

Abstract
Anthrax Vaccine Adsorbed (AVA, BioThrax®) is approved for use in humans as a priming series of 3 intramuscular (i.m.) injections (0, 1, 6 months; 3-IM) with boosters at 12 and 18 months, and annually thereafter for those at continued risk of infection. A reduction in AVA booster frequency would lessen the burden of vaccination, reduce the cumulative frequency of vaccine associated adverse events and potentially expand vaccine coverage by requiring fewer doses per schedule. Because human inhalation anthrax studies are neither feasible nor ethical, AVA efficacy estimates are determined using cross-species bridging of immune correlates of protection (COP) identified in animal models. We have previously reported that the AVA 3-IM priming series provided high levels of protection in non-human primates (NHP) against inhalation anthrax for up to 4 years after the first vaccination. Penalized logistic regressions of those NHP immunological data identified that anti-protective antigen (anti-PA) IgG concentration measured just prior to infectious challenge was the most accurate single COP.
In the present analysis, cross-species logistic regression models of this COP were used to predict probability of survival during a 43 month study in humans receiving the current 3-dose priming and 4 boosters (12, 18, 30 and 42 months; 7-IM) and reduced schedules with boosters at months 18 and 42 only (5-IM), or at month 42 only (4-IM). All models predicted high survival probabilities for the reduced schedules from 7 to 43 months. The predicted survival probabilities for the reduced schedules were 86.8% (4-IM) and 95.8% (5-IM) at month 42 when antibody levels were lowest. The data indicated that 4-IM and 5-IM are both viable alternatives to the current AVA pre-exposure prophylaxis schedule.
Keywords: Bacillus anthracis, Anthrax, Anthrax Vaccine Adsorbed, AVA, Biothrax, Correlates of protection, Clinical trial, Non-clinical trial, Animal model
1. Introduction
The US licensed anthrax vaccine adsorbed (AVA, BioThrax®) was approved in 1970 for prevention of anthrax in humans [1–3]. The primary immunogen in AVA is anthrax toxin protective antigen (PA) [4]. The 1970 regimen for AVA was a subcutaneous (s.c.) six-dose primary schedule at 0, 0.5, 1, 6, 12 and 18 months with subsequent annual boosters. In May 2012, the US Food and Drug Administration (FDA) approved a revised AVA schedule with an intramuscular (i.m.) three-dose primary schedule at months 0, 1, 6 (3-IM), with boosters at months 12 and 18 followed by annual boosters for those at continued risk of infection (http://www.fda.gov/BiologicsBloodVaccines/Vaccines/ApprovedProducts/ucm304758.htm). The public health impact of these changes resides in the significant reduction in the frequency, severity and duration of local adverse events with i.m. administration, the elimination of the injection at week 2 and immunological protection acquired with the administration of the third dose at month 6 rather than after the sixth dose at month 18. Nonetheless, the AVA booster schedule retains a relatively high burden of injections compared to many other vaccines [5,6].
Studies by Pittman and coworkers have demonstrated that increasing the intervals between doses of AVA can increase the antibody response to booster vaccinations [6–8]. Similar conclusions were reported in the CDC Anthrax Vaccine Research Program (AVRP) phase 4 human clinical trial [3,9]. The CDC AVRP demonstrated that study participants receiving the 3-IM priming series with boosters at months 18 and 42 (5-IM) or only at month 42 (4-IM) developed significantly higher levels of anti-PA antibodies at months 19 (5-IM group) and 43 (4-IM and 5-IM groups) compared to those receiving annual boosters (7-IM). These differences were particularly striking in the group receiving only the month 42 booster (4-IM), which achieved post-boost antibody concentrations twice as high as the original licensed 8-SC schedule (433.2 μg/mL vs. 216.8 μg/mL). These data in humans indicated that immunological priming by AVA was long-lasting and robust with the ability to produce a high magnitude anamnestic response up to at least 3 years after 3-IM priming [9].
The 3-IM priming schedule without boosters has also been demonstrated to provide long term protection up to 4 years in rhesus macaques [10]. Chen et al. subsequently applied penalized logistic regression models to the NHP humoral and cellular immunological response profiles to select the most predictive immune correlates of protection (COP) [11]. The most accurate single COP was the serum anti-protective antigen (anti-PA) IgG concentration at the time of infectious challenge. Additional COP with good predictive power included peak anti-PA IgG concentrations and lethal toxin neutralization activity (TNA) titers at month 7, and dual-correlate models that combined one peak measurement (anti-PA IgG or TNA ED50) and anti-PA IgG at challenge [11]. The COP are considered pivotal for cross-species predictions of anthrax vaccine efficacy in humans where clinical efficacy studies are impractical and ethically infeasible ([12,13], http://www.fda.gov/AdvisoryCommittees/CommitteesMeetingMaterials/BloodVaccinesandOtherBiologics/VaccinesandRelatedBiologicalProductsAdvisoryCommittee/ucm239733.htm).
The objective of the current report was to determine the feasibility of reduced AVA booster schedules in humans. We describe bridging of the NHP anti-PA IgG and TNA ED50 COP to the CDC AVRP human study data using logistic regression models to generate predicted survival probabilities provided by annual and reduced booster schedules [13]. Distinct from the previous non-inferiority analysis of peak responses to vaccination, this study provides a specific focus on determining survival probabilities during the periods of receding and lowest antibody levels between the completion of the priming series and the subsequent booster vaccinations. This is the first report of a bridging analysis between the CDC nonhuman primate correlates of protection data with the CDC AVRP human clinical trial.
2. Materials and methods
2.1. Human and NHP data sets
Study schedules for humans and NHP are summarized in Table 1. The human data sets were from the CDC AVRP clinical trial comprising 1563 human participants as previously reported [9]. Three study arms (7-IM, 5-IM, and 4-IM, Table 1) received i.m. priming doses at 0, 1 month, and 6 months, matching the NHP cohort schedule. After completion of priming, the study groups received annual or alternate booster schedules: 7-IM received the complete schedule of boosters at month 12, 18, 30 and 42; 5-IM received booster doses at months 18 and 42; and 4-IM received a single booster at month 42. Serum anti-PA IgG antibodies were quantified in all participants that were According to Protocol (ATP) for immunogenicity [9].TNA ED50 was obtained for a subset of approximately 46% of the ATP participants. The study was sponsored by CDC under an Investigational New Drug (IND) application, was approved by the human investigations committees at participating clinical sites and at CDC, and was conducted according to the International Conference on Harmonization Good Clinical Practices (GCP) (www.clinicaltrials.gov; NCT00119067).
Table 1.
Schedule of intramuscular vaccination for The Human and NHP Study Groups in the AVRP Study.
| Primary series (months) | Booster schedule (months) | Study group | Month 0 | Month 0.5 | Month 1 | Month 6 | Month 12 | Month 18 | Month 30 | Month 42 | Month 52 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Human | 0, 1, 6 | 12, 18, 30, 42 | 7-IM | AVA | Saline | AVA | AVA | AVA | AVA | AVA | AVA | N/A |
| 0, 1, 6 | 18, 42 | 5-IM | AVA | Saline | AVA | AVA | Saline | AVA | Saline | AVA | N/A | |
| 0, 1, 6 | 42 | 4-IM | AVA | Saline | AVA | AVA | Saline | Saline | Saline | AVA | N/A | |
| NHP | 0, 1, 6 | None | Human dose | AVA | No injection | AVA | AVA | No injection | No injection | Challenge (subset) | No injection | Challenge |
| 1:5 | AVA | No injection | AVA | AVA | No injection | No injection | Challenge (subset) | No injection | Challenge | |||
| 1:10 | AVA | No injection | AVA | AVA | Challenge (subset) | No injection | Challenge (subset) | No injection | Challenge | |||
| 1:20 | AVA | No injection | AVA | AVA | Challenge (subset) | No injection | Challenge | NA | NA | |||
| 1:40 | AVA | No injection | AVA | AVA | Challenge | NA | NA | NA | NA |
The human and NHP studies have been described in detail previously [9,10]. At each time point, humans received either a full dose of AVA or a saline placebo. NHP received diluted doses of AVA (undiluted, 1/5, 1/10, 1/20 or 1/40) at months 0, 1 and 6. Subsets of NHP were challenged at months 12, 30 and 52 [10,11].
The NHP study has been reported in detail by Quinn et al. [10] and Chen et al. [11]. Chen et al. identified serum anti-PA IgG concentrations at the time of challenge (last) as the most accurate immunological COP for 3-IM priming in rhesus macaques. Additional correlates with good predictive power included peak anti-PA IgG concentrations and lethal toxin neutralization activity (TNA) titers at month 7 (peak), and dual-correlate models that combined one peak measurement (anti-PA IgG or TNA ED50) and last anti-PA IgG [11].
2.2. Bridging from NHP to humans
The method for bridging a non-human COP to predict survival probability in humans was described by Fay et al. [13] and Kohberger et al. [14] and in http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/BloodVaccinesandOtherBiologics/VaccinesandRelatedBiologicalProductsAdvisoryCommittee/UCM232400.pdf. The method has been applied here to predict survival in humans using the NHP COP models. Using PROC LOGISTIC in SAS® version 9.3.1 the NHP data were fitted to the logistic regression model:
where α is the intercept, β is the vector of slope parameters, and x is the vector of measured correlates. Predicted human survival probability (Pr(survive)) was then calculated for each human individual using the α and β parameters estimated by the NHP survival data and the measured correlates (x) of the humans. The mean of the predicted survival probabilities of all the humans in each booster reduction group was taken as the overall predicted survival probability for that booster schedule (see Fig. 1 for illustration of single-correlate bridging and Fig. 2 for illustration of dual-correlate bridging). Difference tests between groups were performed on the predicted survival probabilities with an alpha of 0.05. Confidence intervals were calculated using a non-parametric double-bootstrap method by resampling both the NHP and human datasets 2000 times, calculating mean predicted survival probability for each resample, sorting these from lowest to highest and taking the 50th and 1950th resamples as the 95% confidence intervals [13,15].
Fig. 1.
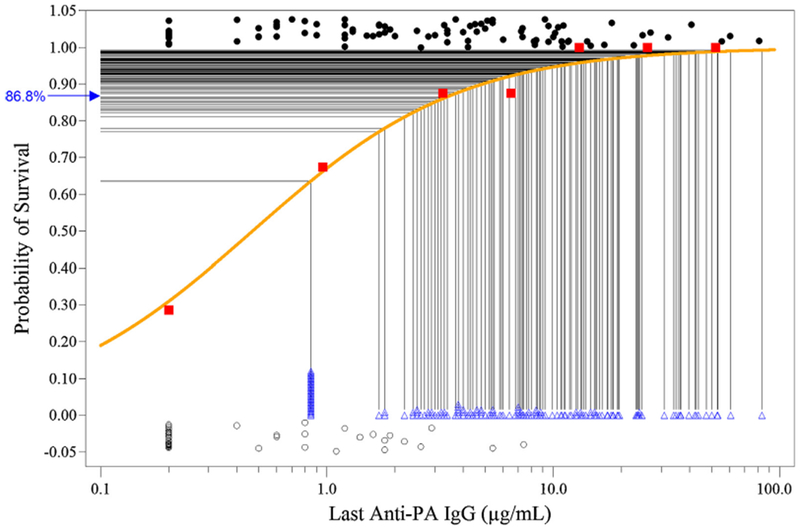
Predicted probability of human survival at month 42 in the Minimum Schedule 4-IM Study Group using a single-correlate model with last anti-PA IgG. NHP survivors (●) (1.0 on the Y-axis) and non-survivors (◯) (0 on the Y-axis) are plotted with slight vertical Y-axis displacements so that overlapping points may be seen. NHP immune response data were binned by anti-PA IgG concentration range; bin 1 > LLOD (0.4 μg/mL); bin 2 LLOD to LLOQ (2.3 μg/mL); remaining bins contain NHP with anti-PA IgG in 2-fold increases above the LLOQ (e.g. ≥2.3 to <4.6, ≥4.6 to <9.2, etc.). Key: (–) logistic regression curve of predicted survival based on the NHP last anti-PA IgG measurements; (∎) mean survival of NHP binned by anti-PA IgG concentration; (Δ) individual human anti-PA IgG responses from the 4IM study group measured at month 42 immediately prior to receiving the booster vaccination, also shown with slight vertical Y-axis displacement so that overlapping points maybe seen; (–) mapping of predicted survival for individual human subjects; (→) mean predicted probability of survival for the human 4-IM cohort at month 42.
Fig. 2.
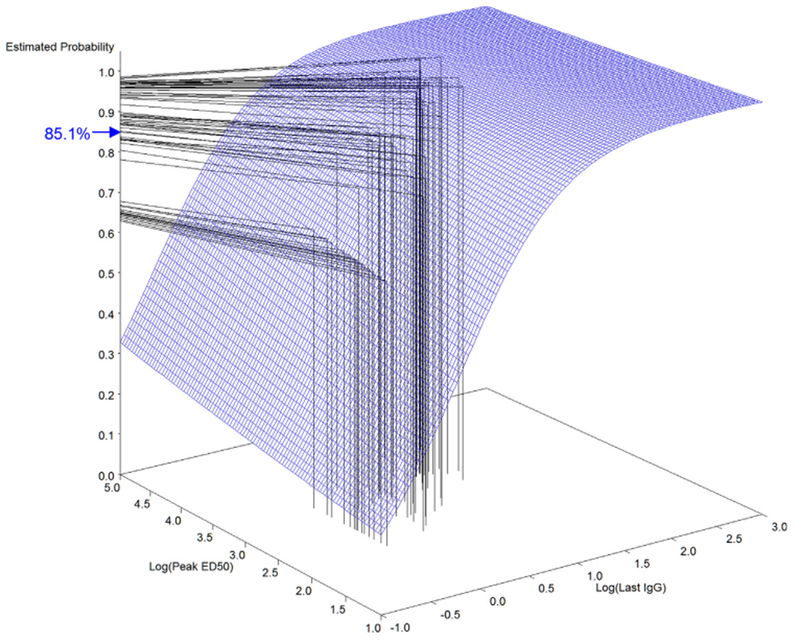
Dual-correlate model surface plot mapping of predicted probability of survival at month 42 in the Human 4-IM Study Group. The surface grid illustrates the relationship among peak ED50, last anti-PA IgG response and survival probability calculated from the NHP survival data. Key: (|) mapping of peak ED50 and last anti-PA IgG response of individual humans from the 4-IM study group measured at month 42; (–) mapping of immune response to predicted survival probability from the surface grid; (→) mean predicted survival probability for the human 4-IM cohort at month 42.
The NHP anti-PA IgG ELISA had a lower limit of detection (LLOD) of 1.7 μg/mL for humans and 0.4 μg/mL for NHP and a lower limit of quantification (LLOQ) of 3.7 μg/mL for humans and 2.3 μg/mL for NHP [10,16]. The TNA assay is considered to be species neutral and had a LLOD ED50 = 11 and lower LLOQED50 = 36 using reference standard AVR801 [17]. For logistic regression analyses using data from these assays all values below the LLOD were masked as 1/2 LLOD for that assay and species.
For all models using a last variable, the result from the time at which survival probability is predicted was used as the last response (e.g. for prediction of survival probability at month 18, the anti-PA IgG measured at month 18 is assigned to the last anti-PA IgG variable in the model, etc.). Within the human data, analyses of single correlate models at the month 7 peak were done by combining the 7-IM, 5-IM and 4-IM study group data since their treatment up to that time point was the same, and no significant differences in anti-PA IgG or TNA ED50 were seen between those groups at that time [9]. For analyses that included later time points when the booster schedules differed, the study groups were analyzed independently.
2.3. Single and dual correlate models
Single correlate models used a single time point in the NHP immunological response time course to calculate probability of survival at specific time points in the human study schedule. Two approaches were applied for single correlate models. In the first approach all of the NHP groups’ data were combined into one dataset. The model used the last measured anti-PA IgG concentration before challenge regardless of challenge time. This approach has the highest accuracy within the NHP dataset [11]. The second approach adopted a more stringent model matching the last anti-PA IgG measurements from NHP at the month 30 and 52 infectious challenge time points with the closest corresponding serological response time points in humans. This approach most closely matches all of the criteria recommended in Fay et al. [13] at the cost of lower statistical power due to the smaller NHP groups.
Dual-correlate models used peak anti-PA IgG or ED50 measured at month 7 with last anti-PA IgG to incorporate both the response to 3-IM priming and the serum antibody levels at the time of challenge.
3. Results
3.1. Single correlate models
Using the combined last anti-PA IgG model (Fig. 1), the 4-IM, 5-IM and 7-IM human study groups all attained similar high levels of predicted survival probability (96.7–97.3%) at month 12, the last time point prior to schedule divergence (Table 2). The subsequent focus of the analyses was on time points immediately prior to injection at which one study schedule received a booster vaccination that a reduced schedule (saline injection) did not. These were study time points at which antibody levels, and consequently the predicted survival probability, were lowest for each schedule. At month 18 the 4-IM and 5-IM study groups (no booster at month 12) maintained a predicted survival probability of 93.3%, compared to 98.3% for the 7-IM group (booster at month 12). At month 30, both 7-IM and 5-IM groups were 1 year post-boost but the 4-IM group had not been boosted (Table 1). At this time point the 5-IM and 7-IM groups attained 97.1% and 97.2% predicted survival probability, respectively, compared to 89.9% for the 4-IM group. At month 42 the 7-IM study group was 1 year post-boost (month 30), the 5-IM group was 2 years post-boost (month 18) and the 4-IM group was 3 years post-priming (month 6). At this study time point, just prior to the last scheduled injection, the 4-IM and 5-IM groups attained 86.8% and 95.8% predicted survival probability, respectively, compared to 98.1% for the 7-IM group. At month 43, the final study time point 4 weeks after all groups received a booster dose, all groups were between 99.6% and 99.8% predicted survival probability.
Table 2.
Human predicted survival probability at key study time points using the combined NHP single correlate last anti-PA IgG model.
| Last anti-PA IgG (combined NHP
groups) |
|||||||
|---|---|---|---|---|---|---|---|
| 4-IM |
5-IM |
7-IM |
|||||
| Time point (months) | NHP N | Human N | % Survival (95% CI) | Human N | % Survival (95% CI) | Human N | % Survival (95% CI) |
| 7 | 137 | 220 | 99.4 (98.0–99.9) | 202 | 99.3 (97.8–99.9) | 214 | 99.5 (98.3–99.9) |
| 12 | 137 | 215 | 96.9 (93.5–99.0) | 197 | 96.7 (93.4–98.8) | 211 | 97.3§ (94.2–99.2) |
| 13 | 137 | 211 | 96.2* (92.6–98.5) | 188 | 96.3* (9254–98.5) | 203 | 99.7 (98.4–100.0) |
| 18 | 137 | 201 | 93.3* (88.6–97.0) | 179 | 93.3§,* (88.3–97.1) | 194 | 98.3§ (95.3–99.6) |
| 19 | 137 | 193 | 93.4*,† (88.6–96.8) | 174 | 99.7 (98.6–100.0) | 192 | 99.6 (98.4–99.9) |
| 30 | 137 | 182 | 89.9*,† (84.7–94.3) | 162 | 97.1 (93.8–98.9) | 179 | 97.2§ (93.7–99.2) |
| 31 | 137 | 179 | 89.4*,† (83.5–94.2) | 153 | 96.9* (93.4–98.9) | 169 | 99.7 (98.8–100.0) |
| 42 | 137 | 161 | 86.8 (80.8–92.1) | 145 | 95.8§,* (91.6–98.5) | 147 | 98.1§ (94.9–99.5) |
| 43 | 137 | 157 | 99.7 (98.7–100.0) | 141 | 99.7 (98.5–100.0) | 139 | 99.7 (98.5–100.0) |
Applying the more stringent models using only the last anti-PA IgG measurements from animals at challenge and the matching time points in humans generated slightly increased predicted survival probabilities at month 30 and 42 (Table 3). At month 12, the 4-IM, 5-IM and 7-IM human study group attained similar high levels of predicted survival probability, from 93.6% to 94.4%. At month 30, the 7-IM human study group attained 99.0% predicted survival probability, the 5-IM attained 98.6%, and the 4-IM attained 93.4%. At month 42, prior to receiving the final booster vaccination, the 7-IM human study group attained 99.5% predicted survival probability, the 5-IM group attained 98.0%, and the 4-IM group attained 90.2%.
Table 3.
Human predicted survival probability at key study time points using the challenge time matched NHP single correlate last anti-PA IgG model.
| Last anti-PA IgG (NHP groups
matched by challenge time) |
|||||||
|---|---|---|---|---|---|---|---|
| 4-IM |
5-IM |
7-IM |
|||||
| Time point (Months) | NHP N | Human N | % Survival (95% CI) | Human N | % Survival (95% CI) | Human N | % Survival (95% CI) |
| 12 | 73 | 215 | 93.8 (81.3–99.0) | 197 | 93.6(81.3–98.7) | 211 | 94.4§ (82.2–99.3) |
| 30 | 58 | 182 | 93.4*,† (86.1–99.6) | 162 | 98.6 (95.5–99.9) | 179 | 99.0§ (95.7–100.0) |
| 42 | 52 | 161 | 90.25§,*,† (81.1–97.9) | 145 | 98.0§ (93.6–99.8) | 147 | 99.5§ (96.8–100.0) |
The human and NHP studies have been described in detail previously [9,10]. Only NHP with matched challenge time plus all controls were used for the model. Confidence intervals (95% CI) are in parentheses.
Time points where boosters were administered.
Statistically significant difference from the 7-IM group.
Statistically significant difference from the 5-IM group.
Predicted survival probabilities calculated using the single-correlate peak anti-PA IgG and ED50 models were the lowest estimates of subsequent protection against inhalation anthrax for all schedules combined (754-IM, Table 4). The peak anti-PA IgG model indicated a predicted survival probability of 83.4%, and the peak TNA model indicated a predicted survival probability of 79.3%.
Table 4.
Human predicted survival probability at month 7 using the single correlate peak anti-PA IgG and ED50 models.
| Peak anti-PA IgG |
Peak ED50 |
||||
|---|---|---|---|---|---|
| 754-IM |
754-IM |
||||
| Time point (Months) | NHP N | Human N | % Survival (95% CI) | Human N | % Survival (95% CI) |
| 7 | 137 | 636 | 83.4 (76.2–89.2) | 289 | 79.3 (72.1–85.7) |
The human and NHP studies have been described in detail previously [9,10]. Predicted % survival in human cohorts based on the NHP peak anti-PA IgG model. The 7-IM, 5-IM and 4-IM groups were combined as 754-IM since they received the same treatment at this month 7 time point. Confidence intervals (95% CI) are in parentheses.
4. Dual correlate models
Predicted survival probability estimates from dual-correlate models ranged from 96.2% to 96.8% using peak anti-PA IgG with last anti-PA IgG and 95.4 to 96.6% using peak ED50 with last anti-PA IgG (Table 5). At month 18, the 4-IM and 5-IM study groups attained predicted survival probabilities of 93.0% and 92.8% using peak anti-PA IgG with last anti-PA IgG and 92.2% and 93.4% using peak ED50 with last anti-PA IgG. The 7-IM study group attained predicted survival probabilities of 97.8% using peak anti-PA IgG with last anti-PA IgG and 97.5% using peak ED50 with last anti-PA IgG. At month 30, the 4-IM study group attained predicted survival probabilities of 89.8% using peak anti-PA IgG with last anti-PA IgG, and 89.0% using peak ED50 with last anti-PA IgG. The 5-IM and 7-IM study groups attained predicted survival probabilities of 96.5% and 96.7% using peak anti-PA IgG with last anti-PA IgG and 96.4% and 96.4% using peak ED50 with last anti-PA IgG. At month 42, the 4-IM study group attained predicted survival probabilities of 87.1% using peak anti-PA IgG with last anti-PA IgG, and 85.1% using peak ED50 with last anti-PA IgG. The 5-IM and 7-IM study groups attained predicted survival probabilities of 95.2% and 97.5% using peak anti-PA IgG with last anti-PA IgG and 95.7% and 97.4% using peak ED50 with last anti-PA IgG. At month 43 (1 month post-boost for all groups), all groups achieved 99.5% predicted survival probability by both models (Table 5).
Table 5.
Human predicted survival probability at key study time points using a NHP dual correlate peak and last model.
| Peak anti-PA IgG, last anti-PA
IgG |
Peak ED50, last anti-PA
IgG |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 4-IM |
5-IM | 7-IM | 4-IM |
5-IM |
7-IM |
||||||||
| Time point (months) | NHP N | Human N | % Survival (95% CI) | Human N | % Survival (95% CI) | Human N | % Survival (95% CI) | Human N | % Survival (95% CI) | Human N | % Survival (95% CI) | Human N | % Survival (95% CI) |
| 7 | 137 | 220 | 99.1 (95.4–99.9) | 202 | 99.0 (95.0–99.8) | 214 | 99.3 (95.9–99.9) | 99 | 98.7 (94.5–99.9) | 88 | 99.2 (95.0–99.9) | 102 | 99.2 (95.4–99.9) |
| 12 | 137 | 210 | 96.3 (91.2–98.8) | 192 | 96.2 (90.9–98.7) | 206 | 96.8§ (91.7–99.1) | 94 | 95.4 (88.7–98.7) | 82 | 96.6 (90.4–99.2) | 98 | 96.4§ (90.2–99.1) |
| 13 | 137 | 206 | 95.7* (90.7–98.3) | 184 | 95.6* (90.1–98.7) | 198 | 99.4(96.4–100.0) | 90 | 95.2* (88.7–98.5) | 79 | 95.8* (89.4–98.8) | 94 | 99.4(95.5–100.0) |
| 18 | 137 | 197 | 93.0* (87.5–96.7) | 175 | 92.8§,* (87.2–96.8) | 189 | 97.8§ (93.2–99.6) | 85 | 92.2* (85.7–96.6) | 73 | 93.4§,* (86.9–97.4) | 89 | 97.5§ (91.7–99.5) |
| 19 | 137 | 189 | 93.0*,† (87.8–96.7) | 170 | 99.5 (96.6–100.0) | 187 | 99.4 (96.4–99.9) | 81 | 92.3*,† (85.4–96.8) | 70 | 99.5 96.2–100.0) | 87 | 99.4 (95.6–100.0) |
| 30 | 137 | 178 | 89.8*,† (84.5–94.4) | 159 | 96.5 (91.4–98.8) | 175 | 96.7§ (91.7–99.1) | 76 | 89.0*,† (82.2–94.5) | 65 | 96.4 (90.5–99.1) | 82 | 96.4§ (90.4–99.1) |
| 31 | 137 | 175 | 89.4*,† (83.6–94.3) | 150 | 96.4* (91.4–98.9) | 166 | 99.5 (97.0–100.0) | 74 | 87.7*,† (81.7–93.6) | 62 | 96.5* (91.0–99.3) | 76 | 99.5 (96.3–100.0) |
| 42 | 137 | 158 | 87.1§,*,† (81.1–92.5) | 142 | 95.2§,* (89.9–98.3) | 143 | 97.5§ (92.8–99.5) | 68 | 85.1§,*,† (78.0–91.5) | 58 | 95.7§ (89.4–98.9) | 64 | 97.4§ (91.1–99.5) |
| 43 | 137 | 154 | 99.5 (96.8–100.0) | 138 | 99.5 (96.6–100.0) | 135 | 99.5 (97.0–100.0) | 64 | 99.5 (96.2–100.0) | 55 | 99.5 (96.0–100.0) | 58 | 99.5 (96.2–100.0) |
Predicted % survival in human cohorts based on the NHP dual-correlate models (peak anti-PA IgG and last anti-PA IgG, or peak ED50 and last anti-PA IgG). All 137 NHP were used for the model. Confidence intervals (95% CI) are in parentheses.
Time points where boosters were administered.
Statistically significant difference from the 7-IM group.
Statistically significant difference from the 5-IM group.
5. Discussion
5.1. Reduced booster schedules
Simpler and better tolerated regimens for AVA vaccination are needed [5,6]. A reduction in the AVA booster schedule would be significant progress toward this goal. The change in route from s.c. to i.m. significantly reduced the rate of adverse reactions, but mild to moderate local adverse reactions still occur in ~50% of the recipients (primarily tenderness and erythema), and mild systemic reactions occur in ~10% of recipients (primarily fatigue and headache) [9]. Human safety and immunogenicity studies have demonstrated that i.m. vaccination and increasing the interval between booster doses have significant potential to improve the clinical profile of the vaccine and reduce the AVA schedule without compromising safety and immunogenicity [6–9]. Reducing the booster schedule is anticipated to reduce the cumulative occurrence of adverse events (AE) per person-year of vaccine coverage due to the reduced number of doses.
Human inhalation anthrax studies for vaccine efficacy are neither feasible nor ethical. Consequently, an assessment of AVA efficacy in humans is dependent on the COP determined from animal studies together with probability of survival determinations from cross-species bridging using statistical models [12,13]. Studies in NHP have demonstrated that a 3-dose i.m. priming series alone (3-IM) provided high levels of protection (60 to 100%) for up to 4 years against high level exposures to B. anthracis Ames strain spores. Exposure to aerosolized spores stimulated rapid and high anamnestic responses in 3-IM vaccinated NHP, indicative of enduring immunological memory [10]. Statistical modeling of NHP data demonstrated that serum anti-PA IgG concentration at the time of challenge was the most accurate single immune COP for determining probability of survival against inhalation anthrax. In the absence of a last anti-PA IgG measurement concurrent with aerosol exposures in NHP, the month 7 anti-PA IgG and TNA responses to 3-IM priming were suitable alternative COP [11].
Fay et al. [13] identified four aspects of vaccine studies that should be matched as closely as possible between human and animal study data when bridging between genera: vaccine formulation, vaccination schedule, the time of immune response measurement and the time of challenge. The AVRP human and NHP studies both used AVA as the vaccine, and both used a 0, 1, 6 month (3-IM) priming schedule [9,10]. The NHP study design included an intentional imbalance of study group assignments and 3 different challenge times (months 12, 30 and 52), with the higher AVA dilution groups (less antigen per vaccine dose) being challenged early and the lower dilution groups being challenged later in the study (Table 1). In order to most closely match all of these aspects, predicted probabilities of human survival at months 12 and 30 were based on the animals challenged at those time points. Predicted probability of human survival at month 42 was based on animals challenged at month 52, the closest matching time point available. Month 18, the time point at which the 7-IM study group received its first boost and the schedule diverges from the 4-IM and 5-IM groups, does not have a corresponding challenge time point in the NHP data set.
In addition to using the more restrictive matching, and in order to make optimal use of the available data, the last measurements before challenge at months 12, 30 and 52 were combined as the last anti-PA IgG variable and used for NHP logistic regressions. This model was based on the hypothesis that the circulating antibody at the time of challenge is the best correlate and was confirmed as the best single COP by the analysis of Chen et al. [11]. The subsequent regression curve was used for all of the human study groups. The human anti-PA IgG measurement at a given time point was used to predict probability of survival at that time point based on the combined last anti-PA IgG NHP regression curve, simulating exposure at that time point. The analysis in Chen et al. demonstrated that this variable performed with the highest accuracy (AUC = 0.821) within the full NHP cohort [11].
The human serum antibody responses to 3-dose i.m. priming with AVA were analogous to those providing long-term protection against inhalation anthrax in rhesus macaques ([9,10], unpublished data of authors). The cross-species meta-analysis by Fay et al. showed that there are differences between species in the amount of protection per unit of antibody measured by the TNA assay, and this likely applies to the highly correlated anti-PA IgG antibody measurement as well. The difference in protection per unit of anti-PA IgG (μg/mL) between human and NHP is unknown; therefore no inter-species adjustment factor has been introduced in these analyses. The peak and last anti-PA IgG and peak TNA COP from NHP were used accordingly to assess the impact on predicted survival probability in humans receiving reduced booster schedules of AVA in a 43 month study.
The validated anti-PA IgG ELISA and TNA assays have both a lower limit of detection (LLOD) and a lower limit of quantification (LLOQ). The analysis used LLOD for the anti-PA IgG ELISA and the TNA assays. The LLOQ is based on pre-defined performance characteristics of accuracy and precision. Application of the lower LLOD masking limit has the effect of distinguishing between low reactivity samples and negative samples, and subsequently stratifies more NHP data at the low end of the range of responses. Use of the LLOD results in a better curve fit–the AUC increases from 0.748 to 0.821 for the last anti-PA IgG model (unpublished data of authors). Only 10 out of 35 NHP below the LLOD survived (28.6%), while 31 out of 46 NHP between the LLOD and the LLOQ survived (67.4%). These survival rates are statistically significantly different (p < 0.0001), therefore it is more appropriate to stratify the data by application of the LLOD mask. While results below the LLOQ may have less precision, these data clearly indicate correlation with protection.
The antibody level ‘trough’ time points prior to administration of booster vaccinations were of primary interest. The key question is how reductions in serum antibody levels affect predicted survival probabilities. Use of a model that includes a last measurement is required to assess and compare protection at those time points. The lowest observed antibody levels were at month 42 in the 4-IM group, but the predicted survival probability for 4-IM remained high, ranging from 85.2% to 90.2% at month 42 contingent on the model applied. In the 7-IM and 5-IM study groups the trough antibody minimum predicted survival probability over the study duration was also high at 97.2% and 93.3%, respectively.
5.2. Predicted survival probability vs. vaccine effectiveness
There are no empirical data on which to verify a quantitative comparison between the predicted survival probability based on the high-dose NHP challenge and human vaccine effectiveness (VE) in naturally occurring, presumed low-dose exposures. Brachman et al. reported 92.5% VE for a predecessor anthrax vaccine under an annual booster schedule. The study evaluated unvaccinated work-forces (samples size range 148 to 655) with ongoing exposure to B. anthracis spores; annual case rates ranged from 0.6 to 1.8% (mean 1.2%) [5]. In persons categorized as ‘high risk’ in the mill environments the infection rate reached 6.1%. Exposures in those environments were estimated at 21–2100 infectious particles per 8 hour day [18,19]. In agreement with the empirical data, modelling of dose-responses for inhalation anthrax in humans by Toth et al. [20] suggested that the low doses in the mill environments had between 1% and 10% probability of infection. Recognizing that predicted survival probability reported here is derived from a cross-species model rather than empirical observations in humans, it is encouraging to note the similarities between the predictions and the reported VE.
There are clear differences between systemic anthrax and the pathogenesis of other vaccine preventable diseases. Nonetheless, the predicted survival probability for the reduced AVA schedules is comparable to VE for several vaccines currently in use. For example, the cholera vaccine has been estimated as having 62% efficacy [21], a large scale rotavirus vaccine trial found an efficacy of 72% [22], and the quadrivalent bacterial meningitis vaccine has a VE estimated at 80–85% [23].
In the quantitative models of the dose-response and time course of inhalation anthrax in humans evaluated by Toth et al. the infection probability approached the 100% asymptote at a single-point total exposure threshold of approximately 1.0 × 105 spores [19]. The logistic curves generated from the current NHP data were from high-dose aerosol challenges (median 504 × LD50 equivalents; 2.8 × 107 spores) [10], over 4 orders of magnitude higher than the mill exposures from which human VE was calculated and over 2 orders of magnitude higher than the 1.0 × 105 spores threshold of Toth et al. [19]. Based on these analyses we propose that the risk of infection from low dose exposures in the environments in which pre-exposure use of AVA is recommended is significantly less than the risk from the extremely high doses in the animal challenge model. COP modelling in NHP high-dose exposures models may therefore overestimate the antibody levels required for protection against naturally occurring anthrax in a pre-exposure prophylaxis scenario. Consequently the predicted probabilities of human survival derived from these data are likely to be significant underestimates of VE.
6. Conclusions
The most accurate correlates of protection identified from non-human primate challenge studies have been used to assess the levels of protection afforded by reduced booster schedules of AVA. Of primary interest are the trough antibody time points just before boost. All schedules provided high probability of survival estimates in humans at all time-points with all models. The lowest estimates were 85.1%–90.2% predicted survival in the 4-IM schedule at month 42; 3 years post-priming with no intervening boosters. The survival predictions match well with the observed annual booster schedule vaccine efficacy of 92.5% [5]. In agreement with previous studies, increasing the interval between boosters generated consistently higher anamnestic responses [6–9]. These enhanced responses to the reduced schedules, together with the sustained predicted survival probability, clearly demonstrate the feasibility of simplifying the AVA booster schedule. We conclude based on these data and analyses that a 3-IM priming series with a 3-year booster (4-IM), or with an initial booster at 1 year and a subsequent 2-year interval (5-IM) are both viable alternatives to the current AVA prime-boost schedule for pre-exposure prophylaxis against anthrax.
Acknowledgments
The authors would like to thank the Anthrax Vaccine Research Program (AVRP) Working Group members: Baylor College of Medicine: W. Keitel, H. El Sahly, N. Bond, D. Nino, C. Rangel, C. Tajonera. WRAIR: J. Babcock, S. Cicatelli, R. Newcomer, R. Nielsen. Mayo Clinic: G. A. Poland, R. M. Jacobson, P. Targonski, I. Ovsyannikova, N. Pinsky. Emory University School of Medicine: H. Keyserling, J. Hilinski, M. Leonard, P. Anderson, V. Grimes, K. Luehrs, P. Newsome, J. Skvarich, K. Stephens. University of Alabama, Birmingham: S. D. Parker, M. Mulligan, F. Johnson, J. Moody, L. Williams, F. Smith. CDC Microbial Pathogenesis & Immune Response (MPIR) Laboratory: V.A. Semenova, H. Li, H. Dababneh, S. K. Martin, D. Boulay, M. Brawner, N. Brown, J. Caba, S. Crenshaw, L. Cronin, R. Desai, L. Foster, J. Lewis, F. Lyde, A. Milton, H. Noland, N. Patel, D. Schmidt, S. Shields, D. Smith, E. Steward-Clark, R. Thompson, J. Walls. CDC, Division of Bacterial Diseases: B. D. Plikaytis, N. Marano, N. E. Messonnier, S. W. Martin, W. Holt, J. Jarrell, F. David, S. Shah; M. McNeil, J. Stamper, J. Wheeling, S. Mohammed.
Ligong Chen and Shannon Dalton were funded by the Atlanta Research and Education Foundation (AREF) through the Department of Veterans Health Administration, Office of Research and Development, Atlanta, GA.
Funding
This study was funded by the Centers for Disease Control and Prevention. Battelle was funded under DHHS CDC contract number 200-2000-10065.
Abbreviations:
- AE
adverse events
- AUC
area under the receiving operator characteristic curve
- AVA
Anthrax Vaccine Adsorbed
- AVRP
Anthrax Vaccine Research Program
- CDC
Centers for Disease Control and Prevention
- COP
correlates of protection
- ED50
effective dilution for 50% neutralization
- FDA
Food and Drug Administration
- GCP
Good Clinical Practices
- IACUC
Institutional Animal Care and Use Committee
- i.m.
intramuscular
- IND
Investigational New Drug
- LLOD
lower limit of detection
- LLOQ
lower limit of quantification
- NHP
non-human primate
- PA
protective antigen
- s.c.
subcutaneous
- TNA
toxin neutralization activity
- VE
vaccine effectiveness
Footnotes
Publisher's Disclaimer: Disclaimer
Publisher's Disclaimer: The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention.
Conflict of interest statement
The authors declare that there are no financial, institutional or other relationships that might lead to bias or a conflict of interest.
References
- [1].Anthrax Vaccine Adsorbed United States patent US 3208909. September 28, 1965.
- [2].Turnbull PC. Anthrax vaccines: past, present and future. Vaccine 1991;9:533–9, 10.1016/0264-410X(91)90237-Z. [DOI] [PubMed] [Google Scholar]
- [3].Wright JG, Quinn CP, Shadomy S, Messonnier N. Use of anthrax vaccine in the United States: recommendations of the Advisory Committee on Immunization Practices (ACIP), 2009. Centers for Disease Control and Prevention (CDC). MMWR Recomm Rep 2010;59(RR06):1–30 (PMID: ). [PubMed] [Google Scholar]
- [4].Saile E, Quinn CP. Bacillus anthracis and Anthrax In: Bergman N, editor. Bacillus anthracis and anthrax. New Jersey: Wiley-Blackwell; 2010. p. 269–93. [Google Scholar]
- [5].Brachman PS, Gold H, Plotkin SA, Fekety FR, Werrin M, sIngraham NR. Field evaluation of a human anthrax vaccine. Am J Public Health 1962;52:632–45, 10.2105/AJPH.52.4.632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Pittman PR, Kim-Ahn G, Pifat DY, Coonan K, Gibbs P, Little S, et al. Anthrax vaccine: immunogenicity and safety of a dose-reduction, route-change comparison study in humans. Vaccine 2002;20:1412–20, 10.1016/S0264-410X(01)00462-5. [DOI] [PubMed] [Google Scholar]
- [7].Pittman PR, Fisher D, Quinn X, Schmader T, Barrera-Oro JG. Effect of delayed anthrax vaccine dose on Bacillus anthracis protective antigen IgG response and lethal toxin neutralization activity. Vaccine 2013;31:5009–14, 10.1016/j.vaccine.2013.08.086. [DOI] [PubMed] [Google Scholar]
- [8].Pittman PR, Mangiafico JA, Rossi CA, Cannon TL, Gibbs PH, Parker GW, et al. Anthrax vaccine: increasing intervals between the first two doses enhances antibody response in humans. Vaccine 2000;19:213–6, 10.1016/S0264-410X(00)00174-2. [DOI] [PubMed] [Google Scholar]
- [9].Wright JG, Plikaytis BD, Rose CE, Parker SD, Babcock J, Keitel W, et al. Effect of reduced dose schedules and intramuscular injection of anthrax vaccine adsorbed on immunological response and safety profile: a randomized trial. Vaccine 2014;32:1019–28, 10.1016/j.vaccine.2013.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Quinn CP, Sabourin CL, Niemuth NA, Li H, Semenova VA, Rudge TL, et al. A three-dose intramuscular injection schedule of anthrax vaccine adsorbed generates sustained humoral and cellular immune responses to protective antigen and provides long-term protection against inhalation anthrax in rhesus macaques. Clin Vaccine Immunol 2012;19:1730–45, 10.1128/CV1.00324-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Chen L, Schiffer JM, Dalton S, Sabourin CL, Niemuth NA, Madigan D, et al. Comprehensive analysis and selection of anthrax vaccine immune correlates of protection in rhesus macaques. Clin Vaccine Immunol 2014;21:1512–20, 10.1128/CV1.00469-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Gronvall GK, Trent D, Borio L, Brey R, Nagao L. The FDA animal efficacy rule and biodefense. Nat Biotechnol 2007;25:1084–7, 10.1038/nbt1007-1084. [DOI] [PubMed] [Google Scholar]
- [13].Fay MP, Follmann DA, Lynn F, Schiffer JM, Stark GV, Kohberger R, et al. Anthrax vaccine-induced antibodies provide cross-species prediction of survival to aerosol challenge. Sci Transl Med 2012;4:151ra126, 10.1126/scitranslmed.3004073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kohberger RC, Jemiolo D, Noriega F. Prediction of pertussis vaccine efficacy using a correlates of protection model. Vaccine 2008;26:3516–21, 10.1016/j.vaccine.2008.04.016. [DOI] [PubMed] [Google Scholar]
- [15].Efron B, Tibshirani J. An introduction to the Bootstrap. New York: Chapman and Hall; 1993. [Google Scholar]
- [16].Semenova VA, Schiffer J, Steward-Clark E, Soroka S, Schmidt DS, Brawner MM, et al. Validation and long term performance characteristics of a quantitative enzyme linked immunosorbent assay (ELISA) for human anti-Pa IgG. J Immunol Methods 2012;376:97–107, 10.1016/j.jim.2011.12.002. [DOI] [PubMed] [Google Scholar]
- [17].Li H, Soroka SD, Taylor TH, Stamey KL, Stinson KW, Freeman AE, et al. Standardized, mathematical model-based and validated in vitro analysis of anthrax lethal toxin neutralization. J Immunol Methods 2008;333:89–106, 10.1016/jjim.2008.01.007. [DOI] [PubMed] [Google Scholar]
- [18].Brachman PS, Plotkin SA, Bumford FH, Atchison MM. An epidemic of inhalation anthrax: the first in the twentieth century. Am J Hyg 1960;72:6–23 (PMID: ). [DOI] [PubMed] [Google Scholar]
- [19].Dahlgren CM, Buchanan LM, Decker HM, Freed SW, Phillips CR, Brachman PS. Bacillus anthracis aerosol in goat hair processing mills. Am J Hyg 1960;72:2–31 (PMID: ). [DOI] [PubMed] [Google Scholar]
- [20].Toth DJA, Gundlapalli AV, Schell WA, Bulmahn K, Walton TE, Woods CW, et al. Quantitative models of the dose response and time course of inhalational anthrax in humans. PLOS Pathog 2013;9(8):1003555 10.1371/journal.ppat.1003555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Masuet Aumatell C, Ramon Torrell JM, Zuckerman JN. Review of oral cholera vaccines: efficacy in young children. Infect Drug Resist 2011;4:155–60, 10.2147/IDR.S10339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Li RC, Huang T, Li Y, Luo D, Tao J, Fu B, et al. Human Rotavirus Vaccine (Rix4414) efficacy in the first two years of life: a randomized, placebo-controlled trial in China. Hum Vaccin Immunother 2013;10:11–8, 10.4161/hv.26319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].MacNeil JR, Cohn AC, Zell ER, Schmink S, Miller E, Clark T, et al. Active Bacterial Core surveillance (ABCs) Team and MeningNet Surveillance Partners. Early estimate of the effectiveness of quadrivalent meningococcal conjugate vaccine. Pediatr Infect Dis J 2011;30:451–5, 10.1097/INF.0b013e31820a8b3c. [DOI] [PubMed] [Google Scholar]