

Abstract
Allogenic graft material and tissue engineering have recently shown promising results for the improvement of both esthetic and functional outcomes in the treatment of large skin defects. We chose human amniotic membrane as a cellular scaffold in order to develop a skin substitute for later in vivo uses. Various methods of de-epithelialization of the human amniotic membrane were evaluated by histological analysis including hematoxylin–eosin and laminin staining, optic coherence tomography, and scanning electron microscopy with 0.25/0.02% trypsin/ethylenediaminetetraacetic acid treatment and mechanical cell removal showing an almost complete loss of the epithelium and a mainly intact basement membrane. Novel examination of human amniotic membrane by optic coherence tomography was feasible, but difficulties were experienced in handling and interpretation of the tissue as no comparable data exist. Subsequently, we developed an air–liquid interface cell culture to cultivate keratinocytes and fibroblasts on the de-epithelialized human amniotic membrane. We achieved a mostly keratinized surface on the epidermal side with a confluent fibroblast network on the chorion side.
Keywords: Tissue engineering, skin graft, basement membrane, human amniotic membrane, de-epithelialization, air–liquid cell culture, optical coherence tomography, electron microscopy, immunohistochemistry
Introduction
The skin acts as an important barrier against noxious agents and helps to maintain a stable water balance.1 Various pathologies, such as burn injuries, tumor resections, and chronic wounds, are often responsible for large skin defects that need to be covered properly and in a timely manner.2–5
Diverse autologous and allogenous grafts are currently in use, although limitations include restricted availability, secondary defects, rejection of the graft, and functional and esthetic problems of the resulting scar.6–9 A promising scaffold for tissue-engineered skin without the limitations of the other types of graft is the human amniotic membrane (hAM). It provides a stable basement membrane for cell culture, expresses anti-immunogenic and anti-inflammatory agents, and shows good results in the treatment of wound defects as a wound coverage.10–12
As for all biological scaffolds, gentle but complete decellularization is a critical step in removing allogenous cells, and various methods have been described.13 In view of the importance of the basement membrane in the process of re-epithelialization and the organization of the dermis, our aim has been primarily to maintain the basement membrane through the decellularization process.14–16 As laminin is an essential component of the basement membrane,17 it was used to verify the integrity of the membrane.
As typical skin shows an orthokeratinized surface, air–liquid interface cultures have been suggested in order to allow air to contact the surface as the typical stimulus for keratinocytes to differentiate into corneocytes, while simultaneously nutrients are provided from the dermal site of the graft.18 Choice of the right culture medium is critical to secure the growth of both the fibroblasts on the dermal side and the keratinocytes on the epidermal side. Although serum-containing media are known to be crucial for the cultivation of fibroblasts, it is also described to inhibit the growth of keratinocytes.19,20 Therefore, we have compared various combinations of serum and keratinocyte medium in a simple trypan blue viability test and by phase contrast microscopy in order to find a suitable air–liquid medium.
Overall, the aim of this study was to generate a skin substitute on base of hAM for further in vivo comparisons. We suggested that detergents would decellularize the hAM better than enzymes with regard to an intact extracellular matrix and a sufficient cell removal. We also compared the resulting hAM for its re-epithelialization properties. In addition, a feasibility part was conducted, where images of the hAM by optical coherence tomography should be achieved.
Materials and methods
Several methods of de-epithelialization of the hAM were compared, and the tissue was subsequently examined (Figure 1). In addition, various cell-culture media were evaluated to ensure fibroblast and keratinocyte viability on the skin graft. De-epithelialized hAM was then cultured with keratinocytes and fibroblasts and shifted to an air–liquid interface cell culture followed by tissue studies.
Figure 1.

Experimental overview: comparison of different de-epithelialization methods and development of an air–liquid interface culture with subsequent tissue studies.
General preparation of hAM and quantitative evaluation
Cryoconserved hAMs (Austrian Cluster for Tissue Regeneration, Linz) were gently thawed at room temperature and rinsed thoroughly in DPBS− (Gibco, Waltham). Once the tissue had been cut into smaller pieces of approximately 5 × 5 cm, they were transferred to the various de-epithelialization methods (Figure 2(a)). After decellularization, the tissue was further rinsed in DPBS+ (Gibco, Waltham) and aprotinin (10 KIU/mL) (Sigma-Aldrich, St. Louis) on a shaker (IKA, Staufen) at 4°C for 48 h, followed by a regular medium change to remove loose epithelial cells and enzyme residues.
Figure 2.

Decellularization of hAM: (a) thawed hAM was cut into smaller pieces while still on the carrier material. (b1–b2) To remove the epithelium mechanically, a cell scraper/ soaked cotton wool tip was gently rubbed over the surface of the hAM. (c) The hAM was fixed onto an insert with a PTFE ring with the epithelial side facing the inside of the insert. (d) The hAM was washed with either SDS/SDC or Triton/SDC on both sides. (e) The hAM was mechanically washed and rinsed on a shaker (200/min) in a closed well.
The decellularization results were quantified by simple cell counting using Image J of randomly chosen decellularized areas (n = 20 for each method) of H/E stained light microscopy images (500 µm of straight membrane each). Cells were counted above and below the basement membrane and separately documented; results were presented with mean value ± standard deviation.
De-epithelialization with trypsin and cell scraper
One method that we evaluated was the use of trypsin and a cell scraper.21 The thawed hAM was transferred into 0.25/0.02% trypsin/ethylenediaminetetraacetic acid (EDTA) (Sigma-Aldrich) at 37°C and incubated for 25 min. Cold DBPS+ was used to stop the enzyme activity. Under regular phase contrast microscopy control, the epithelium was removed by slow and gentle scraping with a cell scraper (Sarsted, Nümbrecht) (Figure 2(b2)). Following this step, the hAM was placed on an insert (Figure 2(c)) in a cell-culture well (Greiner Bio-One, Kremsmünster), fixed with a PTFE ring (Alwin Höfert, Ammersbek) with the epithelial side pointing up, and rinsed on a shaker as described above (Figure 2(e)).
Decellularization with sodium dodecylsulfate/sodium deoxycholate
Following an experiment in which heart valves were decellularized,22 the thawed hAM was placed on an insert as mentioned above. It was then washed with a 0.5% sodium dodecylsulfate (SDS)/sodium deoxycholate (SDC) solution (Sigma-Aldrich) on a shaker (200/min) at room temperature for 48 h (Figure 2(d) and (e)). The solution was changed every 12 h. Subsequently, the tissue was rinsed on a shaker as mentioned above.
De-epithelialization with NaOH
One research group postulated an easy de-epithelialization method involving the use of NaOH and a cotton wool tip.23 A cotton stick soaked in 0.5 M NaOH solution (Roth, Karlsruhe) was gently rubbed over a thawed hAM for over a minute to remove the epithelium (Figure 2(b1)). Subsequently, the hAM was rinsed thoroughly in DPBS+, placed on an insert, and rinsed on a shaker as described above.
Decellularization with Triton/SDC
Although similar to the decellularization of the heart valves with SDS/SDC, another composition of the washing solution has been suggested, namely the use of 0.25% Triton/SDC (Roth, Karlsruhe).24 The thawed hAM was fixed onto an insert and washed for approximately 48 h on a shaker (200/min) at room temperature with a medium change every 12 h. The hAM was then rinsed on the shaker as mentioned above.
Cell isolation and cultures
Excess rat skin was used to isolate keratinocytes and fibroblasts, which subsequently were amplified and cultured on the de-epithelialized hAM until a confluent cell population was reached. The culture was then changed to an air–liquid interface culture until final keratinization.
Isolation of keratinocytes
The isolation of fibroblasts and keratinocytes was performed following the protocol of the isolation of various cell lines from corneal tissue.25 Rat skin was stored in keratinocyte medium (Cellsystems, Troisdorf) for up to 4 h after extraction. After removal of the subcutis, the tissue was then cut into pieces of 5 mm which were then incubated in Thermolysin (50 IU/mL) (Sigma-Aldrich) for 15 h at 4°C. The enzyme activity was subsequently stopped with keratinocyte medium. Using sterile forceps (Aeskulap, Tuttlingen), the epidermal layer was then removed and cut into smaller pieces (Figure 3(a)). These samples were transferred into centrifuge tubes (Greiner Bio-One) filled with TrypLE (Gibco, Waltham) and dissociated by occasional gentle shaking for 30 min at 37°C. Subsequently, the dissociation was stopped with cold keratinocyte medium, and the cell suspension was filtered through a cell strainer (Corning, Corning) and centrifuged. The remaining cell pellet was solved in keratinocyte medium and transferred to cell-culture flasks (Greiner Bio-One) (20 × 103/cm2).
Isolation of fibroblasts
After removal of the epidermis, the remaining pieces of dermis were transferred to fibroblast medium (Gibco, Waltham) and cut into smaller pieces, which were then placed on a cell-culture dish (Greiner Bio-One) (Figure 3(b)). After 4 h, the dish was filled with fibroblast medium with the pieces staying adherent to the dish. After several days with regular medium changes, the pieces were removed when fibroblast colonies were visible.
Figure 3.

From cell isolation to air–liquid interface culture: (a) In the left dish, small skin biopsies are visible, whereas in the right dish, the epithelium is being dissociated with forceps. (b) The remaining dermis is placed in a cell-culture dish. After adherence, the dish is filled with fibroblast medium. (c) 1 mL fibroblast medium with 100,000 fibroblasts is pipetted onto the chorion side of the hAM. After several hours, the insert is placed with the epithelium facing up and is cultured with keratinocytes. (d) On the left side, the insert shows the hAM with the double-sided cell culture visible. After 3–8 days of cultivation, when the cells have reached full confluence, only the basolateral side of the insert is in contact with the modified medium, with the epithelial side being in contact with the air.
Medium changes and cell passages
To amplify the cells, regular medium changes and cell passages had to be performed. The medium was changed every second or third day for keratinocytes or fibroblasts, respectively. For a medium change, the remaining medium was removed, the flask/dish was rinsed with DPBS−, and new medium was administered. To passage the cells, a small amount of TrypLE was used for 15–40 min at 37°C. Dissociation was observed under phase contrast microscopy. When almost all the cells had dissociated, the enzyme was blocked with cold keratinocyte or fibroblast medium. The solution was then centrifuged, and the cell pellet was dispersed in keratinocyte or fibroblast medium and recultured in cell-culture flasks (keratinocytes 10,000/cm2; fibroblasts 4000/cm2).
Comparison of various air–liquid media
In the air–liquid model, only one medium was present on the basolateral side of the insert for both keratinocytes and fibroblasts. Cell cultures of newly passaged fibroblasts and keratinocytes were incubated for several days in fibroblast medium with 10% fetal calf serum (FCS) (Gibco, Waltham) (n = 5), in keratinocyte medium (n = 5), or in keratinocyte medium with 5% FCS (n = 5) or 10% FCS (n = 5). Morphology and growth characteristics were regularly controlled by phase contrast microscopy, and a Trypan Blue exclusion test26 was performed and the viability documented.
Cell culturing on hAM and by air–liquid interface culture
We developed an air–liquid interface culture based on another published model.27 De-epithelialized hAM was placed upside down with the chorion side facing up. Then, 1 mL fibroblast medium with approximately 100,000 fibroblasts were pipetted on this side using the surface tension to avoid run-off (Figure 3(c)). After an incubation time of 6 h, the insert was transferred to a cell-culture well with the epithelial side facing up, and 1.5 mL fibroblast medium was pipetted onto the basolateral side (Figure 3(d)). Subsequently, 0.5 mL keratinocyte medium with 500,000 keratinocytes was placed on the epithelial side of the hAM. Medium changes were performed daily. When confluence of both cell lines was achieved, the cultured tissue was lifted to enable air–liquid interface culture with only keratinocyte medium with 5% FCS being present on the basolateral side of the insert. Medium was changed regularly until final keratinization was accomplished. Similar to protocols used before, using phase contrast microscopy, the re-cellularization for each decellularized hAM (n = 20) of both sides was then semi-quantitatively scored as 0 = absent, 1 = focal (<50%), 2 = non-confluent (>50%), 3 = confluent layer of cells.22 Regarding the keratinization, the following additional score was obtained: A = no keratinization, B = keratinization < 50% of re-cellularized areas, C = keratinization > 50% of re-cellularized areas, D = complete keratinization.
Tissue examination
Hematoxylin and eosin staining, immunohistochemistry
The tissue was fixed in formalin, embedded in paraffin (over 48 h), and subsequently cut into 5-µm-thick sections. Hematoxylin–eosin staining was performed using an established protocol.28 For immunohistochemical analysis, 5-µm-thick sections mounted on slides were deparaffinized using xylene and ethanol. After treatment of the slides with a peroxide and a protein block, they were incubated with primary antibody (polyclonal rabbit antibody against laminin) for 30 min and subsequently with secondary antibody (anti-rabbit antibody) for 45 min. The sections were then treated with streptavidin and counter-stained with hematoxylin
Scanning electron microscopy
The tissue was rinsed in Sörensen buffer, fixed in 2.5% glutaraldehyde (Sigma-Aldrich), rinsed in water, and dried with ethanol and hexamethyldisilazane. It was then sputter-coated and transferred to a scanning electron microscope (Zeiss, Oberkochen).
Optical coherence tomography
As a pilot project, hAM was evaluated using optical coherence tomography (OCT), a non-invasive method that allows images to be taken at the cellular level within the depth of the tissue (millimeters). The hAM, which was fixed in the insert, was simply positioned vertically in front of the OCT apparatus (Heidelberg Engineering, Heidelberg).
Results
De-epithelialization
The membranes treated with trypsin showed good results under phase contrast and light microscopy (Figures 4(b) and 5(a1) and (c1)). The epithelium was almost completely removed with just 3.4 ± 3.3 cells remaining (Figure 6), and the basement membrane was consistent and prominent. The mesenchymal connective tissue showed some loosening compared with the native hAM, while decellularization below the basement membrane was not complete with 16.8 ± 6.3 cells remaining (Figure 6). In addition, some shaded lines were apparent by phase contrast microscopy, although the related layer was difficult to allocate. The lines might represent the effects of the mechanical impact or be a normal aspect of the basement membrane of the hAM. The hAM treated with NaOH presented incomplete de-epithelialization with 9.4 ± 4.1 cells above the basement membrane (Figures 4(d), 5(a3) and (c3), and 6). Furthermore, a loosened mesenchymal connective tissue and a largely consistent but blurred basement membrane were observed following this treatment with no removal of cells below the basement membrane (Figure 6). The two surfactant-containing decellularization solutions also gave incomplete de-epithelialization, but better decellularization below the basement membrane compared to the other methods with 10.8 ± 4.3 remaining cells for SDS/SDC and 7.2 ± 3.2 for Triton/SDC (Figures 4(c) and (e), 5(a2), (a4), (c2) and (c4), and 6). Although the epithelial cell boundaries were no longer visible under phase contrast optics, the epithelium still did not provide a clear view of the mesenchymal connective tissue. By light microscopy, the basement membrane was largely lost and a loosened mesenchymal connective tissue was presented.
Figure 4.

Comparison of de-epithelialization methods. Phase contrast microscopy. 100×. (a) Native hAM with closed hexagonal epithelium. (b) hAM treated with trypsin. No epithelium is visible, but the matrix exhibits some shaded lines as a sign of possible tissue damage. (c) hAM treated with SDS/SDC. The cell boundaries are blurred, and the underlying matrix is not displayed. (d) hAM treated with NaOH. The epithelium shows many gaps, but the partially visible matrix is largely homogeneous. (e) hAM treated with Triton/SDC. The epithelium is blurred without clear cell boundaries, and the underlying matrix is not visible.
Figure 5.

De-epithelialized hAM stained with HE and for laminin. Light microscopy. 20–40×. (a1–a4) HE staining of de-epithelialized hAM. Whereas the hAM treated with trypsin and NaOH shows mostly removed epithelium, the hAM treated with either Triton/SDC or SDS/SDC presents a consistent epithelium. (b1–b2) HE and laminin staining of native hAM, which presents a typical pseudostratified columnar epithelium and a consistent basement membrane. (c1–c4) Laminin staining of de-epithelialized hAM. The hAM treated with trypsin and NaOH present a consistent, strongly stained, basement membrane, whereas the other two groups exhibit no obvious basement membrane.
Figure 6.
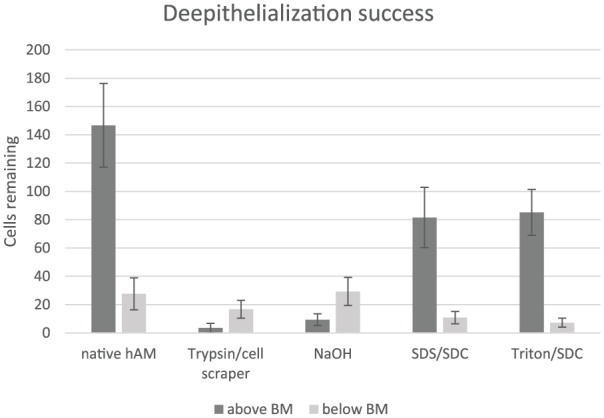
Bar graph of the decellularization success above and below the basement membrane (BM). While hAM processed with trypsin and NaOH showed decent de-epithelialization above the BM, SDS/SDC and Triton/SDC presented worse results. Regarding the decellularization of the stromal side of the BM, the detergential methods showed better cell removal than trypsin and NaOH, with the latter having no significant cell removal at all.
Colonization of hAM—air–liquid culture
Fibroblast growth was completely inhibited when incubated with keratinocyte medium DermaLife K (Figure 7(a1)). When FCS was added, the fibroblasts showed pronounced vital cell growth with 10% FCS showing slightly better results than 5% FCS (Figure 7 (a2) and (a3)). As expected, the best growth conditions were provided by Dulbecco’s Modified Eagle’s Medium (DMEM) with 10% FCS (Figure 7(a4)). The keratinocyte growth under keratinocyte medium showed a typical hexagonal vital appearance of the cell culture (Figure 7(b4)). Almost the same aspect was observed when 5% FCS was added (Figure 7(b3)). With 10% FCS and DMEM, the cells showed typical signs of cell damage, such as a change in shape and a numeral decrease (Figure 7(b1) and (b2)). The trypan blue exclusion test confirmed these findings. Whereas the three groups growing in culture media with added FCS showed a cell viability >85%, the only group without FCS had a viability of only 0%–5%. The opposite effects were presented by the keratinocyte cultures. The viability of the cultures incubated without FCS or with 5% FCS had a viability of 90%–95%. When more FCS was added, the viability decreased to almost 0%. As the keratinocytes and fibroblasts had to be supplied with the same medium for several days while being in the air–liquid culture, the keratinocyte medium with 5% FCS additive, which showed the best results, was therefore used for the air–liquid interface culture.
Figure 7.

Comparison of various culture media on keratinocytes and fibroblasts. Phase contrast microscopy. 100×. Left: Top down (a1–a4), fibroblast cultures are displayed after 5 days incubation in different culture media. The viability increases with the use of FCS. Right: Top (b1–b4) down, keratinocyte cultures are visible after 5 days incubation in media of different compositions. The smaller the amount of FCS added, the higher the viability.
The colonizing of the hAM started with the fibroblasts. The formation of the differentiated cell networks on the chorion side occurred acceptably, no matter which de-epithelialization method was used (Table 1). By contrast, the growth of the keratinocytes on the epithelial side differed greatly between the methods used (Table 1). The culture was harvested 2 days after relocation and at the end of the air–liquid culture. The hAM treated with trypsin presented a dense and confluent epithelial layer after 2 days (Figure 8(a1)). At the end of the air–liquid culture, some areas of the membrane showed keratinized epithelium, while others did not (Figure 8(b1), Table 1). The hAM treated with NaOH lacked the typical confluent epithelium. The cells appeared rounded and overlying, which could be a sign of low adherence and vitality (Figure 8(a2)). At the end of the air–liquid culture, this hAM seemed similar to those treated with trypsin, but with more gaps in the epithelium (Figure 8(b2), Table 1). After 2 days, the membranes washed with SDS/SDC presented a confluent epithelium not different than that after decellularization (Figure 8(a3)) and without signs of keratinization at the end of the air–liquid culture (Figure 8(b3), Table 1). Therefore, almost no keratinocytes adhered on such processed hAM. The hAM treated with Triton/SDC showed occasional round keratinocytes after 2 days (Figure 8(a4)) and some keratinization at the end of the air–liquid culture (Figure 8(b4), Table 1), but to a much lower extent than that in the trypsin group.
Table 1.
Semi-quantitative score of the re-cellularized hAM.
| Keratinocytes | Fibroblasts | Keratinization | |
|---|---|---|---|
| Trypsin/cell scraper | 2-2-2-2-2 | 3-3-3-3-3 | C-C-C-C-C |
| NaOH/cotton pad | 1-2-1-2-1 | 3-3-3-3-3 | B-B-A-B-B |
| SDS/SDC | 1-1-1-1-1 | 3-3-3-3-3 | A-A-A-A-A |
| Triton/SDC | 1-1-1-1-1 | 3-3-3-3-3 | B-A-B-A-A |
SDS: sodium dodecylsulfate; SDC: sodium deoxycholate.
Confluency: 0 = absent, 1 = focal (<50%), 2 = non-confluent (>50%), 3 = confluent layer of cells. Keratinization: A = no keratinization, B = keratinization < 50% of re-cellularized areas, C = keratinization > 50% of re-cellularized areas, D = complete keratinization.
Figure 8.

Colonized epithelial side of de-epithelialized hAM. Phase contrast microscopy. 200×. (a1–a4) Re-epithelialized hAM processed by various de-epithelialization methods after 2 days. After 2 days, only the hAM treated with trypsin showed a confluent gap-less epithelium. Membranes prepared with NaOH and Triton/SDC showed no differentiated epithelial cell complex, and the cells remained round. The hAM treated with SDS/SDC showed no difference from the state after de-epithelialization. (b1–b4) The same membranes at the end of air–liquid culture. The hAM treated with trypsin and NaOH showed incomplete multilayered epithelialization with signs of keratinization. In the membranes washed with Triton/SDC, the epithelium was much more incomplete, whereas the hAM treated with SDS/SDC presented almost no adherent keratinocytes.
Scanning electron microscopy
The native hAM demonstrated a hexagonal growth pattern with tight cell junctions and microvilli on the epithelial side (Figure 9(a1) and (a2)) and loosened irregular collagen fibers on the chorion side (Figure 9(c1)) similar to the findings presented be other authors.29 The trypsinized hAM exhibited the complete removal of the epithelium with only some detritus remaining and a smooth appearance as an indication of an intact basement membrane (Figure 9(b)). On the stromal side of the membrane, some fissures could be found (Figure 9(c2)) but, otherwise, no different morphology compared with the native hAM. The hAM analyzed at the end of the air–liquid culture showed partially single-layered, partially multilayered, or pseudostratified epithelium (Figure 9(d1) and (d2). Similar to the aspect under phase contrast optics, a large variation could be seen beetween raised epithelial cell complexes and areas not covered by any epithelial cells. A confluent fibroblast network on the chorion side also confirmed the findings of the analysis carried out by phase contrast microscopy (Figure 9(c3))
Figure 9.

Various hAM preparations. Scanning electron microscopy. (a1) (1000×) Epithelial side of a native hAM in plain shot. (a2) (5000×) Epithelial side of a native hAM in inclined shot. The hAM showed the typical hexagonal architecture of the epithelium. Marked with red: exemplary cell boundaries. (b) (100×) Epithelial side of a de-epithelialized hAM in plain shot. The hexagonal epithelium was completely removed, although rare detritus remained (arrow). (c1–c3) (1000×) Inclined shot of the stromal side of a native hAM, and plain shots of a de-epithelialized membrane and a recolonized hAM. The native membrane and the de-epithelialized hAM showed no cells, the latter showing occasional fissures (arrows). The re-epithelialized hAM had a confluent typical fibroblast network. (d1–d2) (1000×) Epithelial side of a hAM at the end of the air–liquid culture in plain and inclined shot. The epithelium showed a typical morphology with several rounded cells (arrows) found above.
OCT
Several native, trypsinized, and recolonized hAM were analyzed within a pilot project to test whether the OCT was suitable for visualizing the hAM. In particular, the positioning of the hAM in front of the OCT was difficult and only possible under non-sterile conditions. We were however able to prepare an overview image and a thin-layered series of images (Figure 10(a)–(c)). Whereas the native (Figure 10(a)) and recolonized hAM (Figure 10(c)) were significantly thicker, mostly by >1 mm, the de-epithelialized hAM (Figure 10(b)) was comparatively thin. Exact measurements were not obtained because of possible artifacts from light refraction. The epithelial layers seemed to be well definable with respect to the underlying matrix. Furthermore, the epithelia of the different hAM showed visible differences in thickness with the recolonized hAM seeming to have the thickest epithelium. At various areas of the membranes, we were able to recognize defects in the membrane that were not detectable by phase contrast microscopy
Figure 10.
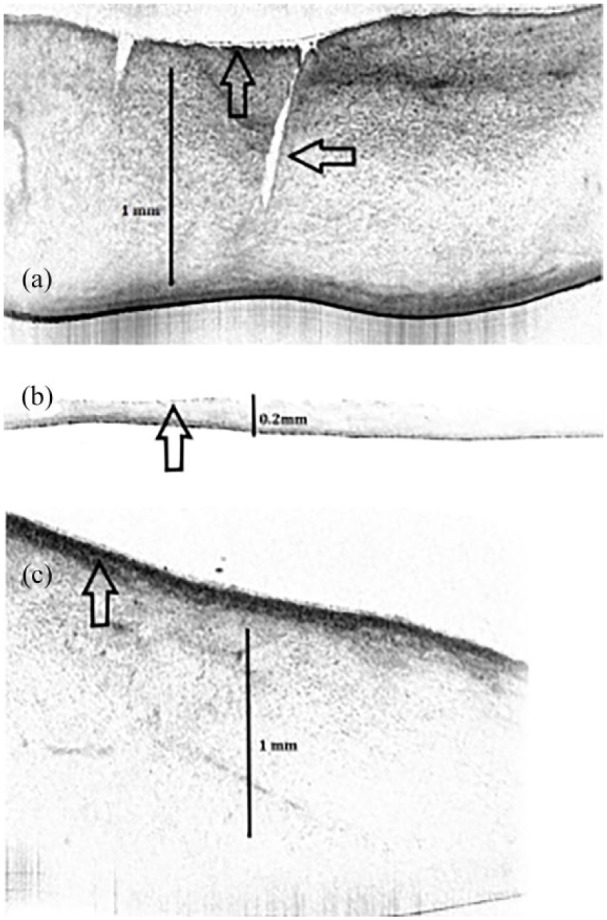
Native and variously processed hAM. OCT. (a) Display of a native hAM with a profound defect (horizontal arrow). The epithelium is thin (vertical arrow). (b) De-epithelialized hAM, which is significantly thinner and virtually without an epithelium (vertical arrow). (c) Recolonized hAM. The membrane is thick with a prominent epithelium (vertical arrow).
Discussion
Various de-epithelialization methods were compared in this study. Trypsin showed the best results with regard to the removal of the epithelium and the integrity of the basement membrane. We also obtained good results with NaOH,23 which until now is not as established as trypsinization and should therefore be further evaluated. The SDS/SDC and Triton/SDC protocols were used because of their effective decellularization of heart valves. Contrary to our expectations that the amniotic epithelium, which has to resist mechanical strains in the uterus,30 would act similarly to the endothelium of the equally mechanically stressed heart valves, the two surfactant-containing decellularization methods seemed to be insufficient to remove the epithelium in contrast to other authors who have presented better results using such methods.31 Often, more rigorous decellularization is accompanied with more damage to the extracellular matrix. Trypsin is well known for its strong collagenase effect, which might however lead to mechanical instability of the stroma.13 Light microscopy (LM) and Scanning electron microscopy (SEM) revealed some tears in membranes treated with trypsin; this indicates that trypsin causes tissue damage. Moreover, the further handling of the de-epithelialized hAM was difficult because some membranes, often when fixed on the insert, showed macroscopic lesions and had to be discarded. Regarding the non-epithelial side, there were differences in the decellularization properties of the different methods with the detergents being more efficient. However, when observing the re-cultivation, there were no differences with complete confluent fibroblast networks regardless of the previous decellularization.
Alternative methods have been published in which only the epithelial side comes into contact with the enzymatic agent.25 These should be further evaluated with regard to possible tissue damage and to the effect on re-cultivation of the non-epithelial side. In addition, considering the wide variance of the amniotic tissue used, a continuous phase contrast microscopy control of the de-epithelialization should be performed to minimize the exposure time to the enzyme. Overall, despite there being many different de-epithelialization methods for amniotic membranes, no ideal method has been established as yet; similar conclusions have been expressed by other authors.32
The isolation and cultivation of the keratinocytes and fibroblasts was largely problem-free. Moreover, primary cultivation with trypsin-de-epithelialized hAM showed confluent results. With regard to the composition of the air–liquid medium, a keratinocyte-based medium with the addition of 5%–10% serum provided both good fibroblast and good keratinocyte viability. Noticeably, the confluence of the recultivated hAM was partially lost with a few areas without any epithelium and many areas with significant multilayered epithelium and keratinization. Other research groups cultivating hAM with non-keratinizing epithelium have not described such findings.21,33 As we did not perform cell viability tests on the hAM, the reason for these findings remains uncertain: Possible explanations are that the remaining trypsin molecules might have caused local cytotoxicity or heterogeneous membrane conditions that might have inhibited nutrition from the chorion side while the tissue was under air–liquid conditions. To date, no investigations have been published regarding the filter characteristics of de-epithelialized hAM, and therefore, it remains uncertain where the medium might have penetrated the membrane sufficiently to achieve confluent keratinization. Other authors have mentioned such perfusion as the most important aspect for obtaining confluent and differentiated keratinocytes.34 Further testing, such as viability essays with the hAM after de-epithelialization should provide further data regarding this issue. Moreover, more air–liquid media should be evaluated to enhance further the keratinocyte viability. Some serum-free synthetic media for fibroblasts are the subject of ongoing research.27,35 They might, with the addition of specific growth factors for keratinocytes, be adapted to the requirement of air–liquid culture. Overall, the re-epithelialized hAM can still be improved regarding gap-less epithelialization. However, they should already be tested in vivo as skin substitutes to look for possible advantages regarding wound healing to justify further research efforts.
The OCT examination was considered as a pilot project. Whereas the presentation of the hAM was feasible, we found it difficult to adjust the hAM in the sterile cell-culture well in front of the OCT apparatus. Furthermore, refraction at the various surfaces complicated the location of the membrane structures. In addition, no comparable images have previously been published for any comparisons to be made. Nonetheless, the advantages of the OCT include the non-invasive fast characterization of the hAM with regard to the thickness of the membrane and the identification of lesions not visible by phase contrast microscopy, both important features for a mechanically stable membrane.36 In the future, a standardized OCT examination might be employed to exclude unsuitable membranes for further experimental and clinical use in order to improve the results obtained. Such standardization should be approached in future studies.
When using hAM, its variable inter-individual conditions should be considered: Many reasons, such as age, general condition, gestational age, and the sex of the fetus, play significant roles in the expression of cytokines, hyaluronic acid, and other factors.37 Furthermore, intra-individual factors such as the sampling location are relevant: hAM from the cervical zone is often thinner with less epithelium than hAM sampled from other zones, and the expression of cytokines also varies.37 In addition, the alignment of the collagen fibers of the hAM is irregular with occasional weak spots.36 For logistical reasons, we used cryoconserved hAM. Other authors have found the low expression of angiogenin, IL-6, and CP-1 in such processed membranes compared with fresh membranes,36 whereas still others could find no such differences.37 As the conservation of the hAM has possible harmful effects, it should be avoided in further studies. These limitations must be considered when using biological membranes, and findings have to be interpreted with great caution.
Conclusion
Treatment with trypsin and a cell scraper showed the best de-epithelialization results in hAM but were accompanied with detectable stromal strains. Nevertheless, with the use of serum-containing keratinocyte-based growth medium, the culturing of keratinocytes and fibroblasts on the de-epithelialized hAM in an air–liquid culture was possible and resulted in a mostly keratinized surface. Whereas tissue-engineered skin grafts on a base of hAM can still be improved with regard to gap-less epithelialization, they should be evaluated in in vivo experiments to justify further research.
Acknowledgments
S.J.: author and scientist involved in all experimental testing, analysis, and interpretation of the acquired data. M.R.K.: involved in the development of the study design and in the interpretation and discussion of the results obtained. P.P.: handling of the SEM, obtaining, and interpreting of the obtained SEM images. M.S.: general advice for all laboratory tasks regarding materials used and performance of histologic and cell culture. A.v.B.: author and scientist involved in all experimental testing and analysis.
Footnotes
Declaration of conflicting interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
ORCID iD: Samuel John  https://orcid.org/0000-0001-6113-238X
https://orcid.org/0000-0001-6113-238X
References
- 1. Presland RB, Jurevic RJ. Making sense of the epithelial barrier: what molecular biology and genetics tell us about the functions of oral mucosal and epidermal tissues. J Dent Educ 2002; 66(4): 564–574. [PubMed] [Google Scholar]
- 2. Prasanna M, Mishra P, Thomas C. Delayed primary closure of the burn wounds. Burns 2004; 30(2): 169–175. [DOI] [PubMed] [Google Scholar]
- 3. Rhee PH, Friedman CD, Ridge JA, et al. The use of processed allograft dermal matrix for intraoral resurfacing: an alternative to split-thickness skin grafts. Arch Otolaryngol Head Neck Surg 1998; 124(11): 1201–1204. [DOI] [PubMed] [Google Scholar]
- 4. Xiao-Wu W, Herndon DN, Spies M, et al. Effects of delayed wound excision and grafting in severely burned children. Arch Surg 2002; 137(9): 1049–1054. [DOI] [PubMed] [Google Scholar]
- 5. Zelen CM, Serena TE, Denoziere G, et al. A prospective randomised comparative parallel study of amniotic membrane wound graft in the management of diabetic foot ulcers. Int Wound J 2013; 10(5): 502–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Moore P, Moore M, Blakeney P, et al. Competence and physical impairment of pediatric survivors of burns of more than 80% total body surface area. J Burn Care Rehabil 1996; 17(6 Pt 1): 547–551. [DOI] [PubMed] [Google Scholar]
- 7. Rosenberg AS, Munitz TI, Maniero TG, et al. Cellular basis of skin allograft rejection across a class I major histocompatibility barrier in mice depleted of CD8+ T cells in vivo. J Exp Med 1991; 173(6): 1463–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tanaka A, Hatoko M, Tada H, et al. An evaluation of functional improvement following surgical corrections of severe burn scar contracture in the axilla. Burns 2003; 29(2): 153–157. [DOI] [PubMed] [Google Scholar]
- 9. Tomasek JJ, Gabbiani G, Hinz B, et al. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol 2002; 3(5): 349–363. [DOI] [PubMed] [Google Scholar]
- 10. Jiang D, Liang J, Noble PW. Hyaluronan in tissue injury and repair. Annu Rev Cell Dev Biol 2007; 23, 435–461. [DOI] [PubMed] [Google Scholar]
- 11. Loeffelbein DJ, Rohleder NH, Eddicks M, et al. Evaluation of human amniotic membrane as a wound dressing for split-thickness skin-graft donor sites. Biomed Res Int 2014; 2014: 572183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. MacNeil S. Progress and opportunities for tissue-engineered skin. Nature 2007; 445(7130): 874–880. [DOI] [PubMed] [Google Scholar]
- 13. Gilbert TW. Strategies for tissue and organ decellularization. J Cell Biochem 2012; 113(7): 2217–2222. [DOI] [PubMed] [Google Scholar]
- 14. Kubota Y, Kleinman HK, Martin GR, et al. Role of laminin and basement membrane in the morphological differentiation of human endothelial cells into capillary-like structures. J Cell Biol 1988; 107(4): 1589–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. LeBleu VS, Macdonald B, Kalluri R. Structure and function of basement membranes. Exp Biol Med 2007; 232(9): 1121–1129. [DOI] [PubMed] [Google Scholar]
- 16. Wood FM, Stoner M. Implication of basement membrane development on the underlying scar in partial-thickness burn injury. Burns 1996; 22(6): 459–462. [DOI] [PubMed] [Google Scholar]
- 17. Shruthy R, Sharada P, Swaminathan U, et al. Immunohistochemical expression of basement membrane laminin in histological grades of oral squamous cell carcinoma: a semiquantitative analysis. J Oral Maxillofac Pathol 2013; 17(2): 185–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Muller L, Brighton LE, Carson JL, et al. Culturing of human nasal epithelial cells at the air liquid interface. J Vis Exp 2013; 80. DOI: 10.3791/50646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Boyce ST, Ham RG. Calcium-regulated differentiation of normal human epidermal keratinocytes in chemically defined clonal culture and serum-free serial culture. J Invest Dermatol 1983; 81(1 Suppl.): 33s–40s. [DOI] [PubMed] [Google Scholar]
- 20. Rittie L, Fisher GJ. Isolation and culture of skin fibroblasts. Methods Mol Med 2005; 117: 83–98. [DOI] [PubMed] [Google Scholar]
- 21. Ahn KM, Lee JH, Hwang SJ, et al. Fabrication of myomucosal flap using tissue-engineered bioartificial mucosa constructed with oral keratinocytes cultured on amniotic membrane. Artif Organs 2006; 30(6), 411–423. [DOI] [PubMed] [Google Scholar]
- 22. Baraki H, Tudorache I, Braun M, et al. Orthotopic replacement of the aortic valve with decellularized allograft in a sheep model. Biomaterials 2009; 30(31): 6240–6246. [DOI] [PubMed] [Google Scholar]
- 23. Saghizadeh M, Winkler MA, Kramerov AA, et al. A simple alkaline method for decellularizing human amniotic membrane for cell culture. PLoS One 2013; 8(11): e79632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang KX, Zhang JF, Zhan QP, et al. Effect of trypsin and Triton-X 100 for decellularization of porcine aortic heart valves. Di Yi Jun Yi Da Xue Xue Bao 2005; 25(1): 22–25. [PubMed] [Google Scholar]
- 25. Mariappan I, Maddileti S, Savy S, et al. In vitro culture and expansion of human limbal epithelial cells. Nat Protoc 2010; 5(8): 1470–1479. [DOI] [PubMed] [Google Scholar]
- 26. Strober W. Trypan blue exclusion test of cell viability. Curr Protoc Immunol 2015; 111: A3.B. 1–A3.B. 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lin H, Li H, Cho HJ, et al. Air-liquid interface (ALI) culture of human bronchial epithelial cell monolayers as an in vitro model for airway drug transport studies. J Pharm Sci 2007; 96(2): 341–350. [DOI] [PubMed] [Google Scholar]
- 28. Cardiff RD, Miller CH, Munn RJ. Manual hematoxylin and eosin staining of mouse tissue sections. Cold Spring Harb Protoc 2014; 2014(6): 655–658. [DOI] [PubMed] [Google Scholar]
- 29. Zhang L, Zou D, Li S, et al. An ultra-thin amniotic membrane as carrier in corneal epithelium tissue-engineering. Sci Rep 2016; 6: 21021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bourne GL. The anatomy of the human amnion and chorion. Proc R Soc Med 1966; 59(11 Part 1): 1127–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Figueiredo GS, Bojic S, Rooney P, et al. Gamma-irradiated human amniotic membrane decellularised with sodium dodecyl sulfate is a more efficient substrate for the ex vivo expansion of limbal stem cells. Acta Biomater 2017; 61: 124–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shortt AJ, Secker GA, Lomas RJ, et al. The effect of amniotic membrane preparation method on its ability to serve as a substrate for the ex-vivo expansion of limbal epithelial cells. Biomaterials 2009; 30(6): 1056–1065. [DOI] [PubMed] [Google Scholar]
- 33. Nakamura T, Endo K, Cooper LJ, et al. The successful culture and autologous transplantation of rabbit oral mucosal epithelial cells on amniotic membrane. Invest Ophthalmol Vis Sci 2003; 44(1): 106–116. [DOI] [PubMed] [Google Scholar]
- 34. Prunieras M, Regnier M, Woodley D. Methods for cultivation of keratinocytes with an air-liquid interface. J Invest Dermatol 1983; 81(1 Suppl.): 28s–33s. [DOI] [PubMed] [Google Scholar]
- 35. Ejiri H, Nomura T, Hasegawa M, et al. Use of synthetic serum-free medium for culture of human dermal fibroblasts to establish an experimental system similar to living dermis. Cytotechnology 2015; 67(3): 507–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schmidt W, Klima G. Experimental and histologic studies on fetal membrane tensility and membrane rupture. Zentralbl Gynakol 1989; 111(3): 129–141. [PubMed] [Google Scholar]
- 37. Litwiniuk M, Grzela T. Amniotic membrane: new concepts for an old dressing. Wound Repair Regen 2014; 22(4): 451–456. [DOI] [PubMed] [Google Scholar]