

Abstract
Aminoacyl-tRNA synthetases (ARSs) are critical for protein translation. Pathogenic variants of ARSs have been previously associated with peripheral neuropathy and multisystem disease in heterozygotes and homozygotes, respectively. We report seven related children homozygous for a novel mutation in tyrosyl-tRNA synthetase (YARS, c.499C > A, p.Pro167Thr) identified by whole exome sequencing. This variant lies within a highly conserved interface required for protein homodimerization, an essential step in YARS catalytic function. Affected children expressed a more severe phenotype than previously reported, including poor growth, developmental delay, brain dysmyelination, sensorineural hearing loss, nystagmus, progressive cholestatic liver disease, pancreatic insufficiency, hypoglycemia, anemia, intermittent proteinuria, recurrent bloodstream infections and chronic pulmonary disease. Related adults heterozygous for YARS p.Pro167Thr showed no evidence of peripheral neuropathy on electromyography, in contrast to previous reports for other YARS variants. Analysis of YARS p.Pro167Thr in yeast complementation assays revealed a loss-of-function, hypomorphic allele that significantly impaired growth. Recombinant YARS p.Pro167Thr demonstrated normal subcellular localization, but greatly diminished ability to homodimerize in human embryonic kidney cells. This work adds to a rapidly growing body of research emphasizing the importance of ARSs in multisystem disease and significantly expands the allelic and clinical heterogeneity of YARS-associated human disease. A deeper understanding of the role of YARS in human disease may inspire innovative therapies and improve care of affected patients.
Introduction
Translation of the genetic code to protein is essential for cell survival in all known organisms. A group of enzymes called aminoacyl-transfer ribonucleic acid (tRNA) synthetases (ARSs) catalyze the covalent attachment of each amino acid to its corresponding tRNA in a two-step aminoacylation process. An amino acid and adenosine triphosphate (ATP) first bind to the ARS, forming an aminoacyl adenylate intermediate. The ARS then binds the cognate tRNA to facilitate transfer of the amino acid to form an acyl tRNA (1,2). Acyl tRNAs subsequently transfer amino acids to growing polypeptide chains and ensure accurate translation of genetic information. Though best known for their role in mRNA translation, ARSs are increasingly recognized to have secondary (‘non-canonical’) roles in transcription regulation, splicing, immune function, angiogenesis, apoptosis and cell stress (3,4).
The ARSs are highly conserved across species and ubiquitously expressed among human tissues, underscoring their critical role in cell function (3). In human cells, most ARSs localize to either the cytoplasm (denoted as ‘ARS’) or mitochondria (‘ARS2’), but a small number are bifunctional (3,5). The ARSs are further classified based on structural characteristics of the catalytic domain. Class I ARSs contain a parallel β-sheet nucleotide-binding (Rossmann) fold, whereas Class II ARSs share a series of homologous amino acid sequences designated motifs 1, 2 and 3 (3).
ARSs were first linked to human disease in 2003, when heterozygosity for variants in glycyl-tRNA synthetase (GARS) was found in subjects with Charcot–Marie–Tooth (CMT) disease type 2D (MIM 601472) and distal spinal muscular atrophy type V (MIM 600794) (6). Subsequent reports described heterozygosity for missense and in-frame deletions in other ARSs genes, including tyrosyl-tRNA synthetase (YARS, MIM 603623), underlying various forms of CMT (6–9). However, the molecular pathophysiology of CMT-associated ARS mutations has not been defined (10).
More recently, diseases beyond the peripheral nervous system have been associated with recessive, loss-of-function variants in multiple ARS-encoding genes (11–26). Nowaczyk et al. described two siblings with compound heterozygosity in YARS catalytic and C-terminal domains and somatic growth failure, hypertriglyceridemia, developmental delay, liver dysfunction, lung cysts and abnormal brain white matter (27). Additionally, homozygosity for mutations just outside the YARS catalytic domain was described in an adult with retinal degeneration, hearing impairment, agenesis of the corpus callosum, primary amenorrhea and liver disease (28). Mutations in mitochondrial YARS2 have been associated with myopathy, lactic acidosis and sideroblastic anemia (29–33). While the above case reports raised interest in the role of YARS mutations in recessive phenotypes, the lack of a strong genetic argument for pathogenicity and the differences in patient phenotypes warrant further assessments of YARS variants in human disease.
Here, we report the natural history of seven related children from an Amish kindred homozygous for a pathogenic missense variant in the YARS catalytic domain (c.499C > A, p.Pro167Thr) identified by whole exome sequencing. Affected children had multisystem disease including sensorineural hearing loss, nystagmus, developmental delay, growth failure, chronic anemia, hypoglycemia, progressive cholestatic liver disease and renal dysfunction. Related adults heterozygous for YARS p.Pro167Thr had no evidence of peripheral neuropathy. Yeast complementation assays demonstrate that the YARS p.Pro167Thr substitution causes reduced gene function and poor growth. Protein–protein interaction studies in human embryonic kidney cells show that YARS p.Pro167Thr results in reduced dimerization, which is essential for YARS catalytic function (34–37).
Results
Identification of YARS c.499C > A, p.Pro167Thr
The proband presented to the Clinic for Special Children in January 2014 and family history revealed three deceased relatives with similar multisystem disease (Fig. 1A). Targeted allele testing was non-diagnostic for population-specific genetic conditions with an overlapping phenotype, including prolidase deficiency [PEPD c.793C > T, (MIM 170100)], pyruvate kinase deficiency [PKLR c.1436G > A, (MIM 266200)], familial intrahepatic cholestasis [ATP8B1 c.923G > T, (MIM 211600)] and Cockayne syndrome type B [ERCC6 IVS14 + 1G > T, (MIM 133540)]. High-resolution chromosomal microarray analysis was uninformative.
Figure 1.
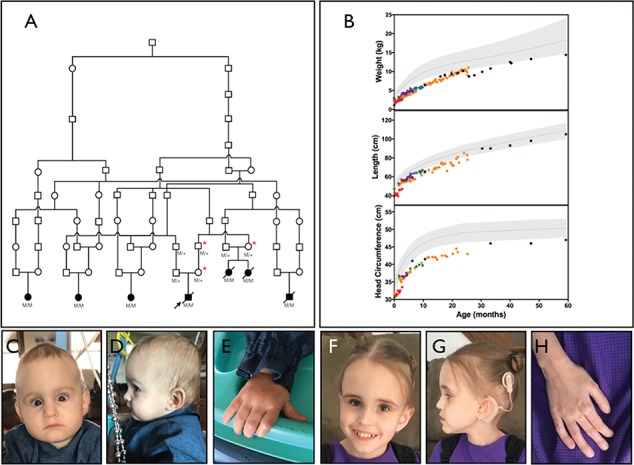
Pedigree, physical features and growth for YARS c.499C > A, p.Pro167Thr homozygotes. (A) Family pedigree for seven related children homozygous for the novel YARS variant. Solid shapes represent YARS p.Pro167Thr homozygotes (M/M) described in the current report. Red asterisks denote related YARS p.Pro167Thr heterozygotes (M/+) who underwent detailed neurologic examination and electrophysiological studies. (B) All children were microcephalic with poor weight gain. Five children had linear growth below the third percentile for age. For growth curves, shaded areas represent reference ranges for age. Colors represent individual children. (C–E) An 18-month-old girl homozygous for YARS p.Pro167Thr with a long forehead, strabismus, deeply set eyes (C) and small hands (E). (F–H) A five-year-old girl homozygous for YARS p.Pro167Thr with a long forehead (F), thin hair, low-set ears (G) and digital clubbing (H).
We then proceeded to whole exome sequencing of the proband and nine related individuals (parents, siblings and maternal grandparents) and identified homozygous candidate variants in two genes: YARS (.g.chr1:33272094(G > T); NM_003680.3:c.499C > A; NP_003671.1:p.Pro167Thr) and KDELC1 (.g.chr13:103449267C > T; NM_024089.2:c.275G > A; NP_076994.2:p.Arg92Gln). The KDELC1 variant was excluded since it did not segregate appropriately in the extended pedigree and genotyping of additional unaffected Amish samples revealed four homozygotes for the variant. Sanger sequencing and high-resolution melt analysis confirmed homozygosity of YARS c.499C > A in the proband, his three deceased relatives and three additional living children with a concordant clinical phenotype (Table 1).
Table 1.
Clinical phenotype
| Number of patients | ||
|---|---|---|
| Clinical finding | N | % |
| Birth history | ||
| Premature birth | 3 | 43 |
| Placental abnormalities | 5 | 71 |
| Physical characteristics | ||
| Deep-set eyes | 7 | 100 |
| Central nervous system | ||
| Microcephaly | 7 | 100 |
| Bilateral sensorineural hearing loss | 7 | 100 |
| Nystagmus | 6 | 86 |
| Gross motor delay | 7 | 100 |
| Expressive language delay | 7 | 100 |
| Cardiopulmonary | ||
| Chronic pulmonary disease | 2 | 29 |
| Ventricular septal defect | 1 | 14 |
| Gastrointestinal | ||
| Poor growth | 7 | 100 |
| Nasogastric or gastrostomy feedings | 7 | 100 |
| Cholestatic liver disease | 7 | 100 |
| Pancreatic insufficiency | 7 | 100 |
| Hematology | ||
| Transfusion dependent anemia | 3 | 43 |
| Renal | ||
| Intermittent proteinuria +/− hematuria | 4 | 57 |
| Endocrine | ||
| Severe hypoglycemia | 5 | 71 |
| Infectious disease | ||
| Frequent bloodstream infections | 3 | 43 |
Summary of clinical findings in seven YARS c.499C > A, p.Pro167Thr homozygotes.
YARS c.499C > A, p.Pro167Thr homozygote clinical phenotype
Early clinical course and physical characteristics
Three affected infants were born prematurely (27–31 weeks gestation) due to placental abruption (n = 1) or preterm labor (n = 2) and two mothers noted decreased fetal movement at the end of gestation. Placental abnormalities were noted for five (71%) infants and included thin umbilical cord (n = 1), cystic mass displacing the cord (n = 1), ‘dried grapes’ appearance with histology showing multiple infarcts (n = 1) and irregularly shaped placenta with friable maternal side and fatty deposits on the infant side (n = 2).
Four (57%) infants had intrauterine growth restriction (0.2–9.7 percentile). Postnatal weight and head circumference remained below the third percentile for all children and length was below third percentile for all but one (Fig. 1B). Fortified formulas, elemental formulas and supplemental oils were used for enteral feeding. All children used nasogastric or gastrostomy tubes for nutritional support and two received chronic parenteral nutrition via central line. Affected infants had deep-set eyes but no other distinctive facial characteristics during infancy and early childhood (Fig. 1C–E). The oldest living child (5 years of age) has a long forehead, thin hair, low-set ears and digital clubbing (Fig. 1F–H).
Development and central nervous system
Development was delayed to variable degrees in all children. The four deceased children died between age 10 and 25 months, and only one was able to sit independently (age 20 months). All cooed but none babbled. The two youngest living children are 18 and 20 months old. Both children roll and the 18-month-old child sits with minimal support of the thighs and crawls. Both children bring hands to face, transfer objects between hands, coo and smile. The oldest living child sat at age 2 years and walked independently by age 4 years. At age 5 years, she uses >30 signs, speaks in short sentences using >100 words and is learning letters and numbers.
Three children had brain magnetic resonance imaging (MRI) between 6 and 19 months of age. All images showed dysmyelination, though subtle in the eldest child. Diffusion-weighted mapping at age 6 months showed restricted diffusion along the white matter of the temporal, occipital and parietal lobes and central tegmental tracts, abnormal T2 hyperintensity involving the optic nerves, tegmental tracts and superior cerebellar peduncles and absent myelination of the splenium of the corpus callosum (Fig. 2A and B). Susceptibility-weighted imaging showed no evidence of calcification or hemorrhage. Magnetic resonance spectroscopy was qualitatively normal.
Figure 2.
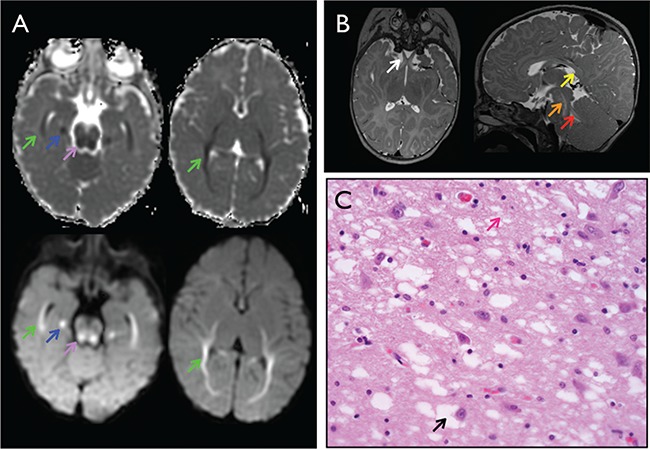
Brain imaging and histology for YARS c.499C > A, p.Pro167Thr homozygotes. (A) Brain MRI at 6 months of age with multiple areas of dysmyelination. Axial diffusion weighted imaging (bottom) and apparent diffusion coefficient (ADC) mapping (top) with restricted diffusion along the white matter of the temporal, occipital and parietal lobes (green arrows), medial temporal lobe (blue arrows) and central tegmental tracts (purple arrows). (B) Abnormal T2 hyperintensity of the optic nerves (white arrow), tegmental tracts (orange arrow) and superior cerebellar peduncles (red arrow) and absent myelination of the corpus callosum (yellow arrow). (C) Hematoxylin and eosin staining of brain biopsy with diffuse vacuolar changes in the gray matter neurons (black arrow) and neurofilament (pink arrow).
An autopsy for one child who died at 10 months of age from liver cirrhosis showed grossly normal brain morphology, but microscopic evidence of chronic cortical neuronal loss, gliosis and diffuse vacuolar changes in both gray and white matter (Fig. 2C).
One child suffered an anoxic brain injury (age 7 months) in the setting of gastroenteritis, dehydration and severe coagulopathy. Seizures developed following this event and electroencephalogram (EEG) showed diffuse encephalopathy. Computerized tomography initially showed loss of gray and white matter density suggestive of hypoperfusion and subsequent imaging showed frontal lobe infarcts and diffuse neuronal injury. None of the six remaining children had seizures but two had EEGs with slow electrical background.
Hearing and vision
All children had bilateral sensorineural hearing loss, congenital for the one child who was screened as a newborn. One affected child had a cochlear implant at 2 years of age with marked improvement in hearing and language development.
Six (86%) children had intermittent and variable nystagmus, usually detected during infancy (n = 5). One child had visual impairment (presumed to be cortical) and wore corrective lenses by age 1 year. Various abnormalities were noted on ophthalmology examination, including underdeveloped retinal vasculature, optic nerve pallor, absent foveal light reflex (n = 1, age 8 months) and fine and scattered retinal pigment (n = 1, age 21 months). Another child (age 8 months) had bilateral optic nerve hypoplasia, blond fundus and peripheral pigmentary changes. Repeat exam at age 5 years showed retinal dystrophy, peripheral vasculopathy with exudation, diffuse pigmentary changes and subretinal hemorrhages in a temporal distribution.
Gastroenterology and hepatology
All children had exocrine pancreatic insufficiency and required oral enzyme replacement. Four (57%) children were treated with gastric motility agents due to persistent vomiting or slowed gastric emptying, five (71%) were diagnosed with gastroesophageal reflux and two (29%) underwent Nissen fundoplication.
All children had chronic cholestatic liver disease detected by age 9 months, but with variable severity. Conjugated hyperbilirubinemia normalized for one child by age 36 months. Children had chronically elevated prothrombin time and occasionally became clinically coagulopathic. Hypoalbuminemia was present in all children and two (29%) required albumin infusions to manage ascites. Alanine aminotransferase (ALT) was modestly elevated in all children, but normalized by age 36 months for one. Gamma-glutamyl transferase (GGT) remained elevated for all patients. Ammonia levels were variable but often elevated (Fig. 3A and B).
Figure 3.
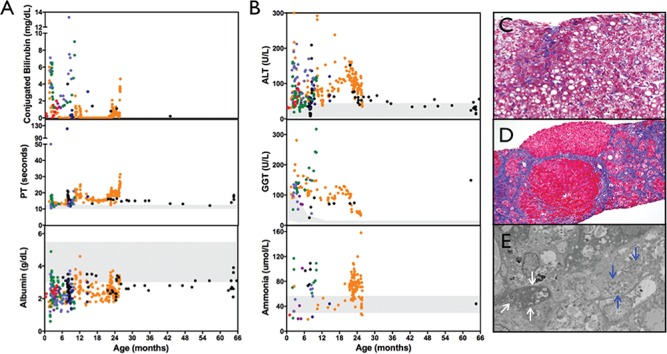
Chronic liver disease in YARS c.499C > A, p.Pro167Thr homozygotes. (A and B) Affected children had chronic direct hyperbilirubinemia, coagulopathy and hypoalbuminemia accompanied by modest elevations in ALT, GGT and ammonia. (C) Trichrome staining of liver biopsy for child (age 3 months) initially revealed diffuse micro- and macrovesicular steatosis but no evidence of fibrosis. (D) Repeat biopsy (age 18 months) showed disrupted liver architecture, bridging fibrosis and nodule formation. (E) Electron microscopy showed poorly defined cristae within the mitochondria (white arrows). Blue arrows identify normal-appearing mitochondria. For laboratory values, colors represent individual children. Shaded areas reflect laboratory reference ranges.
Hepatobiliary scintigraphy of two children showed cholestasis but no evidence of biliary atresia. Five (71%) children had liver biopsies. The earliest biopsies collected at 10 weeks and 3 months of age showed micro- and macrovesicular hepatic steatosis without fibrosis. Repeat biopsy at 18 months of age showed disrupted liver architecture, bridging fibrosis and nodule formation. Electron microscopy from a specimen collected at 10 weeks of age demonstrated poorly defined cristae within mitochondria (Fig. 3C–E). Liver biopsies collected at 8–18 months of age for three additional children consistently showed micro- and macrovesicular steatosis, cholestasis and fibrosis (images not shown). Inflammatory infiltrates [lymphocytes (n = 2) and neutrophils and eosinophils (n = 1)] were noted for three children. Autopsy findings from a child who died at 10 months of age revealed the above findings as well as regenerating nodules consistent with cirrhosis.
Five children had persistent hypoglycemia treated with continuous feeds, IV dextrose, TPN or enteral dextrose (15–20%). One of these children underwent a diagnostic fast at 8 months of age. Prior to fasting, the child was receiving continuous, fortified (27 kcal/oz) enteral feedings and IV dextrose at a glucose infusion rate of 1.7 mg/kg/min. One hour after fasting, two consecutive serum glucose levels were <50 mg/dL. Critical laboratories were inconclusive, showing hypoketosis, undetectable insulin, normal C-peptide and absent response to glucagon. The patient did, however, respond to glucagon during the fed state.
Hematology
Three children had chronic, transfusion-dependent anemia; two were treated with erythropoietin. One child was initially found to have minimal reticulocytes in the setting of anemia. Reticulocyte counts increased over time without erythropoietin treatment, yet anemia persisted without evidence of hemolysis or blood loss. Erythrocyte indices initially showed normal mean corpuscular volume (MCV) which increased to macrocytic range over time, and red cell distribution width (RDW) was chronically elevated (Fig. 4A and B). Blood smears commonly revealed abnormal erythrocyte morphology including target cells, central pallor and teardrop cells (Fig. 4C).
Figure 4.
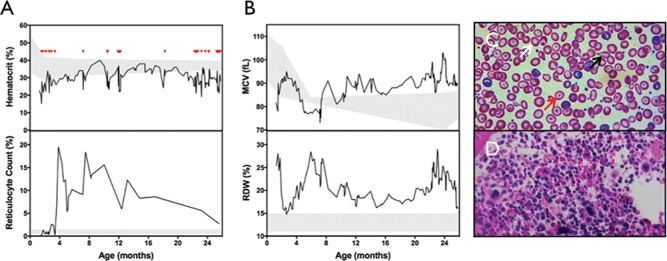
Chronic anemia in YARS c.499C > A, p.Pro167Thr homozygotes. (A) Anemia was noted on presentation (age 5 weeks) for one child and required multiple blood transfusions (red triangles) throughout life. Reticulocyte count was initially low in the setting of anemia, then increased robustly without the use of erythropoietin. (B) MCV was initially low, then increased to macrocytic range around 8 months of age. RDW remained chronically elevated. (C) Blood smear at age 9 months shows abnormal erythrocyte morphology, including central pallor (white arrow), target cells (red arrow) and tear drop cells (black arrow). (D) Bone marrow biopsy at age 3 months with normocellular bone marrow and trilineage hematopoeiesis, but increased myeloid to erythroid ratio with a paucity of erythroid precursors. For laboratory studies, shaded regions represent reference ranges.
Bone marrow biopsy from one child (age 3 months) showed normal trilineage hematopoiesis but increased myeloid to erythroid ratio and a paucity of erythroid precursors (Fig. 4D). Bone marrow biopsy from another child was initially unremarkable (age 2 months), but when repeated at age 8 months, while being treated with erythropoietin, showed prominent erythroid precursors, increased megakaryocytes, numerous focal macrophages, focal increases in iron and an abnormal population of foamy histiocytes (images not available).
Nephrology
All children had normal serum electrolytes and creatinine levels. One child had a horseshoe-shaped kidney and transient hydronephrosis. Intermittent proteinuria was noted between 2 and 22 months of age for four (57%) children, two of whom also had hematuria.
Kidney light and electron microscopy from one child with hematuria and proteinuria (age 2.5 months) was normal and immunostaining was negative for IgG, IgM, IgA, C3, C1q and albumin. Light microscopy from another child (age 8 months) with proteinuria was also normal, but electron microscopy showed effacement of epithelial foot processes occupying less than 50% of glomerular surface area as well as splitting of the glomerular basement membrane in a small number of the capillary loops (images not available). Renal MRI for a child with proteinuria and hematuria (age 23 months) showed decreased corticomedullary differentiation and heterogeneous cortical hyperintensity but normal enhancement, consistent with a chronic inflammatory process.
Infectious disease and immunology
Three children had multiple infections beginning in early infancy, including bacteremia (e.g. group B streptococcus, Staphylococcus aureus, Escherichia coli, Klebsiella pneumonia and Candida parapsilosis), chronic cytomegalovirus infection, enterovirus meningitis, spontaneous bacterial peritonitis (Ralstonia pickettii) and bacterial pneumonia. Two children had indwelling central lines removed due to recurrent bloodstream infections. No child had leukopenia or other laboratory markers of immune deficiency. Immunoglobulins A, G and M (n = 2), flow cytometry (n = 1), lymphocyte enumeration panel (n = 1) and post-vaccine titers (n = 1) were normal.
Cardiopulmonary
Three children had evidence of chronic lung disease despite term delivery. Chest radiographs showed diffuse interstitial prominence by 8–12 months of age. One child was treated with inhaled glucocorticoids.
One child had a ventricular septal defect. No other structural defects or cardiac issues were noted for remaining children. One child had triglyceride levels measured and were normal.
Morbidity and mortality
Four children died between age 10 and 25 months due to end-stage liver disease, complicated by fungal sepsis for one and S. aureus sepsis for the other. Three children are living at the time of manuscript preparation.
YARS c.499C > A, p.Pro167Thr heterozygote clinical findings
All (n = 3, ages 33 to 66 years) adults had normal cranial nerve testing and musculature of the neck, shoulders, arms and legs. None of the adults had evidence of diffuse sensorimotor peripheral neuropathy on electromyography. All had evidence of carpel tunnel syndrome (n = 3), cubital tunnel syndrome (n = 1) or lumbrosacral radiculopathy (n = 3), thought to be related to age rather than genotype.
YARS p.Pro167Thr is a loss-of-function allele in yeast complementation assays
Yeast complementation assays have been previously employed to study the functional consequences of disease-associated ARS mutations (2) including mutations identified in tyrosyl-tRNA synthetase (YARS) (38,39). To determine the functional consequences of YARS p.Pro167Thr, we modeled this mutation in the yeast ortholog of YARS (TYS1) and evaluated the ability of this allele to support yeast cell growth in vivo. Briefly, a haploid yeast strain deleted for the endogenous TYS1 locus—and harboring a maintenance vector with a URA3 gene and a wild-type copy of TYS1—was transfected with a pRS315 experimental vector containing the LEU2 gene and (1) no TYS1 insert (‘Empty’ in Fig. 5); (2) wild-type TYS1; (3) TYS1 p.Pro167Thr; (4) TYS1 p.Gly41Arg (a previously reported and disease-associated null allele) (39); or (5) TYS1 p.Glu196Gln (a previously reported and disease-associated hypomorphic allele) (38). Resulting colonies were grown on solid or liquid media containing 5-fluoroorotic acid (5-FOA) to select for spontaneous loss of the maintenance vector (40). Subsequently, yeast strains were visually inspected or subjected to growth curve analysis to assess viability. Wild-type TYS1 supported robust cellular growth and the vector with no TYS1 insert did not support any cellular growth (Fig. 5A). These data confirm that the experimental vector harbors a functional wild-type copy of TYS1 and that TYS1 is an essential gene. Furthermore, TYS1 p.Gly41Arg did not support any cellular growth and TYS1 p.Glu196Gln supported severely reduced cellular growth (Fig. 5A). These findings indicate that TYS1 p.Gly41Arg is a functional null allele and that TYS1 p.Glu196Gln is a hypomorphic allele, as previously reported (38,39). Finally, TYS1 p.Pro167Thr supported severely reduced cellular growth (Fig. 5A) in a manner similar to TYS1 p.Glu196Gln, indicating that YARS p.Pro167Thr is a loss-of-function, hypomorphic allele, consistent with the recessive phenotype of the patients described here.
Figure 5.
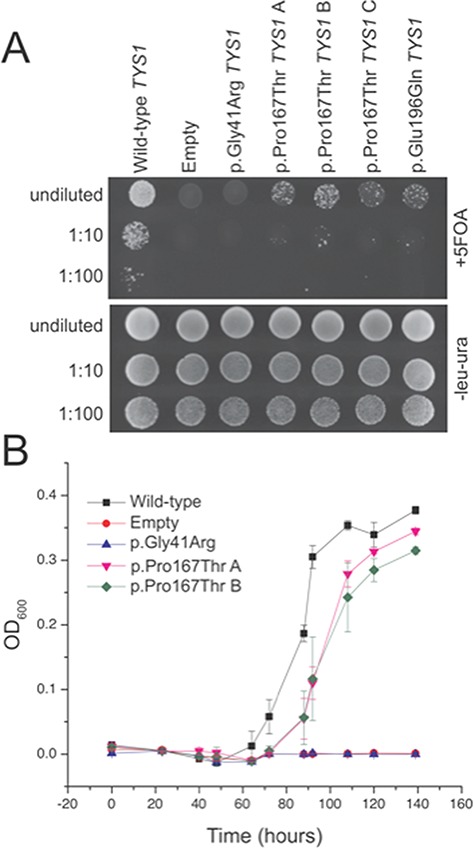
YARS p.Pro167Thr results in a loss-of-function in yeast complementation assays. Yeast lacking endogenous TYS1 (the yeast ortholog of YARS) and harboring a maintenance vector containing wild-type (Wt) TYS1 and URA3 were transformed with an experimental vector containing LEU2 and Wt TYS1, TYS1 p.Gly41Arg (a known null allele), TYS1 p.Glu196Gln (a known hypomorphic allele), TYS1 p.Pro167Thr (in triplicate; A, B, and C) or a vector with no TYS1 insert (`Empty’). (A) All yeast lines grew on solid media deficient in leucine and uracil (lower panel), confirming proper uptake of both vectors. Adding 5-FOA to select against the URA3-containing maintenance vector allowed an assessment of growth supported by the experimental vector alone (upper panel). Wt TYS1 (‘Wt’) supported robust cellular growth while the vector with no TYS1 insert (‘Empty’) did not support any cellular growth. TYS1 p.Gly41Arg did not support any cellular growth and TYS1 p.Glu196Gln supported severely reduced cellular growth, consistent with previous assessments of these alleles. In a manner similar to TYS1 p.Glu196Gln, TYS1 p.Pro167Thr supported severely reduced cellular growth. (B) Yeast carrying the TYS1 p.Gly41Arg allele or an empty vector was unable to grow in liquid 5-FOA media. Note the 15-h lag between mid-log phase of wild-type and TYS1 p.Pro167Thr expressing yeast. Error bars represent standard deviations of the average OD600 of three biological replicates for each strain. TSY1 p.Glu196Gln was excluded from growth curve analysis to simplify the figure.
To quantify the difference in yeast cell growth between wild-type and TYS1 p.Pro167Thr, we performed growth curve analysis of the above yeast strains in liquid media containing 5-FOA. Yeast carrying the TYS1 p.Gly41Arg allele or an empty vector were unable to grow in liquid 5-FOA media (Fig. 5B), as predicted based on the growth on solid media containing 5-FOA (Fig. 5A). Wild-type TYS1 expressing yeast entered mid log phase growth at ∼90 h and, in contrast, TYS1 p.Pro167Thr expressing yeast had a 15-h lag before reaching mid-log phase growth (Fig. 5B). This is consistent with the slower growth observed for TYS1 p.Pro167Thr yeast on solid 5-FOA media; TYS1 p.Glu196Gln was excluded from growth curve analysis to simplify the figure. All of the above yeast strains grew similarly in liquid media lacking uracil and leucine, and not containing 5-FOA (data not shown). Together, our functional studies in yeast indicate that YARS p.Pro167Thr is a loss-of-function, hypomorphic allele.
YARS p.Pro167Thr impairs homodimerization
From prokaryotes to humans, YARS is only catalytically active as a homodimer in which the paired N-terminal catalytic domains undergo a conformational change at the dimer interface required for amino acid activation (Tyr-AMP) (34–36). Monomeric native human YARS is incapable of catalyzing aminoacylation (35). The YARS p.Pro167Thr substitution occurs in the highly conserved dimer interface adjacent to several amino acid residues that stabilize the homodimer (Fig. 6A). We hypothesized that substitution of a polar Thr167 for the non-polar Pro167 destabilizes the YARS homodimeric structure required for amino acid activation and aminoacylation (34–36).
Figure 6.
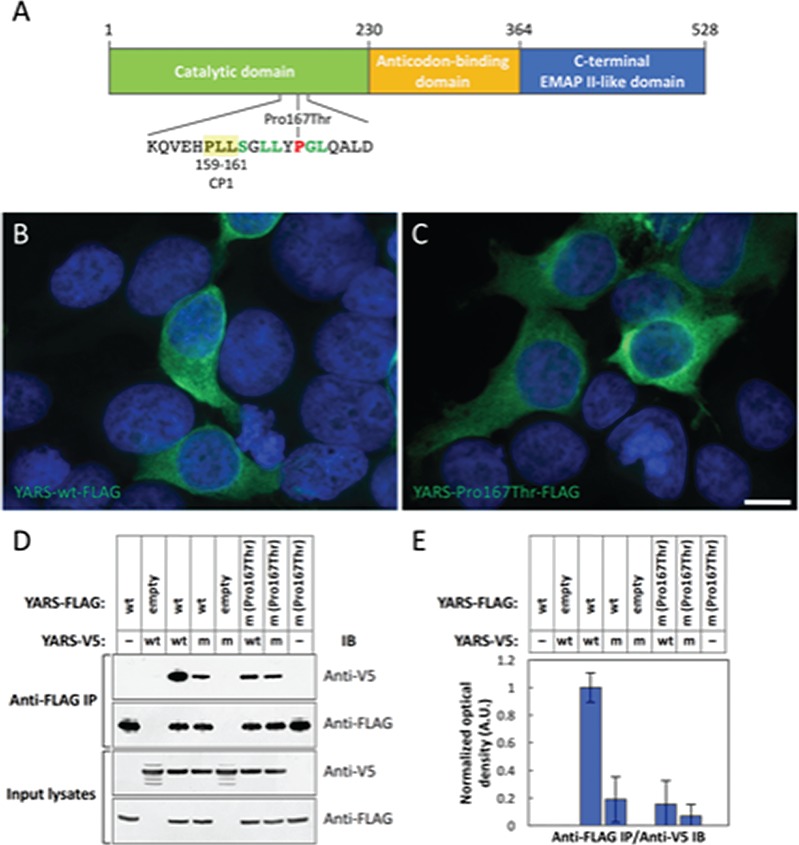
Substitution of threonine for proline impairs YARS homodimerization. (A) Schematic of the functional domains of human TyrRS (35,52). YARS c.499C > A causes a substitution of Thr for Pro at amino acid position 167, within the Rossmann fold necessary for YARS homodimerization. Neighboring amino acids 159–161 of connective polypeptide 1 (CP1) (Pro, Leu, Leu, − PLL; yellow highlight) are critical for dimer formation (35), and the amino acids highlighted in green are highly conserved members of the dimer interface. Recognition of tRNA-Tyr occurs at the anticodon binding domain, and the C-terminus is homologous to the proinflammatory cytokine endothelial-monocyte-activating polypeptide II (EMAP II) (41,42,53). (B and C) Recombinant YARS-FLAG p.Pro167Thr overexpressed in HEK-293 T cells (C) localizes to the cytosol in a manner indistinguishable from that of TyrRS-FLAG wild-type (wt) (B) (n = 3). Scale bar in (C) = 10 μm. Green fluorescence: anti-FLAG immunoreactivity; blue fluorescence: DAPI. (D and E) Human YARS-FLAG wt and p.Pro167Thr constructs were co-overexpressed with human YARS-V5 wt and p.Pro167Thr constructs in HEK-293 T cells (as indicated). YARS-FLAG proteins were immunoprecipitated (IP) from whole cell lysates (input) (n = 5). YARS-FLAG wt co-precipitated YARS-V5 wt abundantly [normalized to 1 in the immunoblotted (IB) protein band optical densities displayed in E]. Co-precipitation of YARS-V5 p.Pro167Thr by YARS-FLAG wt and YARS-V5 wt by YARS-FLAG p.Pro167Thr were reduced roughly 5-fold. Co-precipitation of YARS-V5 p.Pro167Thr by YARS-FLAG p.Pro167Thr was reduced 10-fold. wt: wild type; m: YARS c.499C > A, p.Pro167Thr mutant; empty: ‘empty vector’ encoding the FLAG epitope tag peptide sequence followed by a stop codon. IB protein band optical density (E) was measured from three replicates of the co-immunoprecipitation data shown in D. Error bars represent one standard deviation.
YARS-FLAG wt and p.Pro167Thr variants localized to the cytosol identically in HEK-293T (human embryonic kidney), SH-SY5Y (human neuroblast) and Neuro2a (mouse neuroblast) cells, suggesting that subcellular localization of YARS is unaffected by the p.Pro167Thr substitution. Data from HEK-293 T alone are presented for simplicity (Fig. 6B and C). Immunoprecipitation of YARS-FLAG wt co-precipitated abundant YARS-V5 wt from HEK-293 T cells co-overexpressing the two constructs. However, the abundances of YARS-V5 p.Pro167Thr co-precipitated by YARS-FLAG wt and YARS-V5 wt co-precipitated by YARS-FLAG p.Pro167Thr were reduced 5-fold. Co-precipitation of YARS-V5 p.Pro167Thr by YARS-FLAG p.Pro167Thr was reduced 10-fold (Fig. 6D and E). These results are consistent with a reduced quaternary structure quality estimate (QSQE) score, a measure of protein oligomerization, for YARS p.Pro167Thr versus YARS wt in protein structural modeling (Fig. 7).
Figure 7.
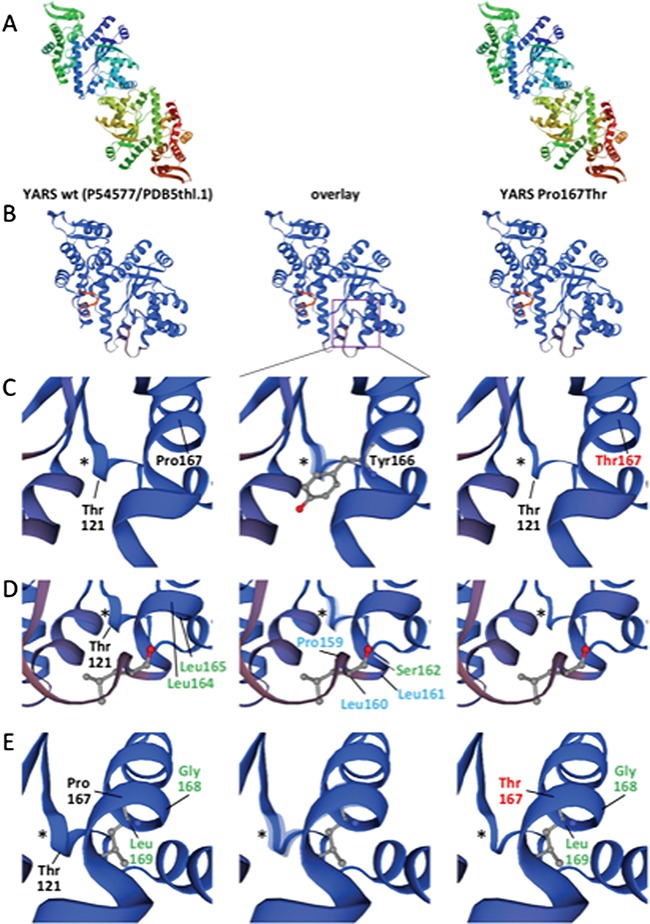
Protein structural modeling of the YARS p.Pro167Thr substitution. (A) Structural models for YARS wt (Uniprot P54577) (left) and YARS p.Pro167Thr (right) generated using SWISS-MODEL/ProMod3 v1.1.0 (54–58) based on template PDB 5thl.1.A (best fit from 72 templates; GMQE = 0.7; identity = 99.73). In both cases, YARS is displayed as a homodimer. The models assume homozygosity for YARS wt (left) and YARS p.Pro167Thr (right). Homodimerization is preserved in the YARS Pro167Thr model, but with a 3.1% reduction of the QSQE oligomerization score (from 0.65 for wt to 0.63 for Pro167Thr). (B–E) Models in B–E are displayed as monomers for greater resolution. ‘Overlay’ (center) highlights differences between the models. Proline 167 (Pro167) is situated within the Rossmann fold necessary for YARS homodimerization. Neighboring amino acids of CP1 (Pro159, Leu160, Leu161; blue labels) are important for dimer formation, although C-terminal amino acids also stabilize the dimer (59). Amino acids labeled in green are highly conserved members of the dimer interface (34). The YARS Pro167Thr substitution induces reorganization of amino acids 119–123 (Lys-Gly-Thr-Asp-Tyr) (purple box in B and asterisk (*) near Thr121 in C–E) which normally folds as a small beta-sheet/alpha-helix/turn motif in close proximity to the dimer interface and tyrosine (Tyr) binding pocket. Tyrosine166 (Tyr166), adjacent to Pro167 and the helix formed by amino acids 120–122, is one of five amino acids in this region that coordinate Tyr binding (60). (C–E) Rotations of the model from the perspectives of amino acids Tyr166 (C), Leu160 (D) and Leu 169 (E) demonstrating the impact of the p.Pro167Leu substitution on local structure within the dimer interface and tyrosine (Tyr) binding pocket.
Discussion
Clinical phenotype
ARSs are critical for protein translation and variants in these enzymes are increasingly recognized in association with multisystem disease (10). Biallelic YARS variants (one in the catalytic domain, the other in the C-terminal domain) were recently reported to cause developmental delay, hypertriglyceridemia, liver disease, cystic lung lesions, thinning of the corpus callosum and cystic changes in the periventricular white matter in two living siblings (27). Likewise, retinal degeneration, hearing impairment, agenesis of the corpus callosum, primary amenorrhea and liver disease were attributed to homozygous variants just outside of YARS catalytic domain in an adult case report (28). In contrast, the seven related children from the currently described cohort share homozygosity for mutations in the catalytic domain of YARS. The children described here share some of the phenotypic features with the children in the prior reports (e.g. deep-set eyes, developmental delay, visual impairment poor growth and liver disease) but also broaden the phenotypic spectrum to include disease manifestations in the auditory, hematologic and renal systems. To the best of the authors’ knowledge, this is the first report of ARS variants causing pancreatic dysfunction.
Even among YARS p.Pro167Thr homozygotes, we observed significant phenotypic heterogeneity. Developmental outcomes and lifespan were limited for the four deceased patients, possibly related to the severity of liver disease and significant disease burden in other organ systems. However, the eldest patient (age 5 years) walks independently and speaks in short sentences. The extent of hematologic disease was also variable, with three children requiring transfusions repeatedly while the remaining four required them only rarely. Likewise, four (57%) children had significant proteinuria and/or hematuria. Such variability in clinical outcomes may represent part of a phenotypic spectrum associated with the YARS p.Pro167Thr variant or the influence of modifier genes not yet identified.
In addition to its canonical aminoacylation activity, YARS can be secreted from the cell and cleaved into an N-terminal fragment (YARSmini) and a C-terminal EMAP II-like domain (Fig. 6A), both of which act as cytokines (41). YARSmini retains its aminoacylation activity as a homodimer, but upon dissociation into monomers acts as an interleukin-8-like cytokine to stimulate polymorphonuclear leukocyte migration (35,41,42). The C-terminal EMAP II-like domain has strong chemotactic activity for leukocytes and monocytes and triggers production of tumor necrosis factor-α, myeloperoxidase and tissue factor (41). The EMAP II-like domain cytokine activity should be unaffected by the p.Pro167Thr substitution. However, catalytically active dimeric wild-type YARSmini antagonizes leukocyte migration whereas monomeric YARSmini promotes leukocyte migration (35). Thus, any shift in monomer:dimer equilibrium that results from altered dimerization of YARS p.Pro167Thr could result in dysregulation of the cytokine function of YARSmini. In support of this hypothesis, YARS c.499C > A, p.Pro167Thr homozygotes have evidence of inflammation in brain tissue, chest radiograph, liver biopsy and renal MRI, suggesting immune dysregulation may play a role in the underlying disease pathophysiology.
Cellular studies
Yeast complementation assays have been previously employed to study the functional consequences of disease-associated ARS mutations, including mutations identified in YARS (2), (38,39). To determine the functional consequences of YARS p.Pro167Thr, we modeled this mutation in TYS1, the yeast ortholog of YARS and evaluated the ability of this allele to support yeast cell growth in vivo. In the current report, the p.Pro167Thr substitution causes dramatically reduced, but not ablated, yeast cell growth, indicating that this is a hypomorphic allele.
YARS is only catalytically active as a homodimer. Paired N-terminal catalytic domains undergo a conformational change at their interface, required for amino acid activation whereas wild-type monomeric human YARS is incapable of catalyzing aminoacylation (34–37). Conservation of Pro167 over eukaryotic evolution and its position within a series of highly conserved amino acid residues that stabilize the dimer interface suggest that Pro167 may be important for YARS homodimer formation (34–36). Likewise, our studies in human embryonic kidney cells demonstrate that substitution of polar threonine for non-polar proline does not impact cellular localization to the cytoplasm, but greatly impairs YARS homodimerization and presumably its aminoacylation activity. Impaired YARS p.Pro167Thr homodimerization is further supported by a decreased QSQE score in protein structural modeling relative to YARS wt (Fig. 7).
Taken together, our functional data suggest that YARS p.Pro167Thr is a hypomorphic allele that reduces but does not eliminate YARS function. Individuals homozygous for YARS c.499C > A, p.Pro167Thr may suffer substantially impaired YARS aminoacylation activity and as a result, significantly reduced incorporation of tyrosine into protein. Impaired protein synthesis, especially for an amino acid residue subject to extensive post-translational modification (e.g. tyrosine phosphorylation in growth factor/morphogen receptor tyrosine kinase signaling) (43–45), during development is consistent with the multisystem impact and growth impairment associated with homozygosity for YARS c.499C > A, p.Pro167Thr. Our findings may also have implications for defining the molecular mechanisms of neuropathy-associated YARS variants (39). None of the heterozygous individuals studied showed evidence of neuropathy; however, age of onset and decreased penetrance may complicate interpretations of our clinical evaluations.
Interestingly, while neomorphic functions have been observed for certain neuropathy-associated ARS alleles in a Drosophila model (46), the lack of a neuropathy phenotype may be due to the reduced dimerization of YARS p.Pro167Thr, which would reduce the proposed dominant-negative effect of neuropathy-associated YARS mutations.
Therapeutic implications
As the number and complexity of ARSopathies continue to grow, so does the need for more effective therapies. For the children in this cohort, medical treatment has been limited to supportive therapies (refractive lenses, hearing aids/cochlear implants, pancreatic enzyme and nutritional supplements, blood transfusions, albumin infusions and therapy services). Although these interventions may promote survival and improve quality of life, they do not alter the underlying disease process.
There is limited evidence that supplementing the cognate amino acid rescues growth in yeast with impaired MARS and VARS2 activity (47,48). Currently, it is unknown if enteral amino acid supplementation in a patient with impaired ARS function will achieve a measurable impact on enzyme activity or prevent/reverse disease manifestations. Our in vitro evidence of impaired YARS homodimerization suggests that the enzyme may remain inactive even in the presence of excess tyrosine, casting doubt on the utility of oral amino acid supplementation, but this hypothesis remains to be tested.
Among YARS p.Pro167Thr homozygotes, cholestatic liver disease appears to be the most severe and life-threatening disease manifestation. This raises the question of whether liver transplant would be an appropriate therapeutic approach for this particular disease. Although liver transplant might restore normal liver function, it would not necessarily alter disease progression in other organ systems. If the underlying disease mechanism in the currently described YARS variant involves immune dysregulation, liver disease could recur in the transplanted liver over time. Moving forward, efforts to achieve a deeper understanding of the molecular mechanisms of disease in patients with ARS mutations could shed light on how to better organize their care.
Materials and Methods
Subjects
We reviewed medical records of seven related children (ages 5 months to 5 years) homozygous for YARS c.499C > A, p.Pro167Thr. Three related adults heterozygous for this variant (two mothers and one grandfather/uncle, ages 33 to 66 years) underwent detailed neurologic examination and electrophysiological studies. Children homozygous for YARS c.499C > A did not undergo detailed neurologic examinations or electrophysiological studies. The Institutional Review Board of Lancaster General Hospital (Lancaster, PA) approved the study and adults consented in writing to participate on behalf of themselves or their affected children.
Whole exome sequencing and variant analysis
We performed whole exome sequencing for the affected male proband, four unaffected female siblings, both unaffected parents and unaffected maternal grandparents. Genomic DNA was extracted from peripheral blood samples and submitted for whole exome sequencing and variant analysis in collaboration with the Regeneron Genetics Center (Tarrytown, NY) as previously described (49).
Yeast complementation assays and growth curves
Yeast complementation assays to study YARS variants were performed as previously described (38). Briefly, the QuickChange II XL Site-Directed Mutagenesis Kit (Stratagene, San Diego, CA) was used to generate the TYS1 p.Pro167Thr mutation in yeast. Gateway cloning technology (Invitrogen, Carlsbad, CA) was employed to clone the open-reading frame into a LEU2-bearing pRS315 vector. The wild-type, TYS1 p.Gly41Arg and TYS1 p.Glu196Gln expression constructs and the ΔTYS1 haploid yeast strain were created as previously described (38). Resulting constructs were purified and sequenced to confirm mutagenesis and exclude PCR-induced errors.
The haploid ΔTYS1 strain, which harbors a maintenance vector to express wild-type TYS1 and URA3, was transformed with a LEU2-bearing pRS315 vector containing either no TYS1 insert (‘Empty’) or the appropriate wild-type or mutant TYS1 locus, and selected on medium lacking uracil and leucine (Teknova, Hollister, CA). Three independent TYS1 p.Pro167Thr pRS315 plasmid preparations were transformed and two colonies from each transformation were selected for additional analyses. Selected colonies were grown to saturation for 2 days at 37°C in liquid media lacking uracil and leucine. A 10-μL aliquot of each culture was spotted undiluted, or diluted 1:10 or 1:100 in H2O on plates containing 0.1% 5-FOA (Teknova, Hollister, CA) or medium lacking uracil and leucine and incubated at 30°C for 72 h (40). Cellular growth was determined by visual inspection. Growth curve analysis was performed in duplicate under similar conditions using liquid growth medium.
Immunofluorescence microscopy and immunoprecipitation assays
Full-length human YARS coding cDNA (GenBank: NM_003680.3) without the stop codon was amplified by polymerase chain reaction (PCR) from cDNA reverse transcribed from human HEK-293T cell mRNA using SuperScript III reverse transcriptase (Thermo Fisher, Waltham, MA). PCR products were ligated into pENTR/D-TOPO (Thermo Fisher, Waltham, MA), and the YARS c.499C > A variant was introduced by site-directed mutagenesis (QuikChange II, Agilent Technologies, Santa Clara, CA). LR recombination (LR Clonase II, Thermo Fisher, Waltham, MA) was used to move YARS cDNA constructs from pENTR clones into pcDNA3.2/FLAG and pcDNA3.2/V5 mammalian expression plasmids to encode YARS-fusion proteins with C-terminal FLAG and V5 epitope tags, respectively. C-terminal epitope-tagged YARS WT retains its canonical aminoacylation activity upon dimerization (50). All clones were verified with Sanger sequencing.
HEK-293T, SH-SY5Y and Neuro2a cells were purchased from the American Type Culture Collection. SH-SY5Y cells were cultured in DMEM:F12 with 10% fetal bovine serum (FBS); HEK-293 T and Neuro2a were cultured in DMEM with 10% FBS. Cells were transfected with FuGENE 6 (Roche, Indianapolis, IN) at a 3:1 FuGENE6:DNA ratio following the manufacturer’s instructions. All cells were used at low passage and maintained at 37°C and 5% CO2.
Cells cultured on uncoated German glass coverslips transfected with pcDNA3.2/YARS/FLAG WT or c.499C > A cDNAs were rinsed with phosphate buffered saline (PBS) and fixed with 4% paraformaldehyde in PBS at 21°C for 15 min. Cells were then prepared for immunofluorescence microscopy and documented as described previously (17). The primary and secondary antibodies used were anti-FLAG M2 (Sigma-Aldrich, St. Louis, MO, 1:1,000) and Alexa-Fluor 488-goat anti-mouse IgG1 (Thermo Fisher, Waltham, MA, 1:400), respectively. Nuclei were stained with 1-μg/mL DAPI (Santa Cruz, Dallas, TX), and coverslips were mounted with Fluoromount-G (Electron Microscopy Sciences, Hatfield, PA).
Co-immunoprecipitation was performed as described previously (17,51). Briefly, pcDNA3.2/YARS/FLAG WT or c.499C > A cDNAs or empty vector were co-transfected with pcDNA3.2/YARS/V5 WT or c.499C > A cDNAs in HEK-293 T cells for 40–44 h. Whole cell lysates were clarified by centrifugation (‘input’) and incubated with anti-FLAG M2 affinity resin (Sigma-Aldrich, St. Louis, MO) for 2 h with end-over-end mixing at 4°C. Resin beads were pelleted and washed 3× in Tris-buffered saline (pH 7.2) with protease inhibitor cocktail (Roche, Indianapolis, IN), phosphatase inhibitor (PhosSTOP, Roche, Indianapolis, IN), 1-mm phenylmethylsulfonyl fluoride (PMSF) and 0.5% Tween-20, before elution with 150 ng/μl 3×-FLAG peptide (Sigma-Aldrich, St. Louis, MO). Input lysates and eluates were denatured in sample buffer containing 50-mm dithiothreitol (DTT; Sigma-Aldrich, St. Louis, MO) for 50 min at 21°C followed by 10 min at 70°C and then immunoblotted with anti-V5 (Thermo Fisher, Waltham, MA, 1:5,000) antibody. Blots were stripped (Restore Plus, Thermo Fisher, Waltham, MA) and reprobed with anti-FLAG M2 antibody (Sigma-Aldrich, St. Louis, MO, 1:1,000). Protein band optical densities were measured using Image J calibrated to a Kodak step tablet.
Acknowledgements
We thank the families who share their stories and inspire our efforts to improve health care for children with special needs.
Conflict of Interest statement. None declared.
Funding
National Institute of General Medical Sciences (GM110184 and GM118647 to A.A.; GM007315, GM007863 and NS092238 to L.G.); Howard Hughes Medical Institute (52006294 and 52007538 to R.J.); Center for Research on Women and Newborn Health and ConnectCare3 [to R.J.]; charitable contributions from Amish and Mennonite communities of Pennsylvania and surrounding states. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or other organizations listed above. The funding sources had no role in the design and conduct of the study, collection, management, analysis and interpretation of the data, preparation, review or approval of the manuscript, or decision to submit the manuscript for publication. All authors discussed and commented on the manuscript.
References
- 1. Delarue M. (1995) Aminoacyl-tRNA synthetases. Curr. Opin. Struct. Biol., 5, 48–55. [DOI] [PubMed] [Google Scholar]
- 2. Oprescu S.N., Griffin L.B., Beg A.A. and Antonellis A. (2017) Predicting the pathogenicity of aminoacyl-tRNA synthetase mutations. Methods, 113, 139–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Antonellis A. and Green E.D. (2008) The role of aminoacyl-tRNA synthetases in genetic diseases. Annu. Rev. Genomics Hum. Genet., 9, 87–107. [DOI] [PubMed] [Google Scholar]
- 4. He X.D., Gong W., Zhang J.N., Nie J., Yao C.F., Guo F.S., Lin Y., Wu X.H., Li F., Li J. et al. (2018) Sensing and transmitting intracellular amino acid signals through reversible lysine aminoacylations. Cell Metab., 27, 151–166e156. [DOI] [PubMed] [Google Scholar]
- 5. Yao P. and Fox P.L. (2013) Aminoacyl-tRNA synthetases in medicine and disease. EMBO Mol. Med., 5, 332–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Antonellis A., Ellsworth R.E., Sambuughin N., Puls I., Abel A., Lee-Lin S.Q., Jordanova A., Kremensky I., Christodoulou K., Middleton L.T. et al. (2003) Glycyl tRNA synthetase mutations in Charcot–Marie–Tooth disease type 2D and distal spinal muscular atrophy type V. Am. J. Hum. Genet., 72, 1293–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gonzalez M., McLaughlin H., Houlden H., Guo M., Yo-Tsen L., Hadjivassilious M., Speziani F., Yang X.L., Antonellis A., Reilly M.M. et al. (2013) Exome sequencing identifies a significant variant in methionyl-tRNA synthetase (MARS) in a family with late-onset CMT2. J. Neurol. Neurosurg. Psychiatry, 84, 1247–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Latour P., Thauvin-Robinet C., Baudelet-Mery C., Soichot P., Cusin V., Faivre L., Locatelli M.C., Mayencon M., Sarcey A., Broussolle E. et al. (2010) A major determinant for binding and aminoacylation of tRNA(Ala) in cytoplasmic Alanyl-tRNA synthetase is mutated in dominant axonal Charcot–Marie–Tooth disease. Am. J. Hum. Genet., 86, 77–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vester A., Velez-Ruiz G., McLaughlin H.M., Program N.C.S., Lupski J.R., Talbot K., Vance J.M., Zuchner S., Roda R.H., Fischbeck K.H. et al. (2013) A loss-of-function variant in the human histidyl-tRNA synthetase (HARS) gene is neurotoxic in vivo. Hum. Mutat., 34, 191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Meyer-Schuman R. and Antonellis A. (2017) Emerging mechanisms of aminoacyl-tRNA synthetase mutations in recessive and dominant human disease. Hum. Mol. Genet., 26, R114–R127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Casey J.P., Slattery S., Cotter M., Monavari A.A., Knerr I., Hughes J., Treacy E.P., Devaney D., McDermott M., Laffan E. et al. (2015) Clinical and genetic characterisation of infantile liver failure syndrome type 1, due to recessive mutations in LARS. J. Inherit. Metab. Dis., 38, 1085–1092. [DOI] [PubMed] [Google Scholar]
- 12. Edvardson S., Shaag A., Kolesnikova O., Gomori J.M., Tarassov I., Einbinder T., Saada A. and Elpeleg O. (2007) Deleterious mutation in the mitochondrial arginyl-transfer RNA synthetase gene is associated with pontocerebellar hypoplasia. Am. J. Hum. Genet., 81, 857–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kopajtich R., Murayama K., Janecke A.R., Haack T.B., Breuer M., Knisely A.S., Harting I., Ohashi T., Okazaki Y., Watanabe D. et al. (2016) Biallelic IARS mutations cause growth retardation with prenatal onset, intellectual disability, muscular hypotonia, and infantile hepatopathy. Am. J. Hum. Genet., 99, 414–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McLaughlin H.M., Sakaguchi R., Liu C., Igarashi T., Pehlivan D., Chu K., Iyer R., Cruz P., Cherukuri P.F., Hansen N.F. et al. (2010) Compound heterozygosity for loss-of-function lysyl-tRNA synthetase mutations in a patient with peripheral neuropathy. Am. J. Hum. Genet., 87, 560–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McMillan H.J., Humphreys P., Smith A., Schwartzentruber J., Chakraborty P., Bulman D.E., Beaulieu C.L., Consortium F.C., Majewski J., Boycott K.M. et al. (2015) Congenital visual impairment and progressive microcephaly due to lysyl-transfer ribonucleic acid (RNA) synthetase (KARS) mutations: the expanding phenotype of aminoacyl-transfer RNA synthetase mutations in human disease. J. Child Neurol., 30, 1037–1043. [DOI] [PubMed] [Google Scholar]
- 16. McMillan H.J., Schwartzentruber J., Smith A., Lee S., Chakraborty P., Bulman D.E., Beaulieu C.L., Majewski J., Boycott K.M. and Geraghty M.T. (2014) Compound heterozygous mutations in glycyl-tRNA synthetase are a proposed cause of systemic mitochondrial disease. BMC Med. Genet., 15, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Puffenberger E.G., Jinks R.N., Sougnez C., Cibulskis K., Willert R.A., Achilly N.P., Cassidy R.P., Fiorentini C.J., Heiken K.F., Lawrence J.J. et al. (2012) Genetic mapping and exome sequencing identify variants associated with five novel diseases. PloS One, 7, e28936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Safka Brozkova D., Deconinck T., Griffin L.B., Ferbert A., Haberlova J., Mazanec R., Lassuthova P., Roth C., Pilunthanakul T., Rautenstrauss B. et al. (2015) Loss of function mutations in HARS cause a spectrum of inherited peripheral neuropathies. Brain, 138, 2161–2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Santos-Cortez R.L., Lee K., Azeem Z., Antonellis P.J., Pollock L.M., Khan S., Irfanullah, Andrade-Elizondo P.B., Chiu I., Adams M.D. et al. (2013) Mutations in KARS, encoding lysyl-tRNA synthetase, cause autosomal-recessive nonsyndromic hearing impairment DFNB89. Am. J. Hum. Genet., 93, 132–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Simons C., Griffin L.B., Helman G., Golas G., Pizzino A., Bloom M., Murphy J.L., Crawford J., Evans S.H., Topper S. et al. (2015) Loss-of-function alanyl-tRNA synthetase mutations cause an autosomal-recessive early-onset epileptic encephalopathy with persistent myelination defect. Am. J. Hum. Genet., 96, 675–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Meel E., Wegner D.J., Cliften P., Willing M.C., White F.V., Kornfeld S. and Cole F.S. (2013) Rare recessive loss-of-function methionyl-tRNA synthetase mutations presenting as a multi-organ phenotype. BMC Med. Genet., 14, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wolf N.I., Salomons G.S., Rodenburg R.J., Pouwels P.J., Schieving J.H., Derks T.G., Fock J.M., Rump P., Beek D.M., Knaap M.S. et al. (2014) Mutations in RARS cause hypomyelination. Ann. Neurol., 76, 134–139. [DOI] [PubMed] [Google Scholar]
- 23. Wolf N.I., Toro C., Kister I., Latif K.A., Leventer R., Pizzino A., Simons C., Abbink T.E., Taft R.J., Knaap M.S. et al. (2015) DARS-associated leukoencephalopathy can mimic a steroid-responsive neuroinflammatory disorder. Neurology, 84, 226–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang X., Ling J., Barcia G., Jing L., Wu J., Barry B.J., Mochida G.H., Hill R.S., Weimer J.M., Stein Q. et al. (2014) Mutations in QARS, encoding glutaminyl-tRNA synthetase, cause progressive microcephaly, cerebral-cerebellar atrophy, and intractable seizures. Am. J. Hum. Geneti., 94, 547–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Karaca E., Harel T., Pehlivan D., Jhangiani S.N., Gambin T., Coban Akdemir Z., Gonzaga-Jauregui C., Erdin S., Bayram Y., Campbell I.M. et al. (2015) Genes that affect brain structure and function identified by rare variant analyses of Mendelian neurologic disease. Neuron, 88, 499–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Taft R.J., Vanderver A., Leventer R.J., Damiani S.A., Simons C., Grimmond S.M., Miller D., Schmidt J., Lockhart P.J., Pope K. et al. (2013) Mutations in DARS cause hypomyelination with brain stem and spinal cord involvement and leg spasticity. Am. J. Hum. Genet., 92, 774–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nowaczyk M.J., Huang L., Tarnopolsky M., Schwartzentruber J., Majewski J., Bulman D.E., FORGE Canada Consortium, Care4Rare Canada Consortium, Hartley T. and Boycott K.M. (2017) A novel multisystem disease associated with recessive mutations in the tyrosyl-tRNA synthetase (YARS) gene. Am. J. Med. Genet. A, 173, 126–134. [DOI] [PubMed] [Google Scholar]
- 28. Tracewska-Siemiatkowska A., Haer-Wigman L., Bosch D.G.M., Nickerson D., Bamshad M.J., University of Washington Center for Mendelian Genomics, Vorst M., Rendtorff N.D., Moller C., Kjellstrom U. et al. (2017) An expanded multi-organ disease phenotype associated with mutations in YARS. Genes, 8, 381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ardissone A., Lamantea E., Quartararo J., Dallabona C., Carrara F., Moroni I., Donnini C., Garavaglia B., Zeviani M. and Uziel G. (2015) A novel homozygous YARS2 mutation in two Italian siblings and a review of literature. JIMD Rep., 20, 95–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nakajima J., Eminoglu T.F., Vatansever G., Nakashima M., Tsurusaki Y., Saitsu H., Kawashima H., Matsumoto N. and Miyake N. (2014) A novel homozygous YARS2 mutation causes severe myopathy, lactic acidosis, and sideroblastic anemia 2. J. Hum. Genet., 59, 229–232. [DOI] [PubMed] [Google Scholar]
- 31. Riley L.G., Cooper S., Hickey P., Rudinger-Thirion J., McKenzie M., Compton A., Lim S.C., Thorburn D., Ryan M.T., Giege R. et al. (2010) Mutation of the mitochondrial tyrosyl-tRNA synthetase gene, YARS2, causes myopathy, lactic acidosis, and sideroblastic anemia—MLASA syndrome. Am. J. Hum. Genet., 87, 52–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Riley L.G., Menezes M.J., Rudinger-Thirion J., Duff R., Lonlay P., Rotig A., Tchan M.C., Davis M., Cooper S.T. and Christodoulou J. (2013) Phenotypic variability and identification of novel YARS2 mutations in YARS2 mitochondrial myopathy, lactic acidosis and sideroblastic anaemia. Orphanet J. Rare Dis., 8, 193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sommerville E.W., Ng Y.S., Alston C.L., Dallabona C., Gilberti M., He L., Knowles C., Chin S.L., Schaefer A.M., Falkous G. et al. (2017) Clinical features, molecular heterogeneity, and prognostic implications in YARS2-related mitochondrial myopathy. JAMA Neurol., 74, 686–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jones D.H., McMillan A.J., Fersht A.R. and Winter G. (1985) Reversible dissociation of dimeric tyrosyl-tRNA synthetase by mutagenesis at the subunit interface. Biochemistry, 24, 5852–5857. [DOI] [PubMed] [Google Scholar]
- 35. Vo M.N., Yang X.L. and Schimmel P. (2011) Dissociating quaternary structure regulates cell-signaling functions of a secreted human tRNA synthetase. J. Biol. Chem., 286, 11563–11568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ward W.H. and Fersht A.R. (1988) Tyrosyl-tRNA synthetase acts as an asymmetric dimer in charging tRNA. A rationale for half-of-the-sites activity. Biochemistry, 27, 5525–5530. [DOI] [PubMed] [Google Scholar]
- 37. Yaremchuk A., Kriklivyi I., Tukalo M. and Cusack S. (2002) Class I tyrosyl-tRNA synthetase has a class II mode of cognate tRNA recognition. EMBO J., 21, 3829–3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gonzaga-Jauregui C., Harel T., Gambin T., Kousi M., Griffin L.B., Francescatto L., Ozes B., Karaca E., Jhangiani S.N., Bainbridge M.N. et al. (2015) Exome sequence analysis suggests that genetic burden contributes to phenotypic variability and complex neuropathy. Cell Rep., 12, 1169–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jordanova A., Irobi J., Thomas F.P., Van Dijck P., Meerschaert K., Dewil M., Dierick I., Jacobs A., De Vriendt E., Guergueltcheva V. et al. (2006) Disrupted function and axonal distribution of mutant tyrosyl-tRNA synthetase in dominant intermediate Charcot–Marie–Tooth neuropathy. Nat. Genet., 38, 197–202. [DOI] [PubMed] [Google Scholar]
- 40. Boeke J.D., LaCroute F. and Fink G.R. (1984) A positive selection for mutants lacking orotidine-5′-phosphate decarboxylase activity in yeast: 5-fluoro-orotic acid resistance. Mol. Gen. Genet., 197, 345–346. [DOI] [PubMed] [Google Scholar]
- 41. Wakasugi K. and Schimmel P. (1999) Two distinct cytokines released from a human aminoacyl-tRNA synthetase. Science, 284, 147–151. [DOI] [PubMed] [Google Scholar]
- 42. Wakasugi K. and Schimmel P. (1999) Highly differentiated motifs responsible for two cytokine activities of a split human tRNA synthetase. J. Biol. Chem., 274, 23155–23159. [DOI] [PubMed] [Google Scholar]
- 43. Brewer J.R., Mazot P. and Soriano P. (2016) Genetic insights into the mechanisms of Fgf signaling. Genes Dev., 30, 751–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Endo M. and Minami Y. (2018) Diverse roles for the ror-family receptor tyrosine kinases in neurons and glial cells during development and repair of the nervous system. Dev. Dyn., 247, 24–32. [DOI] [PubMed] [Google Scholar]
- 45. McDonell L.M., Kernohan K.D., Boycott K.M. and Sawyer S.L. (2015) Receptor tyrosine kinase mutations in developmental syndromes and cancer: two sides of the same coin. Hum. Mol. Genet., 24, R60–R66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Niehues S., Bussmann J., Steffes G., Erdmann I., Kohrer C., Sun L., Wagner M., Schafer K., Wang G., Koerdt S.N. et al. (2015) Impaired protein translation in Drosophila models for Charcot–Marie–Tooth neuropathy caused by mutant tRNA synthetases. Nat Commun, 6, 7520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Diodato D., Melchionda L., Haack T.B., Dallabona C., Baruffini E., Donnini C., Granata T., Ragona F., Balestri P., Margollicci M. et al. (2014) VARS2 and TARS2 mutations in patients with mitochondrial encephalomyopathies. Hum. Mutat., 35, 983–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hadchouel A., Wieland T., Griese M., Baruffini E., Lorenz-Depiereux B., Enaud L., Graf E., Dubus J.C., Halioui-Louhaichi S., Coulomb A. et al. (2015) Biallelic mutations of methionyl-tRNA synthetase cause a specific type of pulmonary alveolar proteinosis prevalent on reunion island. Am. J. Hum. Genet., 96, 826–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Strauss K.A., Gonzaga-Jauregui C., Brigatti K.W., Williams K.B., King A.K., Van Hout C., Robinson D.L., Young M., Praveen K., Heaps A.D. et al. (2018) Genomic diagnostics within a medically underserved population: efficacy and implications. Genet. Med., 20, 31–41. [DOI] [PubMed] [Google Scholar]
- 50. Cao X., Li C., Xiao S., Tang Y., Huang J., Zhao S., Li X., Li J., Zhang R. and Yu W. (2017) Acetylation promotes TyrRS nuclear translocation to prevent oxidative damage. Proc. Natl. Acad. Sci. U.S. A., 114, 687–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jinks R.N., Puffenberger E.G., Baple E., Harding B., Crino P., Fogo A.B., Wenger O., Xin B., Koehler A.E., McGlincy M.H. et al. (2015) Recessive nephrocerebellar syndrome on the Galloway-Mowat syndrome spectrum is caused by homozygous protein-truncating mutations of WDR73. Brain, 138, 2173–2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Fu G., Xu T., Shi Y., Wei N. and Yang X.L. (2012) tRNA-controlled nuclear import of a human tRNA synthetase. J. Biol. Chem., 287, 9330–9334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kleeman T.A., Wei D., Simpson K.L. and First E.A. (1997) Human tyrosyl-tRNA synthetase shares amino acid sequence homology with a putative cytokine. J. Biol. Chem., 272, 14420–14425. [DOI] [PubMed] [Google Scholar]
- 54. Benkert P., Biasini M. and Schwede T. (2011) Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics, 27, 343–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bertoni M., Kiefer F., Biasini M., Bordoli L. and Schwede T. (2017) Modeling protein quaternary structure of homo- and hetero-oligomers beyond binary interactions by homology. Sci. Rep, 7, 10480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bienert S., Waterhouse A., Beer T.A., Tauriello G., Studer G., Bordoli L. and Schwede T. (2017) The SWISS-MODEL Repository—new features and functionality. Nucleic Acids Res., 45, D313–D319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Guex N., Peitsch M.C. and Schwede T. (2009) Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: a historical perspective. Electrophoresis, 30Suppl 1, S162–173. [DOI] [PubMed] [Google Scholar]
- 58. Waterhouse A., Bertoni M., Bienert S., Studer G., Tauriello G., Gumienny R., Heer F.T., Beer T.A.P., Rempfer C., Bordoli L. et al. (2018) SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res., 46, W296–W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yang X.L., Kapoor M., Otero F.J., Slike B.M., Tsuruta H., Frausto R., Bates A., Ewalt K.L., Cheresh D.A. and Schimmel P. (2007) Gain-of-function mutational activation of human tRNA synthetase procytokine. Chem. Biol., 14, 1323–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yang X.L., Otero F.J., Skene R.J., McRee D.E., Schimmel P. and Ribas de Pouplana L. (2003) Crystal structures that suggest late development of genetic code components for differentiating aromatic side chains. Proc. Natl. Acad. Sci. U.S.A., 100, 15376–15380. [DOI] [PMC free article] [PubMed] [Google Scholar]