

Abstract
Tuberculous meningitis (TBM) is a devastating form of tuberculosis (TB), and key TB antimicrobials, including rifampin, have restricted brain penetration. A lack of reliable data on intralesional drug biodistribution in infected tissues has limited pharmacokinetic (PK) modeling efforts to optimize TBM treatments. Current methods to measure intralesional drug distribution rely on tissue resection, which is difficult in humans and generally limited to a single time point even in animals. In this study, we developed a multidrug treatment model in rabbits with experi mentally induced TBM and performed serial noninvasive dynamic 11C-rifampin positron emission tomography (PET) over 6 weeks. Area under the curve brain/plasma ratios were calculated using PET and correlated with postmortem mass spectrometry. We demonstrate that rifampin penetration into infected brain lesions is limited, spa tially heterogeneous, and decreases rapidly as early as 2 weeks into treatment. Moreover, rifampin concentrations in the cerebrospinal fluid did not correlate well with those in the brain lesions. First-in-human 11C-rifampin PET performed in a patient with TBM confirmed these findings. PK modeling predicted that rifampin doses (≥30 mg/kg) were required to achieve adequate intralesional concentrations in young children with TBM. These data demonstrate the proof of concept of PET as a clinically translatable tool to noninvasively measure intralesional antimicrobial distribution in infected tissues.
INTRODUCTION
Tuberculous meningitis (TBM) can be considered the most destructive form of tuberculosis (TB), especially among HIV-infected individuals and young children (1, 2). Not only is it difficult to diagnose and treat, but it is also associated with significant morbidity and mortality even with an appropriate and prolonged (at least months) multidrug treatment (3). The central nervous system (CNS) has multiple compartments [e.g., brain parenchyma and cerebrospinal fluid (CSF)], which are separated from the systemic circulation by the blood-brain barrier (BBB) and blood-CSF barrier (BCSFB) that limit penetration of many antimicrobials. Despite the knowledge that key antimicrobials, including rifampin, do not penetrate into the brain adequately, current TBM treatments are not optimized and are generally based on those used for pulmonary TB (2). The lack of animal models and the difficulties and dangers of sampling brain tissues or performing serial lumbar punctures in patients with TBM have been significant barriers for obtaining reliable data on intralesional concentrations and temporal changes in drug biodistribution. Therefore, knowledge about antimicrobial concentrations in the infected brain tissue is needed to support pharmaco-kinetic (PK) modeling and optimize TBM treatment.
Rifampin is an essential first-line TB drug, with potent sterilizing activity against Mycobacterium tuberculosis. Studies have demonstrated that rifamycins have dose-dependent activity, with the area under the curve (AUC) being a critical pharmacodynamic (PD) parameter required for bacterial killing (4). Moreover, while 10 mg/kg per day of rifampin for adults is currently recommended, daily doses of up to 35 mg/kg of rifampin are well tolerated in adults with pulmonary TB (5) and 50 mg/kg of rifampin was demonstrated to have greater early bactericidal activity in a recent clinical trial simulation (6). Unfortunately, rifampin has limited penetration into the CNS. However, dose optimization could potentially overcome this barrier, as demonstrated by Ruslami et al. (7), where intensified treatment in patients with TBM with high-dose intravenous rifampin during the first 2 weeks of treatment substantially decreased mortality.
We have previously reported the use of 11C-rifampin, a radio-labeled analog of rifampin, for positron emission tomography (PET) in live M. tuberculosis–infected mice to noninvasively assess rifampin biodistribution in the lung (8) using specially designed sealed bio-containment devices (9). In the current study, we developed a multi-drug TB treatment model in rabbits with experimentally induced TBM (10) and performed dynamic 11C-rifampin bioimaging before and during treatment. Imaging data were correlated with postmortem analysis including rifampin concentration by mass spectrometry (MS), and an integrated PK–brain biodistribution model was used to pre-dict brain biodistribution and the changes during treatment. In addition, we performed first-in-human 11C-rifampin bioimaging in a 24-year-old patient with TBM and demonstrated proof of concept and feasibility of PET to noninvasively assess the spatial biodistribution of a key antimicrobial in privileged compartments.
RESULTS
Multidrug treatment
A multidrug treatment model in rabbits with TBM using direct inoculation of M. tuberculosis via the subarachnoid route (10) was developed. Oral treatment with rifampin (30 mg/kg; equipotent to 10 mg/kg in humans) and isoniazid (50 mg/kg; equipotent to ~16 mg/kg in humans) (11) was initiated 3 weeks after infection (Fig. 1A). Cohorts of similarly infected rabbits were either imaged using dynamic PET for 30 min immediately after an intravenous dose of 11C-rifampin or necropsied 30 min after an intravenous dose of unlabeled rifampin (30 mg/kg) for post-mortem MS. Together, the PET and MS data were used for PK modeling (Fig. 1B).
Fig. 1. Experimental timeline and schematic.
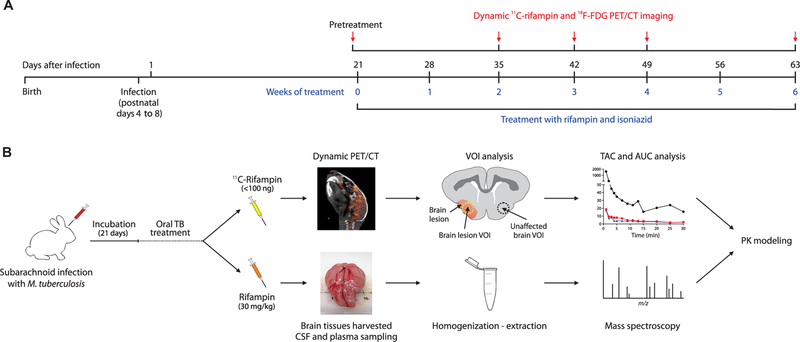
(A) Timeline of infection, antimicrobial treatment, and study procedures in rabbits. (B) 11C-rifampin for PET analysis (top) with three-dimensional (3D) volumes of interest (VOIs) drawn to measure the PET signal to generate time-activity curves (TACs) used to calculate AUC over 30 min (AUC0–30), or intravenous rifampin (bottom) for MS analysis. These results were used to develop a PK model to predict rifampin exposures in brain tissues. m/z, mass/charge ratio.
2-[18F]-fluorodeoxyglucose (18F-FDG) PET, which is a nonspecific but sensitive marker of inflammation, was used in conjunction with computed tomography (CT) to visualize brain lesions (BLs) (Fig. 2A), demonstrating higher uptake in BLs compared with the contralateral unaffected brain (UB) (P < 0.008; Fig. 2B). BLs, shown on postmortem examination (Fig. 2, C to E), were heterogeneous and located in several regions of the brain, including along the meninges and deep within the parenchyma. Histopathologic examination of these lesions demonstrated areas of dense cellularity and few bacteria in the lesions (Fig. 2F). Multidrug treatment led to a progressive decrease in the bacterial burden [colony-forming units (CFU)] (Fig. 2G and fig. S1) and improvement in CSF protein and glucose (Fig. 2, H and I) over several weeks of treatment.
Fig. 2. Multidrug treatment in rabbits with experimentally induced TBM.
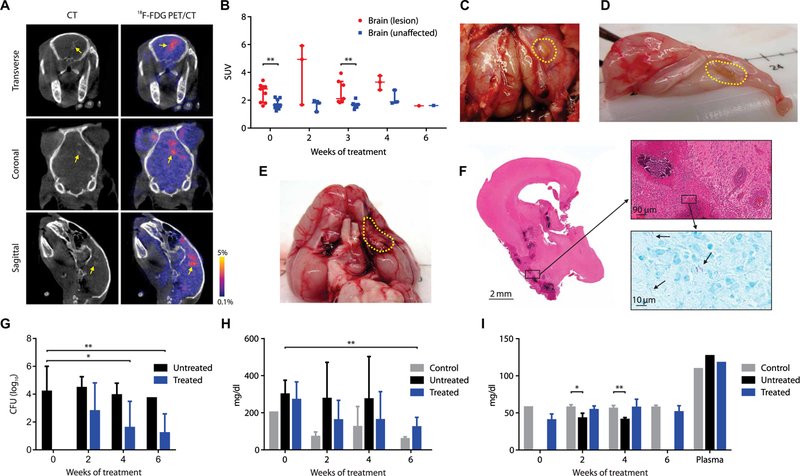
(A) Transverse, coronal, and sagittal images from a representative infected rabbit pretreat-ment (week 0). BL seen on CT (left panels, yellow arrow) and 18F-FDG PET signal (right panels). (B) Standardized uptake values (SUVs) for 18F-FDG PET during treatment (data represent three to nine VOIs from two to six animals at each time point except week 6, where one VOI from a single animal is shown). (C to E) Gross pathology demonstrating BLs (yellow dotted outline) from animals 7 to 9 weeks after infection. (F) Hematoxylin and eosin (magnification, ×1; top, ×20) and acid-fast (bottom inset, ×100) staining of brain tissues [from (E)]. (G) CFU in the brain tissues are shown on a logarithmic scale. At least five animals were used for each group and time point except at weeks 4 (n = 4; treated group) and 6 (n = 1; untreated group). (H) CSF total protein and (I) glucose concentration over the treatment duration are shown. Controls refer to uninfected animals. At least three animals were used for each group and time point except at week 0 for protein and glucose assays in the control group ( n = 2) and plasma glucose assays ( n = 1 per group). CFU and CSF data are presented as means ± SD with statistical comparisons performed using two-tailed Student’s t test. Imaging data are presented as median ± IQR with statistical comparisons performed using two-tailed Mann-Whitney U test. Statistical significance is represented by *P < 0.05 or **P < 0.01.
Spatial heterogeneity of rifampin penetration into the brain and temporal changes during treatment
Dynamic 11C-rifampin PET was performed over 30 min after an intravenous injection, and rifampin rapidly localized to the liver. The paucity of 11C-rifampin signal in the brain suggested limited penetration (Fig. 3A). The 3D VOIs were drawn around TB lesions, and PET activity was measured at many time points over 30 min to generate TACs. Compared with pretreatment (week 0), there was a de-crease in the 11C-rifampin exposures to the infected BL after the initiation of TB treatment (Fig. 3B) with a similar decrease in the contralateral UB (fig. S2). A 3D reconstruction of the 11C-rifampin PET in the infected rabbit is shown (movie S1).
Fig. 3. Dynamic 11C-rifampin PET and rifampin MS.

(A) 3D reconstruction (left) and CT (top), 18 F-FDG PET/CT (middle), and 11C-rifampin PET/CT (bottom) images of the sagittal, coronal, and transverse sections of the brain of a representative infected rabbit at 4 weeks of treatment. Outline of the brain compartment (dotted yellow line), BL (yellow arrow), and site of injection (white arrow) are shown. (B) TACs demonstrating 11C-rifampin exposures (Bq/ml) over 30 min in a BL from one animal imaged pretreatment (week 0) and at 4 and 6 weeks into treatment (corrected for weight and tracer dose). (C) 11C-rifampin brain/plasma AUC0–30 ratios in BLs [data represent three to nine VOIs from two to six ani-mals at each time point except week 6, where one VOI from a single animal is shown]. (D) Rifampin concentration ( g/ml) and (E) tissue/plasma ratios (tissue30min ratio) from MS in postmortem samples at 30 min after rifampin intravenous dose at weeks 0 (n = 3 rabbits), 2 (n = 2), 3 (n = 1), and 4 (n = 3). (F) 11C-rifampin TAC from the same representative rabbit [from (A)]. (G) Rifampin concentration from all infected rabbits sampled (n = 9). Median and IQR are shown. *P < 0.05, **P < 0.01, and ***P < 0.001 by Wilcoxon signed rank test and two-tailed Mann-Whitney U test.
11C-rifampin TACs were used to calculate the AUC over 30 min (AUC0–30) for each VOI and brain/plasma AUC0–30 ratios calculated for all imaged rabbits to measure the penetration of 11C-rifampin over the duration of treatment (Fig. 3C). Penetration of 11C-rifampin into BLs significantly decreased (from 32 to 11%; P = 0.009) at 2 weeks after initiation of treatment and remained low with similar decreases also noted for the contralateral UB (from 29 to 9%; P = 0.009; fig. S3).
To validate the PET data, we also measured rifampin and 25-desacetyl rifampin (active metabolite of rifampin) concentrations using postmortem analyses. Since 11C-rifampin (mass administered <100 ng) was undetectable by standard MS, tissues were obtained after an intravenous dose of rifampin (30 mg/kg; equipotent to 10 mg/kg in humans). 25-Desacetyl rifampin levels were undetectable in CSF and brain tissue and less than 0.22% in the plasma (table S1). To mirror the PK profile of 11C-rifampin injected for PET, blood (plasma) was sampled 10, 20, and 30 min after the dose, although brain tissue and CSF (intracisternal) were only sampled at the time of necropsy (30 min). Unexpectedly, rifampin concentration in the CSF remained significantly lower than those in the BLs (P < 0.05) and did not correlate well with the changes in brain concentration over the duration of treatment (Fig. 3, D and E). Absolute rifampin concentrations from the contralateral UB tissues are shown (fig. S4).
11C-rifampin PET exposures were spatially heterogeneous with inter-subject and intrasubject variations in 11C-rifampin brain/plasma AUC0–30 ratios in infected rabbits (Fig. 3F and table S2). Rifampin concentrations measured postmortem by MS in plasma, BL, and UB were 26.19 g/ml [interquartile range (IQR), 23.07 to 30.36], 1.39 g/ml (IQR, 1.07 to 1.79), and 0.86 g/ml (IQR, 0.81 to 1.40), respectively, at 30 min after the intravenous (therapeutic) dose of rifampin (Fig. 3G). Median rifampin concentration was not higher in the BL compared with the UB (P = 0.39). The CSF concentrations (0.74 g/ml; IQR, 0.46 to 0.88) in the same animals were significantly lower than those in the BLs (P = 0.014). In the uninfected control (sham) animals, the CSF concentration was also lower than the uninfected brain (fig. S5). The 11C-rifampin brain/ plasma AUC0–30 ratios in the uninfected control animals were signifi-cantly lower (P = 0.002) than those in the infected rabbits (fig. S6).
Dynamic 11C-rifampin PET in a patient with TBM
First-in-human 11C-rifampin PET was performed in a 24-year-old female with TBM 2 weeks into TBM treatment (Fig. 4). Brain magnetic resonance imaging (MRI) demonstrated focal areas of white matter and cortical region abnormalities (Fig. 4A, left). Post-gadolinium sequence showed multifocal nodular and meningeal enhancement consistent with active neuroinflammation (Fig. 4A, right). 11C-rifampin PET was safe and well tolerated, and, similar to what was observed in rabbits, limited penetration of rifampin was noted into the brain (Fig. 4, B and C). After completion of imaging, the patient also re-ceived an intravenous dose of rifampin (600 mg over a 30-min infusion in lieu of the daily morning oral dose), and blood samples were collected to determine rifampin and 25-desacetyl rifampin concen-trations (Fig. 4D). Concentrations of 25-desacetyl rifampin remained low (<6%) over this 30-min period.
Fig. 4. Dynamic 11C-rifampin PET in a patient with TBM.
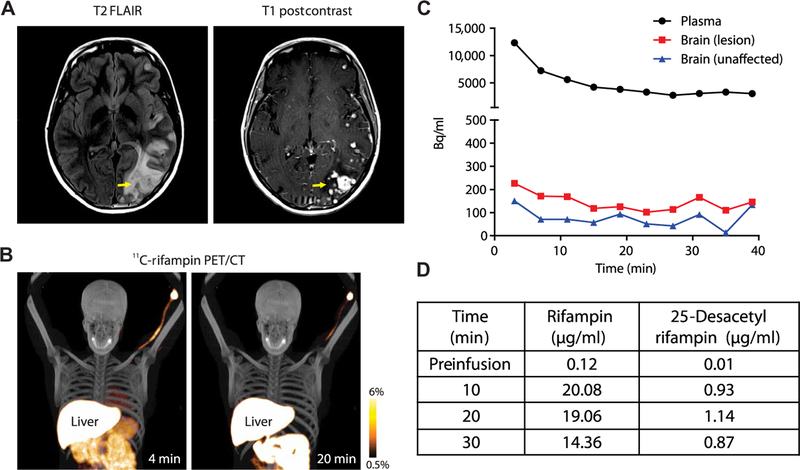
(A) Brain MRI fluid-attenuated inversion recovery (FLAIR) and post-gadolinium sequences. (B) Dynamic 11C-rifampin PET/CT fusion images and (C) TACs demonstrating 11C-rifampin exposures (Bq/ml) over 40 min in the plasma and brain (lesion and unaffected). (D) Mass spectrometry results for plasma rifampin and 25-desacetyl rifampin in this patient.
PK modeling
A previously published PK model for rifampin after oral administration in patients with pulmonary TB (12), where the PK parameters were allometrically scaled down using body weight, was able to predict both rifampin and 11C-rifampin plasma concentrations in rabbits across the 6 weeks of treatment. The model schematics are shown (Fig. 5A), with the parameter estimates listed in table S3.
Fig. 5. Plasma 11C-rifampin and rifampin have sim-ilar PK characteristics.
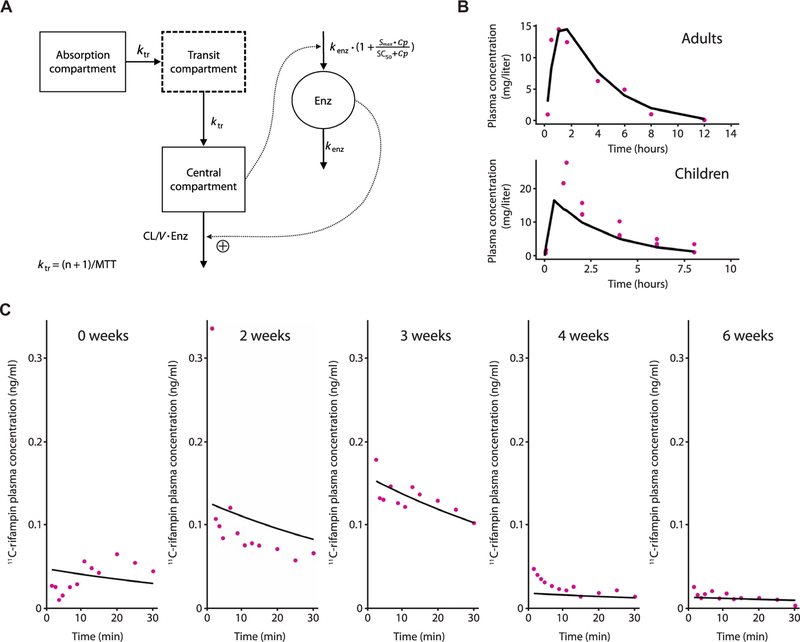
(A) Schematic representation of the PK model of oral rifampin from (12). Mean transit time (MTT), clearance (CL), volume of distribution (V), maximal increase in the enzyme production rate (Smax ), rifampin concentration at which half the Smax is reached (SC50), and rate constant for first-order degradation of the enzyme pool (k enz). (B) Observed (purple dots) and model-predicted (black lines) rifampin plasma concentrations using digitized data from literature (table S4). (C) Ob-served (purple dots) and individual model predicted (black line) 11C-rifampin activity in plasma in represent ative TBM rabbits during treatment.
This model successfully predicted the mean rifampin plasma concentrations in experimentally infected rabbits and the patient with TBM. External validation using literature-based PK data (table S4) demonstrated that the mean prediction of plasma rifampin of this chosen model captures the magnitude and slope of the published data (Fig. 5B). Furthermore, the allometrically scaled PK model successfully fit 11C-rifampin plasma activity in rabbits with TBM during TB treatment (Fig. 5C). Although the observed profiles differ in magnitude due to different 11C-rifampin doses and body weights of representativerabbits, the model captured the slopes. Last, comparison of rifampin measured by MS and dose-normalized 11C-rifampin PET signal in the plasma of a cohort of 10 patients with TB [9 with pulmonary TB and 1 with TBM (shown in Fig. 4)] also demonstrated similar PK characteristics manifested by their slopes (fig. S7).
The observed 11C-rifampin concentrations were used to develop the PK–brain biodistribution model (Fig. 6 and table S5) and describe the dis-tribution of 11C-rifampin into BLs (Fig. 6B) and UB in rabbits (fig. S8). The PK–brain biodistri-bution model was then used to project rifampin exposures [AUC0–24h and peak concentration (Cmax)] in children. Because of the lower rifam-pin exposures reported in children, oral admin-istration was simulated using 50% bioavailability (13, 14). The probability of target attainment in children aged 2 to 11 years for different rifampin doses is shown in Fig. 7. Although the rifampin tissue PD parameters required for optimal bac-terial killing in TBM are not well defined, CSF Cmax has often been quoted as such (7). Of the 1000 simulated subjects, only oral doses of ≥30 mg/kg provided ≥90% probability of target attainment. Thresholds to obtain probability of attainment were defined as Cmax >4 μg/ml (bactericidal concentration (15), assuming M. tuberculosis minimum inhibitory concentration of 1 μg/ml) in the BLs at the initiation of treatment and sustained Cmax >1 μg/ml during the intensive phase of TBM treatment. The projected mean rifampin AUC0–24h and Cmax in BLs in a 2-year-old child (weighing 15 kg) at different rifampin doses are presented in table S6. The rifampin concentrations were also predicted in the patient with TBM imaged in this study (table S7), which demonstrated that by 2 weeks of treatment, both daily oral 600 and 900 mg had similarly low BL Cmax (<1 μg/ml).
Fig. 6. PK–brain biodistribution rifampin model schematic and its goodness of fit in BLs.
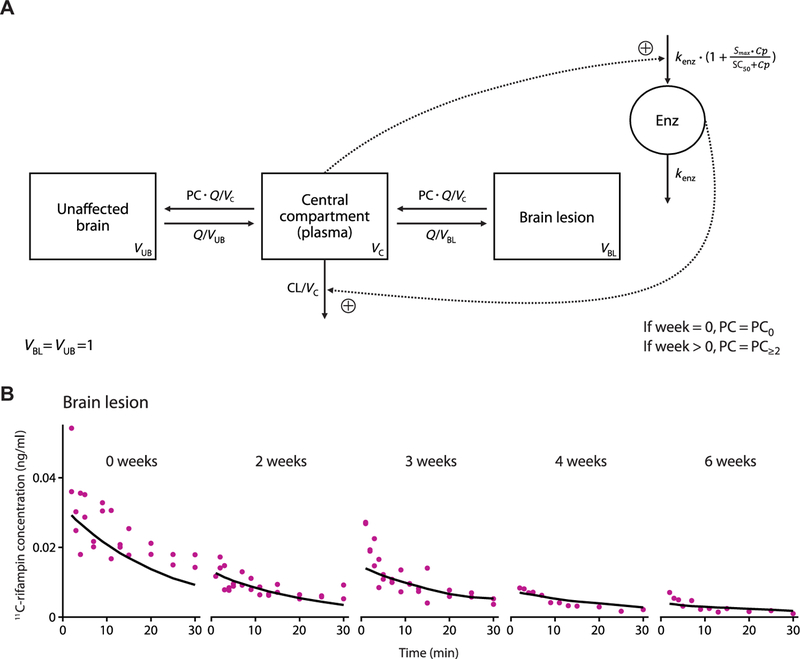
(A) Schematic of the developed PK–brain biodistribution model describing the distribution of rifampin into UB and BLs. Parameters include clearance (CL) from the central compartment, volume of distribution (VC) of the central com-partment, clearance of unbound rifampin from plasma to UB or from plasma to BLs (Q), volume of distribution of UB (VUB), volume of distribution of BLs (VBL), partition/penetration coefficient (PC), partition/penetration coefficient at the start of treatment [week 0 (PC0)], partition/penetration coefficient beyond 2 weeks of treatment (PC≥2), MTT, maximal increase in the enzyme produc-tion rate (Smax), rifampin concentration at which half the Smax is reached (SC50), and rate constant for first-order degradation of the enzyme pool (kenz). (B) Observed (purple dots) and average PK model predicted (black lines) 11C-rifampin expo-sures in BLs in representative rabbits.
Fig. 7. Probability of target attainment for different rifampin doses in children.
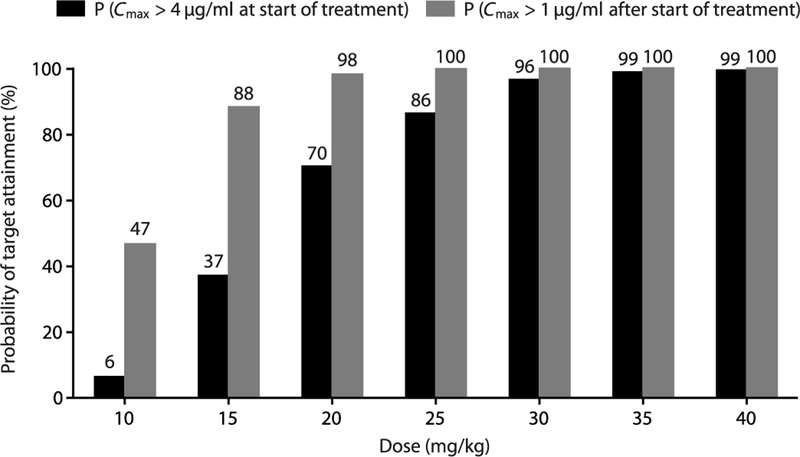
A population of 1000 children (2 to 11 years) between the World Health Organization (WHO) 25th and 75th percentile weight distribution was used to perform Monte Carlo simulations based on the developed PK–brain biodistribution model. Rifampin exposures in the lesions (Cmax) were then used to calculate the probability of target attainment, defined as the percentage that achieved rifampin Cmax values in the lesions above 4 g/ml at the start of treatment or above 1 g/ml subsequently.
DISCUSSION
Historically, dosing recommendations for TB drugs have been based on blood concentrations and traditional measures of efficacy such as bactericidal activity and relapse free cure. However, data on drug concentrations achieved at the target infection sites are still limited, restricting efforts toward optimizing antimicrobial treatments (16). Serious consequences of inadequate drug concentration in target tissues include treatment failure and selection pressure for antimicrobial-resistant organisms (17). Similarly, higher than necessary concentrations of some drugs can be associated with serious toxicities (18). Therefore, a growing number of studies and the U.S. Food and Drug Administration (FDA) support the importance of monitoring drug concentrations in infected tissues (19). Preclinical studies have been useful to model antimicrobial PK in pulmonary TB (20) but may be fundamental for devastating diseases like TBM (3), where infection sites are separated from the systemic circulation.
For deep-seated and localized infections, clinical samples such as blood or CSF often fail to correlate with the pathological changes at the infection sites. Moreover, direct measurement cannot be achieved in humans except in extreme circumstances when tissue biopsy/resection is planned for clinical reasons. Advanced techniques such as matrix-assisted laser desorption ionization (MALDI) MS can provide detailed spatial distribution of TB drugs but rely on invasive resection of tissue (21). However, molecular imaging techniques such as PET can quantify the distribution of radiolabeled compounds with high sensitivity (nano-to picomolar) throughout the body (22). Moreover, dynamic PET can yield continuous data at multiple time points to simultaneously determine AUC in multiple compartments of interest (brain, plasma, infected site, etc.). Furthermore, noninvasive imaging permits longitudinal assessments in the same individual over the course of the disease (e.g., duration of treatment), which is a fundamental advantage over current tools that are often limited to one time point (23). Although PET may not be widely available in the developing world, it can still be performed at most major referral centers and thus enable small phase 0 studies (24), which could provide detailed human data of new or repurposed antimicrobials into diseased tissues without the need of invasive procedures.
In the current study, dynamic PET allowed us to determine the spatial biodistribution and temporal changes in drug exposures over the duration of TBM treatment. Consistent with previous data (8, 25), rifampin penetration into the brain was substantially restricted. However, we also demonstrate that rifampin penetration into the brain decreased substantially by 2 weeks of TB treatment. This finding is important because it suggests that increasing the rifampin dose during the initial phase of treatment could be effective in achieving higher intralesional concentrations in infected BL. In addition, since rifampin is a potent sterilizing agent that is administered for the full 12-month duration of TBM treatment, it is possible that only subtherapeutic intralesional concentrations are achieved after the initial 8-week intensive treatment. The decreased rifampin penetration in the brain with TB treatment is most likely due to progressive repair of the leaky BBB (26), but induction of P-glycoprotein efflux pumps (27) could also have contributed to this effect.
In addition to the intersubject variability, rifampin exposures were also spatially heterogeneous and variable in the BLs within the same host. This is consistent with the concept that different pathological states occur simultaneously in the same host (28), and getting sufficient drug to these different lesions would be essential for optimizing TBM treatment. Rifampin concentrations in BLs were significantly higher than those in the CSF and did not correlate well with the changes in brain concentration over the duration of treatment. While CSF can be sampled more easily (versus brain biopsy) and, therefore, is generally the current standard for assessing antimicrobial penetration into the brain, the finding that the CSF does not necessarily correlate with the concentrations in the infected BLs is an important one. Rohlwink et al. (29) have demonstrated that biochemical characteristics of the CSF obtained via lumbar puncture were different from that obtained from the ventricles in children with TBM, possibly because of barriers that affect the uniform circulation of CSF, reinforcing the need for measuring drug concentrations at the infection site. Using MALDI MS, Prideaux et al. (21) have demonstrated that after administration of multidoses, rifampin accumulates in the necrotic lung tissues of M. tuberculosis–infected rabbits and patients with pulmonary TB. However, MS results from our TBM study did not demonstrate rifampin accumulation in the brain or CSF. Last, we also performed 11C-rifampin in a patient with TBM who had received 2 weeks of TB treatment, including corticosteroids. Consistent with the data from the rabbit model, penetration of rifampin into BLs was significantly restricted.
Imaging data were used to develop an integrated PK–brain biodistribution model scaled to project BL exposures in children at varying doses. External validation was performed on available rifampin PK data in adult and pediatric populations with pulmonary TB re-ceiving varying doses, which demonstrated that this chosen PK mod-el captured the magnitude and slope of the data. Current guidelines for TBM in children suggest an oral dose of 10 to 20 mg/kg (30); however, our model-based Monte Carlo simulations suggest that only oral doses (≥30 mg/kg) would provide ≥90% probability of tar-get attainment. These results are similar to pediatric dosing recommendations from a population PK/PD model using plasma and CSF data from adults and children (31). Similarly, projected BL exposures in the patient with TBM demonstrated that by 2 weeks of treatment, the BL Cmax was well below target. However, our data did not incorporate the concomitant use of corticosteroids, which, by reducing the permeability of the BBB (32), could potentially limit even further the penetration of rifampin into the BLs, thereby reducing Cmax.
Although Ruslami et al. demonstrated that high-dose intravenous rifampin (13 mg/kg) during the first 2 weeks of treatment in adult patients with TBM substantially decreased mortality (7), a subsequent larger trial reported by Heemskerk et al. (33) using oral rifampin (15 mg/kg) for the first 8 weeks of treatment did not show this benefit. Although differences in the study populations (more patients with advanced TBM in the Ruslami et al. trial) and different rifampin drug exposures due to oral versus intravenous dosing may have contributed to the different results, Heemskerk et al. have also pos-tulated that clinical outcomes may be more closely associated with intracerebral inflammatory response rather than rapid bacterial killing. These questions will also need to be addressed in preclinical studies and subsequent clinical trials.
Our study has some limitations. The rabbit model in this study uses direct inoculation, which does not recapitulate the natural aerosol route of infection, but it reliably produces the desired TBM infection with key neuropathological features of human disease. Given high protein binding (34, 35), rifampin concentrations measured by MS (representing bound and unbound rifampin) may overrepresent the active proportion of rifampin in plasma. Total and unbound rifampin should be in equilibrium, but only the un-bound drug is active and diffuses into tissues, and this was therefore accounted for in the modeling. Because of the high specific activity of 11C-rifampin, only nanogram quantities of rifampin are typically administered to each subject in PET studies with no significant therapeutic effects. Although it has been hypothesized that extra polation of data derived from microdosing studies may poorly predict drug disposition when given at therapeutic doses, current evidence suggests that microdosing is a reliable predictor of the biodistribu-tion of therapeutic doses in humans (36). Furthermore, our analyses demonstrated that biodistribution of 11C -rifampin in plasma was similar to that of rifampin administered at therapeutic doses in both rabbits and humans. Moreover, while we noted an initial decrease in rifampin plasma concentrations, no decrease was noted beyond 7 days of treatment, signifying that autoinduction of hepatic metabolism had peaked by this time into treatment. To avoid affecting drug dis-tribution or binding, carbon-11 (or 11C), a radioisotope with a half-life of 20.38 min, was chosen so that it could replace an endogenous carbon atom in rifampin without altering its chemical structure. Rifampin is known to rapidly equilibrate into tissues [equilibration t1/2 ~1 min (37)], and we therefore fully captured it within the 30-to 40-min dynamic imaging window. However, the ability to image for longer periods would indeed be advantageous and could be achieved by introducing longer half-life isotopes such as fluorine-18 in other promising TB drugs that already have fluorine atoms (e.g., oxazol idinone, pretomanid, and delamanid). For 11C-rifampin, the posi-tion of the radiolabel was chosen to ensure that it would be retained on the molecule even after being metabolized in the liver (38). Although PET cannot distinguish the drug from the metabolite, the propor-tion of 25-desacetyl rifampin was low (undetectable in brain tissue and CSF and <0.22% in plasma) in rabbits and patient with TBM (<6%) and thus did not contribute significantly to the PET signal.
While critical for TB treatment, rifampin is also widely used to treat serious infections such as infective endocarditis and infected prostheses, especially due to Staphylococcus aureus (and methicillin-resistant S. aureus), which are similarly plagued with inadequately optimized dosing (39). Therefore, PET could also be used to optimize rifampin dosing in serious nonmycobacterial diseases, which are a cause of significant morbidity and mortality in the United States. Last, while we have focused on rifampin, this technology is broadly applicable and, combined with PK/PD modeling, could be used to study other antimicrobials and enable precision medicine in resource-rich settings.
MATERIALS AND METHODS
Study design
The objective of this study was to use a noninvasive technology in M. tuberculosis–infected subjects to understand the spatial distribution of rifampin at infection sites and how this distribution may change during treatment. CT and 18F-FDG were performed in rabbits to assist with noninvasive visualization of BLs. Dynamic 11C-rifampin PET/CT imaging and MS analysis were performed to follow changes to rifampin exposures with treatment. TACs were used to calculate AUCs in the brain and plasma to determine tissue/plasma AUC0–30 ratios. PET results were corroborated by MS assays for rifampin and 25-desacteyl rifampin analysis in plasma (human and rabbit), CSF (rabbit only), and brain tissue (rabbit only). First-in-human 11C-rifampin PET studies were performed in a cohort of 10 patients with TB to demonstrate the proof of concept and feasibility of PET as a clinically translatable tool. An integrated PK–brain biodistribution model was developed that captured the rifampin plasma and brain distribution and was used to predict brain PK in children. Sample size, selection, and replicates are provided in the figure legends. Although the study was not blinded, a unique identification number was provided to each subject, and VOIs and MS were performed without knowledge of group assignment. All protocols were approved by the Johns Hopkins University Biosafety, Radiation Safety, Animal Care and Use and Institutional Review Board Committees. Primary data are reported in table S8.
Animal infection
M. tuberculosis H37Rv titrated frozen stocks were obtained from the laboratory of S.K.J. Male and female New Zealand White rabbits (Robinson Services Inc.) at postnatal days 4 to 8 were inoculated in their subarachnoid space (10). A 28-gauge insulin syringe was used to inject 20 μl of freshly prepared M. tuberculosis suspension delivering 1.42 ± 0.21 log10 CFU into the brain of each animal. Uninfected control (sham) rabbits were injected with phosphate-buffered saline in the same manner as done in infected animals. In infected rabbits that received treatment, antimicrobials were administered starting 3 weeks after infection. Daily oral doses (5 days per week) of rifampin (30 mg/kg; equipotent to 10 mg/kg in humans; Sigma-Aldrich) and isoniazid (50 mg/kg; equipotent to ~16 mg/kg in humans; Sigma-Aldrich) were administered (11). Animals were euthanized with pentobarbital sodium (120 mg/kg), and organs were aseptically harvested. To enumerate the bacterial burden, organs from infected rabbits were homogenized and plated onto Middlebrook 7H11 selective plates (Becton Dickinson) ( 10). CSF samples were centrifuged, and the supernatant was used to measure protein (BCA protein test, Pierce) and glucose (glucose assay kit, BioVision).
Imaging
11C-rifampin was synthesized as a sterile, pyrogen-free solution of high specific activity (826 ± 198 GBq/ mol) and high radiochemical purity at the Johns Hopkins PET Radiotracer Center using Current Good Manufacturing Practices.
Rabbits
Live M. tuberculosis–infected and uninfected control rabbits were imaged using sealed biocontainment devices compliant with biosafety level 3 (10). Because of the short half-life of 11C-rifampin, “on-table” injections via the ear vein (62.83 ± 8.66 MBq of 11C-rifampin) were administered (8), and dynamic PET was performed over 30 min (except one animal at week 4 that was imaged for 15 min) using the nanoScan PET/CT (Mediso) small animal imager. On the day of imaging, the oral rifampin dose was administered only after the completion of the 11C-rifampin PET. All animals also underwent 18F-FDG PET ~45 min after intravenous injection of 16.41 ± 8.36 MBq of 18F-FDG (Sofie Co.) (40) and ~100 min after 11C-rifampin imag-ing, when 11C would have decayed.
Humans
Ten patients with confirmed TB (using microbiology or molecular testing) receiving daily rifampin-based treatment were recruited under an ongoing study investigating the biodistribution and PK of rifampin. A patient with TBM treated with oral dexamethasone and weeks into treatment was among those recruited. 11C-rifampin was used per the FDA Radioactive Drug Research Committee program guidelines (41). The study team had no role in the diagnosis or clinical management of the patients. The morning dose of rifampin was not administered before imaging. The subject received an intravenous bolus of 370 MBq of 11C-rifampin followed immediately by dynamic PET/CT (Biograph mCT, Siemens) for 40 min using a multi-bed dynamic protocol. After completion of imaging, 600 mg of rifampin was administered intravenously and blood samples were collected from the contralateral arm. Plasma was separated on the same day and stored for analysis.
Image analysis
Images were reconstructed and coregistered using VivoQuant 3.5 (InviCRO). 3D spherical VOIs were drawn to measure 11C-rifampin PET signal in whole blood (carotid in rabbits; left ventricle in humans), BLs [visualized on MRI (human) or CT and/or with 18F-FDG PET (rabbits)], and contralateral UB regions in the infected brain (human and rabbits) and presented as mean Bq/ml. Multiple VOIs were drawn if multiple lesions were visualized in the same subject. Whole-blood VOIs were corrected to plasma using the average hematocrit (50%) in young rabbits (42) and the patient’s measured hematocrit (37.9%). The brain VOIs were corrected for cerebral blood volume (3%) (43).
Mass spectrometry
Blood samples were drawn and collected in tubes with EDTA [BD Vacutainer (human) and BD Microtainer (rabbit), Fisher Scientific Co. LLC] to separate into the plasma sent for measurement by validated ultra performance liquid chromatography/tandem MS (LC-MS/MS) assays of rifampin and 25-desacetyl rifampin at the University of Florida. The standard curves for each compound ranged from 50.00 to 0.05 g/ml. CSF and brain tissues from rab-bits were also analyzed. The assay measured total (free and protein bound) concentrations.
Plasma PK model
A published PK model for rifampin after oral administration was used to describe the PK of both rifampin and 11C-rifampin in plasma across different species and body weights (12). This model was scaled up, and its ability to predict rifampin concentrations in humans was evaluated using both internal and external data digitized from the literature (six adult and eight pediatric populations) (full list in table S4) (13, 44).
PK–brain biodistribution model
11C-rifampin PET data (Bq/ml) were converted to mass (ng/ml) using the molecular weight of 11C-rifampin, specific activity, and injected dose. To capture the observed decrease in rifampin penetration BLs and UB over time, two different penetration/partition coefficients (PCs) were estimated: PC0, at the start of treatment (week 0), and PC≥2, at 2 weeks after treatment initiation and onward. Only the unbound rifampin fraction in plasma (0.25) (35) was assumed to be available to penetrate the brain tissue.
Pediatric dosing simulations
Monte Carlo simulations were performed using the developed PK– brain biodistribution model to predict rifampin exposures in BLs (AUC0–24h and Cmax), assuming that the distribution kinetics of plasma to brain in rabbits is similar and scalable to children. A simulated population of 1000 children (2 to 11 years) with a weight distribution between the 25th and 75th percentiles of the WHO weight-for-age standards (45) was used. Rifampin exposures in the lesions (Cmax) were then used to calculate the probability of target attainment.
Statistical analysis
Data were analyzed using GraphPad Prism (GraphPad Software Inc.). Unpaired two-tailed Student’s t test was used to analyze CFU and CSF indices, with data presented as means ± SD. Bacterial burden (CFU) is presented as means ± SD on a logarithmic scale (base 10). PET and MS data are presented as median ± IQR, and Wilcoxon signed rank test or Mann-Whitney U test was used for comparison. Statistical software package R (v2.15.2) and RStudio (v0.97.248) were used for the analysis of modeling data. PK model activity profiles are presented on a logarithmic scale. P value <0.05 was considered statistically significant.
Supplementary Material
Acknowledgments:
We thank the Maryland Department of Health and Mental Hygiene for their support and help in recruiting patients with TB. We also thank R. Abdullah and C. Voicu at Johns Hopkins Hospital for coordinating the human imaging studies.
Funding: This work was funded by the National Institutes of Health (Director’s Transformative Research Award R01-EB020539 to S.K.J., R01-HL131829 to S.K.J., R01-HD069562 to S.K., and NICHD Pediatric Critical Care and Trauma Scientist Development Program K12-HD047349 to E.W.T.), “Stimulating and Advancing ACCM Research” (StAAR) grant from the Department of Anesthesiology and Critical Care Medicine, Johns Hopkins Medicine (Mentored Training Award to E.W.T.), and Johns Hopkins All Children’s Hospital Foundation Institutional Grant Program to E.W.T.
Footnotes
Competing interests: S.K.J. received consulting fees, unrelated to this work, from Mediso Medical Imaging Systems Ltd.
Data and Materials availability: All data associated with this study are present in the paper or Supplementary Materials.
REFERENCES AND NOTES
- 1.Wilkinson RJ, Rohlwink U, Misra UK, van Crevel R, Mai NTH, Dooley KE, Caws M, Figaji A, Savic R, Solomons R, Thwaites GE, Tuberculous Meningitis International Research Consortium, Tuberculous meningitis. Nat. Rev. Neurol 13, 581–598 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Jain SK, Tobin DM, Tucker EW, Venketaraman V, Ordonez AA, Jayashankar L, Siddiqi OK, Hammoud DA, Prasadarao NV, Sandor M, Hafner R, Fabry Z, NIH Tuberculous Meningitis Writing Group, Tuberculous meningitis: A roadmap for advancing basic and translational research. Nat. Immunol 19, 521–525 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chiang SS, Khan FA, Milstein MB, Tolman AW, Benedetti A, Starke JR, Becerra MC, Treatment outcomes of childhood tuberculous meningitis: A systematic review and meta-analysis. Lancet Infect. Dis 14, 947–957 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Velásquez GE, Brooks MB, Coit JM, Pertinez H, Vargas Vásquez D., Sánchez Garavito E., Calderón RI, Jiménez J, Tintaya K, Peloquin CA, Osso E, Tierney DB, Seung KJ, Lecca L, Davies GR, Mitnick CD, Efficacy and safety of high-dose rifampin in pulmonary tuberculosis: A randomized controlled trial. Am. J. Respir. Crit. Care Med 198, 657–666 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boeree MJ, Heinrich N, Aarnoutse R, Diacon AH, Dawson R, Rehal S, Kibiki GS, Churchyard G, Sanne I, Ntinginya NE, Minja LT, Hunt RD, Charalambous S, Hanekom M, Semvua HH, Mpagama SG, Manyama C, Mtafya B, Reither K, Wallis RS, Venter A, Narunsky K, Mekota A, Henne S, Colbers A, van Balen GP, Gillespie SH, Phillips PPJ, Hoelscher M, High-dose rifampicin, moxifloxacin, and SQ109 for treating tuberculosis: A multi-arm, multi-stage randomised controlled trial. Lancet Infect. Dis 17, 39–49 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Svensson RJ, Svensson EM, Aarnoutse RE, Diacon AH, Dawson R, Gillespie SH, Moodley M, Boeree MJ, Simonsson USH, Greater early bactericidal activity at higher rifampicin doses revealed by modeling and clinical trial simulations. J Infect Dis 218, 991–999 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Ruslami R, Ganiem AR, Dian S, Apriani L, Achmad TH, van der Ven AJ, Borm G, Aarnoutse RE, van Crevel R, Intensified regimen containing rifampicin and moxifloxacin for tuberculous meningitis: An open-label, randomised controlled phase 2 trial. Lancet Infect. Dis 13, 27–35 (2013). [DOI] [PubMed] [Google Scholar]
- 8.DeMarco VP, Ordonez AA, Klunk M, Prideaux B, Wang H, Zhuo Z, Tonge PJ, Dannals RF, Holt DP, Lee CKK, Weinstein EA, Dartois V, Dooley KE, Jain SK, Determination of [11C]rifampin pharmacokinetics within Mycobacterium tuberculosis-infected mice by using dynamic positron emission tomography bioimaging. Antimicrob. Agents Chemother 59, 5768–5774 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weinstein EA, Liu L, Ordonez AA, Wang H, Hooker JM, Tonge PJ, Jain SK, Noninvasive determination of 2-[18F]-fluoroisonicotinic acid hydrazide pharmacokinetics by positron emission tomography in Mycobacterium tuberculosis-infected mice. Antimicrob. Agents Chemother 56, 6284–6290 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tucker EW, Pokkali S, Zhang Z, DeMarco VP, Klunk M, Smith ES, Ordonez AA, Penet MF, Bhujwalla Z, Jain SK, Kannan S, Microglia activation in a pediatric rabbit model of tuberculous meningitis. Dis. Model. Mech 9, 1497–1506 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers (U.S. Food and Drug Administration, 2005); https://www.fda.gov/downloads/Drugs/Guidances/UCM078932.pdf. [Google Scholar]
- 12.Smythe W, Khandelwal A, Merle C, Rustomjee R, Gninafon M, Bocar Lo M, Sow OB, Olliaro PL, Lienhardt C, Horton J, Smith P, McIlleron H, Simonsson USH, A semimechanistic pharmacokinetic-enzyme turnover model for rifampin autoinduction in adult tuberculosis patients. Antimicrob. Agents Chemother 56, 2091–2098 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koup JR, Williams-Warren J, Weber A, Smith AL, Pharmacokinetics of rifampin in children. I. Multiple dose intravenous infusion. Ther. Drug Monit 8, 11–16 (1986). [DOI] [PubMed] [Google Scholar]
- 14.McIlleron H, Hundt H, Smythe W, Bekker A, Winckler J, van der Laan L, Smith P, Zar HJ, Hesseling AC, Maartens G, Wiesner L, van Rie A, Bioavailability of two licensed paediatric rifampicin suspensions: Implications for quality control programmes. Int. J. Tuberc. Lung Dis 20, 915–919 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scheld WM, Sande MA, Bactericidal versus bacteriostatic antibiotic therapy of experimental pneumococcal meningitis in rabbits. J. Clin. Invest 71, 411–419 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Srivastava S, Gumbo T, In vitro and in vivo modeling of tuberculosis drugs and its impact on optimization of doses and regimens. Curr. Pharm. Des 17, 2881–2888 (2011). [DOI] [PubMed] [Google Scholar]
- 17.Pasipanodya JG, Srivastava S, Gumbo T, Meta-analysis of clinical studies supports the pharmacokinetic variability hypothesis for acquired drug resistance and failure of antituberculosis therapy. Clin. Infect. Dis 55 Suppl 2, 169–177 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McKenzie C, Antibiotic dosing in critical illness. J. Antimicrob. Chemother 66 Suppl 2, ii25–ii31 (2011). [DOI] [PubMed] [Google Scholar]
- 19.Muller M, dela Pena A., Derendorf H, Issues in pharmacokinetics and pharmacodynamics of anti-infective agents: Distribution in tissue. Antimicrob. Agents Chemother 48, 1441–1453 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rifat D, Prideaux B, Savic RM, Urbanowski ME, Parsons TL, Luna B, Marzinke MA, Ordonez AA, DeMarco VP, Jain SK, Dartois V, Bishai WR, Dooley KE, Pharmacokinetics of rifapentine and rifampin in a rabbit model of tuberculosis and correlation with clinical trial data. Sci. Transl. Med 10, eaai7786 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prideaux B, Via LE, Zimmerman MD, Eum S, Sarathy J, O’Brien P, Chen C, Kaya F, Weiner DM, Chen P-Y, Song T, Lee M, Shim TS, Cho JS, Kim W, Cho SN, Olivier KN, Barry CE, Dartois V, The association between sterilizing activity and drug distribution into tuberculosis lesions. Nat. Med 21, 1223–1227 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.James ML, Gambhir SS, A molecular imaging primer: Modalities, imaging agents, and applications. Physiol. Rev 92, 897–965 (2012). [DOI] [PubMed] [Google Scholar]
- 23.Rudin M, Weissleder R, Molecular imaging in drug discovery and development. Nat. Rev. Drug Discov 2, 123–131 (2003). [DOI] [PubMed] [Google Scholar]
- 24.Rubinstein LV, Steinberg SM, Kummar S, Kinders R, Parchment RE, Murgo AJ, Tomaszewski JE, Doroshow JH, The statistics of phase 0 trials. Stat. Med 29, 1072–1076 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Te Brake L., Dian S, Ganiem AR, Ruesen C, Burger D, Donders R, Ruslami R, van Crevel R, Aarnoutse R, Pharmacokinetic/pharmacodynamic analysis of an intensified regimen containing rifampicin and moxifloxacin for tuberculous meningitis. Int. J. Antimicrob. Agents 45, 496–503 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Sáez-Llorens X, Jafari HS, Severien C, Parras F, Olsen KD, Hansen EJ, Singer II, McCracken GH Jr., Enhanced attenuation of meningeal inflammation and brain edema by concomitant administration of anti-CD18 monoclonal antibodies and dexamethasone in experimental Haemophilus meningitis. J. Clin. Invest 88, 2003–2011 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nau R, Sorgel F, Eiffert H, Penetration of drugs through the blood-cerebrospinal fluid/ blood-brain barrier for treatment of central nervous system infections. Clin. Microbiol. Rev 23, 858–883 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lenaerts A, Barry CE III, Dartois V, Heterogeneity in tuberculosis pathology, microenvironments and therapeutic responses. Immunol. Rev 264, 288–307 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rohlwink UK, Mauff K, Wilkinson KA, Enslin N, Wegoye E, Wilkinson RJ, Figaji AA, Biomarkers of cerebral injury and inflammation in pediatric tuberculous meningitis. Clin. Infect. Dis 65, 1298–1307 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rapid Advice: Treatment of Tuberculosis in Children (World Health Organization, 2010); http://apps.who.int/iris/handle/10665/44444. [PubMed] [Google Scholar]
- 31.Savic RM, Ruslami R, Hibma JE, Hesseling A, Ramachandran G, Ganiem AR, Swaminathan S, McIlleron H, Gupta A, Thakur K, van Crevel R, Aarnoutse R, Dooley KE, Pediatric tuberculous meningitis: Model-based approach to determining optimal doses of the anti-tuberculosis drugs rifampin and levofloxacin for children. Clin. Pharmacol. Ther 98, 622–629 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Witt KA, Sandoval KE, Steroids and the blood-brain barrier: Therapeutic implications. Adv. Pharmacol 71, 361–390 (2014). [DOI] [PubMed] [Google Scholar]
- 33.Heemskerk AD, Bang ND, Mai NTH, Chau TTH, Phu NH, Loc PP, Chau NVV, Hien TT, Dung NH, Lan NTN, Lan NH, Lan NN, Phong le T, Vien NN, Hien NQ, Yen NTB, Ha DTM, Day JN, Caws M, Merson L, Thinh TTV, Wolbers M, Thwaites GE, Farrar JJ, Intensified antituberculosis therapy in adults with tuberculous meningitis. N. Engl. J. Med 374, 124–134 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Alghamdi WA, Al-Shaer MH, Peloquin CA, Protein binding of first-line antituberculous drugs. Antimicrob. Agents Chemother 62, e00641–18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Woo J, Cheung W, Chan R, Chan HS, Cheng A, Chan K, In vitro protein binding characteristics of isoniazid, rifampicin, and pyrazinamide to whole plasma, albumin, and −1-acid glycoprotein. Clin. Biochem 29, 175–177 (1996). [DOI] [PubMed] [Google Scholar]
- 36.Lappin G, Noveck R, Burt T, Microdosing and drug development: Past, present and future. Expert Opin. Drug Metab. Toxicol 9, 817–834 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kjellsson MC, Via LE, Goh A, Weiner D, Low KM, Kern S, Pillai G, Barry CE III, Dartois V, Pharmacokinetic evaluation of the penetration of antituberculosis agents in rabbit pulmonary lesions. Antimicrob. Agents Chemother 56, 446–457 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu L, Xu Y, Shea C, Fowler JS, Hooker JM, Tonge PJ, Radiosynthesis and bioimaging of the tuberculosis chemotherapeutics isoniazid, rifampicin and pyrazinamide in baboons. J. Med. Chem 53, 2882–2891 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu C, Bayer A, Cosgrove SE, Daum RS, Fridkin SK, Gorwitz RJ, Kaplan SL, Karchmer AW, Levine DP, Murray BE, Raybak MJ, Talan DA, Chambers HF, Infectious Diseases Society of America, Clinical practice guidelines by the Infectious Diseases Society of America for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children. Clin. Infect. Dis 52, e18–55 (2011). [DOI] [PubMed] [Google Scholar]
- 40.Davis SL, Nuermberger EL, Um PK, Vidal C, Jedynak B, Pomper MG, Bishai WR, Jain SK, Noninvasive pulmonary [18F]-2-fluoro-deoxy-d-glucose positron emission tomography correlates with bactericidal activity of tuberculosis drug treatment. Antimicrob. Agents Chemother 53, 4879–4884 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.The Radioactive Drug Research Committee: Human Research Without an Investigational New Drug Application (U.S. Food and Drug Administration, 2010); www.fda.gov/downloads/Drugs/Guidances/UCM163892.pdf. [Google Scholar]
- 42.Abelson JN, Simon MI, Everse J, Vandegriff KD, Winslow RM, Hemoglobins, Part B: Biochemical and Analytical Methods (Academic Press, 1994), vol. 231, pp. 98–99. [Google Scholar]
- 43.Syvänen S, Eriksson J, Advances in PET imaging of P-glycoprotein function at the blood-brain barrier. ACS Chem. Nerosci 4, 225–237 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Loos U, Musch E, Jensen JC, Mikus G, Schwabe HK, Eichelbaum M, Pharmacokinetics of oral and intravenous rifampicin during chronic administration. Klin. Wochenschr 63, 1205–1211 (1985). [DOI] [PubMed] [Google Scholar]
- 45.World Health Organization, Child growth standards (2018); www.who.int/childgrowth/en/.
- 46.Beal S, Sheiner LB, Boeckmann A, Bauer RJ, NONMEM user’s guides (Icon Development Solutions, 2013). [Google Scholar]
- 47.Lindbom L, Pihlgren P, Jonsson N, PsN-toolkit—A collection of computer intensive statistical methods for non-linear mixed effect modeling using NONMEM. Comput. Methods Programs Biomed 79, 241–257 (2005). [DOI] [PubMed] [Google Scholar]
- 48.Keizer RJ, van Benten M, Beijnen JH, Schellens JHM, Huitema ADR, Piraña and PCluster: A modeling environment and cluster infrastructure for NONMEM. Comput. Methods Programs Biomed 101, 72–79 (2011). [DOI] [PubMed] [Google Scholar]
- 49.Baron KT, Gastonguay MR, Simulation from ODE-based population PK/PD and systems pharmacology models in R with mrgsolve. J. Pharmacokinet. Pharmacodyn 23, S84–S85 (2015). [Google Scholar]
- 50.Ruslami R, Nijland HMJ, Alisjahbana B, Parwati I, van Crevel R, Aarnoutse RE, Pharmacokinetics and tolerability of a higher rifampin dose versus the standard dose in pulmonary tuberculosis patients. Antimicrob. Agents Chemother 51, 2546–2551 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schaaf HS, Willemse M, Cilliers K, Labadarios D, Maritz JS, Hussey GD, McIlleron H, Smith P, Donald PR, Rifampin pharmacokinetics in children, with and without human immunodeficiency virus infection, hospitalized for the management of severe forms of tuberculosis. BMC Med 7, 19 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thee S, Seddon JA, Donald PR, Seifart HI, Werely CJ, Hesseling AC, Rosenkranz B, Roll S, Magdorf K, Schaaf HS, Pharmacokinetics of isoniazid, rifampin, and pyrazinamide in children younger than two years of age with tuberculosis: Evidence for implementation of revised World Health Organization recommendations. Antimicrob. Agents Chemother 55, 5560–5567 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ramachandran G, Hemanth Kumar AK, Bhavani PK, Poorana Gangadevi N., Sekar L, Vijayasekaran D, Banu Rekha VV, Ramesh Kumar S., Ravichandran N, Mathevan G, Swaminathan S, Age, nutritional status and INH acetylator status affect pharmacokinetics of anti-tuberculosis drugs in children. Int. J. Tuberc. Lung Dis 17, 800–806 (2013). [DOI] [PubMed] [Google Scholar]
- 54.Hiruy H, Rogers Z, Mbowane C, Adamson J, Ngotho L, Karim F, Gumbo T, Bishai W, Jeena P, Subtherapeutic concentrations of first-line anti-TB drugs in South African children treated according to current guidelines: The PHATISA study. J. Antimicrob. Chemother 70, 1115–1123 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Boeree MJ, Diacon AH, Dawson R, Narunsky K, du Bois J, Venter A, Phillips PPJ, Gillespie SH, McHugh TD, Hoelscher M, Heinrich N, Rehal S, van Soolingen D, van Ingen J, Magis-Escurra C, Burger D, Plemper van Balen G., Aarnoutse RE, PanACEA Consortium, A dose-ranging trial to optimize the dose of rifampin in the treatment of tuberculosis. Am. J. Respir. Crit. Care Med 191, 1058–1065 (2015). [DOI] [PubMed] [Google Scholar]
- 56.Chigutsa E, Pasipanodya JG, Visser ME, van Helden PD, Smith PJ, Sirgel FA, Gumbo T, McIlleron H, Impact of nonlinear interactions of pharmacokinetics and MICs on sputum bacillary kill rates as a marker of sterilizing effect in tuberculosis. Antimicrob. Agents Chemother 59, 38–45 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kwara A, Enimil A, Gillani FS, Yang H, Sarfo AM, Dompreh A, Ortsin A, Osei-Tutu L, Kwarteng Owusu S., Wiesner L, Norman J, Kurpewski J, Peloquin CA, Ansong D, Antwi S, Pharmacokinetics of first-line antituberculosis drugs using WHO revised dosage in children with tuberculosis with and without HIV coinfection. J. Pediatric. Infect. Dis. Soc 5, 356–365 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Swaminathan S, Pasipanodya JG, Ramachandran G, Hemanth Kumar AK, Srivastava S, Deshpande D, Nuermberger E, Gumbo T, Drug concentration thresholds predictive of therapy failure and death in children with tuberculosis: Bread crumb trails in random forests. Clin. Infect. Dis 63, S63–S74 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bekker A, Schaaf HS, Draper HR, van der Laan L, Murray S, Wiesner L, Donald PR, McIlleron HM, Hesseling AC, Pharmacokinetics of rifampin, isoniazid, pyrazinamide, and ethambutol in infants dosed according to revised WHO-recommended treatment guidelines. Antimicrob. Agents Chemother 60, 2171–2179 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.