

Abstract
SHP2 is a ubiquitously expressed protein tyrosine phosphatase, which is involved in many signaling pathways to regulate the skeletal development. In endochondral ossification, SHP2 is known to modify the osteogenic fate of osteochondroprogenitors and to impair the osteoblastic transdifferentiation of hypertrophic chondrocytes. However, how SHP2 regulates osteoblast differentiation in intramembranous ossification remains incompletely understood. To address this question, we generated a mouse model to ablate SHP2 in the Prrx1-expressing mesenchymal progenitors by using “Cre-loxP”-mediated gene excision and examined the development of calvarial bone, in which the main process of bone formation is intramembranous ossification. Phenotypic characterization showed that SHP2 mutants have severe defects in calvarial bone formation. Cell lineage tracing and in situ hybridization data showed less osteoblast differentiation of mesenchymal cells and reduced osteogenic genes expression, respectively. Further mechanistic studies revealed enhanced TGFβ and suppressed BMP2 signaling in SHP2 ablated mesenchymal progenitors and their derivatives. Our study uncovered the critical role of SHP2 in osteoblast differentiation through intramembranous ossification and might provide a potential target to treat craniofacial skeleton disorders.
Keywords: SHP2, intramembranous ossification, mesenchymal progenitors, osteoblast differentiation, TGFβ and BMP2 signaling
1. Introduction
The skull vault of mammals, providing essential protection of the brain and important sensory organs, consists of the frontal, parietal, inter-parietal and occipital bones, which all undergo intramembranous ossification[1, 2]. During development, the cell lineage analysis showed that the frontal bones are mainly contributed from the neural crest cells, while parietal and inter-parietal bones originate from the paraxial mesoderm[3–5]. These cells migrate into defined locations overlying the cerebral hemispheres, and subsequently differentiate into osteogenic mesenchyme, which is characterized by the expression of Runt-related transcription factor 2(RUNX2) and OSTERIX, the earliest molecular determinants of bone formation[6–9]. In contrast to the endochondral ossification that ensues from a cartilaginous anlagen, intramembranous ossification features the direct differentiation of mesenchymal progenitors into osteoblasts, responsible for the synthesis of collagen type 1(COL1α1)-rich bone extracellular matrix and its mineralization.
Protein tyrosine phosphatase non-receptor type 11(PTPN11), also known as SHP2, is a widely expressed SH2 domain-containing non-receptor protein tyrosine phosphatase. Accumulating evidences suggest a crucial role for SHP2 in regulation of skeletal development and homeostasis. SHP2 gain-of-function (GOF) mutations cause Noonan Syndrome (NS), which has calvarial abnormality including macrocephaly, oral malformation and hypertelorism[10–12]. Conversely, SHP2 loss-of-function(LOF) mutation cause benign cartilage tumor metachondromatosis both in human and in mice[13, 14]. Recently, we reported that SHP2 determines the cell fate of osteochondroprogenitors[15] and its specific deletion in collagen type 2(COL2a1) or collagen type 10(COL10a1) expressing chondrocytes impairs the osteoblast transdifferentiation of hypertrophic chondrocyte in endochondral ossification, which indicates SHP2 plays a crucial role in osteoblast differentiation[16]. SHP2 ablation in mesenchymal progenitors cause defects of calvarial bone formation[17], but the cellular and molecular mechanism how SHP2 regulates osteoblast differentiation and bone formation in calvarial development is not fully understood.
Calvarial development is a complex process consisting both endochondral ossification and intramembranous ossification. Here, we specifically delete SHP2 in mesenchymal progenitors through Prrx1-Cre to investigate the role of SHP2 in the development of parietal and inter-parietal bone, which undergo intramembranous ossification. During the early phases of the parietal and inter-parietal bone formation, mesenchymal progenitors first undergo proliferation and differentiation, leading to the formation of several bony anlagen margined by the osteogenic fronts and connected by the fibrous connective tissue sutures, which are rich in mesenchymal progenitors and fibroblasts. After acquiring the basic shape, each of the skull bony elements remains separated by sutures to ensures that calvarial expansion is coordinated with growth of the underlying brain. Synchronized with the osteoblast-mediated bone formation, calvarial bones also undergo dynamic modeling and remodeling through the coordinated resorptive activity of osteoclasts. These developmental processes at last establish the three-dimensional architecture of the skull.
Lines of genetic and molecular evidence suggest multiple signaling pathways and transcription factors as being important regulators of skull development, such as the signaling pathways evoked by BMP/TGFβ[18], FGFs[19], WNT[20] and hedgehog [21] et al. Among these signaling pathways, transforming growth factor beta (TGFβ) and bone morphogenic protein2 (BMP2) are considered as one of the most important signaling pathways to regulate osteogenesis[18, 22], which are both the TGFβ superfamily members. TGFβ ligand binds to TGFβ receptor complex (TGFβR I &TGFβR II) and then phosphorylate SMAD2/3. Similarly, BMP2 ligand binds to BMP receptor complex (BMPR I & BMPR II) and then phosphorylate SMAD1/5/8 [23, 24], respectively. TGFβ signaling was demonstrated to promote proliferation and early differentiation of osteoprogenitor, however, it inhibits the maturation, mineralization and transit of osteoblast to osteocyte[18]. In addition, TGFβ treatment blocks osteoblast mineralization in vitro[25]. In contrast, BMP2 signaling has been proved to promote osteogenic differentiation of mesenchymal progenitors and bone formation [22, 24]. Furthermore, both TGFβ signaling and BMP2 signaling are reported to regulate osteoblast differentiation by modifying the activity of RUNX2 and OSTERIX[18, 26–28], the key osteogenic transcription factors regulating the expression of osteogenic gene programs[29].
In this study, we research on the role of SHP2 in intramembranous ossification of calvarial development at cellular and molecular level. And we demonstrate that SHP2 is critical for osteogenic differentiation of mesenchymal progenitors partially via modifying TGFβ/BMP2 signaling.
2. Materials and Methods
2.1. Animals
Tg(Ptpn11fl/+)[30], Tg(Rosa26ZsG)[31], Tg(Prrx1-Cre)[32] and Tg(Sp7-mCherry) (Sp7mCherry)[33] mice were described previously. PCR genotyping methods for the Ptpn11 floxed allele, Rosa26ZsG and Sp7mCherry reporters, and Cre transgene are reported in the original publications and are available upon request. Mice bearing Ptpn11 floxed alleles were bred to Tg(Prrx1-Cre) mice to generate offspring in which SHP2 was deleted in osteochondroprogenitors (OCPs) that express the paired related homeobox-1 protein PRRX1, and controls. The mice had the following genotypes and nomenclature: Tg(Prrx1-Cre;Ptpn11fl/+) (SHP2Prrx1CTR) and Tg(Prrx1-Cre;Ptpn11fl/fl) (SHP2Prrx1KO). SHP2 Prrx1CTR and SHP2Prrx1KO mice were also bred with Rosa26ZsG and Sp7mCherry reporter mice for experiments designed to trace the fate of Prrx1-expressing cells in vivo. All of the mice were maintained on the C57BL/6J background.
Control and SHP2 mutant animals were sacrificed at the indicated time points and explanted skull tissue was processed for microcomputed tomographic imaging (µCT), histological sectioning, and biochemical and biological analyses. All animal work was reviewed and approved by the Rhode Island Hospital Institutional Animal Care and Use Committee and performed in accordance with PHS policy on the humane care and use of laboratory animals.
2.2. Osteoblast isolation and cultures
Primary osteoblastic cells were isolated from calvarias of the SHP2Prrx1CTR;Rosa26ZSG and SHP2Prrx1KO;Rosa26ZSG newborns. To do so, the parietal bones were collected under sterile condition and digested with 5 ml of Trypsin-EDTA (0.06%), and Collagenase II (285U/ml) for 4 hours. The digestion was halted by adding an equal volume of DMEM with 10% FBS, and the digestion solution was passed through a 40 μm strainer to obtain single cells. Osteoblasts were collected by centrifugation and resuspended in fresh culture medium (DMEM with 10% FBS,1% penicillin and streptomycin). Osteoblast cell preparations were cultured at 37o C under 5% CO2.
Primary osteoblasts were immortalized with retrovirus expressing pBabe(puro)/SV40 large T antigen prepared from 293T cells, as described previously[16], [30, 34]. Infected cells were cultured for 48 hours and then selected with puromycin for 7 days, after which the puromycin-resistant clones were pooled and expanded. Finally, the osteoblastic cells were enriched by FACS sorting based on the Rosa26ZsG GFP fluorophore.
2.3. Reagents
Polyclonal antibodies against murine SHP2 (sc-280) and SMAD1 (sc-7965) were purchased from Santa Cruz Biotechnology, and monoclonal anti-Sp7/OSTERIX (ab22552) was purchased from Abcam. Monoclonal antibodies against murine phospho-SMAD2/3 (8828S) and total SMAD2/3 (3102S) were purchased from Cell Signaling Technology, p-SMAD1/5/8 (AB3848) was purchased from Millipore, and anti-β-ACTIN (ab8226) was purchased from Abcam. Texas Red-X goat anti-rabbit IgG (T6391) was purchased from Invitrogen. 1% Alcian blue and 1% Alizarin Red staining solutions were purchased from Poly Scientific. Click-iT EdU Alexa Fluor 594 Imaging Kit(C10339) is purchased from Life Techonolegy.
2.4. Histological, histochemical and immunostaining analysis
Skulls from P0.5 control and SHP2 mutants were fixed in 4% formaldehyde, soaked in 30% sucrose solution for 24 hours, and embedded in OCT. Frozen sections were stained with hematoxylin and eosin (H&E) and von Kossa (Millipore) to visualize general morphology and bone matrix mineralization, respectively. The fate of Prrx1-expressing mesenchymal cells in vivo was determined by direct visualization of the ZsGreen+ (green), mCherry+ (red), and ZsGreen/mCherry double positive (yellow) fluorophores on frozen sections of parietal bone from SHP2Prrx1CTR;Rosa26ZSG;Sp7mCherry and SHP2Prrx1KO;Rosa26ZSG;Sp7mCherry mice. DAPI was used for nucleus counterstaining. All florescent and phase contrast images were captured using a Nikon digital fluorescence microscope and Aperio slide scanner (Leica Biosystems).
For Immunofluorescent staining, frozen sections were blocked with 0.3%Triton X-100, and 10% goat serum in PBS for 2 hours at room temperature, after which they were incubated with primary antibody overnight (1:100 dilution). After washing, the primary antibodies were localized with Texas Red-X goat anti-rabbit IgG secondary antibody. Nuclei were counter- stained with DAPI.
2.5. In situ hybridization
mRNA expression was determined using RNAscope in situ hybridization, as described previously[16]. Probes against murine Runx2, Sp7, Ctnnb1, Bmp2, Ibsp and Bglap were synthesized and purchased through Advanced Cell Diagnostics (ACD). Visualization of the hybridized probes was achieved using the RNAscope® HD-Brown kit (Advanced Cell Diagnostics). Images of each section were acquired using the Aperio slide scanner.
Osteogenic gene expression was quantified on corresponding 200μm x 100μm areas in the same regions of the parietal bones from SHP2Prrx1CTR and SHP2Prrx1KO mice. The positively-stained area and the total bone tissue area were quantified using NIH ImageJ software.
2.6. Quantitative RT-PCR and micro-CT analyses
Total RNA was extracted from cultured osteoblasts using the RNeasy kit (Qiagen). cDNA was synthesized using 1 µg of total RNA with iScript™cDNA Synthesis Kit (Bio-Rad) and qRT-PCR was performed with RT2SYBR®Green kit on a Bio-Rad CFX machine. All samples were normalized to Gapdh and Actin, and gene expression was presented as fold increases or decreases compared with that of control. High resolution (20 µm isometric voxel size) 3D volume images were generated using a desktop µ-CT40 system (Scanco Medical AG, CH).
2.8. Western blot analysis
Cells were lysed in modified NP-40 lysis buffer (0.5% NP40, 150 mM NaCl, 1 mM EDTA, 50 mM Tris [pH 7.4]) supplemented with a protease inhibitor cocktail (1 mM PMSF, 10 mg/ml aprotinin, 0.5 mg/ml antipain, and 0.5 mg/ml pepstatin)[35]. Cell lysates (20–30 µg) were separated by SDS-PAGE, transferred to PVDF membranes, and incubated with indicated primary antibodies for 2 hours or overnight at 4ºC (primary antibody concentration is 1μg/ml); this was followed by HRP-conjugated secondary antibodies (Bio-Rad, 1:2000 dilution).
2.9. Statistical analysis
Differences between the SHP2 knockouts and controls were evaluated using Student’s t tests, with p values <0.05 considered to be significant. Statistical analyses were performed by using Prism 5.0 (GraphPad, San Diego, CA) and Excel (Microsoft).
3. Results
3.1. SHP2 deletion in the Prrx1-expressing mesenchymal cells impairs calvarial bone ossification.
SHP2Prrx1KO mutants were born at the expected Mendelian ratios, but they were smaller than their control littermates[15]. Most of the SHP2 mutants died within 3 weeks after birth, likely due to difficulty with breathing and feeding, but a few of the mutant mice could survive until 6 weeks[15]. Compared with the SHP2Prrx1CTR mice, the skulls of SHP2Prrx1KO mice were incompletely formed. Alcian blue and Alizarin Red staining revealed marked defects in mineralization of the parietal, interparietal, and occipital bones (Fig. 1A), as well as the tibiae, femur and humerus , which are of endochondral origin[15]. These findings were even more striking in the data from the µCT imaging, where the parietal and interparietal bones were totally absent in the models created from the thresholded µCT images of 6-week-old SHP2Prrx1KO mice (Fig. 1B, bottom), while the skulls of the SHP2Prrx1CTR mice appear entirely normal, with intact cranial vaults and well-defined sutures (Fig. 1B, top).
Figure 1. SHP2 ablation in the Prrx1+ mesenchymal cells impairs intramembranous ossification.
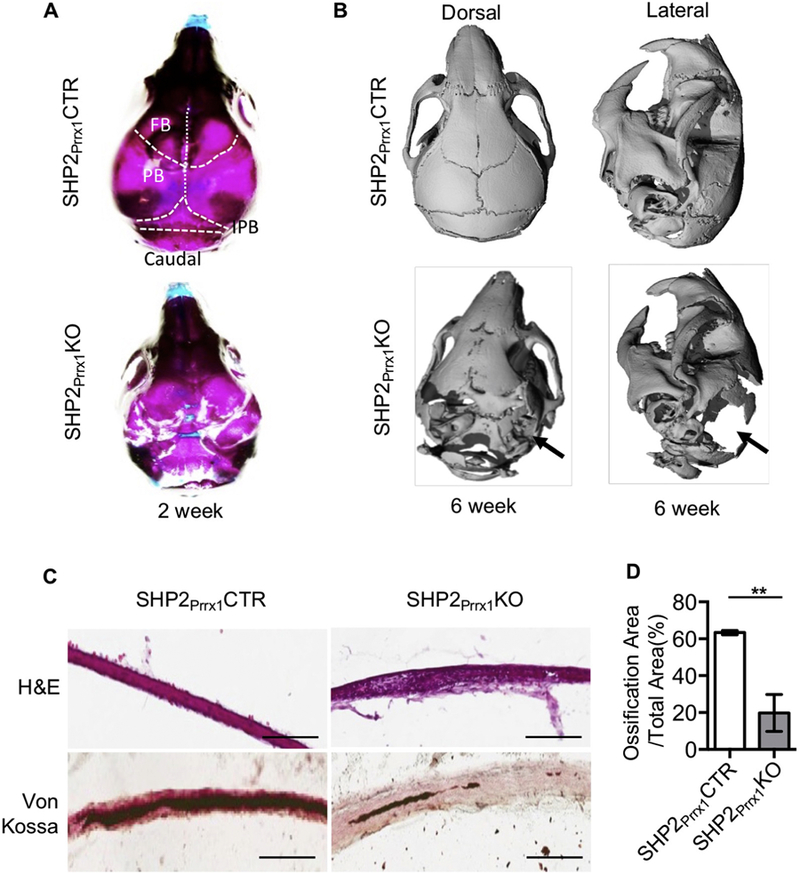
A. Representative skull images of 2-week-old SHP2Prrx1CTR and SHP2Prrx1KO mice stained with Alcian blue and Alizarin Red. n=3. B. μ-CT radiographs demonstrating the skull structure of 6-week-old SHP2Prrx1CTR and SHP2Prrx1KO mice. Note that parietal and interpariental bone ossification was defective (arrows) in the SHP2Prrx1KO mice, compared with age matched SHP2Prrx1CTR mice. n=3. C. Representative H&E (top) and von Kossa (bottom, counterstained by fast red) staining of murine parietal bone coronal frozen sections showing the appearance of a loose mesenchyme tissue and impaired mineralization of the parietal bone in the P0.5 SHP2Prrx1KO mice, compared with SHP2Prrx1CTR mice. Scale bar:100μm. D. Bar graphs show the quantitative data of parietal bone matrix mineralization in SHP2Prrx1CTR and SHP2Prrx1KO mice using NIH ImageJ software (n=3, **P<0.01, Student’s t-test).
Histological analysis of stained frozen sections was performed to evaluate the microarchitecture of the skull elements. H&E and von Kossa staining of the sections from P0.5 SHP2Prrx1CTR mice revealed a sandwich-like morphology in which the fully mineralized parietal bone was covered by two layers of soft periosteal mesenchymal tissue (Fig. 1C left). In contrast, mineralization of the parietal bone was markedly reduced in P0.5 SHP2Prrx1KO mutants as the von Kossa staining image showed (Fig. 1C right). Instead, there was a dense layer of soft connective tissue filled with undifferentiated mesenchymal cells where the parietal bone should have been. Quantitative analysis revealed a ~70% reduction in the portion of the parietal “bone” tissue that was mineralized in the SHP2Prrx1KO mice compared to corresponding regions of SHP2Prrx1CTR mice (19.8±5.7% vs 63.3±0.6%, **p<0.01) (Fig. 1D). And finally, calcified osteogenic fronts appeared clearly in the SHP2Prrx1CTR mice but were absent in the SHP2Prrx1KO mice (Fig S1). Taken together, these data indicate that SHP2 expression in the Prrx1-expressing OCPs is essential for the mineralization of calvarial bones.
3.2. SHP2 is required for the osteogenic differentiation of the Prrx1+ mesenchymal progenitors.
Having established that lack of SHP2 in Prrx1-expressing OCPs inhibits intramembraneous ossification, we next sought to determine whether the cause might be lack of OCP differentiation. To start, we performed a dual fluorescent reporter-based cell lineage tracing study. SHP2Prrx1CTR;Rosa26ZsG and SHP2Prrx1KO;Rosa26ZsG mice were bred with Sp7mCherry reporter mice, with the idea that the resulting SHP2Prrx1CTR;Rosa26ZsG;Sp7mCherry and SHP2Prrx1KO;Rosa26ZsG;Sp7mCherry mice would express ZsGreen in cells that expressed, or had expressed, Prrx1+ (i.e. osteochondral progenitors and their progeny), and mCherry in cells that expressed Sp7 (i.e. osteoblast lineage cells). Importantly, if the Prrx1+ ZsGreen cells differentiated into Sp7-expressing osteoblastic cells, they would co-express the ZsGreen and mCherry reporters, which would appear yellow. As shown in Fig. 2A (left) & D(top), a large number of cells within the osteogenic fronts and the parietal bones fluoresced yellow in the SHP2Prrx1CTR;Rosa26ZsG;Sp7mCherry mice. The yellow cells at the osteogenic fronts were connected by soft tissue bridge rich in ZsGreen+ cells, which indicated they were Prrx1+ mesenchymal progenitors. In contrast, the number of yellow cells in the osteogenic fronts and in the parietal bone were significantly reduced in the SHP2Prrx1KO;Rosa26ZsG;Sp7mCherry mutants (43.19±0.37% vs. 83.24±4.48%, ***p<0.001, Student’s t-test) (Fig. 2A & D). Instead, a large quantity of green cells was observed in the SHP2Prrx1KO;Rosa26ZsG;Sp7mCherry mutants. This suggests that the maturation of Prrx1+ green cells into osteoblasts was compromised in the absence of SHP2. The above observations were bolstered by immunostaining of the parietal bone frozen sections with the Texas-red labeled anti-OSTERIX antibodies (SHP2Prrx1CTR;Rosa26ZsG vs. SHP2Prrx1KO;Rosa26ZsG: mean± SEM of percentage: 70.02±1.75 vs. 42.84±5.17, **p<0.01, Student’s t-test) (Fig. 2B & E). Consistent with these observations, the width of sagittal suture significantly increased in the SHP2 mutants, compared to the controls (186.2±28.1μm vs. 293.9±24.9μm, *p<0.05, Student’s t-test) (Fig. 2C). Taken together, these data suggest that lack of SHP2 in OCPs inhibits their osteogenic differentiation during intramembranous ossification.
Figure 2. SHP2 is required for the osteogenic differentiation of the Prrx1+ mesenchymal cells.

A. Fluorescent images of the sagittal sutures showing the number and location of Prrx1+ (green), Osx+ (red), and Prrx1/Osx double positive cells (yellow) at the osteogenic fronts and sutures. Note that the number of Prrx1 and Osx double positive cells decreased and the width of the sagittal suture (white arrows increased significantly in SHP2Prrx1KO;Rosa26ZsG;Sp7mCherry mice, compared with the SHP2Prrx1CTR;Rosa26ZsG;Sp7mCherry controls. Images at the bottom are the enlarged views of the corresponding boxed areas in the top images. Scale bar: 100μm. B. Sagittal sutures of indicated mice immunostained with anti-OSTERIX antibody (red) showing the number and location of Prrx1+ (green), OSTERIX+ (red), and Prrx1/OSTERIX double positive cells (yellow) at the osteogenic fronts and sutures. Images at the bottom are the enlarged views of the corresponding boxed areas in the top images. Scale bar: 100μm. Scale bar: 100μm. C. Bar graphs showing the quantitative data of sagittal suture width (images of A and B) in the P0.5 SHP2Prrx1CTR and the SHP2Prrx1KO mice (n=6, *P<0.05, Student’s t-test). D-E. Fluorescent images of the parietal bone frozen sections showing the number and location of Prrx1+ (green), Osx+ (red), OSTERIX+ (red), and Prrx1 and Osx (OSTERIX) double positive (yellow) cells in the indicated mice. Quantitative data are shown as bar graphs on the right (n=3, ***p<0.001, Student’s t-test) Scale bar: 100μm.
3.3. SHP2 deletion in the Prrx1-expressing mesenchymal progenitors suppresses osteogenic gene expression.
Calvarial bones ossify through intramembranous ossification in which mesenchymal progenitors undergo progressive differentiation into osteoblasts, which express OSTERIX (Sp7), COL1a1, bone sialoprotein, and OSTEOCALCIN. RUNX2 has been considered the master gene of osteoblast differentiation[6, 8]. RUNX2 is considered essential for the osteogenic differentiation of the Prrx1+ mesenchymal progenitors but it is not required for the terminal differentiation of the Col1a1+ osteoblasts[36]. Having demonstrated that SHP2 is indispensable for the differentiation of OCPs into functioning osteoblasts, we next explored whether the expression of Runx2 and other osteogenic genes were affected in these mutants. RNAScope®-based in situ hybridization revealed significant reductions in Runx2, Sp7, Ctnnb1, Bglap, Bmp2 and Ibsp transcripts in frozen sections of parietal bone from the SHP2Prrx1KO mutants, compared with the SHP2Prrx1CTR controls (Fig. 3.). These data are consistent with the von Kossa staining data showing an impaired mineralization of the parietal bones (Fig. 1C). Collectively, these data indicate that SHP2 regulates the osteogenic differentiation of OCPs in intramembranous ossification by modulating the expression of the osteogenic transcription factors, as well as downstream effecting osteoblastic genes.
Figure 3. SHP2 deletion in the Prrx1-expressing mesenchymal cells impairs osteogenic marker genes and osteogenic transcription factors expression during intramembranous ossification.

A. RNAscope-based in situ hybridization shows the transcript abundance of indicated osteogenic genes in the P0.5 parietal bone of SHP2Prrx1CTR and SHP2Prrx1KO mice. Scale bar: 100μm. B. Enlarged views of the corresponding boxed areas in A. Scale bar: 100μm. C. Bar graphs show the quantitative data of A & B. Data presented as the ratio of the positive staining areas vs. the corresponding total areas using NIH ImageJ (n=3, *p<0.05, **p<0.01, ***p<0.001, Student’s t-test). D. The transcript abundance of indicated osteogenic transcription factors in the P0.5 parietal bone of SHP2Prrx1CTR and SHP2Prrx1KO mice were presented as described in A. Scale bar: 100μm. E. Enlarged views of the corresponding boxed areas in D. Scale bar: 100μm. F. Bar graphs show the quantitative data of D & E. Data presented as the ratio of the positive staining areas vs. the corresponding total areas using NIH ImageJ (n=3, *p<0.05, **p<0.01, ***p<0.001, Student’s t-test).
3.4. SHP2 is required for osteogenic gene expression in the Prrx1+ progenitors and their derivatives
We used cell culture to gain additional insight into the molecular and cellular mechanisms through which SHP2 regulates intramembranous ossification. To do so we isolated ZsGreen+ calvarial cells from the SHP2Prrx1CTR;Rosa26ZSG and SHP2Prrx1KO;R26ZSG mice via FACS and immortalized them via the retroviral expression of SV40 large T antigen (Fig. 4A). Phase contrast images showed that immortalized calvarial cells from both control and SHP2 mutant mice display bipolar or multipolar and elongated shapes, and western blotting confirmed that the SHP2 protein level was significantly reduced in the osteoblastic cells from the SHP2Prrx1KO;Rosa26ZSG mice, compared with SHP2Prrx1CTR;Rosa26ZSG control animals (only about 30% of the controls) (Fig. 4B). qRT-PCR revealed significant reductions in the transcript abundance of osteogenic genes Alp, Col1a1, Ctnnb1, Sp7 and Runx2 in the SHP2Prrx1KO;Rosa26ZSG cells, compared to the SHP2Prrx1CTR;Rosa26ZSG controls (Fig. 4C). ALP activity was also evaluated in the SHP2Prrx1KO;Rosa26ZSG cells and SHP2Prrx1CTR;Rosa26ZSG controls using a colorimetric assay. The number of positively-stained cells and total cells were counted in three fields of view and analyzed. The quantitative data showed that the ALP activity was markedly reduced in the SHP2 mutant cells (mean± SEM of percentage: 38.53± 1.80 vs. 4.00± 0.53, ***p<0.001, Student’s t-test) (Fig. 4D). These data in vitro confirmed our in vivo findings that SHP2 is required for intramembranous ossification by promoting the osteogenic gene expression.
Figure 4. SHP2 deletion in the Prrx1-expressing cells compromises osteogenic genes expression in vitro.
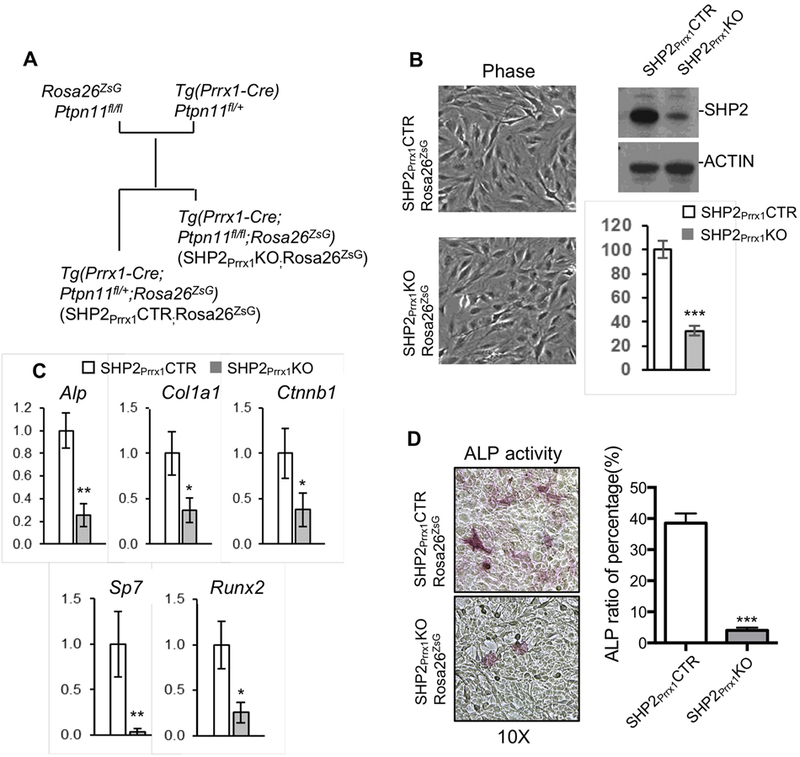
A. Diagram denotes the strategy to generate SV40 large T antigen-immortalized osteoblasts from the SHP2Prrx1CTR;Rosa26ZsG and SHP2Prrx1KO;Rosa26ZsG mice. B. Phase contrast images (left) showing the morphology of immortalized osteoblasts. Western blot and bar graphs [44] demonstrating the level of SHP2 in the immortalized osteoblasts from indicated mice (n=3, ***P<0.001, Student’s t-test). C. Bar graphs showing the relative expression of indicated osteogenic genes in SHP2Prrx1CTR and SHP2Prrx1KO osteoblasts determined by qRT-PCR. (n=3, *p<0.05,**p<0.01, Student’s t-test). D. Phase contrast images demonstrating ALP activity in the immortalized osteoblastic cells from indicated mice. Ratio of ALP positive cells vs total cells in the defined areas are presented as bar graphs on the right (n=3, ***p<0.001, Student’s t-test).
3.5. SHP2 regulates osteogenic gene expression by modifying the TGFβ and BMP2 signaling pathways.
TGFβ and BMP2 signaling has a fundamental role in regulation of embryonic skeletal development and maintaining postnatal bone homeostasis via the regulation of RUNX2 and osteogenic gene expression[18]. Because our data indicated that impaired osteogenic differentiation of SHP2-deficient OCPs involved reductions in Runx2 and other osteogenic genes, we sought to investigate whether SHP2 might influence the TGFβ/BMP2 signaling axis. To do so, calvarial osteoblast cell lines from SHP2Prrx1CTR and SHP2Prrx1KO mice were starved overnight and then harvested 0, 15, 30, 60 and 120, minutes after stimulation with TGFβ (100ng/ml). Western blotting revealed that the SMAD2/3 phosphorylation was significantly increased in SHP2Prrx1KO cells compared to the SHP2Prrx1CTR control cells (Fig. 5A). Moreover, the phosphorylation level of SMAD2/3 returned to basal levels 120mins after stimulation in the SHP2Prrx1CTR cells, but remained high in SHP2Prrx1KO cells (Fig. 5A). We also starved SHP2Prrx1CTR and SHP2Prrx1KO cells overnight, stimulated them with BMP2 (100ng/ml) and then harvested them at 0, 5, 15, 30 and 60 minutes. The western blot data revealed that the phosphorylation level of SMAD1/5/8 was significantly decreased in SHP2Prrx1KO cells, compared to SHP2Prrx1CTR control cells under the same stimulation (Fig. 5B). These data indicate that SHP2 normally represses the phosphorylation of SMAD2/3 by TGFβRI but promotes the phosphorylation of SMAD1/5/8 by BMPR I; these coupled but differential regulation of TGFβ and BMP2 signaling pathway to ensure the maturation and function of osteoblasts.
Figure 5. SHP2 differentially regulates TGFβ and BMP signaling pathways.
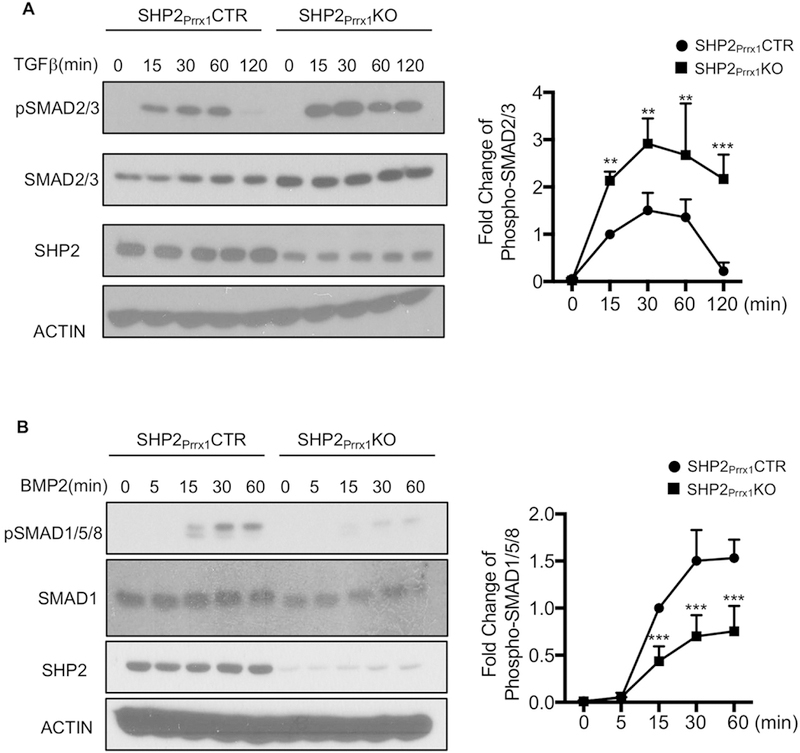
A. Immunoblotting data show that TGFβ-evoked SMAD2/3 phosphorylation was enhanced in the osteoblastic cells from SHP2Prrx1KO mice, compared to that from SHP2Prrx1CTR mice. Cells were starved overnight and stimulated with TGFβ (100ng/ml) for indicated time points. Phosphorylation of SMAD2/3 was quantified using NIH ImageJ software and normalized to the β-ACTIN controls, phosphorylation level of SMAD2/3 was corrected by the total level of SMAD2/3. (n=3, **p<0.01, ***p<0.001, Student’s t-test) B. Immunoblotting data showing decreased SMAD1/5/8 phosphorylation in response to BMP2 in SHP2Prrx1KO osteoblastic cells, compared to SHP2Prrx1CTR osteoblastic cells. Cells were starved overnight and stimulated with BMP2 (100ng/ml) for indicated time points. Phosphorylation of SMAD1/5/8 was quantified as described in A (n=3, **p<0.01, ***p<0.001, Student’s t-test).
4. Discussion
Intramembranous ossification is regulated by a number of signaling pathways, in which protein tyrosine kinases are heavily involved [18, 19]. Increasing evidences suggest that protein tyrosine phosphatases play a crucial role in this process, too. SHP2 is a ubiquitously PTP. Early studies showed that SHP2 is a key modulator of the osteogenic differentiation of OCPs[15] and growth plate hypertrophic chondrocytes by modifying SOX9 abundance during endochondral ossification[16]. Here, we showed that SHP2 is indispensable for the osteogenic differentiation of OCPs in the calvarial bones. SHP2 deficient OCPs were halted at the primitive stages and failed to ossify the matrix of the forming calvarial vault (Fig. 1 & 2), rather than a defect in cell proliferation (Fig S3). Consistent with these morphological defects, transcript abundance of osteogenic transcription factors Runx2 and Osterix and their downstream effectors (Fig. 3 & 4) were found significantly decreased in the SHP2 mutants. Importantly, SHP2 was uncovered to differentially regulate canonical BMP2 (promoting) and TGFβ (suppressing) signaling in OCPs and their derivatives in vitro (Fig. 5). Collectively, our study uncovered an important role for SHP2 in intramembranous ossification by modulating the osteogenic differentiation and maturation of OCPs, these actions, in part, are via the differential regulation of BMP and TGFβ signaling pathways.
BMP and TGFβ signaling pathways are well accepted as one of the most important signaling pathways to regulate the osteoblast differentiation and maturation. TGFβ signaling pathway promotes osteoblastic progenitor proliferation and early differentiation, but suppresses the osteoblastic maturation, mineralization and transit to osteocyte[18]. Upon TGFβ signaling pathway activation, SMAD2/3 is phosphorylated and then recruit class II deacetylases (HDACs 4 and 5) to repress the gene expression and activity of Runx2 [37–40]. In contrast, activation of the BMP signaling pathway regulates osteoblast differentiation. Activated BMPR I phosphorylates SMAD1/5/8, which up-regulates the expression of Runx2 and Osterix and drive osteoblastic differentiation [37–39]. Our immunoblotting data showed that SHP2 deficiency in osteoblast promoted TGFβ-evoked SMAD2/3 phosphorylation and suppressed BMP2-evoked SMAD1/5/8 activation (Fig. 5), indicating that SHP2 differentially regulates BMP and TGFβ signaling pathways and osteoblast differentiation and maturation. This could explain why there was a wider gap between the osteogenic fronts in the sagittal suture of SHP2 mutants and accumulation of primitive Prrx1+ osteochondroprogenitors. Runx2 and Osxterix are considered as the two master transcription factors during osteoblasotgenesis[26–28]. The profound resemblance of the skull phenotype between SHP2 and RUNX2 mutants suggests that RUNX2 functions downstream SHP2 [36]. This notion was further supported by the reduction of RUNX2 responsive osteogenic gene expression (Fig. 4) in SHP2 mutants [18, 27, 41, 42].
TGFβ and BMP2 also signal via SMAD independent non-canonical pathways, in which, they activate TAK1 and TAK1-binding protein1(TAB1) to initiate the MKKs and ERK signaling cascade[22]. TGFβ and BMP2 evoked MKK activation could positively regulate Runx2 expression and promote the differentiation of OCPs[43]. We therefore examined TGFβ and BMP2-evoked ERK activation in osteoblasts and found that they were comparable in the presence or absence of SHP2 (Fig. S2), suggesting that SHP2 regulation of BMP and TGFβ signaling primarily went through the canonical TGFβ/SMAD2/3 and BMP2/SMAD1/5/8 signaling pathways.
BMP and TGFβ signal through BMPR I/II and TGFR I/II respectively to phosphorylate SMAD1/5/8 and SMAD2/3 and activate their downstream transcription programs[44]. Altered SMAD phosphorylation in SHP2 deficient OCPs and their derivatives suggest that SHP2 acts either on a serine/threonine kinase upstream SMADs or a SMAD phosphatases. SHP2 functions a PTP, therefore the effect of SHP2 on BMP2- and TGFβ-evoked SMAD phosphorylation must be mediated via an indirect mechanism. Currently we are investigating whether TGFβ and BMP2 receptor kinase activity is modulated by phosphorylation of tyrosine residues[45].
In sum, SHP2 is found to be essential for the osteogenic differentiation and maturation of OCPs and intramembranous ossification, partially by differentially modifying the TGFβ and BMP signaling pathways. SHP2 and its signaling partner(s) may be potential to serve as pharmacological targets to treat craniofacial disorders, such as craniosynostosis and cleidocranial dysostosis.
Supplementary Material
Highlights.
SHP2 is a key regulator of intramembranous ossification, its deficiency in the Prrx1+ mesenchymal progenitors causes severely developmental defects in the calvaria bones.
SHP2 is required for osteogenic differentiation of mesenchymal progenitors but has minimal effect on their proliferation.
SHP2 is required for BMP2-evoked SMAD1/5/8 activation but suppresses TGFβ-induced SMAD2/3 activation. Its deficiency in the Prrx1+ mesenchymal progenitors compromises the expression of osteogenic transcription factors Runx2, Osterix and other osteogenic genes.
Acknowledgements
This publication was made possible by NIH and the National Institute for General Medicine Sciences (NIGMS) Grant #8P20GM103468 and NIAMS RO1AR066746 (WY). This work was also in part supported by the Rhode Island Hospital Orthopaedic Foundation and Arthritis National Research Foundation (WY). LW is a pilot award recipient from NIGMS1P20 GM119943.
Footnotes
Disclosures: All authors state that they have no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Jin SW, Sim KB, Kim SD, Development and Growth of the Normal Cranial Vault : An Embryologic Review, J Korean Neurosurg Soc 59(3) (2016) 192–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Ishii M, Sun J, Ting MC, Maxson RE, The Development of the Calvarial Bones and Sutures and the Pathophysiology of Craniosynostosis, Curr Top Dev Biol 115 (2015) 131–56. [DOI] [PubMed] [Google Scholar]
- [3].Jiang X, Iseki S, Maxson RE, Sucov HM, Morriss-Kay GM, Tissue origins and interactions in the mammalian skull vault, Dev Biol 241(1) (2002) 106–16. [DOI] [PubMed] [Google Scholar]
- [4].Yoshida T, Vivatbutsiri P, Morriss-Kay G, Saga Y, Iseki S, Cell lineage in mammalian craniofacial mesenchyme, Mech Dev 125(9–10) (2008) 797–808. [DOI] [PubMed] [Google Scholar]
- [5].Deckelbaum RA, Holmes G, Zhao Z, Tong C, Basilico C, Loomis CA, Regulation of cranial morphogenesis and cell fate at the neural crest-mesoderm boundary by engrailed 1, Development 139(7) (2012) 1346–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G, Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation, Cell 89(5) (1997) 747–54. [DOI] [PubMed] [Google Scholar]
- [7].Koga T, Matsui Y, Asagiri M, Kodama T, de Crombrugghe B, Nakashima K,Takayanagi H, NFAT and Osterix cooperatively regulate bone formation, Nat Med 11(8) (2005) 880–5. [DOI] [PubMed] [Google Scholar]
- [8].Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, Shimizu Y,Bronson RT, Gao YH, Inada M, Sato M, Okamoto R, Kitamura Y, Yoshiki S,Kishimoto T, Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts, Cell 89(5) (1997) 755–64. [DOI] [PubMed] [Google Scholar]
- [9].Nakashima K, Zhou X, Kunkel G, Zhang Z, Deng JM, Behringer RR, de Crombrugghe B, The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation, Cell 108(1) (2002) 17–29. [DOI] [PubMed] [Google Scholar]
- [10].Araki T, Mohi MG, Ismat FA, Bronson RT, Williams IR, Kutok JL, Yang W, Pao LI, Gilliland DG, Epstein JA, Neel BG, Mouse model of Noonan syndrome reveals cell type- and gene dosage-dependent effects of Ptpn11 mutation, Nat Med 10(8) (2004) 849–57. [DOI] [PubMed] [Google Scholar]
- [11].Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, Kremer H,van der Burgt I, Crosby AH, Ion A, Jeffery S, Kalidas K, Patton MA, Kucherlapati RS, Gelb BD, Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome, Nat Genet 29(4) (2001) 465–8. [DOI] [PubMed] [Google Scholar]
- [12].Kosaki K, Suzuki T, Muroya K, Hasegawa T, Sato S, Matsuo N, Kosaki R,Nagai T, Hasegawa Y, Ogata T, PTPN11 (protein-tyrosine phosphatase, nonreceptor-type 11) mutations in seven Japanese patients with Noonan syndrome, J Clin Endocrinol Metab 87(8) (2002) 3529–33. [DOI] [PubMed] [Google Scholar]
- [13].Bowen ME, Boyden ED, Holm IA, Campos-Xavier B, Bonafe L, Superti-Furga A, Ikegawa S, Cormier-Daire V, Bovee JV, Pansuriya TC, de Sousa SB, Savarirayan R, Andreucci E, Vikkula M, Garavelli L, Pottinger C, Ogino T, Sakai A, Regazzoni BM, Wuyts W, Sangiorgi L, Pedrini E, Zhu M, Kozakewich HP, Kasser JR, Seidman JG, Kurek KC, Warman ML, Loss-of-function mutations in PTPN11 cause metachondromatosis, but not Ollier disease or Maffucci syndrome, PLoS Genet 7(4) (2011) e1002050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kim HK, Aruwajoye O, Sucato D, Richards BS, Feng GS, Chen D, King PD, Kamiya N, Induction of SHP2 deficiency in chondrocytes causes severe scoliosis and kyphosis in mice, Spine (Phila Pa 1976) 38(21) (2013) E1307–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zuo C, Wang L, Kamalesh RM, Bowen ME, Moore DC, Dooner MS, Reginato AM, Wu Q, Schorl C, Song Y, Warman ML, Neel BG, Ehrlich MG, Yang W, SHP2 regulates skeletal cell fate by modifying SOX9 expression and transcriptional activity, Bone Res 6 (2018) 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wang L, Huang J, Moore DC, Zuo C, Wu Q, Xie L, von der Mark K, Yuan X,Chen D, Warman ML, Ehrlich MG, Yang W, SHP2 Regulates the Osteogenic Fate of Growth Plate Hypertrophic Chondrocytes, Sci Rep 7(1) (2017) 12699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lapinski PE, Meyer MF, Feng GS, Kamiya N, King PD, Deletion of SHP-2 in mesenchymal stem cells causes growth retardation, limb and chest deformity, and calvarial defects in mice, Dis Model Mech 6(6) (2013) 1448–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wu M, Chen G, Li YP, TGF-beta and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease, Bone Res 4 (2016) 16009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ornitz DM, Marie PJ, FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease, Genes Dev 16(12) (2002) 1446–65. [DOI] [PubMed] [Google Scholar]
- [20].Regard JB, Zhong Z, Williams BO, Yang Y, Wnt signaling in bone development and disease: making stronger bone with Wnts, Cold Spring Harb Perspect Biol 4(12) (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Day TF, Yang Y, Wnt and hedgehog signaling pathways in bone development, J Bone Joint Surg Am 90 Suppl 1 (2008) 19–24. [DOI] [PubMed] [Google Scholar]
- [22].Chen G, Deng C, Li YP, TGF-beta and BMP signaling in osteoblast differentiation and bone formation, Int J Biol Sci 8(2) (2012) 272–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Goumans MJ, Mummery C, Functional analysis of the TGFbeta receptor/Smad pathway through gene ablation in mice, Int J Dev Biol 44(3) (2000) 253–65. [PubMed] [Google Scholar]
- [24].Cao X, Chen D, The BMP signaling and in vivo bone formation, Gene 357(1) (2005) 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Matsunobu T, Torigoe K, Ishikawa M, de Vega S, Kulkarni AB, Iwamoto Y, Yamada Y, Critical roles of the TGF-beta type I receptor ALK5 in perichondrial formation and function, cartilage integrity, and osteoblast differentiation during growth plate development, Dev Biol 332(2) (2009) 325–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Lee KS, Kim HJ, Li QL, Chi XZ, Ueta C, Komori T, Wozney JM, Kim EG, Choi JY, Ryoo HM, Bae SC, Runx2 is a common target of transforming growth factor beta1 and bone morphogenetic protein 2, and cooperation between Runx2 and Smad5 induces osteoblast-specific gene expression in the pluripotent mesenchymal precursor cell line C2C12, Mol Cell Biol 20(23) (2000) 8783–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Lee MH, Kwon TG, Park HS, Wozney JM, Ryoo HM, BMP-2-induced Osterix expression is mediated by Dlx5 but is independent of Runx2, Biochem Biophys Res Commun 309(3) (2003) 689–94. [DOI] [PubMed] [Google Scholar]
- [28].Kurata H, Guillot PV, Chan J, Fisk NM, Osterix induces osteogenic gene expression but not differentiation in primary human fetal mesenchymal stem cells, Tissue Eng 13(7) (2007) 1513–23. [DOI] [PubMed] [Google Scholar]
- [29].Nakashima K, de Crombrugghe B, Transcriptional mechanisms in osteoblast differentiation and bone formation, Trends Genet 19(8) (2003) 458–66. [DOI] [PubMed] [Google Scholar]
- [30].Yang W, Wang J, Moore DC, Liang H, Dooner M, Wu Q, Terek R, Chen Q, Ehrlich MG, Quesenberry PJ, Neel BG, Ptpn11 deletion in a novel progenitor causes metachondromatosis by inducing hedgehog signalling, Nature 499(7459) (2013) 491–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, Lein ES, Zeng H, A robust and high-throughput Cre reporting and characterization system for the whole mouse brain, Nat Neurosci 13(1) (2010) 133–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Logan M, Martin JF, Nagy A, Lobe C, Olson EN, Tabin CJ, Expression of Cre Recombinase in the developing mouse limb bud driven by a Prxl enhancer, Genesis 33(2) (2002) 77–80. [DOI] [PubMed] [Google Scholar]
- [33].Strecker S, Fu Y, Liu Y, Maye P, Generation and characterization of Osterix-Cherry reporter mice, Genesis 51(4) (2013) 246–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Hahn WC, Dessain SK, Brooks MW, King JE, Elenbaas B, Sabatini DM, DeCaprio JA, Weinberg RA, Enumeration of the simian virus 40 early region elements necessary for human cell transformation, Mol Cell Biol 22(7) (2002) 2111–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Yang W, Klaman LD, Chen B, Araki T, Harada H, Thomas SM, George EL, Neel BG, An Shp2/SFK/Ras/Erk signaling pathway controls trophoblast stem cell survival, Dev Cell 10(3) (2006) 317–27. [DOI] [PubMed] [Google Scholar]
- [36].Takarada T, Nakazato R, Tsuchikane A, Fujikawa K, Iezaki T, Yoneda Y,Hinoi E, Genetic analysis of Runx2 function during intramembranous ossification, Development 143(2) (2016) 211–8. [DOI] [PubMed] [Google Scholar]
- [37].Li J, Tsuji K, Komori T, Miyazono K, Wrana JL, Ito Y, Nifuji A, Noda M, Smad2 overexpression enhances Smad4 gene expression and suppresses CBFA1 gene expression in osteoblastic osteosarcoma ROS17/2.8 cells and primary rat calvaria cells, J Biol Chem 273(47) (1998) 31009–15. [DOI] [PubMed] [Google Scholar]
- [38].Alliston T, Choy L, Ducy P, Karsenty G, Derynck R, TGF-beta-induced repression of CBFA1 by Smad3 decreases cbfa1 and osteocalcin expression and inhibits osteoblast differentiation, EMBO J 20(9) (2001) 2254–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Hjelmeland AB, Schilling SH, Guo X, Quarles D, Wang XF, Loss of Smad3-mediated negative regulation of Runx2 activity leads to an alteration in cell fate determination, Mol Cell Biol 25(21) (2005) 9460–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kang JS, Alliston T, Delston R, Derynck R, Repression of Runx2 function by TGF-beta through recruitment of class II histone deacetylases by Smad3, EMBO J 24(14) (2005) 2543–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Celil AB, Hollinger JO, Campbell PG, Osx transcriptional regulation is mediated by additional pathways to BMP2/Smad signaling, J Cell Biochem 95(3) (2005) 518–28. [DOI] [PubMed] [Google Scholar]
- [42].Lee MH, Kim YJ, Kim HJ, Park HD, Kang AR, Kyung HM, Sung JH, Wozney JM, Kim HJ, Ryoo HM, BMP-2-induced Runx2 expression is mediated by Dlx5, and TGF-beta 1 opposes the BMP-2-induced osteoblast differentiation by suppression of Dlx5 expression, J Biol Chem 278(36) (2003) 34387–94. [DOI] [PubMed] [Google Scholar]
- [43].Lai CF, Cheng SL, Signal transductions induced by bone morphogenetic protein-2 and transforming growth factor-beta in normal human osteoblastic cells, J Biol Chem 277(18) (2002) 15514–22. [DOI] [PubMed] [Google Scholar]
- [44].Wrighton KH, Lin X, Feng XH, Phospho-control of TGF-beta superfamily signaling, Cell Res 19(1) (2009) 8–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Heldin CH, Miyazono K, ten Dijke P, TGF-beta signalling from cell membrane to nucleus through SMAD proteins, Nature 390(6659) (1997) 465–71. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.