

Abstract
Axonopathies are neurodegenerative disorders caused by axonal degeneration, affecting predominantly the longest neurons. Several of these axonopathies are caused by genetic defects in proteins involved in the shaping and dynamics of the endoplasmic reticulum (ER); however, it is unclear how these defects impinge on neuronal survival. Given its central and widespread position within a cell, the ER is a pivotal player in inter-organelle communication. Here, we demonstrate that defects in the ER fusion protein ATL3, which were identified in patients suffering from hereditary sensory and autonomic neuropathy, result in an increased number of ER–mitochondria contact sites both in HeLa cells and in patient-derived fibroblasts. This increased contact is reflected in higher phospholipid metabolism, upregulated autophagy and augmented Ca2+ crosstalk between both organelles. Moreover, the mitochondria in these cells display lowered motility, and the number of axonal mitochondria in neurons expressing disease-causing mutations in ATL3 is strongly decreased. These results underscore the functional interdependence of subcellular organelles in health and disease and show that disorders caused by ER-shaping defects are more complex than previously assumed.
Introduction
The endoplasmic reticulum (ER) is composed of an intricate network of sheets and tubules that is continuous with the nuclear envelope, maintaining a single lumen. Morphogenesis of this complex organelle is governed by a large number of proteins whose concerted actions determine the overall shape of the ER. Shaping the different ER membrane morphologies is achieved through the competition between curvature inducing or stabilizing mechanisms and mechanisms sustaining flat membranes (1–3). Superimposed on the membrane shape is the dynamic maintenance of the network architecture. These dynamics are a balance between expansions of the network through tubule branching, extension and fusion, opposed by tubule retractions and ring closures (3). The result of these opposing forces is an ER network that is continuously being remodeled while maintaining its overall morphology, allowing the cell to swiftly respond to changing conditions.
In order to sustain this balanced remodelling, homotypic membrane fusion is crucial to maintain a well-spread and highly connected tubular network. In metazoans, homotypic fusion of ER tubules is mediated by a class of membrane-bound dynamin-like GTPases known as atlastins (ATLs) (4,5). While zebrafish and fruit flies have only one ATL isoform, mammals have three highly conserved ATL isoforms (ATL1–3). Mutations in ATL1 and ATL3 have been identified in patients suffering from hereditary spastic paraplegia (HSP) and hereditary sensory and autonomic neuropathy (HSAN) (6–9). In HSP, the motor neurons in the corticospinal tract are affected, leading to progressive spasticity in the lower limbs. In HSAN, predominantly, the sensory peripheral nerves are affected with variable degrees of autonomic involvement, causing loss of pain sensation amongst other symptoms. Common to both diseases is that they are caused by axonal degeneration of neurons with particularly long axons, reaching lengths of up to 1 m in humans. Underscoring the relevance of ER fusion for neuronal functioning, loss of ATLs in flies or zebrafish results in axonal dysfunction and locomotor deficits (10–12).
In the past decades, it has become increasingly evident that organelles are not isolated structures. Rather, different organelles communicate directly with one another through membrane contact sites (MCSs). The ER has been shown to be in contact with most other membrane-bound organelles, including the plasma membrane, endo-lysosomes, peroxisomes and mitochondria (13). MCSs allow these different organelles to coordinate their processes and provide the cell with the means to adapt to local changes in a spatiotemporally controlled fashion. Ca2+ signalling plays a prominent role in these processes (14). Given the extraordinary size and shape of neurons, locally controlled responses to changing conditions are likely to be pivotal for their functioning, particularly within the longest axons.
In particular, MCSs between the ER and mitochondria have been linked to a variety of neurological disorders (15). ER and mitochondrial dynamics are coordinated tightly, and contacts are maintained despite the highly dynamic nature of both organelles (16). Hence, changes in ER network maintenance and motility are likely to affect its communication with mitochondria, and thereby, mitochondrial homeostasis. ER–mitochondria contacts are formed at specialized subdomains of the ER known as mitochondria-associated membranes (MAMs). These MCSs harbor a rich set of proteins that enable essential functions in lipid metabolism, Ca2+ signalling, autophagosome biogenesis, apoptosis induction, redox signalling, mitochondrial metabolism and mitochondrial fission (15,17–21).
Two dominant missense mutations in ATL3, resulting in Y192C and P338R amino acid changes are known to cause HSAN (6,8). We recently showed that both mutations disrupt the fusogenic capacity of ATL3, which not only results in a loss of ER connectivity, but also causes aberrant ER membrane tethering, culminating in tangling of ER tubules (22). We hypothesized that this aberrant tethering could interfere with the dynamics and organization of the different ER subdomains. Because defects in ER shaping and in ER–mitochondria crosstalk are both common features in neurodegenerative disorders, we examined if mutations in ATL3 could also affect ER–mitochondria MCSs formation and crosstalk. Indeed, in this study, we found that the presence of HSAN-causing ATL3 mutations in HeLa cells, patient cells and neurons lead to elevated MAMs formation, which strongly affected mitochondrial function and motility, and suggests a more complex pathogenic mechanism than previously assumed.
Results
Mutations in ATL3 do not affect mitochondrial morphology
To assess whether HSAN-causing mutations in ATL3 might affect mitochondrial homeostasis and functioning, we first studied whether we could observe a direct effect of overexpression of ATL3 on the mitochondrial network. Therefore, we overexpressed Green Fluorescent Protein (GFP)-tagged ATL3 in HeLa cells and stained mitochondria using Mitotracker in live cells (Fig. 1A). As previously reported, wild-type ATL3 (ATL3WT) exclusively localized to the tubular ER network, while the disease-associated ATL3 mutants are mostly enriched in a collapsed ER structure (Fig. 1A) (22). In contrast to the severe morphological defect to the ER, caused by expression of ATL3Y192C or ATL3P338R, the morphology of the mitochondrial network appears not affected (Fig. 1A). In all cell types, the mitochondria form a strongly interconnected network of mostly elongated mitochondria and no significant changes in mitochondrial morphology parameters were detected upon expression of mutant ATL3 (Fig. 1B). High-resolution images were obtained using transmission electron microscopy (TEM). In agreement with the light microscopy analysis, ultrastructural mitochondrial morphology is unaltered in cells expressing mutant ATL3 and mitochondrial surface density within the cytoplasm showed no differences between wild-type (WT) and mutant cells (Supplementary Material, Fig. S1A and B).
Figure 1.
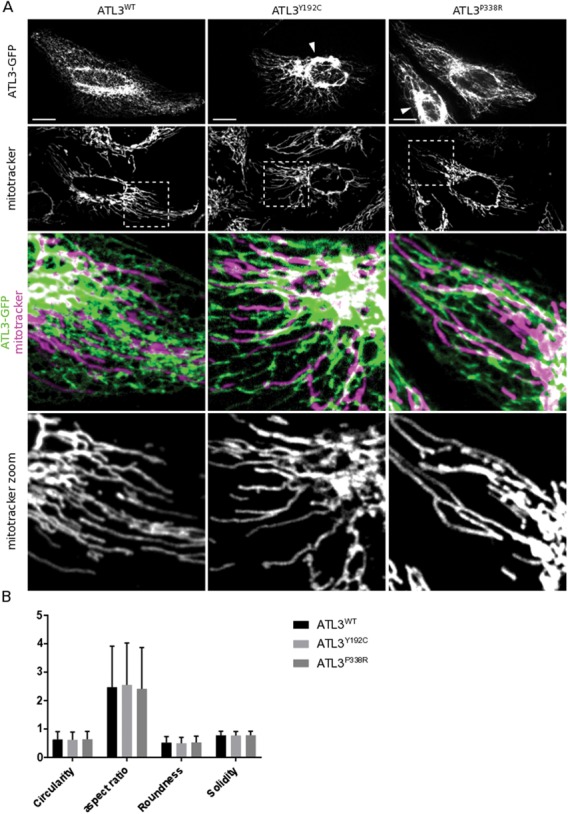
Mitochondrial morphology is unaltered in HeLa cells expressing mutant ATL3. (A) HeLa cells expressing ATL3-GFP were stained with Mitotracker Red. Scale bars, 20 μm; box width, 40 μm. Arrowheads indicate collapsed ER structures. (B) Mitochondrial morphology descriptors reveal no alterations in mitochondrial morphology upon expression of mutant ATL3 in three independent experiments. Mean ± SD from a representative experiment is shown.
Increased number of ER–mitochondria contact sites upon ATL3 mutation
Although the overall mitochondrial morphology is not affected, the disease-causing mutations in ATL3 do have severe effects on ER shaping and dynamics (22). Hence, we wondered whether this could impact the formation of contact sites with mitochondria in these cells. To address this, we used TEM to examine the number and size of ER–mitochondria MCSs in HeLa cells overexpressing ATL3WT or ATL3Y192C (Fig. 2A). Analysis of these images revealed that while the size of these contact sites was similar between WT and mutant-expressing cells (Supplementary Material, Fig. S1C), the number of MCSs was almost double in cells expressing ATL3Y192C (Fig. 2B, left panel). As a consequence, the overall fraction of mitochondrial surface that is in close contact with the ER was increased (Fig. 2B, right panel). This result was confirmed in cells with endogenous expression of ATL3WT or ATL3Y192C, using human skin fibroblasts from a control individual or an HSAN-I patient harboring the ATL3Y192C mutation (Fig. 2C and D). Again, the larger proportion of mitochondrial surface in contact with the ER was due to an increase in the number of contact sites, while the measured size of individual contact sites was similar for both genotypes (Supplementary Material, Fig. S1D).
Figure 2.
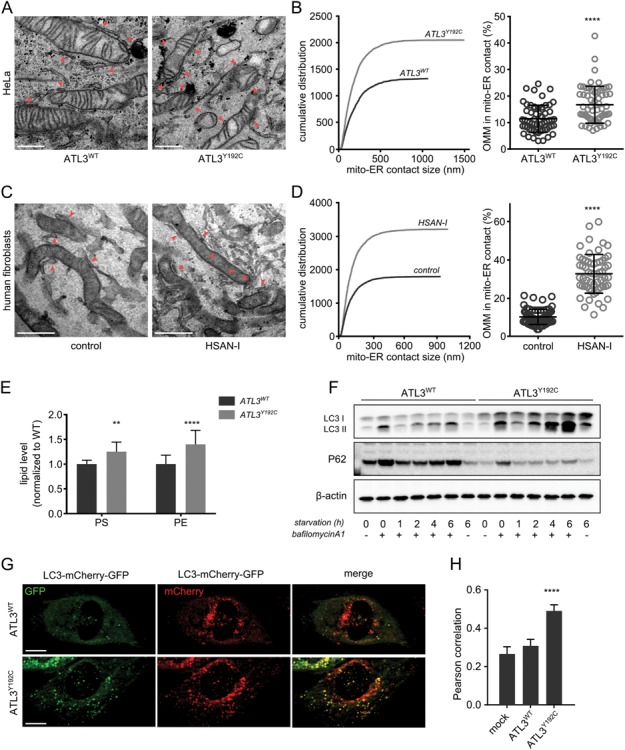
ER–mitochondria contact sites are upregulated by HSAN-causing mutations in ATL3. (A) TEM images of HeLa cells stably expressing WT or mutant ATL3. Examples of ER–mitochondria contact sites that were measured are indicated by red arrowheads. Scale bar, 500 nm. (B) Left: cumulative distribution of the lengths of the MCS boundaries as measured on the sections. Right: percentage of the mitochondrial surface in close contact with the ER (n = 60 cells, 1108 and 1087 mitochondrial profiles analysed in total for ATL3WT and ATL3Y192C, respectively). ****P < 0.0001 in Mann–Whitney test. (C) TEM images of human skin fibroblasts of a control individual (left) or an HSAN-I patient carrying the ATL3Y192C mutation (right). Red arrowheads indicate examples of ER–mitochondria contact sites. Scale bar, 500 nm. (D) As in B, the cumulative distribution of all measured contact sites in the fibroblasts is shown on the left, and the percentage of mitochondrial surface in close contact with the ER is shown on the right (n = 68 cells for control cells and 59 for HSAN-I patient cells; 885 and 763 mitochondrial profiles analysed, respectively). ****P < 0.0001 in a Mann–Whitney test. (E) Normalized steady-state levels of PS and PE in HeLa cells expressing ATL3WT or ATL3Y192C. **P < 0.01; ****P < 0.0001 in two-way ANOVA test. (F) Representative western blot for P62 and LC3 levels in cells expressing ATL3WT or ATL3Y192C. The cells were either untreated, incubated with bafilomycin A1 for 6 h without starvation or starved in the presence/absence of bafilomycin A1, as indicated below the blots. ß-actin blot serves as a loading control. (G) HeLa cells expressing ATL3WT or ATL3Y192C were transfected with double-tagged LC3-GFP-mCherry. Scale bar, 15 μm. (H) Pearson correlation was used to quantify the co-localization of the GFP and mCherry signal of the cells in G. Mock-transfected cells were used as a control. n = 22–48 cells per condition. ****P < 0.0001 in two-way ANOVA test followed by Tukey's multiple comparisons test.
To confirm that these membrane appositions are indeed functional ER–mitochondria MCSs, we assessed the functions known to be localized at these contacts. The first function reported for ER–mitochondria contacts is their role in phospholipid metabolism (15,21). On the ER side of the contact, MAMs are enriched in the enzymes responsible for the synthesis of phosphatidylserine (PS). This PS is transferred from the MAMs to the inner mitochondrial membrane, where it can be converted to phosphatidylethanolamine (PE). In agreement with an increased MAM-dependent phospholipid metabolism, lipidomics analysis revealed that the steady-state levels of both these lipids were higher in HeLa cells expressing ATL3Y192C (Fig. 2E).
A more recently reported function of ER–mitochondria contact sites is their role in autophagosome biogenesis. The MAMs are proposed to form a platform for the initiation of autophagosome formation (19). We monitored macro-autophagy using western blotting for LC3-II and p62 (23) and found that LC3-II levels were higher in ATL3Y192C cells after starvation. This effect was more pronounced in the presence of the lysosomal inhibitor bafilomycin A1, indicating that the higher LC3-II levels are due to an increased autophagy flux rather than a defect in lysosomal degradation (Fig. 2F). Correspondingly, cells expressing mutant ATL3 displayed lower levels of p62, both under control conditions and after induction of autophagy (Fig. 2F). The autophagy flux was also monitored directly through the quantification of double-tagged LC3-mCherry-GFP vesicles in cells expressing mutant or ATL3WT. While the low pH in autolysosomes inhibits GFP, but not mCherry fluorescence, the neutral pH in newly generated autophagosomes allows fluorescence of both GFP and mCherry (23,24). Under non-starved conditions, cells expressing ATL3WT showed very few LC3 vesicles, and most of these vesicles displayed only fluorescence of mCherry, indicating that these are autolysosomes (Fig. 2G). In contrast, in cells expressing ATL3Y192C, many vesicles were observed where GFP and mCherry fluorescence were co-localized (Fig. 2G–H), confirming the previous finding that these cells undergo higher autophagy initiation correlating with a higher number of ER–mitochondria contact sites (Fig. 2A and B). Upregulation of these known functions of MAMs corroborates the finding that ER–mitochondria contact sites are increased and establishes that the membrane appositions observed using TEM are indeed functional signalling platforms.
Increased ER–mitochondria Ca2+ crosstalk upon ATL3 mutation
One of the best characterized modes of ER–mitochondria crosstalk at MCSs is Ca2+ transfer. Upon inositol triphosphate binding, Ca2+ release through the IP3Rs in the MAMs results in high local Ca2+ concentrations, known as microdomains, which allow mitochondrial Ca2+ uptake through the low affinity Mitochondrial Calcium Uniporter (17,25). Given the diverse roles of ER–mitochondria Ca2+ transfer in mitochondrial trafficking and homeostasis, we investigated how mutations in ATL3 influence this crosstalk. First, we determined ER Ca2+ content in HeLa cells stably expressing ATL3WT or ATL3Y192C by quantifying the thapsigargin-induced Ca2+ transients using the cytosolic ratiometric Ca2+ reporter Fura-2. Thapsigargin blocks Serca2 Ca2+ pumps, which are responsible for ER Ca2+ loading, resulting in passive leak of Ca2+ from the ER. Extracellular EGTA was used to prevent Ca2+ entry, ensuring that the Ca2+ signal represents only Ca2+ originating from the ER. The thapsigargin-releasable Ca2+, and thus ER Ca2+ content, did not significantly differ between control cells, cells overexpressing ATL3WT or cells expressing ATL3Y192C (Fig. 3A). Similarly, the total Ca2+ store content as measured by the cytosolic Ca2+ increase upon treatment with the Ca2+ ionophore ionomycin was the same in all conditions (Fig. 3B). Next, we assessed cytosolic Ca2+ signals mediated through IP3Rs by exposing the cells to extracellular agonists that trigger IP3 signalling such as histamine (Fig. 3C) and ATP (Fig. 3D). In both cases, the extracellular agonists resulted in similar Ca2+ responses in the cytosol among control cells and cells overexpressing ATL3WT or ATL3Y192C. These cytosolic Ca2+ measurements indicate that mutant ATL3 expression does not affect the ER Ca2+ store content or the mobilization of Ca2+ from the ER by IP3R channels.
Figure 3.
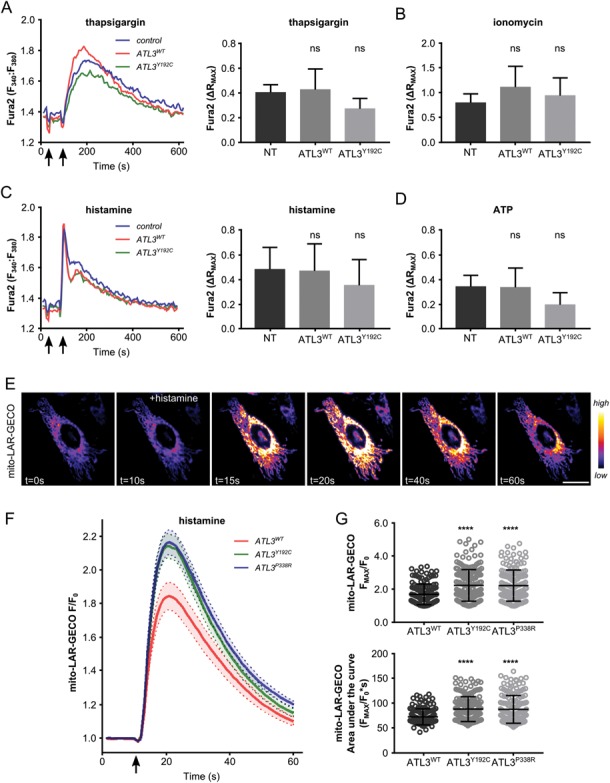
Increased ER–mitochondria Ca2+ transfer in ATL3 mutant cells. (A–D) Fura-2 response in HeLa cells stably expressing ATL3WT (red) or ATL3Y192C (green) and control cells (blue) after indicated treatments. Left: representative trace, whereby the first arrow indicates the addition of EGTA and the second arrow indicates the addition of thapsigargin (A), ionomycin (B), histamine (C) or ATP (D). Right: average ∆RMAX of three independent experiments, error bars show standard deviation. (A) Fura-2 response to passive ER Ca2+ leak upon thapsigargin treatment. (B) Fura-2 response to ionomycin-induced Ca2+ release. (C) Fura-2 response to active ER Ca2+ release upon histamine treatment. (D) Fura-2 response to active ER Ca2+ release upon ATP treatment. ns, not significant in one-way ANOVA test. (E–G) HeLa cells transfected with the mito-LAR-GECO mitochondrial Ca2+ reporter after stimulation with 100 μm histamine. (E) Representative time lapse image sequence. Scale bar, 25 μm. Color code scale on the right indicates fluorescence intensity. (F) Normalized mito-LAR-GECO fluorescence intensity after histamine-induced ER Ca2+ release. Average trace of normalized fluorescence of three independent experiments ± standard error is shown. Arrow indicates the addition of histamine. (G) Peak mitochondrial Ca2+ reporter responses (above, FMAX/F0) and area under the curve (below) were calculated for individual cells from three independent experiments (n = 151–178 cells in total). Mean ± SD is marked in black. *P < 0.05; ***P < 0.001; ****P < 0.0001 in Kruskal–Wallis test followed by Dunn’s multiple comparisons test.
To assess mitochondrial Ca2+ dynamics, ATL3-GFP and mito-LAR-geco, a Ca2+ reporter targeted to the mitochondrial matrix (26) were co-transfected in HeLa cells (Fig. 3E). Using this reporter, we performed live-cell imaging with the cells kept in a Ca2+-free buffer (27). After 10 s, the cells were stimulated by the addition of histamine, which induces IP3-dependent release of Ca2+ from the ER (28). Correspondingly, this release resulted in a transient mitochondrial Ca2+ increase (Fig. 3E), mediated by direct transfer of Ca2+ at the MAMs (17). After background subtraction, fluorescence intensity (F) was normalized to the initial intensity (F0) for each cell. In agreement with increased ER–mitochondria crosstalk (Fig. 2), both mutations in ATL3 resulted in higher Ca2+ peak amplitudes upon mobilization of ER Ca2+ by histamine (Fig. 3F). Indeed, both the maximal peak value (Fmax/F0) and the area under the curve were significantly higher in cells expressing mutant ATL3 (Fig. 3G). Similar results were obtained when cells were stimulated with ATP (Supplementary Material, Fig. S2). These findings confirm the upregulated ER–mitochondria crosstalk in cells expressing HSAN-causing mutations in ATL3, representing a functional readout of the enhanced MCSs formation observed using TEM (Fig. 2).
Mutations in ATL3 interfere with mitochondrial dynamics
Ca2+ transfer to mitochondria serves a variety of functions, including the modulation of mitochondrial trafficking through the mitochondrial Rho GTPases (Miro1 and Miro2), which are Ca2+-sensitive regulators of mitochondrial transport (29). Given the well-known involvement of defects in mitochondrial dynamics in axonopathies, we decided to investigate whether mitochondrial trafficking is affected by mutations in ATL3. Therefore, we performed live cell imaging to analyse mitochondrial motility in HeLa cells (Supplementary Material, Fig. S3 and Video). Whereas, the balance between mitochondrial fusion and fission did not appear to be affected (Fig. 1), the motility of mitochondria within the cytoplasm was found to be reduced in cells expressing mutant ATL3 (Fig. 4A and B). Moreover, a similar decrease in mitochondrial dynamics was also observed in HSAN-I patient-derived fibroblasts (Fig. 4C), indicating that increased ER–mitochondria contact and crosstalk (Figs 2 and 3) may indeed have a negative impact on mitochondrial trafficking.
Figure 4.
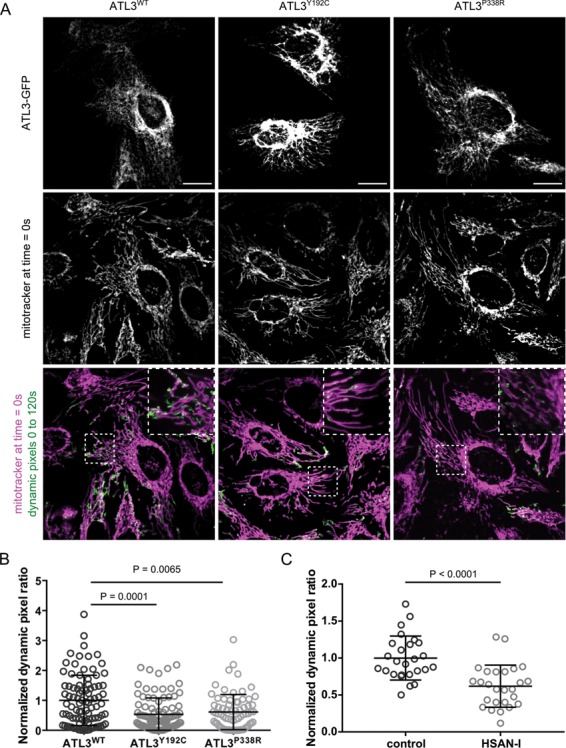
HSAN-causing mutations in ATL3 interfere with mitochondrial trafficking. (A) HeLa cells were transfected with GFP-tagged ATL3WT, ATL3Y192C or ATL3P338R and mitochondria were labelled using Mitotracker Red. Live cell imaging was performed for 2 min with 2 s time intervals. To determine mitochondrial motility, the dynamic pixels in each frame were extracted with the ImageJ Difference tracker plugin (Supplementary Material, Video and Fig. S3). A summed intensity projection of these dynamic pixels over the entire time sequence is shown in green. A zoom of the boxed area is shown. Scale bar and box width, 20 μm. (B) Dynamic pixels were quantified for each cell in three independent experiments, and the normalized data were pooled (mean ± SEM is shown, N = 73–95 cells per genotype). One-way ANOVA and a Kruskal–Wallis test were performed. (C) Control and HSAN-I patient fibroblasts were analysed in three independent experiments as in A and B (Supplementary Material, Video and Fig. S3). N = 36 cells for each genotype. An unpaired t-test was performed.
Mutations in ATL3 disrupt distribution of mitochondria in neurons
To investigate the relevance of this altered mitochondrial trafficking for neurons, we expressed GFP-tagged ATL3WT, ATL3Y192C or ATL3P338R in mouse primary cortical neurons and stained for the outer mitochondrial membrane marker TOM20 (Fig. 5A). Analysis of the distribution of mitochondria within neurites showed that also mitochondria appear retained within the soma, since the density of mitochondria within the neuronal outgrowths was significantly decreased in cells expressing HSAN-causing mutations in ATL3 compared to neurons expressing ATL3WT (Fig. 5B–C). These findings show that a defect in ER membrane fusion in neurons can indirectly affect mitochondrial trafficking. Combined, these data show that defects in ER dynamics resulting in aberrant ER membrane tethering also interfere with the regulation of contact site formation with mitochondria and that increased ER–mitochondria crosstalk due to HSAN-causing mutations in ATL3 can have major implications for mitochondrial trafficking and distribution within neurons.
Figure 5.
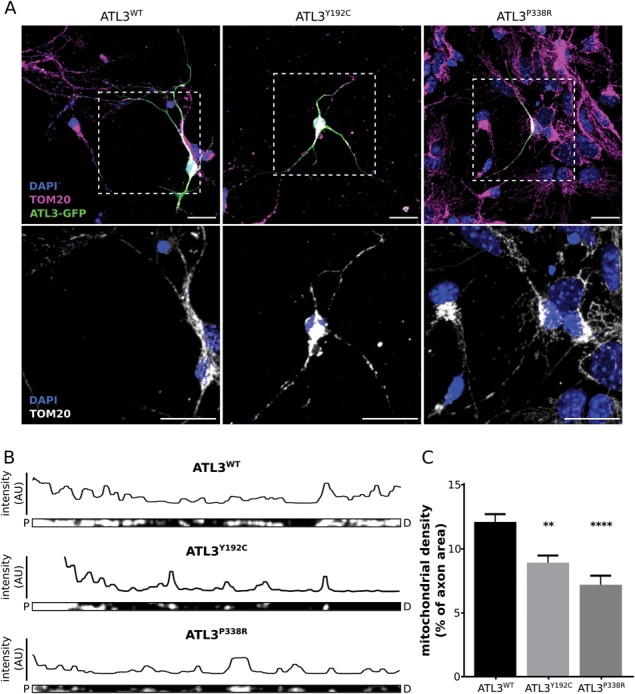
Expression of mutant ATL3 results in altered mitochondrial distribution in primary cortical neurons. (A) Mouse cortical neurons transduced with ATL3-GFP (green) were stained for the mitochondrial marker TOM20 (magenta). Nuclear stain is shown in blue and a zoom of the boxed area is shown below. Scale bars, 20 μm. (B) The neurites from A were straightened in ImageJ using a segmented line selection (P, proximal; D, distal to the soma). Fluorescence intensity of the mitochondrial marker along the tracing is shown above each axon. (C) The mitochondrial density was quantified as the percentage of axonal area occupied by mitochondria. **P < 0.01; ****P < 0.0001 in a one-way ANOVA test followed by Tukey’s multiple comparisons test (n = 39–65 axons in total).
Discussion
Defects in ER network shaping and dynamics have been well established as a common pathogenic mechanism in several neurodegenerative disorders (30). However, how these defects cause axonal degeneration is poorly understood. In previous work, we have shown that HSAN-causing mutations in ATL3 disrupt ER membrane fusion, resulting in aberrant membrane tethering and tangling of ER tubules (22). We now demonstrate that in addition to a defect in ER network shaping, also mitochondrial trafficking is affected, causing altered distribution of mitochondria in cortical neurons expressing mutant ATL3. Whereas, little is known about the dynamics and functioning of the ER within axons, the role of mitochondria is well studied and the importance of maintaining a pool of healthy mitochondria in axons is emphasized by the involvement of defects in mitochondrial dynamics in many neurodegenerative disorders (31–33).
Axonal transport of mitochondria is essential not only to provide healthy mitochondria to sites of high-energy demand such as synapses or the nodes of Ranvier, but also to remove damaged mitochondria from these distal regions and target them for lysosomal degradation. Therefore, the motility of mitochondria is intricately regulated. Mitochondria within neurites display sophisticated motility patterns that involve bi-directional movement over variable distances interspersed with brief pausing, while the majority of axonal mitochondria are stationary for longer periods (34). Moreover, also the sorting and subsequent entry of mitochondria into the axons or dendrites is a highly regulated process that is essential for neuronal development (35). In contrast, slowed axonal transport or failure of mitochondria to enter the axon is detrimental for neuronal functioning (35,36). Interestingly, a P342S mutation in ATL1 affecting the homologous residue of P338 in ATL3, which was identified in an HSP patient, was shown to alter mitochondrial trafficking in neurons derived from induced pluripotent stem cells (37). In fruit flies, the loss of HSPs and HSAN-related ARL6IP1, which modulates the curvature of ER membranes, was found to affect the mitochondrial fission/fusion balance (38). Although these studies demonstrate that a primary defect in the ER can indeed affect mitochondrial dynamics, it is not clear how both organelles influence each other in these models, and direct communication through ER–mitochondria MCSs was not investigated.
Here, we show that while mitochondrial morphology is not affected (Fig. 1 and Supplementary Material, Fig. S1), ER–mitochondria contact site formation is increased both in HeLa cells overexpressing mutant ATL3 and in patient fibroblasts (Fig. 2). This enhanced physical contact is the result of a higher number of contact sites and correlates with upregulated functions of MAMs, including phospholipid metabolism and autophagosome biogenesis (Fig. 2). Moreover, mitochondrial Ca2+ uptake through the release of Ca2+ from the ER via the IP3R channels was found to be significantly increased in HeLa cells expressing mutant ATL3 (Fig. 3). Indirect measurements of the ER and total intracellular Ca2+ store contents showed no effect of ATL3 mutation on these parameters. Furthermore, also IP3R activity per se was not altered upon presence of mutant ATL3, as no significant difference was found in cytosolic Ca2+ transients provoked by the addition of extracellular agonists such as ATP or histamine (Fig. 3). These results excluded that the increased mitochondrial Ca2+ uptake observed in response to histamine or ATP was due to an increased ER Ca2+-store content or an increased Ca2+ release from the ER via IP3Rs. Altogether, these findings indicate that the increase in contact site formation between the ER and mitochondria results in a more efficient flux of Ca2+ from the ER towards mitochondria upon IP3R opening. Interestingly, mitochondrial transport is regulated through the Ca2+-sensitive mitochondria Rho GTPases (Miro1/2) (34,39–41). Binding of Ca2+ to the EF-hands of the Miro GTPases causes mitochondrial transport arrest through the dismantling of the motor complex required for trafficking of mitochondria along the cytoskeletal tracks (29,39,42,43). Thus, high local Ca2+ concentration, e.g. at active synapses, can provide a mechanism for the cell to retain mitochondria in these regions of high energetic demand.
The fact that Miro1/2 was found to be present at ER–mitochondria MCSs, which are known to generate Ca2+ microdomains, could indicate that MAMs also participate in the regulation of mitochondrial trafficking (15,25,44). Although a mechanism for MAM-controlled mitochondrial trafficking has not been shown directly, both organelles do display a coordinated motility (16). In agreement with this, we found that HSAN-causing mutations in ATL3 resulted in disturbed mitochondrial trafficking upon upregulated ER–mitochondria contact site formation (Figs 2–4). Interestingly, similar findings were reported for a mutation causing amyotrophic lateral sclerosis (ALS) in the ER–mitochondria tethering protein Vesicle-associated membrane protein-associated protein B/C (VAPB), which was found to increase the contact and Ca2+ transfer between both organelles (45–47). Mitochondrial trafficking was also found to be reduced in cells expressing mutant VAPB, an effect that was absent when a Ca2+-insensitive mutant of Miro1 was expressed. These findings are in line with the results presented here and indicate that upregulated ER–mitochondria contact may indeed affect mitochondrial trafficking through excessive Ca2+ transfer (46). ER–mitochondria contacts were not examined in the ATL1 P342S patient-derived cells (37); however, also in this model, a primary defect in the ER resulted in a mitochondrial trafficking defect, indicating that our findings for HSAN-causing mutations in ATL3 could also be relevant for genetic defects causing HSP.
Regardless of the role of contact sites with the ER, it is indisputable that a defect in mitochondrial trafficking and distribution within axons will have a strong impact on neuronal functioning and survival. Mitochondrial targeting is crucial, not only for local energy production, but also to influence synaptic activity through the modulation of Ca2+ signalling (32) and during injury repair (36). The critical role of mitochondrial positioning for neuronal survival is underscored by the involvement of defects in mitochondrial fission, fusion, trafficking and turnover in neurodegenerative disorders including Charcot–Marie–Tooth neuropathies, HSPs, Parkinson’s disease and Alzheimer’s dementia (31,34). Therefore, these results underscore the functional interdependence of subcellular organelles in health and disease and provide important insights in the pathogenic mechanism underlying mutations in ATL3 and ER-shaping proteins in general.
Materials and Methods
Cloning, plasmids and cell lines
All ATL3-expressing vectors were created using Gateway recombination system (ThermoFisher Scientific). The WT and Y192C ATL3 cDNA was amplified from a previously reported vector (8) and subsequently cloned into a pDONR221 using the BP clonase (ThermoFisher Scientific). The P338R open reading-frame (ORF) was synthesized by Genscript and cloned in the same way. After sequence validation, expression plasmids were created using the LR clonase (ThermoFisher Scientific) in combination with the pLenti6/V5-Dest (ThermoFisher Scientific) to allow C-terminal V5-tagging or the pLenti6/EGFP-Dest to allow C-terminal GFP-tagging. These constructs were used for transient transfection and/or lentivirus production, as previously described (48). ATL3-mCherry or ATL3-BFP plasmids were created through multisite-gateway cloning. Here, the mCherry or BFP ORFs were amplified and subcloned into the pDONR-P2RP3. Expression vectors were created by combining the aforementioned ATL3 entry clones with the mCherry C-terminal entry clone and a mock N-terminal entry clone in the LR reaction. The mito-LAR-geco construct was a gift from Dr Robert Campbell (49). The pBABE-puro mCherry-EGFP-LC3B construct was a gift from Dr Jayanta Debnath (24). The pTagBFP-N plasmid, from which the BFP ORF was PCR amplified, was a gift from Dr Peter Ponsaerts.
All cell lines were cultured in a 37 C and 5% CO2 atmosphere. HeLa cells were cultured in high-glucose DMEM (ThermoFisher Scientific) supplemented with 10% fetal bovine serum (FBS; ThermoFisher Scientific). Human Skin Fibroblasts, use of which was approved by the local medical ethical committee, were cultured in the same way. Fibroblasts were derived from a 67-year-old female patient harboring the ATL3Y192C mutation. She suffered from adult-onset progressive sensory loss which resulted in foot ulcers, arthropathy and foot bone destruction. Wound healing was delayed. Upper extremities were clinically not affected, and autonomic function was normal. No signs of muscular involvement or affection of upper motor neurons were observed.
Mitotracker live cell imaging
HeLa cells were seeded in a 24-well glass bottom plates (Greiner) and transiently transfected with ATL3-GFP using polyethylenimine. After 24 h, the cells were stained with MitoTracker Red FM (ThermoFisher Scientific) according to the manufacturer’s guidelines and placed in FluoroBrite DMEM (ThermoFisher Scientific) supplemented with 5 mm glutamine and 10% FBS (ThermoFisher Scientific). Fibroblasts were seeded and stained in a similar way and imaged in DMEM +10% FBS. The cells were imaged on the UltraVIEW VoX spinning disk confocal microscope (Perkin Elmer), equipped with a Hamamatsu C9100–50 EMCCD camera and a 60×/1.4 oil immersion objective, controlled through the Volocity software. Individual images (1000 × 1000 pixels) had a pixel dimension of 118 nm. Time lapse imaging was performed for 2 min using a 2 s interval between frames (unless indicated otherwise).
The first frame was used to analyse the mitochondrial morphology using ImageJ/FIJI (50,51). To enhance the mitochondrial signal, Gaussian Blur (1-pixel radius), Mexican Hat (9 × 9 kernel) and background subtraction (sigma, 50 pixels) filtering was applied. Mitochondria were segmented using the intersection of two masks, the first created by global intensity thresholding (Triangle method) and the second by local intensity (Phansalkar, 15-pixel radius). On the mask, a particle analysis was applied to eliminate mitochondrial segments that are too small (<7 pixels) or too large (>100 000 pixels) and to measure the shape descriptors of every remaining mitochondrial element, including circularity [4π × area/(perimeter)^2], aspect ratio (major_axis/minor_axis of fitted ellipse), roundness [4 × area/(π × (major_axis)^2)] and solidity (area/convex area). The complete filtering, segmentation and measurement procedure were executed using an ImageJ macro to process all images in batch (50). To include only HeLa cells containing ATL3-GFP, polygonal regions of interest were used to select GFP-postive cells.
Mitochondrial dynamics were analysed using the Difference tracker plugin for ImageJ (52), after which the dynamic pixels were summed for the entire stack (comprising the 60 frames of the 2 min time lapse sequence) and normalized to the initial Mitotracker signal for each cell (Supplementary Material, Fig. S3 and Video).
Primary cortical neuron cultures
All mouse experiments were carried out with the approval of the Ethical Committee for Laboratory Animals (University of Antwerp). Mice were housed under the care of the Animal Facility Interfaculty Unit, which is accredited by the Association for Assessment and Accreditation of Laboratory Animals. Cortical neurons were extracted from E15.5 C57BL/6 embryos and cultured in neurobasal medium (ThermoFisher Scientific), supplemented with 2% FBS (ThermoFisher Scientific), 2 mm glutamine, 100 μg/ml penicillin–streptomycin, 2% B27 and 4 g/L d-glucose on poly-D-lysine-coated coverslips. After 4 h incubation at 37°C and 7% CO2, appropriate lentivirus was added to the medium. Cultures were kept for at least 5 days before fixation using 2% paraformaldehyde (PFA) and 4% sucrose (Sigma Aldrich) in phosphate buffered saline (PBS). After permeabilization with 0.1% Triton-X-100 in PBS, coverslips were placed in blocking solution (2% FBS and 2% bovine serum albumin in PBS). Cells were incubated overnight at 4°C with the primary rabbit-anti-TOM20 (Abcam) antibody in 10% blocking solution. Goat-anti-rabbit secondary antibody labelled with Alexa Fluor 488 or 594 (ThermoFisher Scientific) was used at a 1:500 dilution. After washing in PBS, cells were incubated with Hoechst for 10 min at room temperature prior to mounting the coverslips using DAKO mounting medium. Imaging was performed on a Zeiss LSM700 confocal microscope using a 40×/1.3 Plan-Neofluar objective. Image analysis was done in ImageJ/FIJI (50,51).
Transmission electron microscopy
In two independent transductions, HeLa cells were infected with lentivirus to allow stable expression of ATL3-GFP or ATL3-V5. Blasticidin-selected HeLa cells were seeded on glass coverslips and allowed at least 24 h to attach. Human skin fibroblasts were seeded in the same way. Cultures were fixed in 4% PFA and 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer, pH 7.2 for 4 h at room temperature followed by fixation overnight at 4°C. After washing, cells were subsequently dehydrated through a graded ethanol series, including a bulk staining with 1% uranyl acetate at the 50% ethanol step followed by embedding in Spurr’s resin.
Ultrathin sections of a gold interference color were cut using an ultra-microtome (Leica EM UC6), followed by a post-staining in a Leica EM AC20 for 40 min in uranyl acetate at 20°C and for 10 min in lead stain at 20°C. Sections were collected on formvar-coated copper slot grids. Grids were viewed with a JEM 1400plus transmission electron microscope operating at 60 kV. Image analysis was done using ImageJ/FIJI (50,51). ER–mitochondria contact site size was determined by measuring the length of mitochondrial boundaries where the ER was closer than 50 nm. To compare mitochondrial morphology and to measure the mitochondrial surface density as the total amount of mitochondrial surface in the cellular volume, a stereological procedure was used. On each image, straight horizontal line probe overlays were generated at 200 nm intervals and all intersections between these stereological line probes and the mitochondrial boundary were counted. Using these intersection counts, the total mitochondrial surface (in 3D) per unit of examined volume is determined by 2× number of intersections per length of the stereological line probes (www.stereology.info). To correct for the amount of cytoplasm that was imaged, this value was divided by the cytoplasm volume per imaged volume, which was measured by and is equal to the area of cytoplasm per image area on 2D images. The resulting mitochondrial surface in 3D per cytoplasm volume is a parameter describing mitochondrial morphology.
Fura-2 measurements
Cytosolic Ca2+ was quantified in HeLa cells stably expressing ATL3-V5 as described previously (53). Briefly, cells grown to 90% confluence in a 96-well plate (Greiner) were loaded with 1 mm Fura-2 AM for 30 min at 25°C in modified Krebs solution (135 mm NaCl, 5.9 mm KCl, 1.2 mm MgCl2, 11.5 mm glucose, 1.5 mm CaCl2 and 11.6 mm HEPES pH 7.3). After a 30 min de-esterification step, Fura-2 fluorescence emission at 510 nm was monitored on a FlexSTation 3 microplate reader (Molecular Devices) in the absence of extracellular Ca2+ (3 mm EGTA was added 30 s after the start of monitoring) by alternately exciting the Ca2+ reporter at 340 nm and 380 nm. Thapsigargin (1 μm), ionomycin (1 μm), histamine (100 μm) or ATP (30 μm) were added as indicated, typically 30 s after the addition of EGTA. All data were obtained in triplicate and are plotted as F340/F380.
Mitochondrial Ca2+ measurements
The mito-LAR-GECO Ca2+ reporter was co-transfected in HeLa cells with ATL3-GFP using linear polyethylenimine MW 25000 (Polysciences). After 24 h, cells were seeded in an 8-well μ-slides (ibidi) and allowed to reach the exponential growth phase for additional 24 h; 50% confluent cells were then placed in a modified Krebs buffer (5 mm KCl, 125 mm NaCl, 5.5 mm glucose, 1 mm Na2HPO4, 1 mm MgSO4 and 20 mm HEPES pH 7.4) and incubated at 37°C on a Zeiss Axiovert 200 M microscope equipped with a 20×/0.8 plan-apochromat objective and an AxioCam MR Rev3 camera. Mitochondrial Ca2+ reporter fluorescence was imaged using 540 nm excitation and a DsRed emission filter at one frame per second for 60 s. After 10 frames, stimulation buffer was added resulting in a final concentration of 10 mm EGTA and 100 μm histamine or 60 μm ATP, as indicated. After background subtraction, average mito-LAR-GECO fluorescence intensity per frame (F) was determined for each cell in ImageJ/FIJI (50,51). In R (http://www.R-project.org/), this intensity was normalized to the initial fluorescence intensity (F0). Data from three independent experiments was pooled for plotting of F/F0 and statistical analysis of maximal F/F0 (Fmax/F0) and area under the curve.
Lipidomics analysis
HeLa cells were lentivirally transduced with ATL3-GFP or ATL3-V5 as described above and kept under blasticidin selection. Cell pellets of 500 000 HeLa cells were used for lipidomic analysis, using seven technical replicates. Total lipid extraction using the Folch method followed by liquid chromatography coupled to high-resolution mass spectrometry for comparative lipidomic analysis was performed by Profilomic, as described (54). This experiment was performed twice in independently transduced cell lines.
Autophagy monitoring
For western blot analysis of autophagic flux, HeLa cells stably transduced with ATL3-GFP or ATL3-V5 were grown to 90% confluent and serum deprived in the presence or absence of bafilomycin A1 (10 nm) as indicated. Protein extraction was done using 0.5% NP40 in PBS supplemented with protease inhibitors (Roche). After 30 min incubation on ice, cell debris was pelleted and the supernatant was diluted in LDS sample buffer (ThermoFisher Scientific) supplemented with 100 mm DTT and boiled for 5 min at 95°C. Samples were run on 18% SDS-PAGE gels or 4–12% Bis-Tris NuPAGE gels (ThermoFisher Scientific), and proteins were transferred to nitrocellulose membrane (GE Healthcare). Blocking of the membrane was performed using 5% milk powder diluted in PBS, supplemented with 0.1% Tween 20. The following primary antibodies were used: rabbit-anti-LC3B (Cell Signaling), mouse IgG1-anti-beta actin (Sigma), mouse-anti-p62 (Santa Cruz). HRP-conjugated goat-anti-rabbit (Jackson), goat-anti-mouse (Jackson) and goat-anti-mouse IgG1 (Southern Biotech) were used as secondary antibodies. Blots were developed using Enhanced Chemiluminescence ECL Plus™ (ThermoFisher Scientific) and imaged with an ImageQuant imager (GE Healthcare).
For immunofluorescence analysis of autophagic flux, HeLa or COS-1 cells were seeded in 35 mm imaging dishes (Mattek) and grown to 50% confluence. The pBABE-puro mCherry-EGFP-LC3B construct was co-transfected with ATL3-BFP expression plasmids using lipofectamine LTX. After 24 h, live cells were imaged on a Zeiss LSM700 microscope equipped with a 37°C and 5% CO2 incubation chamber and a 40×/1.3 Plan-Neofluar objective. The 405 nm laser was used to detect ATL3-BFP. The 488 and 555 nm lasers were used to detect GFP and mCherry, respectively.
Statistical analysis
All statistics were performed using the Graphpad Prism 7 software. Specific tests used are indicated in the figure legends.
Supplementary Material
Acknowledgements
The authors would like to thank Robert Campbell, Peter Ponsaerts and Jayanta Debnath for providing plasmids. The authors would also like to thank Tomas Luyten, Kirsten Welkenhuyzen and Haidar Akl for technical support with FlexStation measurements. Special thanks to Michiel de Bruyne, Peter Verstraelen, Isabel Pintelon and the Antwerp Center for Advanced Microscopy for advice and technical support.
Conflicts of Interest statement. None declared.
Funding
Fund for Scientific Research (PhD fellowship, FWO-Flanders, Belgium, to M.K.); FWO, University of Antwerp, the Hercules Foundation, the ‘Association Belge contre les Maladies Neuro-Musculaires’ (to V.T.), the Medical Foundation Queen Elisabeth (to V.T.), the European Union FP7/2007–2013 NeurOmics project `Integrated European –omics research project for diagnosis and therapy in rare neuromuscular and neurodegenerative diseases' (2012-305121 to V.T.), H2020 project ‘Solving the unsolved rare diseases’ (Solve-RD; 2017-779257 to V.T.); FWO, Ghent University and GROUP-ID (to S.J.); BMBF (13GW0128A and 01GM1513D to F.J.M.), Deutsche Forschungsgemeinschaft (DFG MU 3231/3-1 to F.J.M.); FWO (G0A3416N to G.B.), Research Council - KU Leuven (OT14/101 to G.B.). G.B. is the coordinator of the FWO research community ‘Ca2+ signaling in health, disease & therapy’ (W001917N).
References
- 1. Shemesh T., Klemm R.W., Romano F.B., Wang S., Vaughan J., Zhuang X. et al. (2014) A model for the generation and interconversion of ER morphologies. Proc. Natl. Acad. Sci. U.S.A., 111, E5243–E5251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shibata Y., Shemesh T., Prinz W.A., Palazzo A.F., Kozlov M.M. and Rapoport T.A. (2010) Mechanisms determining the morphology of the peripheral ER. Cell, 143, 774–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Westrate L.M., Lee J.E., Prinz W.A. and Voeltz G.K. (2014) Form follows function: the importance of endoplasmic reticulum shape. Annu. Rev. Biochem., 84, 791–811. [DOI] [PubMed] [Google Scholar]
- 4. Hu J., Shibata Y., Zhu P.-P., Voss C., Rismanchi N., Prinz W.A. et al. (2009) A class of dynamin-like GTPases involved in the generation of the tubular ER network. Cell, 138, 549–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Orso G., Pendin D., Liu S., Tosetto J., Moss T.J., Faust J.E. et al. (2009) Homotypic fusion of ER membranes requires the dynamin-like GTPase atlastin. Nature, 460, 978–983. [DOI] [PubMed] [Google Scholar]
- 6. Fischer D., Schabhüttl M., Wieland T., Windhager R., Strom T.M. and Auer-Grumbach M. (2014) A novel missense mutation confirms ATL3 as a gene for hereditary sensory neuropathy type 1. Brain, 137, e286. [DOI] [PubMed] [Google Scholar]
- 7. Guelly C., Zhu P.-P., Leonardis L., Papić L., Zidar J., Schabhüttl M. et al. (2011) Targeted high-throughput sequencing identifies mutations in atlastin-1 as a cause of hereditary sensory neuropathy type I. Am. J. Hum. Genet., 88, 99–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kornak U., Mademan I., Schinke M., Voigt M., Krawitz P., Hecht J. et al. (2014) Sensory neuropathy with bone destruction due to a mutation in the membrane-shaping atlastin GTPase 3. Brain, 137, 683–692. [DOI] [PubMed] [Google Scholar]
- 9. Zhao J., Matthies D.S., Botzolakis E.J., Macdonald R.L., Blakely R.D. and Hedera P. (2008) Hereditary spastic paraplegia-associated mutations in the NIPA1 gene and its caenorhabditis elegans homolog trigger neural degeneration in vitro and in vivo through a gain-of-function mechanism. J. Neurosci., 28, 13938–13951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fassier C., Hutt J.A., Scholpp S., Lumsden A., Giros B., Nothias F. et al. (2010) Zebrafish atlastin controls motility and spinal motor axon architecture via inhibition of the BMP pathway. Nat. Neurosci., 13, 1380–1387. [DOI] [PubMed] [Google Scholar]
- 11. Lee M., Paik S.K., Lee M.-J., Kim Y.-J., Kim S., Nahm M. et al. (2009) Drosophila atlastin regulates the stability of muscle microtubules and is required for synapse development. Dev. Biol., 330, 250–262. [DOI] [PubMed] [Google Scholar]
- 12. Summerville J.B., Faust J.F., Fan E., Pendin D., Daga A., Formella J. et al. (2016) The effects of ER morphology on synaptic structure and function in Drosophila melanogaster. J. Cell Sci., 129, 1635–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Phillips M.J. and Voeltz G.K. (2015) Structure and function of ER membrane contact sites with other organelles. Nat. Rev. Mol. Cell Biol., 17, 69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Raffaello A., Mammucari C., Gherardi G. and Rizzuto R. (2016) Calcium at the center of cell signaling: interplay between endoplasmic reticulum, mitochondria, and lysosomes. Trends Biochem. Sci., 41, 1035–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Krols M., Isterdael G., Asselbergh B., Kremer A., Lippens S., Timmerman V. et al. (2016) Mitochondria-associated membranes as hubs for neurodegeneration. Acta Neuropathol., 131, 505–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Friedman J.R., Webster B.M., Mastronarde D.N., Verhey K.J. and Voeltz G.K. (2010) ER sliding dynamics and ER-mitochondrial contacts occur on acetylated microtubules. J. Cell Biol., 190, 363–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Csordas G., Thomas A.P. and Hajnoczky G. (1999) Quasi-synaptic calcium signal transmission between endoplasmic reticulum and mitochondria. EMBO J., 18, 96–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Friedman J.R., Lackner L.L., West M., DiBenedetto J.R., Nunnari J. and Voeltz G.K. (2011) ER tubules mark sites of mitochondrial division. Science, 334, 358–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hamasaki M., Furuta N., Matsuda A., Nezu A., Yamamoto A., Fujita N. et al. (2013) Autophagosomes form at ER–mitochondria contact sites. Nature, 495, 389–393. [DOI] [PubMed] [Google Scholar]
- 20. Prudent J., Zunino R., Sugiura A., Mattie S., Shore G.C. and McBride H.M. (2015) MAPL SUMOylation of Drp1 stabilizes an ER/mitochondrial platform required for cell death. Mol. Cell, 59, 941–955. [DOI] [PubMed] [Google Scholar]
- 21. Vance J.E. (1990) Phospholipid synthesis in a membrane fraction associated with mitochondria. J. Biol. Chem., 265, 7248–7256. [PubMed] [Google Scholar]
- 22. Krols M., Detry S., Asselbergh B., Almeida-Souza L., Kremer A., Lippens S., Rycke R., De Winter V., Müller F.-J., Kurth I. et al. (2018) Sensory neuropathy-causing mutations in ATL3 cause aberrant ER membrane tethering. Cell Rep., 23, 2026–2038. [DOI] [PubMed] [Google Scholar]
- 23. Klionsky D.J., Abdelmohsen K., Abe A., Abedin M.J., Abeliovich H., Acevedo Arozena A. et al. (2016) Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy, 12, 1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. N'Diaye E.-N., Kajihara K.K., Hsieh I., Morisaki H., Debnath J. and Brown E.J. (2009) PLIC proteins or ubiquilins regulate autophagy-dependent cell survival during nutrient starvation. EMBO Rep., 10, 173–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Csordás G., Várnai P., Golenár T., Roy S., Purkins G., Schneider T.G. et al. (2010) Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Mol. Cell, 39, 121–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wu J., Prole D.L., Shen Y., Lin Z., Gnanasekaran A., Liu Y. et al. (2014) Red fluorescent genetically encoded Ca2+ indicators for use in mitochondria and endoplasmic reticulum. Biochem. J., 464, 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Naon D., Zaninello M., Giacomello M., Varanita T., Grespi F., Lakshminaranayan S. et al. (2016) Critical reappraisal confirms that mitofusin 2 is an endoplasmic reticulum-mitochondria tether. Proc. Natl. Acad. Sci. U.S.A., 113, 11249–11254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Scorrano L., Oakes S.A., Opferman J.T., Cheng E.H., Sorcinelli M.D., Pozzan T. et al. (2003) BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science, 300, 135–139. [DOI] [PubMed] [Google Scholar]
- 29. Saotome M., Safiulina D., Szabadkai G., Das S., Fransson Å., Aspenström P. et al. (2008) Bidirectional Ca2+−dependent control of mitochondrial dynamics by the Miro GTPase. Proc. Natl. Acad. Sci. U.S.A., 105, 20728–20733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hubner C.A. and Kurth I. (2014) Membrane-shaping disorders: a common pathway in axon degeneration. Brain, 137, 3109–3121. [DOI] [PubMed] [Google Scholar]
- 31. Court F.A. and Coleman M.P. (2012) Mitochondria as a central sensor for axonal degenerative stimuli. Trends Neurosci., 35, 364–372. [DOI] [PubMed] [Google Scholar]
- 32. Vaccaro V., Devine M.J., Higgs N.F. and Kittler J.T. (2017) Miro1-dependent mitochondrial positioning drives the rescaling of presynaptic Ca2+ signals during homeostatic plasticity. EMBO Rep., 18, 231–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Villegas R., Martinez N.W., Lillo J., Pihan P., Hernandez D., Twiss J.L. et al. (2014) Calcium release from intra-axonal endoplasmic reticulum leads to axon degeneration through mitochondrial dysfunction. J. Neurosci., 34, 7179–7189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sheng Z.-H. (2014) Mitochondrial trafficking and anchoring in neurons: new insight and implications. J. Cell Biol., 204, 1087–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ruthel G. and Hollenbeck P.J. (2003) Response of mitochondrial traffic to axon determination and differential branch growth. J. Neurosci., 23, 8618–8624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhou B., Yu P., Lin M.-Y., Sun T., Chen Y. and Sheng Z.-H. (2016) Facilitation of axon regeneration by enhancing mitochondrial transport and rescuing energy deficits. J. Cell Biol., 214, 103–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhu P.-P., Denton K.R., Pierson T.M., Li X.-J. and Blackstone C. (2014) Pharmacologic rescue of axon growth defects in a human iPSC model of hereditary spastic paraplegia SPG3A. Hum. Mol. Genet., 23, 5638–5648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fowler P.C. and O'Sullivan N.C. (2016) ER-shaping proteins are required for ER and mitochondrial network organization in motor neurons. Hum. Mol. Genet., 25, 2827–2837. [DOI] [PubMed] [Google Scholar]
- 39. Fransson Å., Ruusala A. and Aspenström P. (2006) The atypical Rho GTPases Miro-1 and Miro-2 have essential roles in mitochondrial trafficking. Biochem. Biophys. Res. Commun., 344, 500–510. [DOI] [PubMed] [Google Scholar]
- 40. Glater E.E., Megeath L.J., Stowers R.S. and Schwarz T.L. (2006) Axonal transport of mitochondria requires milton to recruit kinesin heavy chain and is light chain independent. J. Cell Biol., 173, 545–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Stowers R.S., Megeath L.J., Górska-Andrzejak J., Meinertzhagen I.A. and Schwarz T.L. (2002) Axonal transport of mitochondria to synapses depends on milton, a novel Drosophila protein. Neuron, 36, 1063–1077. [DOI] [PubMed] [Google Scholar]
- 42. Macaskill A.F., Rinholm J.E., Twelvetrees A.E., Arancibia-Carcamo I.L., Muir J., Fransson Å. et al. (2009) Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron, 61, 541–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang X. and Schwarz T.L. (2009) The mechanism of Ca2+ −dependent regulation of kinesin-mediated mitochondrial motility. Cell, 136, 163–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lee S., Lee K.-S., Huh S., Liu S., Lee D.-Y., Hong S.H. et al. (2016) Polo kinase phosphorylates miro to control ER-mitochondria contact sites and mitochondrial Ca(2+) homeostasis in neural stem cell development. Developmental Cell, 37, 174–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. De Vos K.J., Morotz G.M., Stoica R., Tudor E.L., Lau K.F., Ackerley S. et al. (2012) VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum. Mol. Genet., 21, 1299–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mórotz G.M., De Vos K.J., Vagnoni A., Ackerley S., Shaw C.E. and Miller C.C.J. (2012) Amyotrophic lateral sclerosis-associated mutant VAPBP56S perturbs calcium homeostasis to disrupt axonal transport of mitochondria. Hum. Mol. Genet., 21, 1979–1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Stoica R., De Vos K.J., Paillusson S.E.B., Mueller S., Sancho R.M., Lau K.-F. et al. (2014) ER–mitochondria associations are regulated by the VAPB–PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat. Commun., 5, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Salmon P. and Trono D. (2006) Production and titration of lentiviral vectors. Curr. Protoc. Neurosci, 4.21, 1–24. [DOI] [PubMed] [Google Scholar]
- 49. Wu F., Hu X., Bian X., Liu X. and Hu J. (2014) Comparison of human and Drosophila atlastin GTPases. Protein Cell, 6, 139–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T. et al. (2012) Fiji: an open-source platform for biological-image analysis. Nat. Methods, 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Schneider C.A., Rasband W.S. and Eliceiri K.W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Methods, 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Andrews S., Gilley J. and Coleman M.P. (2010) Difference tracker: ImageJ plugins for fully automated analysis of multiple axonal transport parameters. J. Neurosci. Methods, 193, 281–287. [DOI] [PubMed] [Google Scholar]
- 53. Decuypere J.-P., Welkenhuyzen K., Luyten T., Ponsaerts R., Dewaele M., Molgó J. et al. (2011) Ins(1,4,5)P3 receptor-mediated Ca2+ signaling and autophagy induction are interrelated. Autophagy, 7, 1472–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Seyer A., Boudah S., Broudin S., Junot C. and Colsch B. (2016) Annotation of the human cerebrospinal fluid lipidome using high resolution mass spectrometry and a dedicated data processing workflow. Metabolomics, 12, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.