

Abstract
The PPP1R1B gene is located on chromosome 17q12 (39,626,208–39,636,626[GRCh38/hg38]), which codes for multiple transcripts and two experimentally-documented proteins Darpp-32 and t-Darpp. Darpp-32 (Dopamine and cAMP Regulated Phosphoprotein), discovered in the early 1980s, is a protein whose phosphorylation is upregulated in response to cAMP in dopamine-responsive tissues in the brain. It’s phosphorylation profile modulates its ability to bind and inhibit Protein Phosphatase 1 activity, which, in turn, controls the activity of hundreds of phosphorylated proteins. PPP1R1B knockout mice exhibit subtle learning defects. In 2002, the second protein product of PPP1R1B was discovered in gastric cancers: t-Darpp (truncated Darpp-32). The start codon of t-Darpp is amino acid residue 37 of Darpp-32 and it lacks the domain responsible for modulating Protein Phosphatase 1. Aside from gastric cancers, t-Darpp and/or Darpp-32 is overexpressed in tumor cells from breast, colon, esophagus, lung and prostate tissues. More than one research team has demonstrated that these proteins, through mechanisms that to date remain cloudy, activate AKT, a protein whose phosphorylation leads to cell survival and blocks apoptosis. Furthermore, in Her2 positive breast cancers (an aggressive form of breast cancer), t-Darpp/Darpp-32 overexpression causes resistance to the frequently-administered anti-Her2 drug, trastuzumab (Herceptin), likely through AKT activation. Here we briefly describe how Darpp-32 and t-Darpp were discovered and report on the current state of knowledge of their involvement in cancers. We present a case for the development of an anti-t-Darpp therapeutic agent and outline the unique challenges this endeavor will likely encounter.
Graphical Abstract
1.0. Introduction
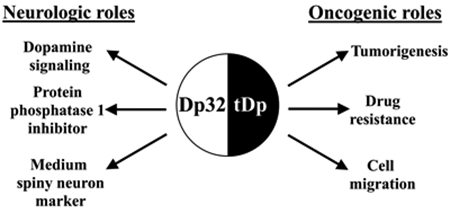
Darpp-32 and t-Darpp are two protein isoforms expressed from the PPP1R1B gene located on chromosome 17q12.1 that have key roles in brain signaling and non-brain cancers (Figure 1). Darpp-32 (Dp32) codes for the 204-residue Dopamine- and cAMP-regulated neuronal phosphoprotein, first characterized in the 1980s by Paul Greengard as an inhibitor of Protein Phosphatase 1 (PP1) [1–3]. During brain development, Dp32 is expressed in medium spiny neurons (MSNs) [4] and in adults it is primarily expressed intracellularly in the brain within regions responsive to dopamine including the caudate nucleus, putamen, nucleus accumbens, cerebral cortex, and cerebellar cortex [5–7]. Dp32 plays a critical role in mediating downstream signaling through dopamine receptor subtype 1 (D1) and is negatively regulated by dopamine through dopamine receptor subtype 2 (D2) as well as glutamate signaling through the N-methyl-D-aspartate receptor (NMDAR), an ionotropic glutamate receptor [8]. Dopamine, glutamate, cocaine, nicotine, ethanol, and amphetamines bind to one or more of these receptors and alter the phosphorylation landscape of Dp32, ultimately modulating its major downstream target PP1 [9].
Figure 1. Gene structure, transcripts and proteins products of PPP1R1B gene.

A. Canonical transcripts (three of the many that have been reported) and two documented protein products. Inclusion of exon 1 creates a transcript that produces Dp32. Inclusion of exon 1b or exon 1c creates a transcript that produces tDp. Also shown are experimentally documented transcription factor binding sites on the gene. B. Schematic diagram showing motifs, phosphorylation sites, and epitopes for commercially available antibodies on Dp32 and tDp. The kinases that phosphorylate Dp32 are: T34: protein kinase A; S45: casein kinase II; T75: cyclin dependent kinases 1 and 5; S102: casein kinase II; S137: casein kinase I [71].
The phosphorylation landscape of Dp32 is composed of multiple phosphorylation sites which, in integrative fashion, control its ability to bind and inhibit PP1 (Figure 2). Most work has concentrated on its Threonine 34 (T34) and Threonine 75 (T75) phosphorylation sites. When Dp32 is phosphorylated at T34 (by protein kinase A [PKA]) it binds to and inhibits PP1 and when T75 is phosphorylated (by cyclin dependent kinase 1/5 [CDK1/5]) PKA’s ability to phosphorylate Dp32 at T34 is blocked, resulting in active PP1. Upon dopamine stimulation of neuronal cells through D1, intracellular cAMP is generated, leading to PKA-mediated phosphorylation of Dp32 (T34), culminating in PP1 inhibition. The T34 phosphorylation site lies within the Dp32 domain that binds to PP1. In sum, Dp32 is a control center that modulates PP1 activity in a phosphorylation-dependent manner in response to extracellular small molecules.
Figure 2. Dopamine and glutamate signaling pathways that modulate Darpp-32’s phosphatase 1 inhibitory activity in neurons.

D1, dopamine receptor 1; D2, dopamine receptor 2, NMDAR, N-methyl-D-aspartate receptor; AC, adenylyl cyclase; Gs, stimulatory G protein; GI, inhibitory G protein; PKA, protein kinase A; CaN, calcineurin; Dp32, Darpp-32; PP1, protein phosphatase 1.
Dp32 action was thought to be limited to neurons until 2002, when Wael El-Rifai, working with gastric cancer cells, discovered another PPP1R1B transcript with a start site within intron 1 that codes for tDp, an N-terminal-truncated version of Dp32, that lacks the first 36 residues [10] (see Figure 1). tDp and/or Dp32 transcripts and proteins have since been shown to be overexpressed in breast, colon, esophageal, gastric, lung and prostate cancer tissues [11–13]. tDp is a calcium-binding protein that, upon phosphorylation by CDK1 or CDK5 at T39 (analogous to T75 in Dp32), activates PKA [14, 15]. In addition, tDp forms a complex with insulin-like growth factor 1 receptor (IGF1R), resulting in increased glucose uptake, glucose catabolism, and robust signaling through the AKT anti-apoptosis pathway [16]. In breast cancer cells, it is hypothesized that these and other tDp-mediated activities lead to cell survival in the presence of the Her2-targeting inhibitor, trastuzumab (Herceptin), making tDp a potential therapeutic target in Her2-type cancers with inherent or acquired resistance to trastuzumab.
2.0. Discovery of Darpp-32 and its signaling pathway in neuronal cells
In 1979, Dp32 was first discovered in rat brain tissue by incubating rat brain lysates with cGMP and the radioisotope [γ32P]ATP. In this autoradiography assay, endogenous kinases activated by cGMP in the lysate utilize [γ32P]ATP to phosphorylate proteins, which are then separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and detected on X-ray-sensitive film (D. Aswad, personal communication). When this assay was performed on rat brain lysates the film (autoradiograph) revealed a prominent protein of approximately 32 kDa phosphorylated in the presence of cGMP; this protein, named Dp32, was later discovered to be more effectively phosphorylated when cAMP replaced cGMP in this assay [1]. Work from several laboratories have shown that Dp32 plays a central role in controlling phosphorylation cascades in neurons [17] and it is a commonly used marker for MSNs [18].
As discussed earlier, Dp32 controls phosphorylation pathways by binding to and inactivating PP1—a phosphatase that removes phosphoryl groups from serine and threonine from hundreds of protein substrates [19]. In neurons, dopamine binds to D1, activates a stimulatory G-protein, which turns on membrane-bound adenylyl cyclase (AC) (Figure 2). AC converts ATP to cAMP, which binds to and activates PKA—the kinase that phosphorylates Dp32 at T34. Phosphorylated Dp32 binds to and inhibits PP1. Thus, modulation of internal cAMP levels controls Dp32 activity. For example, dopamine binding to D2 activates inhibitory G-protein, which inactivates AC, lowering cAMP levels. In addition, some D2 inhibitory activity is due to its ability to release Ca2+ from internal stores, thereby activating calcineurin, a phosphatase that removes the phosphoryl group from T34 on Dp32 and lowers Dp32 activity. Dp32 activity can be down-regulated by yet a third neurotransmitter receptor, N-methyl-D-aspartate receptor (NMDAR). Upon binding glutamate, NMDAR releases Ca2+ from internal stores (similar to D2) where Ca2+ binds to and activates calcineurin to downregulate Dp32 activity [20, 21].
3.0. The cancer connection
3.1. Human studies
The discovery of tDp by El-Rifai’s group and its association with gastric cancer was made 20 years after the initial characterization of Dp32 [10]. The El-Rifai group showed that in primary gastric cancers, xenografts, and cell-lines there is an increased copy number of several genes within the q-arm of chromosome 17 [22]. cDNAs created from the xenografts were used to probe a cDNA microarray built from genes expressed from chromosome 17. One of the highest expressed transcripts from the xenografts hybridized to the 3’ untranslated region of PPP1R1B in the microarray [10]. Careful mapping of the 5’ ends of the transcripts in the xenografts showed that two PPP1R1B transcripts were expressed in gastric cancers, Dp32 and tDp. Analysis of a combination of 50 samples of primary gastric cancers and xenografts showed that more than 50% expressed 20-fold higher levels of PPP1R1B transcripts relative to normal tissue.
Since this discovery, multiple research groups have worked to elucidate the role of tDp and Dp32 in cancer. Increased levels of tDp/Dp32 transcripts in cancer tissue relative to normal tissue have been reported in several cancers including those originating in breast, colon, esophagus, lung, and prostate [11, 12, 16, 23–25]. The 17q12 locus encompassing the PPP1R1B gene has been observed to be frequently amplified in breast and gastric cancers, suggesting that this region contributes to oncogenesis [10, 26–28]. However, in gastric cancers there are frequent instances where PPP1R1B transcript levels are elevated without gene amplification [22]. In breast cancers an analysis of the COSMIC database [29] indicates that the tDp/Dp32 transcripts are overexpressed at 9% frequency (n = 1104) compared to normal breast tissue. This is significant, indicating that overexpression is likely to be associated with breast cancer. On the other hand, the frequency of point mutations in PPP1R1B is only 0.08% (n = 2620) and these mutations are widely dispersed throughout the gene, indicating they are likely to be passenger mutations and not driver mutations, produced by random mutations in the genome during cancer proliferation. This strongly suggests that, unlike KRAS, point mutation-mediated activation of tDp/Dp32 is not likely. Instead, it is the increased level of tDp/Dp32 transcripts in cancer cells that is critical for oncogenesis. Indeed, increased transcripts relative to normal tissue leads to a worse outcome in breast cancer patients [16]. In a study of 513 lung adenocarcinoma patients it was shown that an increased ratio of tDp transcripts to Dp32 transcripts correlates with a poorer outcome (hazard ratio 1.66) and that expression of tDp correlates with later stages of the tumors [12]. Immunohistochemistry analysis confirm the trend of high expression in cancers relative to normal tissue. In breast cancers tDp/Dp32 protein is expressed in 50% of the samples (n = 230) and high expression in the cytoplasm correlates with a hazard ratio of 4.14 [24].
3.2. Transgenic mouse studies
Mouse models of cancer are often used to gain insight into the function of oncogenes. The PPP1R1B-knockout mice were created by targeted disruption of the Darpp-32 translational start site in exon 1 [30] which resulted in undetectable levels of Dp32 and tDp proteins in mouse tissues (brain and mammary tissues were examined) [31]. PPP1R1B-knockout mice show impairment in reversal learning [32], but no reported changes in spontaneous tumorigenesis. To test for PPP1R1B contributions to tumorigenesis a mouse that develops mammary cell tumors, MMTV-PyMT (mouse mammary tumor virus-polyomavirus middle T antigen model), was crossed with PPP1R1B knockout mice. In MMTV-PyMT mice, the long terminal repeat of the mammary tumor virus is engineered to drive the polyomavirus middle T-antigen in mammary cells located in fat pads of female mice resulting in mammary tumors. When this oncogenic mouse is crossed with PPP1R1B−/− mice the age of tumor appearance is not affected relative to MMTV-PyMT controls suggesting that PPP1R1B does not contribute to early tumor onset [31]. However, the net rate of tumor growth in MMTV-PyMT/PPP1R1B+/+ mice is significantly higher compared to MMTV-PyMT/PPP1R1B−/− mice. It should be noted that tumors formed from the control mice (MMTV-PyMT/PPP1R1B+/+) frequently overexpress tDp/Dp32 transcripts in tumor tissue. Thus, PPP1R1B appears to increase tumor growth at some point after initiation.
3.3. Xenograft studies
Another useful tool to aid in the elucidation of oncogenic mechanisms is a xenograft of human tumors or cell-lines implanted in immunocompromised mice. Gastric cancer cells engineered with a tetracycline-regulated system to turn on Dp32 expression were injected subcutaneously into mice flanks and showed high growth rate compared to controls (no tetracycline treatment) [33]. Similarly, lung adenocarcinoma cells genetically engineered to express tDp or Dp32 injected into the thorax of mice showed increased growth rates compared to cells with vector control [12]. In confirmation, lung cancer cells expressing shRNAs directed against endogenous tDp/Dp32 transcripts showed decreased cancer growth compared to cells expressing shRNAs directed against a non-expressed gene. In general, human, mouse and xenograft studies suggest that tDp/Dp32 plays a role in enhancing tumor growth rather than cancer initiation.
4.0. Cellular characteristics and signaling pathways affected by t-Darpp and Darpp-32.
Although there are some reported exceptions [34], both tDp and Dp32 appear to activate cell proliferation pathways, indicating that the cancer-associated domain resides within the subregion of Dp32 that encompasses tDp. We will review the evidence for the role of tDp/Dp32 in several pathways starting with those that have been corroborated by independent research groups (Table I). Note that several pathways lead to AKT activation, which is a focal point in tDp/Dp32-mediated oncogenesis.
Table 1.
Signaling pathways modulated by tDp/Dp32 in oncogenesis-associated activities
| tDp/Dp32 binding partner | Signaling pathways affected | Cell phenotype | Cell type | References |
|---|---|---|---|---|
| ------- | AKT↑* | Resistance to trastuzumab*, tumor growth* | Her2+ breast cancer, gastric cancer | [34, 35, 37, 47] |
| ------- | ------- | Cell migration* | Non-small cell lung cancer; gastric cancer | [12,43] |
| Her2 | AKT↑ | Resistance to trastuzumab | Her2+ cancer | [37] |
| Rlα within PKA | PKA↑ | Resistance to trastuzumab | Her2+ breast cancer | [15] |
| IGF1R | AKT↑, ErbB↑ | High glucose metabolism | Her2+ breast cancer | [16] |
| IKKα | NF-kB2↑ | Cell migration | Non-small cell lung cancer | [12] |
| Unknown | Bcl-2↑ | Anti-apoptosis | Her2+ breast cancer | [35] |
| Unknown | ANGPT2↑, STAT3↑ | Angiogenesis, tumor growth | Gastric cancer | [33] |
| Unknown | Increased interaction between Her1 and Her3; AKT↑ | Resistance to gefitinib (inhibitor of Her1) | Gastric cancer | [46] |
| Unknown | Increased stability of Her1 | Sensitivity to gefitinib, AG1478, and erlotinib (inhibitors of Her1) | Breast cancer | [67] |
| CXC4 | CXC4 increased stability; MT1-MMP/MMP-2 activation | Cell migration | Gastric cancer | [43] |
| SRp20 | CD44E↑ | mRNA splicing | Gastric cancer | [68] |
| Bcl-2 | lnsP3R/Ca2+ | Anti-apoptosis | Leukemia | [69] |
| Unknown | P-catenin↑; Wnt genes↑ | Proliferation | Gastric cancer; esophageal cancer | [70] |
Corroborated by independent groups.
4.1. The AKT pathway.
The central event affected by tDp/Dp32 is AKT activation through phosphorylation. Phosphorylation of AKT at Serine 473 (S473) leads to extended survival of cancer cells. Three research groups have reported that AKT is phosphorylated at S473 when tDp is overexpressed in cancer cells or that AKT is hypophosphorylated when tDp is knocked down [12, 16, 34–36]. In addition, two groups have shown that tDp confers cancer resistance to trastuzumab [34, 35, 37], a drug that binds to Her2 receptor and normally inhibits AKT phosphorylation. Together, these studies suggest that at least part of tDp’s contribution to tumorigenesis and drug resistance phenotype is due to activation of the AKT pathway. Thus, there is some consensus on AKT phosphorylation as an outcome of tDp overexpression. Due to AKT’s central importance in tDp activity and cancer cell survival we will review the AKT signaling pathway from the perspective of its activation by Her2 (Figure 3).
Figure 3. Her2 signaling pathway in breast cancers.

Her2 forms a heterodimer with Her3 (bound to its natural ligand, either heregulin or NRG-2). Productive dimerization causes transphosphorylation of cytoplasmic domains. PI3K binds the cytoplasmic domains and produces phosphatidylinositol (3,4,5)-trisphosphate (PIP3), a small molecule that activates 3-phosphoinositide-dependent protein kinase 1 (PDPK1). PDPK1 phosphorylates AKT at Serine 473, leading to apoptotic evasion and cell survival. t-Darpp and Darpp-32 activates AKT through a mechanism that is not yet clear, likely through PI3K signal strengthening or through another pathway that causes S473 phosphorylation on AKT. Another pathway activated by Her2 is the KRAS pathway. Here GRB2 adaptor protein activates SOS guanine nucleotide exchange protein to replace GDP with GTP on KRAS. KRAS instigates a cascade of phosphorylation reactions starting with RAF and ending in ERK, which binds to transcription factors and activates proliferation genes.
Her2 (ErbB2, neu) forms a dimer with either Her1 (EGFR, ErbB1), Her3 (ErbB3), or Her4 (ErbB4), with Her3 being its common partner [38]. Two signaling pathways emanate from the trans-phosphorylated cytoplasmic domains of Her2 and its receptor binding partner [39]. In one pathway, upon binding to the receptors’ cytoplasmic domains, Grb2 adaptor protein activates SOS to exchange GTP for GDP within Ras. GTP-bound Ras activates a tandem series of Ser/Thr kinases named Raf, Mek and Erk, the last of which activates transcription factors that express proliferation genes.
In the second signaling pathway, which involves AKT, trans-phosphorylation of the cytoplasmic domain of Her2 and its receptor binding partner promotes their binding to phosphoinositide-3 kinase (PI3K) and its activation [39]. Active PI3K phosphorylates phosphatidylinositol (4,5)-bisphosphate (PIP2) and produces phosphatidylinositol 3,4,5-trisphosphate (PIP3). PIP3 associates with AKT to target it to the plasma membrane. PIP3 also binds to 3-phosphoinositide-dependent protein kinase 1 (PDPK1, also known as Protein Kinase B). This binding localizes PDPK1 to the plasma membrane and activates it so that it phosphorylates membrane-bound AKT at S473. Phospho-AKT has several downstream effectors including activation of mTOR (for cell survival) and inhibition of Bad (leading to prevention of apoptosis). Because tDp/Dp32 leads to sustained phosphorylation of AKT in the presence of the Her2 inhibitor trastuzumab [34], it is thought that breast cancer cells are able to survive in the presence of trastuzumab. It bears mentioning that PI3K (upstream of AKT) frequently sustains activating mutations in cancers [38, 40] that lead to phosphorylation of AKT in trastuzumab-resistant cancers; thus it is consistent that tDp-mediated activation of AKT would also lead to resistance. There are other reported tDp-mediated signaling pathways that may or may not be connected to AKT phosphorylation or trastuzumab resistance which we summarize below (see Table I).
4.2. The PKA, IGF1R, and Bcl-2 pathways.
In breast cancer cells tDp activates PKA, a holoenzyme composed of two catalytic subunits and two regulatory subunits [15]. Normally, cAMP binds to the regulatory subunits (RI or RII) and releases the two catalytic subunits that, upon release, phosphorylate several protein substrates. In cells that overexpress tDp, tDp forms a complex with the regulatory subunit RIα and releases active catalytic subunits resulting in activation of the CREB transcription factor [15, 34]. In a second line of inquiry, tDp forms a complex with insulin like growth factor 1 receptor (IGF1R) in breast cancer cells, resulting in increased glucose metabolism (glycolysis) and AKT phosphorylation [16]. Required for increased glycolysis is IGF1Rbinding to members of the ErbB receptor family. In a third line of inquiry, tDp has been shown to increase the level of the anti-apoptosis protein Bcl-2 [35]. In this scenario, tDp leads to increased phosphorylation of AKT, which results in higher Bcl-2 protein levels and increased cancer cell survival in the presence of trastuzumab. Two transcription factors, CREB and ATF1 are required for Bcl-2 protein upregulation.
4.3. The NF-κB2, NF-κB, ANGPT2, CXC4, Her1, and Her2 pathways.
In lung cancer cells tDp increases cell migration (see section 8.0), leading to the suggestion that cell metastasis may be mediated in part by tDp. tDp binds to and activates IκB kinase-α in an NF-κB–inducing kinase-independent manner, resulting in NF-κB2 nuclear translocation to initiate transcription of cell migratory genes [12].
Much work on the role of tDp/Dp32 in gastric cancers has been reported. The cancer-causing bacterium Helicobacter pylori (H. pylori) infects the gastric mucosa, causing chronic inflammation [41]. Cells that survive inflammation sustain mutations that can form gastric cancers. Infection of gastric cancer cells causes NF-κB, commonly activated upon inflammation, to upregulate Dp32 transcription by binding to an NF-κB DNA element upstream of the Dp32 transcriptional start site [42]. In a separate gastric cancer cell study, tDp and Dp32 were shown to increase the angiogenesis-inducing protein angiopoietin 2 (ANGPT2 [33]. ANGPT2 secreted from these gastric cancer cells increased angiogenesis in an endothelial tube formation assay. ANGPT2 level induction was mediated by STAT3 transcription factor and was independent of AKT and NF-κB activation. In a study designed to determine the role of Dp32 in gastric cancer cell invasion it was found that it was critical for Dp32 to form a complex with CXC4 transcription factor, which increases CXC4’s stability [43]. Once activated, CXC4 binds to its responsive element within the matrix metalloproteinase-2 gene thereby upregulating this pathway. In gastric cancer cells Dp32 confers resistance to the Her1-targeting drug gefitinib (Iressa) by blocking drug-induced apoptosis [36]. Dp32 overexpression increases Her1 stability and promotes Her1 dimerization with Her3 resulting in downstream AKT phosphorylation.
In esophageal adenocarcinoma cells tDp has been shown to bind Her2 [37]. Further, expression of tDp leads to increased Her2 half-life and sustained Her2 phosphorylation at Tyrosine 1248 (Y1248) despite the presence of trastuzumab, which normally decreases Her2 phosphorylation.
5.0. Dp32 and tDp structure
Given the clinical importance of tDp/Dp32 in cancers and the role it plays in AKT activation and drug resistance this protein may be drug target candidate. To develop drugs that inhibit tDp/Dp32 activities that lead to poor patient outcomes, it is essential to understand its structure. There are no known tertiary structures reported for tDp/Dp32 presumably due to the presence of a run of glutamates and aspartates in the middle of the protein that constitute the acidic domain (Figure 1). Fluorescence spectroscopy experiments performed on Dp32 suggest that phosphorylation of T34 by PKA or Serine 45 and Serine 102 by casein kinase II (CK2) does not significantly alter Dp32 conformation [44]. It was further determined that Dp32 has at least two distinct structural domains: a highly flexible region near Tryptophan 163 and a more rigid region at Cysteine 72. NMR studies of Dp32 show a propensity for its N-terminal segment (residues 22–29) to form an alpha helix, and that this helix is not significantly altered by phosphorylation at T34 (Figure 1) [45]. Momand’s group has shown that tDp is an elongated monomer (i.e. does not significantly self-oligomerize) that exhibits 12% alpha helix, 29% beta strand, 24% beta turn, and 35% random coil structures [14]. These structures are stable when the protein is briefly heated to 50°C. tDp selectively binds to calcium, likely through its many aspartate and glutamate residues [14]. This run of acidic residues resides within a larger motif that has limited similarity to bone sialoprotein II, a secreted protein in bone that also binds calcium.
6.0. Dp32 fusion proteins
Chimeric transcripts are single mRNAs that arise from two or more genes localized in tandem in the genome. A chimeric transcript that contains PPP1R1B and its immediate 3’ gene on chromosome 17 STARD3 was first identified by the Chinnaiyan research group in breast cancers [46]. The Kim research group further characterized the chimeric transcript as one where exon 7 of PPP1R1B and exon 1 of STARD3 are absent [47]. The transcript translates into a 245 amino acid chimeric protein that lacks the C-terminal 16 amino acids of Dp32 and gains 57 outof-frame amino acids from exon 2 of STARD3. The chimeric transcript is expressed in 21% of primary gastric cancers but not in normal gastric tissues.
Overexpression of the Dp32-STARD3 chimeric protein increased AKT phosphorylation, consistent with the signaling pathway activated by tDp and Dp32 [47]. The chimeric protein also increases tumorigenicity of gastric cancer cells in immunocompromised mice. Importantly, it was demonstrated that inhibitors of PI3K lowered the level of AKT phosphorylation indicating that Dp32 and Dp32-STARD3 fusion protein likely activate PI3K. From this work, we speculate that it is possible that increased Dp32 level may occur through the formation of a stable Dp32-STARD3 fusion protein that is relatively resistant to proteolysis. Experimental validation of this hypothesis is necessary.
7.0. tDp/Dp32 inconsistencies
While the majority of studies show that Dp32 and tDp share the same functions there are a few studies that show that they are not redundant in all activities. One showed that tDp, but not Dp32, confers trastuzumab resistance in Her2+ breast cancer [34]. tDp causes AKT phosphorylation [34] and activation of both PKA [15] and the PKA effector, CREB [34]. However, Dp32 reversed the effect of tDp on all of these activities [15, 34]. In contrast, another research group showed that tDp and Dp32 each confer trastuzumab resistance and AKT phosphorylation in breast cancer cells [24]. In addition, Dp32 was shown to promote gastric cancer cell resistance to another drug doxorubicin (Adriamycin) [48]. Cell context may play a critical role in modulating tDp and Dp32 downstream activities. To investigate the possibility that tDp and Dp32 may alter each other’s activities through heterooligomerization crosslinking experiments were conducted [14]. However, no significant heterooligomerization was detected.
8.0. Cell migration and metastasis
Analysis of tDp/Dp32 in the roles they play in cell migration and metastasis also shows some results that may be explained by cell background differences. Using a wound healing assay to measure cell migration rates in breast cancer cells, one group reported that Dp32 inhibits cell migration when phosphorylated at T34 [49]. Phosphorylation depended on Wnt-5a activation, which triggers increased cAMP levels and PKA-dependent phosphorylation at T34. Dp32-mediated anti-migratory behavior required CREB phosphorylation (triggered by PP1 inactivation or by PKA activation) and Dp32 association with the receptor tyrosine kinase DDR1 [50]. A dominant negative mutant CREB [49] or experimental reduction of DDR1 levels [50] inhibited Dp32-mediated anti-migratory activity.
Interestingly, exploration of Dp32’s role in cell migration activity in lung cancer cells led to a different outcome [12]. Using non-small cell lung adenocarcinoma cells it was shown that Dp32 promoted cell migration. Phosphorylation at T34 is not involved because an Threonine to Alanine substitution at position 34 (T34A mutant) showed no change in promoting cell migration in comparison to wild-type Dp32. In fact, the N-terminal domain of Dp32 does not appear to be involved in lung cancer cell migration because tDp (lacks first 36 amino acids) also promotes cell migration. In this scenario, tDp/Dp32 appears to activate the inhibitor of nuclear factor kappa-B kinase subunit alpha (IKKα) leading to the nuclear translocation of NF-kB2 and subsequent expression of NF-κB2-controlled migratory genes.
Another group, using Dp32, analyzed another cell movement phenotype: cell invasion [43]. In this study, cell invasion assays were used in a series of experiments to demonstrate that Dp32 overexpression significantly increased gastric cancer cell invasion. Dp32 overexpression led to increased levels of membrane-type 1 matrix metalloproteinase (MT-MMP) and CXC-chemokine receptor 4 (CXCR4). Importantly, knockdown of CXCR4 reduced Dp32 effects on cell migration. Evidence suggests that Dp32 enters into a complex with CXCR4 and stabilizes it, which is required for activation of the MT-MMP pathway.
Moving beyond in vitro cell movement studies, clinical evidence also suggests an association of tDp/Dp32 with distant metastasis. Using clinical and histopathological data of 100 patients with colorectal cancer it was demonstrated that tDp/Dp32 was highly expressed in patients with distant metastases and that tDp/Dp32 is a significant predictor of distant metastases and poor prognosis [51].
9.0. Mechanisms of Dp32 upregulation
Up to now we have shown that the PPP1R1B gene has several roles in both neurotransmission and cancer. To date, studies on PPP1R1B gene transcriptional regulation have exclusively focused on the full-length isoform, Dp32, which we review below. While several molecules and signaling pathways have been discovered in transcription control of Dp32, a significant gap in the literature remains regarding transcriptional regulation of tDp.
9.1. Brain-derived neurotrophic factor (BDNF) in medium size spiny neurons
One protein that controls PPP1R1B transcription in neurons is brain-derived neurotrophic factor (BDNF), which is involved in cell maturation and survival. In BDNF null mice the expression of Dp32 was both delayed and lowered compared to wild-type controls [52]. Addition of BDNF to cultured neurons from the null mice rescued this effect, increasing Dp32-immunopositive response. However, as knockout of BDNF did not fully abolish Dp32 expression, it is apparent that other factors can upregulate PPP1R1B. An analysis of the conserved sequences within the PPP1R1B gene between mouse, rat, human, orangutan, dog, and horse yielded potential regulatory sites. Highly conserved sequences in intron 4 correspond to a possible EGR-1 site [53]. Affinity column purification, mass spectrometry, and an electrophoretic mobility shift assay (EMSA) confirmed the presence EGR-1 in complexes with intron 4 in vitro, while chromatin immunoprecipitation (ChIP) assays in mice confirmed EGR-1 binding in vivo [53].
Overexpression of EGR-1 in primary striatal neurons from mice was sufficient to increase Dp32 levels whereas the dominant negative form of EGR-1 significantly reduced BDNF-mediated induction [53]. Treatment with BDNF increases EGR-1 expression, EGR-1 binding to the PPP1R1B intron 4, and Dp32 expression. BDNF also induces the EGR-1 co-activator NAB2, which is found in complex with EGR-1 [54]. However, as EGR-1 (also known as Zif268) null mice maintain Dp32 levels in the striatum there must be redundant transcription factors that can substitute for EGR-1 [55].
Aside from controlling EGR-1-mediated transcription of PPP1R1B, BDNF also induced phosphorylation of AKT and ERK in mouse medium-sized spiny neurons (MSNs) [56]. Activation of these signaling proteins may result from BDNF binding to tropomyosin receptor kinase B (TrkB), which is upstream of the PI3K/AKT and MEK/ERK pathways. To determine if these phosphorylated signaling proteins play a role in Dp32 expression, selective inhibitors of their upstream activating kinases PI3K (upstream to AKT) and MEK (upstream to ERK) were employed. The PI3K inhibitor LY294002, but not the MEK inhibitor UO126, was shown to significantly reduce Dp32 levels. Use of both inhibitors in combination relative to the PI3K inhibitor alone did not show a difference in Dp32 levels suggesting that BDNF can increase Dp32 levels through PI3K/AKT but not MEK/ERK.
Further studies exploring the involvement of the PI3K/AKT pathway in upregulating Dp32 protein levels showed that the constitutively active form of the kinase that activates AKT (catalytic domain, caPI3K), and AKT itself were sufficient to increase Dp32 protein levels in mouse MSNs [57]. Forced expression of a dominant negative mutant of AKT reduced BDNF-induced Dp32 expression, but not below baseline. Expression of a dominant negative mutant of the regulatory PI3K subunit p85 reduced both BDNF-induced and baseline levels of Dp32, suggesting that PI3K is both sufficient and necessary for Dp32 protein level maintenance in MSNs. An increased level of the CDK5 activator, p35, was also noted with both BDNF treatment and caPI3K expression. Use of CDK5 inhibitors as well as expression of the dominant negative mutant of CDK5 diminished BDNF-induced increase of Dp32, however it did not alter baseline levels or have any effect on AKT activation. Thus, it appears that BDNF increases Dp32 expression in MSNs by three mechanisms: 1) EGR-1-mediated transcriptional upregulation of PPP1R1B, 2) Increased phosphorylation of AKT, 3) Increased CDK5 activity. It is not clear how the latter two activities afford increased Dp32 protein levels.
In an attempt to reconcile the data from MSNs showing that AKT is upstream of tDp/Dp32 with the data from cancer cells showing that AKT is downstream of tDp/Dp32 we can speculate that in neurons there is a positive feedback loop of AKT activation. In such a scenario tDp/Dp32 activates AKT and activated AKT, through CDK5, promotes phosphorylation of tDp/Dp32 at T39/T75. The phosphorylated tDp/Dp32, in turn, activates PI3K (the activator of AKT). (There is evidence from CDK5−/− mice that CDK5 is necessary for full activation of AKT in neurons [58], but the mechanism is unclear).
9.2. Retinoic acid (RA) in MSNs
Retinoic acid (RA) has previously been shown to selectively induce Dp32 expression in neurons. In rat lateral ganglionic eminence neurons, treatment with 9-cis RA induced significant increase of Dp32, but not in cortical and medial ganglionic eminence neurons [59]. It was noted that striatal neurons, unlike cortical neurons, uniquely express retinoic acid receptors (RARs) β and γ. Ectopic expression of RARβ1, but not RARγ1, in cortical neurons was sufficient to induce Dp32 suggesting that striatal neurons can regulate Dp32 levels through RARβ1 [60]. It was proposed that Dp32 upregulation in RARβ1 striatal neurons could be through putative DR2 and DR5 retinoic acid response elements (RAREs) located within the 5’ flanking region and intron 1 of the mouse PPP1R1B gene, although this was not experimentally tested. Another group found that RA may increase Dp32 protein levels in striatal neurons through PI3K as treatment with PI3K inhibitor completely abolished RA-induced Dp32 protein level increase [61]. Studies indicate that protein kinase C (PKC) levels correlate with RARβ [62] and that PKC lies downstream of PI3K [61]. PKC isoform-selective inhibitors were used to determine their potential roles in RA-induced Dp32 [61]. It was discovered that inhibition of PKCζ reduced RA-induced Dp32 protein levels by 50%, suggesting a partial role for PKCζ in Dp32 level increase. Interestingly, RA treatment did not result in AKT nor ERK phosphorylation. Furthermore, expression of the dominant negative form of AKT1 or treatment with CDK5 inhibitor failed to prevent RA-induced Dp32 expression. It was concluded that RA does not act through activation of AKT, ERK, or CDK5 in striatal neurons [61]. Taken together, these results suggest that in RARβ-positive striatal neurons RA treatment increases Dp32 through PKC while in RARβ-negative striatal neurons BDNF increases of Dp32 through non-PKC pathways.
9.3. 17β-Estradiol (E2) in MSNs
Another pathway that leads to Dp32 increase appears to proceed through an estrogen receptor. Treatment of mouse medium-sized spiny neurons (MSNs) with the estrogen receptor ligand 17β-estradiol (E2) and the estrogen receptor partial agonist tamoxifen, either alone or in combination, were found to increase Dp32 levels [63]. The estrogen receptor antagonist ICI-182,780 mimicked E2 in inducing Dp32 levels, but when E2 was combined with antagonist Dp32 level did not increase. When isolated neuron cultures derived from female or male embryos were compared it was discovered that female neurons were more responsive to E2 treatment. Surprisingly, only about one-third of E2 matured MSN cultures were Dp32-positive, in contrast to nearly all adult striatal MSNs. The number of MSNs that express the estrogen receptor ß (ERß) in cultures appeared to outnumber the relative number of E2-induced Dp32-positive cells, suggesting that additional regulators of Dp32 are involved. Furthermore, no estrogen response element has been identified in the PPP1R1B gene suggesting that E2 operates on Dp32 indirectly.
9.4. NF-κB in gastric cancer
As previously described Helicobacter pylori (H. pylori) infection is a contributing factor to gastric cancer [64]. H. pylori induces Dp32 transcript which correlates with cell survival and with transcriptional upregulation of RELA (NF-κB-P65, a subunit of NF- κB) and tumor necrosis factor-α (TNF-α), a known NF-κB activator [42]. Treatment of gastric cancer cells with TNF-α significantly increased Dp32 mRNA and protein levels, and treatment of Bay 11–7082 (NF-κB inhibitor) significantly lowered Dp32. Transcriptional activation reporter assays were conducted to map the element responsible for NF-κB responsiveness. The element was located −1008 to −996 upstream of the PPP1R1B transcriptional start site. TNF-α treatment increased transcription when this element was present. A chromatin immunoprecipitation (ChIP) assay using antibody against RELA confirmed binding of the transcription factor to −1008 to −996 in PPP1R1B.
10.0. Future Directions
In this Commentary we have reviewed the roles of tDp/Dp32 in neuronal cells and non-brain cancers. Due to the fact that the PPP1R1B gene is overexpressed at the transcript and protein level in a significant proportion of gastric and breast cancers and its documented involvement in tumorigenesis from xenograft studies and human clinical data analysis its protein products tDp and Dp32 should be considered viable targets for drug development. The best candidate is tDp; tDp is a subsequence of Dp32, so that a drug developed against tDp will likely target Dp32 as well. There are several significant hurdles to anti-tDp drug design. First and foremost, the tertiary structure of tDp is not known. The protein exhibits mostly non-alpha and non-beta sheet secondary structure and few structural insights have been illuminated (reviewed here) likely due to its inability to be crystalized owing to its acidic central domain. Thus, it is probable that high resolution structure analysis will require co-crystallization of tDp with a direct binding partner. However, with advances in single-particle cryo-electron microscopy techniques it may be possible to capture structure information for small proteins such as tDp [65]. A second significant hurdle is the lack of knowledge of proteins that directly bind to tDp. As shown in Table I, there are several proteins that have been demonstrated to form a complex with tDp. However, the methods used to demonstrate complex formation (co-immunoprecipitation and, rarely, proximity ligation assay in cultured mammalian cells) do not preclude the possibility that there may be factors that bridge tDp and the complexing proteins. It is likely that the phosphorylation landscape of tDp influences the composition of these bridging factors, further complicating this area of inquiry.
A third hurdle to drug development is knowledge of precisely how tDp activates AKT. There is strong consensus that tDp activates AKT phosphorylation in cancers. Presumably, knowledge of tDp’s direct binding proteins will point the way toward greater understanding of how tDp mediates AKT phosphorylation on S473; but, given the varied number of pathways that tDp modulates, it would be optimal if the tDp/Dp32/AKT signaling pathway could be reassembled in an in vitro system. Finally, aside from other expected challenges that come with drug design [66], there is the issue of the side effects of tDp/Dp32 inhibition. Since Dp32’s major function is in brain neurons, an anti-cancer tDp-targeting drug that does not cross the blood-brain barrier would likely be optimal.
All of these considerations must be taken into account to develop effective tDp-based anti-cancer therapies designed to extend patient life. What cancers should be targeted? In the future, an anti-tDp drug may prove useful in gastric cancers, a cancer where there is reported to be a high frequency of PPP1R1B overexpression. Another cancer that may stand to benefit from anti-tDp therapy is breast cancer, where overexpression has been shown to be a common occurrence. tDp also mediates trastuzumab-resistance, a persistent problem with most women diagnosed with Her2-type breast cancer. It would be relatively straightforward to test for immunopositivity of tDp at the same time when testing for Her2 immunopositivity on breast cancer biopsies. Patients that test positive for both could be administered anti-Her2 therapy (trastuzumab) plus an anti-tDp drug to achieve greater therapeutic benefit. Given the rapid pace of novel cancer treatments, such a scenario may become a reality in the not-too-distant future.
Acknowledgements
We thank the following for financial support: 1RO1GM105898 (G.L. and J.M.), GM61331 (A.A.). We gratefully acknowledge Dr. Susan Kane for spearheading efforts into the investigation of mechanisms of trastuzumab resistance in breast cancers.
References
- [1].Walaas SI, Aswad DW, Greengard P, A dopamine- and cyclic AMP-regulated phosphoprotein enriched in dopamine-innervated brain regions, Nature 301(5895) (1983) 69–71. [DOI] [PubMed] [Google Scholar]
- [2].Hemmings HC Jr., Greengard P, Tung HY, Cohen P, DARPP-32, a dopamine-regulated neuronal phosphoprotein, is a potent inhibitor of protein phosphatase-1, Nature 310(5977) (1984) 503–5. [DOI] [PubMed] [Google Scholar]
- [3].Bibb JA, Snyder GL, Nishi A, Yan Z, Meijer L, Fienberg AA, Tsai LH, Kwon YT, Girault JA, Czernik AJ, Huganir RL, Hemmings HC Jr., Nairn AC, Greengard P, Phosphorylation of DARPP-32 by Cdk5 modulates dopamine signalling in neurons, Nature 402(6762) (1999) 669–71. [DOI] [PubMed] [Google Scholar]
- [4].Straccia M, Carrere J, Rosser AE, Canals JM, Human t-DARPP is induced during striatal development, Neuroscience 333 (2016) 320–30. [DOI] [PubMed] [Google Scholar]
- [5].Berger B, Febvret A, Greengard P, Goldman-Rakic PS, DARPP-32, a phosphoprotein enriched in dopaminoceptive neurons bearing dopamine D1 receptors: distribution in the cerebral cortex of the newborn and adult rhesus monkey, J Comp Neurol 299(3) (1990) 327–48. [DOI] [PubMed] [Google Scholar]
- [6].Brene S, Lindefors N, Ehrlich M, Taubes T, Horiuchi A, Kopp J, Hall H, Sedvall G, Greengard P, Persson H, Expression of mRNAs encoding ARPP-16/19, ARPP-21, and DARPP-32 in human brain tissue, J Neurosci 14(3 Pt 1) (1994) 985–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Brene S, Hall H, Lindefors N, Karlsson P, Halldin C, Sedvall G, Distribution of messenger RNAs for D1 dopamine receptors and DARPP-32 in striatum and cerebral cortex of the cynomolgus monkey: relationship to D1 dopamine receptors, Neuroscience 67(1) (1995) 37–48. [DOI] [PubMed] [Google Scholar]
- [8].Svenningsson P, Nishi A, Fisone G, Girault JA, Nairn AC, Greengard P, DARPP-32: an integrator of neurotransmission, Annu Rev Pharmacol Toxicol 44 (2004) 269–96. [DOI] [PubMed] [Google Scholar]
- [9].Nairn AC, Svenningsson P, Nishi A, Fisone G, Girault JA, Greengard P, The role of DARPP-32 in the actions of drugs of abuse, Neuropharmacology 47 Suppl 1 (2004) 14–23. [DOI] [PubMed] [Google Scholar]
- [10].El-Rifai W, Smith MF Jr., Li G, Beckler A, Carl VS, Montgomery E, Knuutila S, Moskaluk CA, Frierson HF Jr., Powell SM, Gastric cancers overexpress DARPP-32 and a novel isoform, t-DARPP, Cancer Res 62(14) (2002) 4061–4. [PubMed] [Google Scholar]
- [11].Ebihara Y, Miyamoto M, Fukunaga A, Kato K, Shichinohe T, Kawarada Y, Kurokawa T, Cho Y, Murakami S, Uehara H, Kaneko H, Hashimoto H, Murakami Y, Itoh T, Okushiba S, Kondo S, Katoh H, DARPP-32 expression arises after a phase of dysplasia in oesophageal squamous cell carcinoma, Br J Cancer 91(1) (2004) 119–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Alam SK, Astone M, Liu P, Hall SR, Coyle AM, Dankert EN, Hoffman DK, Zhang W, Kuang R, Roden AC, Mansfield AS, Hoeppner LH, DARPP-32 and t-DARPP promote non-small cell lung cancer growth through regulation of IKKα-dependent cell migration, Communications Biology 1(1) (2018) 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Belkhiri A, Zhu S, El-Rifai W, DARPP-32: from neurotransmission to cancer, Oncotarget 7(14) (2016) 17631–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Momand J, Magdziarz P, Feng Y, Jiang D, Parga E, Celis A, Denny E, Wang X, Phillips ML, Monterroso E, Kane SE, Zhou F, t-Darpp is an elongated monomer that binds calcium and is phosphorylated by cyclin-dependent kinases 1 and 5, FEBS Open Bio 7(9) (2017) 1328–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Theile D, Geng S, Denny EC, Momand J, Kane SE, t-Darpp stimulates protein kinase A activity by forming a complex with its RI regulatory subunit, Cell Signal 40 (2017) 53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lenz G, Hamilton A, Geng S, Hong T, Kalkum M, Momand J, Kane SE, Huss J, t-Darpp activates IGF-1R signaling to regulate glucose metabolism in trastuzumab-resistant breast cancer cells, Clinical Cancer Research (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wang H, Farhan M, Xu J, Lazarovici P, Zheng W, The involvement of DARPP-32 in the pathophysiology of schizophrenia, Oncotarget 8(32) (2017) 53791–53803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ivkovic S, Ehrlich ME, Expression of the striatal DARPP-32/ARPP-21 phenotype in GABAergic neurons requires neurotrophins in vivo and in vitro, J Neurosci 19(13) (1999) 5409–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ceulemans H, Bollen M, Functional diversity of protein phosphatase-1, a cellular economizer and reset button, Physiol Rev 84(1) (2004) 1–39. [DOI] [PubMed] [Google Scholar]
- [20].Halpain S, Girault JA, Greengard P, Activation of NMDA receptors induces dephosphorylation of DARPP-32 in rat striatal slices, Nature 343(6256) (1990) 369–72. [DOI] [PubMed] [Google Scholar]
- [21].Desdouits F, Siciliano JC, Greengard P, Girault JA, Dopamine- and cAMP-regulated phosphoprotein DARPP-32: phosphorylation of Ser-137 by casein kinase I inhibits dephosphorylation of Thr-34 by calcineurin, Proc Natl Acad Sci U S A 92(7) (1995) 2682–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Varis A, Wolf M, Monni O, Vakkari ML, Kokkola A, Moskaluk C, Frierson H Jr., Powell SM, Knuutila S, Kallioniemi A, El-Rifai W, Targets of gene amplification and overexpression at 17q in gastric cancer, Cancer Res 62(9) (2002) 2625–9. [PubMed] [Google Scholar]
- [23].Beckler A, Moskaluk CA, Zaika A, Hampton GM, Powell SM, Frierson HF Jr., El-Rifai W, Overexpression of the 32-kilodalton dopamine and cyclic adenosine 3’,5’-monophosphate-regulated phosphoprotein in common adenocarcinomas, Cancer 98(7) (2003) 1547–51. [DOI] [PubMed] [Google Scholar]
- [24].Hamel S, Bouchard A, Ferrario C, Hassan S, Aguilar-Mahecha A, Buchanan M, Quenneville L, Miller W, Basik M, Both t-Darpp and DARPP-32 can cause resistance to trastuzumab in breast cancer cells and are frequently expressed in primary breast cancers, Breast cancer research and treatment 120(1) (2010) 47–57. [DOI] [PubMed] [Google Scholar]
- [25].Vangamudi B, Peng DF, Cai Q, El-Rifai W, Zheng W, Belkhiri A, t-DARPP regulates phosphatidylinositol-3-kinase-dependent cell growth in breast cancer, Molecular cancer 9 (2010) 240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kauraniemi P, Barlund M, Monni O, Kallioniemi A, New amplified and highly expressed genes discovered in the ERBB2 amplicon in breast cancer by cDNA microarrays, Cancer Res 61(22) (2001) 8235–40. [PubMed] [Google Scholar]
- [27].Kauraniemi P, Kuukasjarvi T, Sauter G, Kallioniemi A, Amplification of a 280-kilobase core region at the ERBB2 locus leads to activation of two hypothetical proteins in breast cancer, Am J Pathol 163(5) (2003) 1979–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Maqani N, Belkhiri A, Moskaluk C, Knuutila S, Dar AA, El-Rifai W, Molecular dissection of 17q12 amplicon in upper gastrointestinal adenocarcinomas, Mol Cancer Res 4(7) (2006) 449–55. [DOI] [PubMed] [Google Scholar]
- [29].Forbes SA, Beare D, Boutselakis H, Bamford S, Bindal N, Tate J, Cole CG, Ward S, Dawson E, Ponting L, Stefancsik R, Harsha B, Kok CY, Jia M, Jubb H, Sondka Z, Thompson S, De T, Campbell PJ, COSMIC: somatic cancer genetics at high-resolution, Nucleic Acids Res 45(D1) (2017) D777–D783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Fienberg AA, Greengard P, The DARPP-32 knockout mouse, Brain Res Brain Res Rev 31(2–3) (2000) 313–9. [DOI] [PubMed] [Google Scholar]
- [31].Christenson JL, Kane SE, Darpp-32 and t-Darpp are differentially expressed in normal and malignant mouse mammary tissue, Molecular cancer 13 (2014) 192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Heyser CJ, Fienberg AA, Greengard P, Gold LH, DARPP-32 knockout mice exhibit impaired reversal learning in a discriminated operant task, Brain Res 867(1–2) (2000) 122–30. [DOI] [PubMed] [Google Scholar]
- [33].Chen Z, Zhu S, Hong J, Soutto M, Peng D, Belkhiri A, Xu Z, El-Rifai W, Gastric tumour-derived ANGPT2 regulation by DARPP-32 promotes angiogenesis, Gut 65(6) (2016) 925–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Gu L, Waliany S, Kane SE, Darpp-32 and its truncated variant t-Darpp have antagonistic effects on breast cancer cell growth and herceptin resistance, PloS one 4(7) (2009) e6220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Belkhiri A, Dar AA, Peng DF, Razvi MH, Rinehart C, Arteaga CL, El-Rifai W, Expression of t-DARPP mediates trastuzumab resistance in breast cancer cells, Clin Cancer Res 14(14) (2008) 4564–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Zhu S, Belkhiri A, El-Rifai W, DARPP-32 increases interactions between epidermal growth factor receptor and ERBB3 to promote tumor resistance to gefitinib, Gastroenterology 141(5) (2011) 1738–48 e1- [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Hong J, Katsha A, Lu P, Shyr Y, Belkhiri A, El-Rifai W, Regulation of ERBB2 receptor by t-DARPP mediates trastuzumab resistance in human esophageal adenocarcinoma, Cancer Res 72(17) (2012) 4504–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Theile D, Lenz G, Momand JA, Kane SE, Resistance to HER2-targeted therapy, in: Prosperi JR (Ed.), Resistance to Targeted Therapies in Breast Cancer, Springer, Cham, 2017, pp. 35–88. [Google Scholar]
- [39].Citri A, Skaria KB, Yarden Y, The deaf and the dumb: the biology of ErbB-2 and ErbB-3, Exp Cell Res 284(1) (2003) 54–65. [DOI] [PubMed] [Google Scholar]
- [40].Chandarlapaty S, Sakr RA, Giri D, Patil S, Heguy A, Morrow M, Modi S, Norton L, Rosen N, Hudis C, King TA, Frequent mutational activation of the PI3K-AKT pathway in trastuzumab-resistant breast cancer, Clin Cancer Res 18(24) (2012) 6784–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Sepulveda AR, Helicobacter, Inflammation, and Gastric Cancer, Curr Pathobiol Rep 1(1) (2013) 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Zhu S, Soutto M, Chen Z, Peng D, Romero-Gallo J, Krishna US, Belkhiri A, Washington MK, Peek R, El-Rifai W, Helicobacter pylori-induced cell death is counteracted by NF-kappaB-mediated transcription of DARPP-32, Gut 66(5) (2017) 761–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zhu S, Hong J, Tripathi MK, Sehdev V, Belkhiri A, El-Rifai W, Regulation of CXCR4-mediated invasion by DARPP-32 in gastric cancer cells, Mol Cancer Res 11(1) (2013) 86–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Neyroz P, Desdouits F, Benfenati F, Knutson JR, Greengard P, Girault JA, Study of the conformation of DARPP-32, a dopamine- and cAMP-regulated phosphoprotein, by fluorescence spectroscopy, J Biol Chem 268(32) (1993) 24022–31. [PubMed] [Google Scholar]
- [45].Dancheck B, Nairn AC, Peti W, Detailed structural characterization of unbound protein phosphatase 1 inhibitors, Biochemistry 47(47) (2008) 12346–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Robinson DR, Kalyana-Sundaram S, Wu YM, Shankar S, Cao X, Ateeq B, Asangani IA, Iyer M, Maher CA, Grasso CS, Lonigro RJ, Quist M, Siddiqui J, Mehra R, Jing X, Giordano TJ, Sabel MS, Kleer CG, Palanisamy N, Natrajan R, Lambros MB, Reis-Filho JS, Kumar-Sinha C, Chinnaiyan AM, Functionally recurrent rearrangements of the MAST kinase and Notch gene families in breast cancer, Nat Med 17(12) (2011) 1646–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Yun SM, Yoon K, Lee S, Kim E, Kong SH, Choe J, Kang JM, Han TS, Kim P, Choi Y, Jho S, Yoo H, Bhak J, Yang HK, Kim SJ, PPP1R1B-STARD3 chimeric fusion transcript in human gastric cancer promotes tumorigenesis through activation of PI3K/AKT signaling, Oncogene 33(46) (2014) 5341–7. [DOI] [PubMed] [Google Scholar]
- [48].Hong L, Zhao Y, Wang J, Han Y, Guo W, Jin H, Zhai H, Bai F, Zhang X, Qiao T, Chen Z, Fan D, Reversal of multidrug resistance of adriamycin-resistant gastric adenocarcinoma cells through the up-regulation of DARPP-32, Dig Dis Sci 53(1) (2008) 101–7. [DOI] [PubMed] [Google Scholar]
- [49].Hansen C, Howlin J, Tengholm A, Dyachok O, Vogel WF, Nairn AC, Greengard P, Andersson T, Wnt-5a-induced phosphorylation of DARPP-32 inhibits breast cancer cell migration in a CREB-dependent manner, J Biol Chem 284(40) (2009) 27533–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Hansen C, Greengard P, Nairn AC, Andersson T, Vogel WF, Phosphorylation of DARPP-32 regulates breast cancer cell migration downstream of the receptor tyrosine kinase DDR1, Exp Cell Res 312(20) (2006) 4011–8. [DOI] [PubMed] [Google Scholar]
- [51].Kopljar M, Patrlj L, Korolija-Marinic D, Horzic M, Cupurdija K, Bakota B, High Expression of DARPP-32 in Colorectal Cancer Is Associated With Liver Metastases and Predicts Survival for Dukes A and B Patients: Results of a Pilot Study, Int Surg 100(2) (2015) 213–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Ivkovic S, Polonskaia O, Farinas I, Ehrlich ME, Brain-derived neurotrophic factor regulates maturation of the DARPP-32 phenotype in striatal medium spiny neurons: studies in vivo and in vitro, Neuroscience 79(2) (1997) 509–16. [DOI] [PubMed] [Google Scholar]
- [53].Keilani S, Chandwani S, Dolios G, Bogush A, Beck H, Hatzopoulos AK, Rao GN, Thomas EA, Wang R, Ehrlich ME, Egr-1 induces DARPP-32 expression in striatal medium spiny neurons via a conserved intragenic element, J Neurosci 32(20) (2012) 6808–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Chandwani S, Keilani S, Ortiz-Virumbrales M, Morant A, Bezdecny S, Ehrlich ME, Induction of DARPP-32 by brain-derived neurotrophic factor in striatal neurons in vitro is modified by histone deacetylase inhibitors and Nab2, PloS one 8(10) (2013) e76842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Valjent E, Aubier B, Corbille AG, Brami-Cherrier K, Caboche J, Topilko P, Girault JA, Herve D, Plasticity-associated gene Krox24/Zif268 is required for long-lasting behavioral effects of cocaine, J Neurosci 26(18) (2006) 4956–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Stroppolo A, Guinea B, Tian C, Sommer J, Ehrlich ME, Role of phosphatidylinositide 3-kinase in brain-derived neurotrophic factor-induced DARPP-32 expression in medium size spiny neurons in vitro, J Neurochem 79(5) (2001) 1027–32. [DOI] [PubMed] [Google Scholar]
- [57].Bogush A, Pedrini S, Pelta-Heller J, Chan T, Yang Q, Mao Z, Sluzas E, Gieringer T, Ehrlich ME, AKT and CDK5/p35 mediate brain-derived neurotrophic factor induction of DARPP-32 in medium size spiny neurons in vitro, J Biol Chem 282(10) (2007) 7352–9. [DOI] [PubMed] [Google Scholar]
- [58].Li BS, Ma W, Jaffe H, Zheng Y, Takahashi S, Zhang L, Kulkarni AB, Pant HC, Cyclin-dependent kinase-5 is involved in neuregulin-dependent activation of phosphatidylinositol 3-kinase and Akt activity mediating neuronal survival, J Biol Chem 278(37) (2003) 35702–9. [DOI] [PubMed] [Google Scholar]
- [59].Wang HF, Liu FC, Regulation of multiple dopamine signal transduction molecules by retinoids in the developing striatum, Neuroscience 134(1) (2005) 97–105. [DOI] [PubMed] [Google Scholar]
- [60].Liao WL, Liu FC, RARbeta isoform-specific regulation of DARPP-32 gene expression: an ectopic expression study in the developing rat telencephalon, Eur J Neurosci 21(12) (2005) 3262–8. [DOI] [PubMed] [Google Scholar]
- [61].Pedrini S, Bogush A, Ehrlich ME, Phosphatidylinositide 3-kinase and protein kinase C zeta mediate retinoic acid induction of DARPP-32 in medium size spiny neurons in vitro, J Neurochem 106(2) (2008) 917–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Ghanooni R, Decaestecker C, Simon P, Gabius HJ, Hassid S, Choufani G, Characterization of patterns of expression of protein kinase C-alpha, -delta, -eta, -gamma and -zeta and their correlations to p53, galectin-3, the retinoic acid receptor-beta and the macrophage migration inhibitory factor (MIF) in human cholesteatomas, Hear Res 214(1–2) (2006) 7–16. [DOI] [PubMed] [Google Scholar]
- [63].Stroppolo A, Tian C, Guinea B, Olm V, Sheffield R, Sommer J, Ehrlich ME, 17beta-Estradiol promotes striatal medium size spiny neuronal maturation in vitro, Neuroendocrinology 79(5) (2004) 259–67. [DOI] [PubMed] [Google Scholar]
- [64].McLean MH, El-Omar EM, Genetics of inflammation in the gastrointestinal tract and how it can cause cancer, Recent Results Cancer Res 185 (2011) 173–83. [DOI] [PubMed] [Google Scholar]
- [65].Liu Y, Gonen S, Gonen T, Yeates TO, Near-atomic cryo-EM imaging of a small protein displayed on a designed scaffolding system, Proc Natl Acad Sci U S A 115(13) (2018) 3362–3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Hoelder S, Clarke PA, Workman P, Discovery of small molecule cancer drugs: successes, challenges and opportunities, Mol Oncol 6(2) (2012) 155–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Denny EC, Kane SE, t-Darpp Promotes Enhanced EGFR Activation and New Drug Synergies in Her2-Positive Breast Cancer Cells, PloS one 10(6) (2015) e0132267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Zhu S, Chen Z, Katsha A, Hong J, Belkhiri A, El-Rifai W, Regulation of CD44E by DARPP-32-dependent activation of SRp20 splicing factor in gastric tumorigenesis, Oncogene 35(14) (2016) 1847–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Chang MJ, Zhong F, Lavik AR, Parys JB, Berridge MJ, Distelhorst CW, Feedback regulation mediated by Bcl-2 and DARPP-32 regulates inositol 1,4,5-trisphosphate receptor phosphorylation and promotes cell survival, Proc Natl Acad Sci U S A 111(3) (2014) 1186–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Vangamudi B, Zhu S, Soutto M, Belkhiri A, El-Rifai W, Regulation of beta-catenin by t-DARPP in upper gastrointestinal cancer cells, Molecular cancer 10 (2011) 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Yger M, Girault JA, DARPP-32, Jack of All Trades… Master of Which?, Front Behav Neurosci 5 (2011) 56. [DOI] [PMC free article] [PubMed] [Google Scholar]