

Abstract
There are increasing demands for informative cancer biomarkers, accessible via minimally invasive procedures, both for initial diagnostics and for follow‐up of personalized cancer therapy, including immunotherapy. Fine‐needle aspiration (FNA) biopsy provides ready access to relevant tissue samples; however, the minute amounts of sample require sensitive multiplex molecular analysis to be of clinical biomarker utility. We have applied proximity extension assays (PEA) to analyze 167 proteins in FNA samples from patients with breast cancer (BC; n = 25) and benign lesions (n = 32). We demonstrate that the FNA BC samples could be divided into two main clusters, characterized by differences in expression levels of the estrogen receptor (ER) and the proliferation marker Ki67. This clustering corresponded to some extent to established BC subtypes. Our analysis also revealed several proteins whose expression levels differed between BC and benign lesions (e.g., CA9, GZMB, IL‐6, VEGFA, CXCL11, PDL1, and PCD1), as well as several chemokines correlating with ER and Ki67 status (e.g., CCL4, CCL8, CCL20, CXCL8, CXCL9, and CXCL17). Finally, we also identified three signatures that could predict Ki67 status, ER status, and tumor grade, respectively, based on a small subset of proteins, which was dominated by chemokines. To our knowledge, expression profiles of CCL13 in benign lesions and BC have not previously been described but were shown herein to correlate with proliferation (P = 0.00095), suggesting a role in advanced BC. Given the broad functional range of the proteins analyzed, immune‐related proteins were overrepresented among the observed alterations. Our pilot study supports the emerging role of chemokines in BC progression. Due to the minimally traumatic sampling and clinically important molecular information for therapeutic decisions, this methodology is promising for future immunoscoring and monitoring of treatment efficacy in BC.
Keywords: breast cancer subtypes, fibroadenomas, fine‐needle aspiration, immune‐related protein biomarker, proximity extension assay
Abbreviations
- BC
breast cancer
- ER
estrogen receptor, ESR1
- ERBB2
receptor tyrosine‐protein kinase erbB‐2
- FNA
fine‐needle aspiration biopsy
- HER
HER2
- HER
HER2‐positive
- IHC
immunohistochemistry
- Ki67
antigen Ki‐67
- LumA
BC subtypes luminal A
- LumB
luminal B
- LumHER
luminal HER2‐positive
- MWL
maximal whisker length
- NPX
normalized protein expression
- PEA
proximity extension assay
- PR
progesterone receptor
- RIPA
radioimmunoprecipitation assay buffer
- TNB
triple‐negative breast cancer
1. Introduction
One of the main current challenges for breast cancer (BC) therapy is to device methods that allow repeated and minimally invasive sampling with the capacity to identify informative molecular signatures. This may provide support for the choice of neoadjuvant and/or immune therapy, and a possibility to monitor the disease characteristics during the course of the disease. In addition to classical BC biomarkers, such as ER, PGR, HER2, and Ki67 (referred to as ‘key properties’), there is a need for biomarkers reflecting the microenvironment and immunodynamics in BC. Therefore, considerable efforts are made to implement measurement of biomarkers via liquid biopsies of peripheral blood. However, tumor sampling via fine‐needle aspiration (FNA) provides direct access to tumor tissue material. FNA sampling is a well‐established cancer diagnostic procedure, where cells, tissue fragments, and/or fluid may be recovered from tumor tissue via a puncture using a very thin needle. The tip of the needle is placed in the center of a lesion, frequently under ultrasound guidance (Ly et al., 2016; Rimsten et al., 1975). Cells can then be aspirated via a syringe during gentle oscillation back and forth. The minimally traumatic FNA is often used for diagnosis of small nonpalpable, deeply located, or otherwise hard‐to‐reach lesions, both for primary tumors and for lymph node and other metastases. Traditionally, these samples have been subjected to cytological analysis with staining for a small number of proteins. By contrast, modern molecular technologies allow more comprehensive analyses of the state of the sampled tissues.
In this pilot study, we have explored one FNA‐compatible technology for multiplex molecular profiling that meets stringent requirements for sensitivity and reproducibility: proximity extension assays (PEA, www.olink.com; Assarsson et al., 2014).
We have previously demonstrated that ‘leftover’ FNA sample material from BC can be analyzed by PEA with high sensitivity, and with results that correlated with routine assessments. In addition, our results revealed a tentative 11‐protein signature that discriminated BC from benign lesions (Franzén et al., 2018).
In this study, we have used the same patient material analyzed with the same protein panels as previously described, to further explore the protein expression in benign lesions in relation to BC subtypes and to several routine parameters such as proliferation and hormonal status. The two protein panels used in these studies—Oncology II and Immuno‐Oncology I (https://www.olink.com/products/immuno-oncology/)—include many proteins that may be relevant for characterization of clinical and biological properties of BC, including proteins involved in immune reactions. Twenty‐seven of the proteins of the ‘Oncology II’ panel and 66 proteins of the ‘Immuno‐Oncology I’ panel are immune‐related according to their annotations [https://www.olink.com/products/]. Due to some overlap between the two panels, the analysis measured levels of a total of 167 different proteins. We applied three complementary statistical approaches to analyze data relative to BC subtypes, and to histological/immunohistochemical (IHC), clinical, and cytology information: (a) hierarchical clustering of protein profiles to examine BC subtypes, (b) multiple‐regression modeling of signatures linking to key properties of BC, and (c) univariate correlations between protein levels and key properties of BC.
The long‐term aim of the study was to explore the potential value for FNA‐based molecular profiling by PEA for diagnosis, therapy selection, and evaluation in an objective and cost‐effective manner for BC patients. Our working hypothesis was that the analysis of key immune signatures in FNA samples using a simplified process may provide a valuable basis for choice of therapy and evaluation of responses to treatment.
2. Materials and methods
The materials and methods have been described previously (Franzén et al., 2018) and are briefly given below.
2.1. Patient samples
The study was approved by the Ethical Committee of the Karolinska Institutet, Stockholm (Dnr 2016/1432‐31/4), and the methodologies conform to the standards set by the Declaration of Helsinki. Female patients from the age of 18 years with mammography‐detectable lesions were invited to participate in the study after taking part of the project information and accepting the informed consent form. The patient cohort was subject to routine diagnostic examination (primary lesions in the breast and axillary metastases), and the material was collected during a period of 6 weeks. All FNA samples were obtained under ultrasound guidance by experienced radiologists using 21‐ to 22‐gauge needles, and after sampling for routine cytology, leftover material from FNA needles was processed immediately. For complete information on all samples, see Tables S1–S3.
2.2. Sample preparation
After sample collection, the FNA needles were rinsed with ice‐cold medium, and cells were pelleted, frozen, and stored at −80 °C until approval and quality control by cytology. Up to three samples were collected per patient, and each sample was assigned a consecutive FD# sample code. Samples approved by cytological examination were lysed in RIPA buffer (Sigma, Stockholm, Sweden R0278), debris was removed by centrifugation at 13 000 g for 15 min, and total protein concentration was determined using a micro‐BCA assay (ThermoFisher, Göteborg, Sweden).
2.3. Subtype classification
Routine core needle biopsy (CNB) tissue samples from primary lesions of BC patients, acquired in parallel with the FNA samples, were used for IHC analysis of estrogen receptor (ER), progesterone receptor (PR), the proliferation marker Ki67, and HER2 (ERBB2) according to routine guidelines. Classification of molecular subtypes was based on recommendations according to the St. Gallen classification system (Goldhirsch et al., 2011) and defined by the Swedish National Guidelines for treatment of BC (‘Nationella Vårdprogrammet för bröstcancer’, version 2.0, SweBCG 2018 [in Swedish; https://www.cancercentrum.se/samverkan/om-oss/nyheter/2018/februari/nationellt-vardprogram-brostcancer-uppdaterat/]) and the Quality and Standardization Committee (KVAST) of the Swedish Society of Pathology (2018, in Swedish).
2.4. Quality control and characterization by cytology
Routine cytology smears were stained (May–Grünwald–Giemsa), scanned at high resolution, and examined by an experienced cytopathologist. Each sample was checked for being representative and of sufficient quality by cytology evaluation. Samples without tumor cells or otherwise nonrepresentative were excluded. At least 20 different areas per preparation were examined for tumor cells and macrophages (MØ). Semiscore evaluation was performed as follows: If no MØ was found in a sample, the MØ score was set to = 0; if only one MØ was found in a sample, the MØ score was set to = 1; and if 2–3 MØ were found, the MØ score was set to = 2. If MØ were found in about 50% of fields examined, the MØ score was set to = 5, and if MØ were found in about > 80% of fields examined, the MØ score was set to = 10.
2.5. Protein profiling by PEA
One microlitre sample (0.5 μg total protein) per panel was analyzed by Olink Multiplex Immuno‐Oncology I and Oncology II panels (Olink Proteomics, Uppsala, Sweden) according to the manufacturer's instructions and as described previously (Larsson et al., 2015). Each panel consists of 92 protein assays and four internal controls. However, due to some overlap between the panels, in total data for 167 proteins were recorded (Table S6). Results were exported from the Biomark reader and normalized using Olink Wizard for GenEx software for further statistical data analysis.
2.6. Statistical analysis
Quality control and data preprocessing (including normalization) of PEA data were done in accordance with the manufacturer's recommended procedures, including the routine Olink normalization into NPX values, and the analysis was performed in R environment. We log2‐transformed the values to render them normally distributed (e.g., for Pearson linear correlation and ANOVA). Since clinical and other phenotypical variables were not necessarily normally distributed, we applied nonparametric statistics, such as chi‐square tests, Fisher's exact test, and Spearman's and Kendall's rank correlation, as indicated for the respective results. The P‐values from the correlation analyses were either adjusted by Benjamini–Hochberg correction (Benjamini and Hochberg, 1995), or by Bonferroni (Tables 2 and S5), or are shown as original unadjusted P‐values (Fig. 3B)—as specified in respective legends. The two‐way hierarchical clustering was performed on normally distributed protein profiles as well. The input to the PEA data analysis were the normalized protein expression values (NPX) as specified by Olink Proteomics. For the lasso/ridge multiple‐regression modeling, we employed package glmnet (available from http://web.stanford.edu/~hastie/glmnet/glmnet_alpha.html). In the latter analysis, parameter alpha equaled 1, while other parameters were set to their defaults. The two‐way hierarchical clustering was also performed on the normally distributed protein profiles. For this hierarchical clustering (shown at the heatmap figures), we employed the function heatmaply (R package heatmaply v. 0.15.12 https://cran.r-project.org/web/packages/heatmaply/) using the default methods for distance matrix calculation (‘euclidean’) and clustering (‘complete linkage’).
Table 2.
Rank correlations and increased (+) or decreased (−) protein levels in given BC subsets. B, B cells; DA, dendritic cells activated; DR, dendritic cells resting; MA, Mast cell activated; MØ, Macrophages; NKA, NK cells activated; NKR, NK cells resting; T4, CD4 naive; T4A, CD4 memory activated; T4R, CD4 memory resting; T8, T cells CD8; Tfh, follicular helper; Tgd, gamma delta; Th, T helper; Treg, regulatory
| Chemokines | Cancer vs benign (1‐way ANOVA) | Grade (Kendall tau) | Ki67 (Spearman rank) | HER2 (Kendall tau) | ER (Spearman rank) | Comments and leukocyte subsetsa |
|---|---|---|---|---|---|---|
| CCL2b | + (P = 0.031) | + (P = 0.017) | + (P = 0.006) | − (P = 0.048) | Cluster. Th2/17/22c | |
| CCL3b | + (P = 0.0082) | + (P = 0.013) | + (P = 0.0099) | − (P = 0.025) | Cluster. Th1c | |
| CCL4b | + (P = 0.018) | + (P = 0.016) | + ( P = 4.0e‐4) * | − (P = 0.0048) | Cluster. NKA, MA, Th1/2/17/22c | |
| CCL7 | + ( P = 0.037) * | + (P = 0.0049) | + (P = 0.037) | M0, Th1, Th2 | ||
| CCL8b | + ( P = 0.037) * | + (P = 0.015) | + ( P = 5.7e‐4) * | − (P = 0.021) | Cluster. M1, M2, DA, Th2 | |
| CCL13 | + (P = 0.039) | + ( P = 9.5e‐4) * | + (P = 0.016) | In model. M2, DR, DA, Th2 | ||
| CCL17 | + (P = 0.036) | DR, DA, Th2/17/22 | ||||
| CCL19b | + (P = 0.0011) | M1, DA | ||||
| CCL20 | + (P = 0.0069) | + ( P = 7.4e‐5) * | − (P = 0.0023) | Cluster. T4A, DA, MA, Th17/22 | ||
| CCL23 | + (P = 0.0048) | In model. M2, Th1 | ||||
| CXCL1 | + (P = 0.021) | + (P = 0.0086) | − (P = 0.042) | Cluster | ||
| CXCL5 | − (P = 0.002)† | In model. M0 | ||||
| CXCL8 (IL‐8) | + (P = 0.0077) | + (P = 0.0063) | − (P = 0.017) | Cluster. Model. Inflammatory cytokine | ||
| CXCL9b | + ( P = 0.015) * | + (P = 0.018) | + ( P = 5.7e‐6) * | + (P = 0.010) | − (P = 0.0014) | Cluster. In model, M1 |
| CXCL10b | + ( P = 0.0068) * | + (P = 0.039) | + ( P = 1.4e‐4) * | + (P = 0.018) | − (P = 0.013) | Cluster. M1, DA, |
| CXCL11b | + ( P = 0.0016) * | + (P = 0.033) | + ( P = 1.3e‐4) * | + (P = 0.029) | − (P = 0.012) | Cluster. M1, DA, |
| CXCL13b | − (P = 0.0018) | + (P = 0.028) | − (P = 0.026) | Cluster. T4A, Tfh, M1c | ||
| CXCL17 | + ( P = 2.2e‐5) * | − ( P = 2.8e‐4) * | In model. | |||
| CX3CL1 | − (P = 0.064) | + (P = 0.020) | + (P = 0.031) | Cluster | ||
| CD markers | ||||||
| CD4 | + (P = 0.033) | + (P = 0.027) | T4R, Treg, M2 | |||
| CD5 | + (P = 0.017) | + ( P = 4.6e‐5) * | + (P = 0.0062) | − (P = 0.006) | Cluster. Treg | |
| CD8A | + (P = 0.016) | + ( P = 1.9e‐4) * | − (P = 0.0022) | Cluster. T8, Tfh | ||
| CD27 | + (P = 0.017) | + (P = 0.0033) | BC, T8, T4, T4R, Tfh, Treg | |||
| CD40 | + (P = 0.0033) | + (P = 0.002) | − (P = 0.013) | M1 | ||
| CD40L | + (P = 0.016) | + (P = 0.020) | − (P = 0.049) | T4, T4R, T4A, Tfh | ||
| CD48 | + (P = 0.0039) | + ( P = 3.1e‐4) * | + (P = 0.015) | − (P = 0.028) | ||
| CD137 (TNFRSF9) | + (P = 0.0055) | − (P = 0.013) | Treg | |||
| CD160 | − ( P = 0.024) * | Tgd, NKR | ||||
| CD208 (LAMP3) | + (P = 0.0025) | + (P = 0.017) | − (P = 0.036) | M1, DCA | ||
| CD244 (CD48L) | + (P = 0.041) | + (P = 0.0088) | − (P = 0.022) | Tgd, NKR, NKA, Treg | ||
| CD229 (LY9) | + (P = 0.0079) | + ( P = 5.0e‐5) * | + (P = 0.031) | − (P = 0.019) | T8, T4, T4R, Tgd | |
| CD258 (TNFSF14) | + (P = 0.0037) | + (P = 0.0017) | + (P = 0.0431) | − (P = 0.025) | NKA, M0, apoptosis related | |
| CD274 (PDL1) | + (P = 0.0029) | + (P = 0.016) | Th1, NK, T8, MØ | |||
| CD279 (PDCD1) | + (P = 0.023) | Tfh | ||||
| CD358 (TNFRSF21) | + (P = 0.045) | + (P = 0.031) | + (P = 0.00044) | − (P = 0.0044) | Apoptosis related (DR6) | |
| Other proteins | ||||||
| IL‐6 | + (P = 6.4e‐4) | + ( P = 1.9e‐4) * | − ( P = 2.0e‐4) * | In model. Inflammatory cytokine | ||
| IL‐18 | + (P = 0.024) | − (P = 0.017) | Inflammatory cytokine | |||
| GZMA | + (P = 0.047) | + ( P = 7.3e‐5) * | − (P = 0.0021) | Cluster. T8, T4R, Tgd, NKR, NKA | ||
| GZMB | + (P = 0.018) | + (P = 0.041) | + ( P = 1.4e‐5) * | − ( P = 1.7e‐4) * | Cluster. T8, T4A, Tgd, NKR, NKA | |
| GZMH | + (P = 0.0059) | + ( P = 3.4e‐5) * | − (P = 0.003) | Cluster. T8, Tgd, NKR, NKA | ||
| CA9 | + (P = 0.0061) | + (P = 0.0021) | − (P = 0.0016) | Hypoxia related | ||
| FASL | + (P = 0.0011) | − (P = 0.013) | Apoptosis related, NK | |||
| MMP12 | + (P = 0.0051) | + (P = 0.0089) | − (P = 0.012) | Cluster. DCR, DCA, M2 | ||
| VEGFA | + ( P = 0.036) * | + (P = 0.035) | + (P = 0.0061) | Angiogenesis related, Treg | ||
| ANG2 (ANGPT2) | + (P = 0.021) | + (P = 0.042) | + (P = 0.009) | − (P = 0.049) | Angiogenesis related, M1, M2 | |
| ESM1 | + (P = 0.0046) | + (P = 0.045) | + (P = 0.0015) | − (P = 0.026) | Angiogenesis related | |
| PDGFB | + (P = 0.0066) | + (P = 0.040) | Platelets, Angiogenesis related | |||
| LYN | + (P = 0.028) | + ( P = 3.8e‐4) * | − ( P = 2.3e‐4) * | Proto‐oncogene, B | ||
| TCL1A | + (P = 0.025) | + (P = 0.024) | B, T8, NK (proteomicsdb.org) | |||
| GAL9 (LGALS9) | + (P = 0.038) | + (P = 0.004) | − (P = 0.015) | B, T4, NK (proteomicsdb.org) | ||
a ‘Cluster’ (see Fig. 1B) or ‘Model’ (see Fig. 2A–C) refers to the analysis where the given protein was found to be altered. In addition, the following reports describe in which subset of leukocytes the respective proteins are expressed at elevated levels: Newman et al. (2015), Lyons et al. (2017), Strazza and Mor (2017), and Prat et al. (2017) and www.proteomicsdb.org. b‘12‐chemokine gene signature’ includes CCL2, CCL3, CCL4, CCL5, CCL8, CCL18, CCL19, CCL21, CXCL9, CXCL10, CXCL11, and CXCL13 (Prabhakaran et al., 2017). cAttract myeloid cells; see Turley et al. (2015). *P‐value in bold text: value after Bonferroni correction. † P‐value from the analysis of ER‐neg vs LumA samples (Table S5).
3. Results
3.1. Patient cohorts and PEA analysis
In total, 38 samples from 33 patients with benign breast lesions and 34 samples (≥ 8 mm in size) from 25 BC patients were analyzed for protein expression by PEA as described previously (Franzén et al., 2018). All information on patients and samples is listed in Tables S1–S3. For an overview of samples and IHC‐based subtype assignment, see Table 1.
Table 1.
Overview of samples from benign lesions and cancers according to subtype. The molecular subtypes were defined according to the following criteria: ‘LumA’, luminal A‐like (ER‐ and/or PR‐positive, i.e., more than 10% positive cells; low Ki67, i.e., < 25% positive cells; and HER2‐negative); ‘LumB’, luminal B‐like HER2‐negative (ER‐positive and/or PR‐positive, and high Ki67, i.e., more than 25% positive cells; and HER2‐negative, i.e., 0 or 1+ according to IHC); ‘LumHER’, luminal B‐like HER2‐positive (ER‐positive and/or PR‐positive, any value for Ki67, and HER2‐positive, i.e., 2+ or 3+); ‘HER’, HER2‐positive, nonluminal (ER‐negative, PR‐negative, any value for Ki67, and HER2‐positive, confirmed by HER2 amplification using routine FISH technology when IHC is 2+ or 3+); and ‘TNB’, triple‐negative (ER‐ and PR‐negative, HER2‐negative, and any Ki67)
| No. of patients | Sample types. Subtype based on IHC (St. Gallen) | Grade | No. of samples for PEA | No. of patients with two samples | No. of patients with three samples |
|---|---|---|---|---|---|
| Total: 32 | Benign lesions | NA | 38 | 6 | |
| Total: 25 | Cancer subtypes | ||||
| 9a | Luminal A (LumA) | I–II | 14 | 3b | 1 |
| 4 | Luminal B (LumB) | II–III | 5 | 1 | |
| 5 | Luminal HER (LumHER) | II–III | 6 | 1b | |
| 3 | Nonluminal HER (HER) | III | 5 | 2b | |
| 4 | TNB (TNBC) | II–III | 4 | 0 | |
| Cancer samples | 34 | 7 | 3 |
aTwo of nine patients were diagnosed with lobular cancer. All other were ductal cancers. bIncludes one patient with samples from both primary tumor and axillary lymph node metastasis.
3.2. Hierarchical clustering of protein profiles
To explore similarities between samples from BC patients, protein profiles were compared by hierarchical clustering (Fig. S1). Pairwise correlations between sample profiles were calculated using PEA values for 134 proteins, that is, after excluding 33 proteins for which > 25% of the sample values were below the protein‐specific limits of detection (LOD; Table S6). The between‐samples clustering showed that the same or similar IHC subtypes clustered closer with each other. Also, when several multifocal (MF) lesions from the same patients were available, these clustered next to each other. There was one exception from the latter: One sample was diagnosed as ductal carcinoma in situ (DCIS, #FD93), while the other sample from the same patient was diagnosed as invasive ductal carcinoma (IDC). There were three cases where both primary tumor (P) and lymph node metastases (M) from the same patient were analyzed. In two out of these three, P and M failed to cluster adjacent to each other. All the three cases of lobular cancer were found in Cluster #2. Clusters #1a and #1b represent 53% ER‐negative samples, and Clusters #1c and #1d represent 100% ER‐positive samples. Thus, the protein‐based clustering indicated consistence with IHC subtypes. Interestingly, the sample #FD11 (Cluster #2) represented a small 12‐mm lesion, mammography code = 2 (i.e., presumably benign), but received triple‐negative breast cancer (TNB) as the final diagnosis. The remaining TNBs had mammography code = 5 (i.e., cancer) and being 25–40 mm in size; all ended up in Clusters #1a and #1b.
To explore how proteins levels could determine sample clustering, we applied a two‐way hierarchical clustering of samples vs protein profiles (Fig. 1). Again, samples tended to cluster according to subtype. One cluster labeled ‘ER Low & Ki67 High’ had only 10% (1/10) samples of LumA type, while another ‘ER High & Ki67 Low’ cluster included 53% (10/19) samples of LumA type. A third cluster (‘Mixed cluster’) contained all three samples with lobular cancers. The first two clusters are significantly associated with different BC subtypes (chi‐square test, P < 0.05). ER and Ki67 expression values as determined by IHC (% positive cells) are significantly different between the first two clusters (t‐test, P < 0.05). In addition, the amount of MØ estimated by cytology was higher in the ‘ER Low & Ki67 High’ vs the ‘ER High & Ki67 Low’ cluster (t‐test, P < 0.05). In order to exclude possible influence of repeated samples available for a few patients in this analysis (i.e., multifocal cancers and metastases), we also performed clustering by leaving only one sample per patient. After reclustering, only two samples shifted cluster location compared to the previous clustering: #FD81 (TNB) and #FD92 (LumHER); both moved from the ‘ER High & Ki67 Low’ cluster to the ‘ER Low & Ki67 High’. Thus, clustering of samples seemed to be robustly correlated with biological features of subtypes and indicated the different biology between ductal and lobular cancers.
Figure 1.
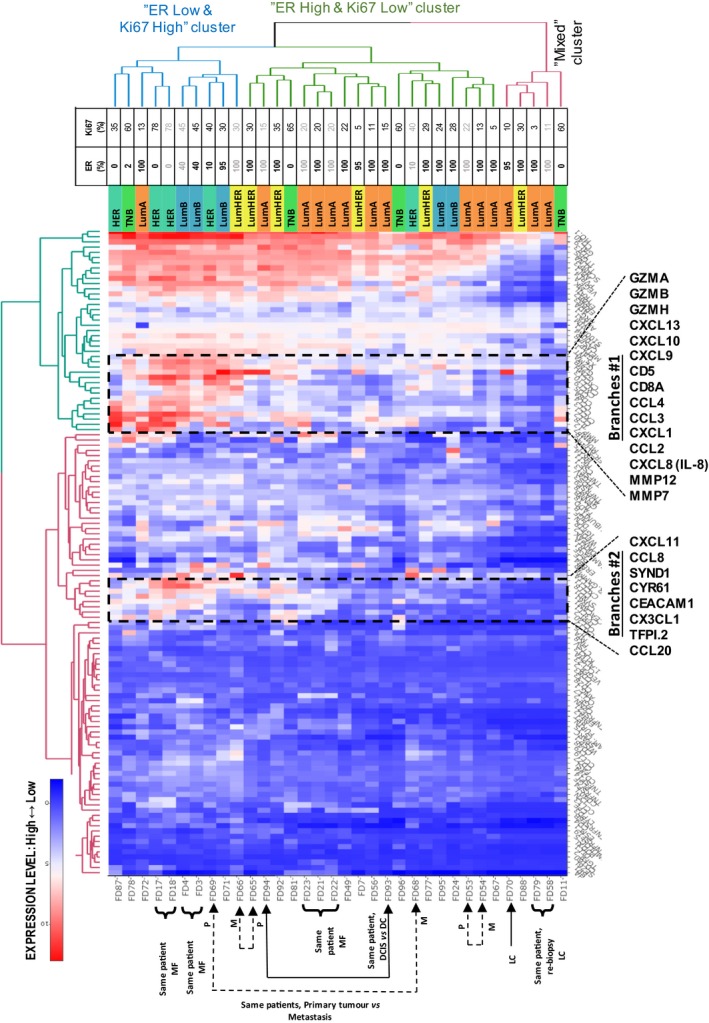
Heatmaps of protein expression for the patient samples: Two‐dimensional hierarchical clustering of samples vs protein profiles. Proteins for which > 25% of the values were below LOD were excluded. Compare Fig. S1 which shows a very similar clustering of samples. Clustering of proteins and samples indicates correlations between subtypes and protein functions. A zoom‐in view of Branches #1 is shown in Fig. S2. An interactive representation providing data values can be explored at http://research.scilifelab.se/andrej_alexeyenko/downloads/PEA/heatmap.PEA.34x124.v10.html.
With respect to the protein dimension, we observed two prominent protein dendrogram branch groups (Branches #1 and Branches #2) that showed elevated expression levels in the ‘ER Low & Ki67 High’ sample cluster compared to the ‘ER High & Ki67 Low’ cluster. Notably, both Branches #1 and #2 were dominated by immune‐related proteins including in total twelve chemokines and three granzymes.
Dendrogram Branches #1 included the MØ/M2‐related protein MMP12 and several protumorigenic chemokines (e.g., CCL2, IL‐8/CXCL8, CXCL1), the T‐cell‐related markers CD8A and CD5, the angiogenic chemokines CXCL9 and CXCL10, and three granzymes known to be produced by cytotoxic T and NK cells (Lyons et al., 2017). A clustering pattern of samples was found, similar to that in Fig. S1. A zoom‐in view of the chemokine rich group of Branches #1 is shown in Fig. S2.
An estimation of the number of tumor‐infiltrating MØ showed that 80% (8/10) of samples in the ‘ER Low/Ki67 High’ cluster contained MØ (average score = 3.1). In contrast, only 31% (5/16) of samples in the ‘ER High/Ki67 Low’ cluster contained MØ (average score = 0.7). The MØ scoring revealed a significant difference between the two clusters (P = 0.011). Thus, high levels of MØ were more associated with the ‘ER Low & Ki67 High’ PEA phenotype and with TNB. All (4/4) TNB and 80% (4/5) HER samples contained MØ, while only 18% (2/11) in the LumA samples did. Cytology image samples from two patients with multifocal HER2 (FD17 and FD18) and multifocal LumA (FD21 and FD22) cancers are shown in Fig. S3. These representative subsets showed few or no MØ in the LumA samples, but greater numbers in the HER2 samples (MØ score = 5).
Given the key role of tumor‐infiltrating lymphocytes (TILs) in BC (Stanton and Disis, 2016), and the clustering neighbors to CD8A (as marker of CD8+ TILs), we performed a correlation analysis using CD8A protein values as a proxy reference of CD8+ TILs (Table S4). A correlation analysis of all protein profiles in all samples using CD8A as a reference confirmed significant correlation between several proteins (CD5, GZMBA, GZMH, CXCL10, CXCL9, GZMB, and CCL4) within Branches #1, increasing the support for higher CD8+ T‐cell‐related activity in samples representing the ‘ER Low & Ki67 High’ cluster (Table S4). The elevated granzyme levels also indicate the presence of activated CD8+ T cells.
The clustering of samples in Fig. 1 indicated clear differences in protein levels between the ‘ER High & Ki67 Low’ vs ‘ER Low & Ki67 High’ clusters. To explore differences specifically related to BC subtypes, the protein levels of ER‐negative (HER and TNB) cancers were compared to those representing the least aggressive IHC subtype, that is, LumA for the complete protein dataset (167 proteins).
There were significant differences (t‐test, P < 0.05) in 53 proteins, with the levels of 50 of these proteins increased in ER‐negative compared to LumA samples. Notably, CA9, IL‐8, CXCL17, CCL20, CCL7, CCl4, and IL‐6 all differed significantly upon Bonferroni correction, and among the top proteins in the fold‐change ranking list, there was an overrepresentation of cytokines (16 of top 34, odds ratio = 9.24, P = 0.000036, Fisher's exact test) demonstrating pronounced immune‐related differences between these subtypes. Twenty out of the top 32 proteins were included within the dendrogram Branches #1 or #2 (Fig. 1). Interestingly, the hypoxia marker CA9 and the MØ‐related protein MMP12 showed the highest fold increases in ER‐negative vs LumA samples (> 36‐fold), followed by the chemokines CXCL8, CCL2, CCL7, CXCL10, and CXCL9. All the latter proteins were > 10‐fold higher in ER‐negative vs LumA samples (Table S5).
3.3. Prediction of Ki67, ER status, and tumor grade by multiple‐regression protein level signatures
The next step in the analysis addressed whether protein profiles could be identified that correlated with key biological properties of BC, as this might reveal important proteins within the tumor microenvironment. Current subtyping of BC depends on subjective IHC‐based microscopic examination by experienced pathologists and, according to the standard protocols, involves binary cutoffs for ER, Ki67, and HER2 levels. Therefore, we explored an alternative approach to determine ER, Ki67, and HER2 levels along a continuous scale via protein signatures, potentially relevant to key biological properties of BC.
We succeeded in producing significant predictive models for the Ki67 and ER IHC clinical variables and for the tumor grade (i.e., malignancy grade according to Nottingham histological grading), but not for the HER2 status (Fig. 2A–C). In these models, the predictive score for a given sample was calculated as a linear sum of protein expression values multiplied by the coefficients for each protein indicated in the plot legends. Surprisingly, these results also revealed that chemokines and/or cytokines strongly contributed to the modeling of all the three (ER, Ki67, and HER2) clinical variables.
Figure 2.
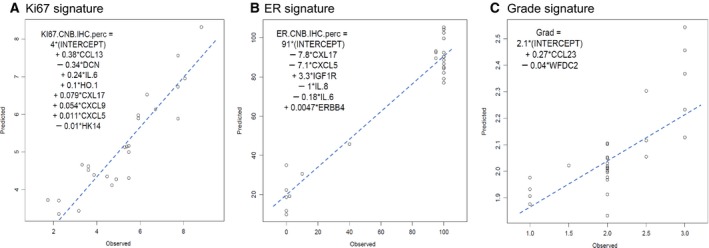
Regression models predictive for IHC‐based observed expression of Ki67 (A), ER (B), and tumor grade (C). Expression levels of chemokines CCL13, CXCL17, CXCL9, CXCL5, CXCL8 (IL‐8), and CCL23 contribute together with other proteins to key properties of BC. ‘Observed’ denotes the IHC values (Ki67 or ER IHC staining positivity) for each of the samples on a continuous scale (0–10, 0–100, or 0–3 for Ki67, ER, and tumor grade, respectively, X‐axis). ‘Predicted’ is the quantitative score assigned by the algorithm along the same continuous range (Y‐axis). Member proteins in the signature are described in the text.
The sets of significant proteins in the prediction models overlapped partially. Notably, the chemokines CXCL17 and CXCL5 and the cytokine IL‐6 contributed significantly to the prediction of both ER and Ki67 status, although with opposite signs of the linear coefficients (Fig. 2A,B). Specifically, CXCL17 and CXCL5 were increased in cancers with high proliferation as assessed by Ki67 and decreased in cancers with high ER levels. The chemokine CCL13 and decorin (DCN) provided strong contributions to the prediction of Ki67 status as previously described (Franzén et al., 2018). IGF1R, known to correlate with ER and good prognosis, contributed positively to the prediction of ER status. Proinflammatory cytokines IL‐6 and IL‐8 showed negative correlation with ER status, while IL‐6 correlated positively with Ki67 status, which indicated increased inflammation in ‘ER Low & Ki67 High’ cancers (Williams et al., 2016).
The regression model for the observed tumor grade used a combination of only two proteins, CCL23 and WFDC2 (Fig. 2C). The chemokine CCL23, also known as MØ inflammatory protein 3 (MIP‐3), is known as highly chemotactic for T cells and monocytes (Zlotnik and Yoshie, 2012). WFDC2 (WAP core domain protein 2), also known as human epididymis protein (HE4), has been proposed as diagnostic biomarker of ovarian cancer (Montagnana et al., 2009, p. 4).
Since the modeling was done on standardized protein values (i.e., expressed on the same scale), the magnitude of coefficients of linear multiple regression was informative regarding the relative contribution of the proteins in the regression models (i.e., the importance in the model). Results show that the top 3 proteins (CCL13, DCN, and IL‐6) contributed > 75% of the predictive value for proliferation, whereas three other proteins (CXCL17, CXCL5, and IGF1R) contributed > 75% of the information for the model predictive of ER status.
Next, we examined the expression levels for the six most prominent protein members in the signatures for the BC subtypes, as well as in the benign lesions representing the baseline. The analysis demonstrated that expression of four of six proteins (CCL13, IL‐6, CXCL17, and DCN) also differed between the benign lesions and cancer (Fig. 3). Despite the obvious need for verification in larger cohorts, this result highlighted the potential roles of the six proteins in BC progression and the potential value of monitoring the balance between key proteins.
Figure 3.
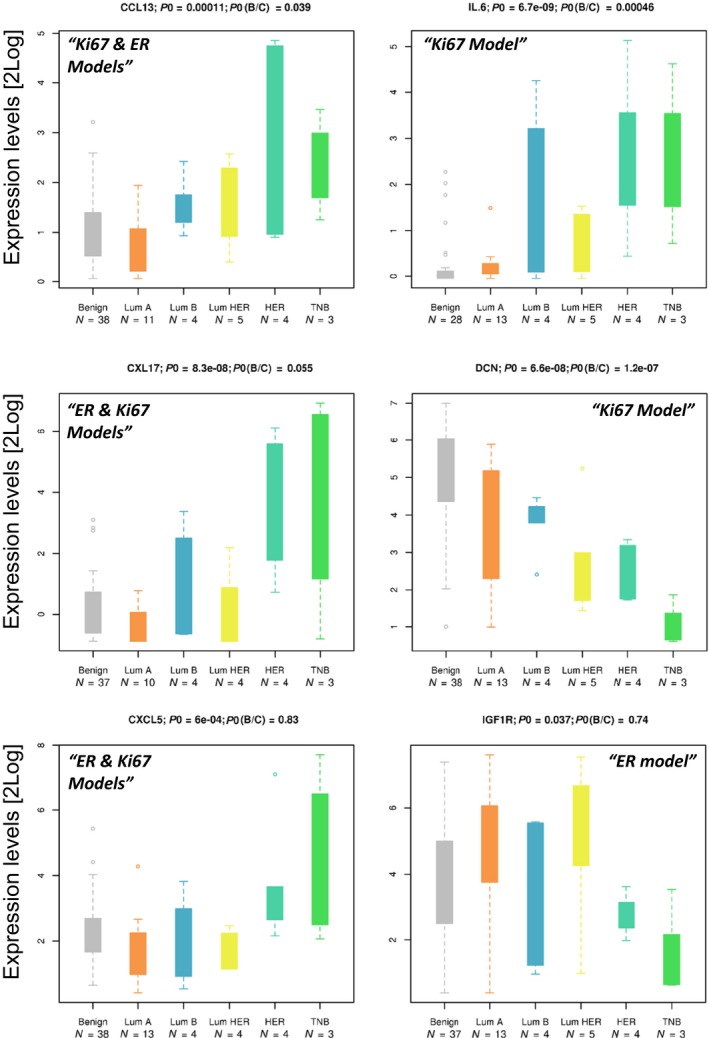
Expression levels of the six most prominent signature protein members in BC subtypes relative to baseline. Histograms show protein levels in benign lesions and cancer subtypes. The significance of differences is indicated by p0 for subtypes and p(B/C) for benign vs cancer. Individual samples are indicated by the FD sample numbers. Protein levels in different sample groups are also presented as boxplots. The protein levels expressed as NPX values are shown on a log2 scale where the boxes contain data points within 25–75th percentile intervals (i.e., between quartiles Q1 and Q3). The maximal whisker length (MWL) is defined as 1.5 times the Q1–Q3 interquartile range (i.e., the box length). Whiskers can extend either to the MWL or to the maximal available data point when the latter is below MWL. Markers thus correspond to data points that extend off the box by more than the MWL value. Sample numbers (FD#) are shown to indicate cases that repeatedly deviate from the main group, for example, the very early TNB case FD11 or the MØ enriched HER2 sample FD17.
3.4. Correlations between immune‐related protein levels and BC key properties
To further explore correlations between protein levels and key properties of BC with respect to a continuous scale of IHC‐measured levels of Ki67, HER2, and ER, we applied a third approach to analyze univariate correlations for each of the 167 PEA assessed proteins to Ki67, HER2, and ER, and the tumor grade. Table 2, using rank correlations, displays proteins that significantly correlated with the aforementioned key properties, and for reference, proteins that also show significantly different levels between cancer and benign lesions (ANOVA). The far‐right column of Table 2 identifies leukocyte subsets associated with each protein, and other relevant information (for references, see Table 2 footnotes). We observed that the levels of 19/20 chemokines in the dataset were altered, a pronounced overrepresentation given that only 12% of all proteins analyzed belonged to this group of proteins (odds ratio = 13.3, P = 0.00097, Fisher's exact test). Interestingly, nine chemokines (CCL2, CCL3, CCL4, CCL8, CCL19, CXCL9, CXCL10, CXCL11, and CXCL13, marked by #) of 19 shown in Table 2 contribute to the 12‐chemokine signature described previously in BC and melanoma progression (Messina et al., 2012; Prabhakaran et al., 2017). Our results suggest that additional chemokines may be involved in the progression of BC, for instance, CCL13, CCL20, and CXCL17. Furthermore, several CD cell surface proteins correlated significantly with many of the key properties analyzed. For instance, the levels of CD8A (cytotoxic T‐cell marker) significantly correlated with tumor grade, Ki67 status, and ER status (P = 0.027, P = 0.00019, and P = 0.0022, respectively). Granzyme B (GZMB), known to be produced by CD8 T cells, showed significant correlation with the same variables. Moreover, several proteins associated with the M2 subset of MØ are positively correlated with the Ki67 status (e.g., CCL8, CCL13, CD4, MMP12, and ANG2).
Complementary to Table 2, the histograms in Fig. 4 illustrate expression levels for a selection of 12 proteins from Table 2. The protein expression levels in BC subtypes are compared to those of benign lesions. All proteins except CD8A showed clear trends of increasing from benign (left bar) to more aggressive subtypes (HER2 and TNB, right bars) as indicated by the arrow at the far bottom of the boxplots. CD8A did not correlate clearly with BC subtype, although in LumA we observed lower levels of CD8A compared to other subtypes and benign lesions.
Figure 4.
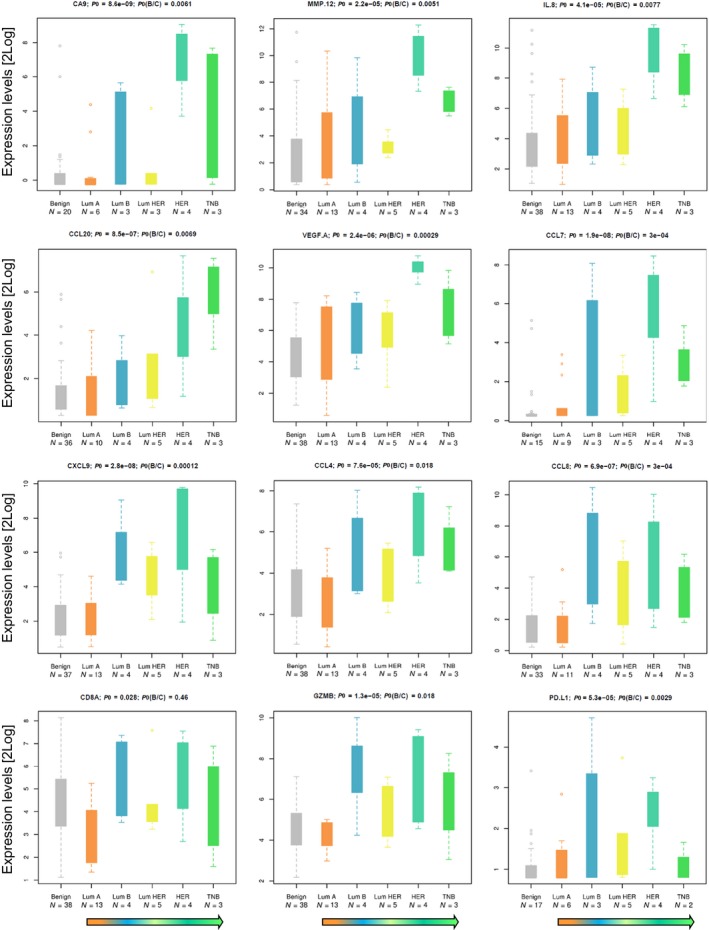
Protein correlations. A selection of proteins with correlation with grade or with the Ki67, HER2, or ER status, or with differences benign vs cancer, and/or LumA vs ER‐negative cancers (compare Fig. 3, Table 2, and Tables S4 and S5). Arrows at the bottom indicate expected decline of prognosis.
4. Discussion
We have previously reported that analysis using proximity extension assays on minute amounts of FNA samples in BC provides data that correlate with routine assessments of key markers such as ER, PGR, HER2, and Ki67. That study demonstrates that one microliter of lysates from FNA samples can be used for extensive targeted protein profiling. We used data from two PEA panels, ‘Oncology II’ and ‘Immuno‐Oncology I’, with a total of 167 unique proteins representing many different biological processes such as immunological mechanisms, cell adhesion, cell differentiation, cell motility, cell proliferation, apoptosis, and metabolic activity. In total, at least 80 of the proteins are directly immune‐related. Results showed that several immune‐related proteins, previously reported as important in therapy and progression of BC, can be quantified in a single FNA sample from minute material of primary lesions as small as 8 mm in size. In addition, several additional immune‐related proteins were found, of which many may be candidates for immune characterization of BC.
In the current study, we have evaluated expression levels of proteins in relation to routine parameters, that is, key properties and subtypes using various statistical approaches. Initially, hierarchical clustering of all protein profiles revealed two sample clusters, ‘ER High & Ki67 Low’ and ‘ER Low & Ki67 High’, with differences in protein levels, that is, proteins that may be related to expected aggressiveness of the tumors (i.e., ‘ER Low & Ki67 High’ cancers are expected to be more aggressive). We note that the difference between the two clusters showed an overrepresentation of immune‐related proteins, including many members from the chemokine family. The two dendrogram branch protein groups #1 and #2 involved 12 of a total of 20 investigated chemokines (60%), corresponding to only 12% of the total number of proteins analyzed. We therefore compared the more contrasting groups with respect to expected aggressiveness of the sampled tumors, namely LumA vs ER‐negative cases. The overrepresentation of chemokines in the protein profiles was confirmed, and in total, 16 chemokines showed significantly higher levels by on average 9.5‐fold in samples from ER‐negative vs LumA tumors. In this analysis, 13 of the top 21 proteins representing the highest fold changes were chemokines, and seven of these were part of the previously reported 12‐chemokine signature in malignant melanomas and BC (Prabhakaran et al., 2017). Our results indicate major differences in the involvement of different entities of the immune response between these subtypes. Given the correlation with clinical outcome, our results may be used to define a signature for future therapy selection in BC.
Interestingly, the MØ score was significantly higher in the ‘ER Low & Ki67 High’ cluster compared to the ‘ER High & Ki67 Low’ sample cluster (see Section 3.2). This difference coincides with the difference in chemokine levels between these two clusters (see Table S5). We observed a 36‐fold higher level of the protein CA9, a well‐established marker for hypoxia, and this may well be linked to several of the altered chemokine levels (Fig. 4). A reasonable hypothesis is that this overexpression is a consequence of recruitment of hypoxia‐driven tumor‐associated MØ (TAMs) and myeloid‐derived suppressor cells (MDSCs) to the tumor microenvironment and that this illustrates the ability of these cell types to differentiate to protumorigenic TAM subtypes. This hypothesis is supported by the observation of a higher level of MMP12 (36‐fold) and > 8‐fold higher levels of CCL2, CCL3, CCL4, and CXCL8, chemokines known to attract myeloid cells that differentiate into TAMs under hypoxia (Van Overmeire et al., 2014). Together with IL‐6, with a fivefold increased expression level, CCL2 may contribute to recruitment and polarization to protumorigenic M2‐like TAM phenotypes. Other chemokines, for example, CCL7, CCL20, CXCL1, and CXCL17, demonstrate similar expression patterns and may also be important contributors to MØ tumor infiltration, MØ differentiation, and a more aggressive phenotype.
Notably, among the top 11 proteins expressed at higher levels in the ‘ER High & Ki67 Low’ sample cluster, seven proteins were also expressed at elevated levels in HPV‐positive tonsillar and base of tongue cancer vs normal adjacent tissues (CA9, CXCL10, MMP12, CCL20, CXCL11, CXCL9, and CXCL8; Ramqvist et al., 2018). This indicates striking similarities in immune activation between the diverse tumor types.
A next level of analysis is provided by multiple‐regression modeling using continuous data for Ki67, ER, and tumor grade, rather than discrete data (i.e., BC subtypes). Three significant models were obtained that revealed protein signatures comprising in total 13 proteins. Here, the predominance of chemokines in the signatures, representing Ki67, ER, and grade, further highlights the apparently central role of chemokines in BC. Signatures show that expression levels of proteins in FNA samples from more aggressive BC were characterized by relatively high levels of CCL13, CXCL17, CXCL5, CCL23, and IL‐6, along with low levels of DCN and IGF1R. Less aggressive BC showed the inverse quantitative relationships for the same proteins. Taken together, the multiple‐regression models may reflect the quantitative balance between proteins in relation to a gradient from less aggressive (high ER, low Ki67, and low grade) to more aggressive (low ER, high Ki67, and high grade) phenotypes. Note also that for example, ERBB2, CD8A, and CD5 are not part of the signatures above. It is possible that the role of immune modulatory proteins has previously been underestimated in BC.
Our results clearly demonstrate that key immune‐related protein markers can be assessed in FNA material, for instance, TIL markers such as CCL2, CX3CL1, CXCL9, CXCL10, GZMA, GZMB, and GZMH (Galon et al., 2013). We confirm here results from previous studies that the expression levels of these markers and thereby TILs tend to be elevated in ER‐negative cancers compared to LumA cancers (Agahozo et al., 2018). However, the classical TIL marker CD8A alone did not differ between cancer and benign lesions and only weakly significant differences were observed between LumA and ER‐negative cancers (P = 0.018, non‐Bonferroni‐corrected P‐value).
Results presented herein also indicate that proteins that are components of the described signatures may play an important role in the biology behind the phenotypes, which traditionally is described in terms of ER, Ki67 expression, and tumor grade. In this context, it is surprising to note the prominent roles that chemokines seem to have in BC. The growth factor inhibitor DCN (decorin), which was previously shown to be decreased in BC compared to benign lesions (Franzén et al., 2018), interacts with IGF1R, EGFR, VEGFR2, ERBB2, and MMP7 (www.proteomicsdb.org), and studies also indicated that DCN may affect signaling via the chemokine receptor CXCR4 and is needed for autophagy.
The cytokine IL‐6 and chemokines CCL20, CXCL8, CXCL9, and CCL8 have previously been linked to overall survival in various cancers (Denkert et al., 2010; Farmaki et al., 2016, p. 8; Knüpfer and Preiß, 2007; Todorović‐Raković and Milovanović, 2013). However, several other chemokines have only to a limited extent been described previously in BC or benign breast lesions, for example, CXCL5, CXCL17, CCL13, CCL23, and CCL4. For instance, one recent report using PEA profiling of the extracellular compartment showed similar profiles in BC and mammographically dense healthy breast tissues compared to nondense healthy breast tissue; for instance, CCL4, CCL7, CCL8, CCL23, CXCL5, CXCL8, CXCL9, and VEGF showed elevated levels (Abrahamsson et al., 2018). Thus, the exact role of these cyto‐ and chemokines as assayed by PEA of FNA extracted tumor material for individual biological characteristics of BC needs further evaluation.
The chemokines CCL4 and CCL20 have been shown to recruit subsets of T cells in esophageal carcinoma, where CCL4 expression was correlated with the expression of CD8 and GZMB. This correlation was also observed in our study, which indicates a similar role in BC (Table S4; Liu et al., 2015). In contrast to CCL4, CCL20 has been shown to have several important functions in BC (Osuala and Sloane, 2014, p. 20).
Only one report so far describes a role for CXCL17 in BC. This is the most recent member of the chemokine family, and it is also known as VEGF coregulated chemokine 1. In this report, CXCL17 was found to be associated with shorter overall survival and thus represents a potential marker of poor prognosis (Guo et al., 2017).
Zhao et al. (2017) found that CXCL5 was overexpressed in tumor tissue and associated with poor prognosis in colorectal cancer patients. We are not aware of any previous reports on CXCL5 expression analysis in BC tissue. This also applies to CCL13 and CCL23 as the expression of these chemokines appears not to have been studied in BC tissue earlier, although CCL23 was found to be involved in angiogenesis (Hwang et al., 2005).
We have shown the correlation between immune markers such as CD8A and granzymes, CCL13, CCL20, CXCL17, CCL8, and CD4. These and others may represent immune contexture markers suggested as substitute to the classification and characterization of colon cancer by Galon et al. (2013). The finding illustrates the increasing potential to use the FNA to analyze predictive biomarkers in validation programs and clinical trials, as demonstrated recently (Foukakis et al., 2018).
Taken together, our data provide increased support for an association between chemokines and BC progression. The expression of cytokines in tumor tissues may be a manifestation of immune oncological host responses to the malignant cells, representing attempts to eradicate the tumor. On the other hand, chemokines, while less potent mitogens than growth factors, may also have a direct role in the proliferation of epithelial cancers. Oncogenes may activate chemokine receptor genes indirectly by regulating transcription factors involved in upregulation of proinflammatory chemokines. For instance, activation of chemokine receptors initiates intracellular signals leading to proliferation via ERK1/2 activation and the PI3K or β‐catenin pathways. Activation of chemokine receptors may also affect proliferation indirectly through transactivation of the epidermal growth factor receptor (EGFR) (Lacalle et al., 2017).
This is to our knowledge the first report on multiplex analysis of key immune‐related proteins in FNA samples from patients with primary BC. Our study demonstrates the feasibility to analyze immune‐related molecular signatures in minimal FNA samples as a possible avenue for future diagnostics, therapy selection, and monitoring of responses to therapy. This approach may also deliver new information about proteins with a role in BC progression and potential markers related to immunotherapy. Therefore, our results may be important for the future development of precision cancer medicine, for example, when considering neoadjuvant therapy or specific types of immunotherapy.
5. Conclusions
Immunological factors have proven significant predictors of neoadjuvant and adjuvant therapy responses in BC. It has previously been shown that several of these factors may be analyzed in FNA samples at the RNA level and that overexpression of immune‐related genes predicts chemosensitivity in patients with luminal advanced BC (Foukakis et al., 2018). However, the ultimate tumor phenotype is driven by the complex tumor microenvironment at the protein level, including products of genes expressed remotely from the sampled tissue and brought there via blood and thus undetectable at the level of mRNA in the tissue sample. Here, we show that a wide range of highly relevant immune‐related proteins can be analyzed in FNA samples, representing a snapshot of the native tumor microenvironment, using a simple sample preparation protocol and semi‐automated PEA technology. This is a pilot study which represents a highly translational approach and which paves the way for a new concept for comprehensive immune scoring and longitudinal monitoring of therapy responses. In conclusion, the approach demonstrated herein offers improved opportunities for individualized therapy selection and immune therapy evaluation.
Conflicts of interest
UL is founder, shareholder, and board member of Olink Proteomics. No other authors declare any conflicts of interest.
Author contributions
BF, MKM, TH, RL, UL, and GA were responsible for project design and infrastructure. JK was responsible for patient selection and sampling of material. LK and BF were responsible for scoring of macrophages by cytology. AA was responsible for data analysis, statistics, and bioinformatics. BF was responsible for writing the manuscript with support from all co‐authors.
Supporting information
Fig. S1. Heatmap representing a two‐dimensional hierarchical clustering performed on a correlation matrix using the protein expression profiles of all cancer patient samples, that is, comparing all cancer samples against each other.
Fig. S2. Zoom‐in figure from Fig. 1b shows the chemokine rich cluster A.
Fig. S3. Example cytology samples from two patients with multifocal HER2 and multifocal luminal A cancers.
Table S1. Listing of all samples subjected to PEA and diagnosis by cytology (FNA material).
Table S2. Benign samples.
Table S3. Cancer samples from a total of 25 patients.
Table S4. Top 22 CD8A correlated proteins with reference to additional analysis results (see 3.3 and 3.4).
Table S5. PEA profiling of FNA samples reveals several significant (P<0.05) differences between samples from ER‐negative (HER2 + TNB) vs luminal A cases.
Table S6. Proteins in the PEA panels used (for more information: www.olink.com).
Acknowledgments
This study received grants from Stockholm County Council Innovation fund (No. 20151043 and No. 20160287), Vinnova (No. 2016‐00595), the Swedish Cancer Society (No. 170246), the Erling‐Persson Family Foundation, the Swedish Foundation for Strategic Research, The Swedish Breast Cancer Association, and the Stockholm Cancer Society. The authors would like to acknowledge support of personnel at Karolinska (Edward Azawedo, Susanne Becker, and Sonja Gustafsson), personnel at BröstCentrum City (Stockholm), personnel at Capio S:t Görans Sjukhus (Lars Löfgren, Margaretha Blåsjö, and Christina Ekström), the Swedish Research Council, the European Community's 7th Framework Program (FP7/2007‐2013) under grant agreements No. 316929 (GastricGlycoExplorer) and No. 294409 (ProteinSeq), and IngaBritt och Arne Lundbergs Forskningsstiftelse. The authors would also like to acknowledge support of the Clinical Biomarker Facility at SciLifeLab Sweden for providing assistance in protein analyses and the National Bioinformatics Infrastructure Sweden for assistance in data analysis, and expertise at KIGene, MMK, Neurogenetics Unit at Karolinska University Hospital for assistance in RNA analyses.
References
- Abrahamsson A, Rzepecka A and Dabrosin C (2018) Equal pro‐inflammatory profiles of CCLs, CXCLs, and matrix metalloproteinases in the extracellular microenvironment in vivo in human dense breast tissue and breast cancer. Front Immunol 8, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agahozo MC, Hammerl D, Debets R, Kok M and van Deurzen CHM (2018) Tumor‐infiltrating lymphocytes and ductal carcinoma in situ of the breast: friends or foes? Mod Pathol 31, 1012–1025. [DOI] [PubMed] [Google Scholar]
- Assarsson E, Lundberg M, Holmquist G, Björkesten J, Bucht Thorsen S, Ekman D and Fredriksson S (2014) Homogenous 96‐Plex PEA immunoassay exhibiting high sensitivity, specificity, and excellent scalability. PLoS One 9, e95192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y and Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc 57, 289–300. [Google Scholar]
- Denkert C, Loibl S, Noske A, Roller M, Müller BM, Komor M, Budczies J, Darb‐Esfahani S, Kronenwett R, Hanusch C et al (2010) Tumor‐associated lymphocytes as an independent predictor of response to neoadjuvant chemotherapy in breast cancer. J Clin Oncol 28, 105–113. [DOI] [PubMed] [Google Scholar]
- Farmaki E, Chatzistamou I, Kaza V and Kiaris H (2016) A CCL8 gradient drives breast cancer cell dissemination. Oncogene 35, 6309–6318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foukakis T, Lövrot J, Matikas A, Zerdes I, Lorent J, Tobin N, Suzuki C, Brage SE, Carlsson L, Einbeigi Z et al (2018) Immune gene expression and response to chemotherapy in advanced breast cancer. Br J Cancer 118, 480–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzén B, Kamali‐Moghaddam M, Alexeyenko A, Hatschek T, Becker S, Wik L, Kierkegaard J, Eriksson A, Muppani NR, Auer G et al (2018) A fine needle aspiration‐based protein signature discriminates benign from malignant breast lesions. Mol Oncol 12, 1415–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galon J, Angell HK, Bedognetti D and Marincola FM (2013) The continuum of cancer immunosurveillance: prognostic, predictive, and mechanistic signatures. Immunity 39, 11–26. [DOI] [PubMed] [Google Scholar]
- Goldhirsch A, Wood WC, Coates AS, Gelber RD, Thurlimann B, Senn H‐J and Panel members (2011) Strategies for subtypes–dealing with the diversity of breast cancer: highlights of the St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2011. Ann Oncol 22, 1736–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo YJ, Zhou YJ, Yang XL, Shao ZM and Ou ZL (2017) The role and clinical significance of the CXCL17‐CXCR8 (GPR35) axis in breast cancer. Biochem Biophys Res Commun 493, 1159–1167. [DOI] [PubMed] [Google Scholar]
- Hwang J, Son K‐N, Kim CW, Ko J, Na DS, Kwon BS, Gho YS and Kim J (2005) Human CC chemokine CCL23, a ligand for CCR1, induces endothelial cell migration and promotes angiogenesis. Cytokine 30, 254–263. [DOI] [PubMed] [Google Scholar]
- Knüpfer H and Preiß R (2007) Significance of interleukin‐6 (IL‐6) in breast cancer (review). Breast Cancer Res Treat 102, 129–135. [DOI] [PubMed] [Google Scholar]
- Lacalle RA, Blanco R, Carmona‐Rodríguez L, Martín‐Leal A, Mira E and Mañes S (2017) Chemokine receptor signaling and the hallmarks of cancer. Int Rev Cell Mol Biol 331, 181–244. [DOI] [PubMed] [Google Scholar]
- Larsson A, Carlsson L, Gordh T, Lind A‐L, Thulin M and Kamali‐Moghaddam M (2015) The effects of age and gender on plasma levels of 63 cytokines. J Immunol Methods 425, 58–61. [DOI] [PubMed] [Google Scholar]
- Liu JY, Li F, Wang LP, Chen XF, Wang D, Cao L, Ping Y, Zhao S, Li B, Thorne SH et al (2015) CTL‐ vs Treg lymphocyte‐attracting chemokines, CCL4 and CCL20, are strong reciprocal predictive markers for survival of patients with oesophageal squamous cell carcinoma. Br J Cancer 113, 747–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ly A, Ono JC, Hughes KS, Pitman MB and Balassanian R (2016) Fine‐needle aspiration biopsy of palpable breast masses: patterns of clinical use and patient experience. J Natl Compr Canc Netw 14, 527–536. [DOI] [PubMed] [Google Scholar]
- Lyons YA, Wu SY, Overwijk WW, Baggerly KA and Sood AK (2017) Immune cell profiling in cancer: molecular approaches to cell‐specific identification. NPJ Precis Oncol 1, 26 10.1038/s41698-017-0031-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messina JL, Fenstermacher DA, Eschrich S, Qu X, Berglund AE, Lloyd MC, Schell MJ, Sondak VK, Weber JS and Mulé JJ (2012) 12‐chemokine gene signature identifies lymph node‐like structures in melanoma: potential for patient selection for immunotherapy? Sci Rep 2, 765 10.1038/srep00765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagnana M, Lippi G, Ruzzenente O, Bresciani V, Danese E, Scevarolli S, Salvagno GL, Giudici S, Franchi M and Guidi GC (2009) The utility of serum human epididymis protein 4 (HE4) in patients with a pelvic mass. J Clin Lab Anal 23, 331–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M and Alizadeh AA (2015) Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 12, 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osuala KO and Sloane BF (2014) Many roles of CCL20: emphasis on breast cancer. Postdoc J 2, 7–16. [PMC free article] [PubMed] [Google Scholar]
- Prabhakaran S, Rizk VT, Ma Z, Cheng C‐H, Berglund AE, Coppola D, Khalil F, Mulé JJ and Soliman HH (2017) Evaluation of invasive breast cancer samples using a 12‐chemokine gene expression score: correlation with clinical outcomes. Breast Cancer Res 19, 71 10.1186/s13058-017-0864-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prat A, Navarro A, Paré L, Reguart N, Galván P, Pascual T, Martínez A, Nuciforo P, Comerma L, Alos L et al (2017) Immune‐Related Gene Expression Profiling After PD‐1 Blockade in Non‐Small Cell Lung Carcinoma, Head and Neck Squamous Cell Carcinoma, and Melanoma. Cancer Res 77, 3540–3550. [DOI] [PubMed] [Google Scholar]
- Ramqvist T, Näsman A, Franzén B, Bersani C, Alexeyenko A, Becker S, Haeggblom L, Kolev A, Dalianis T and Munck‐Wikland E (2018) Protein expression in tonsillar and base of tongue cancer and in relation to human papillomavirus (HPV) and clinical outcome. Int J Mol Sci 19, 978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimsten A, Stenkvist B, Johanson H and Lindgren A (1975) The diagnostic accuracy of palpation and fine‐needle biopsy and an evaluation of their combined use in the diagnosis of breast lesions: report on a prospective study in 1244 women with symptoms. Ann Surg 182, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanton SE and Disis ML (2016) Clinical significance of tumor‐infiltrating lymphocytes in breast cancer. J Immunother Cancer 4, 59 10.1186/s40425-016-0165-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strazza M and Mor A (2017) Consider the chemokines: a review of the interplay between chemokines and T cell subset function. Discov Med 24, 31–39. [PMC free article] [PubMed] [Google Scholar]
- Todorović‐Raković N and Milovanović J (2013) Interleukin‐8 in breast cancer progression. J Interferon Cytokine Res 33, 563–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turley SJ, Cremasco V and Astarita JL (2015) Immunological hallmarks of stromal cells in the tumour microenvironment. Nat Rev Immunol 15, 669–682. [DOI] [PubMed] [Google Scholar]
- Van Overmeire E, Laoui D, Keirsse J, Van Ginderachter JA and Sarukhan A (2014) Mechanisms driving macrophage diversity and specialization in distinct tumor microenvironments and parallelisms with other tissues. Front Immunol 5, 127 10.3389/fimmu.2014.00127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams CB, Yeh ES and Soloff AC (2016) Tumor‐associated macrophages: unwitting accomplices in breast cancer malignancy. NPJ Breast Cancer 2, 15025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Ou B, Han D, Wang P, Zong Y, Zhu C, Liu D, Zheng M, Sun J, Feng H et al (2017) Tumor‐derived CXCL5 promotes human colorectal cancer metastasis through activation of the ERK/Elk‐1/Snail and AKT/GSK3β/β‐catenin pathways. Mol Cancer 16, 70 10.1186/s12943-017-0629-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlotnik A and Yoshie O (2012) The chemokine superfamily revisited. Immunity 36, 705–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Heatmap representing a two‐dimensional hierarchical clustering performed on a correlation matrix using the protein expression profiles of all cancer patient samples, that is, comparing all cancer samples against each other.
Fig. S2. Zoom‐in figure from Fig. 1b shows the chemokine rich cluster A.
Fig. S3. Example cytology samples from two patients with multifocal HER2 and multifocal luminal A cancers.
Table S1. Listing of all samples subjected to PEA and diagnosis by cytology (FNA material).
Table S2. Benign samples.
Table S3. Cancer samples from a total of 25 patients.
Table S4. Top 22 CD8A correlated proteins with reference to additional analysis results (see 3.3 and 3.4).
Table S5. PEA profiling of FNA samples reveals several significant (P<0.05) differences between samples from ER‐negative (HER2 + TNB) vs luminal A cases.
Table S6. Proteins in the PEA panels used (for more information: www.olink.com).