR1 expression was not different in TNAP deficient vs. control cells prior to differentiation (data not shown). In contrast, FGFR2 expression was strongly increased in both TNAP shRNA and TNAP−/− cells prior to differentiation (Fig. 6A). Concordantly, transduction of cells with a TNAP expression adenovirus decreased FGFR2 expression in TNAP deficient cells (Fig. 6B). Together this data indicates that FGFR2 is expressed at higher levels in TNAP deficient cells and that TNAP downregulates FGFR2 expression. Subsequent treatment of cells with the transcription inhibitor actinomycin for six hours lowered both protein and mRNA levels of FGFR2 (Fig. 6C,D), suggesting that the regulation of FGFR2 by TNAP occurs at the transcriptional, as opposed to translational level.
Fig. 6: TNAP Deficiency Promotes FGFR2 Expression via FGF Signaling.
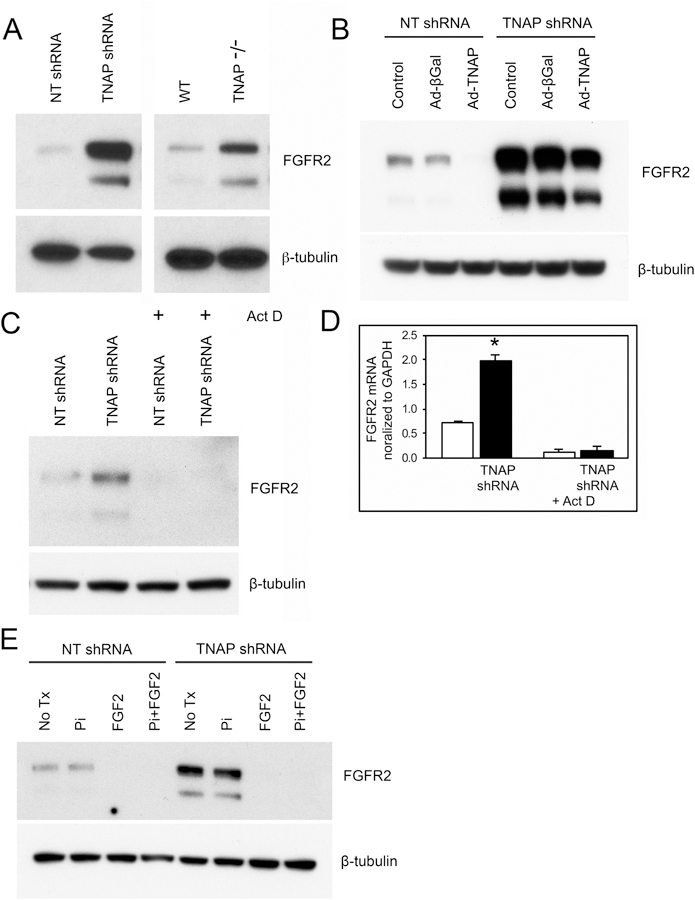
(A) Immunoblotting shows diminished FGFR2 expression in MC3T3E1 cells expressing TNAP shRNA and in calvarial progenitor cells isolated from TNAP−/− mice, as compared to control cells. (B) Transduction of MC3T3E1 cells with a TNAP expression adenovirus diminishes FGFR2 expression in both TNAP shRNA and nontarget shRNA expressing cells. (C) Treatment with the actinomycin D inhibitor of transcription decreases FGFR2 protein expression in both TNAP deficient and control cells. (D) Treatment with the actinomycin D inhibitor of transcription decreases FGFR2 mRNA expression in both TNAP deficient and control cells. (E) FGF2 but not Pi treatment decreases FGFR2 expression in both TNAP deficient and control cells. Quantified cell data are represented as mean ± SD.
Because both Pi and FGF2 can stimulate Erk1/2 and the proliferation of TNAP deficient calvarial cells, we tested if Pi and/or FGF2 treatment could influence FGFR2 expression in TNAP deficient cells. FGF2 but not Pi decreased FGFR2 expression in TNAP deficient and control cells (Fig. 6E). Finally, we stained for FGFR2 protein in mouse cranial tissues. Results showed higher levels of FGFR2 expression in coronal suture tissue of TNAP−/− mice (Fig. 7). Together this data indicates that TNAP deficiency increases FGFR2 expression in calvarial bone progenitor cells, and that the process by which this occurs is not mediated by Pi.
Fig. 7: TNAP Deficiency Increases FGFR2 Expression in in Vivo.
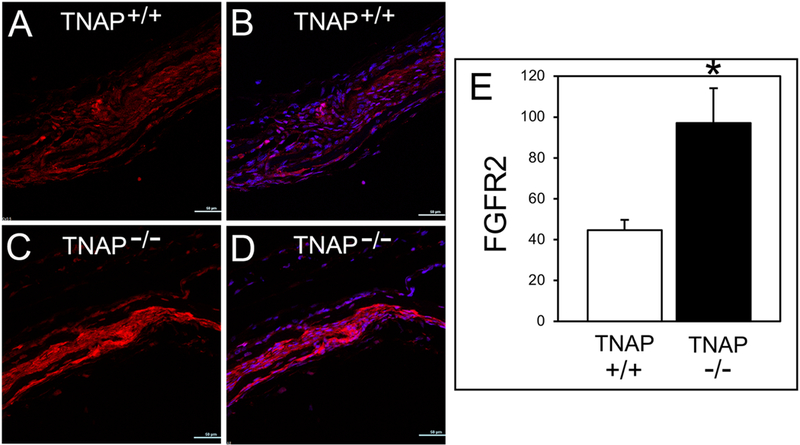
Immunofluorescent stains of frontal and parietal calvarial bones surrounding the coronal suture demonstrate increased FGFR2 levels in the coronal suture tissue of TNAP−/− as compared to TNAP+/+ mice. (A) TNAP+/+ FGFR2 signal (red). (B) TNAP+/+ FGFR2 signal combined with dapi nuclear stain (blue/red merge). (C) TNAP−/− FGFR2 signal. (D) TNAP−/− FGFR2 signal combined with dapi. Quantification of FGFR2 immunofluorescent stain (n=3 per genotype). *p<.05 between genotypes. Quantified cell data are represented as mean ± SD.
1.4. Discussion
Current understanding of the role of TNAP in bone development is limited to its known function in differentiated bone forming cells. TNAP is highly expressed in the extracellular membrane of osteoblasts and their matrix vesicles where it hydrolyzes PPi (inhibitor of mineralization) to Pi (substrate for mineralization) [42]. Because PPi to Pi ratios determine if a tissue will be mineralized, conversion of PPi to Pi by TNAP is essential for promoting bone matrix mineralization [43]. Here we show that TNAP expression is also essential for calvarial progenitor cell cycle progression and cytokinesis. Overall our data indicates that TNAP is essential for proper chromosome condensation, migration and cytokinesis. Immunoblot data demonstrated diminished expression of cyclin B1 and cyclin D1, and unregulated expression of cyclin E1 in synchronized MC3T3E1 TNAP shRNA cells, which indicated a potential defect in cell cycle progression. Cyclin B1 is known to increase during G2 and functions to promote transition from G2 phase to mitosis, while cyclins D1 and E1 are known to peak during G1 to promote transition from G1 to S phase. In addition, we found diminished levels of the active forms of Aurora B and Histone 3, which are known to be essential for chromosome condensation, chromosome migration and cytokinesis [44,45].
Subsequent cell sorting data showed that TNAP deficient calvarial cells are primarily blocked in the G2/M phase of the cell cycle. This data, combined with the fact that the TNAP deficient cells exhibited increased cell size and incidence of binucleation suggests abnormalities in cytokinesis. Immunofluorescent staining of chromosomes and the mitotic spindle subsequently confirmed that TNAP deficient cells exhibited significantly more instances of abnormal chromosome condensation and migration than that seen in control cells. It is worth noting that significantly more, but not all of the TNAP deficient cells exhibited increased cell size, binucleation and abnormal cytokinesis. TNAP appears to promote cell cycle progression and cytokinesis, but not be essential for it. Together, the data presented here indicates that TNAP has a cell cycle function in calvarial bone progenitor cells that is in addition to its previously established function of promoting tissue mineralization when expressed in differentiated osteoblasts. Additional support for the concept that TNAP plays a critical role in progenitor cells comes from studies of pluripotency and embryonic stem cells. TNAP is a marker of migrating primordial germs cells in humans [9], monkey embryonic stem cells [46], and pluripotent cells in embryonic carcinoma [47]. Future studies in which TNAP is conditionally deleted only in calvarial and/or long bone progenitor cells should provide additional insight into how TNAP influences chromosome dynamics and cytokinesis to promote progenitor cell renewal in bone development.
TNAP generates inorganic phosphate (Pi), which was previously shown to stimulate Erk1/2 activity in bone progenitor cells [32,33]. Therefore, we also sought to determine if the effects of TNAP on cell cycle progression were mediated by Erk1/2 activity. As expected, we found that Erk1/2 phosphorylation levels were diminished in TNAP deficient cells when cultured in phosphate-free media. Yet while levels of active Erk1/2 did increase in both wild type and TNAP deficient cells upon Pi treatment, we were surprised to also find Pi induced Erk1/2 phosphorylation levels to be diminished in TNAP deficient as compared to control cells. This data indicates that TNAP stimulates Erk1/2 activity via both Pi dependent and Pi independent mechanisms. The data also suggests that TNAP is involved in or in some manner influences the mammalian sensor for Pi. Cellular systems for sensing and responding to Pi levels are well delineated in bacteria and yeast [48] but remain unknown in mammalian cells. Pi is essential for numerous biochemical and cell signaling pathways [13]. This fact plus the fact that Pi stimulates Erk1/2 activity in mammalian cells suggests that a system for sensing Pi likely exists in mammalian cells. It is possible that TNAP directly interacts with other membrane proteins such as Pi channels, to mediate a Pi response. GPI-anchored alkaline phosphatases are also known to localize to membrane lipid rafts and to alter the solubility of lipid rafts [49–51]. Therefore, it is also possible that TNAP alters solubility of lipid rafts to indirectly influence the mammalian cell signaling response to Pi. Importantly, we also found consistently diminished levels of active Erk1/2 in TNAP−/− developing calvarial tissues. Regardless of the mechanism, TNAP appears to increase levels of active Erk1/2 in calvarial bone progenitor cells.
Because we found diminished induction of Erk1/2 phosphorylation in TNAP deficient cells upon treatment with Pi, we next sought to determine if other known inducers of Erk1/2 signaling could stimulate Erk1/2 phosphorylation in TNAP deficient cells to the same level as that seen in control cells. Results show that FGF2 was able to stimulate Erk1/2 phosphorylation, and in a more sustained manner in TNAP deficient as compared to control cells. Subsequent experiments showed that FGF2 stimulated proliferation of TNAP deficient cells to levels similar to that of control cells. Moreover, FGF2 but not Pi, stimulated TNAP deficient cells to exit G1 and enter G2/M phases. FGF2 also stimulated the cells to enter S phase to a significantly greater extent than Pi. It is worth noting the Pi was able to stimulate cell proliferation and entry into the S phase, but not to stimulate progression through G1 and G2/M. These results together indicate that the influence of TNAP on cell cycle progression and proliferation is mediated by Erk1/2 activity and that FGF stimulation can rescue the abnormalities seen in TNAP deficient cells.
The sustained Erk1/2 signaling upon FGF2 treatment of TNAP deficient cells suggested that TNAP deficiency makes cells more responsive to FGFs. Therefore, we next investigated expression of FGFR1 and FGFR2 in TNAP deficient and control cells before and after osteoblast differentiation. We found that FGFR2 expression was upregulated in TNAP deficient calvarial progenitor cells in culture, and that overexpression of TNAP diminished FGFR2 expression. We also found increased FGFR2 expression in coronal suture tissue of young TNAP−/− mice. From a developmental perspective, it is worth noting that mutations in FGFR2 and mutations in TNAP enzyme both cause craniosynostosis [37–40,52–53]. Mechanisms behind craniosynostosis in hypophosphosphatasia remain unknown. Our data suggests that craniosynostosis downstream of TNAP activity could be mediated by diminished renewal of calvarial progenitor cells causing premature loss of the mesenchymal cranial suture tissue that is normally present between growing calvarials, leading to bone fusion. Based upon our data it is tempting to speculate that changes in FGF signaling mediate the abnormal craniofacial skeletal development seen in humans and mice with TNAP deficiency. We are currently pursuing studies to determine if stimulation of FGF signaling can rescue the abnormal craniofacial and/or long bone phenotypes seen in TNAP−/− mice.
1.5. Conclusions
In summary, here we report a novel role for TNAP in skeletal development (Fig. 8). In calvarial bone progenitor cells, TNAP promotes cell cycle progression and cytokinesis via Erk1/2 signaling. Notably, the stimulation of Erk1/2 signaling by TNAP occurs via both inorganic phosphate dependent and independent mechanisms. In addition, FGFR2 expression is increased in TNAP deficient progenitor cells, making these cells more responsive to FGF stimulation. Together, this data establishes a previously unidentified and essential mechanism by which TNAP influences skeletal development by promoting bone progenitor cell renewal. The data also suggests that pathways downstream of FGF signaling and TNAP activity converge in their influence on craniofacial skeletal development.
Fig. 8: Model of TNAP Function in Bone Progenitor Cells.
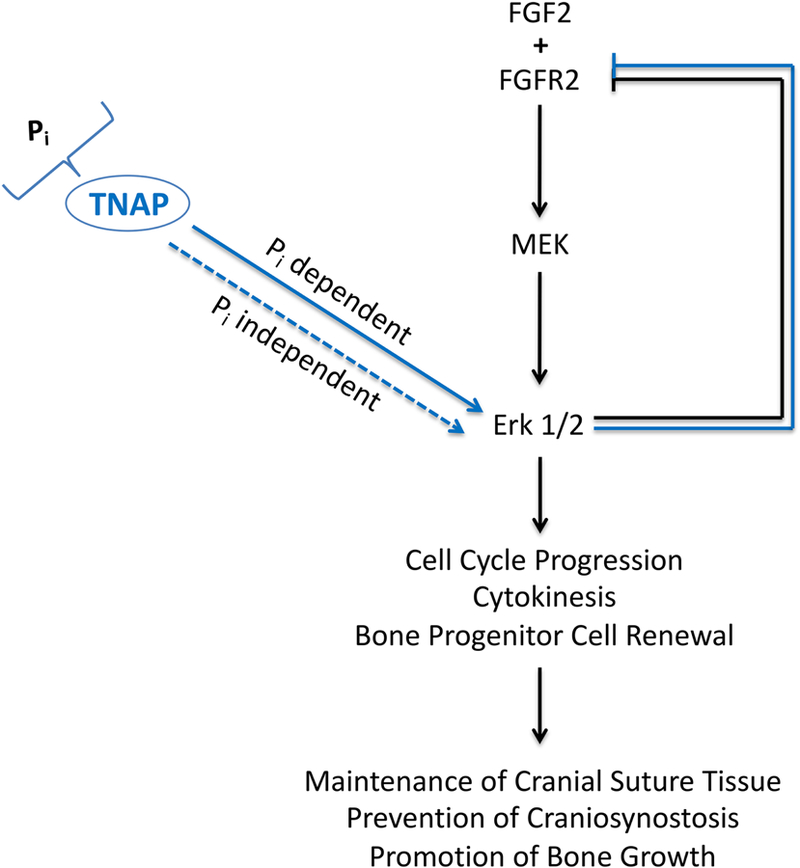
TNAP is an enzymatic generator of inorganic phosphate (Pi). Our data shows that TNAP stimulates Erk1/2 activity via both Pi dependent and independent pathways. Erk1/2 activity promotes cell cycle progression, cytokinesis and bone progenitor cell renewal, which we hypothesize helps to maintain cranial suture mesenchyme tissue between growing calvarials, promote bone growth and prevent craniosynostosis. Erk1/2 activity also feeds back to inhibit FGFR2 expression.
Supplementary Material
Highlights:
TNAP enzyme deficiency causes defects in cranial bone progenitor cell cycle progression and cytokinesis
TNAP stimulates erk1,2 activity
TNAP deficiency increases expression of FGFR2
Stimulation of FGF signaling rescues TNAP deficient cell cycle abnormalities
Acknowledgements
We are extremely grateful to Dr. José Luis Millán who generously provided the TNAP−/− mice.
Funding
This work was supported by a grant from NIH/NIDCR (R01 DE025827 to NEH).
Abbreviations:
- TNAP
Tissue Nonspecific Alkaline Phosphatase
- Pi
inorganic phosphate
- PPi
inorganic pyrophosphate
- FGF2
fibroblast growth factor 2
- FGFR2
fibroblast growth factor receptor 2
Footnotes
Disclosures: The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- 1.Johnson KA, Hessle L, Vaingankar S, Wennberg C, Mauro S, Narisawa S, Goding JW, Sano K, Millan JL, Terkeltaub R. Osteoblast tissue-nonspecific alkaline phosphatase antagonizes and regulates PC-1. Am J Physiol Regul Integr Comp Physiol 2000;279:R1365–1377. [DOI] [PubMed] [Google Scholar]
- 2.Hessle L, Johnson KA, Anderson HC, Narisawa S, Sali A, Goding JW, Terkeltaub R, Millan JL. Tissue-nonspecific alkaline phosphatase and plasma cell membrane glycoprotein-1 are central antagonistic regulators of bone mineralization. Proc Natl Acad Sci 2002:99:9445–9449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Narisawa S, Frohlander N, Millan JL. Inactivation of two mouse alkaline phosphatase genes and establishment of a model of infantile hypophosphatasia. Dev Dyn 1997;208:432–446. [DOI] [PubMed] [Google Scholar]
- 4.Fedde KN, Blair L, Silverstein J, Coburn SP, Ryan LM, Weinstein RS Waymire K, Narisawa S, Millán JL, MacGregor GR, Whyte MP. Alkaline phosphatase knock-out mice recapitulate the metabolic and skeletal defects of infantile hypophosphatasia. J Bone Miner Res 1999;14(12):2015–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu J, Nam HK, Campbell C, Gasque KC, Millán J, Hatch NE. Tissue-nonspecific alkaline phosphatase deficiency causes abnormal craniofacial bone development in the Alpl(−/−) mouse model of infantile hypophosphatasia. Bone 2014:67:81–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu J, Campbell C, Nam HK, Caron A, Yadav MC, Millán JL, Hatch NE. Enzyme replacement for craniofacial skeletal defects and craniosynostosis in murine hypophosphatasia. Bone 2105;78:203–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.MacGregor GR, Zambrowicz BP, Soriano P. Tissue non-specific alkaline phosphatase is expressed in both embryonic and extraembryonic lineages during mouse embryogenesis but is not required for migration of primordial germ cells. Development 1995:121:1487–1496. [DOI] [PubMed] [Google Scholar]
- 8.Lomelí H, Ramos-Mejía V, Gertsenstein M, Lobe CG, Nagy A. Targeted insertion of Cre recombinase into the TNAP gene: excision in primordial germ cells. Genesis 2000;26(2):116–7. [PubMed] [Google Scholar]
- 9.De Felici M Origin, Migration, and Proliferation of Human Primordial Germ Cells. In: Coticchio G, Albertini D, De Santis L, editors. Oogenesis London: Springer; 2013. p. 19–37. [Google Scholar]
- 10.Langer D, Ikehara Y, Takebayashi H, Hawkes R, Zimmermann H. The ectonucleotidases alkaline phosphatase and nucleoside triphosphate diphosphohydrolase 2 are associated with subsets of progenitor cell populations in the mouse embryonic, postnatal and adult neurogenic zones. Neuroscience 2007;150(4):863–79. [DOI] [PubMed] [Google Scholar]
- 11.Kaufman MH. The Atlas of Mouse development The Elsevier Academic Press; 2003. [Google Scholar]
- 12.Sun J, Ishii M, Ting MC, Maxson R. Foxc1 controls the growth of the murine frontal bone rudiment by direct regulation of a Bmp response threshold of Msx2. Development 2013;140(5):1034–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berg JM, Tymoczko LT, Gatto GJ, Styer L. Biochemistry. W.H. Freeman & Company, a Macmillan Education Imprint; 2015.
- 14.Waymire KG, Mahuren JD, Jaje JM, Guilarte TR, Coburn SP, MacGregor GR. Mice lacking tissue non-specific alkaline phosphatase die from seizures due to defective metabolism of vitamin B-6. Nat Genet 1995;11(1):45–51. [DOI] [PubMed] [Google Scholar]
- 15.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 2004;3:Article 3. [DOI] [PubMed] [Google Scholar]
- 16.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J Royal Stat Soc Series B (Methodological) 1995;57(1):289–300. [Google Scholar]
- 17.Yasutis KM, Kozminski KG. Cell cycle checkpoint regulators reach a zillion. Cell Cycle 2013;12(10):1501–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Geng Y, Yu Q, Whoriskey W, Dick F, Tsai KY, Ford HL, Biswas DK, Pardee AB, Amati B, Jacks T, Richardson A, Dyson N, Sicinski P. Expression of cyclins E1 and E2 during mouse development and in neoplasia. Proc Natl Acad Sci U S A 2001;98(23):13138–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dulić V, Lees E, Reed SI. Association of human cyclin E with a periodic G1-S phase protein kinase. Science 1992;257(5078):1958–61. [DOI] [PubMed] [Google Scholar]
- 20.Koff A, Giordano A, Desai D, Yamashita K, Harper JW, Elledge S, Nishimoto T, Morgan DO, Franza BR, Roberts JM. Formation and activation of a cyclin E-cdk2 complex during the G1 phase of the human cell cycle. Science 1992;257(5077):1689–94. [DOI] [PubMed] [Google Scholar]
- 21.Goto H, Tomono Y, Ajiro K, Kosako H, Fujita M, Sakurai M, Okawa K, Iwamatsu A, Okigaki T, Takahashi T, Inagaki M. Identification of a novel phosphorylation site on histone H3 coupled with mitotic chromosome condensation. J Biol Chem 1999;274(36):25543–9. [DOI] [PubMed] [Google Scholar]
- 22.Prigent C, Dimitrov S. Phosphorylation of serine 10 in histone H3, what for? J Cell Sci 2003;116(Pt 18):3677–85. [DOI] [PubMed] [Google Scholar]
- 23.Song L, Li D, Liu R, Zhou H, Chen J, Huang X. Ser-10 phosphorylated histone H3 is involved in cytokinesis as a chromosomal passenger. Cell Biol Int 2007;31(10):1184–90. [DOI] [PubMed] [Google Scholar]
- 24.Goldenson B, Crispino JD. The aurora kinases in cell cycle and leukemia. Oncogene 2015;34(5):537–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hayashi MT, Karlseder J. DNA damage associated with mitosis and cytokinesis failure. Oncogene 2013; 32(39):4593–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Milne LS. The histology of liver tissue regeneration. The Journal of Pathology and Bacteriology 1909;13(1):127–60. [Google Scholar]
- 27.Buchet R, Millán JL, Magne D. Multisystemic functions of alkaline phosphatases. Methods Mol Biol 2013;1053:27–51. [DOI] [PubMed] [Google Scholar]
- 28.Cooper S Rethinking synchronization of mammalian cells for cell cycle analysis. Cell Mol Life Sci 2003;60(6):1099–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hendzel MJ, Wei Y, Mancini MA, Van Hooser A, Ranalli T, Brinkley BR, Bazett-Jones DP, Allis CD. Mitosis-specific phosphorylation of histone H3 initiates primarily within pericentromeric heterochromatin during G2 and spreads in an ordered fashion coincident with mitotic chromosome condensation. Chromosoma 1997;106(6):348–60. [DOI] [PubMed] [Google Scholar]
- 30.Boulton TG, Nye SH, Robbins DJ, Ip NY, Radziejewska E, Morgenbesser SD, DePinho RA, Panayotatos N, Cobb MH, Yancopoulos GD. ERKs: a family of protein-serine/threonine kinases that are activated and tyrosine phosphorylated in response to insulin and NGF. Cell 1991;65(4):663–75. [DOI] [PubMed] [Google Scholar]
- 31.Robbins DJ, Zhen E, Owaki H, Vanderbilt CA, Ebert D, Geppert TD, Cobb MH. Regulation and properties of extracellular signal-regulated protein kinases 1 and 2 in vitro. J Biol Chem 1993;268(7):5097–106. [PubMed] [Google Scholar]
- 32.Beck GR Jr., Knecht N Osteopontin regulation by inorganic phosphate is ERK1/2-, protein kinase C-, and proteasome-dependent. J Biol Chem 2003;278(43):41921–9. [DOI] [PubMed] [Google Scholar]
- 33.Khoshniat S, Bourgine A, Julien M, Weiss P, Guicheux J and Beck L. The emergence of phosphate as a specific signaling molecule in bone and other cell types in mammals. Cell Mol Life Sci 2011;68(2):205–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Julien M, Khoshniat S, Lacreusette A, Gatius M, Bozec A, Wagner EF, Wittrant Y, Masson M, Weiss P, Beck L, Magne D, Guicheux J. Phosphate-dependent regulation of MGP in osteoblasts: role of ERK1/2 and Fra-1. J Bone Miner Res 2009;1: 1856–68. [DOI] [PubMed] [Google Scholar]
- 35.Eswarakumar VP, Monsonego-Ornan E, Pines M, Antonopoulou I, Morriss-Kay GM, Lonai P. The IIIc alternative of Fgfr2 is a positive regulator of bone formation. Development 2002;129:3783–3793. [DOI] [PubMed] [Google Scholar]
- 36.Yu K, Xu J, Liu Z, Sosic D, Shao J, Olson EN, Towler DA, Ornitz DM. Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development 2003;130:3063–3074. [DOI] [PubMed] [Google Scholar]
- 37.Reardon W, Winter RM, Rutland P, Pulleyn LJ, Jones BM, Malcolm S. Mutations in the fibroblast growth factor receptor 2 gene cause Crouzon syndrome. Nat Genet 1994;8:98–103. [DOI] [PubMed] [Google Scholar]
- 38.Wilkie AO, Slaney SF, Oldridge M, Poole MD, Ashworth GJ, Hockley AD, Hayward RD, David DJ, Pulleyn LJ, Rutland P et al. Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome. Nat Genet 1995;9:165–72. [DOI] [PubMed] [Google Scholar]
- 39.Eswarakumar VP, Horowitz MC, Locklin R, Morriss-Kay GM, Lonai P. A gain-of-function mutation of Fgfr2c demonstrates the roles of this receptor variant in osteogenesis. Proc Natl Acad Sci USA 2004;101:12555–12560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fraser D Hypophosphatasia. Am J Med 1957;22(5):730–746. [DOI] [PubMed] [Google Scholar]
- 41.Korsensky L, Ron D. Regulation of FGF signaling: Recent insights from studying positive and negative modulators. Semin Cell Dev Biol 2016;53:101–14. [DOI] [PubMed] [Google Scholar]
- 42.Millan J Mammalian Alkaline Phosphatases: From Biology to Applications in Medicine and Biotechnology Wiley‐VCH; 2006. [Google Scholar]
- 43.Murshed M, Harmey D, Millan JL, McKee M, Karsenty G. Unique coexpression I osteoblasts of broadly expressed genes accounts for the spatial restriction of ECM mineralization to bone. Genes and Dev 2009;19:1093–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Crosio C, Fimia GM, Loury R, Kimura M, Okano Y, Zhou H, Sen S, Allis CD, Sassone-Corsi P. Mitotic phosphorylation of histone H3: spatio-temporal regulation by mammalian Aurora kinases. Mol Cell Biol 2002;22(3):874–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hammond SL, Byrum SD, Namjoshi S, Graves HK, Dennehey BK, Tackett AJ and Tyler JK. Mitotic phosphorylation of histone H3 threonine 80. Cell Cycle 2014;13(3):440–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yasuda SY, Tsuneyoshi N, Sumi T, Hasegawa K, Tada T, Nakatsuji N, Suemori H. NANOG maintains self-renewal of primate ES cells in the absence of a feeder layer. Genes Cells 2006;9:1115–23. [DOI] [PubMed] [Google Scholar]
- 47.Berstine EG, Hooper ML, Grandchamp S, Ephrussi B. Alkaline phosphatase activity in mouse teratoma. Proc Natl Acad Sci USA 1973;70(12):3899–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bergwitz C, Jüppner H. Phosphate sensing. Adv Chronic Kidney Dis 2011;18(2):132–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Morandat S, Bortolato M, Roux B. Cholesterol-dependent insertion of glycosylphosphatidylinositol-anchored enzyme. Biochim Biophys Acta 2002;1564(2):473–78. [DOI] [PubMed] [Google Scholar]
- 50.Morandat S, Bortolato M, Roux B. Role of GPI-anchored enzyme in liposome detergent-resistance. J Membr Biol 2003;191(3):215–21. [DOI] [PubMed] [Google Scholar]
- 51.Giocondi MC, Besson F, Dosset P, Milhiet PE, Le Grimellec C. Temperature-dependent localization of GPI-anchored intestinal alkaline phosphatase in model rafts. J Mol Recognit 2007;20(6):531–7. [DOI] [PubMed] [Google Scholar]
- 52.MP Greenberg CR, Salman NJ, Bober MB, McAlister WH, Wenkert D, Van Sickle BJ, Simmons JH, Edgar TS, Bauer ML, Hamdan MA, Bishop N, Lutz RE, McGinn M, Craig S, Moore JN, Taylor JW, Cleveland RH, Cranley WR, Lim R, Thacher TD, Mayhew JE, Downs M, Millán JL, Skrinar AM, Crine P, Landy H. Enzyme-replacement therapy in life-threatening hypophosphatasia. N Engl J Med 2012;366(10):904–13. [DOI] [PubMed] [Google Scholar]
- 53.Whyte MP, Wenkert D, Zhang F. Hypophosphatasia: Natural history study of 101 affected children investigated at one research center. Bone 2016;93:125–138. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.