

Abstract
The input-output relationships in neural circuits are determined not only by synaptic efficacy but also by neuronal excitability. Activity-dependent alterations of synaptic efficacy have been extensively investigated, but relatively less is known about how the neuronal output is modulated when synaptic efficacy changes are associated with neuronal excitability changes. In this study, we demonstrate that paired pulses of low-frequency stimulation (PP-LFS) induced metabotropic glutamate receptor (mGluR)-dependent LTD at Schaffer collateral (SC)-CA1 synapses in Sprague Dawley rats (both sexes), and this LTD was associated with EPSP to spike (E-S) potentiation, leading to the increase in action potential (AP) outputs. Threshold voltage (Vth) for APs evoked by synaptic stimulation and that by somatic current injection were hyperpolarized significantly after PP-LFS. Blockers of GABA receptors mimicked and occluded PP-LFS effects on E-S potentiation and Vth hyperpolarization, suggesting that suppression of GABAergic mechanisms is involved in E-S potentiation after PP-LFS. Indeed, IPSCs and tonic inhibitory currents were reduced after PP-LFS. The IPSC reduction was accompanied by increased paired-pulse ratio, and abolished by AM251, a blocker for Type 1 cannabinoid receptors, suggesting that PP-LFS suppresses presynaptic GABA release by mGluR-dependent endocannabinoids signaling. By contrast, a Group 1 mGluR agonist, 3, 5-dihydroxyphenylglycine, induced LTD at SC-CA1 synapses but failed to induce significant IPSC reduction and AP output increase. We propose that mGluR signaling that induces LTD coexpression at excitatory and inhibitory synapses regulates an excitation-inhibition balance to increase neuronal output in CA1 neurons.
SIGNIFICANCE STATEMENT Long-lasting forms of synaptic plasticity are usually associated with excitability changes, the ability to fire action potentials. However, excitability changes have been regarded to play subsidiary roles to synaptic plasticity in modifying neuronal output. We demonstrate that, when metabotropic glutamate receptor-dependent LTD is induced by paired pulses of low-frequency stimulation, the action potential output in response to a given input paradoxically increases, indicating that increased excitability is more powerful than synaptic depression. This increase is mediated by the suppression of a presynaptic GABA release via metabotropic glutamate receptor-dependent endocannabinoid signaling. Our study shows that neuronal output changes do not always follow the direction of synaptic plasticity at excitatory synapses, highlighting the importance of regulating inhibitory tone via endocannabinoid signaling.
Keywords: E-I balance, E-S potentiation, endocannabinoid, i-LTD, mGluR-LTD
Introduction
The LTP and LTD of synaptic transmission are considered memory-encoding mechanisms. LTP consists of two independent components: synaptic efficacy potentiation and EPSP to spike (E-S) potentiation, which is the increasing ability of an EPSP to generate a spike (Bliss and Collingridge, 1993; Bear and Malenka, 1994). The involvement of GABAergic mechanisms in E-S potentiation was recognized early (Abraham et al., 1987; Chavez-Noriega et al., 1989, 1990), suggesting that activity-dependent suppression of inhibitory synapses causes increased excitability and significantly influences the neuronal output. Indeed, the calcineurin-mediated inhibition of postsynaptic GABAA receptors (Lu et al., 2000) and the endocannabinoid (eCB)-mediated inhibition of presynaptic GABA release (Chevaleyre and Castillo, 2003; Azad et al., 2004; Jiang et al., 2010) were shown to be involved in the inhibition of GABAergic synapses (i-LTD) to mediate increased excitability associated with LTP in CA1 pyramidal neurons. The possibility that i-LTD is associated with LTD of excitatory synapses (e-LTD) has not been reported yet.
Expression of E-S potentiation or depression was also observed in the presence of GABA receptor (GABAR) blockers (Jester et al., 1995; Daoudal et al., 2002). These results suggest that activity-dependent changes in intrinsic excitability caused by ion channel activity alterations are another important mechanism for modulating E-S coupling (Spitzer, 1999; Beck and Yaari, 2008). The relative contribution of ion channel mechanisms and GABAergic mechanisms to E-S coupling modulation will be important for the comprehensive understanding of neuronal plasticity. Activity-dependent regulation of ion channels, often referred to as intrinsic plasticity, is associated with synaptic plasticity in various ways. Intrinsic plasticity can act synergistically with synaptic plasticity (Daoudal and Debanne, 2003; Wang et al., 2003; Xu et al., 2005), but the opposite has also been reported. When LTP is associated with reduced excitability (Fan et al., 2005; Narayanan and Johnston, 2007; Malik and Johnston, 2017) or LTD is associated with increased excitability (Brager and Johnston, 2007; Gasselin et al., 2017), intrinsic plasticity is regarded as a homeostatic mechanism to prevent an excessive potentiation or depression of neuronal activity.
The activation of Group I metabotropic glutamate receptors (mGluRs) is well known to underlie e-LTD in CA1 pyramidal neurons (Bolshakov and Siegelbaum, 1994; Oliet et al., 1997; Lee et al., 2005). Signaling pathways responsible for mGluR-dependent e-LTD have been extensively investigated, showing the involvement of multiple mechanisms (Oliet et al., 1997; Schnabel et al., 1999; Ireland and Abraham, 2002; Brager and Johnston, 2007; Mockett et al., 2011; Kim et al., 2015). Our recent study shows that paired pulses of low-frequency stimulation (PP-LFS)-induced mGluR-dependent e-LTD are mediated by activation of the phospholipase C (PLC) pathway, whereas 3,5-dihydroxyphenylglycine (DHPG)-induced e-LTD occurs independently of PLC pathways (Kim et al., 2015). On the other hand, Group 1 mGluRs underlie eCB-dependent i-LTD (Liu et al., 1993; Chevaleyre and Castillo, 2003) and the regulation of various ion channels, including HCN channels (Ih) and Ca2+ channels that may potentially contribute to E-S potentiation (Anwyl, 1999; Brager and Johnston, 2007; Park et al., 2010). However, it remains to be elucidated whether mGluR-dependent e-LTD is associated with E-S potentiation and, if it is, how the neuronal output is altered. Possibly, associated E-S coupling changes may be distinct in the two mGluR-dependent e-LTD types that recruit different signaling pathways.
In this study, we found that PP-LFS at the Schaffer collateral (SC) pathway induces mGluR-dependent e-LTD, whereas the input-output (I-O) relationship paradoxically shifts leftward. We provide evidence that PP-LFS activates an mGluR-dependent eCB signaling pathway, which leads to i-LTD, resulting in increased excitability and action potential (AP) output. By contrast, DHPG induces e-LTD without significant i-LTD, leading to no significant change in the I-O relationship. Our results suggest that activity-dependent activation of mGluRs in CA1 neurons induces LTD at both excitatory and inhibitory synapses, the outcome of which is in favor of excitation, resulting in a leftward shift in the I-O relationship.
Materials and Methods
Animals.
All experiments were conducted with the approval of the animal experiment ethics committee at the Seoul National University College of Medicine. Experiments were performed using Sprague Dawley rats of either sex.
Slice preparation.
After decapitation, the whole brain was immediately removed and submerged in an ice-cold aCSF containing the following (in mm): NaCl 116, NaHCO3 26, KCl 3.2, NaH2PO4 1.25, CaCl2 0.5, MgCl2 7, glucose 10, Na-pyruvate 2, and vitamin C 3. Transverse hippocampal slices (300 μm thick) were prepared using a vibratome (VT1200S, Leica Microsystems). Slices were recovered at 32°C for 30 min and then maintained at room temperature in an aCSF containing the following (in mm): NaCl 124, NaHCO3 26, KCl 3.2, NaH2PO4 1.25, CaCl2 2.5, MgCl2 1.3, and glucose 10; we referred to the aCSFs as “recording aCSFs” until they were used for recordings. The recording aCSFs were used during electrophysiological recording. All aCSFs were bubbled with mixture of 95% O2 and 5% CO2 to a final pH of 7.4.
Electrophysiology.
Hippocampal CA1 pyramidal slices were transferred to an immersed recording chamber continuously perfused with oxygenated recording aCSF using a peristaltic pump (Gilson). CA1 pyramidal cells were visualized using an upright microscope equipped with differential interference contrast optics (BX51WI, Olympus). All electrophysiological recordings were made in soma with an EPC-8 amplifier (HEKA Electronik) at a sampling rate of 10 kHz. Data recorded at different sampling rates are indicated. All the recordings were performed at 32 ± 1°C, and the aCSF perfusion rate was maintained at 1–1.5 ml/min. Patch pipettes (3–4 mΩ) and monopolar stimulator pipettes (1–2 mΩ) were made from glass capillaries (Bo-glasscapollaries) using a puller (PC-10, Narishige). The pipettes were filled with internal solutions containing the following (in mm): potassium gluconate 130, KCl 7, NaCl 2, MgCl2 1, EGTA 0.1, ATP-Mg 2, Na-GTP 0.3, and HEPES 10, pH 7.3 (KOH, 295 mosmol/L with sucrose). A stimulator (Stimulus Isolator A360; WPI) connected to a monopolar electrode filled with recording aCSF was placed in the stratum radiatum layer (horizontally and vertically ∼120–150 μm away from the soma) of the CA1 field to evoke SC stimulation induced-synaptic responses. For EPSC and IPSC recordings, the stimulator intensity (100 μs duration; 9–31.5 V) of extracellular stimulation was adjusted to evoke current amplitudes between 100 pA and 300 pA for the baseline. EPSCs and IPSCs were recorded from CA1 pyramidal neurons in a whole-cell configuration at a holding potential of −63 and 10 mV, respectively. We confirmed that EPSCs recorded at −63 mV in the presence of APV were completely abolished by blocking AMPA receptors using NBQX, and that IPSCs recorded at 10 mV were completely abolished by blocking GABAA receptors using bicuculline. Immediately after whole-cell configuration, the pipette series resistance and capacitance were compensated manually and checked throughout the experiment. Cells in which the series resistance exceeded 20 mΩ and changed >15% during the experiment were discarded.
The mGluR-LTD was induced by PP-LFS (1 Hz, 15 min), and pharmacological mGluR-LTD was induced by DHPG application (7 min). The PP-LFS was performed in current-clamp mode at resting membrane potential (RMP) holding with the same stimulus intensity used for baseline EPSC or IPSC recordings. All recordings were performed in the presence of an NMDAR antagonist (APV 50 μm) to block NMDAR-dependent LTD.
To measure tonic GABAergic currents, we applied voltage steps from the holding potential of −60 to −120 mV (5 ms duration) with a 10 s interval, and plotted the changes in instantaneous currents measured at −60 mV (I−60 mV) and −120 mV (I−120 mV) before Ih activation. The difference in I−120 mV before and after GABAR antagonists was measured.
Data analysis.
Somatic (positive or negative) current injections were done at the holding potential −65 mV unless otherwise indicated. Input resistance (Rin) was calculated with the voltage deflection (ΔV = Vmax − Vbaseline) by a −25 pA hyperpolarization current injection. Voltage sag in response to the hyperpolarization current was compared with ΔV (Vmax − Vss) versus Vmax. Rin and voltage sag were also compared at RMP holding, and the results were similar with those compared at the −65 mV holding. Experiments conducted with ZD7288 application were done at RMP holding. Experiments performed with synaptic stimulation were done at RMP. The threshold potential for APs (Vth) evoked by ramp current injection (Vth_ramp) or synaptic stimulation (Vth_SC) was determined by the potential where dV/dt exceeds 10 V/s. We confirmed that Vth was not significantly affected by the SC stimulation intensity. We obtained Vth from at least five APs, and the averaged value was regarded as the Vth of the cell in each experimental condition. Recordings to compare Vth were done at a sampling rate of 10–50 kHz. The AP generation probability was measured by the success or failure of AP generation during the trials [n/10 × 100(%)]. From synaptic-stimuli trials with various stimulus intensity, EPSP slopes were measured from the first EPSP during the first 2 ms. Plotted AP probability along stimulus intensity or the first EPSP slope was fitted with sigmoid function.
Drugs.
AM-251, APV, bicuculline, CGP52432, DHPG, MPEP, NNC-711, picrotoxin, tetrodotoxin, and ZD7288 were purchased from Tocris Bioscience. Stock solutions of these drugs were made by dissolution in deionized water or DMSO and were stored at −20°C. During the experiment, one aliquot was thawed and used. The DMSO concentration in solutions was maintained <0.1%.
Experimental design and statistical analysis.
Experiments were conducted in rats at postnatal days 16–20, and the total number of animals used was 105 of either sex.
The data were analyzed using IgorPro (version 4.1, WaveMetrics) and OriginPro (version 8.0, Microcal) software and presented as mean ± SEM, where n represents the number of cells studied. The statistical significance of differences was evaluated using a Student's t test and the precise p number is written in Results. Statistical correlations were tested using Pearson test.
Results
E-S potentiation associated with PP-LFS-induced LTD leads to increased AP output
The activation of Group 1 mGluRs induces not only e-LTD (Lüscher and Huber, 2010; Kim et al., 2015) but also increased excitability (Ireland and Abraham, 2002). We investigated how the I-O relationship is affected during mGluR-dependent e-LTD in CA1 pyramidal neurons. The e-LTD was induced by PP-LFS (paired pulse in 50 ms interpulse interval delivered at 1 Hz for 15 min) at an SC-CA1 synapse (Fig. 1A), as previously reported (Kim et al., 2015). We recorded PP-LFS-induced e-LTD (e-LTDPP-LFS) for at least 30 min. The relative amplitude of EPSCs recorded at 25–30 min after PP-LFS was 0.59 ± 0.07 (n = 11) of the baseline amplitude obtained before PP-LFS (Fig. 1A). No significant change was detected in the paired-pulse ratio (PPR) during e-LTDPP-LFS (Control, 1.37 ± 0.05; post-PP-LFS [pPP-LFS], 1.36 ± 0.1; n = 11, p = 0.9), suggesting that PP-LFS induces synaptic depression via postsynaptic mechanism, not affecting glutamate release at presynaptic terminals. In accordance with EPSC depression, EPSP amplitudes were also reduced. In current-clamp experiments, we applied a burst synaptic stimulation (5 stimuli, 50 Hz, delivered every 2 s) to record summated EPSPs (Fig. 1B). The amplitude of the first EPSP was significantly reduced after PP-LFS (0.56 ± 0.06, n = 8, p = 0.1), but the summation ratio (summated EPSP amplitude/first EPSP amplitude) was unchanged (Control, 2.25 ± 0.2; pPP-LFS, 2.30 ± 0.4; n = 7, p = 0.9) (Fig. 1B). The decrement in EPSP amplitude after PP-LFS obtained from an individual cell was equivalent to that in its EPSC amplitude (Fig. 1C), suggesting that EPSP changes are parallel to EPSC changes, which indicates no significant change in dendritic excitability during e-LTDPP-LFS. In the presence of the metabotropic glutamate receptor 5 (mGluR5) blocker MPEP (25 μm), EPSCs (1.00 ± 0.05, n = 7) and EPSPs (0.98 ± 0.08, n = 7) remained unchanged after PP-LFS, indicating that e-LTDPP-LFS is predominantly mediated by mGluR5 (Fig. 1D). We also confirmed that MPEP alone does not affect the basal synaptic transmission (EPSC amplitude: Control, 156.4 ± 16.0 pA; +MPEP, 152.1 ± 23.9 pA; n = 5, p = 0.7) or electrical properties, such as RMP (Control, −58.1 ± 0.5 mV; +MPEP, −58.7 ± 0.5 mV; n = 6, p = 0.1) and Rin (Control, 136.7 ± 16.0 mΩ; +MPEP, 130.0 ± 17.6 mΩ; n = 6, p = 0.5).
Figure 1.

Induction of E-S potentiation during e-LTDPP-LFS at SC-CA1 synapses. A, PP-LFS at SC induced long-term change in EPSC amplitude. Top, Representative EPSCs recorded before PP-LFS induction (a, Control) and 25–30 min after PP-LFS (b, pPP-LFS). B, Top, Example traces of summated EPSPs by 50 Hz SC stimulation. Bottom, Traces scaled to first EPSP peak are superimposed. Bar graphs represent no significant change in EPSP summation ratio by PP-LFS. C, Ratio of first EPSC change (EPSCPP-LFS/EPSCControl) is plotted against EPSP change (EPSPPP-LFS/EPSPControl). Fitted line of data is presented with solid line (black), and a linear correlation is observed (r = 0.62). Gray dashed line indicates linear relationship between EPSP and EPSC changes. D, The presence of MPEP throughout the experiment prevented the induction of eLTDPP-LFS. E, Raw voltage traces as a result of 10 trials of SC stimulation (27 V, 5 stimuli, 50 Hz, 2 s interval) before (Control) and 30 min after PP-LFS (pPP-LFS). After PP-LFS induction, the summated EPSP triggered firing in all trials. F, Graph showing distribution of firing probability as a function of stimulus voltage before (Control) and 30 min after PP-LFS (pPP-LFS). Firing probability was obtained from 10 trials at each stimulus intensity. Bar graphs represent that SI50 is significantly decreased after PP-LFS, but no significant change occurred in the presence of MPEP. G, Plot of firing probability as a function of first EPSP slope before (Control) and 30 min after PP-LFS (pPP-LFS). Summary data showing significant decrease of ES50 after PP-LFS. F, G, Light colors represent sigmoid fitted lines. F, G, Filled circles represent where the SI50 and the ES50 are acquired, respectively. H, Left, Indicated traces marked with a and b shown at E are superimposed and enlarged, showing hyperpolarized Vth of APs after PP-LFS. Middle, Phase-plane plots of dV/dt versus membrane potential of first AP induced by SC stimulation. Right, Bar graphs representing significant Vth hyperpolarization that AP induced by SC stimulation (Vth_SC). *p < 0.05. **p < 0.01. n.s., Not significant (p > 0.05). Error bars indicate SEM.
Despite a significant decrease in EPSPs during e-LTDPP-LFS, surprisingly, AP generation significantly increased after PP-LFS (Fig. 1E). We applied 10 burst stimulations at each stimulation intensity to measure the AP generation probability. Figure 1E shows exemplary traces that were obtained at a stimulus intensity of 27 V before PP-LFS induction (Control) and 30 min after LTD induction (pPP-LFS). To plot I-O relationships, we varied the stimulus intensity in a wide range from subthreshold to suprathreshold and found that the I-O relationship shifted leftward after PP-LFS. The magnitude of the leftward shift after PP-LFS was quantified by comparing the stimulus intensity required for 50% AP probability (SI50) in pPP-LFS with that in Control (Fig. 1F). SI50 significantly decreased during e-LTDPP-LFS (SI50 = 0.78 ± 0.05 in pPP-LFS, n = 6, p = 0.02), and this decrease was abolished in the presence of MPEP (SI50 = 1.01 ± 0.06 in pPP-LFS, n = 7, p = 0.9) (Fig. 1F), suggesting that the leftward shift in the I-O relationship during e-LTDPP-LFS is dependent on mGluR5. The leftward shift in the I-O relationship despite synaptic depression may suggest the occurrence of E-S potentiation during e-LTDPP-LFS. Therefore, we obtained the E-S curve by converting the x axis into an EPSP slope (Fig. 1G). The EPSP slope to attain 50% AP probability (ES50) after PP-LFS was 0.75 ± 0.03 (n = 6, p = 0.0006) of Control (Fig. 1G), indicating that PP-LFS induces a significant E-S potentiation.
The increase in AP firing probability at the same EPSP slope implies Vth changes during e-LTDPP-LFS. We determined Vth as the membrane potential where dV/dt was >10 V/s in the phase-plane plots and found that Vth was significantly hyperpolarized after PP-LFS (Control, −40.8 ± 0.8 mV; pPP-LFS, −46.0 ± 1.5 mV; n = 8, p = 0.0006) (Fig. 1H). These results suggest that Vth hyperpolarization contributes to E-S potentiation during e-LTDPP-LFS, which is powerful enough to induce a leftward shift in the I-O relationships despite synaptic depression.
The LTD of GABAergic synapses mediated by eCB underlies E-S potentiation during e-LTDPP-LFS
The next question we addressed was the mechanism underlying E-S potentiation during e-LTDPP-LFS. Disinhibition, an increase in excitability due to a decrease in GABAergic inhibition, was suggested to participate in E-S potentiation during tetanus-induced LTP (Chavez-Noriega et al., 1989, 1990). To investigate whether disinhibition is involved in E-S potentiation during e-LTDPP-LFS, we first examined whether IPSCs are reduced after PP-LFS. IPSCs evoked by stimulating the SC pathway were recorded at a holding potential of 10 mV. We monitored changes of IPSCs after PP-LFS and found that the amplitude of IPSCs was reduced significantly after PP-LFS (0.38 ± 0.05 of baseline at 25–30 min after PP-LFS, n = 5) (Fig. 2A). Because the decrease in IPSCs persisted >30 min, we referred to it as i-LTD. The IPSC amplitude reduction was accompanied by an increase in PPR (Control, 0.84 ± 0.06; 25–30 min after PP-LFS, 1.38 ± 0.3; n = 5, p = 0.04), suggesting that i-LTD is attributable to the decrease in GABA release from presynaptic terminals (Fig. 2A). Furthermore, we confirmed that i-LTD was abolished by MPEP (IPSC: 0.95 ± 0.09 of baseline at 25–30 min after PP-LFS; PPR: Control, 1.07 ± 0.09; 25–30 min after PP-LFS, 1.1 ± 0.08; n = 5, p = 0.3) (Fig. 2B). These results suggest that mGluR5-dependent mechanisms activated during PP-LFS affect presynaptic GABAergic terminals, possibly via retrograde signaling to cause i-LTD.
Figure 2.
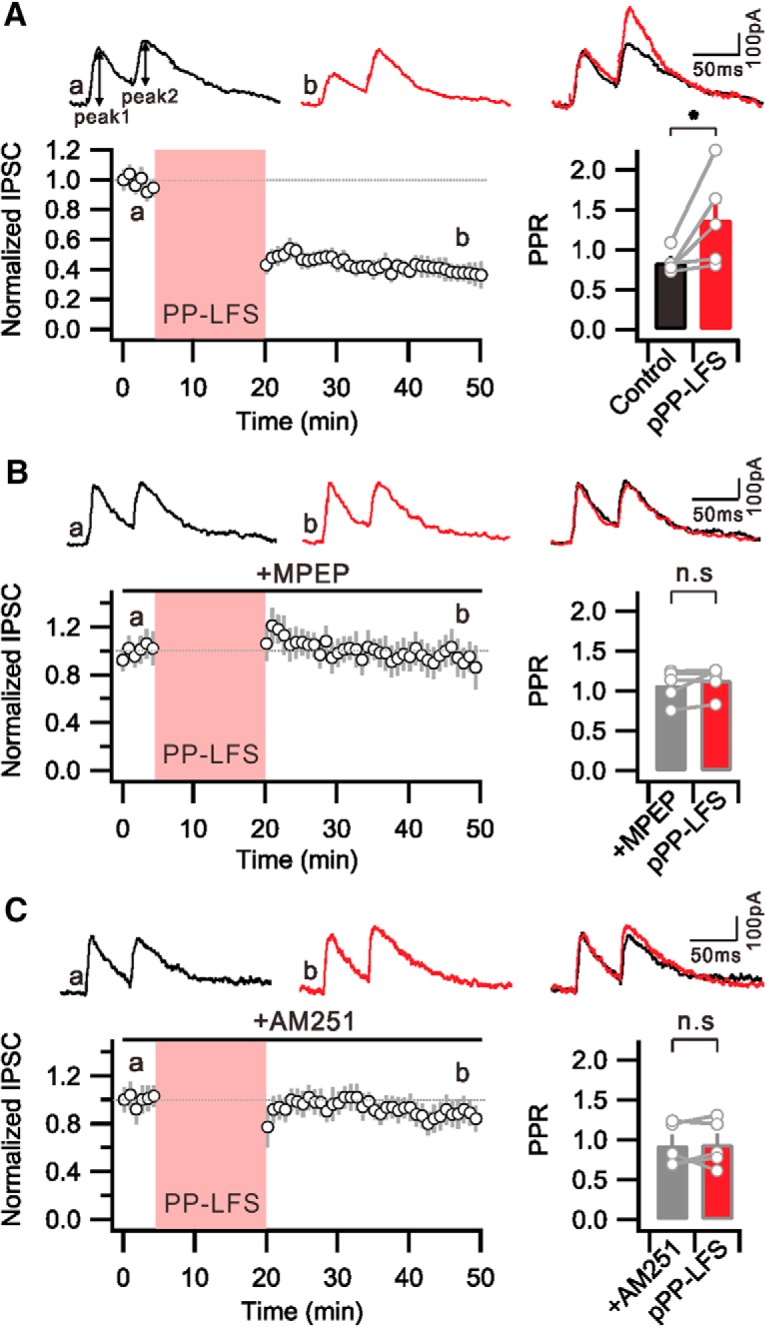
PP-LFS-induced i-LTD is mediated by mGluR/eCB signaling. A, Graph showing PP-LFS at SC fibers induced LTD of IPSC magnitude. Bar graphs representing that PPR of IPSC is significantly increased after PP-LFS. B, PP-LFS-induced i-LTD is not expressed in the presence of MPEP. The PPR of IPSC is not increased after PP-LFS. C, Graph showing that the PP-LFS-induced i-LTD is significantly blocked by inhibiting CB1Rs. The PPR of IPSC in the presence of AM251 remains unchanged after PP-LFS. A–C, Insets, Top, Representative IPSC traces before PP-LFS (a, black) and 25–30 min after PP-LFS (b, red). *p < 0.05. n.s., Not significant (p > 0.05). Error bars indicate SEM.
eCBs, such as 2-AG, have been suggested as retrograde signaling molecules to mediate high-frequency stimulation-induced i-LTD in the SC-CA1 synapse (Chevaleyre and Castillo, 2003, 2004; Younts et al., 2013). The activation of Group 1 mGluRs triggers eCB mobilization upon PLC and diacylglycerol lipase α activation (Castillo et al., 2012). To assess a possible role of eCB signaling in i-LTD, we examined the effects of AM251, a Type 1 cannabinoid receptor (CB1R) antagonist, on i-LTD. In the presence of AM251 (2 μm), PP-LFS no longer induced i-LTD, indicating that i-LTD depends on CB1Rs (IPSC: 0.89 ± 0.08 of baseline at 25–30 min after PP-LFS; PPR: Control, 0.93 ± 0.1; 25–30 min after PP-LFS, 0.92 ± 0.1; n = 5, p = 0.9) (Fig. 2C). These results suggest that PP-LFS at the SC pathway activates mGluR5/PLC pathways in postsynaptic CA1 neurons, which mobilizes eCBs that activate CB1Rs in presynaptic GABAergic interneuron terminals, resulting in reduced GABA release.
To further verify whether disinhibition indeed contributes to E-S potentiation during e-LTDPP-LFS, we examined whether blocking GABARs can mimic the PP-LFS effects. When we blocked GABARs by applying a mixture of a GABAA receptor antagonist (bicuculline 20 μm or picrotoxin 100 μm) and a GABAB receptor antagonist (CGP52432 1 μm) (anti-GABARs), the I-O relationship shifted leftward significantly (SI50 = 0.69 ± 0.03 with anti-GABARs, n = 8, p = 0.00003) (Fig. 3A,B). However, the amplitude of EPSPs (Control, 3.7 ± 0.6 mV; +anti-GABARs, 4.0 ± 0.8 mV; n = 8, p = 0.6) and the EPSP slope (Fig. 3C) were not significantly affected by anti-GABARs, suggesting that IPSPs do not significantly affect EPSPs under our experimental conditions. Nevertheless, E-S coupling was significantly potentiated by anti-GABARs (ES50 = 0.84 ± 0.03, n = 7, p = 0.002) (Fig. 3D), and this potentiation was associated with a significant Vth hyperpolarization (Control, −40.1 ± 1.0 mV; +anti-GABARs, −42.2 ± 1.0 mV; n = 8, p = 0.0007) (Fig. 3E). On the other hand, significant changes were not observed when we applied bicuculline alone (SI50 = 1.01 ± 0.1 with bicuculline, n = 7, p = 0.9; Vth: Control, −41.3 ± 0.8 mV; +bicuculline, −41.8 ± 1.2 mV; n = 6, p = 0.6) or CGP52432 alone (SI50 = 1.06 ± 0.1 with CGP52432, n = 9, p = 0.6; Vth: Control, −40.5 ± 1.4 mV; +CGP52432, −40.8 ± 0.8 mV; n = 5, p = 0.9). These results suggest that suppression of both GABAA and GABAB receptors is required for E-S potentiation in CA1 neurons, and that Vth hyperpolarization is a major mechanism for E-S potentiation, which is consistent with the E-S potentiation mechanism during LTDPP-LFS.
Figure 3.

GABAR blockers mimic and occlude E-S potentiation associated with Vth hyperpolarization during e-LTDPP-LFS. A, Example traces of the voltage responses to 10 trials of suprathreshold SC stimulation obtained before (Control) and after GABAR antagonist application (+anti-GABARs). Inset, Superimposed voltage traces indicating hyperpolarized Vth. B, Graph showing distribution of firing probability as a function of stimulus voltage before (Control) and after anti-GABAR application (+anti-GABARs). Bar graph represents significant decrease of SI50 by GABAR blockade. C, No significant change is detected in the ratio of the first EPSP slope to stimulus intensity after the anti-GABAR application. D, Plot of firing probability as a function of first EPSP slope. Bar graphs represent a significant decrease in ES50 by GABAR inhibition. E, Summary data showing significant Vth hyperpolarization that APs induced by SC stimulation (Vth_SC). F, The e-LTDPP-LFS is still induced in the presence of anti-GABARs. Inset, EPSC traces obtained before (a) and 25–30 min after PP-LFS (b). G, Voltage responses to 10 trials of suprathreshold SC stimulation before (+anti-GABARs) and after PP-LFS (pPP-LFS) in the presence of anti-GABARs during entire recordings. Inset, No significant change in Vth. H, Distribution of firing probability as a function of stimulus voltage before (+anti-GABARs) and after PP-LFS (pPP-LFS) in the presence anti-GABARs. Right, Bar graphs represent significant increase in SI50. I, No significant change is detected in AP probability versus first EPSP slope. Bar graphs represent no significant change in ES50. J, Vth shift by PP-LFS is occluded in the presence of anti-GABARs. *p < 0.05. **p < 0.01. n.s., Not significant (p > 0.05). Error bars indicate SEM.
We then examined whether anti-GABARs occlude PP-LFS effects. The anti-GABARs did not affect the expression of e-LTDPP-LFS (0.61 ± 0.07 of baseline at 25–30 min after PP-LFS, n = 6) (Fig. 3F) but inhibited the effects of PP-LFS on the I-O relationship or E-S coupling (Fig. 3G–I). Indeed, AP generation decreased significantly after PP-LFS (SI50 = 1.21 ± 0.05 in pPP-LFS, n = 8, p = 0.003) (Fig. 3H), whereas E-S coupling (ES50 = 0.98 ± 0.02, n = 8, p = 0.4) (Fig. 3I) and Vth (+anti-GABARs, −42.8 ± 1.3 mV; pPP-LFS, −43.9 ± 0.9 mV; n = 9, p = 0.3) (Fig. 3J) remained unchanged after PP-LFS in the presence of anti-GABARs. The SI50 increase after PP-LFS in the presence of anti-GABARs can be explained by a reduced EPSP amplitude with unchanged Vth. These results are consistent with the hypothesis that E-S potentiation associated with the Vth hyperpolarization after PP-LFS is attributable to decreased GABAergic inputs.
Reduction of tonic GABA currents underlies increased AP output with Vth hyperpolarization during e-LTDPP-LFS
GABAergic inhibition is generally thought to occur via shunting effects. In this regard, decreased GABAergic inputs are expected to increase EPSPs, but we noted that anti-GABARs do not increase the amplitude or slope of EPSPs (Fig. 3C). To understand this observation, we examined effects of anti-GABARs on Rin changes with RMP changes and found that anti-GABARs did not significantly affect Rin (Control, 111.6 ± 8.4 mΩ; +anti-GABARs, 117.7 ± 8.1 mΩ; n = 9, p = 0.1) (Fig. 4A) or RMP (Control, −59.4 ± 0.3 mV; +anti-GABARs, −58.5 ± 0.8 mV; n = 9, p = 0.3). On the contrary, Rin was increased significantly after PP-LFS (Control, 117.7 ± 8.8 mΩ; pPP-LFS, 164.0 ± 14.2 mΩ; n = 8, p = 0.0005) (Fig. 4A), whereas RMP was unchanged (Control, −59.4 ± 0.6 mV; pPP-LFS, −60.1 ± 0.6 mV; n = 14, p = 0.2). However, PP-LFS-induced increases in Rin were unaffected by anti-GABARs (+anti-GABARs, 132.1 ± 8.1 mΩ; pPP-LFS, 164.1 ± 18.9 mΩ; n = 9, p = 0.05) (Fig. 4A), suggesting that decreased GABAergic function does not cause a significant increase in Rin and that the increased Rin after PP-LFS is not related to decreased GABAergic inputs.
Figure 4.

Vth hyperpolarization for APs evoked by somatic depolarization during e-LTDPP-LFS is dependent on GABAergic mechanisms. A, Left, Representative traces showing the voltage responses to a −25 pA square current injection. Traces obtained in condition before (Control) and after anti-GABAR application (+anti-GABARs); before (Control) and 30 min after PP-LFS (pPP-LFS); before (+anti-GABARs) and after PP-LFS (pPP-LFS) in the presence of anti-GABARs during the entire recordings are superimposed. Vbaseline, Baseline of voltage deflection; Vmax, maximum point of voltage deflection. Right, Summary bar graphs of the Rin profile obtained under described conditions. B, Representative voltage responses to a ramp (250 pA/1 s) current injection to soma before (Control) and 30 min after PP-LFS (pPP-LFS). Inset, Superimposed traces of significant Vth hyperpolarization of first AP. Bar graphs represent significant Vth hyperpolarization (Vth_ramp) and increase in number of AP (AP #). Presence of MPEP throughout the experiment prevented the changes in Vth and number of AP. C, The same ramp protocol is performed before (Control) and after perfusion of anti-GABARs (+anti-GABARs). D, The same ramp protocol is performed in the presence of anti-GABARs (+anti-GABARs) and compared with the subsequent PP-LFS induction (pPP-LFS). *p < 0.05. **p < 0.01. n.s., Not significant (p > 0.05). Error bars indicate SEM.
Among PP-LFS-induced effects and anti-GABAR effects, Vth hyperpolarization was commonly found and considered relevant as a mechanism for E-S potentiation. To further characterize the Vth change, we examined Vth hyperpolarization for APs evoked by somatic depolarization. APs were evoked by injecting depolarizing ramp currents (250 pA/s) to the soma under current-clamp conditions, and the Vth for the first AP and the number of APs were measured before and 30 min after PP-LFS. After PP-LFS, Vth was hyperpolarized (Control, −41.4 ± 0.8 mV; pPP-LFS, −47.4 ± 1.9 mV; n = 6, p = 0.04) and more APs were evoked (Control, 8.7 ± 1.5; pPP-LFS, 12.3 ± 1.4; n = 6, p = 0.005) (Fig. 4B). The magnitude of Vth hyperpolarization for APs evoked by direct somatic stimulation was not significantly different from that evoked by SC stimulation (6.0 mV vs 5.2 mV). The Vth values (Control, −39.8 ± 0.7 mV; pPP-LFS, −40.0 ± 2.0 mV; n = 6, p = 0.9) and AP numbers (Control, 7.7 ± 1.1; pPP-LFS, 7.3 ± 1.5; n = 6, p = 0.7) were unchanged after PP-LFS in the presence of MPEP (Fig. 4B), confirming the involvement of mGluR5 activation in these effects.
We then examined whether the inhibition of GABARs can mimic and occlude PP-LFS effects. When we applied anti-GABARs, the AP generation evoked by ramp current injection to the soma significantly increased (Control, 8.7 ± 2.0; +anti-GABARs, 12.0 ± 1.6; n = 6, p = 0.03), and this increase was associated with a significant Vth hyperpolarization (Control, −40.4 ± 1.1 mV; +anti-GABARs, −43.2 ± 1.5 mV; n = 6, p = 0.045) (Fig. 4C). Furthermore, in the presence of anti-GABARs, PP-LFS did not affect AP generation (+anti-GABARs, 10.7 ± 1.4; pPP-LFS, 11.0 ± 1.9; n = 9, p = 0.7) or Vth (+anti-GABARs, −42.3 ± 1.4 mV; pPP-LFS, −43.0 ± 1.3 mV; n = 9, p = 0.5), indicating the occlusion of PP-LFS effects by anti-GABARs (Fig. 4D). Applying GABAA or GABAB receptor antagonists alone did not show significant effects, suggesting the involvement of both GABAA and GABAB receptors in the increased AP firing associated with Vth hyperpolarization after PP-LFS.
A possible mechanism for the effects of anti-GABARs and PP-LFS on APs evoked by somatic depolarization is that tonic GABA currents at an ambient GABA level have inhibitory roles for AP generation by increasing Vth. Ambient GABA levels are expected to decrease when vesicular GABA release is reduced after PP-LFS, which in turn hyperpolarizes Vth and increases AP generation. To verify this hypothesis, identifying tonic GABA currents using the same internal solution used for AP recordings is a prerequisite. The conventional method for recording tonic GABA currents is to measure the change in holding current level by GABAA receptor antagonists under the condition that the driving force for Cl− is maximized using a high Cl− internal solution or ambient GABA concentration is maximized using GABA transporter inhibitors (Dalby, 2003; Glykys and Mody, 2007; Bright and Smart, 2013; Razik et al., 2013). Because detecting holding current level changes by applying anti-GABARs was not successful with the low Cl− internal solution used in the present study (calculated ECl −65.9 mV), we recorded changes in the current level by anti-GABARs at −120 mV by applying voltage steps from the holding potential of −60 to −120 mV (5 ms duration) (Fig. 5A). Although GABAA or GABAB receptor antagonists alone did not induce significant changes, we could observe a small but significant decrease in the current when anti-GABARs were applied. The different currents before and after applying anti-GABARs recorded at −120 mV were regarded as GABAR-mediated currents (IGABARs) (Control, −39.1 ± 8.0 pA, n = 9) (Fig. 5A,E). The current level at −120 mV was decreased by TTX (0.5 μm) (21.6 ± 5.3 pA, n = 5); and thus, the IGABAR amplitude was reduced significantly in the presence of TTX (+TTX, −6.6 ± 4.5 pA, n = 5, p = 0.0008) (Fig. 5C,E). On the other hand, the current level at −120 mV was increased by NNC-711 (5 μm), a GABA uptake inhibitor (24.1 ± 9.3 pA, n = 7). The IGABAR amplitude in the presence of NNC-711 was −46.2 ± 7.7 pA (n = 6) (Fig. 5D,E), slightly larger than that obtained from Control, although the difference was not statistically significant. These results support that IGABAR is the current activated by ambient GABA. Importantly, anti-GABARs had no effect on currents after PP-LFS, suggesting that IGABARs became negligible after PP-LFS (pPP-LFS, −6.3 ± 2.5 pA, n = 7, p = 0.0003) (Fig. 5B,E). These results support that IGABARs at the basal state, which has an inhibitory role in the AP firing of CA1 neurons by Vth modulation, is reduced by PP-LFS, resulting in the AP firing increase during e-LTDPP-LFS.
Figure 5.

PP-LFS inhibits tonic GABA currents. A, Graph showing recorded currents at −60 mV (I−60 mV, gray) or −120 mV (I−120 mV, black) holding. Consistent reduction of currents by anti-GABAR perfusion (+anti-GABARs) is well detected at −120 mV holding. B, IGABAR was no longer detected after pPP-LFS induction (pPP-LFS). C, I−120 mV is reduced by TTX perfusion (+TTX) and is not further reduced by subsequent anti-GABAR treatment (+anti-GABARs). D, I−120 mV is slightly increased by NNC-711 perfusion (+NNC-711) and reduced by subsequent anti-GABAR treatment (+anti-GABARs). E, Bar graphs represent IGABARs in conditions described in A–D. *p < 0.05. **p < 0.01. Error bars indicate SEM.
The PP-LFS-induced reduction of tonic inhibition is mediated by eCB/CB1R activation
We have shown that i-LTD is mediated by eCB/CB1Rs (Fig. 2C). Considering that the main source of ambient GABA responsible for tonic inhibition in the hippocampus is the vesicular GABA release responsible for activating phasic inhibition (Glykys and Mody, 2007), we examined whether eCB/CB1R activation is involved in the reduction of tonic currents after PP-LFS. In the presence of AM251, we found that IGABARs was well detected after PP-LFS, and no significant difference was determined between the IGABAR amplitudes obtained before and after PP-LFS (no PP-LFS, −22.3 ± 2.6 pA at −120 mV, n = 6; pPP-LFS, −20.8 ± 1.2 pA at −120 mV, n = 5; p = 0.6) (Fig. 6A–C). In agreement of this finding, PP-LFS-induced Vth hyperpolarization was abolished in the presence of AM251 (Vth_ramp: +AM251, −39.0 ± 0.4 mV; pPP-LFS, −40.4 ± 0.4 mV; n = 9, p = 0.1; Vth_SC: +AM251, −39.8 ± 0.7 mV; pPP-LFS, −40.8 ± 0.6 mV; n = 6, p = 0.2) (Fig. 6D,E). Collectively, these results suggest that the PP-LFS-induced increases in somatic excitability and E-S potentiation are mediated by the CB1R-dependent inhibition of the inhibitory input.
Figure 6.

PP-LFS-induced reduction of tonic inhibition is mediated by eCB/CB1R activation. A, Graph showing that IGABAR is detected at −120 mV holding in the presence of AM251 (+AM251) during entire recordings. B, In the presence of AM251 (+AM251), IGABAR is still detected at −120 mV holding after PP-LFS (pPP-LFS). C, Bar graph represents measured IGABARs without PP-LFS (no PP-LFS) or after PP-LFS induction (pPP-LFS) in the presence of AM251. D, Ramp current induced voltage traces with CB1Rs blocking (+AM251) and 30 min after PP-LFS (pPP-LFS). Summarized data showing no significant change in Vth_ramp. E, Suprathreshold SC stimulation induced voltage traces before (+AM251) and 30 min after PP-LFS (pPP-LFS) with AM251 perfusion. Bar graphs representing no significant change in Vth_SC. Insets, Superimposed and expanded traces from the boxed area. Arrows indicate no significant change of Vth. n.s., Not significant (p > 0.05). Error bars indicate SEM.
DHPG does not induce the leftward shift in I-O relationship during e-LTD
Next, we investigated whether mGluR-LTD at excitatory synapses induced by the Group 1 mGluR agonist DHPG (e-LTDDHPG) also leads to a leftward shift in I-O relationship and E-S coupling. A bath application of DHPG (100 μm) for 7 min reliably induced synaptic depression in our experimental condition to a similar extent induced by PP-LFS (0.56 ± 0.08 of baseline at 25–30 min after DHPG, n = 6) (Fig. 7A). As were the changes during e-LTDPP-LFS, EPSC changes during e-LTDDHPG were parallel to EPSP changes (0.71 ± 0.06, n = 7) (Fig. 7B). However, the I-O relationship during e-LTDDHPG was different from that during e-LTDPP-LFS (Fig. 7C,D). The AP output during e-LTDDHPG in response to burst synaptic stimulation was not consistently increased or decreased but showed a variable response. As a result, the mean value for SI50 during e-LTDDHPG was not significantly different from that obtained before DHPG application (SI50 = 1.02 ± 0.08, n = 11, p = 0.9) (Fig. 7D). To understand the I-O response variability during e-LTDDHPG, we compared the magnitudes of output changes (1 − normalized SI50) and synaptic weight changes (EPSPDHPG/EPSPControl) and found a linear relationship between these two parameters (Fig. 7D), suggesting that synaptic weight is a major factor for determining AP outputs. Interestingly, the relationship crosses zero when EPSPs were reduced by ∼20%, suggesting that 20% of the EPSP reduction was compensated by another mechanism that increases excitability. The analysis of the E-S coupling revealed a somewhat variable response in that most cells showed E-S potentiation whereas a few cells showed E-S depression. The mean value for ES50 decreased significantly after DHPG (ES50, 0.93 ± 0.03, n = 10, p = 0.01) (Fig. 7E), but the decrease was smaller than that observed after PP-LFS. To examine the involvement of i-LTD in DHPG-induced E-S potentiation, we measured changes in IPSCs after DHPG (0.82 ± 0.06 of baseline at 25–30 min after DHPG, n = 10) (Fig. 7F). In contrast to the expression of significant i-LTD after PP-LFS, only 3 of 10 cells showed IPSC decreases after DHPG by >20% (Fig. 7G, box). There was no correlation between the magnitudes of i-LTD and E-S potentiation (Fig. 7G) or Vth changes (Fig. 7H: number indicates 3 cells that show significant i-LTD in Fig. 7G), suggesting that the increased excitability underlying E-S potentiation during e-LTDDHPG is not mediated by disinhibition (Fig. 7G). The Vth showed variable responses with no significant change in the mean value for Vth after DHPG (Control, −41.0 ± 0.6 mV; pDHPG, −41.5 ± 2.0 mV; n = 11, p = 0.4), but the Vth changes of individual cells correlated with E-S coupling changes after DHPG (Fig. 7H). A correlation between E-S coupling changes and Rin changes was not observed (Fig. 7I). These results suggest that the E-S coupling changes after DHPG involve an intrinsic plasticity mechanism that regulates Vth. Considering that Vth hyperpolarization by anti-GABARs (2.1 mV) was smaller than that by PP-LFS (5.2 mV), intrinsic plasticity mechanisms may also be involved in PP-LFS effects.
Figure 7.

DHPG induce e-LTD with weak E-S potentiation, but no significant i-LTD. A, Time course and magnitude of synaptic depression by bath application of DHPG. B, Ratio of first EPSP change (EPSPDHPG/EPSPControl) is plotted against first EPSC change (EPSPDHPG/EPSPControl). Fitted line of data is presented with solid line (black), and a linear correlation is observed (r = 0.89). Right, Representative traces of EPSCs (A) and EPSPs (B) during the baseline period (0–5 min) (Control, black) and 25–30 min after DHPG application (pDHPG, light blue). C, Representative voltage traces by suprathreshold SC stimulation before (Control) and after DHPG application (pDHPG). Insets, DHPG-induced variable results in depolarization (left) or hyperpolarization (right) of Vth. D, Bar graphs represent no significant changes in SI50. Graph shows strong linear relationship between 1-SI50 and EPSP weight (r = 0.89). E, Distribution of firing probability as a function of EPSP slope before (Control) and 30 min after DHPG application (pDHPG). Light colors represent sigmoid fitted line. Bar graphs represent significant decrease in ES50. F, Time course and magnitude of i-LTD by DHPG application. Inset, Representative IPSC traces obtained before (a) and 25–30 min after DHPG application (b). G, No correlation is detected between 1-ES50 and IPSC weight (r = −0.13). Samples that show IPSC decreases >20% after DHPG are numbered and indicated with box. H, Graph showing a linear correlation between 1-ES50 and ΔVth_SC (r = −0.47). Numbers indicate 3 cells that show significant i-LTD in Figure 7G. I, No correlation is detected between 1-ES50 and ΔRin (r = −0.18). D, G, H, I, Closed circles (light blue) and gray solid lines at indicate averaged number and fitted line of data, respectively. Error bars indicate SEM. *p < 0.05. n.s., Not significant (p > 0.05).
No evidence for the involvement of Ih inhibition in the increased excitability during mGluR-dependent LTD
Increased excitability due to Ih downregulation during mGluR-dependent LTD was reported previously (Brager and Johnston, 2007). To investigate the involvement of Ih inhibition in the increased excitability during e-LTDPP-LFS, we analyzed excitability parameter changes induced by Ih blockade using ZD7288 (20 μm) and compared them with changes induced by PP-LFS. We confirmed that three typical features for Ih inhibition (Maccaferri et al., 1993; Biel et al., 2009), RMP hyperpolarization (Control, −58.8 ± 0.8 mV; +ZD7288, −67.5 ± 1.7 mV; n = 9, p = 0.0003), Rin increase (Control, 131.3 ± 11.1 mΩ; +ZD7288, 250.0 ± 34.6 mΩ; n = 5, p = 0.01), and sagging amplitude decrease (Fig. 8A), were well observed in the presence of ZD7288, whereas PP-LFS did not significantly affect RMP or sagging (Fig. 8B). Increased Rin due to Ih inhibition is expected to increase EPSP decay time and temporal summation (Magee, 1999; Brager and Johnston, 2007), but the summation ratio for EPSPs in response to 50 Hz burst synaptic stimulation was not affected by ZD7288 (Control, 2.73 ± 0.3; +ZD7288, 2.80 ± 0.3; n = 6, p = 0.8) (Fig. 8C). As a result, the I-O relationship obtained from APs evoked by synaptic stimulation was shifted rightward by ZD7288 (SI50, 1.65 ± 0.1, n = 5, p = 0.01) (Fig. 8D) with no significant changes in Vth (Control, −38.0 ± 1.4 mV; +ZD7288, −36.0 ± 1.9 mV; n = 5, p = 0.09) (Fig. 8E). These results showed that Ih inhibition effects on intrinsic excitability and E-S coupling are quite different from PP-LFS effects. We also analyzed excitability changes during e-LTDDHPG and found no significant changes in RMP (Control, −59.5 ± 0.4 mV; pDHPG, −61.0 ± 0.9 mV; n = 14, p = 0.1), Rin (Control, 159.0 ± 3.8 mΩ; pDHPG, 173.9 ± 8.5 mΩ; n = 10, p = 0.2), or sagging, which is also very different from Ih inhibition effects. We further investigated the effects of ZD7288 on intrinsic excitability by analyzing AP firing in response to somatic depolarization. The number of spikes induced by somatic depolarization was reduced by ZD7288 (100 pA injection; Control, 4.4 ± 1.3; +ZD7288, 2.9 ± 1.1, n = 8, p = 0.01), but the reduction was reversed when the hyperpolarized RMP was corrected by current injection (+ZD7288, RMP corrected, 8.3 ± 1.7, n = 8, p = 0.05) (Fig. 8F), suggesting that the increased excitability due to Rin increase was masked by RMP hyperpolarization effects. Importantly, Vth was not affected by ZD7288 (Control, −38.6 ± 0.6 mV; +ZD7288, −36.6 ± 2.5 mV, n = 5, p = 0.4) or changing RMP (+ZD7288, RMP corrected, −36.4 ± 2.6 mV, n = 5, p = 0.3) (Fig. 8G), suggesting that Vth hyperpolarization, which is the most important mechanism for E-S potentiation during e-LTD, is not attributable to Ih inhibition.
Figure 8.

Effects of ZD7288, Ih blocker, on intrinsic excitability and E-S coupling in CA1 neurons. A, Left, Voltage deflections in response to a series of current steps (−200 to 0 pA, 25 pA increment) before (Control) and after ZD7288 application (+ZD7288). Vmax, Maximum point of voltage deflection; Vss: steady state of voltage deflection. Bottom, Voltage response to −200 pA current injection in Control and the similar level of Vmax shown at +ZD7288 are superimposed. Right, ΔV (Vmax − Vss) is plotted against Vmax before (Control) and after ZD7288 application (+ZD7288). B, Left, Representative voltage deflections to current steps before (Control) and 30 min after PP-LFS (pPP-LFS). Right, ΔV (Vmax − Vss) versus Vmax in Control and after PP-LFS are plotted with black and red colors, respectively. C, Left, Example traces of EPSP before (Control) and after ZD7288 application (+ZD7288) by subthreshold synaptic stimulation at 50 Hz. Bar graphs represent no significant changes in EPSP summation ratio by ZD7288 application. D, Left, Plot of firing probability as a function of stimulus intensity before (Control) and after ZD7288 application (+ZD7288). Middle, Example traces of voltage changes in response to a same intensity of synaptic stimulation. Note the decrease in AP firing with the Ih inhibition. Right, Summary graphs of increased SI50 by Ih inhibition. E, Bar graphs represent no significant changes in Vth_SC in the presence of ZD7288. F, Left, Representative voltage traces to current step (100 pA) before (Control), after ZD7288 application (+ZD7288), and RMP corrected with current injection (+ZD7288, RMP corrected). Right, summary data showing significant decrease in AP firing in the presence of ZD7288. There was an increase in AP firing by correcting RMP. G, Voltage responses to ramp current injection before (Control) and after ZD7288 application (+ZD7288). Inset, Bottom, Traces at boxed area are superimposed. Bar graphs represent no significant changes in Vth_ramp. F, *p < 0.05. **p < 0.01. n.s., Not significant (p > 0.05). Error bars indicate SEM.
Discussion
This study identifies that the postsynaptic mGluR5 activation by PP-LFS at the SC produces a sustained and stable form of e-LTD and i-LTD, resulting in an E-S potentiation in CA1 pyramidal neurons. This opposite expression of synaptic and E-S coupling plasticity leads to increased neuronal output, implying that E-S coupling alteration is not just a complementary mechanism to synaptic plasticity but can play a dominant role in regulating the I-O relationship. Figure 9 summarizes the data for alterations of synaptic weights, E-S coupling, and I-O relationship obtained in different experimental conditions, demonstrating that I-O relationship alterations, referred to as I-O plasticity, during mGluR-dependent LTD can be understood by the integration of changes in synaptic and E-S coupling components.
Figure 9.

Understanding I-O plasticity during e-LTD by integration of synaptic plasticity and E-S potentiation. A, Summarized data of changes in synaptic weight (left), E-S coupling (middle), and I-O change (right) in each condition. B, mGluR5-dependent downstream signaling mechanism induced by synaptic stimulation (top) or DHPG application (bottom) in CA1 pyramidal neuron. Gq, Gq protein; L-VGCC, L-type voltage-gated Ca2+ channel; AMPAR, AMPA receptor; IP3, inositol trisphosphate; IN, interneuron; PN, pyramidal neuron. C, Schematic diagram of I-O plasticity. *p < 0.05. **p < 0.01. n.s., Not significant (p > 0.05). Error bars indicate SEM.
Changes in Rin versus Vth involved in E-S potentiation
The increase in intrinsic excitability during e-LTD has been demonstrated previously (Brager and Johnston, 2007; Campanac et al., 2008; Gasselin et al., 2017). Increases in AP firing during e-LTD were associated with Rin increases (Brager and Johnston, 2007) and Rin changes correlated closely with the magnitude of LTD (Gasselin et al., 2017). The increase in EPSP decay time caused by Rin increases was suggested to cause an increase in EPSP summation (Brager and Johnston, 2007). Based on these reports, the Rin increase is considered as an indication of an increase in intrinsic excitability during e-LTD, but direct experiments to show whether it contributes to E-S potentiation have not been conducted. Indeed, increases in EPSP summation were observed at frequencies <20 Hz (Brager and Johnston, 2007), but we used burst synaptic stimulation at 50 Hz to evoke APs with mild stimulation. Under this condition, the temporal summation of EPSPs was not affected by PP-LFS (Fig. 1B), suggesting that the Rin increase may not contribute to E-S potentiation. We searched for another mechanism underlying E-S potentiation changes during e-LTD and found that it has a strong correlation with Vth hyperpolarization (Figs. 1, 3).
Relative contribution of disinhibition and intrinsic plasticity to E-S potentiation
We demonstrate that anti-GABARs mimic and occlude PP-LFS effects on E-S potentiation and Vth without significant effects on Rin (Figs. 3, 4), supporting the idea that decreased GABAR function is responsible for Vth hyperpolarization and serves as a key mechanism underlying E-S potentiation during e-LTDPP-LFS, whereas Rin increases play a minor role in our experimental condition. However, our study does not exclude the possible contribution of intrinsic plasticity to E-S potentiation during e-LTDPP-LFS. The E-S potentiation and Vth hyperpolarization induced by anti-GABARs were indeed smaller than those induced by PP-LFS (ES50, 75% after PP-LFS vs 84% after anti-GABARs; Vth hyperpolarization, 5.2 mV after PP-LFS vs 2.1 mV after anti-GABARs). Furthermore, E-S potentiation associated with Vth hyperpolarization also occurred during e-LTDDHPG where i-LTD was insignificant (Fig. 7), although the magnitude was smaller than that during e-LTDPP-LFS. These results may suggest that intrinsic excitability changes independent of the GABAergic mechanism also contribute to E-S potentiation during e-LTD.
Ih inhibition is well known as a mechanism for an increased intrinsic excitability (Brager and Johnston, 2007; Gasselin et al., 2017). We demonstrate that increased Rin by ZD7288 is accompanied by a huge RMP hyperpolarization (Fig. 8), whereas increased Rin during e-LTDPP-LFS is not accompanied by RMP changes. Furthermore, sagging, a manifestation of Ih activation, was not significantly affected by PP-LFS (Fig. 8B). These differences between Ih inhibition effects and PP-LFS effects suggest that Ih inhibition may not contribute significantly to intrinsic excitability changes. In the present study, we could not identify ion channels that cause increased excitability, but our results suggest that changes in ion channel activity leading to Vth hyperpolarization are most likely to contribute to increased excitability.
Contribution of tonic GABA currents to Vth and Rin
The main source of ambient GABA responsible for tonic inhibitory currents in the hippocampus is the vesicular release responsible for phasic inhibition (Glykys and Mody, 2007). The contribution of reduced tonic inhibition to tetanus-induced E-S potentiation was suggested previously (Chavez-Noriega et al., 1990), but the relative contribution of phasic and tonic inhibition has not been investigated. We found that PP-LFS-induced Vth hyperpolarization to synaptic stimulation (Fig. 1F) is similar to direct somatic activation (Fig. 4B) and that both changes are occluded by anti-GABARs (Figs. 3J, 4D). These results suggest that the reduction of tonic inhibitory currents is mainly responsible for Vth hyperpolarization and E-S potentiation during e-LTDPP-LFS.
It is generally understood that the mechanism underlying increased excitability by suppressing GABAergic inputs is a reduction in shunting effects, which is expected to increase Rin and EPSP amplitude. However, when we calculated from our experimental results the contribution of tonic GABA conductance to resting conductance (−39 pA at −120 mV with ECl = −66 mV), it was only ∼8% (0.7 of 9 nS). Indeed, we did not find significant changes in Rin and EPSP amplitude by anti-GABARs, but the mean value for Rin increased by 5.5% (from 111 to 117 mΩ), which is not greatly different from the value expected from a conductance change by a reduction in tonic GABA currents. Despite a minor effect of tonic GABA currents on the whole-cell conductance, tonic GABA currents have a great influence on AP output by hyperpolarizing Vth. One possible explanation for these findings is that the shunting effect of tonic GABA currents is not global but local.
It was reported that tonic inhibitory currents in the CA1 area are mostly mediated by GABAA receptors (Stell and Mody, 2002; Glykys and Mody, 2006, 2007). However, applying GABAA or GABAB receptor antagonists individually did neither significantly affect Vth and E-S potentiation nor occlude PP-LFS effects, suggesting that not only GABAA receptors, but also GABAB receptors, underlie the tonic inhibition. GABAB receptor-mediated tonic inhibition was reported in locus ceruleus neurons, playing a role in the regulation of spontaneous activity (Wang et al., 2015). An enhancement of GABAA receptor function by postsynaptic GABAB receptors was reported in dentate gyrus granule cells (Ransom et al., 2013) and thalamocortical neurons (Connelly et al., 2013). The roles for GABAB receptors in the tonic inhibition in CA1 neurons remain to be investigated.
Difference in signaling mechanism underlying e-LTDPP-LFS and e-LTDDHPG
Different stimulation methods to activate mGluRs recruit different signaling pathways. DHPG application induces a PLC/PKC-independent increase in intrinsic excitability (Ireland and Abraham, 2002), whereas synaptic stimulation induces a PKC-dependent increase in intrinsic excitability (Brager and Johnston, 2007). We previously showed that the activation of mGluRs alone by DHPG fails to induce robust PLC activation, but glutamate, which activates both iGluRs and mGluRs, is able to activate PLC with the concomitant activation of L-type Ca2+ channels (Kim et al., 2015) (Fig. 9B). Considering that PLC is required for the DAG generation, which is a prerequisite to generating endocannabinoids, such as 2-AG, which is crucial for i-LTD (Edwards et al., 2008; Heifets and Castillo, 2009; Castillo, 2012), the failure of i-LTD induction by DHPG is consistent with our previous study that mGluRs activation alone is insufficient to activate PLC (Kim et al., 2015). Other studies consistently demonstrate that DHPG alone is insufficient to induce eCB-dependent i-LTD, but DHPG in combination with an intracellular Ca2+ increase induces i-LTD (Edwards et al., 2008; Heifets et al., 2008). However, some studies show that DHPG alone induces stable eCB-dependent i-LTD (Chevaleyre and Castillo, 2003; Pan et al., 2008). We also found i-LTD induction by DHPG in 3 of 10 cells (Fig. 7G). Because PLC activation is Ca2+-dependent (Rebecchi and Pentyala, 2000; Hashimotodani et al., 2005), the robustness of DHPG-induced PLC activation may be affected by resting Ca2+ levels, which may vary among different cells.
The physiological significance of the association of i-LTD with e-LTD after PP-LFS
The mechanism of i-LTD associated with e-LTD presented in this study shares common characteristics with that associated with synaptic LTP in previous studies (Lu et al., 2000; Chevaleyre and Castillo, 2003; Azad et al., 2004; Jiang et al., 2010). For instance, both are dependent on mGluR5 and mediated by retrograde CB1R-dependent mechanisms. Thus, it appears that activity-dependent changes in GABAergic mechanisms by mGluR/CB1R signaling are always inhibitory, leading to E-S potentiation. Association of i-LTD with excitatory synaptic LTP was regarded to play a synergistic role to increase potentiation (Bliss and Lomo, 1973; Andersen et al., 1980; Chavez-Noriega et al., 1990; Daoudal et al., 2002), while we show that the effect of GABAergic inhibition during e-LTD is stronger than that of synaptic depression on AP outputs, thereby paradoxically shifting the I-O relationship to the left. These results give new insights into the importance of mGluR/CB1R signaling in the regulation of the excitation-inhibition (E-I) balance, which modulates neuronal outputs. Furthermore, we found that the most frequently used protocols for Group 1 mGluR activation, PP-LFS and DHPG, have different impacts on E-I balance, revealing the complexity of mGluR signaling. Our study highlights the mechanisms underlying the activity-dependent regulation of the E-I balance, which may play an important role in information processing in the brain.
Footnotes
This work was supported by the Korean Ministry of Science, Information and Communication Technology (National Research Foundation Grant 2010-002791 and 2017R1A2B2010186 to W.-K.H.).
The authors declare no competing financial interests.
References
- Abraham WC, Gustafsson B, Wigström H (1987) Long-term potentiation involves enhanced synaptic excitation relative to synaptic inhibition in guinea-pig hippocampus. J Physiol 394:367–380. 10.1113/jphysiol.1987.sp016875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen P, Sundberg SH, Sveen O, Swann JW, Wigström H (1980) Possible mechanisms for long-lasting potentiation of synaptic transmission in hippocampal slices from guinea-pigs. J Physiol 302:463–482. 10.1113/jphysiol.1980.sp013256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anwyl R. (1999) Metabotropic glutamate receptors: electrophysiological properties and role in plasticity. Brain Res Brain Res Rev 29:83–120. 10.1016/S0165-0173(98)00050-2 [DOI] [PubMed] [Google Scholar]
- Azad SC, Monory K, Marsicano G, Cravatt BF, Lutz B, Zieglgänsberger W, Rammes G (2004) Circuitry for associative plasticity in the amygdala involves endocannabinoid signaling. J Neurosci 24:9953–9961. 10.1523/JNEUROSCI.2134-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear MF, Malenka RC (1994) Synaptic plasticity: LTP and LTD. Curr Opin Neurobiol 4:389–399. 10.1016/0959-4388(94)90101-5 [DOI] [PubMed] [Google Scholar]
- Beck H, Yaari Y (2008) Plasticity of intrinsic neuronal properties in CNS disorders. Nat Rev Neurosci 9:357–369. 10.1038/nrn2371 [DOI] [PubMed] [Google Scholar]
- Biel M, Wahl-Schott C, Michalakis S, Zong X (2009) Hyperpolarization-activated cation channels: from genes to function. Physiol Rev 89:847–885. 10.1152/physrev.00029.2008 [DOI] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL (1993) A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361:31–39. 10.1038/361031a0 [DOI] [PubMed] [Google Scholar]
- Bliss TV, Lomo T (1973) Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol 232:331–356. 10.1113/jphysiol.1973.sp010273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolshakov VY, Siegelbaum SA (1994) Postsynaptic induction and presynaptic expression of hippocampal long-term depression. Science 264:1148–1152. 10.1126/science.7909958 [DOI] [PubMed] [Google Scholar]
- Brager DH, Johnston D (2007) Plasticity of intrinsic excitability during long-term depression is mediated through mGluR-dependent changes in Ih in hippocampal CA1 pyramidal neurons. J Neurosci 27:13926–13937. 10.1523/JNEUROSCI.3520-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bright DP, Smart TG (2013) Methods for recording and measuring tonic GABAA receptor-mediated inhibition. Front Neural Circuits 7:193. 10.3389/fncir.2013.00193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campanac E, Daoudal G, Ankri N, Debanne D (2008) Downregulation of dendritic Ih in CA1 pyramidal neurons after LTP. J Neurosci 28:8635–8643. 10.1523/JNEUROSCI.1411-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo PE. (2012) Presynaptic LTP and LTD of excitatory and inhibitory synapses. Cold Spring Harb Perspect Biol 4:a005728. 10.1101/cshperspect.a005728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo PE, Younts TJ, Chávez AE, Hashimotodani Y (2012) Endocannabinoid signaling and synaptic function. Neuron 76:70–81. 10.1016/j.neuron.2012.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez-Noriega LE, Bliss TV, Halliwell JV (1989) The EPSP-spike (E-S) component of long-term potentiation in the rat hippocampal slice is modulated by GABAergic but not cholinergic mechanisms. Neurosci Lett 104:58–64. 10.1016/0304-3940(89)90329-7 [DOI] [PubMed] [Google Scholar]
- Chavez-Noriega LE, Halliwell JV, Bliss TV (1990) A decrease in firing threshold observed after induction of the EPSP-spike (E-S) component of long-term potentiation in rat hippocampal slices. Exp Brain Res 79:633–641. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE (2003) Heterosynaptic LTD of hippocampal GABAergic synapses: a novel role of endocannabinoids in regulating excitability. Neuron 38:461–472. 10.1016/S0896-6273(03)00235-6 [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE (2004) Endocannabinoid-mediated metaplasticity in the hippocampus. Neuron 43:871–881. 10.1016/j.neuron.2004.08.036 [DOI] [PubMed] [Google Scholar]
- Connelly WM, Fyson SJ, Errington AC, McCafferty CP, Cope DW, Di Giovanni G, Crunelli V (2013) GABAB receptors regulate extrasynaptic GABAA receptors. J Neurosci 33:3780–3785. 10.1523/JNEUROSCI.4989-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalby NO. (2003) Inhibition of gamma-aminobutyric acid uptake: anatomy, physiology and effects against epileptic seizures. Eur J Pharmacol 479:127–137. 10.1016/j.ejphar.2003.08.063 [DOI] [PubMed] [Google Scholar]
- Daoudal G, Debanne D (2003) Long-term plasticity of intrinsic excitability: learning rules and mechanisms. Learn Mem 10:456–465. 10.1101/lm.64103 [DOI] [PubMed] [Google Scholar]
- Daoudal G, Hanada Y, Debanne D (2002) Bidirectional plasticity of excitatory postsynaptic potential (EPSP)-spike coupling in CA1 hippocampal pyramidal neurons. Proc Natl Acad Sci U S A 99:14512–14517. 10.1073/pnas.222546399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards DA, Zhang L, Alger BE (2008) Metaplastic control of the endocannabinoid system at inhibitory synapses in hippocampus. Proc Natl Acad Sci U S A 105:8142–8147. 10.1073/pnas.0803558105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y, Fricker D, Brager DH, Chen X, Lu HC, Chitwood RA, Johnston D (2005) Activity-dependent decrease of excitability in rat hippocampal neurons through increases in Ih. Nat Neurosci 8:1542–1551. 10.1038/nn1568 [DOI] [PubMed] [Google Scholar]
- Gasselin C, Inglebert Y, Ankri N, Debanne D (2017) Plasticity of intrinsic excitability during LTD is mediated by bidirectional changes in h-channel activity. Sci Rep 7:14418. 10.1038/s41598-017-14874-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glykys J, Mody I (2006) Hippocampal network hyperactivity after selective reduction of tonic inhibition in GABAA receptor α5 subunit-deficient mice. J Neurophysiol 95:2796–2807. 10.1152/jn.01122.2005 [DOI] [PubMed] [Google Scholar]
- Glykys J, Mody I (2007) The main source of ambient GABA responsible for tonic inhibition in the mouse hippocampus. J Physiol 582:1163–1178. 10.1113/jphysiol.2007.134460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimotodani Y, Ohno-Shosaku T, Tsubokawa H, Ogata H, Emoto K, Maejima T, Araishi K, Shin HS, Kano M (2005) Phospholipase Cβ serves as a coincidence detector through its Ca2+ dependency for triggering retrograde endocannabinoid signal. Neuron 45:257–268. 10.1016/j.neuron.2005.01.004 [DOI] [PubMed] [Google Scholar]
- Heifets BD, Castillo PE (2009) Endocannabinoid signaling and long-term synaptic plasticity. Annu Rev Physiol 71:283–306. 10.1146/annurev.physiol.010908.163149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heifets BD, Chevaleyre V, Castillo PE (2008) Interneuron activity controls endocannabinoid-mediated presynaptic plasticity through calcineurin. Proc Natl Acad Sci U S A 105:10250–10255. 10.1073/pnas.0711880105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ireland DR, Abraham WC (2002) Group I mGluRs increase excitability of hippocampal CA1 pyramidal neurons by a PLC-independent mechanism. J Neurophysiol 88:107–116. 10.1152/jn.2002.88.1.107 [DOI] [PubMed] [Google Scholar]
- Jester JM, Campbell LW, Sejnowski TJ (1995) Associative EPSP-spike potentiation induced by pairing orthodromic and antidromic stimulation in rat hippocampal slices. J Physiol 484:689–705. 10.1113/jphysiol.1995.sp020696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang B, Huang S, de Pasquale R, Millman D, Song L, Lee HK, Tsumoto T, Kirkwood A (2010) The maturation of GABAergic transmission in visual cortex requires endocannabinoid-mediated LTD of inhibitory inputs during a critical period. Neuron 66:248–259. 10.1016/j.neuron.2010.03.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HH, Lee KH, Lee D, Han YE, Lee SH, Sohn JW, Ho WK (2015) Costimulation of AMPA and metabotropic glutamate receptors underlies phospholipase C activation by glutamate in hippocampus. J Neurosci 35:6401–6412. 10.1523/JNEUROSCI.4208-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Min SS, Gallagher M, Kirkwood A (2005) NMDA receptor-independent long-term depression correlates with successful aging in rats. Nat Neurosci 8:1657–1659. 10.1038/nn1586 [DOI] [PubMed] [Google Scholar]
- Liu YB, Disterhoft JF, Slater NT (1993) Activation of metabotropic glutamate receptors induces long-term depression of GABAergic inhibition in hippocampus. J Neurophysiol 69:1000–1004. 10.1152/jn.1993.69.3.1000 [DOI] [PubMed] [Google Scholar]
- Lu YM, Mansuy IM, Kandel ER, Roder J (2000) Calcineurin-mediated LTD of GABAergic inhibition underlies the increased excitability of CA1 neurons associated with LTP. Neuron 26:197–205. 10.1016/S0896-6273(00)81150-2 [DOI] [PubMed] [Google Scholar]
- Lüscher C, Huber KM (2010) Group 1 mGluR-dependent synaptic long-term depression: mechanisms and implications for circuitry and disease. Neuron 65:445–459. 10.1016/j.neuron.2010.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccaferri G, Mangoni M, Lazzari A, DiFrancesco D (1993) Properties of the hyperpolarization-activated current in rat hippocampal CA1 pyramidal cells. J Neurophysiol 69:2129–2136. 10.1152/jn.1993.69.6.2129 [DOI] [PubMed] [Google Scholar]
- Magee JC. (1999) Dendritic lh normalizes temporal summation in hippocampal CA1 neurons. Nat Neurosci 2:508–514. 10.1038/9158 [DOI] [PubMed] [Google Scholar]
- Malik R, Johnston D (2017) Dendritic GIRK channels gate the integration window, plateau potentials, and induction of synaptic plasticity in dorsal but not ventral CA1 neurons. J Neurosci 37:3940–3955. 10.1523/JNEUROSCI.2784-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mockett BG, Guévremont D, Wutte M, Hulme SR, Williams JM, Abraham WC (2011) Calcium/calmodulin-dependent protein kinase II mediates group I metabotropic glutamate receptor-dependent protein synthesis and long-term depression in rat hippocampus. J Neurosci 31:7380–7391. 10.1523/JNEUROSCI.6656-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan R, Johnston D (2007) Long-term potentiation in rat hippocampal neurons is accompanied by spatially widespread changes in intrinsic oscillatory dynamics and excitability. Neuron 56:1061–1075. 10.1016/j.neuron.2007.10.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliet SH, Malenka RC, Nicoll RA (1997) Two distinct forms of long-term depression coexist in CA1 hippocampal pyramidal cells. Neuron 18:969–982. 10.1016/S0896-6273(00)80336-0 [DOI] [PubMed] [Google Scholar]
- Pan B, Hillard CJ, Liu QS (2008) Endocannabinoid signaling mediates cocaine-induced inhibitory synaptic plasticity in midbrain dopamine neurons. J Neurosci 28:1385–1397. 10.1523/JNEUROSCI.4033-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JY, Remy S, Varela J, Cooper DC, Chung S, Kang HW, Lee JH, Spruston N (2010) A post-burst after depolarization is mediated by group i metabotropic glutamate receptor-dependent upregulation of Cav2.3 R-type calcium channels in CA1 pyramidal neurons. PLoS Biol 8:e1000534. 10.1371/journal.pbio.1000534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransom CB, Tao W, Wu Y, Spain WJ, Richerson GB (2013) Rapid regulation of tonic GABA currents in cultured rat hippocampal neurons. J Neurophysiol 109:803–812. 10.1152/jn.00460.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razik DS, Hawellek DJ, Antkowiak B, Hentschke H (2013) Impairment of GABA transporter GAT-1 terminates cortical recurrent network activity via enhanced phasic inhibition. Front Neural Circuits 7:141. 10.3389/fncir.2013.00141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebecchi MJ, Pentyala SN (2000) Structure, function, and control of phosphoinositide-specific phospholipase C. Physiol Rev 80:1291–1335. 10.1152/physrev.2000.80.4.1291 [DOI] [PubMed] [Google Scholar]
- Schnabel R, Palmer MJ, Kilpatrick IC, Collingridge GL (1999) A CaMKII inhibitor, KN-62, facilitates DHPG-induced LTD in the CA1 region of the hippocampus. Neuropharmacology 38:605–608. 10.1016/S0028-3908(98)00229-9 [DOI] [PubMed] [Google Scholar]
- Spitzer NC. (1999) New dimensions of neuronal plasticity. Nat Neurosci 2:489–491. 10.1038/9132 [DOI] [PubMed] [Google Scholar]
- Stell BM, Mody I (2002) Receptors with different affinities mediate phasic and tonic GABAA conductances in hippocampal neurons. J Neurosci 22:RC223. 10.1523/JNEUROSCI.22-10-j0003.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HY, Kuo ZC, Fu YS, Chen RF, Min MY, Yang HW (2015) GABAB receptor-mediated tonic inhibition regulates the spontaneous firing of locus coeruleus neurons in developing rats and in citalopram-treated rats. J Physiol 593:161–180. 10.1113/jphysiol.2014.281378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Xu NL, Wu CP, Duan S, Poo MM (2003) Bidirectional changes in spatial dendritic integration accompanying long-term synaptic modifications. Neuron 37:463–472. 10.1016/S0896-6273(02)01189-3 [DOI] [PubMed] [Google Scholar]
- Xu J, Kang N, Jiang L, Nedergaard M, Kang J (2005) Activity-dependent long-term potentiation of intrinsic excitability in hippocampal CA1 pyramidal neurons. J Neurosci 25:1750–1760. 10.1523/JNEUROSCI.4217-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younts TJ, Chevaleyre V, Castillo PE (2013) CA1 pyramidal cell theta-burst firing triggers endocannabinoid-mediated long-term depression at both somatic and dendritic inhibitory synapses. J Neurosci 33:13743–13757. 10.1523/JNEUROSCI.0817-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]