

Abstract
5-Fluorouracil (5-FU), in combination with other cytotoxic drugs, is commonly used to treat a variety of cancers. Dihydropyrimidine dehydrogenase (DPD) catalyzes the first catabolic step of the 5-FU degradation pathway, converting 80% of 5-FU to its inactive metabolite. Approximately 0.3% of the population demonstrate complete DPD deficiency, translating to extreme toxicity of 5-FU. Here we present a case of a patient who had a fatal outcome after treatment with 5-FU who was found to have an unknown DPD deficiency discovered at autopsy.
Keywords: Dihydropyrimidine Dehydrogenase Deficiency, Drug-Related Side Effects and Adverse Reactions, Genetic Testing, Fluorouracil, Pancytopenia
CASE REPORT
We present the autopsy findings of a 54-year-old male with past medical history of smoking, hypothyroidism, and newly diagnosed T2N2bM0 (clinically staged) human papillomavirus positive (HPV+) squamous cell carcinoma (SCC) of the left base of tongue with metastases to the cervical lymph nodes. The patient was enrolled in the OPTIMA trial, which entailed dose reduced/deintensified chemoradiotherapy for HPV+ oropharyngeal SCC. Prior to the hospital admission, the patient had received carboplatin/abraxane induction chemotherapy (cycle 1) and had good imaging response with complete resolution of his primary tongue tumor accompanied by a decrease in the size of the cervical lymph nodes, with minimal side effects from chemotherapy. The patient was then admitted for inpatient chemoradiotherapy with 5-FU, paclitaxel, and hydroxyurea (part of cycle 1). During the treatment, the patient developed fever in the setting of pancytopenia, followed by intractable diarrhea and vomiting. His pancytopenia was refractory, which, at the time, was attributed to hydroxyurea. Therefore, hydroxyurea was initially dose-decreased and then held. However, the patient’s clinical course continued to worsen. He subsequently developed septic shock, progressive desquamative skin lesions, acute kidney failure, and sustained pancytopenia. Due to persistent hemodynamic instability requiring multiple vasopressors and hypoxia, he was transferred to the intensive care unit and intubated. Shortly thereafter, was transitioned to comfort care and expired 12 days after inpatient admission. A complete unrestricted autopsy was requested and performed.
AUTOPSY FINDINGS
Autopsy findings showed skin lesions involving head, chest, back, and right arm. Histologic examination of these was consistent with toxic epidermal necrolysis (Figures 1A and 1B).
Figure 1. Photomicrograph of the skin with A – toxic epidermal necrolysis (H&E, 4x) and B – toxic epidermal necrolysis. Note dyskeratinocytes with arrows. (H&E, 20x).

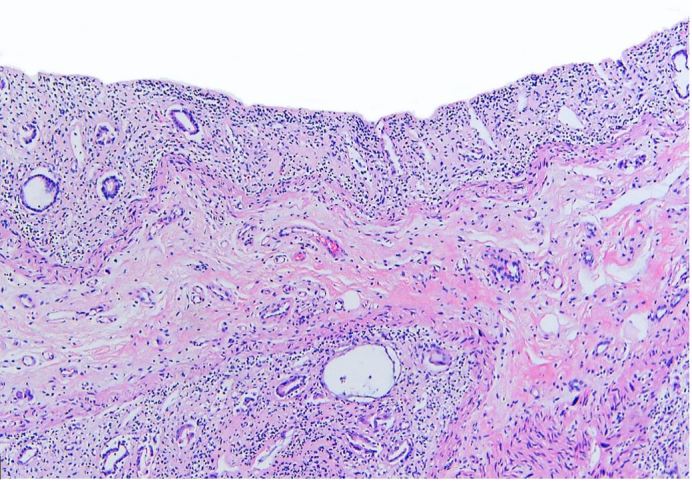
Bone marrow examination showed markedly hypocellular (<5%) bone marrow with diminished trilineage hematopoiesis and virtually no megakaryocytes (Figure 2). Kidneys had diffuse acute tubular necrosis. The gastrointestinal tract was grossly pale-appearing with severely damaged mucosa (Figure 3). Post-mortem blood and bilateral lung cultures were positive for Stenotrophomonas maltophilia.
Figure 2. Photomicrograph of the bone marrow showing Markedly hypocellular (<5%) bone marrow with severe stromal damage and pigmented histiocytes (H&E, 20x).
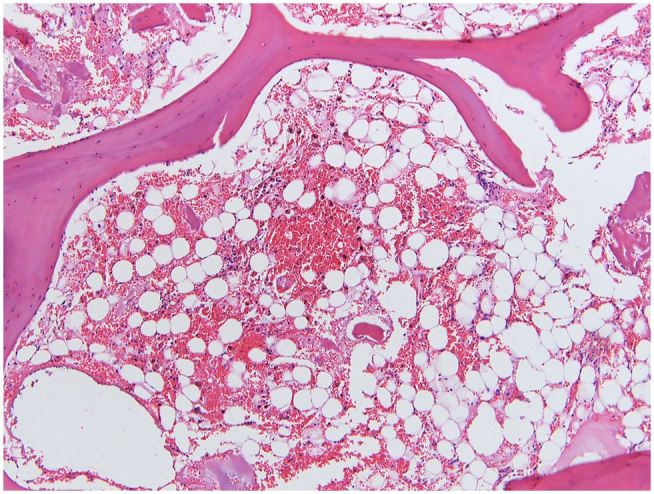
Figure 3. Photomicrograph of the rectum showing severely damaged mucosa, consistent with chemotherapy toxicity (H&E, 4x).
Due to the precipitous clinical deterioration and autopsy findings, DNA analysis by next generation sequencing was performed on formalin-fixed, paraffin-embedded liver sample to evaluate for pathogenic DPYD variants. Sequencing revealed a point mutation one base downstream of exon 14 (DPYD*2A/c.1905+1G>A) at approximately 100% variant allele frequency (Figure 4). Additionally, specific copy-number variant analysis was performed which revealed no loss of genetic material on 1p21.3, confirming homozygosity.
Figure 4. Next Generation Sequencing reads show point mutation one base downstream of exon 14, approximately 100% variant allele frequency, read depth 700, as seen in Integrative Genomics Viewer (IGV).

DISCUSSION
In recent years, the usage of molecular genetics has had a profound impact on cancer treatment.1 The role of molecular genetics has changed from being of just a diagnostic and prognostic utility, to providing tailored treatment on an individual, patient-by-patient basis. Besides allowing patients to be eligible for certain therapeutic regimens and clinical trials, cancer treatment has become more personalized due to the detection of inter-individual differences. Currently, the treatment outcomes and drug toxicities due to personal variations in pharmacokinetics and pharmacodynamics can be foreseen in some cases.2 These differences in pharmacokinetics and pharmacodynamics may result from drug–drug interactions, ethnicity, age, renal and liver function, comorbidities, nutritional status, smoking, and alcohol consumption. More importantly, genetic factors that have a high impact on the drug efficacy and toxicity are being discovered, emphasizing the increasing need to utilize pharmacogenetics testing in cancer treatment.2
Drug metabolism is traditionally divided into modification (phase I), conjugation (phase II), and elimination (via urine or bile).2 Cytochrome P450 (CYP) family of enzymes are responsible for the phase I modifications (oxidation, reduction, and hydrolysis) of the drugs.3 The phase II conjugation reactions, which lead to activation or inactivation of the drugs, are performed by enzymes such as glutathione S-transferases (GSTs) and uridine diphosphate glucuronosyltransferases (UGTs).4 The enzymes responsible for the purine and pyrimidine analog metabolism, thiopurine S-methyltransferase (TPMT) and dihydropyrimidine dehydrogenase (DPD) respectively, have various known polymorphisms in their encoding genes, which ultimately affect their enzymatic activity.2
5-FU and its oral prodrug capecitabine, are one of the most frequently prescribed chemotherapeutic drugs for the treatment of patients with cancers of the gastrointestinal tract, breast, and head and neck.5,6 5-FU undergoes enzymatic activation to a cytotoxic fluoropyrimidine nucleotide for its therapeutic effect, in which it is incorporated into both nuclear and cytoplasmic RNA species affecting the synthesis, stability, processing, and methylation of RNA.7 Similarly, another is the degradation of 5-FU is an important consideration. A majority (80%) of 5-FU is catabolized in the liver by DPD enzyme, which encoded by the DPYD gene; the deficiency of DPD can lead to toxic levels of unmetabolized 5-FU.8 5-FU toxicity was first reported in 1985 in a patient with familial pyrimidinemia who had experienced severe, almost lethal, toxicity upon treatment with 5-FU, providing evidence that a genetic defect in pyrimidine catabolism could be associated with fluoropyrimidine-associated toxicity.9 Although considered a safe treatment in a majority of the patients, up to 30% of patients can experience severe treatment-related toxicity which can be fatal in up to 1% of cases.7,9
The extent of toxicity is determined by the individuals’ functionality of the DPYD polymorphism. There are currently three clinically recognized common non-functional DPYD variants: DPYD*2A/c.1905+1G>A (as seen in this case), *13/c.1679T>G, and rs67376798/c.2846A>T.8 Homozygous or compound heterozygous nonfunctional alleles can cause severe or even fatal toxicity. Homozygotes for nonfunctional variants may have either very low enzymatic activity or be completely DPD deficient,10 and might therefore require selection of an alternative drug.8 The DPYD (NM_000110.3): c.1905+1G>A variant (as seen in this case), also known as rs3918290 (dbSNP) and DPYD*2A (ClinVar Database Allele ID: 15471) with an alternative nomenclature of IVS14+1G>A, results in a pathogenic alteration of the donor site at the splice site of intron 14.11 Recently, two additional DPYD variants, c.1679T>G and c.1236G>A/HapB3 have also been systematically evaluated for their clinical impact on 5-FU metabolism and might require dose adjustment to prevent toxicity.12 Lastly, the absence of these polymorphisms does not equate to absent risk of toxicity, since DPYD is highly polymorphic and additional uncommon variants may result in toxicity.8
In a recent series of 243 patients (88.5% of which were on monotherapy) studied for toxicity from capecitabine (the oral prodrug of 5-FU), a wide range of toxic effects were seen. These included gastrointestinal, hematologic, and neurologic toxicities with moderate-to-severe toxicities in 12.4% patients and severe toxicities in 2.1% of patients. There was only one case with drug-related fatal outcome characterized by thrombocytopenia, diarrhea, renal failure, dyspnea and hypovolemic shock,11 similar to our patient. Other toxic effects included hand-foot syndrome. These toxic effects occurred irrespective of the patient’s performance status, age, renal function, and the line of capecitabine treatment. In this study, a total of 65 DPYD variants were identified, of which, variants D949V and DPYD*2A/c.1905+1G>A were significantly associated with an increased risk of toxicity.11
Along with patient safety, the effectiveness and cost-saving benefits of upfront screening for clinically relevant non-functional variants must also be considered. In a study of 2,038 patients intended to be treated with fluoropyrimidine based therapy, the patients were prospectively screened for DPYD*2A. If they were found to have heterozygous polymorphism, they were given a reduced median dose-intensity of 50%, followed by titration based on tolerance. These patients were found to have their risk of toxicity significantly reduced from 73% (historical controls) to 28% by genotype-guided dosing. Additionally, the average total treatment cost per patient was lower for screening than for non-screening, outweighing the screening costs.13
CONCLUSION
DPD-deficiency is known to be a leading cause of severe fluoropyrimidine toxicity,7,9 culminating in fatality,11 as was observed in our case. In patients who are expected to make a full recovery and are expected to have a good prognosis after their cancer treatment, this is a very unfortunate yet possibly preventable outcome. Although not part of the routine clinical practice, prospective screening for DPYD*2A polymorphism leading to genotype-guided dosing has been successful at significantly reducing toxicity, while at the same time being cost-efficient,13 posing a valid argument of upfront screening which would positively impact both patient safety and treatment efficacy.
Consent was granted for a complete unrestricted autopsy by the corresponding family member.
Footnotes
How to cite: Fidai SS, Sharma AE, Johnson DN, Segal JP, Lastra RR. Dihydropyrimidine Dehydrogenase deficiency as a cause of fatal 5-Fluorouracil toxicity. Autops Case Rep [Internet]. 2018;8(4):e2018049. https://doi.org/10.4322/acr.2018.049
Financial support: None
REFERENCES
- 1.Ashfaq R. Molecular profiling for personalized cancer care. Clin Exp Metastasis. 2012;29(7):653-5. 10.1007/s10585-012-9483-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bertholee D, Maring JG, van Kuilenburg ABP. Genotypes affecting the pharmacokinetics of anticancer drugs. Clin Pharmacokinet. 2017;56(4):317-37. 10.1007/s40262-016-0450-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lamb DC, Waterman MR, Kelly SL, Guengerich FP. Cytochromes P450 and drug discovery. Curr Opin Biotechnol. 2007;18(6):504-12. 10.1016/j.copbio.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 4.Jancova P, Anzenbacher P, Anzenbacherova E. Phase II drug metabolizing enzymes. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2010;154(2):103-16. 10.5507/bp.2010.017. [DOI] [PubMed] [Google Scholar]
- 5.Peyrade F, Cupissol D, Geoffrois L, et al. . Systemic treatment and medical management of metastatic squamous cell carcinoma of the head and neck: Review of the literature and proposal for management changes. Oral Oncol. 2013;49(6):482-91. 10.1016/j.oraloncology.2013.01.005. [DOI] [PubMed] [Google Scholar]
- 6.Meyerhardt JA, Mayer RJ. Systemic therapy for colorectal cancer. N Engl J Med. 2005;352(5):476. [DOI] [PubMed] [Google Scholar]
- 7.van Kuilenburg AB. Dihydropyrimidine dehydrogenase and the efficacy and toxicity of 5-fluorouracil. Eur J Cancer. 2004;40(7):939-50. 10.1016/j.ejca.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 8.Falvella FS, Caporale M, Cheli S, et al. . Undetected toxicity risk in pharmacogenetic testing for Dihydropyrimidine Dehydrogenase. Int J Mol Sci. 2015;16(4):8884-95. 10.3390/ijms16048884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meulendijks D, Cats A, Beijnen JH, Schellens JH. Improving safety of fluoropyrimidine chemotherapy by individualizing treatment based on dihydropyrimidine dehydrogenase activity – Ready for clinical practice? Cancer Treat Rev. 2016;50:23-34. 10.1016/j.ctrv.2016.08.002. [DOI] [PubMed] [Google Scholar]
- 10.van Kuilenburg ABP, Häusler P, Schalhorn A, et al. . Evaluation of 5-Fluorouracil pharmacokinetics in cancer patients with a C.1905+1G>A Mutation in DPYD by means of a bayesian limited sampling strategy. Clin Pharmacokinet. 2012;51(3):163-74. 10.1007/BF03257473. [DOI] [PubMed] [Google Scholar]
- 11.Etienne-Grimaldi M-C, Boyer J-C, Beroud C, et al. . New advances in DPYD genotype and risk of severe toxicity under capecitabine. Plos One. 2017;12;(5)1-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meulendijks D, Henricks LM, Sonke GS, et al. . Clinical relevance of DPYD variants c.1679T>G, c.1236G>A/HapB3, and c.1601G>A as predictors of severe fluoropyrimidine-associated toxicity: a systematic review and meta-analysis of individual patient data. Lancet Oncol. 2015;16(16):1639-50. 10.1016/S1470-2045(15)00286-7. [DOI] [PubMed] [Google Scholar]
- 13.Deenen MJ, Meulendijks D, Cats A, et al. . Upfront Genotyping of DPYD * 2A to individualize fluoropyrimidine therapy: a safety and cost analysis. J Clin Oncol. 2016;34(3):227-34. 10.1200/JCO.2015.63.1325. [DOI] [PubMed] [Google Scholar]