

Abstract
Methotrexate has been used in treatment of rheumatoid arthritis (RA) since the 1980s and to this day is often the first line medication for RA treatment. In this review, we examine multiple hypotheses to explain the mechanism of methotrexate efficacy in RA. These include folate antagonism, adenosine signaling, generation of reactive oxygen species (ROS), decrease in adhesion molecules, alteration of cytokine profiles, and polyamine inhibition amongst some others. Currently, adenosine signaling is probably the most widely accepted explanation for the methotrexate mechanism in RA given that methotrexate increases adenosine levels and on engagement of adenosine with its extracellular receptors an intracellular cascade is activated promoting an overall anti-inflammatory state. In addition to these hypotheses, we examine the mechanism of methotrexate in RA from the perspective of its adverse effects and consider some of the newer genetic markers of methotrexate efficacy and toxicity in RA. Lastly, we briefly discuss the mechanism of additive methotrexate in the setting of TNF-a inhibitor treatment of RA. Ultimately, finding a clear explanation for the pathway and mechanism leading to methotrexate efficacy in RA, there may be a way to formulate more potent therapies with fewer side effects.
Keywords: Methotrexate, rheumatoid arthritis, adenosine signaling, methotrexate efficacy, methotrexate toxicity, adenosine A2A receptor
Introduction
Although there has been development of numerous new targeted therapies for rheumatoid arthritis (RA) in recent years, methotrexate has remained the “anchor drug” for most patients since the late 1980s (1). However, despite its long term and widespread use for RA, the precise mechanism of this drug remains elusive.
Methotrexate was originally designed as a folate pathway antagonist by inhibiting dihydrofolate reductase (DHFR) when given at very high doses for leukemia (as high as 1 gram in a single dose), but it was found that at much lower doses (15–25mg weekly) the drug was effective in RA patients (2). The oncologic mechanism of action involves inhibition of purine synthesis and thus arrest in the S phase of the cell cycle eventually leading to apoptosis of cells (3). The clinical effects of high dose methotrexate used in cancer including the adverse effects can be reversed with high doses of calcium or folinic acid (3). On the other hand, the efficacy of low doses of methotrexate used in RA patients is unaffected by the administration of folic acid and it is in fact almost invariably part of the RA medication regimen to minimize the unwanted methotrexate side effects (4). This indicates that inhibition of purine metabolism is unlikely to be the major mechanism of methotrexate in RA and that another element must be accounting for efficacy of methotrexate in RA patients.
Pharmacokinetics
Methotrexate in RA is usually effective at doses ranging from 15–25mg and it is often initiated as monotherapy. It can also be used with other disease modifying anti-rheumatic drugs (DMARDs) such as hydroxychloroquine and sulfasalazine. Oftentimes when this is ineffective, a biologic DMARD may be used often in conjunction with methotrexate for improved efficacy. Of note, oral methotrexate has highly variable bioavailability and splitting the weekly dose or switching to a subcutaneous mode of delivery can improve this (5). Absorption of oral methotrexate is generally via the protein-coupled folate transporter in the small intestine (6) and it is mainly renally excreted through glomerular filtration and active tubular secretion (7). Some of the drug is also metabolized in the liver and about 10% of excretion is biliary with some enterohepatic recycling (8). Peak plasma concentrations occur at 1–2 hours after ingestion of low dose methotrexate and most of the drug disappears from circulation at 24 hours (9).
Even though methotrexate disappears from circulation fairly quickly, its cellular uptake in inflamed joints via the folate transporter 1 (FOLT) allows the drug to be polyglutamated intracellularly and leads to a steady state of intracellular methotrexate (10). Export of methotrexate is via ATP-binding cassette proteins (ABCC1-ABCC5 and ABCG1) (6). There is usually a time lag in efficacy of low dose methotrexate in clinical practice as accumulation of intracellular polyglutamated methotrexate is slow process (11). It is thought that the polyglutamated form of methotrexate is responsible for its DMARD activity and the following sections will highlight some of the different hypotheses regarding the methotrexate mechanism with variable amounts of evidence.
Folate antagonism
Methotrexate was originally designed in the 1940s as a folate antagonist for treatment of various cancers, and thus there has long been consideration that this mechanism is also related to treatment of RA. In addition to blocking the enzyme 5-aminoimidazole-4- carboxamide ribonucleotide (AICAR) transformylase (ATIC) which converts AICAR to formyl AICAR (FAICAR), methotrexate also inhibits dihydrofolate reductase (DHFR) which catalyzes reduction of dihydrofolate (DHF) to tetrahyrofolate (THF) and it inhibits thymidylate synthetase (TYMS) which catalyzes the formation of thymidine residues.
It has been demonstrated that methotrexate reduces the level of both purine and pyridine pools in primary human T cells (12). Furthermore, low-dose methotrexate reduced the levels of ATP and GTP while increasing levels of UTP inducing reduction in T cell proliferation and increase in apoptosis (13).
While the antagonism of the folate pathway by methotrexate in RA makes sense theoretically, there is not much proof clinically that it is a central mechanism by which methotrexate. Patients on methotrexate are always given daily folate to reduce adverse effects of methotrexate and this has been shown to reduce adverse events and liver function abnormalities (4). However, one might expect that administration of folate with methotrexate would reduce the clinical benefits of methotrexate if it works through this pathway and it has not been demonstrated that this is the case.
Adenosine signaling
The adenosine signaling pathway is currently the leading hypothesis to explain the efficacy of methotrexate in RA patients. Adenosine can be formed intracellularly from ATP and exported from the cell, but the majority of adenosine is formed extracellularly by sequential dephosphorylation of ATP to adenosine in the extracellular space (14). Intracellularly, adenosine was found to inhibit ATIC, leading to increase in intracellular AICAR and extracellular adenosine levels (15) (Fig. 1). AICAR is known to inhibit both adenosine deaminase and hence prevent breakdown of adenosine to inosine. When AICAR level increase, the adenosine level is also increased in the cell and transported extracellularly by extracellular nucleoside transporter (Ent1) (14). The majority of the extracellular adenosine is in fact formed by extracellular transport of ATP from the cell via the ATP transporter and cleavage by the ectoenzyme nucleoside triphosphate phosphohydrolase (NTPP or CD39), which dephosphorylates ATP and subsequently ADP to produce AMP (14). The ecto-5’nucelotidase (CD73) then cleaves AMP to adenosine, which can then activate intracellular signaling via the adenosine receptors or be converted to inosine by adenosine deaminase.
Figure 1. Methotrexate’s effect on adenosine formation.
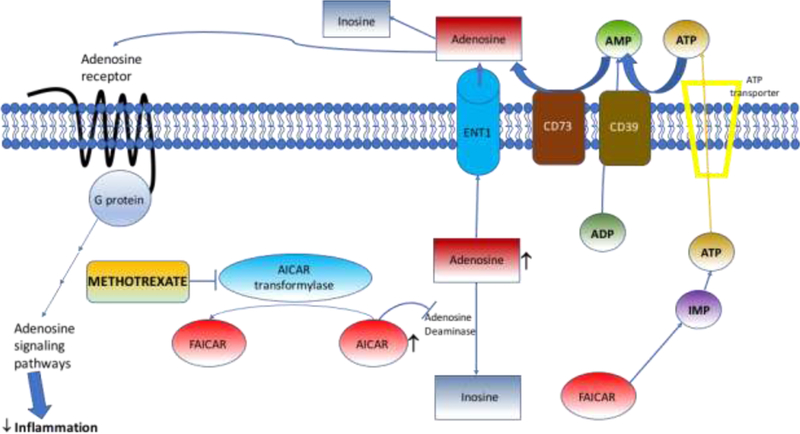
Methotrexate blocks AICAR leading to accumulation of AICAR, which blocks adenosine deaminase. Adenosine formed intracellularly is transported out of the cell by ENT1. ATP and ADP in the extracellular space are dephosphorylated sequentially by transmembrane CD39 to form AMP and AMP is converted to adenosine by transmembrane CD73 and adenosine can be converted to inosine or act via the adenosine receptors to activate various downstream pathways. Adapted from Cronstein and Sitkovsky (2017) (14). Abbreviations: AICAR, amidoimidazolecarboxamidoribonucleotide; ENT1, extracellular nucleoside transporter; ATP, adenosine triphosphate; ADP, adenosine diphosphate; CD39, nucleoside triphosphate phophohydrolase; AMP, adenosine monophosphate; CD73, ecto-5’nucleotidase (CD73).
Adenosine is a paracrine signaling molecule and can bind to 4 different G-protein coupled receptors known as adenosine receptor A1 (A2AR1 or ADORA1), adenosine receptor A2a (A2AR2a or ADORA2a), adenosine receptor A2b (A2AR2b or ADORA2b), and adenosine receptor A3 (A2AR3 or ADORA3) (16). The A1 and A2A receptors have high affinity for adenosine while the A2B and A3 receptors have low affinity. The A2A and A2B receptors both signal via Gs protein (and A2B also signals through a Gq protein), the A1 receptor is associated with a Gi and Gα proteins and the A3 receptor is linked to Gi and Gq proteins (14) (Fig. 2).
Figure 2. Adenosine receptors and signaling pathways.
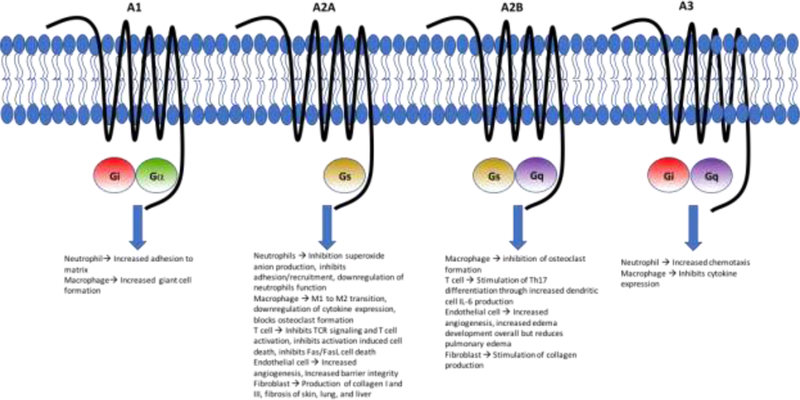
The A1 and A2A receptors are the high affinity adenosine receptors while the A2AB and A3 receptors are lower affinity receptors. Demonstrated in the figure are the different coupled G-proteins with each adenosine receptor and some of the effects this signaling has on certain immune cells. Adapted from Cronstein andSitkovsky (2017) (14).
It has been noted that in patients with RA there is an overexpression of adenosine receptors on immune cells (17–19). Therefore, it is possible that RA patients with overexpression of adenosine receptors will be more responsive to a drug like methotrexate. An in vitro study with methotrexate showed that the treated cells had reduced adherence of neutrophils to fibroblasts and increased release of adenosine from fibroblasts and this effect was blocked with the addition of adenosine deaminase showing adenosine specificity (20). Additionally, using mouse air-pouch models, methotrexate was shown to reduce accrual of leukocytes in the pouch and this effect was reversed by an A2A receptor antagonist but not by an A1 receptor antagonist suggesting importance of the A2A receptor in adenosine signaling (21). A2A knockout mice were shown to have increased adenosine levels in inflammatory exudates after methotrexate treatment in a mouse peritonitis model, however these effects were not observed in the A3 receptor knockouts or wildtype (22). Additionally, CD73 knockout mice, which have impaired ability to convert AMP to adenosine, showed less of a response to methotrexate compared to wildtype indicating the relative importance of extracellular generation of adenosine compared to intracellular release of adenosine (23). In humans, a case series of 17 patients with RA showed that the level of synovial cyclic AMP (cAMP) was inversely correlated with disease activity as well as peripheral markers of inflammation including erythrocyte sedimentation rate (ESR) and IL-18 levels (24). It is known that signaling through the A2A receptor will increase cAMP levels possibly indicating an activation of this receptor by methotrexate within the joint.
As previously mentioned there are four known adenosine G-protein coupled receptors that result in different responses the adenosine (Fig. 2). Based on aforementioned studies, it is currently thought that the majority of the clinical effect observed by adenosine signaling is through the A2A receptor (although the other receptors do play a role). Adenosine signaling through the A2A receptor has numerous effects on different types of inflammatory cells. In neutrophils, it inhibits superoxide anion generation, inhibits adhesion and recruitment, increases engulfment-mediated neutrophil function (14). In macrophages, it promotes transition from M1 to M2 macrophages (M1 macrophages are pro-inflammatory and M2 macrophages have an anti-inflammatory effect), inhibits cytokine expression, and inhibits osteoclast formation and thus bone degradation (14). Adenosine is thought to downregulate T cell activation and proliferation by inhibiting T cell receptor (TCR) triggered activation, inhibiting T cell activation induced cell death and inhibiting Fas and Fas ligand mediated cell death in CD4 cells (25, 26). Additionally, A2A signaling may lead to development of T regulatory cells (Tregs) known to express both CD39 and CD73 that may decrease T cell activation (11). Notably patients that were unresponsive to methotrexate were found to have a lower pre-treatment level of CD39 on Treg cells (27). A2A receptor signaling in endothelial cells increases angiogenesis but also increases barrier integrity to prevent edema formation (28). A2AB receptors similarly increase angiogenesis but they lead to increased edema in arthritis (29). Thus, adenosine signaling effect on angiogenesis and development of joint effusions is complex. Methotrexate may have variable effects on this process in RA patients given it may lead to activation of multiple adenosine receptors that lead to variable effects on prevention or increase of edema in arthritis. Fibroblasts increase production of type I and III collagen through A2A receptor signaling and can also promotes skin, lung, and liver fibrosis (14). Given these effects on inflammatory cells, it is evident that A2A receptor signaling is generally beneficial in reducing inflammation and this may be the major signaling mechanism by which methotrexate works. Briefly, the A1 receptor has been shown to increase neutrophil adhesion to the matrix and macrophage giant cell formation. The A2B receptor leads to inhibition of osteoclast formation, stimulates differentiation of T cells into pro-inflammatory Th17 cells, causes increased angiogenesis and edema formation (except decrease in pulmonary edema) in endothelial cells, and stimulates collagen production in fibroblasts. Lastly in the A3 receptor, neutrophil chemotaxis is increased to the sites of inflammation and macrophages can inhibit certain cytokine expression via A3 signaling (14).
Generation of reactive oxygen species
Increase in reactive oxygen species (ROS) after methotrexate treatment may be a result of the increase in apoptosis of transformed T cells (30). Tetrahydrobiopterin is a cofactor and ligand of endothelial nitric oxide synthease (eNOS) that can be recycled by conversion of oxidized dihydrobiopterin back to tetrahydrobiopterin. Tetrahydrobiopterin leads to nitric oxide production while dihydrobiopterin uncouples eNOS and induces superoxide generation (11). Treatment with methotrexate in mice with constitutively low tetrahydrobiopterin reduces tetrahydrobiopterin levels and thus increase in superoxide generation compared to placebo and this difference is not seen in wildtype mice (11). Furthermore, the increase in ROS by methotrexate leads to apoptosis in T cells is associated with increase in c-Jun N-terminal kinase signaling and this was reversed by addition of tetrahydrobiopterin (31).
Adhesion molecules
Methotrexate may also function by decreasing chemotaxis and adhesion of Inflammatory cells. Synovial biopsies in patients treated with methotrexate for 16 weeks demonstrated reduction compared to pre-treatment levels in both E-selectin and vascular cell adhesion protein 1 (VCAM1) as were levels of IL-1, TNF, and total inflammatory cells in the joint (32). Moreover, methotrexate treatment of peripheral blood mononuclear cells (PBMCs) led to an adenosine and folate dependent decrease in intercellular adhesion molecule 1 (ICAM1) which is involved in the migration of leukocytes (33). Levels of serum soluble adhesion molecules including sICAM1, sVCAM1, and sE-selectin were found to be elevated in RA patients and reduced upon treatment with methotrexate (34). Activation of ADORA2A expression or adenosine administration in vitro has been shown to reduce levels of E-selectin, VCAM1, and ICAM1 (35).
Alteration of the cytokine profile
It has been shown that methotrexate can inhibit the production of proinflammatory cytokines. For example, T cells isolated and activated ex vivo from RA patients treated with methotrexate have diminished capability to produce IFNγ, IL-4, IL-3, TNF, and granulocyte macrophage colony-stimulating factor (36). Given decrease in some of these cytokines it is thought that T cell differentiation into Th1 or Th2 helper cells is decreased compared to RA patients not on methotrexate. Methotrexate treatment has also been noted to reduce the number of TNF positive CD4 T cells and increase the number of IL-10 CD4 T cells (37). Culture of fibroblasts and T cells from RA patients shows less of an increase in IFNγ, IL-6, and IL-17 likely by disrupting cell adhesion (38).
Polyamine inhibition
RA patients accumulate the polyamines spermine and spermidine in synovial tissue, synovial fluid, PBMCs, and urine. The polyamines can be converted by monocytes into lymphotoxins including hydrogen peroxidase and ammonia. By inhibiting DHFR, methotrexate depletes levels of tetrahydrofolate and methyltetrahydrofolate (5-CH3-THF) (Fig. 3). These two compounds normally act as methyl donors to form methionine and S-adenosylmethionine (SAM). These two molecules are also methyl group donors and they lead to the formation of polyamines (39). This particular mechanism suggests that methotrexate, by inhibiting DHFR, decreases downstream mediators including methionine and SAM, thus reducing methylation and subsequent formation of polyamines and therefore lymphotoxins. However, there are some studies that show polyamine inhibition is less likely to explain the efficacy of methotrexate in RA. 3-deazaadenosine, a transmethylation inhibitor, demonstrated no clinical efficacy in RA. Moreover, it is known that methotrexate inhibits chemotaxis and superoxide production in monocytes and this is actually enhanced by methionine but reversed by folinic acid plus SAM (40). Hence, polyamine inhibition could possibly contribute to methotrexate efficacy in some way, but does not appear to be the major mechanism.
Figure 3. Methotrexate inhibition of polyamine and lymphotoxin formation.
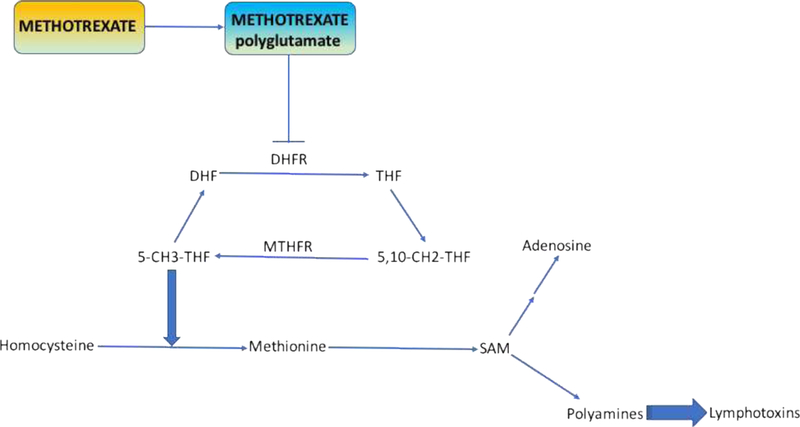
As demonstrated in the figure methotrexate is polyglutamated and then works upstream in the folate reduction pathway to block dihydrofolate reductase. This in effect decreases the levels of 5-CH3-THF and therefore there is less conversion of homocysteine to methionine. Lower levels of methionine decreases formation of SAM and hence methotrexate may be able to decrease polyaminelevels. Adapted from Chan and Cronstein (2010) (39).
Mechanism of methotrexate toxicity
While methotrexate remains first line treatment and is effective in mainly RA patient it has certain side effects that can actually be helpful to further explain its mechanistic effects. Methotrexate can lead to hepatic fibrosis in some cases and this may be secondary to reduction in hepatic folate stores and accumulation of polyglutamated methotrexate (41). Additionally ethanol ingestion (known as a fairly common cause of cirrhosis) has been shown to induce release of plasma levels of adenosine (42). In ex vivo hepatic slices, methotrexate lead to increased adenosine levels; adenosine can stimulate matrix protein production by stellate cells and notably mice deficient for the A2A receptor but not wildtype or A3 knockouts are more resistant to developing fibrosis in a carbon tetrachloride challenge (43). One positive side effect of increased matrix protein formation is that adenosine can promote excisional wound healing by increasing interstitial matrix (44). Methotrexate induced nodules in RA patients are formed from monocyte differentiation into multinucleated giant cells. In vitro monocytes can be induced to form giant cells by phorbol myristate acetate stimulating both differentiation and fusion of the giant cells. It was shown that adenosine acting via the A1 receptor does lead to formation of multinucleated giant cells (45).
There have been numerous studies over the years regarding the use of folic acid or folinic acid supplementation to reduce methotrexate toxicity and its effect on methotrexate efficacy in RA. A meta-analysis in 2013 demonstrated that supplementation with either daily folic acid or weekly folinic acid significantly reduced the incidence of gastrointestinal side effects, elevation in serum transaminase levels, and reduction in discontinuation of methotrexate for any reason (46). Many physicians who use methotrexate as a treatment will begin with folic acid and potentially attempt to switch to folinic acid to reduce adverse effects, however there is no clear evidence indicating that folinic acid is superior to folic acid in this regard (46). Folic acid, unlike weekly folinic acid, is given daily and does not compete with methotrexate cellular uptake via the reduced folate carrier. Folinic acid is generally dosed weekly on the same day as weekly methotrexate and is given approximately 10–12 hours after methotrexate to allow for unopposed uptake of methotrexate. A number of studies have shown that supplementation with folic acid decreases toxicity yet does not alter the efficacy of methotrexate based on RA disease activity parameters including tender or swollen joint count or physician’s global assessment score (47). This continued efficacy of methotrexate for RA in the presence of folic or folinic acid is one reason that alternate mechanisms for RA efficacy such as increased adenosine signaling are favored by many over the inhibition of folate metabolism.
Genetic markers of methotrexate efficacy and toxicity
There have recently been a number of studies evaluating efficacy and also toxicity in RA patients on methotrexate using genetics SNPs and peripheral or synovial biomarkers. One study found that a particular missense SNP in ATIC was a significant predictor of methotrexate response whereas there were no significant differences in SNPs found in DHFR and folylpolyglutamate synthase (FPGS) genes (48). A meta-analysis examining polymorphisms in the ATIC gene also found that certain SNPs in this enzyme were associated with both response to methotrexate as well as toxicity of methotrexate particularly in Caucasian RA patients (49). There is also a SNP found in the reduced folate carrier 1 (RFC1), which results in variable intracellular levels of methotrexate-polyglutamate, was found to be associated with methotrexate efficacy in Asian RA patients but not Caucasians and no association with methotrexate toxicity (50). In a cohort of 223 RA patients, methotrexate nonresponse was associated with eight genotypes of 3 haplotypes in gene encoding MS (methionine synthase), MTRR (methionine synthase reductase), and ATIC. In the same cohort, methotrexate non-response was associated with five genotypes and two haplotypes in the ATIC and adenosine 2A receptor genes (51). In the current literature, there are no clearly apparent human SNPs identified in the A2A receptor gene itself that are associated with methotrexate efficacy or inefficacy, although one study did demonstrate five SNPs in the gene that may be predictive of predominantly gastrointestinal adverse events in RA patients treated with methotrexate leading to treatment discontinuation (52). Potentially with more research, genetic markers may become helpful in stratification of patients who will respond and/or have toxic effects from methotrexate. Furthermore, the understanding of the functions of these markers may help to further elucidate the methotrexate mechanism in RA.
Methotrexate mechanism of efficacy in conjunction with TNF-α inhibitors
While methotrexate is frequently the first line treatment of RA, many patients with erosive disease on presentation or with inadequate response to methotrexate or other DMARDs require treatment with biologic therapies - most notably TNF-α inhibitors (anti-TNFs). However, an issue that may occur with anti-TNF therapy in treatment of RA and other types of inflammatory conditions is the development of anti-drug antibodies that has been observed especially with infliximab and adalimumab among others potentially leading to reduced efficacy of the medication and discontinuation of treatment secondary to treatment failure (53, 54). In studies of adalimumab plus methotrexate treatment (with methotrexate often given as a lower dose than when used as monotherapy), authors have found that development of anti-drug antibodies against the anti-TNFs were significantly reduced with addition of methotrexate along with improved efficacy of therapy (53, 55, 56). Similar results were obtained in studies of methotrexate plus infliximab, whereas this effect was not observed with other DMARDs such as sulfasalazine, azathioprine, cyclosporine, hydroxychloroquine as well as with corticosteroids (57, 58). While the mechanism by which methotrexate decreases these antibodies is not fully apparent, the ability of methotrexate to suppress early T and B cell expansion may explain its role as an effective immunomodulator when given concomitantly with anti-TNF or biologic therapy (59). It is also possible that methotrexate may work synergistically with anti-TNFs independent of the reduction of anti-drug antibodies by preventing synovial inflammation by affecting an alternate pathway than the anti-TNF (60). The ability for methotrexate to prevent anti-drug antibodies and/or synergy of methotrexate and biologics reducing inflammation via two pathways are both viable hypotheses for efficacy in RA and it is very possible that both work in tandem to produce better patient responses.
Conclusion
As one can see from this review there are a number of theories behind the mechanism of methotrexate in RA, each potentially playing some sort of role in its efficacy. Further elucidation of the mechanism of methotrexate to treat RA in these patients may lead to more targeted therapies down the road. An example of this in the adenosine signaling pathway would be stimulation of individual receptors such as the A2A receptor as opposed to the general increase in adenosine that methotrexate induces. Methotrexate remains the mainstay of RA treatment and it will be important to identify the patients in whom the drug is effective and those in which toxicity may be an issue. Genetic and biomarkers may help to stratify patients and possibly shed light on the mechanism for efficacy and for toxicity of methotrexate for RA.
Footnotes
Disclosure of interest: None of the authors has any conflicts of interest to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Weinblatt ME, Coblyn JS, Fox DA, Fraser PA, Holdsworth DE, Glass DN, et al. Efficacy of low-dose methotrexate in rheumatoid arthritis. N Engl J Med. 1985;312(13):818–22. [DOI] [PubMed] [Google Scholar]
- 2.Visser K, van der Heijde D. Optimal dosage and route of administration of methotrexate in rheumatoid arthritis: a systematic review of the literature. Ann Rheum Dis. 2009;68(7):1094–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Visentin M, Zhao R, Goldman ID. The antifolates. Hematol Oncol Clin North Am. 2012;26(3):629–48, ix. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Whittle SL, Hughes RA. Folate supplementation and methotrexate treatment in rheumatoid arthritis: a review. Rheumatology (Oxford). 2004;43(3):267–71. [DOI] [PubMed] [Google Scholar]
- 5.Pichlmeier U, Heuer KU. Subcutaneous administration of methotrexate with a prefilled autoinjector pen results in a higher relative bioavailability compared with oral administration of methotrexate. Clin Exp Rheumatol. 2014;32(4):563–71. [PubMed] [Google Scholar]
- 6.Desmoulin SK, Hou Z, Gangjee A, Matherly LH. The human proton-coupled folate transporter: Biology and therapeutic applications to cancer. Cancer Biol Ther. 2012;13(14):1355–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seideman P, Beck O, Eksborg S, Wennberg M. The pharmacokinetics of methotrexate and its 7-hydroxy metabolite in patients with rheumatoid arthritis. Br J Clin Pharmacol. 1993;35(4):409–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nuernberg B, Koehnke R, Solsky M, Hoffman J, Furst DE. Biliary elimination of low-dose methotrexate in humans. Arthritis Rheum. 1990;33(6):898–902. [DOI] [PubMed] [Google Scholar]
- 9.Godfrey C, Sweeney K, Miller K, Hamilton R, Kremer J. The population pharmacokinetics of long-term methotrexate in rheumatoid arthritis. Br J Clin Pharmacol. 1998;46(4):369–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Angelis-Stoforidis P, Vajda FJ, Christophidis N. Methotrexate polyglutamate levels in circulating erythrocytes and polymorphs correlate with clinical efficacy in rheumatoid arthritis. Clin Exp Rheumatol. 1999;17(3):313–20. [PubMed] [Google Scholar]
- 11.Brown PM, Pratt AG, Isaacs JD. Mechanism of action of methotrexate in rheumatoid arthritis, and the search for biomarkers. Nat Rev Rheumatol. 2016;12(12):731–42. [DOI] [PubMed] [Google Scholar]
- 12.Budzik GP, Colletti LM, Faltynek CR. Effects of methotrexate on nucleotide pools in normal human T cells and the CEM T cell line. Life Sci. 2000;66(23):2297–307. [DOI] [PubMed] [Google Scholar]
- 13.Fairbanks LD, Ruckemann K, Qiu Y, Hawrylowicz CM, Richards DF, Swaminathan R, et al. Methotrexate inhibits the first committed step of purine biosynthesis in mitogen-stimulated human T-lymphocytes: a metabolic basis for efficacy in rheumatoid arthritis? Biochem J. 1999;342 ( Pt 1):143–52. [PMC free article] [PubMed] [Google Scholar]
- 14.Cronstein BN, Sitkovsky M. Adenosine and adenosine receptors in the pathogenesis and treatment of rheumatic diseases. Nat Rev Rheumatol. 2017;13(1):41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baggott JE, Vaughn WH, Hudson BB. Inhibition of 5-aminoimidazole-4-carboxamide ribotide transformylase, adenosine deaminase and 5’-adenylate deaminase by polyglutamates of methotrexate and oxidized folates and by 5-aminoimidazole-4-carboxamide riboside and ribotide. Biochem J. 1986;236(1):193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hasko G, Linden J, Cronstein B, Pacher P. Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat Rev Drug Discov. 2008;7(9):759–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Varani K, Padovan M, Vincenzi F, Targa M, Trotta F, Govoni M, et al. A2A and A3 adenosine receptor expression in rheumatoid arthritis: upregulation, inverse correlation with disease activity score and suppression of inflammatory cytokine and metalloproteinase release. Arthritis Res Ther. 2011;13(6):R197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Varani K, Massara A, Vincenzi F, Tosi A, Padovan M, Trotta F, et al. Normalization of A2A and A3 adenosine receptor up-regulation in rheumatoid arthritis patients by treatment with anti-tumor necrosis factor alpha but not methotrexate. Arthritis Rheum. 2009;60(10):2880–91. [DOI] [PubMed] [Google Scholar]
- 19.Nguyen MT, Lue H, Kleemann R, Thiele M, Tolle G, Finkelmeier D, et al. The cytokine macrophage migration inhibitory factor reduces pro-oxidative stress-induced apoptosis. J Immunol. 2003;170(6):3337–47. [DOI] [PubMed] [Google Scholar]
- 20.Cronstein BN, Eberle MA, Gruber HE, Levin RI. Methotrexate inhibits neutrophil function by stimulating adenosine release from connective tissue cells. Proc Natl Acad Sci U S A. 1991;88(6):2441–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cronstein BN, Naime D, Ostad E. The antiinflammatory mechanism of methotrexate. Increased adenosine release at inflamed sites diminishes leukocyte accumulation in an in vivo model of inflammation. J Clin Invest. 1993;92(6):2675–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Montesinos MC, Desai A, Cronstein BN. Suppression of inflammation by low-dose methotrexate is mediated by adenosine A2A receptor but not A3 receptor activation in thioglycollate-induced peritonitis. Arthritis Res Ther. 2006;8(2):R53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Montesinos MC, Takedachi M, Thompson LF, Wilder TF, Fernandez P, Cronstein BN. The antiinflammatory mechanism of methotrexate depends on extracellular conversion of adenine nucleotides to adenosine by ecto-5’-nucleotidase: findings in a study of ecto-5’-nucleotidase gene-deficient mice. Arthritis Rheum. 2007;56(5):1440–5. [DOI] [PubMed] [Google Scholar]
- 24.Morabito L, Montesinos MC, Schreibman DM, Balter L, Thompson LF, Resta R, et al. Methotrexate and sulfasalazine promote adenosine release by a mechanism that requires ecto- 5’-nucleotidase-mediated conversion of adenine nucleotides. J Clin Invest. 1998;101(2):295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang S, Apasov S, Koshiba M, Sitkovsky M. Role of A2a extracellular adenosine receptor-mediated signaling in adenosine-mediated inhibition of T-cell activation and expansion. Blood. 1997;90(4):1600–10. [PubMed] [Google Scholar]
- 26.Koshiba M, Kojima H, Huang S, Apasov S, Sitkovsky MV. Memory of extracellular adenosine A2A purinergic receptor-mediated signaling in murine T cells. J Biol Chem. 1997;272(41):25881–9. [DOI] [PubMed] [Google Scholar]
- 27.Peres CA, Emilio T, Schietti J, Desmouliere SJ, Levi T. Dispersal limitation induces long-term biomass collapse in overhunted Amazonian forests. Proc Natl Acad Sci U S A. 2016;113(4):892–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feoktistov I, Biaggioni I, Cronstein BN. Adenosine receptors in wound healing, fibrosis and angiogenesis. Handb Exp Pharmacol. 2009(193):383–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Teramachi J, Kukita A, Li YJ, Ushijima Y, Ohkuma H, Wada N, et al. Adenosine abolishes MTX-induced suppression of osteoclastogenesis and inflammatory bone destruction in adjuvant-induced arthritis. Lab Invest. 2011;91(5):719–31. [DOI] [PubMed] [Google Scholar]
- 30.Phillips DC, Woollard KJ, Griffiths HR. The anti-inflammatory actions of methotrexate are critically dependent upon the production of reactive oxygen species. Br J Pharmacol. 2003;138(3):501–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spurlock CF 3rd, Aune ZT, Tossberg JT, Collins PL, Aune JP, Huston JW 3rd, et al. Increased sensitivity to apoptosis induced by methotrexate is mediated by JNK. Arthritis Rheum. 2011;63(9):2606–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dolhain RJ, Tak PP, Dijkmans BA, De Kuiper P, Breedveld FC, Miltenburg AM. Methotrexate reduces inflammatory cell numbers, expression of monokines and of adhesion molecules in synovial tissue of patients with rheumatoid arthritis. Br J Rheumatol. 1998;37(5):502–8. [DOI] [PubMed] [Google Scholar]
- 33.Johnston A, Gudjonsson JE, Sigmundsdottir H, Ludviksson BR, Valdimarsson H. The antiinflammatory action of methotrexate is not mediated by lymphocyte apoptosis, but by the suppression of activation and adhesion molecules. Clin Immunol. 2005;114(2):154–63. [DOI] [PubMed] [Google Scholar]
- 34.Klimiuk PA, Fiedorczyk M, Sierakowski S, Chwiecko J. Soluble cell adhesion molecules (sICAM-1, sVCAM-1, and sE-selectin) in patients with early rheumatoid arthritis. Scand J Rheumatol. 2007;36(5):345–50. [DOI] [PubMed] [Google Scholar]
- 35.Sands WA, Martin AF, Strong EW, Palmer TM. Specific inhibition of nuclear factor- kappaB-dependent inflammatory responses by cell type-specific mechanisms upon A2A adenosine receptor gene transfer. Mol Pharmacol. 2004;66(5):1147–59. [DOI] [PubMed] [Google Scholar]
- 36.Gerards AH, de Lathouder S, de Groot ER, Dijkmans BA, Aarden LA. Inhibition of cytokine production by methotrexate. Studies in healthy volunteers and patients with rheumatoid arthritis. Rheumatology (Oxford). 2003;42(10):1189–96. [DOI] [PubMed] [Google Scholar]
- 37.Rudwaleit M, Yin Z, Siegert S, Grolms M, Radbruch A, Braun J, et al. Response to methotrexate in early rheumatoid arthritis is associated with a decrease of T cell derived tumour necrosis factor alpha, increase of interleukin 10, and predicted by the initial concentration of interleukin 4. Ann Rheum Dis. 2000;59(4):311–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miranda-Carus ME, Balsa A, Benito-Miguel M, Perez de Ayala C, Martin-Mola E. IL-15 and the initiation of cell contact-dependent synovial fibroblast-T lymphocyte cross-talk in rheumatoid arthritis: effect of methotrexate. J Immunol. 2004;173(2):1463–76. [DOI] [PubMed] [Google Scholar]
- 39.Chan ES, Cronstein BN. Methotrexate--how does it really work? Nat Rev Rheumatol. 2010;6(3):175–8. [DOI] [PubMed] [Google Scholar]
- 40.Nesher G, Moore TL, Dorner RW. In vitro effects of methotrexate on peripheral blood monocytes: modulation by folinic acid and S-adenosylmethionine. Ann Rheum Dis. 1991;50(9):637–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kremer JM, Galivan J, Streckfuss A, Kamen B. Methotrexate metabolism analysis in blood and liver of rheumatoid arthritis patients. Association with hepatic folate deficiency and formation of polyglutamates. Arthritis Rheum. 1986;29(7):832–5. [DOI] [PubMed] [Google Scholar]
- 42.Puig JG, Fox IH. Ethanol-induced activation of adenine nucleotide turnover. Evidence for a role of acetate. J Clin Invest. 1984;74(3):936–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chan ES, Montesinos MC, Fernandez P, Desai A, Delano DL, Yee H, et al. Adenosine A(2A) receptors play a role in the pathogenesis of hepatic cirrhosis. Br J Pharmacol. 2006;148(8):1144–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Montesinos MC, Desai A, Chen JF, Yee H, Schwarzschild MA, Fink JS, et al. Adenosine promotes wound healing and mediates angiogenesis in response to tissue injury via occupancy of A(2A) receptors. Am J Pathol. 2002;160(6):2009–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Merrill JT, Shen C, Schreibman D, Coffey D, Zakharenko O, Fisher R, et al. Adenosine A1 receptor promotion of multinucleated giant cell formation by human monocytes: a mechanism for methotrexate-induced nodulosis in rheumatoid arthritis. Arthritis Rheum. 1997;40(7):1308–15. [DOI] [PubMed] [Google Scholar]
- 46.Shea B, Swinden MV, Tanjong Ghogomu E, Ortiz Z, Katchamart W, Rader T, et al. Folic acid and folinic acid for reducing side effects in patients receiving methotrexate for rheumatoid arthritis. Cochrane Database Syst Rev. 2013(5):CD000951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morgan SL, Baggott JE, Vaughn WH, Austin JS, Veitch TA, Lee JY, et al. Supplementation with folic acid during methotrexate therapy for rheumatoid arthritis. A double-blind, placebocontrolled trial. Ann Intern Med. 1994;121(11):833–41. [DOI] [PubMed] [Google Scholar]
- 48.Kurzawski M, Malinowski D, Szarmach N, Nowak A, Goryniak A, Pawlik A, et al. ATIC missense variant affects response to methotrexate treatment in rheumatoid arthritis patients. Pharmacogenomics. 2016;17(18):1971–8. [DOI] [PubMed] [Google Scholar]
- 49.Lee YH, Bae SC. Association of the ATIC 347 C/G polymorphism with responsiveness to and toxicity of methotrexate in rheumatoid arthritis: a meta-analysis. Rheumatol Int. 2016;36(11):1591–9. [DOI] [PubMed] [Google Scholar]
- 50.Li X, Hu M, Li W, Gu L, Chen M, Ding H, et al. The association between reduced folate carrier-1 gene 80G/A polymorphism and methotrexate efficacy or methotrexate related-toxicity in rheumatoid arthritis: A meta-analysis. Int Immunopharmacol. 2016;38:8–15. [DOI] [PubMed] [Google Scholar]
- 51.Lima A, Bernardes M, Azevedo R, Seabra V, Medeiros R. Moving toward personalized medicine in rheumatoid arthritis: SNPs in methotrexate intracellular pathways are associated with methotrexate therapeutic outcome. Pharmacogenomics. 2016;17(15):1649–74. [DOI] [PubMed] [Google Scholar]
- 52.Hider SL, Thomson W, Mack LF, Armstrong DJ, Shadforth M, Bruce IN. Polymorphisms within the adenosine receptor 2a gene are associated with adverse events in RA patients treated with MTX. Rheumatology (Oxford). 2008;47(8):1156–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bartelds GM, Krieckaert CL, Nurmohamed MT, van Schouwenburg PA, Lems WF, Twisk JW, et al. Development of antidrug antibodies against adalimumab and association with disease activity and treatment failure during long-term follow-up. JAMA. 2011;305(14):1460–8. [DOI] [PubMed] [Google Scholar]
- 54.Pascual-Salcedo D, Plasencia C, Ramiro S, Nuno L, Bonilla G, Nagore D, et al. Influence of immunogenicity on the efficacy of long-term treatment with infliximab in rheumatoid arthritis Rheumatology (Oxford). 2011;50(8):1445–52. [DOI] [PubMed] [Google Scholar]
- 55.Bartelds GM, Wijbrandts CA, Nurmohamed MT, Stapel S, Lems WF, Aarden L, et al. Clinical response to adalimumab: relationship to anti-adalimumab antibodies and serum adalimumab concentrations in rheumatoid arthritis. Ann Rheum Dis. 2007;66(7):921–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bartelds GM, Wijbrandts CA, Nurmohamed MT, Stapel S, Lems WF, Aarden L, et al. Antiinfliximab and anti-adalimumab antibodies in relation to response to adalimumab in infliximab switchers and anti-tumour necrosis factor naive patients: a cohort study. Ann Rheum Dis. 2010;69(5):817–21. [DOI] [PubMed] [Google Scholar]
- 57.Maini RN, Breedveld FC, Kalden JR, Smolen JS, Davis D, Macfarlane JD, et al. Therapeutic efficacy of multiple intravenous infusions of anti-tumor necrosis factor alpha monoclonal antibody combined with low-dose weekly methotrexate in rheumatoid arthritis. Arthritis Rheum. 1998;41(9):1552–63. [DOI] [PubMed] [Google Scholar]
- 58.Bendtzen K, Geborek P, Svenson M, Larsson L, Kapetanovic MC, Saxne T. Individualized monitoring of drug bioavailability and immunogenicity in rheumatoid arthritis patients treated with the tumor necrosis factor alpha inhibitor infliximab. Arthritis Rheum. 2006;54(12):3782–9. [DOI] [PubMed] [Google Scholar]
- 59.Krieckaert CL, Bartelds GM, Lems WF, Wolbink GJ. The effect of immunomodulators on the immunogenicity of TNF-blocking therapeutic monoclonal antibodies: a review. Arthritis Res Ther. 2010;12(5):217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jani M, Barton A, Warren RB, Griffiths CE, Chinoy H. The role of DMARDs in reducing the immunogenicity of TNF inhibitors in chronic inflammatory diseases. Rheumatology (Oxford). 2014;53(2):213–22. [DOI] [PMC free article] [PubMed] [Google Scholar]