

Abstract
Gene expression can be regulated at multiple levels, but it is not known if and how there is broad coordination between regulation at the transcriptional and post-transcriptional levels. Transcription factors and chromatin regulate gene expression transcriptionally, whereas microRNAs (miRNAs) are small regulatory RNAs that function post-transcriptionally. Systematically identifying the post-transcriptional targets of miRNAs and the mechanism of transcriptional regulation of the same targets can shed light on regulatory networks connecting transcriptional and post-transcriptional control. We used individual-nucleotide resolution UV crosslinking and immunoprecipitation (iCLIP) for the RNA-induced silencing complex (RISC) component AGO2 and global miRNA depletion to identify genes directly targeted by miRNAs. We found that Polycomb repressive complex 2 (PRC2) and its associated histone mark, H3K27me3, is enriched at hundreds of miRNA-repressed genes. We show that these genes are directly repressed by PRC2 and constitute a significant proportion of direct PRC2 targets. For just over half of the genes corepressed by PRC2 and miRNAs, PRC2 promotes their miRNA-mediated repression by increasing expression of the miRNAs that are likely to target them. miRNAs also repress the remainder of the PRC2 target genes, but independently of PRC2. Thus, miRNAs post-transcriptionally reinforce silencing of PRC2-repressed genes that are inefficiently repressed at the level of chromatin, by either forming a feed-forward regulatory network with PRC2 or repressing them independently of PRC2.
Transcription factors (TFs) and miRNAs together form the largest components of gene regulatory networks and can regulate gene expression through both distinct and coordinated regulatory mechanisms. One of the ways in which TFs can coordinate their regulatory impact with miRNAs is by forming a feed-forward regulatory network. In such a network, a TF regulates a miRNA and both coregulate a common target. In a coherent feed-forward regulatory network, the outcomes of both direct and indirect regulation by a TF are consistent. Many such TF-miRNA feed-forward regulatory networks have been shown to functionally impact several processes in development and disease (O'Donnell et al. 2005; Tsang et al. 2007; Hobert 2008; Polioudakis et al. 2013; Gerloff et al. 2015; Lin et al. 2015).
One potential function of coherent TF-miRNA feed-forward regulatory networks is to reinforce transcriptional regulation at the post-transcriptional level. In particular, this can help suppress residual transcripts produced from leaky transcription of transcriptionally silenced genes. This is most crucial during switches in transcriptional states in response to stress, developmental transitions, cell cycle stages, or other external stimuli (Farh et al. 2005; Wu et al. 2015; del Rosario et al. 2016).
PRC2 is an epigenetic regulator complex that transcriptionally silences genes through modification of histone H3 with trimethylation at lysine 27 (H3K27me3). PRC2 plays a critical role in maintaining the silenced state of genes involved in development and several cancers (Di Croce and Helin 2013; Aranda et al. 2015). In this study, we provide data that points to a broad role of miRNAs in which they independently strengthen and also reinforce the silencing of PRC2-repressed genes post-transcriptionally.
Results
Transcriptome-wide identification of miRNA targets
miRNAs primarily target mRNAs through interactions between their 5′ seed region and the 3′ untranslated region (3′ UTR) of the mRNA, mediated by the RNA-induced silencing complex (RISC). To identify the transcriptome-wide targets of miRNAs, we first performed RNA individual-nucleotide resolution UV crosslinking and immunoprecipitation (iCLIP) for the RISC component AGO2 in glioblastoma multiforme (GBM) cells (König et al. 2010). Several observations indicate that the AGO2-RNA interactions we identified using iCLIP were direct and specific. First, AGO2-RNA complexes were crosslinking- and RNase-treatment–specific (Fig. 1A). Second, reads from iCLIP were enriched at 3′ UTRs as expected from the binding of RISC (Fig. 1B). Third, reads mapping to genes showed high correlation and were reproducible across independent biological replicate experiments (Fig. 1C). We identified 45,362 peaks mapping to 5896 protein-coding genes that were common across two independent biological replicate experiments.
Figure 1.

Identification of miRNA targets by AGO2 iCLIP. (A) Autoradiograph showing AGO2-crosslinked ribonucleoprotein complexes from two replicates. The red box represents the region that was processed to make cDNA libraries. (B) Genomic enrichment of AGO2-crosslinked reads mapping to mRNAs. The plot shows total reads mapped to a genomic feature, normalized to the total length of the feature in the genome. (C) Correlation of the total reads mapping to genes between the two independent replicates of AGO2 iCLIP (left). Genome Browser images showing AGO2 iCLIP peaks at 3′ UTRs for two protein-coding genes (right). (D) AGO2-interacting miRNAs plotted in descending order of read coverage (left). Sequences enriched proximal to AGO2 iCLIP peaks on mRNAs that significantly match the binding sequence for AGO2-interacting miRNAs. Corresponding miRNA seed sequences are also shown (right). (E) PPIF transcript levels in U87MG cells treated with negative control inhibitor or miR-23a-3p inhibitor. Error bars represent standard error across four replicates (two biological and two technical). (*) P < 0.05, paired t-test. (F) PPIF protein levels in U87MG cells treated with negative control inhibitor or miR-23a-3p inhibitor. The immunoblot showing two independent replicates (left) was quantified using ImageJ (right).
In addition to mRNA targets, the AGO2 iCLIP experiment also recovered miRNAs that AGO2 interacts with, allowing for more detailed analysis of functional miRNA–mRNA interactions. Although enrichment of miRNAs in AGO2 iCLIP showed high correlation to their expression levels; miRNAs with the highest expression were not necessarily the most enriched (Supplemental Fig. S1A). We found that the region spanning 200 bases around AGO2 iCLIP peaks was significantly enriched for the seed sequences of several of the top AGO2-interacting miRNA families (Fig. 1D; Supplemental Fig. S1A). In addition, the predicted mRNA target sites of the top 10 highly enriched miRNAs were significantly more likely to occur proximal to AGO2 iCLIP peaks than background mRNAs (Supplemental Fig. S1B). This suggests that the miRNAs associated with AGO2 bind to a significant proportion of the AGO2-enriched mRNAs, further attesting to the ability of the iCLIP experiment to broadly identify the targets of active miRNAs.
To further support the miRNA–mRNA interactions we identified, we validated the regulation of PPIF by miR-23a-3p. miR-23a-3p was among the most enriched miRNAs in our AGO2 iCLIP data set. We selected PPIF as a target of miR-23a-3p because it had a predicted binding site close to an AGO2 iCLIP peak (Supplemental Fig. S2A). Several lines of evidence show that miR-23a-3p directly represses PPIF expression. First, treatment of cells with an inhibitor of miR-23a-3p led to an induction of both PPIF transcript and protein levels (Fig. 1E,F). Second, we checked if the repression of PPIF was through a direct interaction between the 3′ UTR of PPIF and the miR-23a-3p seed region. We performed luciferase reporter assays in which we cloned the 3′ UTR of PPIF and compared luciferase expression to constructs that were mutated at the sites of interaction. We found that the effect of inhibitor treatment on regulation of the PPIF 3′ UTR was abolished upon deletion of the miR-23a-3p binding sites (Supplemental Fig. S2B). Third, both deletion of the miRNA binding sites and disruption of miRNA–mRNA interactions through base substitutions led to a similar rescue in luciferase activity (Supplemental Fig. S2C).
miRNA-repressed genes are enriched for PRC2 binding and H3K27me3
Although AGO2 iCLIP identified all the interaction targets of RISC, the AGO2-RISC complex can regulate gene expression in both a miRNA-dependent and -independent manner (Leung et al. 2011). To specifically identify miRNA-dependent genes, we adopted a strategy to globally deplete miRNAs, followed by expression profiling. Ectopic overexpression of the Vaccinia Virus protein VP55 has been previously shown to polyadenylate and degrade miRNAs, causing a global loss of miRNAs. Transcriptome changes in cells overexpressing VP55 closely match that of cells in response to loss of DICER, indicating that VP55 overexpression does not cause major transcriptional perturbations independent of its effect on miRNAs (Backes et al. 2012; Aguado et al. 2015). We cloned VP55 into a Sleeping Beauty transposon system under a doxycycline-inducible promoter and stably integrated this construct into T98G cells to generate a GBM cell line capable of doxycycline-responsive loss of miRNAs (Kowarz et al. 2015). miR-21, the most abundant miRNA in these cells, was almost completely depleted within 24 h of doxycycline treatment (Fig. 2A). PCR-amplified cDNA libraries prior to size-selection for miRNA-seq showed a significant depletion of miRNAs (Supplemental Fig. S3A). miRNA-seq showed a global depletion of ∼90% of all cellular miRNAs (Fig. 2B). To identify the genes regulated by miRNAs genome-wide, we performed mRNA-seq from doxycycline treated (+ Dox) and untreated cells (− Dox); 3844 protein-coding transcripts were up-regulated and thus repressed by miRNAs, and 3668 transcripts were down-regulated upon loss of miRNAs (Supplemental Fig. S3B). Some of the most strongly miRNA-repressed genes included several genes from the histone gene family. This is consistent with VP55-responsive genes identified previously in HEK293T cells (Aguado et al. 2015).
Figure 2.

miRNA-repressed genes are enriched for PRC2 binding and H3K27me3. (A) miR-21 expression levels in response to VP55 induction. (Dox) doxycycline. (B) Volcano plot of differentially expressed miRNAs in response to VP55 induction. The x-axis represents log2 fold difference between expression of miRNAs in VP55-induced and noninduced cells. The y-axis represents −log2 P-value of the expression difference calculated using DESeq2. (C,D) Enrichment for H3K27me3 (C) and SUZ12 (D) at genes derepressed in miRNA-depleted cells (miRNA-repressed genes). Enrichments were calculated using Enrichr (Chen et al. 2013).
Of the miRNA-repressed transcripts, 37% interacted directly with AGO2 based on iCLIP, suggesting that these were direct functional targets of miRNAs. Thus, miRNA-repressed genes include direct as well as indirect targets that could be regulated at the level of transcription (Gosline et al. 2016). To check if the miRNA-repressed genes were also regulated by specific transcription factors, we checked for the enrichment of TF binding sites proximal to their promoters. We used Enrichr to detect TF regulatory signatures in the ranked list of miRNA-repressed genes (Chen et al. 2013). We found a significant enrichment for H3K27me3 (Fig. 2C) and a corresponding enrichment for SUZ12, a member of the PRC2 complex which adds H3K27me3 (Fig. 2D), at miRNA-repressed genes. This suggested the possibility that many miRNA-repressed genes could additionally be regulated at the level of chromatin by PRC2 and H3K27 methylation. As a control, we also performed Enrichr analysis using a set of genes expressed in the same range as the miRNA-repressed genes, but not repressed by miRNAs. The control set showed no enrichment for any specific TF but did show an enrichment for H3K9me3 and H3K27me3, as would be expected from low-expressed genes (Supplemental Fig. S4A). To determine if this could be a general mode of dual regulatory control in other cell types, we analyzed data from mouse embryonic stem cells (mESCs) with a knockout of the miRNA-processing factor DICER, which also results in loss of miRNAs (Zheng et al. 2014). Similar to GBM cells, we found that genes repressed by miRNAs in mESCs also showed enrichment for H3K27me3 and PRC2 (Supplemental Fig. S4B,C).
miRNAs repress hundreds of genes directly repressed by PRC2
Based on the above results, we hypothesized that many genes that are post-transcriptionally repressed by miRNAs are also transcriptionally repressed by PRC2. To test this hypothesis, we generated a CRISPR-Cas9 knockout of EZH2 in GBM cells. H3K27me3 levels, as well as levels of SUZ12, were reduced in EZH2−/− cells, signifying loss of the PRC2 complex (Supplemental Fig. S5A; Pasini et al. 2004). Upon loss of EZH2, 1519 protein-coding genes were down-regulated, and 1834 protein-coding genes were up-regulated (derepressed) (Fig. 3A). A significant number of the EZH2-repressed genes (444 genes, P = 3.5 × 10−8 by hypergeometric test) were also repressed by miRNAs. Consistent with our Enrichr analysis, this data suggests that the genes repressed by EZH2 are also repressed by miRNAs. Alternatively, low-expressed genes could be regulated by miRNAs irrespective of them being repressed by PRC2. To test this alternative possibility, we utilized a bootstrapping approach (Methods) in which we tested the overlap of the 3844 miRNA-repressed genes with random sets of 1834 genes expressed at similar levels to those of the EZH2-repressed genes. This bootstrap analysis showed that the number of genes corepressed by EZH2 and miRNAs was indeed highly significant (P < 1 × 10−4). Thus, the overlap we observed between EZH2 and miRNA-repressed genes is not driven by the fact that both sets of genes are low-expressed genes.
Figure 3.

miRNAs repress hundreds of genes directly repressed by PRC2. (A) Heat map showing differential expression of protein-coding genes in response to EZH2 knockout. Genes are ranked in increasing order of log2 fold change (≤−0.5 and ≥0.5 log2 fold change) in WT/EZH2−/−. (B) Heat map showing enrichment scores (−log10 Q-value calculated using MACS2) for EZH2, H3K27me3, and H3K4me3 for genes plotted in and ordered as in A. (C) Average EZH2, H3K27me3, and H3K4me3 ChIP-seq signal for genes plotted in A. (D, left) Heat map plotted as in B for genes corepressed by EZH2 and miRNAs. (Right) Overlap between genes directly repressed by EZH2 and gene repressed by miRNAs. Significance of overlap was calculated using the hypergeometric test. (E) qRT-PCR validation of genes repressed by both EZH2 and miRNAs, plotted as in Figure 1E. (*) P < 0.05, paired t-test.
To determine if the genes corepressed by EZH2 and miRNAs were directly regulated by EZH2, we performed ChIP-seq for EZH2, as well as for H3K27me3. We detected EZH2 binding proximal to the transcription start site (TSS) of 6132 genes where the level of H3K27me3 positively correlated with that of EZH2 binding (Supplemental Fig. S5B,C). Genes derepressed upon loss of EZH2 (EZH2-repressed genes) showed EZH2 binding and H3K27me3 (Fig. 3B; Supplemental Fig. S6A). Consistent with previous studies, EZH2-repressed genes also showed occupancy by H3K4me3 (Fig. 3B; Supplemental Fig. S6A), with the enrichment for EZH2 and H3K27me3 showing a reciprocal relationship to H3K4me3 at EZH2-repressed genes (Fig. 3C; Abou El Hassan et al. 2015; Jadhav et al. 2016). Of the 1834 EZH2-repressed genes, 959 had an EZH2 chromatin peak close to their TSS and were thus likely directly repressed by EZH2.
We then checked if the 444 genes corepressed by EZH2 and miRNAs were directly regulated by EZH2. Of these 444 genes, 213 genes showed EZH2 binding proximal to their TSS. These 213 genes that were jointly and directly repressed by EZH2 and by miRNAs constituted 22% of all direct EZH2 targets (P = 0.01 by hypergeometric test) (Fig. 3D). This overlap was also significant (P = 0.0002) based on a bootstrapping approach to control for the low expression of the genes (Methods). We verified derepression upon loss of miRNAs or upon loss of EZH2 of multiple genes using qRT-PCR (Fig. 3E). Of the genes corepressed by EZH2 and miRNAs, 34.7% also showed direct interaction with AGO2 by iCLIP (Supplemental Fig. S6B,C). miRNAs thus post-transcriptionally reinforce transcriptional repression by PRC2 of at least one-fifth of all PRC2 targets.
miRNAs repress PRC2 target genes through a feed-forward regulatory network with PRC2
PRC2 and miRNAs were recently shown to independently corepress endocytosis genes in mESCs (Mote et al. 2017). To test if miRNAs and PRC2 repressed a common set of genes independently in GBM cells, we depleted miRNAs globally by overexpressing VP55 in EZH2−/− T98G cells and performed miRNA-seq and mRNA-seq. Most miRNAs were depleted within 24 h of doxycycline treatment (Fig. 4A; Supplemental Fig. S7A). Both WT and EZH2−/− cells showed similar responses to doxycycline treatment in terms of the number of miRNAs depleted and the extent of depletion, indicating that VP55 overexpression was equally effective in WT and EZH2−/− cells (Supplemental Fig. S7B,C). If miRNAs and EZH2 repressed a common set of genes independently of one another, we would expect that these corepressed genes would be repressed by miRNAs even in the absence of EZH2, and therefore show derepression upon miRNA depletion even in EZH2−/− cells. Of the 213 genes corepressed by EZH2 and miRNAs, 116 genes (P = 8.17 × 10−12) showed reduced derepression in response to miRNA depletion in EZH2−/− cells compared to WT cells (Fig. 4B–D), suggesting that PRC2 and miRNAs work coordinately, rather than independently, to repress these genes. Consistent with the heat map (Fig. 4B), these feed-forward regulated genes showed derepression in response to miRNA depletion in WT but not in EZH2−/− cells (Fig. 4C). We confirmed this finding by qRT-PCR for several genes (Fig. 4E). The impaired derepression upon miRNA depletion in the absence of EZH2 is also not due to genes attaining maximally derepressed expression levels in EZH2−/− cells. The remaining 47% of the genes corepressed by EZH2 and miRNAs that were not part of the feed-forward network showed significant derepression in response to miRNA loss both in WT and EZH2−/− cells, indicating that multiple levels of repression are detectable for genes that are not part of the coordinated feed-forward regulatory network (Supplemental Fig. S8).
Figure 4.

miRNAs repress PRC2 target genes through a feed-forward regulatory network with PRC2. (A) Depletion of miR-21 in response to VP55 induction in EZH2−/− cells. (B) Heat map showing derepression of gene expression in response to miRNA loss (+/− Dox) in WT (WT, +/− Dox), and EZH2−/− cells (EZH2−/−, +/− Dox) for genes corepressed by EZH2 and miRNAs. Only 146 of the 213 corepressed genes that had an average of 0.5 log2 fold difference in derepression between WT and EZH2−/− cells are shown. Genes are ordered in decreasing order of WT, +/− Dox/EZH2−/−, +/− Dox values. (C) Box plot comparing expression of feed-forward regulated genes in miRNA-depleted (+ Dox) and nondepleted (− Dox) WT and EZH2−/− cells. (*) P < 0.05; (NS) not significant. (D) Overlap between genes corepressed by EZH2 and miRNAs, and genes showing lower derepression upon miRNA depletion in EZH2−/− cells compared to WT cells. Significance of overlap was calculated using the hypergeometric test. (E) qRT-PCR showing transcript levels in miRNA-depleted WT and EZH2−/− cells, plotted as in Figure 1E.
This coordination between PRC2 and miRNAs can be accounted for by a feed-forward regulatory network in which EZH2 promotes the miRNA-mediated repression of a subset (54%) of its direct targets, by activating miRNAs that repress those targets. Alternatively, it is possible that miRNAs somehow promote PRC2 function, perhaps by promoting the binding of PRC2 to its repression targets (Graham et al. 2016). To distinguish between these possibilities, we performed EZH2 ChIP-seq in cells overexpressing VP55 (+ Dox) and untreated (−Dox) controls. EZH2 binding in the untreated cells (VP55 integrated cell line) was similar to T98G WT cells (Supplemental Fig. S9A–C). If miRNAs promoted PRC2 binding at the feed-forward regulated genes, we would expect EZH2 binding to decrease in response to loss of miRNAs (+ Dox). However, there was no reduction in the level of EZH2 binding in Dox-treated relative to untreated cells at the feed-forward regulated genes (Fig. 5A,B). We used qRT-PCR to verify derepression of key genes in response to miRNA depletion after Dox treatment in the same cultures of cells used for this ChIP-seq experiment (Supplemental Fig. S9D). These results confirm that miRNAs do not promote PRC2 binding or repression activity at the genes that are coordinately repressed by both miRNAs and PRC2, indicating instead that PRC2 promotes the miRNA-mediated repression of many of its targets.
Figure 5.

(A) Normalized density of EZH2 ChIP enrichment scores (−log10 Q-value calculated using MACS2, left) and EZH2 ChIP-seq reads (right) in the 10-kb region spanning the TSS of the feed-forward regulated genes in cells treated with Dox (+ Dox) and untreated (− Dox). (B) Genome Browser view of FRMD4B showing tracks for EZH2 ChIP-seq in WT +Dox and WT −Dox cells containing VP55. (C) Heat map showing differential expression of miRNAs in response to EZH2 knockout. Rows are ordered from low to high log2 fold change between WT and EZH2−/− miRNA counts. (D) Predicted interactions between 1000 random sets of 14 miRNAs and feed-forward regulated genes. The average across 1000 intersections is marked “Average.” The number of observed interactions between EZH2-activated miRNAs and feed-forward regulated genes is marked “Observed.” P-value = 0.024 was calculated using the bootstrap method (Methods). (E) Luciferase assay comparing miRNA inhibition of WT 3′ UTR transfected into WT and EZH2−/− cells.
Our data thus suggests a model in which PRC2 transcriptionally represses hundreds of genes in GBM cells and for a significant fraction of these genes, it further promotes additional repression by activating miRNAs that post-transcriptionally repress those genes. To identify miRNAs regulated by PRC2, we performed miRNA-seq in WT and EZH2−/− cells and identified 31 miRNAs that were activated and 84 miRNAs that were repressed by EZH2 (Fig. 5C). We found no EZH2 binding to chromatin around the EZH2-activated miRNAs, suggesting that EZH2 activated them indirectly.
To determine whether the feed-forward-regulated genes as a group were likely to be targeted by EZH2-activated miRNAs, we tested whether the number of predicted interactions between them were higher than expected by random chance. There were 342 unique interactions predicted between 14 EZH2-activated miRNAs (conserved miRNAs with mRNA target predictions) and 85 of the 116 feed-forward regulated genes. This observed number of interactions was significantly higher than expected by random chance, which we estimated using a bootstrapping approach (P = 0.024) (Methods; Fig. 5D). To verify that EZH2 promoted post-transcriptional repression of its direct targets, we performed luciferase reporter assays in EZH2−/− and WT cells where we cloned the 3′ UTR spanning the AGO2 binding sites in two feed-forward regulated genes, FRMD4B and SLCO5A1. We detected lower luciferase activity in WT cells compared to EZH2−/− cells for both genes, pointing to higher post-transcriptional repression in the presence of EZH2 (Fig. 5E). This implies that EZH2 indirectly promotes expression of the miRNAs that are likely to post-transcriptionally regulate its direct targets.
PRC2 regulates FRMD4B expression through a feed-forward regulatory network with let-7i
To verify the feed-forward regulatory network for a selected example in detail, we analyzed the repression of FRMD4B by PRC2. Consistent with direct transcriptional regulation by PRC2, both H3K27me3 and EZH2 were enriched near the promoter of FRMD4B (Fig. 6A). To verify that PRC2 promoted miRNA-mediated repression of FRMD4B, we measured its transcript levels in WT and EZH2−/− cells, with and without miRNA depletion. Loss of EZH2 caused derepression of FRMD4B, whereas depletion of miRNAs caused derepression of FRMD4B expression in WT but not in EZH2−/− cells (Fig. 6B), verifying that PRC2 promotes miRNA-mediated regulation of FRMD4B. To test if PRC2 promotes expression of a specific miRNA that targets FRMD4B, we first identified let-7i as an EZH2-activated miRNA with a predicted interaction site near an AGO2 iCLIP site on the FRMD4B transcript (Fig. 6C; Supplemental Fig. S10A). We then analyzed the regulation of FRMD4B by let-7i using luciferase assays with a WT or mutant FRMD4B 3′ UTR in WT and EZH2−/− cells. Deletion of the let-7i binding site led to a marked increase in luciferase signal in WT but a much smaller increase in EZH2−/− cells (P = 0.0098, paired t-test) (Fig. 6D). Thus, FRMD4B is more strongly repressed by let-7i in the presence of its activator EZH2.
Figure 6.

PRC2 regulates FRMD4B expression through a feed-forward regulatory network with let-7i. (A) Genome Browser view of FRMD4B showing tracks for EZH2 and H3K27me3 ChIP-seq. (B) qRT-PCR showing expression changes of FRMD4B in miRNA-depleted (+ Dox) and nondepleted (− Dox) WT and EZH2−/− cells, plotted as in Figure 1E. (*) P < 0.05. (C) Genome Browser view of let-7i in two replicates of WT and EZH2−/− cells. (D) Luciferase assay comparing miRNA inhibition between WT and mutant FRMD4B 3′ UTR (seed deletion) transfected WT and EZH2−/− cells. (E) Feed-forward regulatory network formed by PRC2, LIN28B, let-7i, and FRMD4B.
To address how EZH2 indirectly activates let-7i, we searched for likely repressors of let-7i among the genes directly repressed by EZH2. We found that LIN28B, a known repressor of the let-7 family of miRNAs, showed chromatin marks characteristic of EZH2 activity (Supplemental Fig. S10B). Our RNA-seq data showed that LIN28B was significantly derepressed upon loss of EZH2 (FDR-corrected P < 0.05), suggesting that it was directly repressed by EZH2. LIN28B is an RNA-binding protein that inhibits the biogenesis of let-7 miRNAs (Balzeau et al. 2017). Because LIN28B is known to be a common post-transcriptional repressor of the let-7 family, EZH2 repression of LIN28B might be expected to activate multiple members of the let-7 family. Indeed, several other let-7 miRNAs also showed derepression in EZH2−/− cells, although only let-7i and let-7f-3p met the stringent threshold for statistical significance we used earlier to identify derepressed miRNAs (Supplemental Fig. S10C). These data suggest that EZH2 activates let-7i and other let-7 miRNAs by directly repressing LIN28B (Fig. 6E). Taken together, these results confirm that PRC2 represses FRMD4B expression through a feed-forward regulatory network with let-7i.
Discussion
Transcription factors and miRNAs work in a highly coordinated manner, forming feed-back and feed-forward networks to regulate diverse processes. Using a combination of crosslinking based immunoprecipitation (iCLIP) and global miRNA depletion followed by RNA-seq, we identified miRNA-regulated genes genome-wide in GBM cells. This approach, however, only identified genes whose transcript levels are lowered due to miRNA function and thus likely underestimates the number of miRNA-repressed genes. Hundreds of miRNA-repressed genes nonetheless showed enrichment for PRC2 binding and H3K27me3 marks. We found that miRNAs post-transcriptionally reinforce the transcriptional repression of a significant fraction of PRC2 target genes, either independently of PRC2, or coordinately, by forming a feed-forward regulatory network with PRC2.
Overall, genes repressed by miRNAs and by PRC2 constituted 22% of all direct PRC2-repressed genes; 54% of these genes (feed-forward regulated genes) showed much less derepression in response to miRNA depletion in the absence of EZH2, indicating that PRC2 contributed to the repression function of some of these miRNAs. Although our data suggests miRNA-mediated post-transcriptional repression of PRC2 direct targets, we could verify AGO2 binding by iCLIP for only 34.7% of the genes corepressed by EZH2 and miRNAs (Supplemental Fig. S6B). This can be attributed to miRNA-independent binding of AGO2 and/or to technical limitations of iCLIP in capturing low-abundance transcripts (Leung et al. 2011; Müller-McNicoll et al. 2016; Bieniasz and Kutluay 2018). The difference in derepression in EZH2−/− cells was not due to reduced miRNA depletion by VP55 in EZH2−/− cells, because the extent and the number of miRNAs depleted in WT and EZH2−/− cells were almost identical (Supplemental Fig. S7B,C). The coordinated feed-forward repression by miRNAs and PRC2 was not due to a mechanism in which miRNAs promote PRC2 function or binding (Graham et al. 2016), because loss of miRNAs had no effect on PRC2 binding at the feed-forward regulated genes (Fig. 5A,B).
We propose that many genes that are transcriptionally repressed by PRC2 continue to produce transcripts at low levels due to inefficient repression by PRC2 at the chromatin and transcriptional level. These transcripts are then further repressed or destabilized by miRNAs, which thus reinforce the repressive function of PRC2 in one of two modes. In the coordinated mode, miRNAs that repress PRC2 targets are themselves indirectly activated by EZH2, forming feed-forward regulatory networks. In the independent mode, miRNAs and PRC2 corepress a common set of target genes independently (Fig. 7). This model would be in agreement with the notion that miRNAs are broadly involved in reinforcing transcriptional programs (Ebert and Sharp 2012). We found that PRC2 binds and trimethylates H3K27 to repress genes that also harbor active H3K4me3 marks. These genes could produce residual transcripts as a result of leaky transcription, and indeed we found this to be the case. Genes corepressed by PRC2 and miRNAs showed significantly higher expression upon depletion of miRNAs than genes silenced by PRC2 alone, even though PRC2 showed equal occupancy at both sets of genes (Supplemental Fig. S11).
Figure 7.
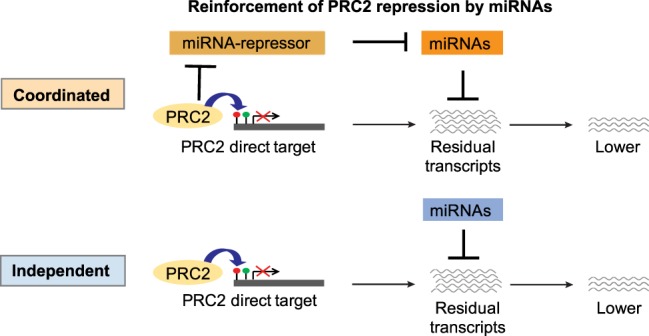
Model for reinforcement of PRC2 repression by miRNAs. Corepression of target genes occurs transcriptionally by PRC2 and post-transcriptionally by miRNAs, either in a coordinated or independent manner.
It is likely that this coordination between PRC2 and miRNA repression occurs in other cellular contexts, given the significant overlap in the genes regulated by PRC2 and miRNAs in mESCs (Supplemental Fig. S4). Our findings not only uncover a novel type of coordination between an epigenetic and post-transcriptional regulator of gene expression but could also provide insight into PRC2's regulatory function in a wide range of disease and developmental pathways.
Methods
Cell lines and reagents
GBM cell lines T98G and U87MG (ATCC-CRL-1690 and ATCC-HTB14) were grown in EMEM with 10% FBS (Gibco). All cell lines were maintained at 37°C and 5% CO2. Antibodies used were EZH2 (Abcam, 186006), AGO2 (Abcam, ab 57113), SUZ12 (Active motif 39877), H3K27me3 (Millipore 07-449), and PPIF (Abcam, ab 110324). pCMV(CAT)T7-SB100 was a gift from Zsuzsanna Izsvak (Addgene plasmid #34879). pSBtet-GP was a gift from Eric Kowarz (Addgene plasmid #60495).
RNA-seq, miRNA-seq and iCLIP
RNA-seq experiments were performed on poly(A) selected mRNA using NEXTFLEX Poly(A) Beads (Bioo 512980) as previously described (Hall et al. 2018). AGO2 iCLIP experiments were performed as previously described (König et al. 2010). Small RNA-seq libraries were prepared using NEBNext small RNA library preparation kit (NEB E7330) as previously described (Shivram and Iyer 2018).
Quantitative reverse transcription PCR (qRT-PCR)
Total RNA was extracted using TRIzol and reverse transcribed by SuperScript III reverse transcriptase (Thermo Fisher Scientific 18080085) using random hexamer primers. PCR was then performed on cDNA using Power SYBR Green master mix (Thermo Fisher Scientific 4367659). Transcript levels were normalized to 18S RNA levels, and fold changes were quantified using the ΔΔCT method (Livak and Schmittgen 2001).
Chromatin immunoprecipitations
Cells were crosslinked with 1% formaldehyde for 20 min and quenched with 125 mM glycine. The rest of the protocol was performed as previously described (Hall et al. 2018).
mRNA-seq and miRNA-seq analysis
Reads were aligned to the human genome (UCSC version hg38) using HISAT (Kim et al. 2015). featureCounts (Liao et al. 2014) was then used to count reads mapping to genes, and differential gene expression analysis was performed using DESeq2 (Love et al. 2014) with default parameters. Significantly differentially expressed genes were identified at a threshold of FDR-corrected P < 0.05 and log2 fold change ≥0.5 or ≤−0.5. For miRNA-seq, reads mapping to mature miRNAs (miRBase hg38) were counted using BEDTools (Quinlan and Hall 2010). Differentially expressed miRNAs were then identified using DESeq2 as described above, using the same thresholds (Love et al. 2014). Feed-forward target genes are those that show at least log2 fold 0.5 higher miRNA-mediated repression in T98G WT cells compared to EZH2−/−.
ChIP-seq analysis
ChIP-seq reads were aligned to the human genome (UCSC version hg38) using BWA (Li and Durbin 2009). MACS2 was then used to call peaks at the cutoff of P-value <0.01 (Zhang et al. 2008). Genes containing an EZH2 peak within 10 kb around the TSS were used as EZH2-bound genes for downstream analysis. For the heat maps plotted in Figure 3, B and D, and Supplemental Figure S5B, the signal represents enrichment scores (−log10 Q-value) output by MACS2. ChIP-seq data for H3K4me3 in GBM cells was obtained from our recently published study (Hall et al. 2018).
iCLIP analysis
iCLIP reads were first processed to remove duplicate reads using FASTX Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/) and then aligned to the human genome (UCSC version 19) using Bowtie 2 (Langmead and Salzberg 2012). Reads mapping to repeat regions were filtered out using RepeatMasker (Smit et al. 2013–2015). Peaks were then called using CLIPper using default parameters and filtered based on reproducibility across replicates (Lovci et al. 2013). miRNA seed enrichments in sequences spanning 200 bases around peaks were determined using HOMER software (Heinz et al. 2010). Prior to intersections with other data sets from RNA-seq, peak coordinates were converted to hg38 from hg19 using liftOver.
CRISPR-Cas9 knockout of EZH2
EZH2 sgRNA (sequence TTATCAGAAGGAAATTTCCG) was designed using http://crispr.mit.edu, and EZH2−/− T98G cells were generated using the GeneArt CRISPR-Cas kit (Thermo Fisher Scientific A21175) following the manufacturer's instructions. Knockout clones were verified by immunoblots.
Generation of VP55-expressing stable cell line
The VP55 coding sequence was PCR-amplified from a vector containing the codon-optimized VP55 sequence (a gift from Christopher Sullivan and Benjamin tenOever) and cloned into pSBtet-GP, a tetracycline/doxycycline-inducible expression vector containing the Sleeping Beauty transposase-specific inverted terminal repeats flanking the cloning site. Introduction of pSBtet-GP-VP55 and the Sleeping Beauty transposase expressing vector (SB100X) into cells allows the transposase-mediated genomic integration of the DNA sequence from pSBtet-GP-VP55 that is flanked by the inverted terminal repeats. SB100X and pSBtet-GP-VP55 constructs were transfected into T98G cells using Lipofectamine 2000 according to the manufacturer's instructions (Thermo Fisher Scientific 11668030). Clonal cells were then selected through puromycin selection at 2.5 µg/mL (Gibco A1113803). Expression of VP55 was induced with doxycycline (Sigma-Aldrich D3072) at 5 µg/mL.
Luciferase reporter assays
The 3′ UTR of target mRNAs spanning at least 500 bp around the predicted binding site of selected miRNAs were cloned into the psi-CHECK2 vector downstream from the Renilla luciferase gene. Mutagenesis was performed using the QuikChange II site directed Mutagenesis Kit (Agilent, 200523). Cells were transfected with WT or mutant 3′ UTR with or without miRNA inhibitors and harvested after 48 h. Luciferase activity was then measured using the Promega Dual-Luciferase kit and calculated as the ratio of luminescence from Renilla to Firefly.
miRNA target enrichment analysis
One thousand random sets of 14 miRNAs were generated from among miRNAs expressed in T98G cells. For each set, the number of predicted interactions with 116 feed-forward related genes were determined using the DIANA miRNA–mRNA interaction annotations (Paraskevopoulou et al. 2013). P-value was estimated from the fraction of iterations with 342 or more interactions that were observed for EZH2-activated miRNAs.
Gene set overlaps using bootstrapping
Ten thousand random sets of 1834 or 959 genes were generated from a pool of genes not repressed by EZH2 but expressed at similar levels as the EZH2-repressed genes. Overlaps were then calculated between each of the 10,000 sets and 3844 miRNA-repressed genes. P-value was estimated from the fraction of iterations with 444 or more genes.
Data access
Primary sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE112242. Scripts used in this manuscript are available as Supplemental Code and at GitHub (https://github.com/haridh/PRC2-miRNA-manuscript).
Supplementary Material
Acknowledgments
We thank Anna Battenhouse for assistance with aligning sequencing data and ChIP-seq analysis; Alan Lambowitz for the use of their UV254 crosslinker; Benjamin tenOever for the VP55 construct; Chris Sullivan for suggesting the use of VP55, providing constructs, and for discussions; Luiz Penalva and Suzanne Burns for advice on the iCLIP experiments; and the Genomic Sequencing and Analysis Facility at UT Austin and the MD Anderson Cancer Center–Science Park NGS Facility for Illumina sequencing. The Science Park NGS Facility was supported by CPRIT Core Facility Support Grants RP120348 and RP170002. We also thank the Texas Advanced Computing Center (TACC) at UT Austin for the use of computational facilities. This work was funded in part by grants from the Cancer Prevention and Research Institute of Texas (RP120194) and the National Institutes of Health (NIH) (CA198648) to V.R.I.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.238311.118.
References
- Abou El Hassan M, Huang K, Eswara MB, Zhao M, Song L, Yu T, Liu Y, Liu JC, McCurdy S, Ma A, et al. 2015. Cancer cells hijack PRC2 to modify multiple cytokine pathways. PLoS One 10: e0126466 10.1371/journal.pone.0126466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguado LC, Schmid S, Sachs D, Shim JV, Lim JK, tenOever BR. 2015. microRNA function is limited to cytokine control in the acute response to virus infection. Cell Host Microbe 18: 714–722. 10.1016/j.chom.2015.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aranda S, Mas G, Di Croce L. 2015. Regulation of gene transcription by Polycomb proteins. Sci Adv 1: e1500737 10.1126/sciadv.1500737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backes S, Shapiro JS, Sabin LR, Pham AM, Reyes I, Moss B, Cherry S, tenOever BR. 2012. Degradation of host microRNAs by poxvirus poly(A) polymerase reveals terminal RNA methylation as a protective antiviral mechanism. Cell Host Microbe 12: 200–210. 10.1016/j.chom.2012.05.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balzeau J, Menezes MR, Cao S, Hagan JP. 2017. The LIN28/let-7 pathway in cancer. Front Genet 8: 31 10.3389/fgene.2017.00031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieniasz PD, Kutluay SB. 2018. CLIP-related methodologies and their application to retrovirology. Retrovirology 15: 35 10.1186/s12977-018-0417-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles G, Clark NR, Ma'ayan A. 2013. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14: 128 10.1186/1471-2105-14-128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Rosario RC, Damasco JR, Aguda BD. 2016. MicroRNA inhibition fine-tunes and provides robustness to the restriction point switch of the cell cycle. Sci Rep 6: 32823 10.1038/srep32823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Croce L, Helin K. 2013. Transcriptional regulation by Polycomb group proteins. Nat Struct Mol Biol 20: 1147–1155. 10.1038/nsmb.2669 [DOI] [PubMed] [Google Scholar]
- Ebert MS, Sharp PA. 2012. Roles for microRNAs in conferring robustness to biological processes. Cell 149: 515–524. 10.1016/j.cell.2012.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farh KK, Grimson A, Jan C, Lewis BP, Johnston WK, Lim LP, Burge CB, Bartel DP. 2005. The widespread impact of mammalian microRNAs on mRNA repression and evolution. Science 310: 1817–1821. 10.1126/science.1121158 [DOI] [PubMed] [Google Scholar]
- Gerloff D, Grundler R, Wurm AA, Bräuer-Hartmann D, Katzerke C, Hartmann JU, Madan V, Müller-Tidow C, Duyster J, Tenen DG, et al. 2015. NF-κB/STAT5/miR-155 network targets PU.1 in FLT3-ITD-driven acute myeloid leukemia. Leukemia 29: 535–547. 10.1038/leu.2014.231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosline SJ, Gurtan AM, JnBaptiste CK, Bosson A, Milani P, Dalin S, Matthews BJ, Yap YS, Sharp PA, Fraenkel E. 2016. Elucidating microRNA regulatory networks using transcriptional, post-transcriptional, and histone modification measurements. Cell Rep 14: 310–319. 10.1016/j.celrep.2015.12.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham B, Marcais A, Dharmalingam G, Carroll T, Kanellopoulou C, Graumann J, Nesterova TB, Bermange A, Brazauskas P, Xella B, et al. 2016. MicroRNAs of the miR-290–295 family maintain bivalency in mouse embryonic stem cells. Stem Cell Rep 6: 635–642. 10.1016/j.stemcr.2016.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall AW, Battenhouse AM, Shivram H, Morris AR, Cowperthwaite MC, Shpak M, Iyer VR. 2018. Bivalent chromatin domains in glioblastoma reveal a subtype-specific signature of glioma stem cells. Cancer Res 78: 2463–2474. 10.1158/0008-5472.CAN-17-1724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38: 576–589. 10.1016/j.molcel.2010.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobert O. 2008. Gene regulation by transcription factors and microRNAs. Science 319: 1785–1786. 10.1126/science.1151651 [DOI] [PubMed] [Google Scholar]
- Jadhav U, Nalapareddy K, Saxena M, O'Neill NK, Pinello L, Yuan GC, Orkin SH, Shivdasani RA. 2016. Acquired tissue-specific promoter bivalency is a basis for PRC2 necessity in adult cells. Cell 165: 1389–1400. 10.1016/j.cell.2016.04.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Langmead B, Salzberg SL. 2015. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12: 357–360. 10.1038/nmeth.3317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- König J, Zarnack K, Rot G, Curk T, Kayikci M, Zupan B, Turner DJ, Luscombe NM, Ule J. 2010. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat Struct Mol Biol 17: 909–915. 10.1038/nsmb.1838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowarz E, Löscher D, Marschalek R. 2015. Optimized Sleeping Beauty transposons rapidly generate stable transgenic cell lines. Biotechnol J 10: 647–653. 10.1002/biot.201400821 [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung AK, Young AG, Bhutkar A, Zheng GX, Bosson AD, Nielsen CB, Sharp PA. 2011. Genome-wide identification of Ago2 binding sites from mouse embryonic stem cells with and without mature microRNAs. Nat Struct Mol Biol 18: 237–244. 10.1038/nsmb.1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25: 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W. 2014. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30: 923–930. 10.1093/bioinformatics/btt656 [DOI] [PubMed] [Google Scholar]
- Lin Y, Zhang Q, Zhang HM, Liu W, Liu CJ, Li Q, Guo AY. 2015. Transcription factor and miRNA co-regulatory network reveals shared and specific regulators in the development of B cell and T cell. Sci Rep 5: 15215 10.1038/srep15215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔC(T) method. Methods 25: 402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- Lovci MT, Ghanem D, Marr H, Arnold J, Gee S, Parra M, Liang TY, Stark TJ, Gehman LT, Hoon S, et al. 2013. Rbfox proteins regulate alternative mRNA splicing through evolutionarily conserved RNA bridges. Nat Struct Mol Biol 20: 1434–1442. 10.1038/nsmb.2699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mote RD, Mahajan G, Padmanabhan A, Ambati R, Subramanyam D. 2017. Dual repression of endocytic players by ESCC microRNAs and the Polycomb complex regulates mouse embryonic stem cell pluripotency. Sci Rep 7: 53 10.1038/s41598-017-17828-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller-McNicoll M, Botti V, de Jesus Domingues AM, Brandl H, Schwich OD, Steiner MC, Curk T, Poser I, Zarnack K, Neugebauer KM. 2016. SR proteins are NXF1 adaptors that link alternative RNA processing to mRNA export. Genes Dev 30: 553–566. 10.1101/gad.276477.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. 2005. c-Myc-regulated microRNAs modulate E2F1 expression. Nature 435: 839–843. 10.1038/nature03677 [DOI] [PubMed] [Google Scholar]
- Paraskevopoulou MD, Georgakilas G, Kostoulas N, Vlachos IS, Vergoulis T, Reczko M, Filippidis C, Dalamagas T, Hatzigeorgiou AG. 2013. DIANA-microT web server v5.0: service integration into miRNA functional analysis workflows. Nucleic Acids Res 41: W169–W173. 10.1093/nar/gkt393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasini D, Bracken AP, Jensen MR, Denchi EL, Helin K. 2004. Suz12 is essential for mouse development and for EZH2 histone methyltransferase activity. EMBO J 23: 4061–4071. 10.1038/sj.emboj.7600402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polioudakis D, Bhinge AA, Killion PJ, Lee BK, Abell NS, Iyer VR. 2013. A Myc–microRNA network promotes exit from quiescence by suppressing the interferon response and cell-cycle arrest genes. Nucleic Acids Res 41: 2239–2254. 10.1093/nar/gks1452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivram H, Iyer VR. 2018. Identification and removal of sequencing artifacts produced by mispriming during reverse transcription in multiple RNA-seq technologies. RNA 24: 1266–1274. 10.1261/rna.066217.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit AFA, Hubley R, Green P. 2013–2015. RepeatMasker Open-4.0. http://www.repeatmasker.org.
- Tsang J, Zhu J, van Oudenaarden A. 2007. MicroRNA-mediated feedback and feedforward loops are recurrent network motifs in mammals. Mol Cell 26: 753–767. 10.1016/j.molcel.2007.05.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q, Qin H, Zhao Q, He XX. 2015. Emerging role of transcription factor-microRNA-target gene feed-forward loops in cancer. Biomed Rep 3: 611–616. 10.3892/br.2015.477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. 2008. Model-based Analysis of ChIP-Seq (MACS). Genome Biol 9: R137 10.1186/gb-2008-9-9-r137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng GX, Do BT, Webster DE, Khavari PA, Chang HY. 2014. Dicer-microRNA-Myc circuit promotes transcription of hundreds of long noncoding RNAs. Nat Struct Mol Biol 21: 585–590. 10.1038/nsmb.2842 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.