

Abstract
Background
Recognition is growing that social anxiety disorder (SAnD) is a chronic and disabling disorder, and data from early trials demonstrate that medication may be effective in its treatment. This systematic review is an update of an earlier review of pharmacotherapy of SAnD.
Objectives
To assess the effects of pharmacotherapy for social anxiety disorder in adults and identify which factors (methodological or clinical) predict response to treatment.
Search methods
We searched the Cochrane Common Mental Disorders Controlled Trials Register (CCMDCTR‐Studies and CCMDCTR‐References) to 17 August 2015. The CCMDCTR contains reports of relevant RCTs from MEDLINE (1950‐), Embase (1974‐), PsycINFO (1967‐) and CENTRAL (all years). We scanned the reference lists of articles for additional studies. We updated the search in August 2017 and placed additional studies in Awaiting Classification, these will be incorporated in the next version of the review, as appropriate.
Selection criteria
We restricted studies to randomised controlled trials (RCTs) of pharmacotherapy versus placebo in the treatment of SAnD in adults.
Data collection and analysis
Two authors (TW and JI) assessed trials for eligibility and inclusion for this review update. We extracted descriptive, methodological and outcome information from each trial, contacting investigators for missing information where necessary. We calculated summary statistics for continuous and dichotomous variables (if provided) and undertook subgroup and sensitivity analyses.
Main results
We included 66 RCTs in the review (> 24 weeks; 11,597 participants; age range 18 to 70 years) and 63 in the meta‐analysis. For the primary outcome of treatment response, we found very low‐quality evidence of treatment response for selective serotonin reuptake inhibitors (SSRIs) compared with placebo (number of studies (k) = 24, risk ratio (RR) 1.65; 95% confidence interval (CI) 1.48 to 1.85, N = 4984). On this outcome there was also evidence of benefit for monoamine oxidase inhibitors (MAOIs) (k = 4, RR 2.36; 95% CI 1.48 to 3.75, N = 235), reversible inhibitors of monoamine oxidase A (RIMAs) (k = 8, RR 1.83; 95% CI 1.32 to 2.55, N = 1270), and the benzodiazepines (k = 2, RR 4.03; 95% CI 2.45 to 6.65, N = 132), although the evidence was low quality. We also found clinical response for the anticonvulsants with gamma‐amino butyric acid (GABA) analogues (k = 3, RR 1.60; 95% CI 1.16 to 2.20, N = 532; moderate‐quality evidence). The SSRIs were the only medication proving effective in reducing relapse based on moderate‐quality evidence. We assessed the SSRIs and the serotonin and norepinephrine reuptake inhibitor (SNRI) venlafaxine on the basis of treatment withdrawal; this was higher for medication than placebo (SSRIs: k = 24, RR 2.59; 95% CI 1.97 to 3.39, N = 5131, low‐quality evidence; venlafaxine: k = 4, RR 3.23; 95% CI 2.15 to 4.86, N = 1213, moderate‐quality evidence), but there were low absolute rates of withdrawal for both these medications classes compared to placebo. We did not find evidence of a benefit for the rest of the medications compared to placebo.
For the secondary outcome of SAnD symptom severity, there was benefit for the SSRIs, the SNRI venlafaxine, MAOIs, RIMAs, benzodiazepines, the antipsychotic olanzapine, and the noradrenergic and specific serotonergic antidepressant (NaSSA) atomoxetine in the reduction of SAnD symptoms, but most of the evidence was of very low quality. Treatment with SSRIs and RIMAs was also associated with a reduction in depression symptoms. The SSRIs were the only medication class that demonstrated evidence of reduction in disability across a number of domains.
We observed a response to long‐term treatment with medication for the SSRIs (low‐quality evidence), for the MAOIs (very low‐quality evidence) and for the RIMAs (moderate‐quality evidence).
Authors' conclusions
We found evidence of treatment efficacy for the SSRIs, but it is based on very low‐ to moderate‐quality evidence. Tolerability of SSRIs was lower than placebo, but absolute withdrawal rates were low.
While a small number of trials did report treatment efficacy for benzodiazepines, anticonvulsants, MAOIs, and RIMAs, readers should consider this finding in the context of potential for abuse or unfavourable side effects.
Plain language summary
Medication for social anxiety disorder (SAnD): a review of the evidence
Why is this review important?
Individuals with SAnD often experience intense fear, avoidance, and distress in unfamiliar social situations. There is evidence that medications are useful in minimising these symptoms.
Who will be interested in this review?
‐ People with SAnD.
‐ Families and friends of people who suffer from anxiety disorders.
‐ General practitioners, psychiatrists, psychologists, and pharmacists.
What questions does this review aim to answer?
‐ Is pharmacotherapy an effective form of treatment for SAnD in adults?
‐ Is medication effective and tolerable for people in terms of side effects?
‐ Which factors (methodological or clinical) predict response to pharmacotherapy?
Which studies were included in the review?
We included studies comparing medication with placebo for the treatment of SAnD in adults.
We included 66 trials in the review, with a total of 11,597 participants.
What does the evidence from the review tell us?
There was evidence of benefit that selective serotonin reuptake inhibitors (SSRIs) were more effective than placebo, although the evidence was of very low quality. There was also evidence of benefit for monoamine oxidase inhibitors (MAOIs), reversible inhibitors of monoamine oxidase A (RIMAs), and benzodiazepines, even though the evidence was low in quality. The anticonvulsants gabapentin and pregabalin also showed moderate‐quality evidence of a clinical response. We did not observe this effect for the remaining medication classes. The SSRIs were the only medication proving effective in reducing relapse based on moderate‐quality evidence. There was low‐quality evidence that more people taking SSRIs and SNRIs dropped out due to side effects than those taking placebo, but absolute withdrawal rates were low.
For the outcome of SAnD symptom severity, there was evidence of benefit for SSRIs, the serotonin and norepinephrine reuptake inhibitor (SNRI) venlafaxine, MAOIs, RIMAs, benzodiazepines, the antipsychotic olanzapine, and the noradrenergic and specific serotonergic antidepressant (NaSSA) atomoxetine, but most of the evidence was of very low quality. SSRIs and RIMAs reduced depression symptoms, and SSRIs reduced functional disability across all domains.
We also observed response to long‐term treatment with SSRIs (based on low‐quality evidence), MAOIs (based on very low‐quality evidence), and RIMAs (based on moderate‐quality evidence).
What should happen next?
Most evidence for treatment efficacy is related to SSRIs. Nevertheless, SSRI trials were associated with very low‐quality evidence and high risk of publication bias. It would be useful for future studies to evaluate the treatment of SAnD in people with comorbid disorders, including substance use disorders. Trials that provide adequate information on randomisation and allocation concealment are needed.
Summary of findings
Background
Description of the condition
Although the symptoms of social anxiety disorder (SAnD) have long been recognised (Marks 1966), the disorder only appeared within the official psychiatric nomenclature relatively recently (DSM‐III 1980). Diagnostic criteria for SAnD in the third edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM‐III) encouraged research on its epidemiology, psychobiology, and treatment. Subsequent epidemiological research determined that the disorder is highly prevalent in a wide range of settings, that it is characterised by significant chronicity and comorbidity, and that it is associated with marked functional impairment, including academic, occupational, marital, and social dysfunction (Stein 2008). Such data allay skepticism about whether SAnD is really a medical disorder and support the importance of pharmacotherapy for its treatment.
SAnD usually begins in childhood or adolescence, with studies reporting lifetime prevalence rates of between 3% and 16% (Kessler 1994; Kessler 2005). Some European studies describe a one‐year prevalence of 2% to 5% (Wancata 2009). Typically, individuals with SAnD experience severe, intense fear of drawing attention to themselves in unfamiliar social situations or negative evaluation in situations that are potentially embarrassing or humiliating. This in turn results in avoiding the phobic situation or tolerating it with extreme distress. The affected individual recognises this fear as unreasonable, excessive, and more than mere shyness. These symptoms may take the form of a situationally predisposed panic attack and always impair functioning on a number of levels (DSM‐IV 2004).
There is a growing body of work demonstrating that SAnD is mediated by specific neurocircuitry, with serotonergic and dopaminergic systems particularly relevant (Stein 2002d), providing a rationale for the use of pharmacotherapy. The glutamatergic and noradrenergic systems, as well as substance P, may also be implicated in the neurological basis of SAnD, suggesting a role for agents such as gabapentin, pregabalin, and neurokinin‐1 receptor antagonists (Pande 1999; Pande 2004, Tauscher 2010). Indeed there is accumulating evidence that particular medications are effective in the management of SAnD (Stein 2004).
The effectiveness of cognitive‐behavioural therapy (CBT) in managing SAnD is also receiving increasing empirical support (Dorrepaal 2014; Fedoroff 2001; Gould 1997), and current expert consensus is that both pharmacotherapy and psychotherapy have a role in the management of this disorder (Ballenger 1998; Bandelow 2002; Bandelow 2012; Bandelow 2015). Interestingly, both CBT and pharmacotherapy are able to normalise functional neuroanatomical abnormalities in SAnD (Furmark 2002). Nevertheless, this review focuses exclusively on pharmacotherapy interventions.
Description of the intervention
Research has evaluated a range of medications for SAnD. Early reports noted the potential value of irreversible monoamine oxidase inhibitors (MAOIs) (Fahlen 1995; Van Vliet 1992), beta‐blockers (Gorman 1987; Turner 1994), reversible inhibitors of monoamine oxidase A (RIMAs) (Tyrer 1973; Versiani 1992), and high potency benzodiazepines (Davidson 1991). More recent studies have focused on the selective serotonin reuptake inhibitors (SSRIs) (Van der Linden 2000), plus other newer agents such as GW876008 (NCT00397722). Randomised controlled trials (RCTs) have also assessed buspirone (Davidson 1993), the noradrenergic and specific serotonergic antidepressant (NaSSA) mirtazepine (Muehlbacher 2005), the new generation antipsychotic olanzapine (Barnett 2002), the highly selective noradrenaline reuptake inhibitor (NARI) atomoxetine (Ravindran 2009), the serotonin antagonist and reuptake inhibitor (SARI) nefazodone (Van Ameringen 2007), the serotonin and norepinephrine reuptake inhibitor (SNRI) venlafaxine (Rickels 2004), and certain anticonvulsants/gamma‐amino butyric acids (GABAs) (Pande 1999; Pande 2004).
How the intervention might work
Although the pathophysiology of SAnD is incompletely understood, various mono‐aminergic systems, such as the serotonergic, dopaminergic, and noradrenergic systems, may play a role in mediating SAnD symptoms. Strongly serotonergic drugs such as SSRIs have specific activity in the inhibition of serotonin reuptake, with minimal direct effects on norepinephrine and dopamine reuptake. This inhibition of reuptake increases the bio‐available concentration of serotonin, which then binds to and activates various receptors. Clinical efficacy is observed with 70% to 80% occupancy of serotonin transporters (Stahl 2008). SNRIs are potent inhibitors of the reuptake of catecholamines but weak inhibitors of dopamine reuptake (Rickels 2004; Stahl 2008). The putative effects of the selective NARI atomoxetine on the noradrenergic neurotransmitter system in adults and children with attention‐deficit/hyperactivity disorder (ADHD), reported by Chamberlain 2007 and Michelson 2001, suggest potential use for anxiety and mood disorders (Ravindran 2009). There is evidence to suggest that some serotonin‐dopamine antagonists (SDAs) such as quetiapine and olanzapine may possess anxiolytic properties, and they may potentially have a role in treatment‐resistant SAnD (Barnett 2002; Vaishnavi 2007). Nefazodone is an agent with both pre‐ and postsynaptic serotonin reuptake inhibition, but concerns about hepatotoxicity limit its use in clinical practice (Van Ameringen 2007).
MAOIs increase biogenic amine neurotransmitter levels by inhibiting their degradation, facilitating presynaptic reuptake of these chemicals through specific transporter molecules and inhibiting their de‐amination in mitochondria by the enzyme MAO. This inhibition may be either reversible or irreversible (Stahl 2008). RIMAs are generally safer and better tolerated than MAOIs and have shown efficacy in treating SAnD (Versiani 1992). The drug functions by selectively binding to a specific isoenzyme of monoamine oxidase (Davidson 2006).
Beta‐adrenoreceptor antagonists block catecholamines released in the stress response, thus potentially reducing the physiological symptoms associated with SAnD. This may then enable the individual to function with fewer objective signs of anxiety (Liebowitz 1992; Stahl 2008; Turner 1994). The NaSSA mirtazepine has potent action on central adrenergic receptors, leading to a net increase in noradrenaline and serotonin, without the unwanted activation of cholinergic receptors (Muehlbacher 2005).
The efficacy of benzodiazepines in the treatment of many anxiety disorders is consistent with the hypothesized role of GABA in these conditions. In low doses, benzodiazepines act as anxiolytic agents and may be especially useful in providing rapid control of anxiety symptoms (Gorman 2003; Pecknold 1997; Stahl 2008). Repeated buspirone treatment may desensitise inhibitory 5‐HT1A receptors and this, together with its modest postsynaptic 5‐HT1A receptor agonist actions, could lead to an overall increase in 5‐HT neurotransmission as well (Cowen 1997).
The anticonvulsant pregabalin reduces the release of norepinephrine, glutamate, and substance P from the brain and spinal cord and may have a benefit in anxiety reduction (Pande 2004). Gabapentin may be similarly effective for these conditions (Stephen 2013).
Newer medications such as GW876008 have corticotropin‐releasing factors (CRFs) that target behavioural, autonomic, and neurochemical responses linked to a variety of anxiety and stress‐related disorders (Hubbard 2011). GW876008 also plays a role in the activation of the hypothalamus in patients with anxiety (Hubbard 2011).
Why it is important to do this review
A systematic review of pharmacotherapy studies may be useful in tackling several questions for the field. First, is pharmacotherapy in fact an effective form of treatment in SAnD? Given scepticism about SAnD diagnosis and the importance of psychological models and psychotherapy studies for the disorder, the role of pharmacotherapy remains moot for some. For those who accept the role of pharmacotherapy, questions remain about the appropriate dose and duration of treatments. Although in the late 1990s expert consensus suggested continuing pharmacotherapy for at least a year, there was arguably relatively little data to support this conclusion at the time (Ballenger 1998).
Second, are particular medication classes more effective in treating symptoms, more tolerable to the patient in terms of adverse events, or both? The use of recently introduced antidepressants (e.g. SSRIs) for SAnD has raised the question of how these agents compare with older medications (e.g. MAOIs). Current expert consensus has suggested that in view of their efficacy and tolerability, SSRIs should be considered as first‐line medications for the treatment of SAnD, while beta‐blockers have a role in the management of performance anxiety (Ballenger 1998; Bandelow 2002; Bandelow 2012; Bandelow 2015); it is important to determine whether such recommendations are supported by evidence from RCTs.
Third, can a systematic review of RCTs provide information about the most important variables affecting pharmacotherapy response? Methodological factors such as the number of participating centres or the duration of the trial may affect treatment outcomes (Stein 2002c). Some authors have also suggested that clinical factors such as the nature of the SAnD sub‐type present (e.g. generalised versus non‐generalised), and the severity of baseline symptoms, may play a role (Stein 2002c). The body of evidence from RCTs may provide more conclusive information about the predictors of pharmacotherapy response in this disorder.
Indeed, a series of reviews of the pharmacotherapy of SAnD has been published (Blanco 2002; Blanco 2013; Curtiss 2017; De Menezes 2011). These reviews have been useful in summarising the existing research, pointing to methodological flaws, and outlining areas for future research. A number of systematic reviews also exist (Blanco 2003; Davis 2014; Fedoroff 2001; Gould 1997; Van der Linden 2000), and these have provided useful lessons for both clinicians and researchers. Further reviews in this area, from Cochrane or elsewhere, need to adhere to guidelines for the systematic identification of trials, investigation of sources of heterogeneity, measurement of methodological quality, and estimation of the effects of intervention (Moher 1999; Mulrow 1997).
The authors updated the systematic review of RCTs of the pharmacotherapy of SAnD, following Cochrane guidelines and software (Higgins 2011; RevMan 2014).
Objectives
To assess the effects of pharmacotherapy for social anxiety disorder in adults and identify which factors (methodological or clinical) predict response to treatment.
Methods
Criteria for considering studies for this review
Types of studies
We considered all RCTs, irrespective of publication status, methodological differences, or language. We only included trials comparing multiple forms of medication if one of the comparison groups was a placebo group. Because publication is not necessarily related to study quality and indeed may imply certain biases (Dickersin 1992; Easterbrook 1991; Scherer 1994), we also considered unpublished reports, abstracts, and brief and preliminary reports. We also included cluster‐randomised controlled trials, cross‐over trials and multiple treatment trials in the analysis.
Types of participants
Participant characteristics
We included adult participants diagnosed with SAnD irrespective of diagnostic criteria and measure, duration and severity of SAnD symptoms, age, and sex. We did, however, tabulate these descriptors in order to address the question of their possible impact on the effects of medication.
Comorbidities
We placed no restrictions on comorbid psychopathological disorders secondary to SAnD.
Setting
We placed no restrictions on setting.
Subsets of participants
We did not include trials that only included a subset of participants that met the review inclusion criteria in the analysis. None of the trials provided such information, so randomisation was preserved.
Types of interventions
We considered any medication administered to treat SAnD versus an active or non‐active placebo. We also included trials with multiple treatment arms if the comparator was a placebo, as well as placebo‐controlled trials studying multimodal treatments (cognitive behavioural therapy), if an active drug was a comparator.
Experimental interventions
We grouped specific pharmacological interventions according to medication class. We added anticonvulsants/GABAs, the anticonvulsant levetiracetam, NARIs, NaSSAs, and SARIs post hoc (see Differences between protocol and review). The medication classes are listed below.
5HT1A partial agonists (e.g. buspirone).
Anticonvulsants/gamma‐amino butyric acids (GABAs, e.g. gabapentin and pregabalin).
The anticonvulsant levetiracetam.
Antipsychotics (e.g. olanzapine).
Benzodiazepines (e.g. clonazepam and bromazepam).
Beta‐blockers (e.g. atenolol).
Mono‐amine oxidase inhibitors (MAOIs, e.g. brofaromine and moclobemide).
Noradrenaline reuptake inhibitors (NARIs, e.g. atomoxetine and mirtazepine).
Noradrenergic and specific serotonergic antidepressants (NaSSAs, e.g. mirtazepine).
Reversible inhibitors of monoamine oxidase A (RIMAs, e.g. phenelzine).
Serotonin antagonist and reuptake inhibitors (SARIs, e.g. nefazodone).
Serotonin and norepinephrine reuptake inhibitors (SNRIs, e.g. venlafaxine).
Selective serotonin reuptake inhibitors (SSRIs, e.g. paroxetine, fluvoxamine, sertraline, fluoxetine and citalopram).
Comparator interventions
Placebo (active or non‐active).
We placed no restrictions on timing, dose, duration, or co‐interventions.
Types of outcome measures
Primary outcomes
Treatment efficacy:
Clinical Global Impressions Improvement scale or similar: we determined treatment efficacy from the number of participants with SAnD who responded to treatment, as assessed by the Clinical Global Impressions Improvement scale (CGI‐I) or a closely related measure or definition (Guy 1976). The CGI ranges from 1 (normal, not at all ill) to 7 (among the most extremely ill patients). We defined responders as having a change item score of 1 ('very much') improved and 2 ('much') improved. Given its wide use and its reliability as a robust measure of the clinical value of treatment in SAnD, the CGI‐I served as a primary outcome measure for comparisons of both short‐ and long‐term trials in this review.
Relapse rate: The number of treatment responders who subsequently relapsed, according to investigator‐defined criteria, was compared between the medication and control groups.
2.Treatment tolerability: we included the total proportion of participants who withdrew from the RCTs due to treatment‐emergent side effects in the analysis as a surrogate measure of treatment tolerability, in the absence of other more direct indicators.
Secondary outcomes
Reduction in SAnD symptoms: we assessed symptom severity and, where available, symptom cluster response, using the Liebowitz Social Anxiety Scale (LSAS), a validated, commonly used, and psychometrically sound instrument (Liebowitz 1987). The LSAS is a 24‐item scale that provides separate scores for fear and avoidance in social and performance situations, with higher scores representing increased social anxiety. The LSAS contains three total scores: total fear score (0 to 72), total avoidance score (0 to 72), and total overall score (0 to 144). Suggested interpretations are that total scores of 55 to 65 indicate moderate social phobia; 65 to 80, marked social phobia; 80 to 95, severe social phobia; and greater than 95, very severe social phobia.
Reduction in depressive symptoms: we determined comorbid depressive symptoms using standardised scales such as the Hamilton Depression scale (HAM‐D) (Hamilton 1959) or Hamilton Rating Scale for Depression (HDRS), the Beck Depression Inventory (BDI) (Beck 1961), the Montgomery–Åsberg Depression Rating Scale (MADRS) or similar. The Hamilton Depression scale (HAM‐D) is a multiple item questionnaire with 17 to 29 items (depending on the version). Patients are rated on a 3 or 5 point scale. A score of 0‐7 is considered to be normal and a score of 20 or higher moderate, severe, or very severe. The Beck Depression Inventory (BDI) is a 21‐question multiple‐choice self‐report, one of the most widely used psychometric tests for measuring the severity of depression. A score of 0–9 indicates minimal depression, 10–18 mild depression, 19–29 moderate depression and 30–63 severe depression. The MADRS is a ten‐item diagnostic questionnaire which psychiatrists use to measure the severity of depressive episodes in patients with mood disorders. A higher MADRS score indicates more severe depression, and each item yields a score of 0 to 6. The overall score ranges from 0 to 60. Usual cutoff points are 0 to 6 – normal/symptom absent; 7 to 19 – mild depression; 20 to 34 – moderate depression; and > 34 – severe depression.
Functional disability: we considered measures such as the Sheehan Disability Scale (SDS) (Sheehan 1996). The Sheehan Disability Scale is a composite of three self‐rated items designed to measure the extent to which three major sectors in the patient’s life are impaired by panic, anxiety, phobic, or depressive symptoms. The patient rates the extent to which his or her 1) work, 2) social life or leisure activities, and 3) home life or family responsibilities are impaired by his or her symptoms on a 10‐point visual analogue scale. The numerical ratings of 0‐10 can be translated into a percentage if desired. The three items may be summed into a single dimensional measure of global functional impairment that ranges from 0 (unimpaired) to 30 (highly impaired).
Dropout rates: we compared all‐cause dropout rates for both short‐ and long‐term trials in order to provide some indication of treatment effectiveness.
Timing of outcome assessment
For studies that assessed outcomes at multiple time points (e.g. Baldwin 1999; Blomhoff 2001; Feltner 2011), we synthesised data from the last assessment within a 16‐week, postbaseline period to estimate the acute effects of medication, with the final assessment in longer trials providing data for the assessment of maintenance effects (20 to 24 weeks).
Hierarchy of outcome measures
If several measures for one outcome were reported, we selected the measures or scales laid out in the Methods. We used both clinician‐rated scales and self‐reported scales.
Search methods for identification of studies
Cochrane Specialised Register (CCMDCTR)
The Cochrane Common Mental Disorders Group maintained a comprehensive, specialised register of randomized controlled trials, the CCMDCTR (to June 2016). The register contains over 39,000 reference records (reports of RCTs) for anxiety disorders, depression, bipolar disorder, eating disorders, self‐harm and other mental disorders within the scope of this Group. The CCMDCTR is a partially studies based register with >50% of reference records tagged to c12,500 individually PICO coded study records. Reports of trials for inclusion in the register were collated from (weekly) generic searches of Medline (1950‐), Embase (1974‐) and PsycINFO (1967‐), quarterly searches of the Cochrane Central Register of Controlled Trials (CENTRAL) and review specific searches of additional databases. Reports of trials were also sourced from international trial registries, drug companies, the hand‐searching of key journals, conference proceedings and other (non‐Cochrane) systematic reviews and meta‐analyses. Details of CCMD's core search strategies (used to identify RCTs) can be found on the Group's website with an example of the core Medline search displayed in Appendix 1.
Electronic searches
The CCMD Group's information specialist searched the CCMDCTR (studies and references) registers on condition alone, due to concerns regarding multiple searches, change of authorship, and broad scope of this review. The latest version of the review incorporates results of searches to 17 August 2015 (Appendix 2). The information specialist ran a pre‐publication search (2 August 2017) (Appendix 3) and we have placed two additional studies in Awaiting Classification. These will be incorporated in the next version of the review, as appropriate.
The review author team also conducted earlier searches on PubMed, PsycINFO and clinicaltrials.gov (1966 to 2011), using terms 'social phobia OR social anxiety disorder' and 'medication OR pharmacotherapy OR treatment'. We undertook an initial broad search to find RCTs and open‐label trials, as well as journal and chapter reviews of the pharmacotherapy of SAnD.
The review authors also searched the ICTRP (apps.who.int/trialsearch) using the terms 'social phobia' or 'social anxiety' as search queries for this database (August 2012). We repeated this search on 4 November 2015 and 15 March 2017.
A 2016 Google Scholar search also yielded an additional included study by Nordahl 2016. We repeated this search on 15 March 2017.
Searching other resources
Reference lists
We scanned the bibliographies of all identified trials for additional studies.
Personal communication
We obtained published and unpublished trials from key researchers, who we identified by the frequency with which they were cited in the bibliographies of RCTs and open‐label studies.
Data collection and analysis
With the exception of the Egger test of funnel plot asymmetry, and generation of the contour enhanced funnel plots (created using the metafor package of the R statistical computing platform (Viechtbauer 2010), we used Review Manager 5 (RevMan 5) to perform all analyses reported in this review (RevMan 2014).
Selection of studies
Two authors (TW and JI) independently assessed the title and abstract of RCTs identified from the search. We subsequently scanned full‐text articles agreed upon as potentially eligible. The authors independently collated the data listed under Data extraction and management that satisfied the inclusion criteria specified in the Criteria for considering studies for this review section. We listed studies for which we need additional information to determine eligibility in the Studies awaiting classification table, pending the availability of this information. We resolved any disagreements in the trial assessment and data collation procedures by discussion with a third review author (DS).
Data extraction and management
We obtained the following information from each trial.
Description of the trials, including the year of publication, diagnostic instrument used (e.g. the Structured Clinical Interview for DSM‐IV (SCID)), use of placebo run‐in, use of a minimal severity criterion, number of participating centres, presence of support from the pharmaceutical industry, and methodological quality.
Characteristics of participants, including the diagnostic criteria met (e.g. DSM‐IV), subtype of SAnD (e.g. generalised SAnD), duration of symptoms, presence of comorbid depression, mean age, age range, and sex distribution.
Characteristics of the intervention, including its duration, the class of medication used, and the doses employed.
Outcome measures employed (primary and secondary), and summary continuous (means and standard deviations) and dichotomous (number of responders) data. We included additional information, such as the number of dropouts per group as well as the number that dropped out due to treatment‐emergent side effects.
Main comparisons
We planned to compare the following medications (grouped according to medication class) against placebo for treating SAnD.
5HT1A partial agonists.
Anticonvulsants/GABAs.
The anticonvulsant levetiracetam.
Antipsychotics.
Benzodiazepines.
Beta‐blockers.
MAOIs.
NARIs.
NaSSAs.
RIMAs.
SARIs.
SNRIs.
SSRIs.
Other medications.
Subgroup analyses included the following comparisons.
Multicentre compared to single‐centre trials.
Generalised SAnD compared to inclusive SAnD.
Industry funding compared to no industry funding.
Inclusion versus exclusion of participants diagnosed with major depressive disorder (MDD).
Assessment of risk of bias in included studies
We assessed the risk of bias of each included study using the Cochrane 'Risk of bias' tool (Higgins 2011). We considered the following six domains.
Random sequence generation: did investigators use a random number table or a computerised random number generator?
Allocation concealment: was the medication sequentially numbered, sealed, and placed in opaque envelopes?
Blinding of participants, personnel, and outcome assessors for each main outcome or class of outcomes: was knowledge of the allocated treatment or assessment adequately prevented during the study?
Incomplete outcome data for each main outcome or class of outcomes: were missing or excluded outcome data adequately addressed?
Selective outcome reporting: were the reports of the study free of suggestion of selective outcome reporting? We could only make such a judgement based on the availability of the protocol.
Other sources of bias: was the study apparently free of other problems that could put it at a high risk of bias?
We extracted relevant information from each study report, where provided. We made a judgement on the risk of bias for each domain within and across studies, based on the following three categories: 'low' risk of bias, 'unclear' risk of bias, and 'high' risk of bias. Two independent review authors (TW and JI) assessed the risk of bias for the included studies. We discussed any disagreements with a third review author (DS). Where necessary, we contacted the authors of the studies for further information. We present all risk of bias data graphically and describe them in the text.
Measures of treatment effect
Categorical data
We calculated the risk ratio (RR) of response to treatment for the dichotomous outcomes of interest. We used RR instead of odds ratio (OR), as ORs are more difficult to interpret. ORs also tend to overestimate the size of the treatment effect relative to RRs, especially when the occurrence of the outcome of interest is common (as anticipated in this review, with an expected response greater than 20%) (Deeks 2011).
Continuous data
In cases in which studies used a range of scales for each outcome, such as the assessment of comorbid depressive symptoms on the Hamilton Depression scale (HAM‐D) and the Beck Depression Inventory (BDI), we calculated treatment outcome using the standardised mean difference (SMD). The SMD standardises the differences between the means of the treatment and control groups in terms of the variability observed across all participants in the trial.
Unit of analysis issues
Cluster‐randomised trials
In cluster‐randomised trials, groups of individuals are randomised to different interventions rather than individuals themselves. Analysing treatment response in cluster‐randomised trials without taking these groupings into account is potentially problematic, as participants within any one cluster often tend to respond in a similar manner, and thus analyses cannot assume that participants' data are independent of the rest of the cluster. Cluster‐randomised trials also face additional risk of bias issues including recruitment bias, baseline imbalance, loss of clusters, and non‐comparability with trials randomising individuals (Higgins 2011). No cluster‐randomised trials were eligible for inclusion in this review. To prevent unit of analysis errors in future updates of this review, we plan to divide the effective sample size of each comparison group in trials that do not adjust for clustering by the design effect metric (Higgins 2011). For these analyses the intraclass correlation coefficient (ICC) that is incorporated within the design effect will be set equivalent to the median ICC from published cluster‐randomised pharmacotherapy RCTs for anxiety disorders.
Cross‐over trials
We only included cross‐over trials in the calculation of summary statistics when it was possible to extract medication and placebo/comparator data from the first treatment period or when the inclusion of these data from both treatment periods was justified by a washout period of sufficient duration that minimised the risk of carry‐over effects. In the latter case, we included data from both periods only when it was possible to determine the correlation between participants' responses to the interventions in the different phases (Elbourne 2002). A washout period of at least two weeks was necessary in the case of trials assessing the efficacy of agents with extended half‐lives, such as the SSRI fluoxetine (Gury 1999).
Multiple treatment groups
A number of the trials included in this review compared more than two intervention groups or multiple doses of the same medication against placebo. Including data from the same placebo group for these studies repeatedly in the same comparison would result in a unit of analysis error (Higgins 2011). To prevent these errors for trials comparing multiple dosages of the same agent to placebo, we averaged the mean and standard deviation of the outcome of interest across dosage groups. We included outcome data from multiple treatment arms in the same comparison if the agents tested were from different medication classes. We turned off the subtotals of the outcome if the placebo group appeared twice in the analysis to accommodate for the second medication. In the case of trials testing multiple agents from the same classes, and in calculating the total effect across all medication classes, we restricted data from multi‐arm RCTs to the agent that was least represented in the database.
We will circumvent unit‐of‐analysis bias resulting from the simultaneous comparison of multiple arms from the same trial in future updates of this review by means of a multiple‐treatments meta‐analysis (MTM) (Lumley 2002). An MTM allows the assessment of treatment efficacy through the combination of both direct and indirect comparisons of all interventions on a specific outcome. We can subsequently assess potential unit‐of‐analysis bias in a sensitivity analysis in which we compare the results obtained with those from a meta‐analysis restricted to data from direct comparisons of interventions.
Dealing with missing data
All analyses of dichotomous data were intention‐to‐treat (ITT). We only included data from trials providing information on the original group size (prior to dropouts) in the analyses of treatment efficacy. We gave preference within studies to the inclusion of summary statistics for continuous outcome measures derived from mixed‐effects models, followed by last observation carried forward (LOCF) and observed cases (OC) summary statistics (in that order). This is in line with evidence that mixed‐effects methods are more robust to bias than LOCF analyses (Verbeke 2000).
Assessment of heterogeneity
We assessed heterogeneity by means of the Chi2 test for heterogeneity to assess whether observed differences in results were compatible with chance alone. A low P value (or a large Chi2 test relative to its degree of freedom (df)) provides evidence of heterogeneity of intervention effects (variation in effect estimates beyond chance). If the Chi2 test had a P value of less than 0.10, we interpreted it as evidence of heterogeneity, given the low power of the Chi2 test when the number of trials is small (Deeks 2011). We also used the Deeks' stratified test of heterogeneity (Deeks 200), as implemented in RevMan 5, to assess differences by means of the Qb metric in treatment response between subgroups, by subtracting the sum of the Chi2 statistic for the subgroups from the total Chi2 statistic for those subgroups.
In addition, we used the I2 statistic reported by RevMan 5 to quantify the inconsistency of the trial results within each analysis (Higgins 2003). Thresholds for the interpretation of I2 can be misleading, since the importance of inconsistency depends on several factors. We followed a rough guide for interpretation.
0% to 40%: might not be important.
30% to 60%: may represent moderate heterogeneity.
50% to 90%: may represent substantial heterogeneity.
75% to 100%: considerable heterogeneity.
Assessment of reporting biases
Funnel plots provide a graphic illustration of the effect estimates of an intervention from individual studies against some measure of the precision of that estimate. We visually inspected publication bias from the funnel plot for treatment efficacy, with the consideration of confounding selection bias, poor methodological quality, true heterogeneity, artefact, and chance. We also calculated Eggers' regression tests as a more objective quantitative measure of funnel plot asymmetry using the Dersimonian and Laird estimator of heterogeneity (Egger 1997). Given that both tests are dependent on having 10 trials per outcome, we could only calculate this for SSRIs. Irwig 1998 and others have voiced concerns that the statistical phenomenon increasing the likelihood of falsely detecting publication bias when applying the Egger test to odds ratio effect estimates may extend to risk ratio effect estimates (Sterne 2011). Accordingly, we generated contour enhanced funnel plots, which explicitly illustrate the relationship between missing studies and the statistical significance of study findings at various statistical thresholds (e.g. alpha = 0.1, 0.05, and 0.01) (Peters 2008). We estimated the position of missing studies in these plots using the trim and fill method.
Data synthesis
We obtained binary and continuous treatment effects from a random‐effects model. Random‐effects analytic models include both within‐study sampling error and between‐study variation in determining the precision of the confidence interval around the overall effect size, whereas fixed‐effect modelling approaches take only within‐study variation into account. In recognition of the possibility of differential effects for different types of medication, such as the SSRIs and the MAOIs, we stratified all of the comparisons by medication class. We expressed the outcomes of these comparisons in terms of an average effect and 95% confidence interval (CI) for each subgroup.
Subgroup analysis and investigation of heterogeneity
We conducted the following subgroup analyses to assess the degree to which methodological differences between trials might have systematically influenced differences observed in the primary treatment outcomes (Thompson 1994).
We compared multicentre versus single‐centre trials. The latter are more likely to be associated with lower sample size but will tend to have less variability in clinician ratings.
We compared trials including only participants diagnosed with the generalised form of SAnD versus those including both the generalised and specific subtypes of SAnD. Specialists recognise generalised SAnD as often representing a more severe form of the disorder.
In addition, we assessed the following.
Whether or not trials were industry funded. In general, published trials sponsored by pharmaceutical companies appear more likely to report positive findings than trials that are not supported by profit companies (Als‐Nielsen 2003; Baker 2003).
Whether or not the sample included patients diagnosed with major depression. Such an analysis might assist in determining the extent to which the efficacy of a medication agent in treating SAnD is independent of its ability to reduce symptoms of depression, an important consideration given the classification of many of these medications as anti‐depressants.
Sensitivity analysis
Sensitivity analyses determine the robustness of the review authors' conclusion to methodological assumptions made in conducting the meta‐analysis. We planned to compare the effect of assessing interventions in terms of clinical treatment response versus non‐response in light of evidence that treatment response may result in less consistent outcome statistics than non‐response (Deeks 2002). However, reports on this potential difference in outcome consistency have arisen only when the control group event rate was above 50%, far higher than the baseline proportion of response observed across trials in this review (30%). Accordingly, we did not conduct this analysis.
In addition, we performed a 'worst case/best case' analysis to determine whether the the coding of participants who were lost to follow‐up influenced the findings of treatment efficacy (Deeks 2011). In this analysis, we recorded all the missing data for the treatment group as responders in the worst case scenario, whereas in the best case, we coded all missing data for the control group as responders. If the conclusions regarding treatment efficacy did not differ between these two comparisons, we assumed that missing data in trial reports did not have a significant influence on this outcome.
Summary of findings tables
We compiled 'Summary of findings' tables to summarise the best evidence for all relevant outcomes (i.e. experimental versus comparator interventions), reporting the following six elements according to a fixed format (Higgins 2011).
A list of all important outcomes, both desirable and undesirable.
A measure of the typical burden of these outcomes (e.g. illustrative risk, or illustrative mean, on control intervention).
Absolute and relative magnitude of effect (if both are appropriate).
Numbers of participants and studies addressing these outcomes.
A grade of the overall quality of the body of evidence for each outcome.
Space for comments.
We based downgrading of the evidence rating for outcomes on five factors. We classified reasons for downgrading the evidence as 'serious' (downgrading the quality rating by one level) or 'very serious' (downgrading the quality grade by two levels).
Limitations in the design and implementation of the trial.
Indirectness of evidence.
Unexplained heterogeneity or inconsistency of results.
Imprecision of results.
High probability of publication bias.
We classified the quality of evidence for each outcome according to the following categories.
High quality: further research is very unlikely to change our confidence in the estimate of effect.
Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate.
Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate.
Very low quality: we are very uncertain about the estimate.
Main comparisons
We planned the following outcomes and grouped specific pharmacological interventions according to medication class.
5HT1A partial agonists versus placebo.
Anticonvulsants/GABAs versus placebo.
The anticonvulsant levetiracetam versus placebo.
Antipsychotics versus placebo.
Benzodiazepines versus placebo.
Beta‐blockers versus placebo.
MAOIs versus placebo.
NARIs versus placebo.
NaSSAs versus placebo.
RIMAs versus placebo.
SARIs versus placebo.
SNRIs versus placebo.
SSRIs versus placebo.
Other medication versus placebo.
Results
Description of studies
Results of the search
We found a total of 2611 study reports through the search process (CCMDCTR 2162; ICTRP 449). We scanned each title and abstract (if provided) for eligibility. Three hundred and two studies initially seemed relevant, but after further inspection we excluded 166 of these, leaving 136 studies that potentially met the inclusion criteria (we found the additional study by Nordahl 2016 from a search conducted in 2016). After independent review of the full‐text studies, we found that 70 failed to meet inclusion criteria, leaving 66 RCTs eligible for inclusion in the review (see Characteristics of included studies and Figure 1). Of the 66 trials, we included 63 RCTs in the meta‐analysis. Eleven studies are awaiting classification, and five studies are ongoing (see Studies awaiting classification and Ongoing studies).
1.
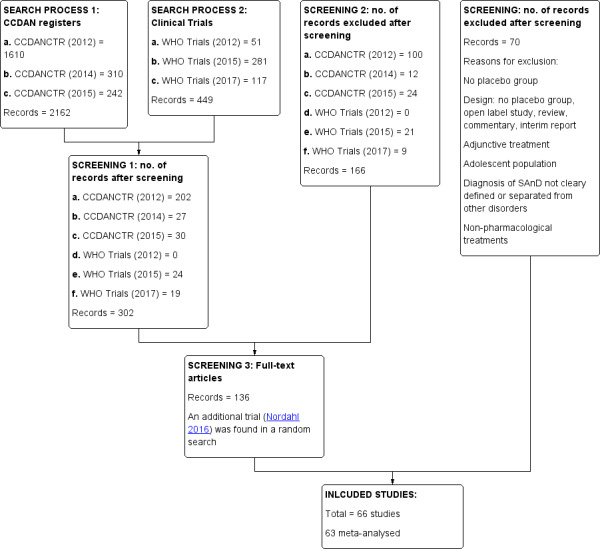
Study flow diagram.
The update search performed by CCMD's information specialist retrieved 754 records (de‐duplicated), and after screening the abstracts, we identified an addition two studies which we have added to awaiting classification. We will incorporate these into the next version of the review as appropriate.
Included studies
Design
The review includes 66 RCTs treating participants for SAnD from 1 to 24 weeks. Of these, 56 of the trials were short term (14 weeks or less), while 7 trials included a maintenance component, and 4 trials a relapse component (14 to 24 weeks or less). This update includes 29 new studies. All trials included a placebo comparison group, with eight studies having two medication arms (i.e. two trials assessing paroxetine and venlafaxine, and one trial each assessing citalopram and NK1 antagonist GR205171, paroxetine and escitalopram, phenelzine and atenolol, paroxetine and neurokinin‐1 (NK‐1) antagonist LY686017, phenelzine and moclobemide, and GW876008 and paroxetine) (Allgulander 2004; Furmark 2005; Lader 2004; Liebowitz 1992; Liebowitz 2005b; NCT00397722; Tauscher 2010; Versiani 1992). Katzelnick 1995 employed a a cross‐over design in testing the efficacy of sertraline. All trials were published in English, and pharmaceutical companies contributed funding for 41.
Participants
The 66 eligible trials included 11,597 participants aged 18 to 70, most of whom were outpatients. The average sample size was 176 and ranged from 12 in Barnett 2002 and Katzelnick 1995 to 839 in Lader 2004. Sixty trials involved both men and women, while Muehlbacher 2005 included only women and Moghadam 2015 only men. Adult participants were diagnosed with SAnD according to DSM criteria (DSM‐IV‐TR, DSM‐IV, DSM‐III‐R), assessed by means of a variety of instruments, including the Structured Clinical Interview for DSM (SCID/SCID‐I) (e.g. Moghadam 2015; NCT00470483), the Mini International Neuropsychiatric Interview (MINI) (e.g. Furmark 2005; Lepola 2004; NCT00403962; Stein 2002b; Zhang 2005), and the Initial Evaluation Form and the Anxiety Disorders Interview Schedule‐Revised (ADIS‐R) (e.g. Turner 1994).
Twenty‐one studies included individuals with major depressive disorder (MDD), while MDD was an exclusion criterion for 41. Four studies did not specify, so we were unable to determine whether or not individuals with MDD could take part (Moghadam 2015; NCT00470483; Nordahl 2016; Tauscher 2010). We also reported other comorbidities related to functional disability and avoidant personality where provided.
Setting
Most eligible RCTs took place in the USA (number of studies (k) = 58), while others were in Europe (k = 10), Japan (k = 2), South Africa (k = 6), and Iran (k = 1). Of the trials included in the analysis, 24 studies were single‐centre trials, and 42 studies took place in multiple centres. Participants were recruited via telephone, newspaper, radio, or clinical referral. Recruitment settings included university‐based hospitals and centres, medical centres, and research and private clinics.
Interventions
Approximately half of the included RCTs tested the efficacy of selective serotonin reuptake inhibitors (SSRIs; k = 34), including 19 studies of paroxetine, of which two studies had a third arm investigating venlafaxine, one study had a third arm investigating escitalopram, one had a third arm investigating LY686017 and another a third arm investigating GW876008 (Allgulander 1999; Allgulander 2004;Baldwin 1999; Book 2008; Lader 2004; Lepola 2004; Liebowitz 2002; Liebowitz 2005b; NCT00273039; NCT00318669; NCT00403962; NCT00397722; NCT00470483; Nordahl 2016; Randall 2001; Stein 1996; Stein 1998; Stein 2003; Tauscher 2010). In addition, the SSRI RCTs included five trials of fluvoxamine (Asakura 2007; Davidson 2004a; Stein 1999; Van Vliet 1994; Westenberg 2004), six trials of sertraline (Blomhoff 2001; Katzelnick 1995; Liebowitz 2003; Moghadam 2015; Van Ameringen 2001a; Walker 2000), two trials of fluoxetine (Davidson 2004b; Kobak 2002), and single RCTs of citalopram (Furmark 2005, with a third arm investigating NK1 antagonist GR205171) and escitalopram (Kasper 2005). The lowest and highest dosage for paroxetine ranged from 7.5 mg/d (NCT00403962) to 60 mg/d (Book 2008; NCT00397722; Nordahl 2016); for sertraline from 50 mg/d (Blomhoff 2001; Katzelnick 1995) to 200 mg/d (Katzelnick 1995; Liebowitz 2003; Van Ameringen 2001a; Walker 2000); for fluoxetine from 20 mg/d to 60 mg/d (Davidson 2004b); for fluvoxamine from 50 mg/d (Asakura 2007; Stein 1999; van Vliet 1997) to 300 mg/d (Asakura 2007; Davidson 2004a; Stein 1999; Stein 2003; Westenberg 2004); for citalopram from 20 mg/d to 40 mg/d (Furmark 2005); and for escitalopram from 10 mg/d to 20 mg/d (Kasper 2005).
Eight trials studied reversible inhibitors of monoamine oxidase A (RIMAs), including three RCTs of brofaromine (Fahlen 1995; Lott 1997; Van Vliet 1992), plus five of moclobemide (Katschnig 1997; Noyes 1997; Oosterbaan 2001; Schneier 1998; Stein 2002a). For brofaromine, the daily dosage ranged from 50 mg/d to 150 mg/d in both Lott 1997 and Van Vliet 1992, and for moclobemide, it ranged from 75 mg/d in Noyes 1997 to 750 mg/d in Stein 2002a.
Four trials evaluated the mono‐amine oxidase inhibitor (MAOI) phenelzine, one of which had a third arm investigating atenolol and another investigating moclobemide (Blanco 2010; Heimberg 1998; Liebowitz 1992; Versiani 1992). Dosage for the phenelzine ranged from 15 mg/d in Blanco 2010 and Heimberg 1998 to 100 mg/d in Liebowitz 1992.
There were four trials of the serotonin and norepinephrine reuptake inhibitor (SNRI) venlafaxine (Liebowitz 2005a; Liebowitz 2005b; Stein 2005; Rickels 2004). Dosage ranged from 75 mg/d to 225 mg/d in all of these trials.
Three trials focused on anticonvulsants comprising gamma‐amino butyric acid (GABA) analogues (including two studies of pregabalin, Feltner 2011 and Pande 2004, and one of gabapentin, Pande 1999). Two trials investigated levetiracetam, another anticonvulsant (Stein 2010; Zhang 2005).
Three trials studied benzodiazepines, including two studies of clonazepam in doses ranging from 0.25 mg/d in both Connor 1998 and Davidson 1993 to 2.5 mg/d in Connor 1998, and one of bromazepam (Versiani 1997), administered in doses of 3 mg/d to 9 mg/d.
There were two trials on antipsychotics, including one study of olanzapine, with dosage ranging from 5 mg/d to 20 mg/d (Barnett 2002), and another study on quetiapine, with dosage of 25 mg/d to 300 mg/d (Vaishnavi 2007).
Two trials evaluated the noradrenergic and specific serotonergic antidepressant (NaSSA) mirtazepine, in doses ranging from 30 mg/d in Muehlbacher 2005 and Schutters 2010 to 45 mg/d in Schutters 2010.
Turner 1994 assessed the beta‐blocker atenolol, administered in doses of 25 mg/d to 100 mg/d; Ravindran 2009 assessed the noradrenaline reuptake inhibitor (NARI) atomoxetine, administered in doses of 20 mg/d to 100 mg/d; Van Ameringen 2007 looked at the serotonin antagonist and reuptake inhibitor (SARI) nefazodone, with dosage of 100 mg/d to 600 mg/d; NCT00397722 studied GW876008, at doses of 25 mg/d to 50 mg/d; Tauscher 2010 studied LY686017 at 50 mg/d; Furmark 2005 investigated the NK1 antagonist GR205171, at 4 mL to 100 mL; and Van Vliet 1997 the 5HT1A partial agonist buspirone, at doses of 15 mg/d to 30 mg/d.
Psychiatrists, pharmacists, mental health professionals, and multi‐disciplinary teams of investigators administered the interventions.
Outcomes
Twenty‐three trials assessed the primary efficacy outcome – number of participants with SAnD who responded to treatment – using the Clinical Global Impression – Global Improvement (CGI–I) Scale, whilst 27 other trials assessed this outcome as a secondary measure using the same scale. Forty‐two trials used the Liebowitz Social Anxiety Scale (LSAS) to assess the reduction of SAnD symptoms, while eight used the Clinical Global Impression ‐ Severity scale (CGI‐S). The most common measures used to assess depression were the Beck Depression Inventory (BDI; k = 2), the the Hamilton Rating Scale for Depression (HAM‐D; k = 12), and the Montgomery–Åsberg Depression Rating Scale (MADRS) (k = 10). Thirty trials assessed functional disability using the Sheehan Disability Scale (SDS). Post‐treatment follow‐up assessments ranged from two weeks in Tauscher 2010 to 15 months in Oosterbaan 2001. See Characteristics of included studies for more details.
Excluded studies
We excluded 70 studies from the review (see Characteristics of excluded studies). The most common reason we considered trials ineligible was the absence of a placebo comparator (Allsopp 1984; ACTRN12608000363381; Atmaca 2002; Blank 2006; Bystritsky 2005; Clark 2003; Dunlop 2007; Falloon 1981; Gelernter 1991; EUCTR2004‐001894‐24‐DE; Guastella 2009; Hofmann 2006; Krishman 1976; Liappas 2003; Liebowitz 1999; Oosterbaan 1997; Otto 2000; Pecknold 1982; Rickels 2004; Seedat 2003; Simon 2010; Tubaki 2012; ACTRN12609000091202). We also excluded studies with participants under the age of 18 and those with combined populations and certain comorbidities (Dempsey 2009; Grosser 2012; Ionescu 2013). We excluded Dempsey 2009, Gale 2007, and Mangano 2003 because these trials were secondary analyses of previous studies. We also excluded open‐label studies that did not employ a randomised double‐blind placebo‐control study design (Angelini 1989). Twelve early trials with anxiety disorders did not report data separately for patients with SAnD, so we could not include them (Angelini 1989; Coupland 2000; NCT00248612; Greenhill 1999; Heun 2013; Malcolm 1992; Mountjoy 1977; Pine 2001; Schuurmans 2004; Solyom 1973; Solyom 1981; Tyrer 1973). One study focused on neuroendocrine and behavioural responses of SAnD (Shlik 2002). Four studies on brain functioning, with specific reference to the modulation of amygdala‐frontal reactivity and connectivity, were ineligible for inclusion (Dodhia 2014; Gorka 2015; NCT00332046; Phan 2015). A number of trials assessed the effect of medication on individuals with performance anxiety who were not diagnosed with SAnD (Brantigan 1982; Clark‐Elford 2015; Fang 2014; Gates 1985; Gorka 2015; Hartley 1983; James 1977; James 1984; James 1983; Liebowitz 2014; Liden 1974; NCT00308724; NCT00343707; Neftel 1982; Siitonen 1976; Wardle 2012). One excluded study only measured treatment‐emergent side effects (Rynn 2008). Finally, we excluded studies that combined pharmacotherapy and psychotherapy (Donahue 2009; Feifel 2011; Haug 2003; Mortberg 2007; NCT00308724; Prasko 2004; Ravindran 2014; Silverstone 1973; Wardle 2012), as well as studies using herbal treatments (NCT00118833). See Characteristics of excluded studies for more details.
Studies awaiting classification
Thirteen trials are awaiting classification (Asakura 2016; Careri 2015; De la Barquera 2008; Frick 2015; Krylov 1996; NCT00114127; NCT00208741; NCT00215254; NCT00246441; NCT00294346; NCT00485888; NCT00612859; NCT01316302). NCT00294346 conducted a study on the effectiveness of an investigational drug AV608 in subjects with social anxiety disorder (SAnD), using the Liebowitz Social Anxiety Scale (LSAS) to assess reductions of anxiety symptoms. De la Barquera 2008 assessed clonazepam compared to placebo in patients with social phobia, and Frick 2015 citalopram, GR205171, or placebo. Frick 2015 involved 18 SAnD patients and used the Liebowitz Social Anxiety Scale (LSAS) to assess symptom severity and positron emission tomography (PET) imaging to assess brain function. Krylov 1996 assessed alprazolam, buspirone, or placebo in 66 patients with social phobia. NCT00485888 investigated the effects of cipralex on the reduction of social phobia, as measured by the LSAS and various other secondary scales, in 71 outpatients aged 18 to 75 years and diagnosed with social phobia. NCT01316302 studied desvenlafaxine (Pristiq) compared to matching placebo over 12 weeks. Sixty‐three patients enrolled, and the primary outcome of symptom severity was measured with the LSAS. Similarly, NCT00208741 investigated Gabitril compared to placebo over 24 weeks with 50 patients using the LSAS and CGI‐C scales as primary outcome measures; NCT00246441 compared paroxetine to placebo over 16 weeks with 42 patients; and NCT00114127 compared duloxetine to placebo for 18 weeks with 28 patients whose symptoms were assessed with the LSAS. The additional trial by NCT00612859 assessed the efficacy and safety of levetiracetam versus placebo for the treatment of generalised SAnD measured by the LSAS. See Characteristics of studies awaiting classification and Figure 1 for more details.
We identified two studies from the update search performed by CCMD's information specialist (Asakura 2016; Careri 2015). We will incorporate these into the next version of the review as appropriate.
Ongoing studies
Five studies are ongoing (NCT00182533; NCT01712321; NCT02083926; NCT02294305; NCT02432703). NCT02083926 will compare ketamine compared placebo (saline) using the Beck Anxiety Inventory (BAI), the CGI scale, and the LSAS to determine levels of anxiety severity in SAnD patients, whereas NCT02432703 will compare JNJ‐42165279 to placebo for 12 weeks on the LSAS. NCT00182533 will assess the efficacy of sertraline, and NCT01712321 and NCT02294305 will compare vortioxetine to placebo for treating generalised social anxiety disorder in outpatients. The most common secondary outcome measures include the assessment of depression (NCT00182533; NCT01712321; NCT02294305), anxiety (NCT01712321; NCT02294305), functional disability, and quality of life (NCT00182533). See Characteristics of ongoing studies and Figure 1 for more details.
Risk of bias in included studies
We assessed the risk of bias using the Cochrane 'Risk of bias' tool for allocation concealment, blinding, incomplete outcome data, selective reporting and other potential sources of bias. We classified twelve of the included studies as being at high risk for at least one type of bias (see Figure 2 and Figure 3).
2.
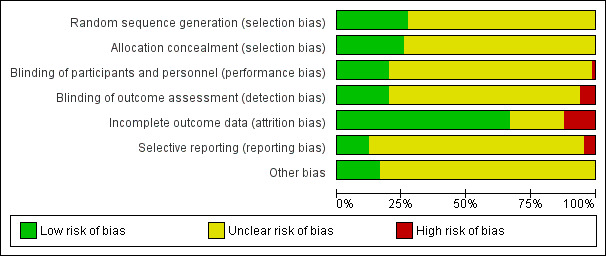
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
3.
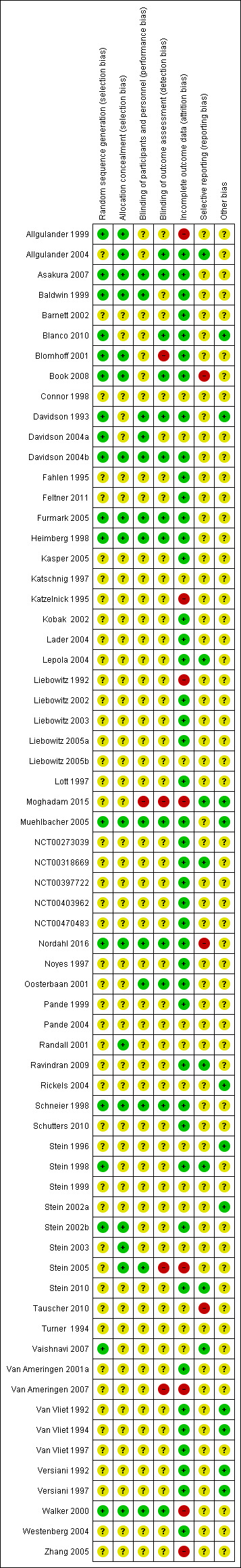
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
Randomisation
Seventeen of the included studies described the sequence generation as randomised. Allgulander 1999, Blanco 2010, Heimberg 1998, Muehlbacher 2005, and Walker 2000 used tabulated random numbers to randomly assign participants to two groups. In other studies, group assignment was via a computer‐generated urn program (Book 2008; Vaishnavi 2007), block program or randomisation (Baldwin 1999; Davidson 2004b; Furmark 2005; Nordahl 2016; Stein 1998), or a computer‐generated randomisation list (Blomhoff 2001; Davidson 1993; Stein 2002b). Asakura 2007 used a randomisation scheme by double‐dummy method, whereas Davidson 2004a and Schneier 1998 determined randomisation through the sponsor of the central source (e.g. pharmacy) or through a data manager with no patient contact. We classified all these studies as being at low risk for selection bias, while we designated the risk of bias as unclear in the remaining studies.
Allocation concealment
We could only classify 17 of the 66 trials included in the review as being at low risk for allocation concealment from their description of the method of allocation. A pharmacist or sponsor who prepared and supplied the study medication maintained allocation concealment in nine trials (Allgulander 1999; Allgulander 2004; Asakura 2007; Baldwin 1999; Blomhoff 2001; Book 2008; Nordahl 2016; Randall 2001; Walker 2000). Other studies maintained concealment with the use of sealed envelopes (Blomhoff 2001; Furmark 2005; Schneier 1998), double‐blind packaging (Asakura 2007; Davidson 2004b; Stein 2002b; Stein 2003; Walker 2000), label coding for each participant or numbered box (Davidson 2004b; Muehlbacher 2005; Schneier 1998; Stein 2005; Stein 2003), and cohorts of patients randomly intermixed (Heimberg 1998).
Blinding
Blinding of participants and personnel
We classified 13 studies included in the review as being at low risk of performance bias, as participants and personnel were explicitly described as blinded in the study report (Asakura 2007; Baldwin 1999; Davidson 1993; Davidson 2004a; Davidson 2004b; Furmark 2005; Heimberg 1998; Muehlbacher 2005; Nordahl 2016; Oosterbaan 2001; Schneier 1998; Stein 2005; Walker 2000). In Oosterbaan 2001 and Schneier 1998, participants and therapists or independent evaluators guessed post‐test whether medication or placebo had been administered. Correct classifications did not differ from chance. We classified one trial as being at high risk because it provided no information on blinding participants or personnel, nor did investigators describe the study as double‐blind (Moghadam 2015). We classified risk of bias for the remaining trials as unclear, despite investigators describing them in the study reports as double‐blinded, as they did not specify the actual parties blinded.
Blinding of outcome assessors
We classified 13 of the studies included in the review as being at low risk of detection bias, as the study reports explicitly described outcome assessors as blinded in the study report (Allgulander 2004; Asakura 2007; Blanco 2010; Book 2008; Davidson 1993; Davidson 2004b; Furmark 2005; Heimberg 1998; Muehlbacher 2005; Nordahl 2016; Oosterbaan 2001; Schneier 1998; Walker 2000). We classified Blomhoff 2001, Stein 2005, and Van Ameringen 2007 as being at high risk, as these studies reported that outcome assessors were not blinded to treatment, and Moghadam 2015 because authors provided no information to determine if outcome assessors were blinded, nor if the study was double blind. We classified the remaining studies as being at unclear risk.
Incomplete outcome data
Fourteen studies failed to provide sufficient information to determine whether the medication and placebo groups were comparable with respect to dropout proportions, or in terms of the demographic and clinical characteristics of those who withdrew (Connor 1998; Davidson 2004a; Katschnig 1997; Liebowitz 2005b; Pande 2004; Randall 2001; Rickels 2004; Stein 1996; Stein 1999; Stein 2002a; Stein 2003; Tauscher 2010; Turner 1994; Vaishnavi 2007). We rated the risk of attrition bias as unclear for these trials. We observed substantial differences in the proportion of study dropouts between the medication and placebo groups in Allgulander 1999, Katzelnick 1995, Liebowitz 1992, Moghadam 2015; Stein 2005, Van Ameringen 2007, and in the maintenance treatment design that Walker 2000 and Zhang 2005 employed, justifying a rating of high risk. We rated the remaining 44 studies as being at low risk for this domain, on the basis of comparable dropout rates in each comparison group and no difference in the demographic characteristics. Overall, the total dropout rate was 25% across all medications compared to placebo (with the exclusion of one trial of paroxetine, to give preference to the experimental drug that is least represented in the data set). We assessed intention‐to‐treat data for all outcomes in 11 studies and used LOCF (k = 46), observed cases (k = 9) and mixed‐effect models (k = 3) to account for missing data in the rest of the trials. Thirteen studies did not specify how they addressed the issue of missing data.
Selective reporting
It was unclear whether selective reporting took place in 55 trials because the study protocols were not available for the study. We rated eight studies as being at low risk for selective reporting because authors reported on all outcomes as specified in their respective trial protocols (Allgulander 2004; Lepola 2004; Moghadam 2015; NCT00318669; Ravindran 2009; Stein 1998; Stein 2010; Vaishnavi 2007). In Book 2008, Nordahl 2016, and Tauscher 2010, the pre‐specified secondary outcomes in the protocol did not appear in the study report, meriting a classification of high risk.
Other potential sources of bias
We classified 11 studies as being at low risk for other potential sources of bias (Blanco 2010; Davidson 1993; Moghadam 2015; Muehlbacher 2005; Rickels 2004; Stein 1996; Stein 2002a; Van Vliet 1992; Van Vliet 1994; Versiani 1992; Versiani 1997). We rated the risk of other bias for the remaining studies as unclear due to industry involvement, whether through funding or provision of study medication.
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4; Table 5; Table 6; Table 7; Table 8; Table 9; Table 10; Table 11; Table 12; Table 13; Table 14; Table 15; Table 16
Summary of findings for the main comparison. Comparison 1: 5HT1A partial agonists versus placebo for social anxiety disorder (SAnD).
| Comparison 1: 5HT1A partial agonists versus placebo for social anxiety disorder (SAnD) | ||||||
| Patient or population: adults with SAnD Settings: outpatient settings Intervention: 5HT1A partial agonists Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| With placebo | With 5HT1A partial agonists | |||||
| Global Impressions scale change item (CGI‐I or similar): no. of responders (acute phase) | Study population | RR 1.00 (0.07 to 14.55) | 30 (1 study) | ⊕⊝⊝⊝ Very lowa,b | There was no evidence of an effect on the number of participants in the 5HT1A partial agonist group compared to the placebo group who responded 'Very Much Improved' or 'Much Improved' on the CGI‐I scale (P = 1.00). | |
| 67 per 1000 | 67 per 1000 (5 to 970) | |||||
| Moderate | ||||||
| 67 per 1000 | 67 per 1000 (5 to 975) | |||||
| Dropouts due to adverse events (acute phase) | Study population | RR 3.00 (0.13 to 68.26) | 30 (1 study) | ⊕⊝⊝⊝ Very lowa,b | Dropout rates due to adverse events were low in the 5HT1A partial agonist group (1/30, 3%). No participants withdrew from the placebo group. | |
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Moderate | ||||||
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS avoidance subscale | The mean anxiety score for the control group was 24.3 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS avoidance) in the intervention groups was 1.4 points lower (11.61 lower to 8.81 higher) | 30 (1 study) | ⊕⊝⊝⊝ Very lowa,c | The mean LSAS avoidance anxiety score for the 5HT1A partial agonist intervention group was 22.9 which suggests 'low' social phobia. | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; CGI‐I: Clinical Global Impressions Improvement scale; LSAS: Liebowitz Social Anxiety Scale;RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: we are very uncertain about the estimate. | ||||||
aDowngraded one level due to serious risk of bias (concerns with randomisation procedures). bDowngraded two levels due to very serious imprecision (small sample size, few events and wide confidence interval). cDowngraded two levels due to very serious imprecision (small sample size and wide confidence interval). dResponse is defined as the number of participants with SAnD who responded to treatment, as assessed by the CGI‐I or similar.
Summary of findings 2. Comparison 2: anticonvulsants/GABAs versus placebo for social anxiety disorder (SAnD).
| Comparison 2: anticonvulsants/GABAs versus placebo for social anxiety disorder (SAnD) | ||||||
| Patient or population: adults with SAnD Settings: outpatient settings Intervention: anticonvulsants/GABAs Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| With placebo | With anticonvulsants/GABAs | |||||
| Global Impressions scale change item (CGI‐I or similar): no. of responders (acute phase) | Study population | RR 1.60 (1.16 to 2.20) | 532 (3 studies) | ⊕⊕⊕⊝ Moderatea | There was evidence of benefit on the number of participants with SAnD who responded to treatment (P = 0.004). A RR score greater than 1 and 95% CI that does not overlap with 1 indicates that there were a statistically significantly greater number of people in the anticonvulsant/GABA groups compared to the placebo groups who responded 'Very Much Improved' or 'Much Improved' on the CGI‐I scale. | |
| 221 per 1000 | 353 per 1000 (256 to 486) | |||||
| Moderate | ||||||
| 217 per 1000 | 347 per 1000 (252 to 477) | |||||
| Dropouts due to adverse events (acute phase) | Study population | RR 2.90 (0.92 to 9.14) | 532 (3 studies) | ⊕⊝⊝⊝ Very lowa,b,c | Dropout rates due to adverse events were high in the anticonvulsant/GABA groups (64/369, 17%) relative to placebo (9/163, 6%). | |
| 55 per 1000 | 160 per 1000 (51 to 505) | |||||
| Moderate | ||||||
| 87 per 1000 | 252 per 1000 (80 to 795) | |||||
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS total score | The mean anxiety score for the control group was 71.8 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS total score) in the intervention groups was 11.50 lower (25.20 lower to 2.20 higher) | 69 (1 study) | ⊕⊕⊝⊝ Lowa,d | The mean LSAS total anxiety score for the anticonvulsant/GABA intervention group was 60.3 which suggests 'moderate' social phobia. | |
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS avoidance subscale | The mean anxiety score for the control group was 33.9 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS avoidance) in the intervention groups was 4.60 points lower (11.88 lower to 2.68 higher) | 69 (1 study) | ⊕⊕⊝⊝ Lowa,d | The mean LSAS avoidance anxiety score for the anticonvulsant/GABA intervention group was 29.3 which suggests 'low' social phobia. | |
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS fear subscale | The mean anxiety score for the control group was 37.9 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS fear) in the intervention groups was 6.90 points lower (13.65 lower to 0.15 lower) | 69 (1 study) | ⊕⊕⊝⊝ Lowa,d | The mean LSAS fear anxiety score for the anticonvulsant/GABA intervention group was 31.0 which suggests 'low' social phobia. | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; CGI‐I: Clinical Global Impressions Improvement scale; LSAS: Liebowitz Social Anxiety Scale;RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: we are very uncertain about the estimate. | ||||||
aDowngraded one level due to serious risk of bias (concerns with randomisation procedures). bDowngraded two levels due to moderate heterogeneity (I2 of 56%). cDowngraded two levels due to very serious imprecision (few events and wide confidence interval). dDowngraded two levels due to very serious imprecision (wide confidence interval). eResponse is defined as the number of participants with SAnD who responded to treatment, as assessed by the CGI‐I or similar.
Summary of findings 3. Comparison 3: anticonvulsant levetiracetam versus placebo for social anxiety disorder (SAnD).
| Comparison 3: levetiracetam versus placebo for social anxiety disorder (SAnD) | ||||||
| Patient or population: adults with SAnD Settings: outpatient settings Intervention: other anticonvulsants Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| With placebo | Wtih levetiracetam | |||||
| Global Impressions scale change item (CGI‐I or similar): no. of responders (acute phase) | Study population | RR 0.98 (0.70 to 1.37) | 228 (2 studies) | ⊕⊕⊕⊝ Moderatea | There was no evidence of an effect on the number of participants in the anticonvulsant levetiracetam groups compared to the placebo groups who responded 'Very Much Improved' or 'Much Improved' on the CGI‐I scale (P = 0.90). | |
| 373 per 1000 | 365 per 1000 (261 to 511) | |||||
| Moderate | ||||||
| 266 per 1000 | 261 per 1000 (186 to 364) | |||||
| Dropouts due to adverse events (acute phase) | Study population | RR 2.00 (0.81 to 4.94) | 235 (2 studies) | ⊕⊝⊝⊝ Very lowa,b | The proportion of dropouts due to adverse events was high in participants receiving the anticonvulsant levetiracetam (15/122, 12%) relative to placebo (6/113, 5%). There was no evidence of a difference between the number of participants that dropped out due to adverse events (P = 0.14). | |
| 53 per 1000 | 106 per 1000 (43 to 262) | |||||
| Moderate | ||||||
| 28 per 1000 | 56 per 1000 (23 to 138) | |||||
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS total score | The mean anxiety score ranged across control groups from 62.4 to 75.4 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS total score) in the intervention groups was 0.24 points lower (15.69 lower to 15.21 higher) | 228 (2 studies) | ⊕⊝⊝⊝ Very lowa,c | The mean LSAS total anxiety score for the anticonvulsant levetiracetam intervention groups ranged from 55 to 65 which suggests 'moderate' social phobia. | |
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS avoidance subscale | The mean anxiety score for the control group was 36.71 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS avoidance) in the intervention groups was 9.15 points lower (26.86 lower to 8.56 higher) | 16 (1 study) | ⊕⊝⊝⊝ Very lowa,d | The mean LSAS avoidance anxiety score for the anticonvulsant levetiracetam intervention group was 27.56 which suggests 'low' social phobia. | |
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS fear subscale | The mean anxiety score for the control group was 38.71 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS fear) in the intervention groups was 8.71 points lower (26.02 lower to 8.60 higher) | 16 (1 study) | ⊕⊝⊝⊝ Very lowa,d | The mean LSAS fear anxiety score for the anticonvulsant levetiracetam intervention group was 30.0 which suggests 'low' social phobia. | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; CGI‐I: Clinical Global Impressions Improvement scale; LSAS: Liebowitz Social Anxiety Scale;RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: we are very uncertain about the estimate. | ||||||
aDowngraded one level due to serious risk of bias (concerns with randomisation procedures). bDowngraded two levels due to very serious imprecision (few events and wide confidence interval). cDowngraded two levels due to very serious imprecision (wide confidence interval). dDowngraded two levels due to very serious imprecision (small sample size, few events and wide confidence interval). eResponse is defined as the number of participants with SAnD who responded to treatment, as assessed by the CGI‐I or similar.
Summary of findings 4. Comparison 4: antipsychotics versus placebo for social anxiety disorder (SAnD).
| Comparison 4: antipsychotics versus placebo for social anxiety disorder (SAnD) | ||||||
| Patient or population: adults with SAnD Settings: outpatient settings Intervention: antipsychotics Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| With placebo | With antipsychotics | |||||
| Global Impressions scale change item (CGI‐I or similar): no. of responders (acute phase) | Study population | RR 7.00 (0.45 to 108.26) | 10 (1 study) | ⊕⊝⊝⊝ Very lowa,b | There was no evidence of an effect on the number of participants in the antipsychotic group compared to the placebo group who responded 'Very Much Improved' or 'Much Improved' on the CGI‐I scale (P = 0.16). | |
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Moderate | ||||||
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Dropouts due to adverse events (acute phase) | Study population | RR 0.71 (0.06 to 8.90) | 12 (1 study) | ⊕⊝⊝⊝ Very lowa,c | There was no difference on the number of participants who withdrew due to adverse events (P = 0.79). | |
| 200 per 1000 | 142 per 1000 (12 to 1000) | |||||
| Moderate | ||||||
| 200 per 1000 | 142 per 1000 (12 to 1000) | |||||
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS total score | The mean anxiety score for the control group was 86.0 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS total score) in the intervention groups was 37.80 points lower (74.22 lower to 1.38 lower) | — | 9 (1 study) | ⊕⊝⊝⊝ Very lowa,b | The mean LSAS total anxiety score for the antipsychotic intervention group was 48.2 which suggests 'low' social phobia. |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; CGI‐I: Clinical Global Impressions Improvement scale; LSAS: Liebowitz Social Anxiety Scale;RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: we are very uncertain about the estimate. | ||||||
aDowngraded one level due to serious risk of bias (concerns with randomisation procedures). bDowngraded two levels due to very serious imprecision (few events and wide confidence interval). cDowngraded two levels due to very serious imprecision (small sample size, few events, and wide confidence interval). dResponse is defined as the number of participants with SAnD who responded to treatment, as assessed by the CGI‐I or similar.
Summary of findings 5. Comparison 5: benzodiazepines versus placebo for social anxiety disorder (SAnD).
| Comparison 5: benzodiazepines versus placebo for social anxiety disorder (SAnD) | ||||||
| Patient or population: adults with SAnD Settings: inpatient and outpatient settings Intervention: benzodiazepines Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| With placebo | With benzodiazepines | |||||
| Global Impressions scale change item (CGI‐I or similar): no. of responders (acute phase) | Study population | RR 4.03 (2.45 to 6.65) | 132 (2 studies) | ⊕⊕⊝⊝ Lowa,b | There was evidence of a large effect on the number of participants with SAnD who responded to treatment (P < 0.00001). A RR score greater than 1 and 95% CI that does not overlap with 1 indicates that there were a statistically significantly greater number of people in the benzodiazepine groups compared to the placebo groups who responded 'Very Much Improved' or 'Much Improved' on the CGI‐I scale. | |
| 200 per 1000 | 806 per 1000 (490 to 1000) | |||||
| Moderate | ||||||
| 200 per 1000 | 806 per 1000 (490 to 1000) | |||||
| Relapse rate ‐ no. relapsed | Study population | RR 0.12 (0.01 to 2.14) | 36 (1 study) | ⊕⊕⊝⊝ Lowa,c | There was no evidence on the number of treatment responders who subsequently relapsed (P = 0.15). | |
| 211 per 1000 | 25 per 1000 (2 to 451) | |||||
| Moderate | ||||||
| 211 per 1000 | 25 per 1000 (2 to 452) | |||||
| Dropouts due to adverse events (acute phase) | Study population | RR 1.68 (0.21 to 13.13) | 96 (2 studies) | ⊕⊝⊝⊝ Very lowa,d | Dropout rates due to adverse events were low in the benzodiazepine groups (2/47; 4%) and did not differ from the placebo groups (P = 0.62). | |
| 20 per 1000 | 34 per 1000 (4 to 268) | |||||
| Moderate | ||||||
| 17 per 1000 | 29 per 1000 (4 to 223) | |||||
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS total score | The mean anxiety score ranged across control groups from 61.7 to 82.2 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS total score) in the intervention groups was 39.75 points lower (71.11 lower to 8.39 lower) | 135 (2 studies) | ⊕⊝⊝⊝ Very lowa,b,e | The mean LSAS total anxiety score for the benzodiazepine intervention groups ranged from 26.6 to 38.1 which suggests 'low' social phobia. | |
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS avoidance subscale | The mean anxiety score for the control group was 30.2 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS avoidance) in the intervention groups was 10.40 points lower (16.08 lower to 4.72 lower) | 75 (1 study) | ⊕⊕⊝⊝ Lowa,b | The mean LSAS avoidance anxiety score for the benzodiazepine intervention group was 19.8 which suggests 'low' social phobia. | |
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS fear subscale | The mean anxiety score for the control group was 31.9 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS fear) in the intervention groups was 10.80 points lower (16.62 lower to 4.98 lower) | 75 (1 study) | ⊕⊕⊝⊝ Lowa,b | The mean LSAS fear anxiety score for the benzodiazepine intervention group was 21.1 which suggests 'low' social phobia. | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; CGI‐I: Clinical Global Impressions Improvement scale; LSAS: Liebowitz Social Anxiety Scale;RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: we are very uncertain about the estimate. | ||||||
aDowngraded one level due to serious risk of bias (concerns with randomisation procedures). bDowngraded two levels due to very serious imprecision (wide confidence interval). cDowngraded two levels due to very serious imprecision (small sample size and wide confidence interval). dDowngraded two levels due to very serious imprecision (few events and wide confidence interval). eDowngraded two levels due to considerable heterogeneity (I2 of 94%). fResponse is defined as the number of participants with SAnD who responded to treatment, as assessed by the CGI‐I or similar.
Summary of findings 6. Comparison 6: beta‐blockers versus placebo for social anxiety disorder (SAnD).
| Comparison 6: beta‐blockers versus placebo for social anxiety disorder (SAnD) | ||||||
| Patient or population: adults with SAnD Settings: inpatient and outpatient settings Intervention: beta‐blockers Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| With placebo | With beta‐blockers | |||||
| Global Impressions scale change item (CGI‐I or similar): no. of responders (acute phase) | Study population | RR 1.09 (0.63 to 1.88) | 97 (2 studies) | ⊕⊝⊝⊝ Very lowa,b | There was no evidence of an effect on the number of participants in the beta‐blocker groups compared to the placebo groups who responded 'Very Much Improved' or 'Much Improved' on the CGI‐I scale (P = 0.75). | |
| 312 per 1000 | 341 per 1000 (197 to 587) | |||||
| Moderate | ||||||
| 332 per 1000 | 362 per 1000 (209 to 624) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; CGI‐I: Clinical Global Impressions Improvement scale; LSAS: Liebowitz Social Anxiety Scale;RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: we are very uncertain about the estimate. | ||||||
aDowngraded one level due to serious risk of bias (concerns with randomisation procedures). bDowngraded two levels due to very serious imprecision (wide confidence interval). cResponse is defined as the number of participants with SAnD who responded to treatment, as assessed by the CGI‐I or similar.
Summary of findings 7. Comparison 7: MAOIs versus placebo for social anxiety disorder (SAnD).
| Comparison 7: MAOIs versus placebo for social anxiety disorder (SAnD) | ||||||
| Patient or population: adults with SAnD Settings: inpatient and outpatient settings Intervention: MAOIs Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| With placebo | With MAOIs | |||||
| Global Impressions scale change item (CGI‐I or similar): no. of responders (acute phase) | Study population | RR 2.36 (1.48 to 3.75) | 235 (4 studies) | ⊕⊕⊝⊝ Lowa,b,c | There was evidence of an effect on the number of participants with SAnD who responded to treatment (P = 0.0003). A RR score greater than 1 and 95% CI that does not overlap with 1 indicates that there were a statistically significantly greater number of people in the MAOI groups compared to the placebo groups who responded 'Very Much Improved' or 'Much Improved' on the CGI‐I scale. | |
| 263 per 1000 | 621 per 1000 (389 to 987) | |||||
| Moderate | ||||||
| 274 per 1000 | 647 per 1000 (406 to 1000) | |||||
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS total score | The mean anxiety score ranged across control groups from 4.05 to 63.29 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS total score) in the intervention groups was 16.39 points lower (32.27 lower to 0.51 lower) | 218 (4 studies) | ⊕⊝⊝⊝ Very lowa,c,d | The mean LSAS total anxiety score for the MAOI intervention groups ranged from 14.0 to 47.8 which suggests 'low' social phobia. | |
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS avoidance subscale | The mean anxiety score for the control group was 24.54 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS avoidance) in the intervention groups was 5.42 points lower (14.69 lower to 3.85 higher) | 51 (1 study) | ⊕⊝⊝⊝ Very lowa,c | The mean LSAS avoidance anxiety score for the MAOI intervention group was 19.12 which suggests 'low' social phobia. | |
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS fear subscale | The mean anxiety score for the control group was 27.31 | The mean reduction of of anxiety symptoms (clinician‐rated: LSAS fear) in the intervention groups was 5.23 points lower (13.97 lower to 3.51 higher) | 51 (1 study) | ⊕⊝⊝⊝ Very lowa,c | The mean LSAS fear anxiety score for the MAOI intervention group was 22.08 which suggests 'low' social phobia. | |
|
Clinical Global Impressions scale change item (CGI‐I):no. of responders (long term) Post‐treatment: 6 months |
Study population | RR 1.84 (1.02 to 3.33) | 113 (2 studies) | ⊕⊝⊝⊝ Very lowa,c | There was data indicating greater efficacy over the long term on the number of participants with SAnD who responded to treatment (P = 0.04). A RR score greater than 1 and 95% CI that does not overlap with 1 indicates that there were a statistically significantly greater number of people in the MAOI groups compared to the placebo groups who responded 'Very Much Improved' or 'Much Improved' on the CGI‐I scale. | |
| 264 per 1000 | 486 per 1000 (269 to 880) | |||||
| Moderate | ||||||
| 263 per 1000 | 484 per 1000 (268 to 876) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; CGI‐I: Clinical Global Impressions Improvement scale; LSAS: Liebowitz Social Anxiety Scale;RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: we are very uncertain about the estimate. | ||||||
aDowngraded one level due to serious risk of bias (concerns with randomisation procedures). bDowngraded two levels due to moderate heterogeneity (I2 of 44%). cDowngraded two levels due to very serious imprecision (wide confidence interval). dDowngraded two levels due to considerable heterogeneity (I2 of 93%). eResponse is defined as the number of participants with SAnD who responded to treatment, as assessed by the CGI‐I or similar.
Summary of findings 8. Comparison 8: NARIs versus placebo for social anxiety disorder (SAnD).
| Comparison 8: NARIs versus placebo for social anxiety disorder (SAnD) | ||||||
| Patient or population: adults with SAnD Settings: outpatient settings Intervention: NARIs Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| With placebo | With NARIs | |||||
| Global Impressions scale change item (CGI‐I or similar): no. of responders (acute phase) | Study population | RR 0.70 (0.19 to 2.54) | 27 (1 study) | ⊕⊝⊝⊝ Very lowa,b | There was no evidence of an effect on the number of participants in the NARI group compared to the placebo group who responded 'Very Much Improved' or 'Much Improved' on the CGI‐I scale (P = 0.58). | |
| 308 per 1000 | 215 per 1000 (58 to 782) | |||||
| Moderate | ||||||
| 308 per 1000 | 216 per 1000 (59 to 782) | |||||
| Dropouts due to adverse events (acute phase) | Study population | RR 2.80 (0.12 to 63.20) | 27 (1 study) | ⊕⊝⊝⊝ Very lowa,c | Only 1 of 14 (7%) participants withdrew during 10 weeks of NARI treatment. There was no evidence for a difference in the number of participants who withdrew due to adverse events (P = 0.52). | |
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Moderate | ||||||
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS total score | The mean anxiety score for the control group was 70.8 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS total score) in the intervention groups was 2.60 points higher (15.43 lower to 20.63 higher) | 26 (1 study) | ⊕⊝⊝⊝ Very lowa,b | The mean LSAS total anxiety score for the NARI intervention group was 73.4 which suggests 'marked' social phobia. | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; CGI‐I: Clinical Global Impressions Improvement scale; LSAS: Liebowitz Social Anxiety Scale;RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: we are very uncertain about the estimate. | ||||||
aDowngraded one level due to serious risk of bias (concerns with randomisation procedures). bDowngraded two levels due to very serious imprecision (small sample size and wide confidence interval). cDowngraded two levels due to very serious imprecision (small sample size, few events and wide confidence interval). dResponse is defined as the number of participants with SAnD who responded to treatment, as assessed by the CGI‐I or similar.
Summary of findings 9. Comparison 9: NaSSAs versus placebo for social anxiety disorder (SAnD).
| Comparison 9: NaSSAs compared to placebo for social anxiety disorder (SAnD) | ||||||
| Patient or population: adults with SAnD Settings: outpatient settings Intervention: NaSSAs Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| With placebo | With NaSSAs | |||||
| Global Impressions scale change item (CGI‐I or similar): no. of responders (acute phase) | Study population | RR 1.00 (0.28 to 3.63) | 60 (1 study) | ⊕⊝⊝⊝ Very lowa,b | An equal number of participants (4/30, 13%) responded to treatment in both the NaSSA and placebo groups, however this difference was not statistically significant (P = 1.00). | |
| 133 per 1000 | 133 per 1000 (37 to 484) | |||||
| Moderate | ||||||
| 133 per 1000 | 133 per 1000 (37 to 483) | |||||
| Dropouts due to adverse events (acute phase) | Study population | RR 5.00 (0.25 to 99.95) | 60 (1 study) | ⊕⊝⊝⊝ Very lowa,b | A small proportion of participants receiving NaSSAs dropped out due to adverse events (2/30, 7%). Although, the difference was not statistically significant (P = 0.29). | |
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Moderate | ||||||
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS total score | The mean anxiety score ranged across control groups from 62.4 to 67.1 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS total score) in the intervention groups was 15.37 points lower (28.10 lower to 2.63 lower) | 126 (2 studies) | ⊕⊝⊝⊝ Very lowa,c,d | The mean LSAS total anxiety score for the NaSSA intervention groups ranged from 46.3 to 54.8 which suggests 'low' social phobia. | |
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS avoidance subscale | The mean anxiety score for the control group was 28.9 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS avoidance) in the intervention groups was 3.90 points lower (9.90 lower to 2.10 higher) | 60 (1 study) | ⊕⊕⊝⊝ Lowa,c | The mean LSAS avoidance anxiety score for the NaSSA intervention group was 25.0 which suggests 'low' social phobia. | |
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS fear subscale | The mean anxiety score for the control group was 33.5 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS fear) in the intervention groups was 3.70 points lower (9.42 lower to 2.02 higher) | 60 (1 study) | ⊕⊕⊝⊝ Lowa,c | The mean LSAS fear anxiety score for the NaSSA intervention group was 29.8 which suggests 'low' social phobia. | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; CGI‐I: Clinical Global Impressions Improvement scale; LSAS: Liebowitz Social Anxiety Scale;RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: we are very uncertain about the estimate. | ||||||
aDowngraded one level due to serious risk of bias (concerns with randomisation procedures). bDowngraded two levels due to very serious imprecision (few events and wide confidence interval). cDowngraded two levels due to very serious imprecision (wide confidence interval). dDowngraded two levels due to substantial heterogeneity (I2 of 79%). eResponse is defined as the number of participants with SAnD who responded to treatment, as assessed by the CGI‐I or similar.
Summary of findings 10. Comparison 10: RIMAs versus placebo for social anxiety disorder (SAnD).
| Comparison 10: RIMAs versus placebo for social anxiety disorder (SAnD) | ||||||
| Patient or population: adults with SAnD Settings: inpatient and outpatient settings Intervention: RIMAs Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| With placebo | With RIMAs | |||||
| Global Impressions scale change item (CGI‐I or similar): no. of responders (acute phase) | Study population | RR 1.83 (1.32 to 2.55) | 1270 (8 studies) | ⊕⊕⊝⊝ Lowa,b | There was evidence of benefit on the number of participants with SAnD who responded to treatment (P = 0.0003). A RR score greater than 1 and 95% CI that does not overlap with 1 indicates that there were a statistically significantly greater number of people in the RIMA groups compared to the placebo groups who responded 'Very Much Improved' or 'Much Improved' on the CGI‐I scale. | |
| 254 per 1000 | 465 per 1000 (335 to 647) | |||||
| Moderate | ||||||
| 207 per 1000 | 379 per 1000 (273 to 528) | |||||
| Dropouts due to adverse events (acute phase) | Study population | RR 1.42 (0.86 to 2.34) | 1305 (8 studies) | ⊕⊝⊝⊝ Very lowa,c | Dropout rates due to adverse events were low in the RIMA groups and equivalent to those observed in the placebo groups (72/83; 9%). | |
| 49 per 1000 | 69 per 1000 (42 to 114) | |||||
| Moderate | ||||||
| 32 per 1000 | 45 per 1000 (28 to 75) | |||||
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS total score | The mean anxiety score ranged across control groups from 54.4 to 79.3 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS total score) in the intervention groups was 12.17 points lower (23.51 lower to 0.84 lower) | 1163 (6 studies) | ⊕⊝⊝⊝ Very lowa,d,e | The mean LSAS total anxiety score for the RIMA intervention groups ranged from 27.0 to 62.6 which suggests low to 'moderate' social phobia. | |
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS avoidance subscale | The mean anxiety score ranged across control groups from 23.3 to 39.2 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS avoidance) in the intervention groups was 5.05 points lower (7.91 lower to 2.18 lower) | 695 (5 studies) | ⊕⊕⊝⊝ Lowa,e | The mean LSAS avoidance anxiety score for the RIMA intervention groups ranged from 15.3 to 30.7 which suggests 'low' social phobia. | |
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS fear subscale | The mean anxiety score ranged across control groups from 29.4 to 40.1 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS fear) in the intervention groups was 5.40 points lower (8.92 lower to 1.88 lower) | 724 (6 studies) | ⊕⊝⊝⊝ Very lowa,e,f | The mean LSAS fear anxiety score for the RIMA intervention groups ranged from 19.1 to 33.3 which suggests 'low' social phobia. | |
|
Clinical Global Impressions scale change item (CGI‐I): no. of responders (long term) Post‐treatment: 1‐15 months |
Study population | RR 1.50 (1.12 to 2.00) | 90 (1 study) | ⊕⊕⊕⊝ Moderatea | There was evidence of a long‐term effect on treatment efficacy in participants with SAnD who responded to treatment (P = 0.006). A RR score greater than 1 and 95% CI that does not overlap with 1 indicates that there were a statistically significantly greater number of people in the RIMA group compared to the placebo group who responded 'Very Much Improved' or 'Much Improved' on the CGI‐I scale. | |
| 575 per 1000 | 862 per 1000 (644 to 1000) | |||||
| Moderate | ||||||
| 575 per 1000 | 862 per 1000 (644 to 1000) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; CGI‐I: Clinical Global Impressions Improvement scale; LSAS: Liebowitz Social Anxiety Scale;RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: we are very uncertain about the estimate. | ||||||
aDowngraded one level due to serious risk of bias (concerns with randomisation procedures). bDowngraded two levels due to substantial heterogeneity (I2 of 70%). cDowngraded two levels due to very serious imprecision (few events and wide confidence interval). dDowngraded two levels due to considerable heterogeneity (I2 of 94%). eDowngraded two levels due to very serious imprecision (wide confidence interval). fDowngraded two levels due to moderate heterogeneity (I2 of 55%). gResponse is defined as the number of participants with SAnD who responded to treatment, as assessed by the CGI‐I or similar.
Summary of findings 11. Comparison 11: SARIs versus placebo for social anxiety disorder (SAnD).
| Comparison 11: SARIs versus placebo for social anxiety disorder (SAnD) | ||||||
| Patient or population: adults with SAnD Settings: outpatient settings Intervention: SARIs Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| With placebo | With SARIs | |||||
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS total score | The mean anxiety score for the control group was 71.2 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS total score) in the intervention groups was 6.10 points lower (16.55 lower to 4.35 higher) | 102 (1 study) | ⊕⊝⊝⊝ Very lowa,b | The mean LSAS total anxiety score for the SARI intervention group was 65.1 which suggests 'marked' social phobia. | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; CGI‐I: Clinical Global Impressions Improvement scale; LSAS: Liebowitz Social Anxiety Scale;RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: we are very uncertain about the estimate. | ||||||
aDowngraded one level due to serious risk of bias (concerns with randomisation procedures). bDowngraded two levels due to very serious imprecision (wide confidence interval).
Summary of findings 12. Comparison 12: SNRIs versus placebo for social anxiety disorder (SAnD).
| Comparison 12: SNRIs compared to placebo for social anxiety disorder (SAnD) | ||||||
| Patient or population: adults with SAnD Settings: outpatient settings Intervention: SNRIs Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| With placebo | With SNRIs | |||||
| Global Impressions scale change item (CGI‐I or similar): no. of responders (acute phase) | Study population | RR 1.30 (0.85 to 1.99) | 1173 (4 studies) | ⊕⊕⊝⊝ Lowa,b | There was no evidence of an effect on the number of participants in the SNRI groups compared to the placebo groups who responded 'Very Much Improved' or 'Much Improved' on the CGI‐I scale (P = 0.22). | |
| 374 per 1000 | 486 per 1000 (460 to 610) | |||||
| Moderate | ||||||
| 347 per 1000 | 451 per 1000 (465 to 617) | |||||
| Dropouts due to adverse events (acute phase) | Study population | RR 3.23 (2.15 to 4.86) | 1213 (4 studies) | ⊕⊕⊕⊝ Moderatea | The proportion of dropouts due to adverse events was more than three times as high in participants receiving SNRIs (109/663, 16%) compared to placebo (27/550, 5%), a statistically significant difference (P < 0.00001). | |
| 49 per 1000 | 159 per 1000 (106 to 239) | |||||
| Moderate | ||||||
| 49 per 1000 | 158 per 1000 (105 to 238) | |||||
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS total score | The mean anxiety score ranged across control groups from ‐22.2 to 66.0 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS total score) in the intervention groups was 11.91 points lower (16.06 lower to 7.76 lower) | 902 (3 studies) | ⊕⊕⊕⊝ Moderatea | The mean LSAS total anxiety score for the SNRI intervention groups ranged from ‐35.0 to 57.7 which suggests 'low' to 'moderate' social phobia. | |
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS avoidance subscale | The mean anxiety score for the control group was 32.1 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS avoidance) in the intervention groups was 4.30 points lower (8.14 lower to 0.46 lower) | 261 (1 study) | ⊕⊕⊝⊝ Lowa,c | The mean LSAS avoidance anxiety score for the SNRI intervention group was 27.8 which suggests 'low' social phobia. | |
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS fear subscale | The mean anxiety score for the control group was 33.9 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS fear) in the intervention groups was 4.00 points lower (7.68 lower to 0.32 lower) | 261 (1 study) | ⊕⊕⊝⊝ Lowa,c | The mean LSAS fear anxiety score for the SNRI intervention group was 29.9 which suggests 'low' social phobia. | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; CGI‐I: Clinical Global Impressions Improvement scale; LSAS: Liebowitz Social Anxiety Scale;RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: we are very uncertain about the estimate. | ||||||
aDowngraded one level due to serious risk of bias (concerns with randomisation procedures). bDowngraded two levels due to considerable heterogeneity (I2 of 89%). cDowngraded two levels due to very serious imprecision (wide confidence interval). dResponse is defined as the number of participants with SAnD who responded to treatment, as assessed by the CGI‐I or similar.
Summary of findings 13. Comparison 13: SSRIs versus placebo for social anxiety disorder (SAnD).
| Comparison 13: SSRIs versus placebo for social anxiety disorder (SAnD) | ||||||
| Patient or population: adults with SAnD Settings: inpatient and outpatient settings Intervention: SSRIs Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| With placebo | With SSRIs | |||||
| Global Impressions scale change item (CGI‐I or similar): no. of responders (acute phase) | Study population | RR 1.65 (1.48 to 1.85) | 4984 (24 studies) | ⊕⊝⊝⊝ Very lowa,b,c | There was evidence of benefit on the number of participants with SAnD who responded to treatment (P < 0.00001). A RR score greater than 1 and 95% CI that does not overlap with 1 indicates that there were a statistically significantly greater number of people in the SSRI groups compared to the placebo groups who responded 'Very Much Improved' or 'Much Improved' on the CGI‐I scale. | |
| 317 per 1000 | 523 per 1000 (469 to 587) | |||||
| Moderate | ||||||
| 290 per 1000 | 476 per 1000 (478 to 537) | |||||
| Relapse rate ‐ no. relapsed | Study population | RR 0.34 (0.22 to 0.5) | 389 (3 studies) | ⊕⊕⊕⊝ Moderatea | There was evidence that SSRIs prevented relapse compared to placebo (P < 0.00001). | |
| 397 per 1000 | 135 per 1000 (87 to 198) | |||||
| Moderate | ||||||
| 391 per 1000 | 133 per 1000 (86 to 195) | |||||
| Dropouts due to adverse events (acute phase) | Study population | RR 2.59 (1.97 to 3.39) | 5131 (24 studies) | ⊕⊕⊝⊝ Lowa,c | The proportion of dropouts due to adverse events was approximately three times higher amongst participants receiving SSRIs compared to placebo ((P < 0.00001). | |
| 40 per 1000 | 105 per 1000 (80 to 137) | |||||
| Moderate | ||||||
| 37 per 1000 | 96 per 1000 (73 to 125) | |||||
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS total score | The mean anxiety score ranged across control groups from ‐7.8 to 69.88 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS total score) in the intervention groups was 10.14 points lower (14.05 to 6.22 lower) | 1990 (14 studies) | ⊕⊕⊝⊝ Lowa,d | The mean LSAS total anxiety score for the SSRI intervention groups ranged from 14.7 to 60.3 which suggests 'low' to 'moderate' social phobia. | |
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS avoidance subscale | The mean anxiety score ranged across control groups from 24.2 to 34.8 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS avoidance) in the intervention groups was 7.01 points lower (10.21 to 3.80 lower) | 1173 (7 studies) | ⊕⊕⊝⊝ Lowa,e | The mean LSAS avoidance anxiety score for the SSRI intervention groups ranged from 17.09 to 26.11 which suggests 'low' social phobia. | |
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS fear subscale | The mean anxiety score ranged across control groups from 29.83 to 37.4 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS fear) in the intervention groups was 7.28 points lower (10.86 to 3.71 lower) | 1173 (7 studies) | ⊕⊕⊝⊝ Lowa,f | The mean LSAS fear anxiety score for the SSRI intervention groups ranged from 19.84 to 32.79 which suggests 'low' social phobia. | |
|
Clinical Global Impressions scale change item (CGI‐I): no. of responders (long term) Post‐treatment: 1‐4 months |
Study population | RR 1.27 (1.07 to 1.51) | 806 (4 studies) | ⊕⊕⊝⊝ Lowa,g | There was evidence for a response to long‐term treatment compared to placebo in participants with SAnD (P = 0.007). A RR score greater than 1 and 95% CI that does not overlap with 1 indicates that there were a statistically significantly greater number of people in the SSRI groups compared to the placebo groups who responded 'Very Much Improved' or 'Much Improved' on the CGI‐I scale. | |
| 579 per 1000 | 735 per 1000 (619 to 874) | |||||
| Moderate | ||||||
| 584 per 1000 | 742 per 1000 (625 to 882) | |||||
|
Dropouts due to adverse events (long term) Post‐treatment: 1‐4 months |
Study population | RR 1.17 (0.43 to 3.18) | 1274 (3 studies) | ⊕⊝⊝⊝ Very lowa,h,i | We found no difference in dropout rates due to adverse events between the SSRI and control groups (P = 0.76). | |
| 52 per 1000 | 61 per 1000 (23 to 166) | |||||
| Moderate | ||||||
| 50 per 1000 | 58 per 1000 (22 to 159) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; CGI‐I: Clinical Global Impressions Improvement scale; LSAS: Liebowitz Social Anxiety Scale;RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: we are very uncertain about the estimate. | ||||||
aDowngraded one level due to serious risk of bias (concerns with randomisation procedures). bDowngraded two levels due to moderate heterogeneity (I2 of 50%). cDowngraded two levels due to very serious publication bias (t = 2.6426, df = 22, p = 0.015). dDowngraded two levels due to substantial heterogeneity (I2 of 67%). eDowngraded two levels due to substantial heterogeneity (I2 of 65%). fDowngraded two levels due to substantial heterogeneity (I2 of 75%). gDowngraded two levels due to moderate heterogeneity (I2 of 60%). hDowngraded two levels due to moderate heterogeneity (I2 of 57%). iDowngraded two levels due to very serious imprecision (few events and wide confidence interval). jResponse is defined as the number of participants with SAnD who responded to treatment, as assessed by the CGI‐I or similar.
Summary of findings 14. Comparison 14: GW876008 versus placebo for social anxiety disorder (SAnD).
| Comparison 14: GW876008 versus placebo for social anxiety disorder (SAnD) | ||||||
| Patient or population: adults with SAnD Settings: outpatient settings Intervention: GW876008 Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control | Comparison 14: GW876008 versus placebo | |||||
| Global Impressions scale change item (CGI‐I or similar): no. of responders (acute phase) | Study population | RR 0.83 (0.58 to 1.19) | 250 (1 study) | ⊕⊕⊕⊝ moderate1 | There was no evidence of an effect on the number of participants in the GW876008 group compared to the placebo group who responded 'Very Much Improved' or 'Much Improved' on the CGI‐I scale (P = 0.32). | |
| 364 per 1000 | 302 per 1000 (211 to 433) | |||||
| Moderate | ||||||
| 364 per 1000 | 302 per 1000 (211 to 433) | |||||
| Dropouts due to adverse events (acute phase) | Study population | RR 2.95 (0.67 to 13.02) | 252 (1 study) | ⊕⊕⊝⊝ low1,2 | The proportion of dropouts due to adverse events was approximately three times higher amongst participants receiving GW876008 (11/164, 7%) compared to placebo (2/88, 2%), though this difference was not statistically significant (P = 0.15). | |
| 23 per 1000 | 67 per 1000 (15 to 296) | |||||
| Moderate | ||||||
| 23 per 1000 | 68 per 1000 (15 to 299) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
a Downgraded one level due to serious risk of bias (concerns with randomisation procedures). b Downgraded one level due to serious imprecision (wide confidence intervals).
c Response is defined as the number of participants with SAnD who responded to treatment, as assessed by the CGI‐I or similar.
Summary of findings 15. Comparison 15: NK1 receptor antagonist GR205171 versus placebo for social anxiety disorder (SAnD).
| Comparison 15: NK1 receptor antagonist GR205171 versus placebo for social anxiety disorder (SAnD) | ||||||
| Patient or population: adults with SAnD Settings: outpatient settings Intervention: NK1 receptor antagonist GR205171 Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control | Comparison 15: NK1 receptor antagonist GR205171 versus placebo | |||||
| Global Impressions scale change item (CGI‐I or similar): no. of responders (acute phase) | Study population | RR 5 (0.68 to 36.66) | 24 (1 study) | ⊕⊝⊝⊝ very low1,2 | There was no evidence of an effect on the number of participants in the NK1 receptor antagonist GR205171 group compared to the placebo group who responded 'Very Much Improved' or 'Much Improved' on the CGI‐I scale (P = 0.11). | |
| 83 per 1000 | 417 per 1000 (57 to 1000) | |||||
| Moderate | ||||||
| 83 per 1000 | 415 per 1000 (56 to 1000) | |||||
| Dropouts due to adverse events (acute phase) | See comment | See comment | Not estimable | 24 (1 study) | ⊕⊕⊕⊝ moderate1 | No participants withdrew due to adverse events. |
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS total score | The mean anxiety score for the control group was 4.1 | The mean clinician‐rated: liebowitz social anxiety scale (lsas): reduction of anxiety symptoms ‐ lsas total score in the intervention groups was 0.5 points lower (1.35 lower to 0.35 higher) | 24 (1 study) | ⊕⊝⊝⊝ very low2 | The mean LSAS total anxiety score for the NK1 receptor antagonist GR205171 intervention group was 3.6 which suggests 'low' social phobia. | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
a Downgraded one level due to serious risk of bias (concerns with randomisation procedures). b Downgraded two levels due to very serious imprecision (small sample size and wide confidence intervals).
c Response is defined as the number of participants with SAnD who responded to treatment, as assessed by the CGI‐I or similar.
Summary of findings 16. Comparison 16: LY686017 versus placebo for social anxiety disorder (SAnD).
| Comparison 16: LY686017 versus placebo for social anxiety disorder (SAnD) | ||||||
| Patient or population: adults with SAnD Settings: outpatient settings Intervention: LY686017 Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control | Comparison 16: LY686017 versus placebo | |||||
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS total score | The mean anxiety score for the control group was ‐22.59 | The mean clinician‐rated: liebowitz social anxiety scale (lsas): reduction of anxiety symptoms ‐ lsas total score in the intervention groups was 1.8 points higher (6.92 lower to 10.52 higher) | 99 (1 study) | ⊕⊝⊝⊝ very low1,2 | The mean LSAS total anxiety score for the LY686017 intervention group was ‐20.79 which suggests 'low' social phobia. | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
a Downgraded two levels due to serious risk of bias (concerns with randomisation procedures). b Downgraded one level due to serious imprecision (wide confidence intervals).
See 'Summary of findings' tables for the main comparisons of medication classes compared to placebo for treating SAnD (where provided): Table 1; Table 2; Table 3; Table 4; Table 5; Table 6; Table 7; Table 8; Table 9; Table 10; Table 11; Table 12; Table 13; Table 14; Table 15; Table 16. Please refer to Types of outcome measures for a description of the scoring systems used in included studies.
Comparison 1: 5HT1A partial agonists versus placebo
Primary outcome measures
There was no evidence of an effect of buspirone on treatment efficacy in participants with SAnD compared to placebo (k = 1, risk ratio (RR) 1.00, 95% confidence interval (CI) 0.07 to 14.55, N = 30; see Analysis 1.1;Van Vliet 1997). In addition, the quality of the evidence for this outcome is very low. Dropout rates due to adverse events were low in the medication arm of one RCT of buspirone (1/30, 3%) (see Analysis 1.2); again, however, this conclusion is based on very low‐quality evidence.
1.1. Analysis.
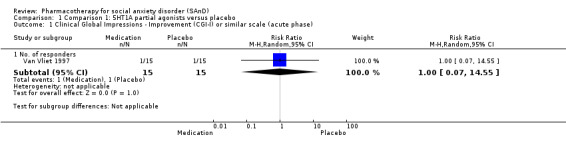
Comparison 1 Comparison 1: 5HT1A partial agonists versus placebo, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase).
1.2. Analysis.
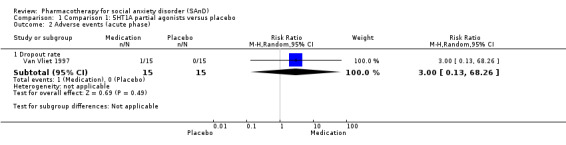
Comparison 1 Comparison 1: 5HT1A partial agonists versus placebo, Outcome 2 Adverse events (acute phase).
Secondary outcomes measures
Buspirone was not superior to placebo on the avoidance subscale of the LSAS for the reduction of anxiety symptoms (k = 1, mean difference (MD) −1.40 points, 95% CI −11.61 to 8.81; N = 30; Analysis 1.3; Van Vliet 1997).The evidence that was available for this outcome was of very low quality. There was no evidence of an effect of buspirone on depression symptoms on the HAM‐D scale in participants with SAnD compared to placebo (k = 1, MD −0.60 points, 95% CI −2.86 to 1.66, N = 27; see Analysis 1.4; Van Vliet 1997).
1.3. Analysis.
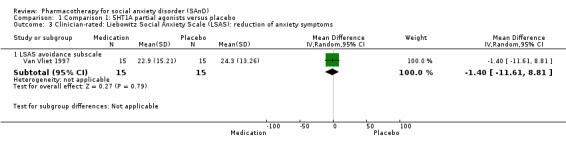
Comparison 1 Comparison 1: 5HT1A partial agonists versus placebo, Outcome 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms.
1.4. Analysis.
Comparison 1 Comparison 1: 5HT1A partial agonists versus placebo, Outcome 4 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale).
More participants withdrew from the placebo group (3/15; 20%) compared to the buspirone group (0/15; 0%). Nevertheless, the total proportion of dropouts (20%) is relatively low and is confirmed by the lack of evidence of a difference between the two groups (k = 1, RR 0.14; 95% CI 0.01 to 2.55, N = 30; see Analysis 1.5).
1.5. Analysis.
Comparison 1 Comparison 1: 5HT1A partial agonists versus placebo, Outcome 5 All‐cause dropouts (acute phase).
Comparison 2: anticonvulsants/GABAs versus placebo
Primary outcome measures
There was evidence of benefit for gabapentin and pregabalin compared to placebo on the number of participants with SAnD who responded to treatment (k = 3, RR 1.60 ; 95% CI 1.16 to 2.20, N = 532; see Analysis 2.1; Feltner 2011; Pande 2004; Pande 1999), with moderate‐quality evidence. The proportion of dropouts due to adverse events was approximately three times as high in participants receiving gabapentin or pregabalin (64/369, 17%) relative to placebo (9/163, 6%) (k = 3, RR 2.90; 95% CI 0.92 to 9.14; see Analysis 2.2). The higher dropout rate associated with medication was particularly apparent in the Feltner 2011 trial of pregabalin, with the proportion of discontinuations due to adverse events ranging between 14% and 20.5% for the three dosages tested (300 mg/d, 450 mg/d, 600 mg/d), versus 1.2% in the placebo group (RR 14.00; 95% CI 1.96 to 100.12).
2.1. Analysis.
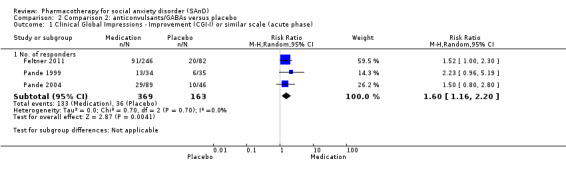
Comparison 2 Comparison 2: anticonvulsants/GABAs versus placebo, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase).
2.2. Analysis.
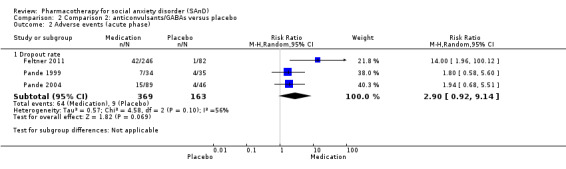
Comparison 2 Comparison 2: anticonvulsants/GABAs versus placebo, Outcome 2 Adverse events (acute phase).
The GABA anticonvulsants were significantly more efficacious than the anticonvulsant levetiracetam (Chi2 = 4.25, df = 1, P = 0.04; I2 = 76.5%).
Secondary outcomes measures
There was no evidence that gabapentin reduced total SAnD symptom severity scores on the LSAS (k = 1, MD −11.50 points, 95% CI −25.20 to 2.20, N = 69; see Analysis 2.3; Pande 1999). In addition, there was no evidence of a difference on either the avoidance (MD −4.60 points, 95% CI −11.88 to 2.68, see Analysis 2.3) or fear subscale of the LSAS (MD −6.90 points, 95% CI −13.65 to −0.15, see Analysis 2.3). The evidence for the three outcomes was low in quality. We did not find an effect for medication in reducing depression symptoms on the HAM‐D scale in a single RCT of gabapentin compared to placebo (MD −2.30 points, 95% CI −4.78 to 0.18, N = 69; see Analysis 2.4; Pande 1999).
2.3. Analysis.
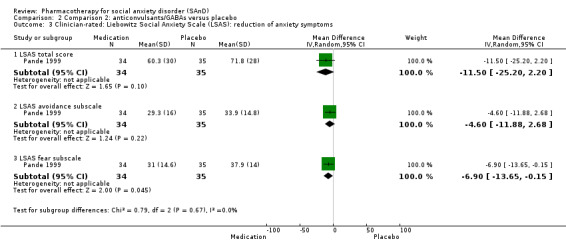
Comparison 2 Comparison 2: anticonvulsants/GABAs versus placebo, Outcome 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms.
2.4. Analysis.
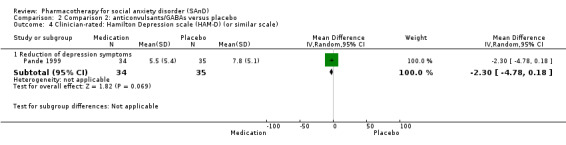
Comparison 2 Comparison 2: anticonvulsants/GABAs versus placebo, Outcome 4 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale).
Three studies with 488 participants reported similar proportions of dropouts due to any cause in the medication and placebo groups (RR 1.16; 95% CI 0.78 to 1.70; see Analysis 2.5; Feltner 2011; Pande 2004; Pande 1999).
2.5. Analysis.
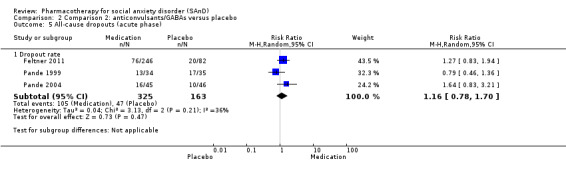
Comparison 2 Comparison 2: anticonvulsants/GABAs versus placebo, Outcome 5 All‐cause dropouts (acute phase).
Comparison 3: anticonvulsant levetiracetam versus placebo
Primary outcome measures
There was no evidence of an effect of levetiracetam on treatment efficacy in participants with SAnD compared to placebo (k = 2, RR 0.98; 95% CI 0.70 to 1.37, N = 228; see Analysis 3.1; Stein 2010; Zhang 2005). The evidence that was available for this outcome was of moderate quality. There was evidence of a response to treatment with the anticonvulsant levetiracetam compared to anticonvulsants with gamma‐amino butyric acid (GABA) analogues (Chi2 = 4.25, df = 1, P = 0.04; I2 = 76.5%), the benzodiazepines (Chi2 = 21.10, df = 1, P < 0.00001; I2 = 95.3%), the MAOIs (Chi2 = 5.86, df = 1, P = 0.02; I2 = 82.9%), the RIMAs (Chi2 = 6.76, df = 1, P = 0.009; I2 = 85.2%), and the SSRIs (Chi2 = 8.02, df = 1, P = 0.005; I2 = 87.5%).
3.1. Analysis.
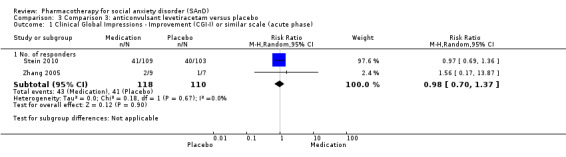
Comparison 3 Comparison 3: anticonvulsant levetiracetam versus placebo, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase).
The proportion of dropouts due to adverse events was more than twice as high in participants receiving levetiracetam (15/122, 12%) relative to placebo (6/113, 5%). There was no evidence of a difference between the number of participants that dropped out due to treatment‐emergent side effects (k = 2, RR 2.00; 95% CI 0.81 to 4.94; see Analysis 3.2), but the quality of the evidence is very low.
3.2. Analysis.
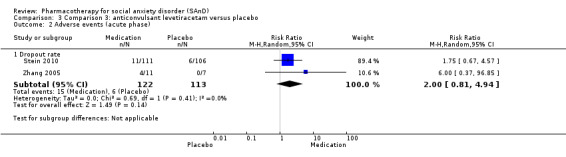
Comparison 3 Comparison 3: anticonvulsant levetiracetam versus placebo, Outcome 2 Adverse events (acute phase).
Secondary outcome measures
There was no evidence of a difference for a reduction of SAnD symptom severity in participants treated with levetiracetam compared to placebo on the LSAS (k = 2, MD −0.24 points, 95% CI −15.69 to 15.21, N = 228; Analysis 3.3; Stein 2010; Zhang 2005). In addition, there was no evidence of a difference between these groups in Zhang 2005 (N = 16) on either the avoidance (MD −9.15 points, 95% CI −26.86 to 8.56, Analysis 3.3) or fear subscale of the LSAS (MD −8.71 points, 95% CI −26.02 to 8.60, Analysis 3.3). The evidence that was available for the three outcomes was of very low quality. We found no evidence of an effect for levetiracetam of a reduction in depression symptoms on the HDRS compared to placebo (k = 1, MD 0.20 points, 95% CI −1.33 to 1.73, N = 212; Analysis 3.4; Stein 2010).
3.3. Analysis.
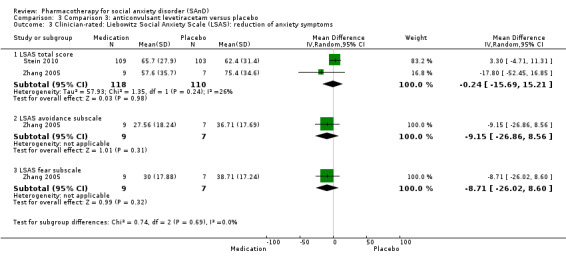
Comparison 3 Comparison 3: anticonvulsant levetiracetam versus placebo, Outcome 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms.
3.4. Analysis.
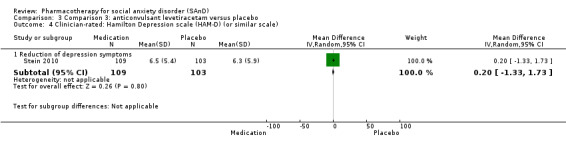
Comparison 3 Comparison 3: anticonvulsant levetiracetam versus placebo, Outcome 4 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale).
The proportion of participants dropping out due to any cause from the anticonvulsants and placebo groups was comparable (31% in both groups). We found no evidence of a difference for levetiracetam compared to placebo (k = 2, RR 1.42; 95% CI 0.30 to 6.76, N = 235; see Analysis 3.5; Stein 2010; Zhang 2005).
3.5. Analysis.
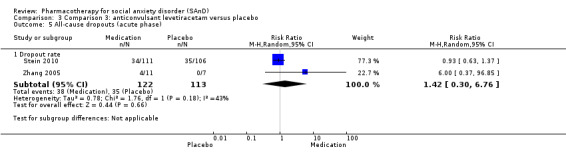
Comparison 3 Comparison 3: anticonvulsant levetiracetam versus placebo, Outcome 5 All‐cause dropouts (acute phase).
Comparison 4: antipsychotics versus placebo
Primary outcome measures
In a small RCT with 10 participants, there was no evidence of an effect for olanzapine compared to placebo on the number of participants who responded to treatment (k = 1, RR 7.00; 95% CI 0.45 to 108.26, Analysis 4.1; Barnett 2002). The data provided for this outcome was of very low quality.
4.1. Analysis.
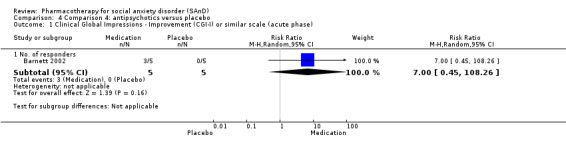
Comparison 4 Comparison 4: antipsychotics versus placebo, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase).
Of the 12 participants randomised to 8 weeks of treatment with olanzapine or placebo, there was no difference in the number of participants who withdrew due to treatment‐emergent side effects (k = 1, RR 0.71; 95% CI 0.06 to 8.90; see Analysis 4.2). The evidence that was available for this outcome was of very low quality.
4.2. Analysis.
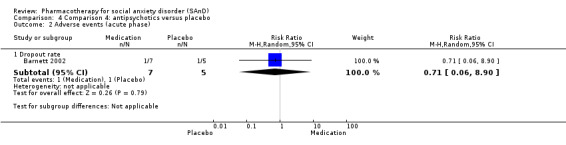
Comparison 4 Comparison 4: antipsychotics versus placebo, Outcome 2 Adverse events (acute phase).
Secondary outcome measures
There was evidence of benefit for olanzapine compared to placebo in the reduction of SAnD symptoms on the LSAS (k = 1, MD −37.80 points, 95% CI −74.22 to −1.38, N = 9; see Analysis 4.3; Barnett 2002), though the evidence is of very low quality.
4.3. Analysis.
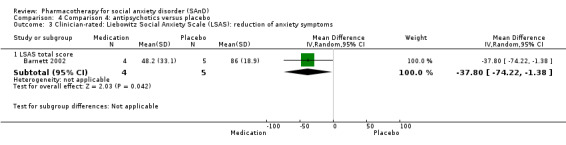
Comparison 4 Comparison 4: antipsychotics versus placebo, Outcome 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms.
A similar proportion of participants withdrew from the olanzapine group (43%) compared to the placebo group (40%) (k = 1, RR 1.07; 95% CI 0.27 to 4.23, N = 12; see Analysis 4.4; Barnett 2002).
4.4. Analysis.
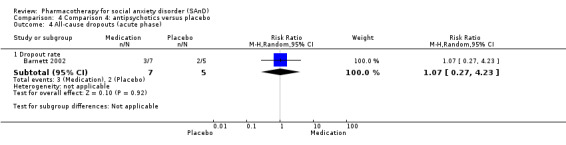
Comparison 4 Comparison 4: antipsychotics versus placebo, Outcome 4 All‐cause dropouts (acute phase).
Comparison 5: benzodiazepines versus placebo
Primary outcome measures
There was evidence of a large effect of clonazepam and bromazepam on treatment efficacy in participants with SAnD compared to placebo (k = 2, RR 4.03; 95% CI 2.45 to 6.65, N = 132; see Analysis 5.1; Davidson 1993; Versiani 1997), although the evidence was of low quality. The benzodiazepines demonstrated superiority with respect to treatment response relative to all the other medication classes that contained data from more than a single study, with the exception of the MAOI phenelzine (Chi2 = 1.67, df = 1, P = 0.20; I2 = 40.3%).
5.1. Analysis.
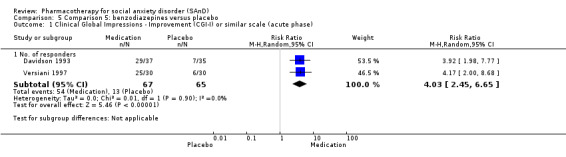
Comparison 5 Comparison 5: benzodiazepines versus placebo, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase).
There was no evidence that clonazepam prevented relapse in SAnD participants more than placebo (k = 1, RR 0.12; 95% CI 0.01 to 2.14, N = 36; see Analysis 5.2; Connor 1998).
5.2. Analysis.
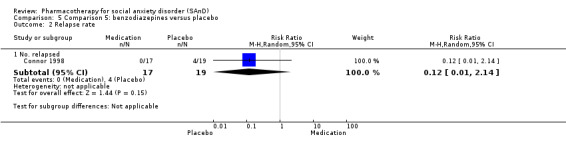
Comparison 5 Comparison 5: benzodiazepines versus placebo, Outcome 2 Relapse rate.
The dropout rate due to adverse events was low in RCTs of clonazepam and bromazepam (2/47; 4%) and did not differ appreciably from that observed in the placebo groups (k = 2, RR 1.68; 95% CI 0.21, 13.13, N = 96; see Analysis 5.3; Connor 1998; Versiani 1997). The evidence that was available for this outcome was of very low quality.
5.3. Analysis.
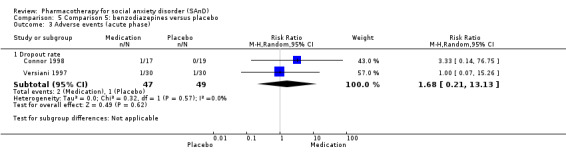
Comparison 5 Comparison 5: benzodiazepines versus placebo, Outcome 3 Adverse events (acute phase).
Secondary outcome measures
There was evidence of a reduction of SAnD symptoms following treatment with benzodiazepines compared to placebo on the LSAS (k = 2, MD −39.75 points, 95% CI −71.11 to −8.39, N = 135, see Analysis 5.4; Davidson 1993; Versiani 1997) although the evidence was very low in quality. There was considerable heterogeneity in effect size estimates for this outcome (P < 0.001; I2 = 94%), while effect estimates from both of the included RCTs demonstrated a beneficial effect of benzodiazepines relative to placebo. Clonazepam was superior to placebo on both the avoidance (MD −10.40 points, 95% CI −16.08 to −4.72, see Analysis 5.4) and fear subscale of the LSAS (MD −10.80 points, 95% CI −16.62 to −4.98, see Analysis 5.4) in one trial with 75 participants (Davidson 1993). The quality of evidence was very low for both outcomes, however. We found no evidence for an effect of clonazepam compared to placebo (k = 1, MD −1.60 points, 95% CI −3.96 to 0.76, N = 75; see Analysis 5.5; Davidson 1993) on the HDRS for depression symptoms.
5.4. Analysis.
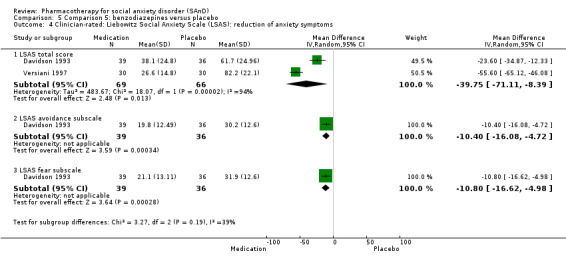
Comparison 5 Comparison 5: benzodiazepines versus placebo, Outcome 4 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms.
5.5. Analysis.
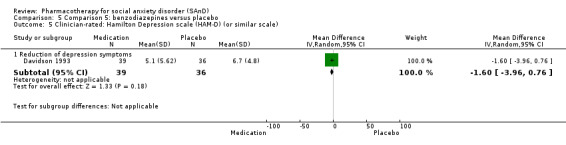
Comparison 5 Comparison 5: benzodiazepines versus placebo, Outcome 5 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale).
There was evidence of an effect of clonazepam and bromazepam compared to placebo on the reduction of associated disability on the work (MD −3.58 points, 95% CI −6.39 to −0.78, N = 135; see Analysis 5.6) and social subscale of the SDS (MD −2.31 points, 95% CI 3.79 to −0.83, N = 135; see Analysis 5.6) in two trials (Davidson 1993; Versiani 1997). We found no evidence of an overall effect for the benzodiazepines in reducing levels of associated disability on the family subscale of the SDS (k = 2, MD −2.02 points, 95% CI −4.26 to 0.22, N = 135; see Analysis 5.6). There was considerable heterogeneity in effect size estimates for the outcomes of disability (P < 0.001, I2 = 93%; P = 0.07, I2 = 69%; P = 0.04, I2 = 77%, respectively).
5.6. Analysis.
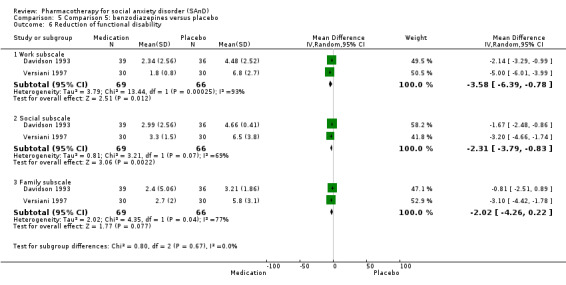
Comparison 5 Comparison 5: benzodiazepines versus placebo, Outcome 6 Reduction of functional disability.
Similar dropout rates were observed in the medication and placebo arms for the benzodiazepines (k = 3, RR 0.79; 95% CI 0.41 to 1.52, N = 171; see Analysis 5.7; Davidson 1993; Connor 1998; Versiani 1997).
5.7. Analysis.
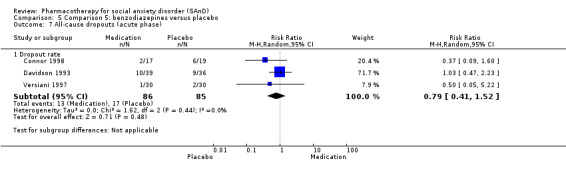
Comparison 5 Comparison 5: benzodiazepines versus placebo, Outcome 7 All‐cause dropouts (acute phase).
Comparison 6: beta‐blockers versus placebo
Primary outcome measures
There was no evidence of an effect of atenolol on treatment efficacy in participants with SAnD compared to placebo (k = 2, RR 1.09; 95% CI 0.63 to 1.88, N = 97; see Analysis 6.1; Liebowitz 1992; Turner 1994). The evidence that was available for this outcome was of very low quality. We observed a treatment response for the beta‐blockers compared to the benzodiazepines, in favour of the benzodiazepines (Chi2 = 12.02, df = 1, P = 0.0005; I2 = 91.7%).
6.1. Analysis.
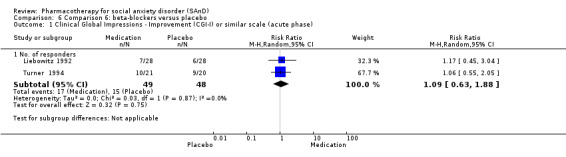
Comparison 6 Comparison 6: beta‐blockers versus placebo, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase).
Secondary outcome measures
We found no evidence of an effect for atenolol versus placebo in the single trial that provided data for the reduction of depression symptoms (MD 1.82 points, 95% CI −1.38 to 5.02, N = 46; Analysis 6.2; Liebowitz 1992) on the HDRS. There was also no evidence of an effect of atenolol compared to placebo on the reduction of associated disability on the work (k = 1, MD −0.05 points, 95% CI −1.80 to 1.70, N = 42; see Analysis 6.3), social (k = 1, MD 0.07 points, 95% CI −1.71 to 1.85, N = 42; see Analysis 6.3) and family subscales of the SDS (MD −0.11 points, 95% CI −1.74 to 1.52, N = 42; see Analysis 6.3) in one trial that reported this outcome (Liebowitz 1992).
6.2. Analysis.
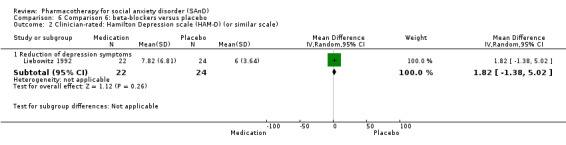
Comparison 6 Comparison 6: beta‐blockers versus placebo, Outcome 2 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale).
6.3. Analysis.
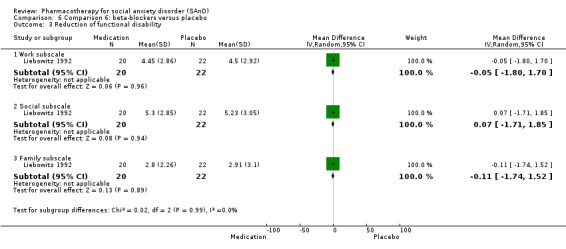
Comparison 6 Comparison 6: beta‐blockers versus placebo, Outcome 3 Reduction of functional disability.
The proportion of dropouts was more than three times higher in participants receiving atenolol (4/25, 16%) than placebo (1,21, 5%) in a single trial, though this difference was not statistically significant (RR 3.36; 95% CI 0.41 to 27.80; N = 46; Analysis 6.4; Turner 1994).
6.4. Analysis.
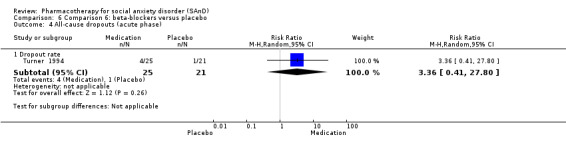
Comparison 6 Comparison 6: beta‐blockers versus placebo, Outcome 4 All‐cause dropouts (acute phase).
Comparison 7: MAOIs versus placebo
Primary outcome measures
There was evidence of an effect of phenelzine on acute treatment efficacy in participants with SAnD compared to placebo (k = 4, RR 2.36; 95% CI 1.48 to 3.75, N = 235; see Analysis 7.1; Blanco 2010; Heimberg 1998; Liebowitz 1992; Versiani 1992), though the evidence was low in quality. There were data indicating greater efficacy over the long term for phenelzine compared to placebo in participants with SAnD (k = 2, RR 1.84; 95% CI 1.02 to 3.33, N = 113; see Analysis 7.6; Blanco 2010; Liebowitz 1992). This conclusion was based on very low‐quality evidence, however. Treatment efficacy was greater for the MAOIs compared to the anticonvulsant levetiracetam (Chi2 = 5.86, df = 1, P = 0.02; I2 = 82.9%) and the GW876008 (Chi2 = 7.97, df = 1, P = 0.005; I2 = 87.5%).
7.1. Analysis.
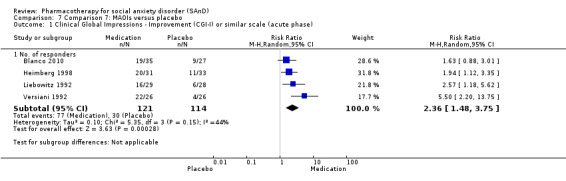
Comparison 7 Comparison 7: MAOIs versus placebo, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase).
7.6. Analysis.
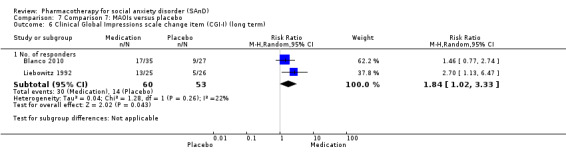
Comparison 7 Comparison 7: MAOIs versus placebo, Outcome 6 Clinical Global Impressions scale change item (CGI‐I) (long term).
Secondary outcome measures
We assessed data on the reduction of SAnD symptoms, measured by the LSAS, in four studies comparing phenelzine to placebo (MD −16.39 points, 95% CI −32.27 to −0.51, N = 218; see Analysis 7.2; Blanco 2010; Heimberg 1998; Liebowitz 1992; Versiani 1992). There was evidence of benefit even though the data were based on low‐quality evidence. Considerable heterogeneity on this outcome (Chi2 = 45.00, df = 3, P < 0.001; I2 = 93%) partially reflected the unusually large medication effect observed in Versiani 1992 (MD −42.20 points, 95% CI −55.66 to −28.74). Removing this trial resulted in a substantially reduced and statistically non‐significant treatment effect (MD −7.39 points, 95% CI −16.77 to 1.99), with considerable heterogeneity still evident across the effects for the remaining trials (Chi2 = 8.94, df = 2, P = 0.01; I2 = 78%). Phenelzine was not superior to placebo on either the avoidance (MD −5.42 points, 95% CI −14.69 to 3.85; see Analysis 7.2) or fear subscale of the LSAS (MD −5.23 points, 95% CI −13.97 to 3.51; see Analysis 7.2) in one trial with 51 participants (Liebowitz 1992). These outcomes were also based on very low‐quality evidence, however.
7.2. Analysis.
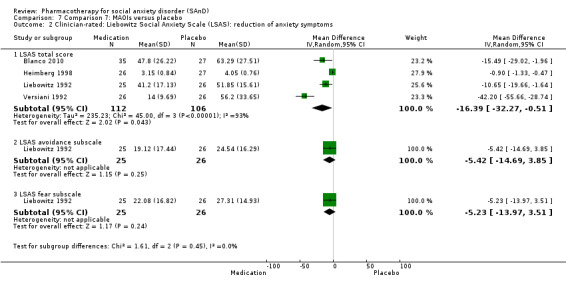
Comparison 7 Comparison 7: MAOIs versus placebo, Outcome 2 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms.
There was no evidence of an effect of phenelzine in the reduction of depression symptoms compared to placebo (k = 4, SMD −0.40 ; 95% CI −1.11 to 0.31, N = 216; see Analysis 7.3; Blanco 2010; Heimberg 1998; Liebowitz 1992; Versiani 1992) on the HAM‐D. Moreover, there was considerable heterogeneity in effect size estimates for this outcome (Chi2 = 33.26, df = 3, P < 0.001; I2 = 85%). This was partly due to large medication effects of one study (Versiani 1992). However, after the removal of this study from the analysis, no evidence for efficacy of the MAOIs was still apparent (k = 3, SMD −0.51; 95% CI −1.49 to 0.47, N = 167). There was an effect of phenelzine in the reduction of associated disability compared to placebo on the work subscale of the SDS (MD −2.84 points, 95% CI −4.40 to −1.28; see Analysis 7.4) in two trials with 95 participants (Liebowitz 1992; Versiani 1992). There was no evidence that phenelzine reduced associated disability on the social (MD −3.26 points, 95% CI −7.25 to 0.72, N = 94; see Analysis 7.4) or family subscale of the SDS (k = 2, MD −2.20 points, 95% CI −5.34 to 0.95, N = 95; see Analysis 7.4) in these trials. There was also considerable heterogeneity in effect size estimates for the outcomes of disability (P = 0.06, I2 = 72%; P < 0.001, I2 = 92%; P = 0.002, I2 = 90%, respectively).
7.3. Analysis.
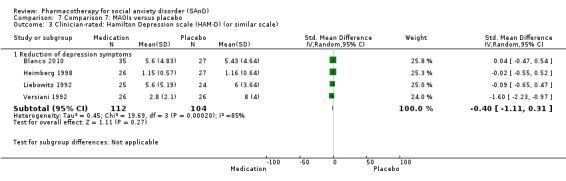
Comparison 7 Comparison 7: MAOIs versus placebo, Outcome 3 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale).
7.4. Analysis.
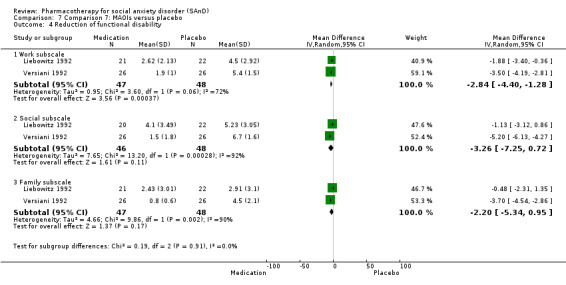
Comparison 7 Comparison 7: MAOIs versus placebo, Outcome 4 Reduction of functional disability.
Similar dropout rates were observed in the medication and placebo arms for the MAOIs (RR 1.33; 95% CI 0.71 to 2.48; see Analysis 7.5) in four trials with 235 participants (Blanco 2010; Heimberg 1998; Liebowitz 1992; Versiani 1992).
7.5. Analysis.
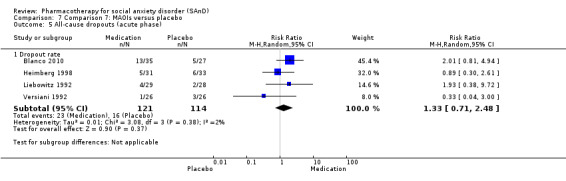
Comparison 7 Comparison 7: MAOIs versus placebo, Outcome 5 All‐cause dropouts (acute phase).
Comparison 8: NARIs versus placebo
Primary outcome measures
There was no evidence of an effect for atomoxetine on treatment efficacy in participants with SAnD compared to placebo (RR 0.70; 95% CI 0.19 to 2.54, see Analysis 8.1) in one trial with 27 participants (Ravindran 2009). The evidence that was available for this outcome was of very low quality. There was evidence of an effect for the NARIs compared to the benzodiazepines, in favour of the benzodiazepines (Chi2 = 6.17, df = 1, P = 0.01; I2 = 83.8%).
8.1. Analysis.
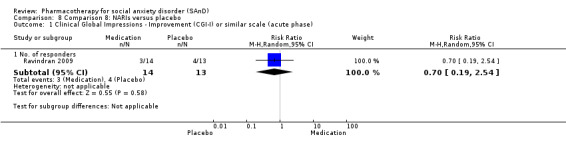
Comparison 8 Comparison 8: NARIs versus placebo, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase).
Only 1 of 14 (7%) participants withdrew during 10 weeks of treatment with atomoxetine (see Analysis 8.2); there was no evidence for a difference in the number of participants who withdrew due to treatment‐emergent side effects. The evidence that was available for this outcome was of very low quality.
8.2. Analysis.
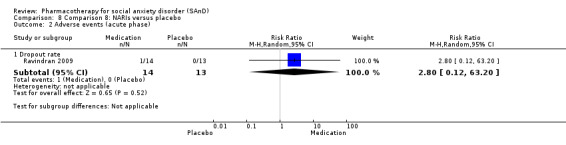
Comparison 8 Comparison 8: NARIs versus placebo, Outcome 2 Adverse events (acute phase).
Secondary outcome measures
We found no evidence for an effect of the single trial that provided data for the reduction of SAnD symptoms (i.e. LSAS total symptom severity) when comparing atomoxetine to placebo (MD 2.60 points, 95% CI −15.43 to 20.63, N = 26; see Analysis 8.3). The evidence that was available for this outcome was of very low quality. Furthermore, we found no evidence of an effect for the reduction of depression symptoms on the HAM‐D when comparing atomoxetine to placebo (k = 1, MD −0.10 points, 95% CI −2.73 to 2.53, N = 26; see Analysis 8.4).
8.3. Analysis.
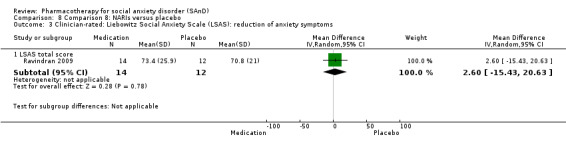
Comparison 8 Comparison 8: NARIs versus placebo, Outcome 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms.
8.4. Analysis.
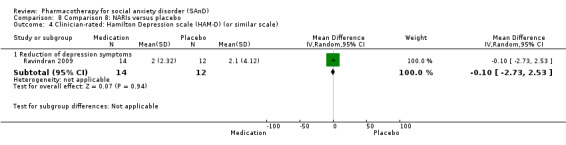
Comparison 8 Comparison 8: NARIs versus placebo, Outcome 4 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale).
Similar dropout rates were observed in the medication and placebo arms for the NARIs (k = 1, RR 0.93; 95% CI 0.23 to 3.81, N = 27; see Analysis 8.5; Ravindran 2009).
8.5. Analysis.
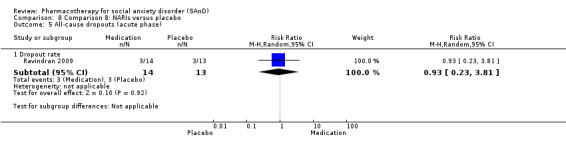
Comparison 8 Comparison 8: NARIs versus placebo, Outcome 5 All‐cause dropouts (acute phase).
Comparison 9: NaSSAs versus placebo
Primary outcome measures
An equal number of participants (4/30, 13%) responded to treatment in both the medication and placebo arms of a trial of mirtazepine (k = 1, RR 1.00; 95% CI 0.28 to 3.63; N = 60, see Analysis 9.1; Schutters 2010). The evidence that was available for this outcome was of very low quality.
9.1. Analysis.
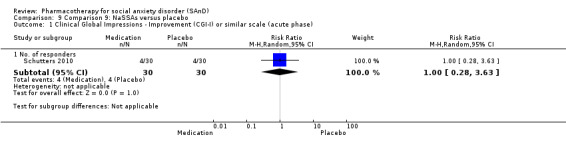
Comparison 9 Comparison 9: NaSSAs versus placebo, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase).
A small proportion of participants receiving mirtazepine dropped out due to adverse events (2/30, 7%) in one trial with 60 participants (Schutters 2010). Although this proportion was larger than that observed for placebo (0/30, 0%), the difference was not statistically significant (P = 0.29, see Analysis 9.2). The evidence that was available for this outcome was of very low quality.
9.2. Analysis.
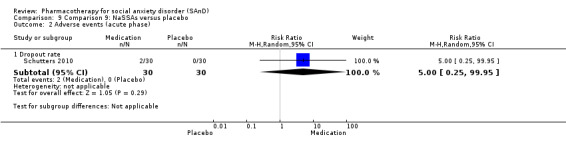
Comparison 9 Comparison 9: NaSSAs versus placebo, Outcome 2 Adverse events (acute phase).
Secondary outcome measures
Evidence of an effect was found for mirtazepine compared to placebo in the reduction of total SAnD symptom severity on the LSAS (k = 2, MD −15.37 points, 95% CI −28.10 to −2.63, N = 126; see Analysis 9.3; Schutters 2010; Muehlbacher 2005), even though the evidence was very low in quality. Mirtazepine was not superior to placebo on either the avoidance (MD −3.90 points, 95% CI −9.90 to 2.10, see Analysis 9.3) or fear subscale of the LSAS (MD −3.70 points, 95% CI −9.42 to 2.02, see Analysis 9.3) in one trial with 60 participants (Schutters 2010). This conclusion was based on low‐quality evidence.
9.3. Analysis.
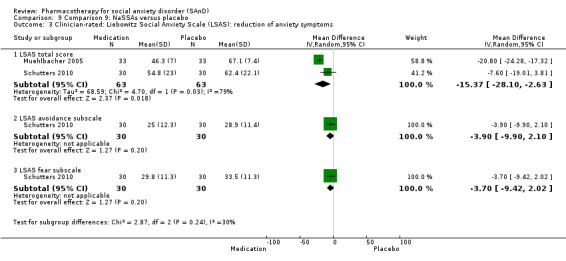
Comparison 9 Comparison 9: NaSSAs versus placebo, Outcome 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms.
A low proportion of participants withdrew for any cause during 12 weeks of treatment with mirtazepine (2/30, 7%), with overall dropout rates similar to those observed following treatment with placebo (1/30, 3%) (k = 1, RR 2.00; 95% CI 0.19 to 20.90; see Analysis 9.4; Schutters 2010).
9.4. Analysis.
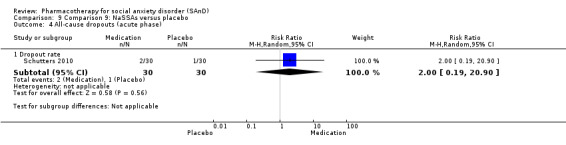
Comparison 9 Comparison 9: NaSSAs versus placebo, Outcome 4 All‐cause dropouts (acute phase).
Comparison 10: RIMAs versus placebo
Primary outcome measures
There was evidence of benefit for brofaromine and moclobemide on treatment efficacy in participants with SAnD compared to placebo (k = 8, RR 1.83; 95% CI 1.32 to 2.55, N = 1270; see Analysis 10.1; Fahlen 1995; Katschnig 1997; Lott 1997; Noyes 1997; Oosterbaan 2001; Schneier 1998; Stein 2002a; Van Vliet 1992), even though the evidence was low quality. There was substantial heterogeneity in effect size estimates for this outcome (Chi2 = 23.57, df = 7, P = 0.001; I2 = 70%). This was partly due to large medication effects for earlier, small studies (i.e. Fahlen 1995; Van Vliet 1992), with moderate heterogeneity evident after excluding these trials (Chi2 = 8.86, df = 5, P = 0.11; I2 = 44%). Evidence for efficacy of the RIMAs was still apparent after removing these two studies (k = 6, RR 1.49; 95% CI 1.17 to 1.90, N = 1165).
10.1. Analysis.
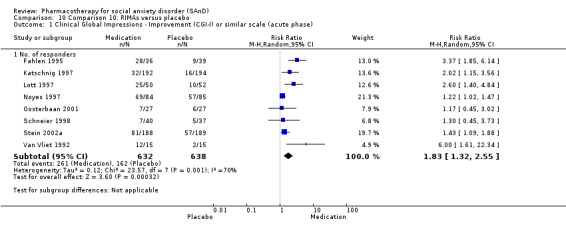
Comparison 10 Comparison 10: RIMAs versus placebo, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase).
Given differences in the mechanism of action for moclobemide and brofaromine and the lack of availability for the latter, we conducted a post hoc analysis for moclobemide only. There was evidence that this medication was efficacious compared to placebo (k = 5, RR 1.32; 95% CI 1.14 to 1.52, N = 1063; P < 0.001). Furthermore, there was moderate‐quality evidence of a long‐term effect for moclobemide on treatment efficacy in participants with SAnD compared to placebo (k = 1, RR 1.50; 95% CI 1.12 to 2.00, N = 90; see Analysis 10.7; Stein 2002a).
10.7. Analysis.
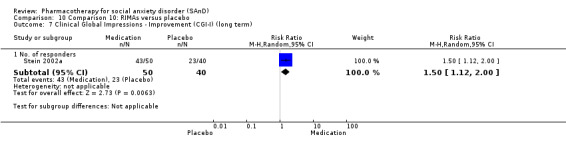
Comparison 10 Comparison 10: RIMAs versus placebo, Outcome 7 Clinical Global Impressions ‐ Improvement (CGI‐I) (long term).
Comparisons between medication classes revealed that response to the RIMAs was smaller than that observed for the benzodiazepines (Chi2 = 6.63, df = 1, P = 0.01; I2 = 84.9%).
Dropout rates due to adverse events were low in RCTs of brofaromine and moclobemide and equivalent to those observed in the placebo arms (72/83; 9%) (k = 8, RR 1.42; 95% CI 0.86 to 2.34, N = 1305; see Analysis 10.2). The evidence that was available for this outcome was of very low quality.
10.2. Analysis.
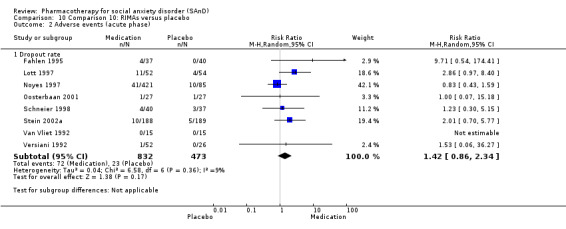
Comparison 10 Comparison 10: RIMAs versus placebo, Outcome 2 Adverse events (acute phase).
Secondary outcome measures
There was evidence for a reduction of SAnD symptom severity on the LSAS in six studies comparing brofaromine or moclobemide to placebo (MD −12.17 points, 95% CI −23.51, −0.84 , N = 1163; see Analysis 10.3; Katschnig 1997; Lott 1997; Noyes 1997; Schneier 1998; Stein 2002a; Versiani 1992), although the quality of evidence was very low. We observed considerable heterogeneity for this outcome (Chi2 = 77.55, df = 5, P < 0.001; I2 = 94%), but most disappeared after removing Versiani 1992 (Chi2 = 3.14, df = 4, P = 0.53; I2 = 0%), with data from the remaining five trials still supporting the efficacy of this class of medications (MD = −7.59 points, 95% CI −11.35 to −3.84). Brofaromine and moclobemide were superior to placebo on both the avoidance (k = 5, MD −5.05 points, 95% CI −7.91 to −2.18, N = 695; see Analysis 10.3; Fahlen 1995; Katschnig 1997; Lott 1997; Oosterbaan 2001; Schneier 1998) and fear subscale of the LSAS (k = 6, MD −5.40 points, 95% CI −8.92 to −1.88, N = 724; see Analysis 10.3; Fahlen 1995; Katschnig 1997; Lott 1997; Oosterbaan 2001; Schneier 1998; Van Vliet 1992). Evidence from the social subscale was low in quality. We observed moderate to substantial variability in the effects of these agents on the fear subscale (Chi2 = 11.13, df = 5, P = 0.05; I2 = 55%), and the evidence was very low in quality.
10.3. Analysis.
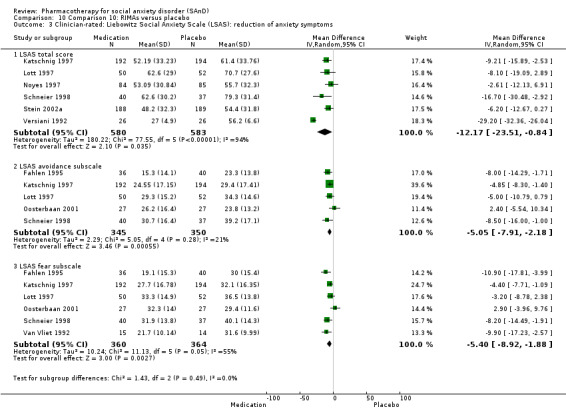
Comparison 10 Comparison 10: RIMAs versus placebo, Outcome 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms.
We observed evidence of an effect for the reduction of depression symptoms when comparing brofaromine and moclobemide to placebo (SMD −0.28; 95% CI −0.55 to 0.00, see Analysis 10.4) in seven trials with 765 participants (Fahlen 1995; Katschnig 1997; Lott 1997; Oosterbaan 2001; Schneier 1998; Van Vliet 1992; Versiani 1992) on the HDRS or the MADRS. There was substantial heterogeneity in effect size estimates for this outcome (Chi2 = 16.21, df = 6, P = 0.01; I2 = 63%). This effect was partly due to large medication effects of two studies (Fahlen 1995; Versiani 1992). Evidence for efficacy of the RIMAs was not apparent after removing these studies (k = 5, RR −0.08; 95% CI −0.24 to 0.07, N = 637).
10.4. Analysis.
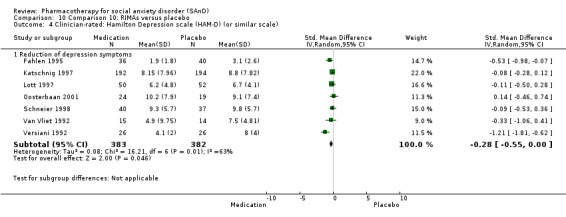
Comparison 10 Comparison 10: RIMAs versus placebo, Outcome 4 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale).
There was no evidence of an effect for brofaromine or moclobemide compared to placebo for reducing associated disability on the work (MD −0.61 points, 95% CI −1.89 to 0.68, see Analysis 10.5), social (MD −1.14 points, 95% CI −2.32 to 0.05, see Analysis 10.5), or family subscale of the SDS (MD −0.51 points, 95% CI −1.45 to 0.44, see Analysis 10.5) in five trials with 660 participants (Katschnig 1997; Lott 1997; Oosterbaan 2001; Schneier 1998; Versiani 1992). There was substantial and considerable heterogeneity in effect size estimates for these three scales (P < 0.001, I2 = 88%; P < 0.001, I2 = 83%; P = 0.002, I2 = 65%, respectively). When we removed Versiani 1992 from this outcome, the findings were non‐significant for reduction of associated disability on the work, social, and family subscale of the SDS.
10.5. Analysis.
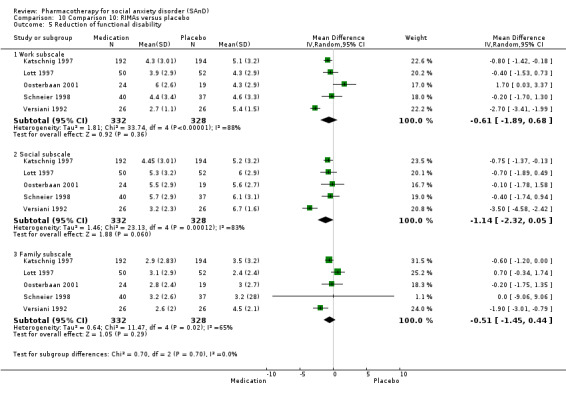
Comparison 10 Comparison 10: RIMAs versus placebo, Outcome 5 Reduction of functional disability.
We observed similar dropout rates in the medication and placebo arms for the RIMAs (k = 6, RR 0.80; 95% CI 0.60 to 1.08, N = 512; see Analysis 10.6; Fahlen 1995; Katschnig 1997; Lott 1997; Oosterbaan 2001; Schneier 1998; Van Vliet 1992).
10.6. Analysis.
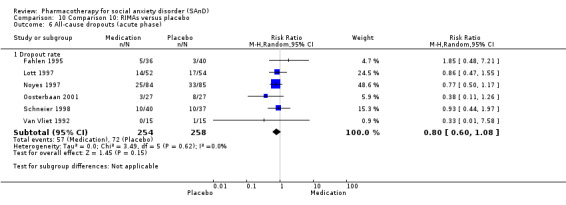
Comparison 10 Comparison 10: RIMAs versus placebo, Outcome 6 All‐cause dropouts (acute phase).
Comparison 11: SARIs versus placebo
Primary outcome measures
There were no data available to assess treatment efficacy and dropouts due to adverse events.
Secondary outcome measures
There was no evidence of an effect of nefazodone compared to placebo on the reduction of total SAnD symptom severity on the LSAS (MD −6.10 points, 95% CI −16.55 to 4.35, see Analysis 11.1) in one trial with 102 participants (Van Ameringen 2007). The evidence that was available for this outcome was of very low quality. There was also no evidence of an effect of nefazodone for reducing depression symptoms compared to placebo (k = 1, MD 0.80 points, 95% CI −2.10 to 3.70, N = 102; see Analysis 11.2) on the BDI.
11.1. Analysis.
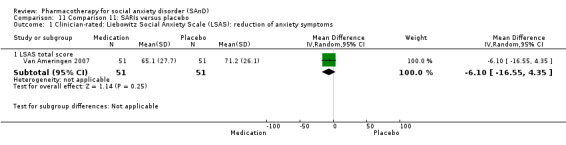
Comparison 11 Comparison 11: SARIs versus placebo, Outcome 1 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms.
11.2. Analysis.
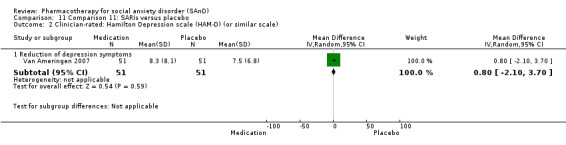
Comparison 11 Comparison 11: SARIs versus placebo, Outcome 2 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale).
Furthermore, there was evidence that nefazodone reduced social disability compared to placebo (k = 1, MD −1.00 point, 95% CI −1.97 to −0.03, N = 102; Analysis 11.3), with no evidence of similar reductions on the work (k = 1, MD −0.90 points, 95% CI −1.87 to 0.07, N = 102; see Analysis 11.3) or family subscale of the SDS (k = 1, MD −0.20 points, 95% CI −1.06 to 0.66, N = 102; see Analysis 11.3; Van Ameringen 2007). The data for the disability subscales were of low quality.
11.3. Analysis.
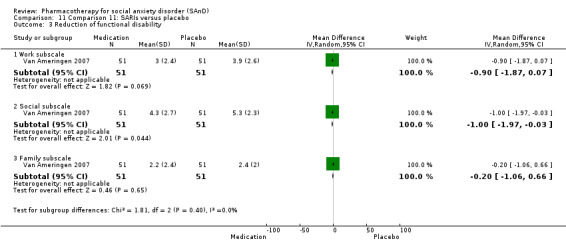
Comparison 11 Comparison 11: SARIs versus placebo, Outcome 3 Reduction of functional disability.
The proportion of dropouts was more than twice as high in participants receiving nefazodone (15/52, 29%) compared to the placebo group (7/53, 13%), though this difference was not statistically significant (k = 1, RR 2.18; 95% CI 0.97 to 4.92; see Analysis 11.4; Van Ameringen 2007).
11.4. Analysis.
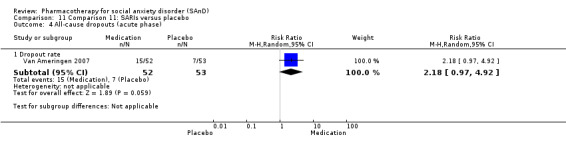
Comparison 11 Comparison 11: SARIs versus placebo, Outcome 4 All‐cause dropouts (acute phase).
Comparison 12: SNRIs compared to placebo for the treatment of SAnD
Primary outcome measures
There was no evidence of an effect of venlafaxine on treatment efficacy in participants with SAnD compared to placebo (RR 1.30; 95% CI 0.85 to 1.99, see Analysis 12.1) in four trials with 1173 participants (Liebowitz 2005a; Liebowitz 2005b; Rickels 2004; Stein 2005). The evidence that was available for this outcome was of low quality. We observed considerable heterogeneity for this outcome (Chi2 = 26.70, df = 3, P < 0.001; I2 = 89%). There was evidence of an effect of the SNRIs compared to the benzodiazepines (Chi2 = 11.39, df = 1, P = 0.0007; I2 = 91.2%) however, favouring the benzodiazepines.
12.1. Analysis.
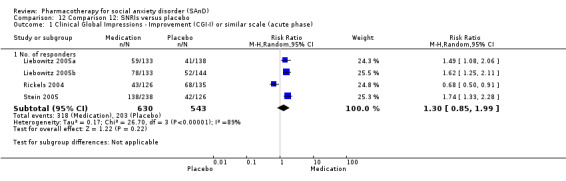
Comparison 12 Comparison 12: SNRIs versus placebo, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase).
The proportion of dropouts due to adverse events was more than three times as high in participants receiving venlafaxine (109/663, 16%) compared to placebo (27/550, 5%), a statistically significant difference (k = 4, RR 3.23, 95% CI 2.15 to 4.86; Analysis 12.2). The evidence that was available for this outcome was of moderate quality.
12.2. Analysis.
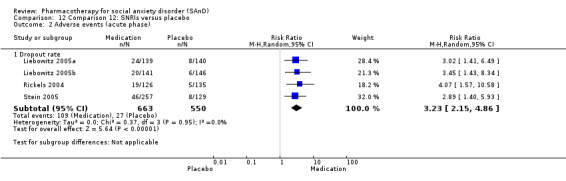
Comparison 12 Comparison 12: SNRIs versus placebo, Outcome 2 Adverse events (acute phase).
Secondary outcome measures
There was evidence of benefit for venlafaxine compared to placebo on the reduction of total SAnD symptom severity on the LSAS (k = 3; MD −11.91 points, 95% CI −16.06 to −7.76) based on moderate‐quality evidence. This outcome was calculated using a combination of endpoint (for Rickels 2004) and change‐from‐baseline scores (from Liebowitz 2005b and Stein 2005) across a total of 903 participants. Venlafaxine was superior to placebo on both the avoidance (k = 1, MD −4.30 points, 95% CI −8.14 to −0.46, N = 261; see Analysis 12.3) and fear subscale of the LSAS (k = 1, MD −4.00 points, 95% CI −7.68 to −0.32, N = 261; see Analysis 12.3) for a single trial providing data on these outcomes (Rickels 2004). The evidence on the remaining outcomes of avoidance and fear was low in quality.
12.3. Analysis.
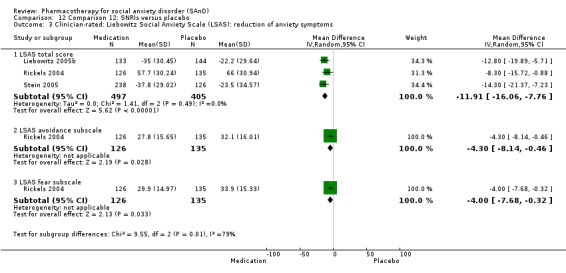
Comparison 12 Comparison 12: SNRIs versus placebo, Outcome 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms.
We found similar dropout rates in the medication and placebo arms for the SNRIs (k = 4, RR 0.90; 95% CI 0.76 to 1.07, N = 1224; see Analysis 12.4; Allgulander 2004; Liebowitz 2005a; Liebowitz 2005b; Stein 2005).
12.4. Analysis.
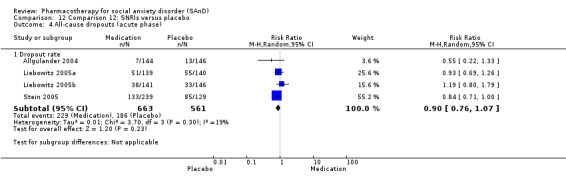
Comparison 12 Comparison 12: SNRIs versus placebo, Outcome 4 All‐cause dropouts (acute phase).
Comparison 13: SSRIs versus placebo
Primary outcome measures
There was evidence of treatment efficacy for paroxetine, fluvoxamine, sertraline, fluoxetine, and citalopram compared to placebo in participants with SAnD (k = 24, RR 1.65; 95% CI 1.48 to 1.85, N = 4984; see Analysis 13.1; Allgulander 1999; Asakura 2007; Baldwin 1999; Blomhoff 2001; Book 2008; Davidson 2004a; Davidson 2004b; Furmark 2005; NCT00318669; NCT00403962; NCT00397722; Kasper 2005; Kobak 2002; Lader 2004; Lepola 2004; Liebowitz 2002; Liebowitz 2003; Liebowitz 2005b; Nordahl 2016; Randall 2001; Stein 1998; Stein 1999; Van Ameringen 2001a; Westenberg 2004), though the evidence was very low in quality. There was evidence for a response to long‐term treatment with paroxetine and fluvoxamine compared to placebo in participants with SAnD (k = 4, RR 1.27; 95% CI 1.07 to 1.51, N = 806; see Analysis 13.8; Lader 2004; Stein 2002b; Stein 2003), though again this conclusion was based on low quality evidence. There was moderate heterogeneity in effect size estimates for long‐term treatment response (Chi2 = 7.54, df = 3, P = 0.06; I2 = 60%). This was partly due to large medication effects of one study (Stein 2002b). However, evidence for efficacy of the SSRIs was still apparent after removing this study from the analysis (k = 3, RR 1.18; 95% CI 1.05 to 1.32, N = 483).
13.1. Analysis.
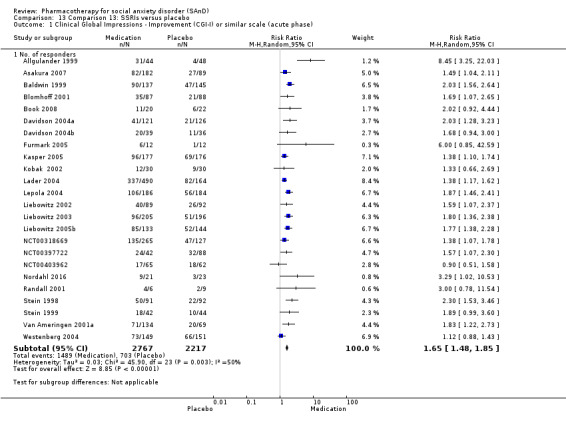
Comparison 13 Comparison 13: SSRIs versus placebo, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase).
13.8. Analysis.
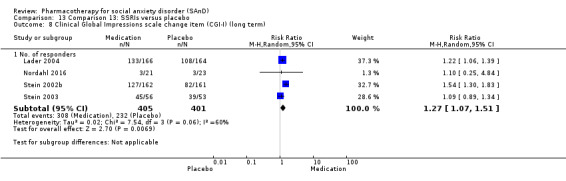
Comparison 13 Comparison 13: SSRIs versus placebo, Outcome 8 Clinical Global Impressions scale change item (CGI‐I) (long term).
There was evidence for a smaller response to treatment with the anticonvulsant levetiracetam (Chi2 = 8.02, df = 1, P = 0.005; I2 = 87.5%) compared to the benzodiazepines (Chi2 = 11.58, df = 1, P = 0.0007; I2 = 91.4%) and the GW876008 (Chi2 = 12.28, df = 1; P = 0.0005, I2 = 91.9%), although the response favoured the benzodiazepines.
There was evidence that paroxetine and sertraline prevented relapse compared to placebo (k = 3, RR 0.34; 95% CI 0.22 to 0.50, N = 389; see Analysis 13.2; Stein 1996; Stein 2002b; Walker 2000).
13.2. Analysis.
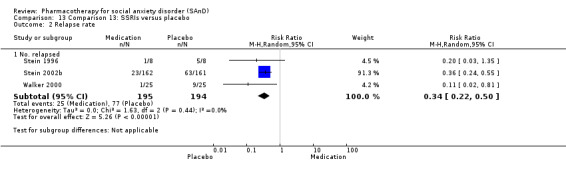
Comparison 13 Comparison 13: SSRIs versus placebo, Outcome 2 Relapse rate.
The proportion of dropouts due to adverse events was approximately three times higher amongst participants receiving SSRIs compared to placebo. This difference was statistically significant (RR 2.59; 95% CI 1.97 to 3.39, N = 5131; see Analysis 13.3). Twelve per cent of participants across 24 RCTs who received medication withdrew prior to study endpoint. The evidence that was available for this outcome was of low quality. We found no difference in dropout rates due to treatment‐emergent side effects between the medication and control groups (RR 1.17; 95% CI 0.43 to 3.18, see Analysis 13.9) in three long‐term trials with 1274 participants (Lader 2004; Stein 2002b; Stein 2003). The evidence that was available for this outcome was of very low quality.There was moderate heterogeneity in effect size estimates for this outcome (Chi2 = 4.60, df = 2, P = 0.10; I2 = 57%).
13.3. Analysis.
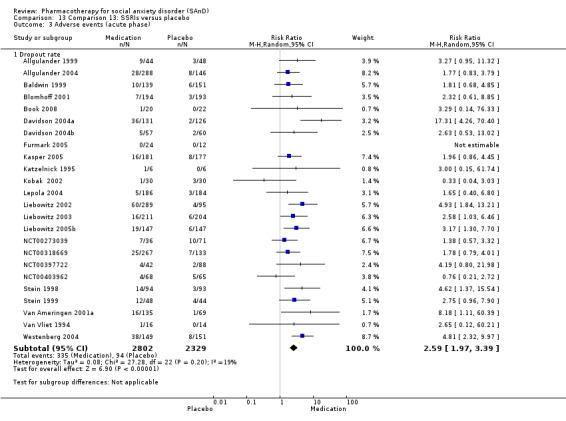
Comparison 13 Comparison 13: SSRIs versus placebo, Outcome 3 Adverse events (acute phase).
13.9. Analysis.
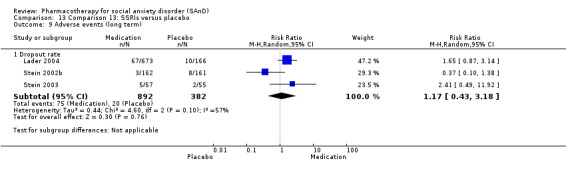
Comparison 13 Comparison 13: SSRIs versus placebo, Outcome 9 Adverse events (long term).
Secondary outcome measures
There was evidence of benefit for the SSRIs compared to placebo for reducing total SAnD symptoms on the LSAS (k = 14, MD −10.14 points, 95% CI −14.05 to −6.22, N = 1990; see Analysis 13.4; Allgulander 1999; Asakura 2007; Baldwin 1999; Book 2008; Davidson 2004a; Furmark 2005; Katzelnick 1995; Kobak 2002; Liebowitz 2002; Liebowitz 2003; Nordahl 2016; Stein 1998; Stein 1999; Tauscher 2010), though the evidence was low in quality. There was considerable heterogeneity in effect size estimates for this outcome (Chi2 = 39.61, df = 13, P < 0.001; I2 = 67%). This was partly due to large medication effects of Allgulander 1999. Evidence for efficacy of the SSRIs was still apparent after removing this study (k = 13, RR −8.69; 95% CI −11.83 to −5.54, N = 1898). The SSRIs were superior to placebo on both the avoidance (MD −7.01 points, 95% CI −10.21 to −3.80; see Analysis 13.4) and fear subscale of the LSAS (MD −7.28 points, 95% CI −10.86 to −3.71, see Analysis 13.4) in seven trials with 1173 participants (Allgulander 1999; Asakura 2007; Furmark 2005; Liebowitz 2002; Liebowitz 2003; Stein 1998; Van Vliet 1994). The evidence that was available for the avoidance and fear outcomes was of low quality. There was considerable to substantial heterogeneity in effect size estimates of the avoidance and fear subscales of the LSAS (Chi2 = 17.31, df = 6, P = 0.008; I2 = 65%; Chi2 = 24.25, df = 6, P < 0.001; I2 = 75%, respectively).
13.4. Analysis.
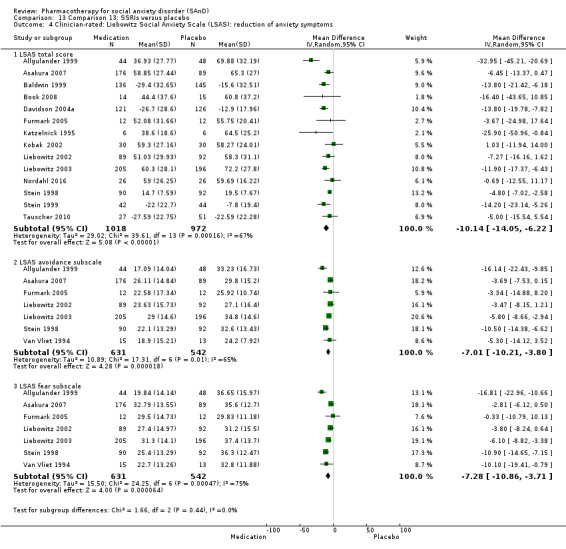
Comparison 13 Comparison 13: SSRIs versus placebo, Outcome 4 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms.
We found evidence of an effect for reducing depression symptoms when comparing paroxetine, sertraline, fluoxetine, and fluvoxamine to placebo on the HAM‐D/HDRS or MADRS (k = 6, SMD −0.26; 95% CI −0.48 to −0.03, N = 960; see Analysis 13.5; Baldwin 1999; Katzelnick 1995; Kobak 2002; Liebowitz 2002; Liebowitz 2003; Van Vliet 1994). There was moderate heterogeneity in effect size estimates for this outcome (Chi2 = 11.05, df = 5, P = 0.05; I2 = 55%), which was partly due to large medication effects of two studies (Baldwin 1999; Van Vliet 1994). Evidence for efficacy of the SSRIs was not apparent after removing these studies (k = 4, RR −0.13; 95% CI −0.28 to 0.03, N = 652).
13.5. Analysis.
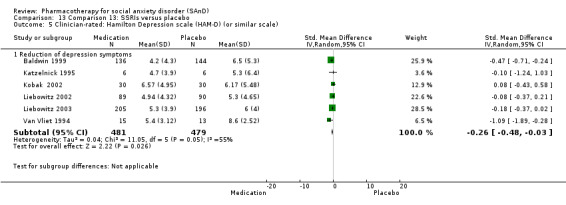
Comparison 13 Comparison 13: SSRIs versus placebo, Outcome 5 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale).
Futhermore, we found evidence of an effect for reducing associated disability when comparing paroxetine, sertraline, fluvoxamine and citalopram to placebo on the work (MD −0.81 points, 95% CI −1.18 to −0.45; see Analysis 13.6) and family subscale of the SDS (MD −0.45 points, 95% CI −0.75, −0.15, see Analysis 13.6), with evidence of an effect on the social subscale (MD −0.87 points, 95% CI −1.26 to −0.47, see Analysis 13.6) in five trials with 854 participants (Furmark 2005; Liebowitz 2002; Liebowitz 2003; Stein 1998; Stein 1999).
13.6. Analysis.
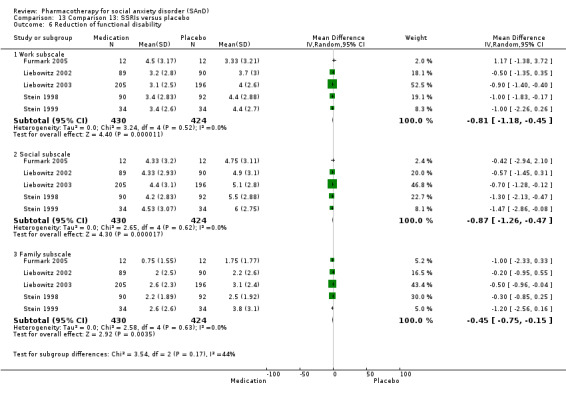
Comparison 13 Comparison 13: SSRIs versus placebo, Outcome 6 Reduction of functional disability.
We observed similar dropout rates in the medication and placebo arms for the SSRIs (k = 26, RR 1.01; 95% CI 0.90 to 1.14, N = 5208; see Analysis 13.7). Findings were similar for participants who dropped out after long‐term treatment due to any cause (k = 1, RR 1.00; 95% CI 0.37 to 2.70, N = 52; see Analysis 13.10).
13.7. Analysis.
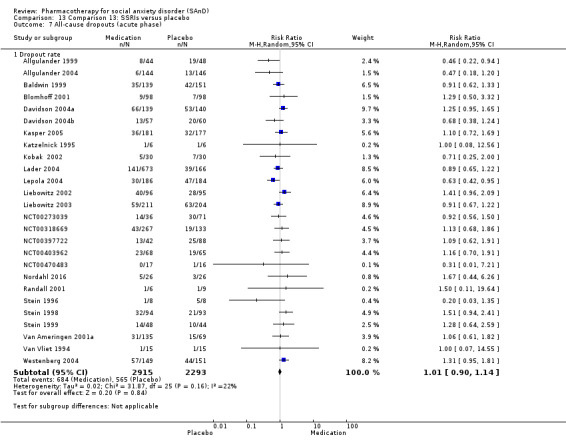
Comparison 13 Comparison 13: SSRIs versus placebo, Outcome 7 All‐cause dropouts (acute phase).
13.10. Analysis.
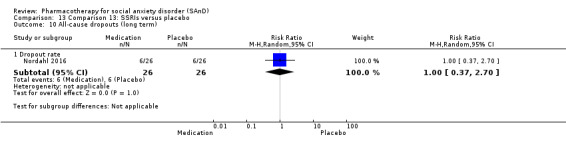
Comparison 13 Comparison 13: SSRIs versus placebo, Outcome 10 All‐cause dropouts (long term).
Comparison 14: GW876008 versus placebo
Primary outcome measures
There was no evidence of treatment efficacy for GW876008 versus placebo (RR 0.83; 95% CI 0.58 to 1.19, see Analysis 14.1) in one trial with 250 participants (NCT00397722), based on moderate quality evidence.
14.1. Analysis.
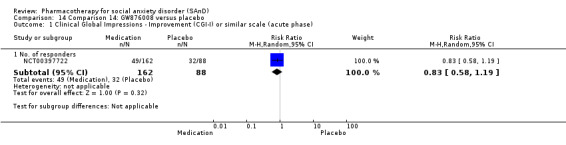
Comparison 14 Comparison 14: GW876008 versus placebo, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase).
Treatment dropouts were three times as high in participants receiving GW876008 (11/164, 7%) relative to placebo (2/88, 2%) (NCT00397722), though this difference was not statistically significant. There was no evidence of a difference between the number of participants that dropped out due to treatment‐emergent side effects (k = 1, RR 2.95; 95% CI 0.67 to 13.02, N = 252; see Analysis 16.2) in the RCT of GW876008. The evidence that was available for this outcome was of low quality.
The GW876008 demonstrated superiority with respect to treatment response relative to all the other medication classes that contained data from more than a single study, with the exception of the SNRI venlafaxine (Chi2 = 2.49, df = 1, P = 0.11; I2 = 59.8%).
Secondary outcome measures
We observed similar dropout rates in the medication and placebo arms in the trial of GW876008 compared to placebo (RR 0.77; 95% CI 0.50 to 1.20, N = 252; see Analysis 14.3; NCT00397722).
14.3. Analysis.
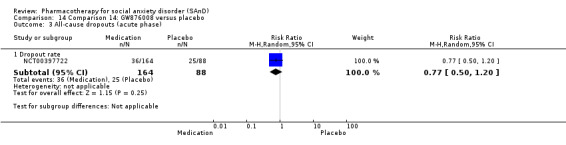
Comparison 14 Comparison 14: GW876008 versus placebo, Outcome 3 All‐cause dropouts (acute phase).
Comparison 15: NK1 receptor antagonist GR205171 versus placebo
Primary outcome measures
There was no evidence of a superior treatment response to the NK1 receptor antagonist GR205171 than placebo in a single trial with 24 participants (RR 5.00; 95% CI 0.68 to 36.66, see Analysis 15.1; Furmark 2005), based on very low quality evidence.
15.1. Analysis.
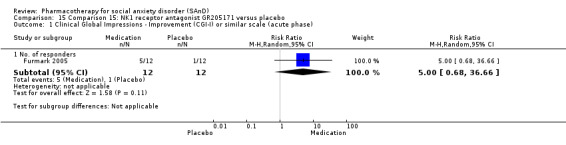
Comparison 15 Comparison 15: NK1 receptor antagonist GR205171 versus placebo, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase).
No participants withdrew due to adverse events from treatment during the six‐week RCT of the NK1 receptor antagonist GR205171 (Furmark 2005; Analysis 15.2). The evidence that was available for this outcome was of moderate quality.
15.2. Analysis.
Comparison 15 Comparison 15: NK1 receptor antagonist GR205171 versus placebo, Outcome 2 Adverse events (acute phase).
Secondary outcome measures
We observed no evidence for an effect of the NK1 antagonist GR205171 on total LSAS symptom severity (k = 1, MD ‐0.50 points, 95% CI ‐1.35 to 0.35, N = 24; see Analysis 15.3; Furmark 2005). This conclusion was based on data of very low quality.
15.3. Analysis.
Comparison 15 Comparison 15: NK1 receptor antagonist GR205171 versus placebo, Outcome 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms.
No participants withdrew from treatment during the six‐week RCT of the NK1 receptor antagonist GR205171 (Furmark 2005; Analysis 15.4).
15.4. Analysis.
Comparison 15 Comparison 15: NK1 receptor antagonist GR205171 versus placebo, Outcome 4 All‐cause dropouts (acute phase).
Comparison 16: LY686017 versus placebo
Primary outcome measures
There were no data available to assess treatment efficacy and dropouts due to adverse events.
Secondary outcome measures
We observed no evidence for an effect of the LY686017 on total LSAS symptom severity (k = 1, MD 1.80 points, 95% CI ‐6.92 to 10.52, N = 99; see Analysis 16.1; Furmark 2005; Tauscher 2010). This conclusion was based on data of very low quality.
16.1. Analysis.
Comparison 16 Comparison 16: LY686017 versus placebo, Outcome 1 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms.
Comparison 17: total effect of medication versus placebo
There was evidence of an effect across all medication classes (k = 51, RR 1.62; 95% CI 1.46 to 1.81, N = 8564), with substantial heterogeneity across subgroups (Chi2 = 42.93, df = 13, P < 0.0001; I2 = 69.7%). For this analysis we excluded data from two studies with additional arms of paroxetine to give preference to the least‐represented intervention in the review (i.e. GW876008 and GR205171; Furmark 2005; NCT00397722). We also excluded data from Liebowitz 1992, a phenelzine study, and from Liebowitz 2005b, a paroxetine study, to give preference to atenolol and venlafaxine (see Analysis 17.1). There was evidence that medication class explained a substantial proportion of variability in treatment efficacy when assessed across all trials providing data on this outcome (Chi2 = 143.17, df = 50, P < 0.00001; I2 = 65%; see Analysis 17.1).
17.1. Analysis.
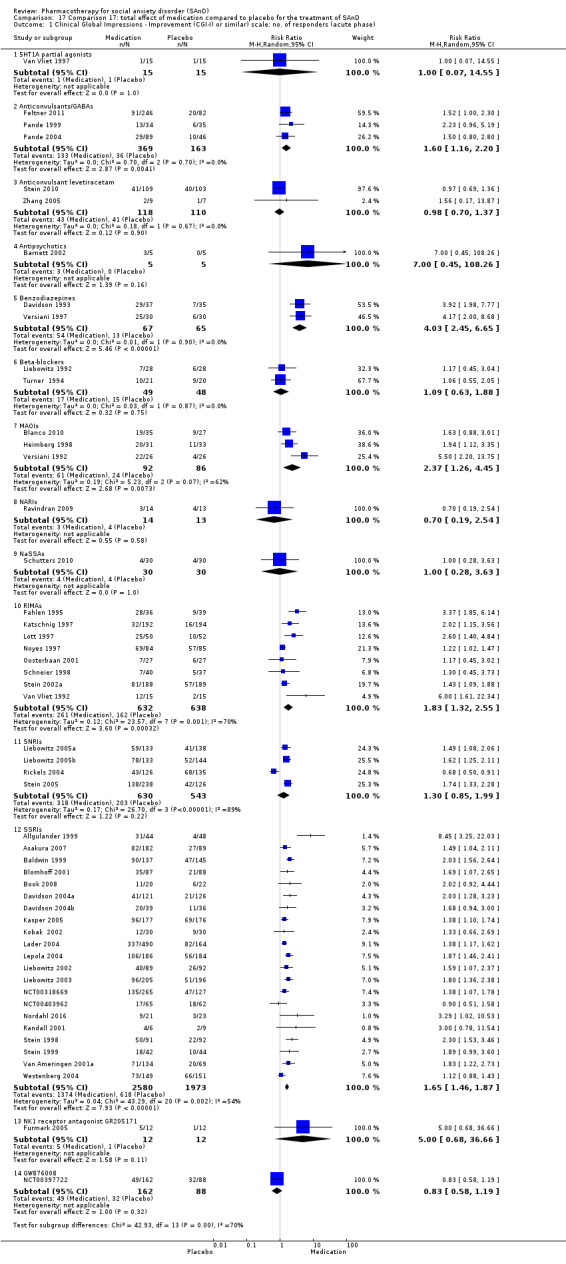
Comparison 17 Comparison 17: total effect of medication compared to placebo for the treatment of SAnD, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar) scale: no. of responders (acute phase).
The proportion of dropouts due to adverse events was two to three times higher across all medication classes compared to placebo (k = 45, RR 2.41; 95% CI 1.99 to 2.91, N = 8751), a statistically significant difference (P < 0.001; Chi2 = 49.24, df = 44, P = 0.27; I2 = 11%). Nevertheless, the total proportion of dropouts due to adverse events in the medication arms (12%) is relatively low, suggesting that dropout rates may not have biased the outcomes. For this analysis we excluded data from two studies reporting data for paroxetine, as these studies also report data for the GW876008 and the NK1 receptor antagonist GR205171 (Furmark 2005; NCT00397722).
In addition, the proportion of dropouts due to any cause across both the medication and placebo groups was similar (25%) and was comparable across subgroups (Chi2 = 13.31, df = 13, P = 0.42; I2 = 2.3%). For this analysis we excluded data from one study with an additional arm of paroxetine to give preference to the experimental intervention, GR205171 (NCT00397722; see Analysis 17.3).
17.3. Analysis.
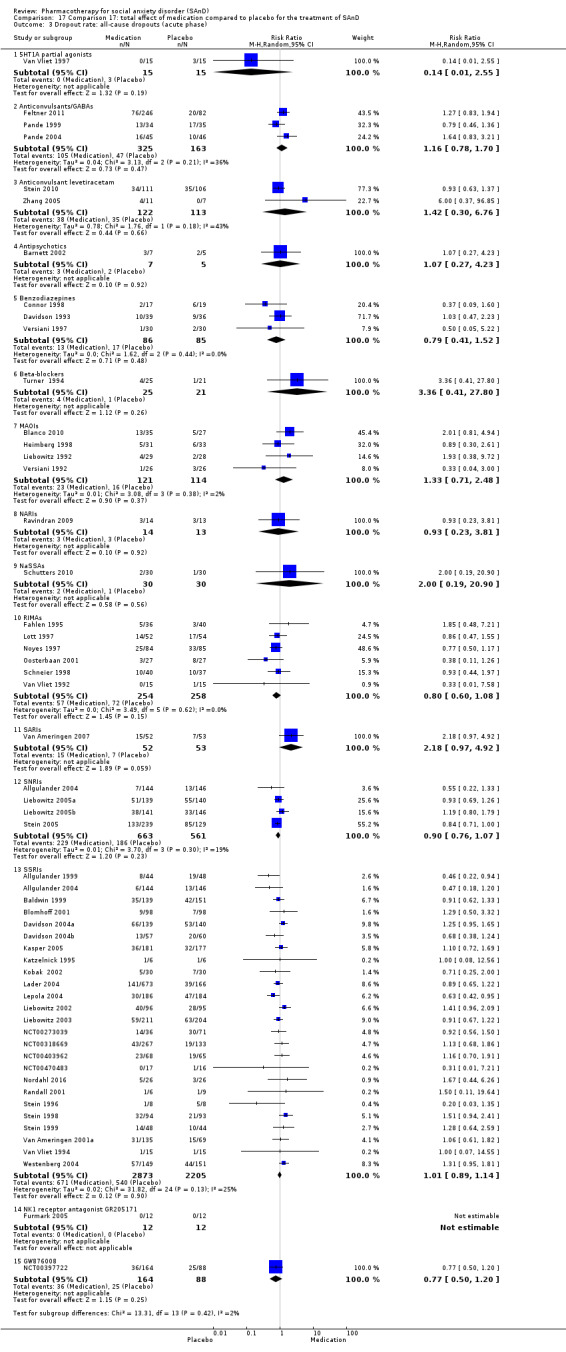
Comparison 17 Comparison 17: total effect of medication compared to placebo for the treatment of SAnD, Outcome 3 Dropout rate: all‐cause dropouts (acute phase).
18. Subgroup analyses
There was evidence of differences between RCTs conducted at multiple centres versus single centres (Chi2 = 7.05, df = 1, P = 0.008; I2 = 85.8%; see Analysis 18.1), with larger treatment effects reported for studies conducted at single centres (k = 20, RR 2.24, 95% CI 1.67 to 3.02, N = 1332).
18.1. Analysis.
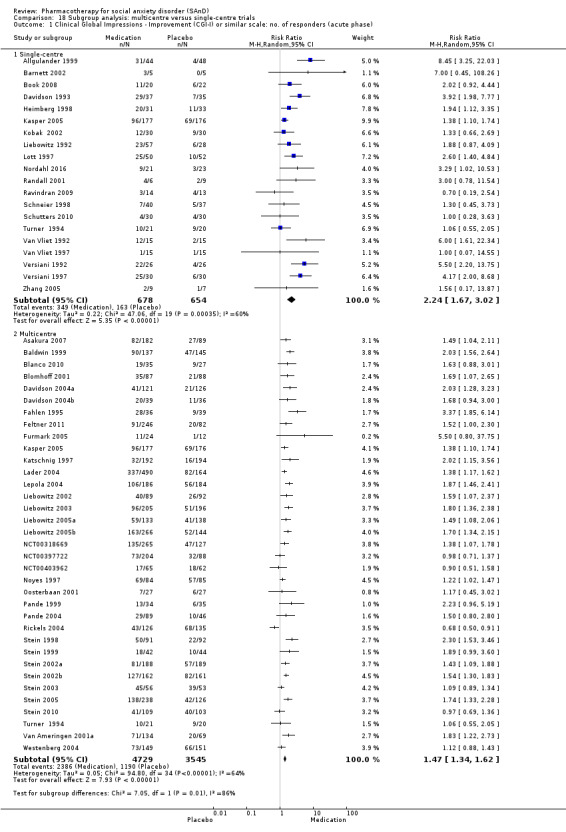
Comparison 18 Subgroup analysis: multicentre versus single‐centre trials, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale: no. of responders (acute phase).
There was no evidence of a difference in treatment response based on source of funding (Chi2 = 1.52, df = 1, P = 0.22; I2 = 34.1%; see Analysis 20.1) or the inclusion of participants diagnosed with major depressive disorder (Chi2 = 1.70, df = 1, P = 0.19; I2 = 41.1%; Analysis 21.1).
20.1. Analysis.
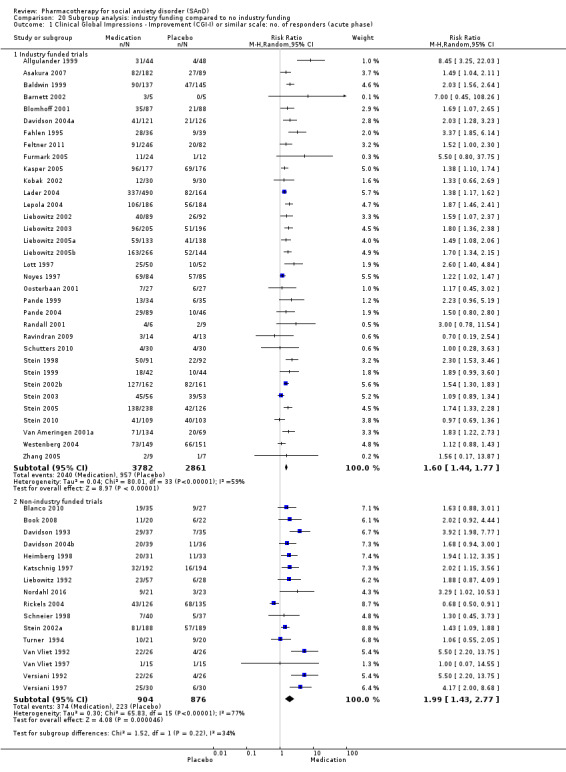
Comparison 20 Subgroup analysis: industry funding compared to no industry funding, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale: no. of responders (acute phase).
21.1. Analysis.
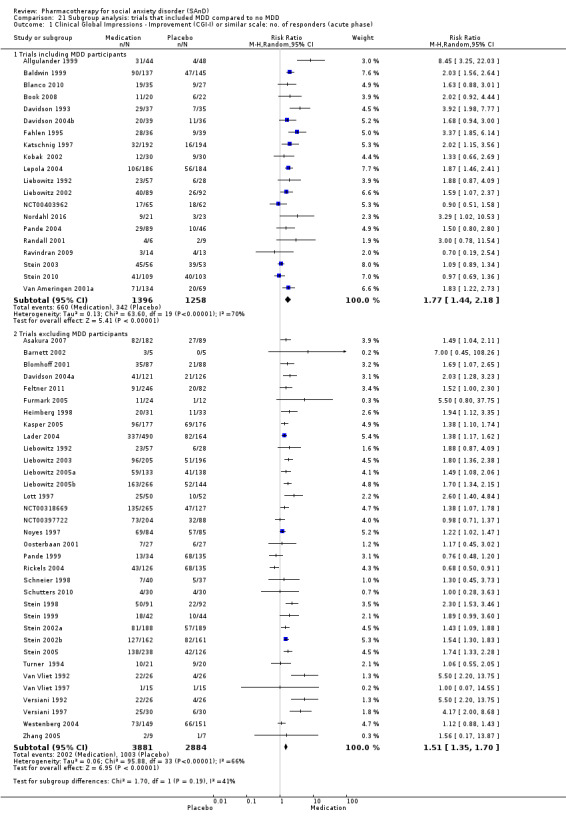
Comparison 21 Subgroup analysis: trials that included MDD compared to no MDD, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale: no. of responders (acute phase).
Studies that involved only participants diagnosed with generalised SAnD reported smaller treatment effects (k = 26, RR 1.49, 95% CI 1.31 to 1.69, N = 5522) than trials that did not necessarily restrict inclusion to the generalised subtype (k = 27, RR 1.83, 95% CI 1.54 to 2.18, N = 3712; see Analysis 19.1). This difference was statistically significant (P < 0.001).
19.1. Analysis.
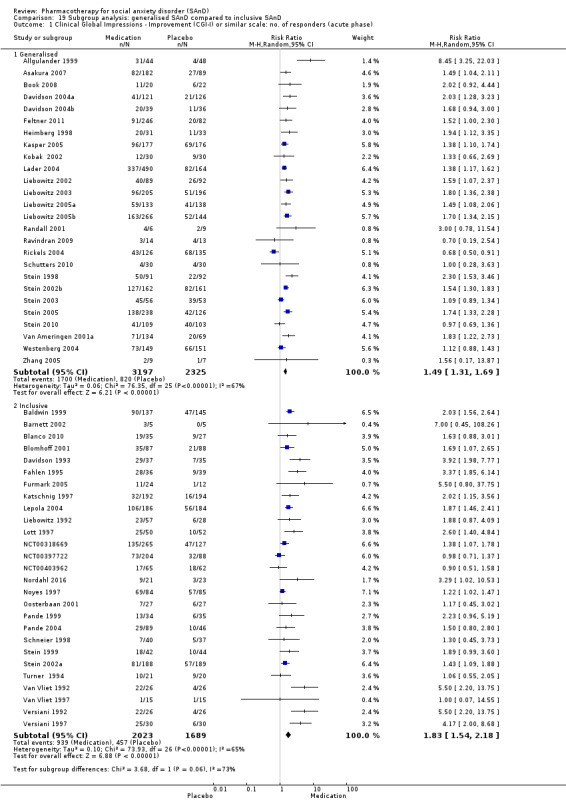
Comparison 19 Subgroup analysis: generalised SAnD compared to inclusive SAnD, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale: no. of responders (acute phase).
19. Sensitivity analyses
Twenty‐four parallel RCTs failed to include all of the participants randomised to the treatment groups in the responder analysis. The comparison of separate efficacy analyses in which the omitted participants were regarded as responders in either the medication or placebo groups supported the robustness of the evidence of this agent's efficacy in treating social anxiety disorder (respective results: RR 1.59, 95% CI 1.41 to 1.80, P < 0. 001, N = 5437, Analysis 22.1; and RR 1.63, 95% CI 1.44 to 1.85, P < 0. 001, N = 5215; Analysis 22.2). We therefore consider that missing data in these trial reports did not have a significant influence on this outcome.
22.1. Analysis.
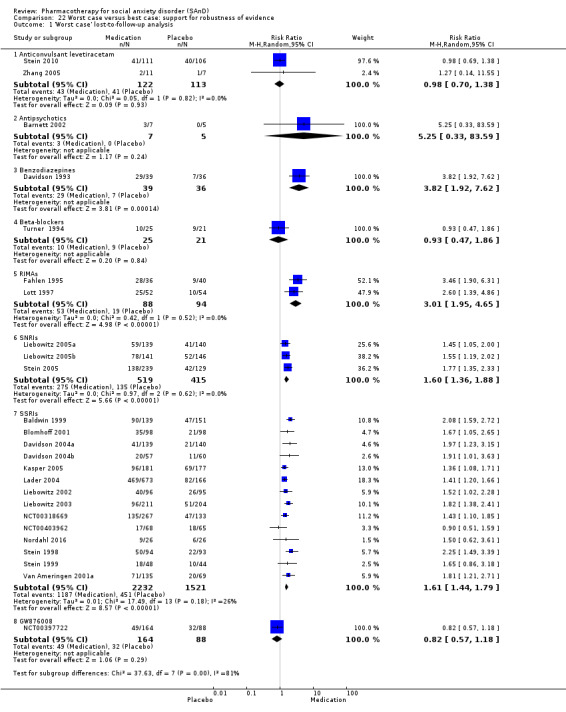
Comparison 22 Worst case versus best case: support for robustness of evidence, Outcome 1 'Worst case' lost‐to‐follow‐up analysis.
22.2. Analysis.
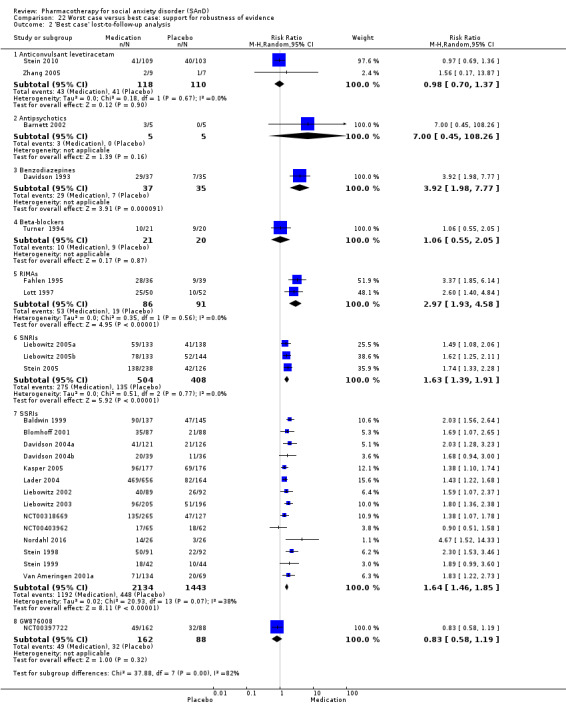
Comparison 22 Worst case versus best case: support for robustness of evidence, Outcome 2 'Best case' lost‐to‐follow‐up analysis.
20. Publication bias
There is evidence of possible funnel plot asymmetry providing data on response to short‐term medication treatment, both for the SSRIs and all medications combined. Inspection of the contour enhanced funnel plots for the SSRIs (Figure 4) and all of the trials (Figure 5) suggests that this asymmetry is due to publication bias, as trials with less precise treatment response outcomes are more likely than their higher precision counterparts to be missing from regions of the plot representing statistically non‐significant treatment effects. Egger regression tests quantitatively confirmed this visual impression, providing evidence of possible publication bias for all of the medication trials (t = 2.8226, df = 49, P = 0.0069) and for the SSRIs (t = 2.6426, df = 22, P = 0.015). Nevertheless, trim and fill imputation of missing effect estimates from small studies with negative results continue to provide support for the efficacy of both the SSRIs (RR 1.52, 95% CI 1.35 to 1.71) and all medications (RR = 1.48, 95% CI 1.32 to 1.66) in treating SAnD. There were insufficient data to assess publication bias for the other medication classes.
4.
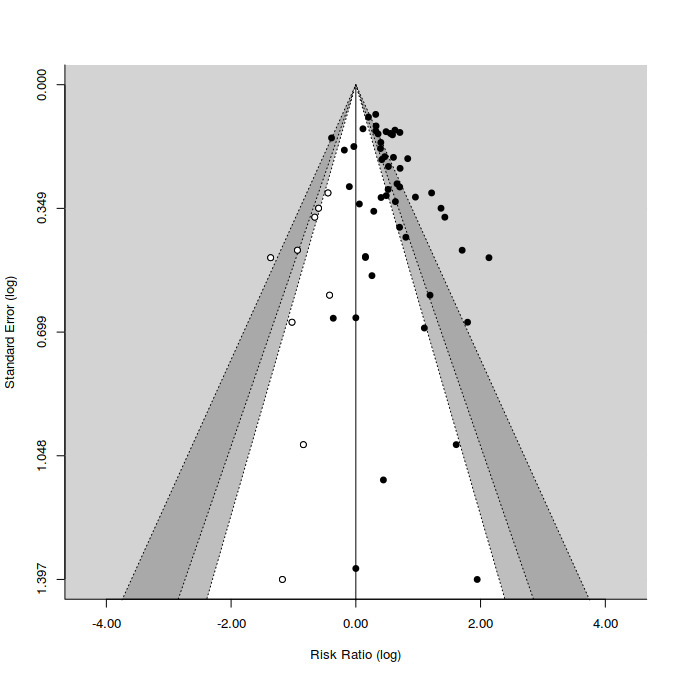
Contour enhanced funnel plot for treatment response on the CGI‐I for all medication trials Filled circles = trial effect estimates; empty circles = trim and fill generated missing study estimates Contours in light‐gray, gray and dark gray correspond to increasing stringent statistical boundaries (alpha = 0.1, 0.05, and 0.01, respectively). White corresponds to regions of statistical non‐significance.
5.
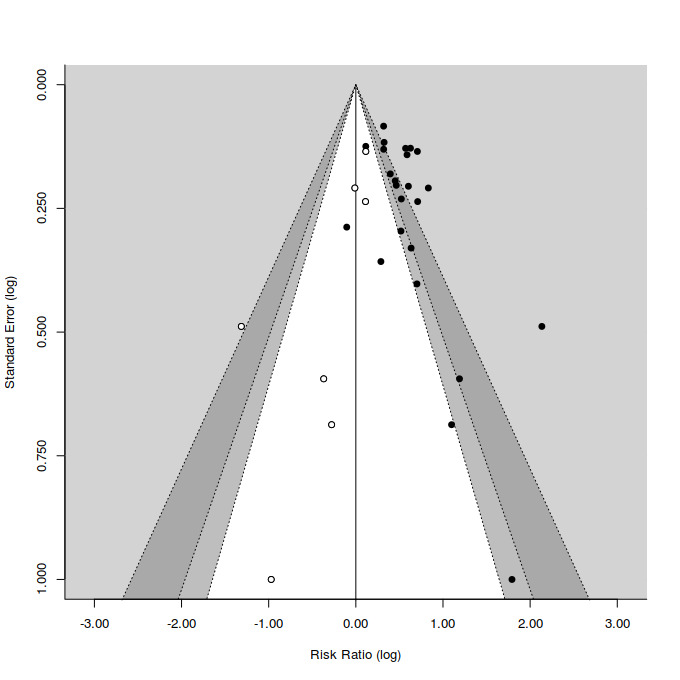
Contour enhanced funnel plot for treatment response on the CGI‐I for SSRI trials Filled circles = trial effect estimates; empty circles = trim and fill generated missing study estimates Contours in light‐gray, gray and dark gray correspond to increasing stringent statistical boundaries (alpha = 0.1, 0.05, and 0.01, respectively). White corresponds to regions of statistical non‐significance.
Discussion
Summary of main results
We found evidence of an overall benefit of medication for treating SAnD in this review. Most evidence was for the SSRIs (i.e. paroxetine, fluvoxamine, sertraline, fluoxetine, and citalopram) and was very low in quality. Similarly, we found low‐quality evidence of efficacy for the MAOI phenelzine, the RIMAs brofaromine and moclobemide, and the benzodiazepines clonazepam and bromazepam, with moderate‐quality evidence of efficacy observed for the anticonvulsant/GABAs gabapentin and pregabalin. Conversely, there was no evidence in this review of a global clinical response to treatment for certain medication classes, including the 5HT1A partial agonists, the anticonvulsant levetiracetam, the antipsychotic olanzapine, the beta‐blockers, the NARIs, the NaSSAs, or the SNRI venlafaxine, as well as for the experimental agents GW876008 and NK1 receptor antagonist GR205171.
This review also found evidence for an effect of the SSRIs in preventing relapse. Individuals diagnosed with SAnD who were treated with either SSRIs or the SNRI venlafaxine were more likely to withdraw from treatment due to treatment‐related adverse events. Nevertheless, the absolute proportion of individuals dropping out from treatment with these medications due to adverse events over the short term (14 weeks or less) was low (12% for SSRIs and 16% for venlafaxine). There was little evidence that any of the SSRI agents were less tolerable than any other (I2 = 23%), as confirmed by a post hoc comparisons of dropout rates due to adverse events by agent (Chi2 = 31.82, df = 24, P = 0.13; I2 = 25%). The MAOIs and benzodiazepines appeared to be relatively well tolerated and also showed the largest treatment response (see Table 5; Table 7).
We found sufficient evidence of efficacy for reducing depression symptoms only observed for the SSRIs and RIMAs. Nevertheless, the possibility that response to treatment with these agents is entirely accounted for by their anti‐depressant effect is countered by evidence (albeit of very low quality) in this review that the SSRIs and RIMAs are effective in reducing SAnD symptoms (in addition to the MAOIs, the benzodiazepines, the SNRI venlafaxine, the antipsychotic olanzapine, and the NaSSA mirtazapine). Moreover, although trials of the MAOIs, SARIs, and benzodiazepines demonstrated evidence of efficacy of these agents in improving functioning in some domains, the SSRIs were the only medications showing a treatment effect on all three subscales (i.e. work, social, and family) of the SDS. It was on the basis of evidence such as this that the US FDA approved the SSRIs paroxetine, sertraline, and fluvoxamine for treating SAnD – the only agents that currently enjoy this status (Blanco 2013).
We found a response to long‐term treatment (20 to 24 weeks) with medication for the SSRIs (based on low‐quality evidence), the MAOIs (based on very low‐quality evidence), and for the RIMAs (based on moderate‐quality evidence). A similar proportion of participants withdrew due to treatment‐emergent side effects from the paroxetine, fluvoxamine, and placebo groups after long‐term treatment. Readers should exercise great caution in any inferences about the experimental agents (GW876008, the NK1 antagonist GR20517 and LY686017), more trials with each of these agents in the future may allow better comparison of classes of medication.
For the subgroup outcomes, there was evidence that treatment response was influenced by whether RCTs took place at single or multiple centres. The size of the effect observed was also dependent on participant characteristics, with a smaller response to treatment observed in studies that restricted inclusion to participants diagnosed with generalised SAnD. Nevertheless, there was no evidence that treatment response across all medication classes varied as a function of whether trials were funded by industry or included participants diagnosed with major depressive disorder. There is also reason to believe that the effect sizes reported across all medication classes, as well as for the SSRIs, may be inflated, based on properties of the data that are consistent with publication bias. Nevertheless, the effects of medication in general and the SSRIs in particular on treatment response persist even after adjustments to account for the effect of the increased likelihood of publishing small trials if they report a positive effect of medication.
Overall completeness and applicability of evidence
Completeness of evidence
Reviews of the body of evidence for pharmacotherapy of SAnD have designated SSRIs as first‐line medication agents for treating SAnD (Baldwin 2014; Ballenger 1998; Bandelow 2002; NICE 2011). The current review supports the efficacy of SSRIs for short‐term treatment of SAnD. Similarly, maintenance studies demonstrate some further consolidation of clinical response to SSRIs over time.
Importantly, although there was evidence for treatment withdrawal owing to drug‐related adverse events in the SNRI and SSRI trials, dropout rates were comparable between these medication classes. This contradicts findings in a meta‐analysis of the depression literature, which found venlafaxine to be more efficacious than SSRIs in increasing remission rates, but with a higher rate of withdrawal owing to treatment‐related adverse events (Nemeroff 2008). Given the frequent prescription of beta‐blockers for performance anxiety, the relative lack of empirical support for this intervention is surprising. We also found evidence of heterogeneity for the MAOIs, the RIMAs, and the SSRIs when investigating the efficacy of treatment in participants with SAnD. These differences may be partly accounted for by methodological and clinical variance but may also reflect real differences in efficacy across different medications. None of the four trials of the MAOI phenelzine provide information on the number of participants who withdrew from treatment as a result of drug‐related adverse events, against which to weigh the observed evidence for a response to treatment with this agent. There has been no investigation into the efficacy of other medication classes that may be considered for treating SAnD, such as the tricyclic antidepressants (TCAs).
Although the SSRI trials under review used medication doses that are consistent with those advised in recent expert consensus recommendations (Ballenger 1998), there are few rigorous dose‐finding studies using fixed‐dose comparisons. The paroxetine database suggests that the dose‐response curve of SSRIs is relatively flat in SAnD, but that some participants may require higher doses (Ballenger 1998). Most SSRI studies lasted at least 12 weeks. The finding from the paroxetine database that a significant proportion of non‐responders at week 8 become responders by week 12 suggests that patients in clinical practice should be treated for a minimum period of 12 weeks before modifying treatment regimens due to medication non‐response (Stein 2002c). Similarly, the maintenance studies demonstrate some further consolidation of clinical response over time in SAnD.
The negative 14‐week trial of fluoxetine raises the question of whether all SSRIs are equally effective (Kobak 2002). The fluoxetine study included in this review had a relatively high placebo response rate and was not consistent with several early open‐label reports of the value of this medication in SAnD. Further work is needed to understand the differences in placebo response across the database of SAnD studies (Oosterbaan 1997). In a study by Katschnig 1997, for example, placebo response was higher in participants without avoidant personality disorder, in participants with a short duration of illness, and in participants with less severe illness. Kobak 2002 found that placebo‐responders had an earlier response than medication responders and that lower severity of illness was associated with increased placebo response. These findings were similar to a recent report on two meta‐analyses where bias (i.e. reporting bias) appeared to have led to significant increases in the number of positive findings in the literature (Roest 2015).
Applicability of evidence
The outcomes of this review may be generalisable to a diverse range of settings. Studies were conducted in the USA, Europe, Japan, South Africa, and Iran, in both outpatient and inpatient settings (mainly outpatient settings). The interventions targeted both men and women across a wide age range. Differences in treatment delivery, as well as the background and training of study investigators and outcome assessors, increases the likelihood that the findings of this review are applicable to a range of countries in different income brackets. However, careful consideration should be given to the specific contexts in which the trials took place, given the lack of evidence on the availability and cost‐effectiveness of individual medications. The findings of this review will not apply equally to the full range of individuals presenting at clinics for the treatment of SAnD. RCTs included in this review were also restricted to those assessing the effectiveness of pharmacotherapy for SAnD in adults. The finding in this review that clinicians should consider medication, and in particular the SSRIs, for treating SAnD, may not apply to children. Readers are referred in this regard to another Cochrane Review on the efficacy of pharmacotherapy for anxiety disorders in children and adolescents (Ipser 2009a). Most published trials of pharmacotherapy for SAnD exclude individuals with concurrent psychopathology, including substance use disorders, which are highly comorbid with SAnD. Although the limited RCT database suggests that medication may be equally effective in people with SAnD who are dependent on alcohol (Book 2008), and certain forms of comorbidity may actually be prognostic of greater treatment response (Stein 2002a), firm evidence supporting conclusions regarding whether medication is indicated for these populations is currently lacking.
Quality of the evidence
Twelve of the included RCTs possessed a high risk of bias related to at least one aspect of study design, with the most commonly observed weaknesses having to do with attrition bias (Allgulander 1999; Book 2008; Katzelnick 1995; Liebowitz 1992; Moghadam 2015; Nordahl 2016; Stein 2005; Van Ameringen 2007; Walker 2000; Zhang 2005). Lack of blinding for outcome assessors may also have influenced the detection of treatment effects (Blomhoff 2001; Moghadam 2015; Stein 2005; Van Ameringen 2007). There was also evidence for selective reporting bias in Book 2008, Nordahl 2016 and Tauscher 2010. Even though the SSRIs were the best‐represented medication class, with 24 of the 51 RCTs contributing to the outcome of clinical response, most of these trials scored unclear for random sequence generation as well as for allocation concealment.
Judgements of response to treatment with the SSRIs were based on evidence rated as being of very low quality. Therefore, we are very uncertain about the estimate (see Table 13). The RIMAs, MAOIs, and benzodiazepines were based on evidence rated as being of low quality (see Table 5; Table 7), indicating that additional data from further studies may change the size of the treatment effect estimate for these medication classes, as well as our confidence in that estimate of effect. The anticonvulsants/GABAs, on the other hand, were based on evidence rated as being of moderate quality (see Table 2; Table 12). Further research is very unlikely to change our confidence in the estimate of treatment effect for these medication classes. The most common reasons for downgrading studies were imprecise effect estimates, low response rates, small sample size, a rating of high or unclear bias for study design (based on 50% attrition rates across groups, detection, and selective reporting bias), and inconsistency between studies (indicated by heterogeneity between the medication and placebo group).
Variability in the number of sites contributing data reported in included studies could also have biased the results of the review. A recent meta‐analysis demonstrated that the size of treatment effect estimates are larger in single‐centre than multicentre trials, even after adjusting for sample size and various bias risk factors (Dechartres 2011). It is notable that approximately a third of the trials providing data on treatment response for this review were in single centres. Moreover, those trials were over‐represented amongst RCTs of SSRIs and in studies reporting the largest treatment effects, including more than half of the MAOI trials and both benzodiazepine RCTs. Additionally, there was a smaller treatment response in RCTs that explicitly described limiting study inclusion to people diagnosed with generalised SAnD. This finding is in agreement with the perception that the generalised SAnD subtype is more difficult to treat. It may once again have resulted in a positive bias for the MAOI, RIMA, and benzodiazepine studies in particular, given that with the exception of a single trial of phenelzine (Heimberg 1998), all of the included RCTs for these medication classes failed to specify that they restricted inclusion to the generalised subtype.
The size of the response to acute treatment with any medication and specifically with the SSRIs may be exaggerated, as possible funnel plot asymmetry for this outcome suggests (Figure 4; Figure 5). Egger regression tests quantitatively confirmed the presence of small sample bias. Although small sample bias may arise from other factors besides publication bias, the absence of smaller studies with statistically non‐significant effects of treatment, as illustrated by contour‐enhanced funnel plots (Figure 4; Figure 5), does suggest that investigators are not publishing smaller studies that fail to detect treatment effects.
Potential biases in the review process
We conducted an extensive search for studies meeting rigorous inclusion criteria and repeatedly attempted to obtain missing data from the trial investigators. Nevertheless, there were insufficient data available to assess the extent to which selective publication may have introduced bias in the findings for medication classes besides the SSRIs. Furthermore, the post hoc addition of comparisons of the efficacy of 5HT partial agonists, anticonvulsants with or without GABAs, antipsychotics, NARIs, NaSSAs, and SARIs may have also introduced bias as a result of the small number of eligible RCTs for these medication classes (1 to 3 studies).
The reliance on the LSAS, while considered the gold standard for measurement of SAnD symptom severity in clinical trials, represents another potential source of bias. Treatment outcomes not assessed by standardised scales such as the LSAS also deserve more attention. For example, physiological symptoms are often a source of concern for people (symptoms such as excessive sweating may for example lead to a request for surgery), and the LSAS does not specifically record these (Davidson 1998).
Meta‐analysis also has inherent problems (Bailar 1999); although indirect comparison of competing interventions usually agree with direct ones (Song 2003), meta‐analysis is by no means a substitute for direct clinical research. Furthermore, the context of general clinical practice differs from RCTs in specialised centres in many respects, not the least being the need to treat more complex participants. In particular, conclusions about the relative efficacy of different agents from trials with different participants require confirmation in head‐to‐head comparisons. An early head‐to‐head study of clomipramine versus diazepam suggested superiority of clomipramine, but there were relatively few participants with social phobia (Allsopp 1984). The only head‐to‐head comparison of an SSRI versus moclobemide did not include a placebo control but did suggest equal efficacy and tolerability, lending caution to the findings of the current review (Atmaca 2002).
Agreements and disagreements with other studies or reviews
The finding in this review of moderate to substantial variability in the response of participants with SAnD to pharmacotherapy is likely to reflect real differences in efficacy across different medication and differences brought on by trial methodology and the clinical characteristics of patients. In comparing medication classes in terms of beneficial and adverse responses to medication, the data included in this review are consistent with findings from other systematic reviews and meta‐analyses in identifying the SSRIs as first line agents for the treatment of SAnD (Ballenger 1998; Bandelow 2002; Bandelow 2012; Bandelow 2015; Blanco 2003; Van der Linden 2000). The finding of efficacy for the SSRIs reported in this review is based on a considerably larger database of randomised controlled trials than in previous reviews and reports.
This review classified medications based on their putative mechanisms of action (taken from CCMD antidepressant classification map) (Davies 2015), and they do not necessarily map onto the drug classification schemes employed in other reviews. Nevertheless, we are able to confirm the efficacy of the SSRIs, SNRIs, the RIMAs, and the anticonvulsants/GABAs.
MAOIs, in the form of phenelzine, as well as certain benzodiazepines, are also effective in SAnD, but in view of concerns about ease of administration (e.g. MAOIs require dietary and medication restrictions) and side effects (risk of abuse with benzodiazepines seems to be highest in individuals with a history of substance abuse (Licata 2008)), it does not seem reasonable to view these drugs as second‐line agents.
Authors' conclusions
Implications for practice.
Medication can be effective for the treatment of SAnD, with higher rates of treatment response compared to placebo, as well as reductions in core SAnD symptoms and associated disability. Most evidence of efficacy in this review was for the SSRIs. Although there was evidence that treatment with SSRIs was less tolerable than placebo, the absolute proportions of participants withdrawing due to drug‐related adverse events after treatment with these agents were small (< 17%). Moreover, the SSRIs were the only medication class that demonstrated consistent evidence of reductions in functional disability across a number of domains. The possible influence of publication bias on the validity of conclusions regarding the efficacy overall, and the SSRIs in particular, in treating SAnD, as well as the potential risk of bias for this outcome, needs to be acknowledged. This review also found preliminary evidence to support the use of the anticonvulsants/GABAs in treating SAnD. Readers should exercise great caution in making any inferences about the experimental agents GW876008 and NK1 receptor antagonist GR205171; more trials with each of these agents in the future may allow better comparison of classes of medication.
Treatment efficacy of medication was significantly larger in trials conducted at single centres than multiple centres, as well as when administered to participants described as being diagnosed with generalised SAnD. The findings that both RIMAs and benzodiazepines were efficacious and tolerable in treating SAnD therefore needs to be interpreted in light of the observation that none of the included trials for these medications reported restricting inclusion to the generalised SAnD subtype. Moreover, the RIMAs are not available in clinical practice, while findings from the small number of trials of benzodiazepines must be weighed against concerns regarding their adverse effects and their lack efficacy for common comorbid conditions that occur with SAnD (Ford 2014; Licata 2008).
Trials took place in a diverse range of settings and with heterogenous samples and should therefore currently be considered in a broad spectrum of SAnD participants. Nevertheless, we recognise that certain forms of the disorder, such as those consisting of 'pure' public speaking fears, were not well represented in this review. We observed continued response to medication in trials extending beyond 12 weeks in duration, supporting the consensus that medication responders should continue to be treated over the long term. Although beta‐blockers are often recommended for the treatment of performance anxiety, there was insufficient evidence in this review to support the use of this medication class for people without the generalised type of SAnD.
Implications for research.
Differences in the efficacy of different medications deserve more attention; there are few studies directly comparing modern agents with one another. In addition, there is a paucity of RCTs evaluating the efficacy of medications for treating SAnD in people with comorbid disorders, including substance use disorders, and in people in general psychiatric and medical settings. Studies of these populations could address the question of whether early and robust pharmacotherapy of SAnD can prevent subsequent morbidity and comorbidity. Further attention should also be paid to people who fall on a putative SAnD spectrum of disorders, including people with performance anxiety. Finally, commonly used symptom outcomes in SAnD have limitations, with more refined measures of response to treatment warranting further investigation. This would help inform the concept of remission in SAnD.
Additional work is also needed to determine the best approach to people who fail to respond to pharmacotherapy; there are relatively few studies of treatment augmentation (Ipser 2009b; Pecknold 1982; Van Ameringen 1996; Stein 2001; Aarre 2003) or pharmacotherapy switching (Kelsey 1995). The current review also does not directly address the question of whether pharmacotherapy or psychotherapy has a larger effect size in SAnD (Gould 1997). In clinical practice, clinicians often use exposure instructions (Gelernter 1991), while theoretically, modern understanding of SAnD as involving psychobiological dysfunctions would indicate that it is unnecessary to institute false dichotomies between brain and mind, and that both kinds of intervention might be useful (Furmark 2002). Therefore, another research priority revolves around the combined use of pharmacotherapy with psychotherapy and when and if these treatments should be combined or sequenced.
Feedback
Formal mistake
Summary
The word 'bias' is missing in line 4 of the section 'Authors' conclusions' in the abstract.
I certify that I have no affiliations with or involvement in any organisation or entity with a direct financial interest in the subject matter of my criticisms.
Reply
We thank Dr Mate for his feedback and have amended the abstract accordingly.
Contributors
Dr. Christian Maté A user Email christan.mate@netcare.at Date Received 21/10/2004 19:35:10
Feedback on Pharmacotherapy for Social Phobia , 17 August 2015
Summary
Although the authors caution against "the possibility of publication bias", they conclude that drugs, in particular SSRIs, appear effective. I don't agree. The trials were of very poor quality, and the effect decreased so dramatically with the number of patients in the trial that any meta‐analysis of these data would be grossly unreliable. The authors nonetheless meta‐analysed their data and reported a relative risk of non‐response of 0.64 (95% CI 0.57 to 0.73) on the Global Impression Scale. But they also showed in a figure that the largest trials found an effect close to zero.
Another problem is that all the scales appear to have been rated by the clinicians and not the patients, which is known to create a large bias in trials of SSRIs. For depression trials, the standardised mean difference in trials that had both psychiatrists and patients as observers was around 0.25 when the psychiatrists evaluated the effect but only 0.05 when the patients were their own judges (1).
The review also reported an effect in relapse prevention studies, but such trials are highly unreliable because abstinence symptoms are introduced in the placebo group when the patients come off their drug cold turkey (1). An additional problem is that many trials use the last observation carried forward, which would be expected to bias these trials further since the patients' abstinence symptoms can be depression and anxiety, causing them to drop out, against which the effect of the active drug is judged. This is an unfair comparison.
In my opinion, neither SSRIs, nor benzodiazepines should be used for social phobia. Psychotherapy works and the patients need to learn how to cope with their anxiety rather than being emotionally numbed by drugs that many have difficulty stopping again, as they become dependent on them (1).
I agree with the conflict of interest statement below:
I certify that I have no affiliations with or involvement in any organization or entity with a financial interest in the subject matter of my feedback.
1. Gøtzsche PC. Deadly psychiatry and organised denial. Copenhagen: People's Press; 2015.
Reply
Prof. Gøtzsche recently commented on our Cochrane review of pharmacotherapy for social phobia, published in 2006. While we have responded to his comments below, we would like to note that the review in question is currently in the process of being updated to conform to the most recent Cochrane standards regarding assessment of trial quality using the risk of bias tool and GRADE quality ratings.
It is true that the quality of the reporting of some of the included studies was poor, as assessed using the CCMD Quality of Research Scale. This was particularly apparent amongst the earlier and smaller studies, the same studies that contributed towards evidence for possible publication bias on the CGI‐I. It should be noted, however, that the weight assigned to data from individual studies in deriving the overall treatment effect estimate for CGI‐I non‐response is proportional to the size of their samples. As a result, the less reliable effect estimates from smaller studies have a relatively minor influence on the overall treatment effect. This can be appreciated when one considers that removing the half of the 26 studies contributing towards this outcome with the smallest sample sizes has minimal effect on the total effect estimate (Risk Ratio = 0.68, 95% CI = 0.60 to 0.78). It should also be noted that the majority of the remaining trials (9/13), each of which contain at least 40 participants in each treatment arm, compared the efficacy of SSRIs to placebo. These findings suggest that evidence for the efficacy of medication in treating social anxiety disorder is relatively robust.
The peer‐reviewed literature suggests that self‐rated and clinician rated SAD symptom severity measures are highly concordant (1), and that these measures might perhaps best be thought of as providing different kinds of data with respect to treatment response, at least within the depression literature (2). Alluding to lower outcome effect estimates as indicative of less susceptibility to bias presumes a level of knowledge about the true efficacy of medication in treating psychiatric disorders that we do not currently possess. Indeed, one of the primary motivations for conducting our review in the first place was to arrive at a best estimate of the effect of medication in treating SAD, using data from trials that are optimally designed to be informative with respect to their efficacy. Preference was given to clinician‐rated instruments in our review, as they have been more consistently employed across treatment studies, thereby facilitating meta‐analytic synthesis of data provided by these studies.
Three of the four relapse prevention trials included in the analysis referred to by Prof. Gøtzsche employed gradual step‐wise down‐titration of doses to minimise the possibility of abstinence symptoms. For instance, Connor et al. (1998) describe employing "a fixed‐dose taper of 0.25 mg every 2 weeks" of clonazepam, so that it took between 6 and 18 weeks to take participants completely off medication. In the single study where participants were abruptly discontinued from sertraline (Walker et al. 2000), the authors report that only "two patients (8%) in the Placebo‐Switch group discontinued in the 2 weeks following abrupt discontinuation of sertraline because of adverse events that may have been attributable to discontinuation reactions". While we do not agree that relapse prevention studies are highly unreliable, it is certainly important that withdrawal effects be considered and addressed.
The evidence indicates that certain forms of psychotherapy are effective in social anxiety disorder, although they arguably involve a good deal more than learning to cope. We are not aware of a great deal of evidence that medications lead to emotional numbing in social anxiety disorder. While benzodiazepines can certainly be associated with withdrawal effects, the relapse prevention studies in SAnD indicate relatively few adverse events with SSRI withdrawal. We would note that in settings such as our own, it was estimated by the World Health Organization that in 2011 there was one psychologist for roughly 300 000 people (http://www.who.int/mental_health/evidence/atlas) and that even in well‐resourced developed nations, individuals with SAnD face barriers in accessing or engaging with psychotherapy.
1. Fresco, DM.; Coles, ME.; Heimberg, RG.; Liebowitz, MR.; Hami, S.; Stein, MB. & Goetz, D. (2001). The Liebowitz Social Anxiety Scale: a comparison of the psychometric properties of self‐report and clinician‐administered formats. Psychol Med, 31, 1025‐1035
2. Uher R, Perlis RH, Placentino A, Dernovšek MZ, Henigsberg N, Mors O, Maier W, McGuffin P, Farmer A. (2012). Self‐report and clinician‐rated measures of depression severity: can one replace the other? Depress Anxiety, 29(12):1043‐9
Contributors
Feedback submitted by: Peter C Gøtzsche, Nordic Cochrane Centre
Response submitted by: Jonathan C Ipser, Dan J Stein, Taryn Amos
Feedback submitted, 24 April 2018
Summary
There seems to be a mistake in the following paragraph of the abstract:
'We assessed tolerability of SSRIs and the serotonin and norepinephrine reuptake inhibitor (SNRI) venlafaxine on the basis of treatment withdrawal; this was higher for medication than placebo (SSRIs: k = 24, RR 2.59; 95% CI 1.97 to 3.39, N = 5131,low‐quality evidence; venlafaxine: k = 4, RR 3.23; 95% CI 2.15 to 4.86, N = 1213, moderate‐quality evidence), but there were low absolute rates of withdrawal for both these medications classes compared to placebo.'
According to the data presented, the tolerability of the medication is not higher but lower compared with placebo.
Reply
We would like to thank Hilde Habraken for this helpful feedback on our Cochrane review. We have amended this paragraph in the abstract to avoid any ambiguity.
Contributors
Feedback submitted by: Hilde Habraken, Researcher at the Belgian Center for Pharmacotherapeutic Information (BCFI)
Response: Taryn Williams and Dan Stein (Authors)
What's new
| Date | Event | Description |
|---|---|---|
| 25 January 2019 | Feedback has been incorporated | Feedback has been added, along with a response from the authors |
| 25 January 2019 | Amended | Small change made to the abstract in response to feedback to avoid ambiguity |
History
Protocol first published: Issue 1, 1998 Review first published: Issue 4, 2004
| Date | Event | Description |
|---|---|---|
| 2 August 2017 | New citation required and conclusions have changed | Search updated August 2017; 37 RCTs added. Summary of findings tables added. |
| 14 September 2015 | Amended | Feedback incorporated |
| 4 November 2008 | Amended | Converted to new review format. |
| 20 July 2000 | New citation required and conclusions have changed | Substantive amendment |
Acknowledgements
We would like to thank the Medical Research Council (Cape Town, South Africa) for its financial support, Lize van der Merwe for statistical guidance, as well as Satoshi Asakura, David Baldwin, Jonathan RT Davidson, Tomas Furmark, Richard G Heimberg, Moritz Muehlbacher, Franklin R Schneier, and John R Walker for additional trial data. The authors would also like to thank the Cochrane internal and external reviewers for comments on an earlier draft and for providing advice on the process of a Cochrane Review. We are also grateful to Dr Tamara Kredo for her continuous support.
CRG Funding Acknowledgement: the National Institute for Health Research (NIHR) is the largest single funder of the Cochrane Depression, Anxiety and Neurosis Group.
Disclaimer: the views and opinions expressed therein are those of the authors and do not necessarily reflect those of the NIHR, NHS or the Department of Health.
Appendices
Appendix 1. CCMDCTR core MEDLINE search
Core Ovid MEDLINE search used to inform the Cochrane Common Mental Disorders Specialised Register (CCMD‐CTR). A weekly search alert based on Condition + RCT filter only. 1. [MeSH Headings]: eating disorders/ or anorexia nervosa/ or binge‐eating disorder/ or bulimia nervosa/ or female athlete triad syndrome/ or pica/ or hyperphagia/ or bulimia/ or self‐injurious behavior/ or self mutilation/ or suicide/ or suicidal ideation/ or suicide, attempted/ or mood disorders/ or affective disorders, psychotic/ or bipolar disorder/ or cyclothymic disorder/ or depressive disorder/ or depression, postpartum/ or depressive disorder, major/ or depressive disorder, treatment‐resistant/ or dysthymic disorder/ or seasonal affective disorder/ or neurotic disorders/ or depression/ or adjustment disorders/ or exp antidepressive agents/ or anxiety disorders/ or agoraphobia/ or neurocirculatory asthenia/ or obsessive‐compulsive disorder/ or obsessive hoarding/ or panic disorder/ or phobic disorders/ or stress disorders, traumatic/ or combat disorders/ or stress disorders, post‐traumatic/ or stress disorders, traumatic, acute/ or anxiety/ or anxiety, castration/ or koro/ or anxiety, separation/ or panic/ or exp anti‐anxiety agents/ or somatoform disorders/ or body dysmorphic disorders/ or conversion disorder/ or hypochondriasis/ or neurasthenia/ or hysteria/ or munchausen syndrome by proxy/ or munchausen syndrome/ or fatigue syndrome, chronic/ or obsessive behavior/ or compulsive behavior/ or behavior, addictive/ or impulse control disorders/ or firesetting behavior/ or gambling/ or trichotillomania/ or stress, psychological/ or burnout, professional/ or sexual dysfunctions, psychological/ or vaginismus/ or Anhedonia/ or Affective Symptoms/ or *Mental Disorders/
2. [Title/ Author Keywords]: (eating disorder* or anorexia nervosa or bulimi* or binge eat* or (self adj (injur* or mutilat*)) or suicide* or suicidal or parasuicid* or mood disorder* or affective disorder* or bipolar i or bipolar ii or (bipolar and (affective or disorder*)) or mania or manic or cyclothymic* or depression or depressive or dysthymi* or neurotic or neurosis or adjustment disorder* or antidepress* or anxiety disorder* or agoraphobia or obsess* or compulsi* or panic or phobi* or ptsd or posttrauma* or post trauma* or combat or somatoform or somati#ation or medical* unexplained or body dysmorphi* or conversion disorder or hypochondria* or neurastheni* or hysteria or munchausen or chronic fatigue* or gambling or trichotillomania or vaginismus or anhedoni* or affective symptoms or mental disorder* or mental health).ti,kf.
3. [RCT filter]: (controlled clinical trial.pt. or randomized controlled trial.pt. or (randomi#ed or randomi#ation).ab,ti. or randomly.ab. or (random* adj3 (administ* or allocat* or assign* or class* or control* or determine* or divide* or distribut* or expose* or fashion or number* or place* or recruit* or subsitut* or treat*)).ab. or placebo*.ab,ti. or drug therapy.fs. or trial.ab,ti. or groups.ab. or (control* adj3 (trial* or study or studies)).ab,ti. or ((singl* or doubl* or tripl* or trebl*) adj3 (blind* or mask* or dummy*)).mp. or clinical trial, phase ii/ or clinical trial, phase iii/ or clinical trial, phase iv/ or randomized controlled trial/ or pragmatic clinical trial/ or (quasi adj (experimental or random*)).ti,ab. or ((waitlist* or wait* list* or treatment as usual or TAU) adj3 (control or group)).ab.)
4. (1 and 2 and 3)
Records were screened for reports of RCTs within the scope of the Cochrane Common Mental Disorders Group. Secondary reports of RCTs are tagged to the appropriate study record.
Similar weekly search alerts were also conducted on OVID EMBASE and PsycINFO, using relevant subject headings (controlled vocabularies) and search syntax, appropriate to each resource.
A quaterly search of the Cochrane Central Register of Controlled Trials (CENTRAL) was conducted c/o the Cochrane Register of Studies Online (CRSO).
Appendix 2. CCMD searches to 2015
#1 (social* NEAR2 (anxious or anxiety or phobi*)) or "phobic avoidance" or "social avoidance" or "interpersonal anxiety"
#2 "social* inhib*" or "social* stress*" or heterosocial* or "taijin kyofusho"
#3 (#1 or #2)
Appendix 3. CCMD update search 2017
In August 2017 the CCMD Group's information specialist ran an update search to identify new studies published or registered since the date of the last search. Records were de‐duplicated, screened and x new studies placed in awaiting classification. These will be incorporated in the next version of this review.
As the CCMD Group's specialised register was out of date at this point, the information specialist ran searches on the following databases:
1. Cochrane Central Register of Controlled Trials (CENTRAL) (c/o Cochrane Register of Studies Online (CRSO)) Search All Fields [condition only]: (“social anxiety” or “social phobia”) AND 31/01/2015 to 31/08/2017:DL 2. CCMDCTR (studies and references) (“social anxiety” or “social phobia”) 18/08/2015 to 14/06/2016 3. OVID Cross‐search Databases: PsycINFO <1806 to July Week 4 2017>, Embase <1974 to 2017 Week 31>, Ovid MEDLINE(R) Epub Ahead of Print, In‐Process & Other Non‐Indexed Citations, Ovid MEDLINE(R) Daily and Ovid MEDLINE(R) <1946 to 2‐Aug‐2017> Search Strategy: ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ 1 (social* adj2 (phobi* or anxi*)).ab,kf,id,hw. 2 trial.ti. 3 (randomi#ed or randomi#ation or randomi#ing).ti,ab,kf,kw,id. 4 (RCT or at random or (random* adj3 (assign* or allocat* or control* or crossover or cross‐over or design* or divide* or division or number))).ti,ab,kf,kw,id. 5 placebo.hw,ti,ab,kf,kw,id. 6 (control* adj2 (trial or group?)).ab. 7 Randomized Controlled Trial.sh,pt. 8 Double Blind Procedure/ 9 Double Blind Method/ 10 Controlled Clinical Trial and placebo.af. 11 (clinical trial or empirical study).md. 12 ((single or double or triple) adj2 (blind* or mask* or dummy)).ti,ab,kf,kw,id. 13 or/2‐12 14 placebo.af. 15 drug therapy.fs. 16 exp Central Nervous System Agents/ 17 drug literature index.ec. 18 drug activity/ or drug effect/ or drug efficacy/ 19 "Clinical Psychopharmacology ".cc. 20 drug therapy/ 21 exp drugs/ 22 or/15‐21 23 (2015* or 2016* or 2017*).yr. 24 (2015* or 2016* or 2017*).dd. 25 (2015* or 2016* or 2017*).dc,ed. and (medline* or pubmed* or in‐data‐review or in‐process or publisher).st. 26 (2015* or 2016* or 2017*).an. and PsycINFO database record.ab. 27 or/23‐26 28 (1 and 13 and 14 and 27) 29 (1 and 13 and 22 and 27) 30 (28 or 29) 31 remove duplicates from 30 4. International Trial Registers ClinicalTrials.gov Advanced Search: Interventional Studies | social phobia OR social anxiety | Studies received from 01/01/2015 to 02/08/2017 WHO ICTRP Advanced Search: All Studies social phobia OR social anxiety | Studies received from 01/01/2015 to 02/08/2017 5. PubMed (not MEDLINE) 2‐Aug‐2017 #1 Search ((publisher[sb] OR inprocess[sb] OR pubmednotmedline[sb])) #2 Search (social anxiety[Title] OR social phobia[Title]) #3 Search RCT OR random* OR placebo #4 Search (#1 AND #2 AND #3) Sort by: PublicationDate Filters: Publication date from 2015/01/01 to 2017/12/31
Data and analyses
Comparison 1. Comparison 1: 5HT1A partial agonists versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 No. of responders | 1 | 30 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.07, 14.55] |
| 2 Adverse events (acute phase) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Dropout rate | 1 | 30 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 68.26] |
| 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.1 LSAS avoidance subscale | 1 | 30 | Mean Difference (IV, Random, 95% CI) | ‐1.40 [‐11.61, 8.81] |
| 4 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 4.1 Reduction of depression symptoms | 1 | 27 | Mean Difference (IV, Random, 95% CI) | ‐0.60 [‐2.86, 1.66] |
| 5 All‐cause dropouts (acute phase) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 Dropout rate | 1 | 30 | Risk Ratio (M‐H, Random, 95% CI) | 0.14 [0.01, 2.55] |
Comparison 2. Comparison 2: anticonvulsants/GABAs versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase) | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 No. of responders | 3 | 532 | Risk Ratio (M‐H, Random, 95% CI) | 1.60 [1.16, 2.20] |
| 2 Adverse events (acute phase) | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Dropout rate | 3 | 532 | Risk Ratio (M‐H, Random, 95% CI) | 2.90 [0.92, 9.14] |
| 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.1 LSAS total score | 1 | 69 | Mean Difference (IV, Random, 95% CI) | ‐11.5 [‐25.20, 2.20] |
| 3.2 LSAS avoidance subscale | 1 | 69 | Mean Difference (IV, Random, 95% CI) | ‐4.60 [‐11.88, 2.68] |
| 3.3 LSAS fear subscale | 1 | 69 | Mean Difference (IV, Random, 95% CI) | ‐6.90 [‐13.65, ‐0.15] |
| 4 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 4.1 Reduction of depression symptoms | 1 | 69 | Mean Difference (IV, Random, 95% CI) | ‐2.3 [‐4.78, 0.18] |
| 5 All‐cause dropouts (acute phase) | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 Dropout rate | 3 | 488 | Risk Ratio (M‐H, Random, 95% CI) | 1.16 [0.78, 1.70] |
Comparison 3. Comparison 3: anticonvulsant levetiracetam versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase) | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 No. of responders | 2 | 228 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.70, 1.37] |
| 2 Adverse events (acute phase) | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Dropout rate | 2 | 235 | Risk Ratio (M‐H, Random, 95% CI) | 2.00 [0.81, 4.94] |
| 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.1 LSAS total score | 2 | 228 | Mean Difference (IV, Random, 95% CI) | ‐0.24 [‐15.69, 15.21] |
| 3.2 LSAS avoidance subscale | 1 | 16 | Mean Difference (IV, Random, 95% CI) | ‐9.15 [‐26.86, 8.56] |
| 3.3 LSAS fear subscale | 1 | 16 | Mean Difference (IV, Random, 95% CI) | ‐8.71 [‐26.02, 8.60] |
| 4 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 4.1 Reduction of depression symptoms | 1 | 212 | Mean Difference (IV, Random, 95% CI) | 0.20 [‐1.33, 1.73] |
| 5 All‐cause dropouts (acute phase) | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 Dropout rate | 2 | 235 | Risk Ratio (M‐H, Random, 95% CI) | 1.42 [0.30, 6.76] |
Comparison 4. Comparison 4: antipsychotics versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 No. of responders | 1 | 10 | Risk Ratio (M‐H, Random, 95% CI) | 7.0 [0.45, 108.26] |
| 2 Adverse events (acute phase) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Dropout rate | 1 | 12 | Risk Ratio (M‐H, Random, 95% CI) | 0.71 [0.06, 8.90] |
| 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.1 LSAS total score | 1 | 9 | Mean Difference (IV, Random, 95% CI) | ‐37.8 [‐74.22, ‐1.38] |
| 4 All‐cause dropouts (acute phase) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 Dropout rate | 1 | 12 | Risk Ratio (M‐H, Random, 95% CI) | 1.07 [0.27, 4.23] |
Comparison 5. Comparison 5: benzodiazepines versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase) | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 No. of responders | 2 | 132 | Risk Ratio (M‐H, Random, 95% CI) | 4.03 [2.45, 6.65] |
| 2 Relapse rate | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 No. relapsed | 1 | 36 | Risk Ratio (M‐H, Random, 95% CI) | 0.12 [0.01, 2.14] |
| 3 Adverse events (acute phase) | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 Dropout rate | 2 | 96 | Risk Ratio (M‐H, Random, 95% CI) | 1.68 [0.21, 13.13] |
| 4 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 4.1 LSAS total score | 2 | 135 | Mean Difference (IV, Random, 95% CI) | ‐39.75 [‐71.11, ‐8.39] |
| 4.2 LSAS avoidance subscale | 1 | 75 | Mean Difference (IV, Random, 95% CI) | ‐10.40 [‐16.08, ‐4.72] |
| 4.3 LSAS fear subscale | 1 | 75 | Mean Difference (IV, Random, 95% CI) | ‐10.80 [‐16.62, ‐4.98] |
| 5 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 5.1 Reduction of depression symptoms | 1 | 75 | Mean Difference (IV, Random, 95% CI) | ‐1.60 [‐3.96, 0.76] |
| 6 Reduction of functional disability | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 6.1 Work subscale | 2 | 135 | Mean Difference (IV, Random, 95% CI) | ‐3.58 [‐6.39, ‐0.78] |
| 6.2 Social subscale | 2 | 135 | Mean Difference (IV, Random, 95% CI) | ‐2.31 [‐3.79, ‐0.83] |
| 6.3 Family subscale | 2 | 135 | Mean Difference (IV, Random, 95% CI) | ‐2.02 [‐4.26, 0.22] |
| 7 All‐cause dropouts (acute phase) | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 Dropout rate | 3 | 171 | Risk Ratio (M‐H, Random, 95% CI) | 0.79 [0.41, 1.52] |
Comparison 6. Comparison 6: beta‐blockers versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase) | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 No. of responders | 2 | 97 | Risk Ratio (M‐H, Random, 95% CI) | 1.09 [0.63, 1.88] |
| 2 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2.1 Reduction of depression symptoms | 1 | 46 | Mean Difference (IV, Random, 95% CI) | 1.82 [‐1.38, 5.02] |
| 3 Reduction of functional disability | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.1 Work subscale | 1 | 42 | Mean Difference (IV, Random, 95% CI) | ‐0.05 [‐1.80, 1.70] |
| 3.2 Social subscale | 1 | 42 | Mean Difference (IV, Random, 95% CI) | 0.07 [‐1.71, 1.85] |
| 3.3 Family subscale | 1 | 42 | Mean Difference (IV, Random, 95% CI) | ‐0.11 [‐1.74, 1.52] |
| 4 All‐cause dropouts (acute phase) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 Dropout rate | 1 | 46 | Risk Ratio (M‐H, Random, 95% CI) | 3.36 [0.41, 27.80] |
Comparison 7. Comparison 7: MAOIs versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase) | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 No. of responders | 4 | 235 | Risk Ratio (M‐H, Random, 95% CI) | 2.36 [1.48, 3.75] |
| 2 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms | 4 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2.1 LSAS total score | 4 | 218 | Mean Difference (IV, Random, 95% CI) | ‐16.39 [‐32.27, ‐0.51] |
| 2.2 LSAS avoidance subscale | 1 | 51 | Mean Difference (IV, Random, 95% CI) | ‐5.42 [‐14.69, 3.85] |
| 2.3 LSAS fear subscale | 1 | 51 | Mean Difference (IV, Random, 95% CI) | ‐5.23 [‐13.97, 3.51] |
| 3 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale) | 4 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.1 Reduction of depression symptoms | 4 | 216 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.40 [‐1.11, 0.31] |
| 4 Reduction of functional disability | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 4.1 Work subscale | 2 | 95 | Mean Difference (IV, Random, 95% CI) | ‐2.84 [‐4.40, ‐1.28] |
| 4.2 Social subscale | 2 | 94 | Mean Difference (IV, Random, 95% CI) | ‐3.26 [‐7.25, 0.72] |
| 4.3 Family subscale | 2 | 95 | Mean Difference (IV, Random, 95% CI) | ‐2.20 [‐5.34, 0.95] |
| 5 All‐cause dropouts (acute phase) | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 Dropout rate | 4 | 235 | Risk Ratio (M‐H, Random, 95% CI) | 1.33 [0.71, 2.48] |
| 6 Clinical Global Impressions scale change item (CGI‐I) (long term) | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 No. of responders | 2 | 113 | Risk Ratio (M‐H, Random, 95% CI) | 1.84 [1.02, 3.33] |
Comparison 8. Comparison 8: NARIs versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 No. of responders | 1 | 27 | Risk Ratio (M‐H, Random, 95% CI) | 0.70 [0.19, 2.54] |
| 2 Adverse events (acute phase) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Dropout rate | 1 | 27 | Risk Ratio (M‐H, Random, 95% CI) | 2.8 [0.12, 63.20] |
| 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.1 LSAS total score | 1 | 26 | Mean Difference (IV, Random, 95% CI) | 2.60 [‐15.43, 20.63] |
| 4 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 4.1 Reduction of depression symptoms | 1 | 26 | Mean Difference (IV, Random, 95% CI) | ‐0.10 [‐2.73, 2.53] |
| 5 All‐cause dropouts (acute phase) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 Dropout rate | 1 | 27 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.23, 3.81] |
Comparison 9. Comparison 9: NaSSAs versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 No. of responders | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.28, 3.63] |
| 2 Adverse events (acute phase) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Dropout rate | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 5.0 [0.25, 99.95] |
| 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.1 LSAS total score | 2 | 126 | Mean Difference (IV, Random, 95% CI) | ‐15.37 [‐28.10, ‐2.63] |
| 3.2 LSAS avoidance subscale | 1 | 60 | Mean Difference (IV, Random, 95% CI) | ‐3.90 [‐9.90, 2.10] |
| 3.3 LSAS fear subscale | 1 | 60 | Mean Difference (IV, Random, 95% CI) | ‐3.70 [‐9.42, 2.02] |
| 4 All‐cause dropouts (acute phase) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 Dropout rate | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 2.0 [0.19, 20.90] |
Comparison 10. Comparison 10: RIMAs versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase) | 8 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 No. of responders | 8 | 1270 | Risk Ratio (M‐H, Random, 95% CI) | 1.83 [1.32, 2.55] |
| 2 Adverse events (acute phase) | 8 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Dropout rate | 8 | 1305 | Risk Ratio (M‐H, Random, 95% CI) | 1.42 [0.86, 2.34] |
| 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms | 9 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.1 LSAS total score | 6 | 1163 | Mean Difference (IV, Random, 95% CI) | ‐12.17 [‐23.51, ‐0.84] |
| 3.2 LSAS avoidance subscale | 5 | 695 | Mean Difference (IV, Random, 95% CI) | ‐5.05 [‐7.91, ‐2.18] |
| 3.3 LSAS fear subscale | 6 | 724 | Mean Difference (IV, Random, 95% CI) | ‐5.40 [‐8.92, ‐1.88] |
| 4 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale) | 7 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 4.1 Reduction of depression symptoms | 7 | 765 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.28 [‐0.55, ‐0.00] |
| 5 Reduction of functional disability | 5 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 5.1 Work subscale | 5 | 660 | Mean Difference (IV, Random, 95% CI) | ‐0.61 [‐1.89, 0.68] |
| 5.2 Social subscale | 5 | 660 | Mean Difference (IV, Random, 95% CI) | ‐1.14 [‐2.32, 0.05] |
| 5.3 Family subscale | 5 | 660 | Mean Difference (IV, Random, 95% CI) | ‐0.51 [‐1.45, 0.44] |
| 6 All‐cause dropouts (acute phase) | 6 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 Dropout rate | 6 | 512 | Risk Ratio (M‐H, Random, 95% CI) | 0.80 [0.60, 1.08] |
| 7 Clinical Global Impressions ‐ Improvement (CGI‐I) (long term) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 No. of responders | 1 | 90 | Risk Ratio (M‐H, Random, 95% CI) | 1.50 [1.12, 2.00] |
Comparison 11. Comparison 11: SARIs versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.1 LSAS total score | 1 | 102 | Mean Difference (IV, Random, 95% CI) | ‐6.10 [‐16.55, 4.35] |
| 2 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2.1 Reduction of depression symptoms | 1 | 102 | Mean Difference (IV, Random, 95% CI) | 0.80 [‐2.10, 3.70] |
| 3 Reduction of functional disability | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.1 Work subscale | 1 | 102 | Mean Difference (IV, Random, 95% CI) | ‐0.90 [‐1.87, 0.07] |
| 3.2 Social subscale | 1 | 102 | Mean Difference (IV, Random, 95% CI) | ‐1.0 [‐1.97, ‐0.03] |
| 3.3 Family subscale | 1 | 102 | Mean Difference (IV, Random, 95% CI) | ‐0.20 [‐1.06, 0.66] |
| 4 All‐cause dropouts (acute phase) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 Dropout rate | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 2.18 [0.97, 4.92] |
Comparison 12. Comparison 12: SNRIs versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase) | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 No. of responders | 4 | 1173 | Risk Ratio (M‐H, Random, 95% CI) | 1.30 [0.85, 1.99] |
| 2 Adverse events (acute phase) | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Dropout rate | 4 | 1213 | Risk Ratio (M‐H, Random, 95% CI) | 3.23 [2.15, 4.86] |
| 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms | 3 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.1 LSAS total score | 3 | 902 | Mean Difference (IV, Random, 95% CI) | ‐11.91 [‐16.06, ‐7.76] |
| 3.2 LSAS avoidance subscale | 1 | 261 | Mean Difference (IV, Random, 95% CI) | ‐4.30 [‐8.14, ‐0.46] |
| 3.3 LSAS fear subscale | 1 | 261 | Mean Difference (IV, Random, 95% CI) | ‐4.0 [‐7.68, ‐0.32] |
| 4 All‐cause dropouts (acute phase) | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 Dropout rate | 4 | 1224 | Risk Ratio (M‐H, Random, 95% CI) | 0.90 [0.76, 1.07] |
Comparison 13. Comparison 13: SSRIs versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase) | 24 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 No. of responders | 24 | 4984 | Risk Ratio (M‐H, Random, 95% CI) | 1.65 [1.48, 1.85] |
| 2 Relapse rate | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 No. relapsed | 3 | 389 | Risk Ratio (M‐H, Random, 95% CI) | 0.34 [0.22, 0.50] |
| 3 Adverse events (acute phase) | 24 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 Dropout rate | 24 | 5131 | Risk Ratio (M‐H, Random, 95% CI) | 2.59 [1.97, 3.39] |
| 4 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms | 15 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 4.1 LSAS total score | 14 | 1990 | Mean Difference (IV, Random, 95% CI) | ‐10.14 [‐14.05, ‐6.22] |
| 4.2 LSAS avoidance subscale | 7 | 1173 | Mean Difference (IV, Random, 95% CI) | ‐7.01 [‐10.21, ‐3.80] |
| 4.3 LSAS fear subscale | 7 | 1173 | Mean Difference (IV, Random, 95% CI) | ‐7.28 [‐10.86, ‐3.71] |
| 5 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale) | 6 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 5.1 Reduction of depression symptoms | 6 | 960 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.26 [‐0.48, ‐0.03] |
| 6 Reduction of functional disability | 5 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 6.1 Work subscale | 5 | 854 | Mean Difference (IV, Random, 95% CI) | ‐0.81 [‐1.18, ‐0.45] |
| 6.2 Social subscale | 5 | 854 | Mean Difference (IV, Random, 95% CI) | ‐0.87 [‐1.26, ‐0.47] |
| 6.3 Family subscale | 5 | 854 | Mean Difference (IV, Random, 95% CI) | ‐0.45 [‐0.75, ‐0.15] |
| 7 All‐cause dropouts (acute phase) | 26 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 Dropout rate | 26 | 5208 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.90, 1.14] |
| 8 Clinical Global Impressions scale change item (CGI‐I) (long term) | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 8.1 No. of responders | 4 | 806 | Risk Ratio (M‐H, Random, 95% CI) | 1.27 [1.07, 1.51] |
| 9 Adverse events (long term) | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 9.1 Dropout rate | 3 | 1274 | Risk Ratio (M‐H, Random, 95% CI) | 1.17 [0.43, 3.18] |
| 10 All‐cause dropouts (long term) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 10.1 Dropout rate | 1 | 52 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.37, 2.70] |
Comparison 14. Comparison 14: GW876008 versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 No. of responders | 1 | 250 | Risk Ratio (M‐H, Random, 95% CI) | 0.83 [0.58, 1.19] |
| 2 Adverse events (acute phase) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Dropout rate | 1 | 252 | Risk Ratio (M‐H, Random, 95% CI) | 2.95 [0.67, 13.02] |
| 3 All‐cause dropouts (acute phase) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 Dropout rate | 1 | 252 | Risk Ratio (M‐H, Random, 95% CI) | 0.77 [0.50, 1.20] |
14.2. Analysis.
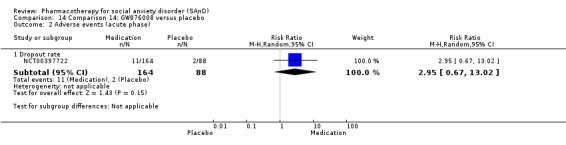
Comparison 14 Comparison 14: GW876008 versus placebo, Outcome 2 Adverse events (acute phase).
Comparison 15. Comparison 15: NK1 receptor antagonist GR205171 versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 No. of responders | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 5.0 [0.68, 36.66] |
| 2 Adverse events (acute phase) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Dropout rate | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.1 LSAS total score | 1 | 24 | Mean Difference (IV, Random, 95% CI) | ‐0.50 [‐1.35, 0.35] |
| 4 All‐cause dropouts (acute phase) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 Dropout rate | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 16. Comparison 16: LY686017 versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.1 LSAS total score | 1 | 99 | Mean Difference (IV, Random, 95% CI) | 1.80 [‐6.92, 10.52] |
Comparison 17. Comparison 17: total effect of medication compared to placebo for the treatment of SAnD.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar) scale: no. of responders (acute phase) | 51 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 5HT1A partial agonists | 1 | 30 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.07, 14.55] |
| 1.2 Anticonvulsants/GABAs | 3 | 532 | Risk Ratio (M‐H, Random, 95% CI) | 1.60 [1.16, 2.20] |
| 1.3 Anticonvulsant levetiracetam | 2 | 228 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.70, 1.37] |
| 1.4 Antipsychotics | 1 | 10 | Risk Ratio (M‐H, Random, 95% CI) | 7.0 [0.45, 108.26] |
| 1.5 Benzodiazepines | 2 | 132 | Risk Ratio (M‐H, Random, 95% CI) | 4.03 [2.45, 6.65] |
| 1.6 Beta‐blockers | 2 | 97 | Risk Ratio (M‐H, Random, 95% CI) | 1.09 [0.63, 1.88] |
| 1.7 MAOIs | 3 | 178 | Risk Ratio (M‐H, Random, 95% CI) | 2.37 [1.26, 4.45] |
| 1.8 NARIs | 1 | 27 | Risk Ratio (M‐H, Random, 95% CI) | 0.70 [0.19, 2.54] |
| 1.9 NaSSAs | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.28, 3.63] |
| 1.10 RIMAs | 8 | 1270 | Risk Ratio (M‐H, Random, 95% CI) | 1.83 [1.32, 2.55] |
| 1.11 SNRIs | 4 | 1173 | Risk Ratio (M‐H, Random, 95% CI) | 1.30 [0.85, 1.99] |
| 1.12 SSRIs | 21 | 4553 | Risk Ratio (M‐H, Random, 95% CI) | 1.65 [1.46, 1.87] |
| 1.13 NK1 receptor antagonist GR205171 | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 5.0 [0.68, 36.66] |
| 1.14 GW876008 | 1 | 250 | Risk Ratio (M‐H, Random, 95% CI) | 0.83 [0.58, 1.19] |
| 2 Dropout rate: adverse events (acute phase) | 45 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 5HT1A partial agonists | 1 | 30 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 68.26] |
| 2.2 Anticonvulsants/GABAs | 3 | 532 | Risk Ratio (M‐H, Random, 95% CI) | 2.90 [0.92, 9.14] |
| 2.3 Anticonvulsant levetiracetam | 2 | 235 | Risk Ratio (M‐H, Random, 95% CI) | 2.00 [0.81, 4.94] |
| 2.4 Antipsychotics | 1 | 12 | Risk Ratio (M‐H, Random, 95% CI) | 0.71 [0.06, 8.90] |
| 2.5 Benzodiazepines | 2 | 96 | Risk Ratio (M‐H, Random, 95% CI) | 1.68 [0.21, 13.13] |
| 2.6 NARIs | 1 | 27 | Risk Ratio (M‐H, Random, 95% CI) | 2.8 [0.12, 63.20] |
| 2.7 NaSSAs | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 5.0 [0.25, 99.95] |
| 2.8 RIMAs | 8 | 1305 | Risk Ratio (M‐H, Random, 95% CI) | 1.42 [0.86, 2.34] |
| 2.9 SNRIs | 4 | 1213 | Risk Ratio (M‐H, Random, 95% CI) | 3.23 [2.15, 4.86] |
| 2.10 SSRIs | 22 | 4965 | Risk Ratio (M‐H, Random, 95% CI) | 2.56 [1.94, 3.38] |
| 2.11 NK1 receptor antagonist GR205171 | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 2.12 GW876008 | 1 | 252 | Risk Ratio (M‐H, Random, 95% CI) | 2.95 [0.67, 13.02] |
| 3 Dropout rate: all‐cause dropouts (acute phase) | 54 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 5HT1A partial agonists | 1 | 30 | Risk Ratio (M‐H, Random, 95% CI) | 0.14 [0.01, 2.55] |
| 3.2 Anticonvulsants/GABAs | 3 | 488 | Risk Ratio (M‐H, Random, 95% CI) | 1.16 [0.78, 1.70] |
| 3.3 Anticonvulsant levetiracetam | 2 | 235 | Risk Ratio (M‐H, Random, 95% CI) | 1.42 [0.30, 6.76] |
| 3.4 Antipsychotics | 1 | 12 | Risk Ratio (M‐H, Random, 95% CI) | 1.07 [0.27, 4.23] |
| 3.5 Benzodiazepines | 3 | 171 | Risk Ratio (M‐H, Random, 95% CI) | 0.79 [0.41, 1.52] |
| 3.6 Beta‐blockers | 1 | 46 | Risk Ratio (M‐H, Random, 95% CI) | 3.36 [0.41, 27.80] |
| 3.7 MAOIs | 4 | 235 | Risk Ratio (M‐H, Random, 95% CI) | 1.33 [0.71, 2.48] |
| 3.8 NARIs | 1 | 27 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.23, 3.81] |
| 3.9 NaSSAs | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 2.0 [0.19, 20.90] |
| 3.10 RIMAs | 6 | 512 | Risk Ratio (M‐H, Random, 95% CI) | 0.80 [0.60, 1.08] |
| 3.11 SARIs | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 2.18 [0.97, 4.92] |
| 3.12 SNRIs | 4 | 1224 | Risk Ratio (M‐H, Random, 95% CI) | 0.90 [0.76, 1.07] |
| 3.13 SSRIs | 25 | 5078 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.89, 1.14] |
| 3.14 NK1 receptor antagonist GR205171 | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.15 GW876008 | 1 | 252 | Risk Ratio (M‐H, Random, 95% CI) | 0.77 [0.50, 1.20] |
17.2. Analysis.
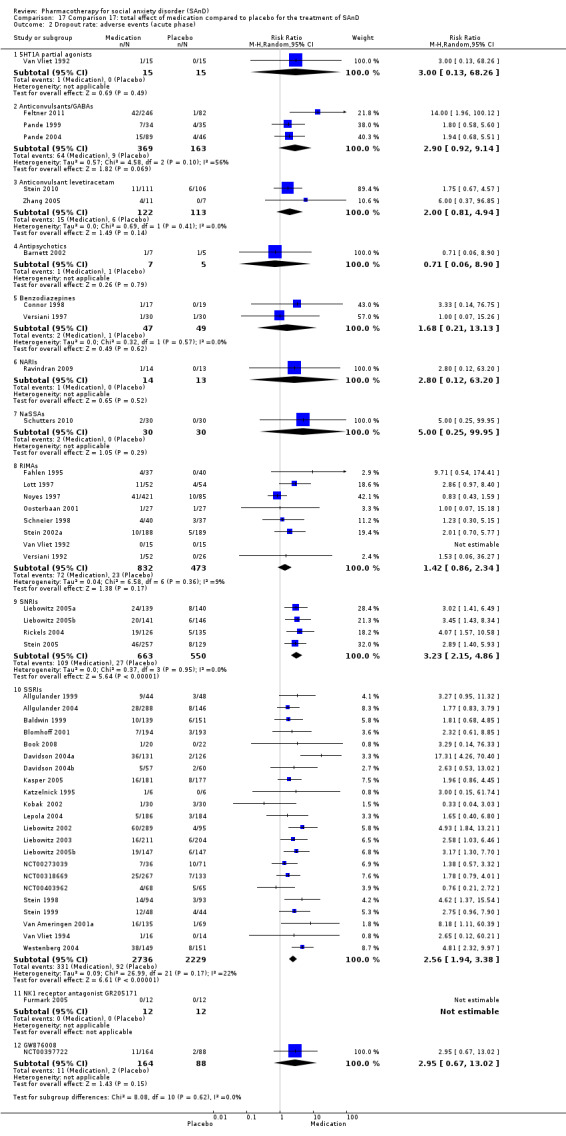
Comparison 17 Comparison 17: total effect of medication compared to placebo for the treatment of SAnD, Outcome 2 Dropout rate: adverse events (acute phase).
Comparison 18. Subgroup analysis: multicentre versus single‐centre trials.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale: no. of responders (acute phase) | 53 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Single‐centre | 20 | 1332 | Risk Ratio (M‐H, Random, 95% CI) | 2.24 [1.67, 3.02] |
| 1.2 Multicentre | 35 | 8274 | Risk Ratio (M‐H, Random, 95% CI) | 1.47 [1.34, 1.62] |
Comparison 19. Subgroup analysis: generalised SAnD compared to inclusive SAnD.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale: no. of responders (acute phase) | 53 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Generalised | 26 | 5522 | Risk Ratio (M‐H, Random, 95% CI) | 1.49 [1.31, 1.69] |
| 1.2 Inclusive | 27 | 3712 | Risk Ratio (M‐H, Random, 95% CI) | 1.83 [1.54, 2.18] |
Comparison 20. Subgroup analysis: industry funding compared to no industry funding.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale: no. of responders (acute phase) | 50 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Industry funded trials | 34 | 6643 | Risk Ratio (M‐H, Random, 95% CI) | 1.60 [1.44, 1.77] |
| 1.2 Non‐industry funded trials | 16 | 1780 | Risk Ratio (M‐H, Random, 95% CI) | 1.99 [1.43, 2.77] |
Comparison 21. Subgroup analysis: trials that included MDD compared to no MDD.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale: no. of responders (acute phase) | 53 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Trials including MDD participants | 20 | 2654 | Risk Ratio (M‐H, Random, 95% CI) | 1.77 [1.44, 2.18] |
| 1.2 Trials excluding MDD participants | 34 | 6765 | Risk Ratio (M‐H, Random, 95% CI) | 1.51 [1.35, 1.70] |
Comparison 22. Worst case versus best case: support for robustness of evidence.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 'Worst case' lost‐to‐follow‐up analysis | 25 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Anticonvulsant levetiracetam | 2 | 235 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.70, 1.38] |
| 1.2 Antipsychotics | 1 | 12 | Risk Ratio (M‐H, Random, 95% CI) | 5.25 [0.33, 83.59] |
| 1.3 Benzodiazepines | 1 | 75 | Risk Ratio (M‐H, Random, 95% CI) | 3.82 [1.92, 7.62] |
| 1.4 Beta‐blockers | 1 | 46 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.47, 1.86] |
| 1.5 RIMAs | 2 | 182 | Risk Ratio (M‐H, Random, 95% CI) | 3.01 [1.95, 4.65] |
| 1.6 SNRIs | 3 | 934 | Risk Ratio (M‐H, Random, 95% CI) | 1.60 [1.36, 1.88] |
| 1.7 SSRIs | 14 | 3753 | Risk Ratio (M‐H, Random, 95% CI) | 1.61 [1.44, 1.79] |
| 1.8 GW876008 | 1 | 252 | Risk Ratio (M‐H, Random, 95% CI) | 0.82 [0.57, 1.18] |
| 2 'Best case' lost‐to‐follow‐up analysis | 25 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Anticonvulsant levetiracetam | 2 | 228 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.70, 1.37] |
| 2.2 Antipsychotics | 1 | 10 | Risk Ratio (M‐H, Random, 95% CI) | 7.0 [0.45, 108.26] |
| 2.3 Benzodiazepines | 1 | 72 | Risk Ratio (M‐H, Random, 95% CI) | 3.92 [1.98, 7.77] |
| 2.4 Beta‐blockers | 1 | 41 | Risk Ratio (M‐H, Random, 95% CI) | 1.06 [0.55, 2.05] |
| 2.5 RIMAs | 2 | 177 | Risk Ratio (M‐H, Random, 95% CI) | 2.97 [1.93, 4.58] |
| 2.6 SNRIs | 3 | 912 | Risk Ratio (M‐H, Random, 95% CI) | 1.63 [1.39, 1.91] |
| 2.7 SSRIs | 14 | 3577 | Risk Ratio (M‐H, Random, 95% CI) | 1.64 [1.46, 1.85] |
| 2.8 GW876008 | 1 | 250 | Risk Ratio (M‐H, Random, 95% CI) | 0.83 [0.58, 1.19] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Allgulander 1999.
| Methods | Design: single‐centre, randomised, placebo‐controlled, parallel, flexible dose, double‐blind Duration of intervention: 12 weeks and 6 days of dose‐tapering Post‐treatment: no follow‐up Placebo run‐in: 1‐week assessment period without medication |
|
| Participants | Sample size: 92 randomised to paroxetine and placebo Mean age: 41 years Sex: 48 men and 44 women Diagnostic measure: DSM‐IV Inclusion criteria: quote: "Aged 18‐65 years with previously untreated and incapacitating social anxiety; DSM‐IV social anxiety disorder causing substantial impairment and with a duration of at least 1 year; DSM‐IV diagnoses of generalised anxiety, dysthymia or a cluster C personality disorder were the only concurrent psychiatric disorders allowed". Exclusion criteria: quote: "No psychoactive medications were permitted, including beta‐receptor‐blocking agents; the blood and urine of all subjects was screened for substance abuse" Dropouts: 27/92 (8/44 in the paroxetine group and 19/48 in the placebo group) |
|
| Interventions | Pharmacological intervention: quote: "The subjects were randomly allocated at baseline to double‐blind treatment for three months with paroxetine 20‐50 mg daily administered in 10‐mg weekly increments, or placebo. One dose reduction was allowed in case of adverse events." | |
| Outcomes | Primary outcomes: LSAS (for reduction of anxiety) and CGI (for treatment efficacy) Secondary outcomes: BSPS (for reduction of anxiety), SDI (for reduction of functional disability), FNES (for reduction of anxiety) and VAS scores (reflecting self‐confidence in social interactions, anticipatory anxiety, acute anxiety reactions in social situations, and dysphoria following anxiety reactions) Time points: Quote: "Assessments were made after 1, 2, 4, 6, 8 and 12 weeks, and after 6 days of dose‐tapering" |
|
| Notes | Industry funded: yes. Quote: "This study was funded by Novo Nordisk Pharma, Sweden." Medication provided by industry: unclear Any of the authors work for industry: no |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomisation was performed at the hospital pharmacy using tabulated random numbers" |
| Allocation concealment (selection bias) | Low risk | Author correspondence: quote: "The randomization and blinding and packaging of study materials were undertaken by our hospital pharmacy and by Wyeth" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No description of blinding is provided in the study report. Quote: "Patients were randomized to double‐blind treatment with paroxetine 20‐50 mg daily or placebo for 3 months" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No mention is made of whether the outcome assessors were indeed blinded and independent. Quote: "To reduce variability in assessments, all subjects were treated and assessed in one centre by the author and a research nurse" |
| Incomplete outcome data (attrition bias) All outcomes | High risk | A larger proportion of participants discontinued the study in the paroxetine (8/44; 18%) group compared to the placebo group (19/48; 39%). Reasons for treatment withdrawal were provided for the treatment group with only reasons given for 5 participants in the placebo group. No information was provided on sample characteristics at endpoint. Overall 29% of the participants dropped out of the study. All analyses were intention‐to‐treat (ITT) |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. Quote: "Comparatively high response rate of subjects on paroxetine and the low response rate of those on placebo in this study may be due to the low variability in assessments in a single centre, the use of self‐rating instruments, or the fact that only previously untreated cases were included" |
Allgulander 2004.
| Methods | Design: multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group trial Duration of intervention: 12 weeks Post‐treatment: no follow‐up Placebo run‐in: 1‐week, single‐blind, placebo lead‐in period |
|
| Participants | Sample size: 434 were randomised to venlafaxine, paroxetine, or placebo (2 individuals excluded; 389 ITT population) Mean age (SD): 38.8 (10.97) years Sex: 183 men and 206 women Diagnostic measure: DSM‐IV Inclusion criteria: quote: "Study participants were adult (18 years of age) outpatients who met DSM‐IV criteria for generalized SAD for at least 6 months prior to study day 1. Participants were eligible if they had a score 54 on item 1 (severity of illness) of the clinical global impression severity (CGI‐S) scale; a minimum total score of at least 50 on the Liebowitz social anxiety scale (LSAS), with 430% decrease between the prestudy and baseline visits (i.e. during the placebo lead‐in period); a prestudy Raskin depression total score 49, and a 17‐item Hamilton rating scale for depression (HAM‐D17) score <15; and provided informed consent". Exclusion criteria: quote: "Patients were excluded if they had been treated with venlafaxine immediate release or venlafaxine ER within 6 months of study day 1 or had concurrent disorders that confounded the evaluation of treatment, including substance use disorders, personality disorders (except avoidant personality disorder), depression or other primary anxiety disorders, diagnosed by clinical interview. While patients who had not responded to previous treatment with paroxetine were not prohibited from participating in the study, ongoing psychotherapy and recent treatment with psychoactive medications precluded entry into the study". Dropouts: 26/434 (6/144 in the paroxetine group, 7/144 in the venlafaxine group, and 13/146 in the placebo group) |
|
| Interventions | Pharmacological intervention: quote: "After a 1‐week, single‐blind, placebo lead‐in period to eliminate subjects with situational anxiety and ascertain generalized social anxiety disorder, patients symptomatic at baseline were randomly assigned to receive flexible doses of venlafaxine ER (75–225 mg/day), paroxetine (20–50 mg/day), or placebo for up to 84 days". | |
| Outcomes | Primary outcome: LSAS (for reduction of anxiety) Secondary outcomes: CGI‐S (for reduction of anxiety), CGI‐I (for treatment efficacy), SPIN (for reduction of anxiety), the fear/anxiety and avoidance subscales of the LSAS (for reduction of anxiety), SDI (for reduction of functional disability) and the WPAI questionnaire (for reduction of functional disability) Time points: Quote: "Patient evaluations occurred at baseline and on days 7, 14, 21, 28, 42, 56, 70 and 84. Final efficacy evaluations were performed on the last day that the patient received a full dose of study medication or within 3 days thereafter" |
|
| Notes | Industry funded: yes. Quote: "Contract/grant sponsor: Wyeth Research" Medication provided by industry: unclear Any of the authors work for industry: yes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Authors report that participants were randomised; however, no mention is made of the method of randomisation. Quote: "Adult outpatients with generalized SAD (n=434) were randomized to receive capsules of venlafaxine ER 75 mg to 225 mg/day, paroxetine 20 mg to 50 mg/day, or placebo for 12 weeks ... At the baseline visit, after the investigator had ascertained that the patient was qualified to enter the study, the patient was also given a randomization number and the accompanying treatment supplies". |
| Allocation concealment (selection bias) | Low risk | Author correspondence: quote: "The randomization and blinding and packaging of study materials were undertaken by our hospital pharmacy and by Wyeth". |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Medication and placebo was provided in identical capsules. However, it is not clear whether both participants and personnel were blinded. Quote: "study medication was provided in identically appearing capsules containing venlafaxine ER 75 mg, paroxetine 10 mg, paroxetine 20 mg, or placebo, and the number of capsules was identical for all treatments". |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "In order to ensure that the assessor (i.e. the investigator) was unaware of the treatment group to which a patient was assigned, study medication was provided in identically appearing capsules containing venlafaxine ER 75 mg, paroxetine 10 mg, paroxetine 20 mg, or placebo, and the number of capsules was identical for all treatments". |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | More participants withdrew from the placebo group (13/132; 10%) compared to the paroxetine (6/128; 5%) and venlafaxine (7/129; 5%) group. The most common reasons for withdrawal were adverse events and unsatisfactory response. No information was provided on whether participants differed in terms of characteristics by group at week 12, however. Nevertheless, the total proportion of dropouts (7%) is relatively low, suggesting that dropout rates may not have biased the outcomes. Quote: "A total of 363 (84%) patients completed the 12‐week double‐blind treatment period (119 in the placebo group, 122 in the venlafaxine ER group and 122 in the paroxetine group) ... the most common reasons for withdrawal were adverse events and unsatisfactory response. Significantly more participants in the placebo group withdrew due to unsatisfactory response than in the venlafaxine ER group or the paroxetine group". Overall 7% of the participants dropped out of the study. Baseline analysis were intention‐to‐treat (ITT) whereas the analysis of the primary and secondary outcomes were last observation carried forward (LOCF). |
| Selective reporting (reporting bias) | Low risk | All outcomes were reported as specified in the protocol. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias was identified. |
Asakura 2007.
| Methods | Design: multicentre, randomised, double‐blind, placebo‐controlled study Duration of intervention: 10 weeks Post‐treatment: no follow‐up Placebo run‐in: no |
|
| Participants | Sample size: 273 randomised to fluvoxamine or placebo (273 randomised: 2 excluded; 271 ITT population) Mean age (SD): 38.6 (11.25) years Sex: 179 men and 86 women (265 randomised in the efficacy analysis population) Diagnostic measure: DSM‐IV Inclusion criteria: quote: "Eligible patients were aged 18–65 yr and were required to meet the DSM‐IV criteria for GSAD, have a minimum score of > 60 on the Liebowitz Social Anxiety Scale – Japanese Version, have no serious medical history, and to have taken no psychotropic medications for at least 14 d prior to randomization. The diagnosis of GSAD was made according to DSM‐IV criteria by well‐trained research psychiatrists. Patients were required, in addition to meeting DSM‐IV criteria for SAD, to exhibit fear and/or avoidance of at least four social situations (at least two involving interpersonal interactions)". Exclusion criteria: quote: "Patients were excluded if they had any Axis I psychiatric disorder (e.g. schizophrenia, bipolar disorder, major depressive disorder, dysthymic disorder, panic disorder, alcohol abuse/dependence), or medical or neurological disorder. Other exclusion criteria were any clinically significant abnormal laboratory or electrocardiogram (ECG) findings at the screening visit. Women who were pregnant, lactating, or not using an acceptable method of contraception were also ineligible". Dropouts: 6/271 (4/93 and 2/89 in the fluvoxamine groups and 0/89 in the placebo group; the additional two were excluded prior to the allocation of treatment). |
|
| Interventions | Pharmacological intervention: quote: "Eligible patients were randomly assigned to either fluvoxamine (at an initial dose of 50 mg/d fluvoxamine in two divided doses) or placebo in a 2:1 ratio. Fluvoxamine‐treated patients were randomly divided into two subgroups; a daily dose was increased by 50mg increments per week to a maintenance dose of 150 mg/d in one subgroup and to that of 300 mg/d in the other subgroup". | |
| Outcomes | Primary outcome: LSAS‐J (for reduction of anxiety) Secondary outcomes: CGI (for treatment efficacy) and SDS (for reduction of functional disability) Time points: Quote: "Patients were evaluated at nine study visits (baseline and weeks 1, 2, 3, 4, 5, 6, 8, and 10)" |
|
| Notes | Industry funded: yes. Quote: "This study was sponsored by Solvay Seiyaku K.K. and Meiji Seika Kaisha, Ltd". Medication provided by industry: unclear Any of the authors work for industry: no |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Author correspondence: quote: "This was a double‐blind study, meaning both the subjects and the investigators were blinded to the randomization scheme by double‐dummy method". |
| Allocation concealment (selection bias) | Low risk | Author correspondence: quote: "An independent third party (CRO) randomly allocated the packages of investigational drug using SAS procedure, as a set consisted of investigational drug for 4 cases for fluvoxamine group and 2 cases for placebo group, and sealed the packages. They held the key code during the course of the study and were to break the blind after all CRFs were collected and all CRF data had been entered into the database and the database locked". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Author correspondence: quote: "This was a double‐blind study, meaning both the subjects and the investigators were blinded to the randomization scheme by double‐dummy method". |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Author correspondence: quote: "This was a double‐blind study, meaning both the subjects and the investigators were blinded to the randomization scheme by double‐dummy method". |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | A small proportion of participants withdrew from the fluvoxamine groups (4/93, 2/89) with no reported dropouts for the placebo group (0/89). Patients withdrew due to adverse events and protocol deviations. No information was provided on whether participants differed by group characteristics at at week 10, however. Nevertheless, the total proportion of dropouts (2%) is relatively low, suggesting that dropout rates may not have biased the outcomes. Quote: "The efficacy analysis population was composed of 265 patients (176 receiving fluvoxamine and 89 receiving placebo), excluding six patients for whom no valid post‐baseline efficacy evaluation was obtained due to premature discontinuation [four withdrew due to adverse events and two withdrew due to protocol deviations (inappropriate concomitant medications)]". Overall 2% of the participants dropped out of the study. Quote: "Efficacy data are presented for the last observation carried forward (LOCF) dataset. The LOCF dataset used the last available on‐treatment observation for each patient to estimate missing data‐points". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. There was a difference in gender proportions, but there is no reason to believe that this may have biased the study. Quote: "Our findings on the gender ratio confirmed that men were predominant in this study. However, it is unknown whether the finding represents the status of gender ratio in Japanese SAD patients. There seems to be no clear sex predominance for this disorder". |
Baldwin 1999.
| Methods | Design: multicentre, randomised, placebo‐controlled, parallel, flexible dose, double‐blind Duration of intervention: 12 weeks Post‐treatment: no follow‐up Placebo run‐in: one‐week, single blind, placebo run‐in phase |
|
| Participants | Sample size: 290 randomised to paroxetine or placebo Mean age (SD): 36 (11.5) years Sex: 133 men and 157 women Diagnostic measure: DSM‐IV Inclusion criteria: quote: "Male or female out‐patients, aged 18 years or over, patients with a primary diagnosis of social phobia according to the DSM‐IV criteria were included in the study following the provision of written informed consent". Exclusion criteria: quote: "Patients were excluded at the screening visit if they had a primary diagnosis of any other Axis I disorder within the past six months, if they had been diagnosed as having body dysmorphic disorder or if they had a history of schizophrenia or bipolar affective disorder. Patients were excluded if they had a past history of seizure disorders or any serious medical disorder that could preclude the administration of paroxetine. In addition, patients requiring concomitant therapy with beta‐adrenergic blockers, monoamine oxidase inhibitors, benzodiazepines or other psychoactive medications were not included. Patients were also not included if: they had taken psychotropic drugs or antidepressants within the past two weeks or depot neuroleptics within 12 weeks; they had been previously unresponsive or intolerant to paroxetine, or they had used an investigational drug during the past month; they had undergone previous treatment for social phobia with an SSRI at a dose and duration that would have been adequate to show a response, or undergone electroconvulsive therapy (within three months) or psychotherapy (except ongoing stabilised therapies of six months or more). Other exclusion criteria included pregnancy (or a likelihood of becoming pregnant), lactation and alcohol substance misuse (within the past three months) or dependence (within the past six months). Patients were also excluded if they posed a current serious risk of suicide or homicide". Dropouts: 77/290 (35/139 in the paroxetine and 42/151 in the placebo group) |
|
| Interventions | Pharmacological intervention: quote: "Patients initially received 20 mg/day paroxetine or placebo for two weeks, followed by 10 mg/day at weekly intervals to a maximum dose of 50 mg/day according to clinical response and tolerability". | |
| Outcomes | Primary outcomes: LSAS (for reduction of anxiety) and CGI‐I (for treatment efficacy) Secondary outcomes: SADS (for reduction of anxiety), SDS (for reduction of functional disability) and CGI‐S (for reduction of anxiety) Time points: Quote: "Efficacy and safety assessments were made at weeks 1,2,3,4,6,8 and 12 and additional, further safety assessments were made at week 15. At week 12, or on early withdrawal from the study, a physical examination, laboratory tests, body weight determination and HAM‐D (17‐item) assessments were performed. After the week 12 visit, the dose of study medication was reduced during a thno week tapering period; safety assessments (but not efficacy assessments) were made during this period. Patients also attended a follow‐up visit when safety pammetcrs were assessed if they had withdrawn from the study prematurely owing to an adverse event, or if they had completed the study with an ongoing adverse event" |
|
| Notes | Industry funded: yes. Quote: "Smith Kline Beecharn Pharmaceuticals provided financial support for this study". Medication provided by industry: unclear Any of the authors work for industry: no |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Author correspondence: quote: "Block randomisation" |
| Allocation concealment (selection bias) | Low risk | Author correspondence: quote: "Randomisation was performed in the pharmacy at a distant site to the clinical site where the research team were based". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Author correspondence: quote: "All patients and research staff were blinded to treatment allocation at all centres throughout the study". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | A similar proportion of participants withdrew from the paroxetine group (35/139, 25%) compared to the placebo group (42/151; 28%). Common withdrawals in the paroxetine group were adverse experience and lack of efficacy in the placebo group. The 2 groups did not differ by sample characteristics at baseline. Quote: "There was no overall difference between the treatment groups in the number of patients who withdrew during the study: 35 (25%) patients in the paroxetine group v. 42 (28%) in the placebo group ... The number of patients lost to follow‐up, although comparable between the groups was high and is probably characteristic of the patient population under study; owing to the nature of the disorder". Overall 27% of the participants dropped out of the study. Quote: "Outcome measures were performed on the intent‐to‐treat (lTT) efficacy population. Last on‐therapy observations were carried forward for patients with missing data points". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias were identified. |
Barnett 2002.
| Methods | Design: single‐centre, randomised, placebo‐controlled, parallel, flexible dose, double‐blind Duration of intervention: 8 weeks Post‐treatment: no follow‐up Placebo run‐in: 1‐week single‐blind placebo lead‐in |
|
| Participants | Sample size: 12 randomised to olanzapine or placebo Participant age range: 18‐65 years Sex: not specified Diagnostic measure: DSM‐IV Inclusion criteria: quote: "Subjects included men and women aged 18–65 years with a diagnosis of SAD and a minimum Brief Social Phobia Scale (BSPS) score of 20". Exclusion criteria: quote: "At the initial visit, blood samples were obtained from eligible subjects for serum chemistry, haematology, and serum beta‐human chorionic gonadotropin for women of childbearing potential ... No concomitant psychotropic medications were permitted during the study". Dropouts: 5/12 (3/7 in the olanzapine group and 2/5 in the placebo group) |
|
| Interventions | Pharmacological intervention: quote: "Olanzapine was begun at a dose of 5 mg/day (or placebo equivalent) and was titrated upwards as tolerated and clinically indicated at the rate of 5 mg per week to a maximum of 20 mg/day". | |
| Outcomes | Primary and secondary outcomes: LSAS (for reduction of anxiety), BSPS (for reduction of anxiety), SPIN (for reduction of anxiety), SDS (for reduction of functional disability), CGI‐I (for treatment efficacy) and BAS, AIMS and SSS (side effects) Time points: Quote: "The BSPS, SPIN and SDS were performed at weeks 2, 3, 4, 6 and 8, and the LSAS at weeks 4 and 8. Global improvement was measured by the Clinical Global Impression‐Improvement scale (CGI‐I) at all post‐baseline visits (including week 1). Safety was assessed by recording adverse events using the Severity of Symptoms Scale, weight and vital signs, the Barnes Akathisia Scale (BAS) and the Abnormal Involuntary Movements Scale (AIMS)" |
|
| Notes | Industry funded: yes. Quote: "This study was supported by a grant from Eli Lilly and Company to Dr R. T. Davidson" Medication provided by industry: unclear Any of the authors work for industry: yes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The method of randomisation was not reported. Quote: "Subjects were then randomised in a 2:1 ratio to receive flexible‐dose olanzapine or placebo, respectively, for 8 weeks". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | A similar proportion of participants withdrew from the olanzapine group (3/7; 43%) compared to the placebo group (2/5; 40%). Participants withdrew due to adverse experience or were lost to follow‐up. The 2 groups did not differ by sample characteristics at baseline. Quote: "Data analysis was performed on the intent‐to‐treat (ITT) population using the last‐observation‐carried‐forward (LOCF) method for missing data ... Missing data has been imputed using appropriate methods ... Of the 12 randomized subjects, seven received olanzapine and five received placebo. Demographic characteristics did not differ significantly between groups. Seven subjects (four olanzapine and three placebo) completed the study through week 8 ... Reasons for early discontinuation were similar in both groups and included loss to follow‐up and adverse experience. Adverse experiences associated with subject discontinuation included gastrointestinal distress (placebo) and sedation (olanzapine). Overall 42% of the subjects dropped out of the study." |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias was identified. |
Blanco 2010.
| Methods | Design: multicentre, randomised, double‐blind, placebo‐controlled trial, maintenance study Duration of intervention: 12 weeks Post‐treatment: 6 months follow‐up Placebo run‐in: no |
|
| Participants | Sample size: 84 randomised to phenelzine sulfate or placebo (166 randomised: 45 phenelzine sulfate, 40 cognitive behavioural group therapy, 42 combination therapy, 39 placebo) Mean age (SD): 31.35 (8.36) years Sex: 63 men and 21 women Diagnostic measure: DSM‐IV Inclusion criteria: quote: "The inclusion criteria were (1) a primary DSM‐IV diagnosis of SAD and (2) age 18 to 65 years". Exclusion criteria: quote: "The exclusion criteria were (1) a comorbid anxiety disorder more clinically salient for the patient; (2) a lifetime history of schizophrenia, bipolar disorder, or mental disorder due to a general medical condition; (3) major depressive disorder or substance use disorder in the past 6 months; (4) previous failure of treatment with phenelzine or CBT, defined as nonresponse to 60 mg or more of phenelzine (or the equivalent dose of another monoamine oxidase inhibitor) for at least 4 weeks or to 6 sessions of CBT for SAD; (5) concurrent psychiatric or psychological treatment; and (6) pregnancy, lactation, or inability or unwillingness to use contraceptive measures for the duration of the study". Dropouts: 18/84 (13/45 in the phenelzine sulfate group and 5/39 in the placebo group; 40 dropouts across all 4 groups: 22 participants withdrew before receiving treatment, 18 withdrew after receiving treatment) |
|
| Interventions | Pharmacological intervention: quote: "Pharmacotherapy patients began with phenelzine sulfate, 15 mg/d, or matching placebo for 3 days, then 30 mg/d for 4 days, 45 mg/d for week 2, and 60 mg/d for weeks 3 and 4. Depending on clinical progress and adverse effects, the dosage could be raised to 75 mg for week 5 and to 90 mg for weeks 6 to 12". | |
| Outcomes | Primary and secondary outcomes: LSAS (for reduction of anxiety), ADIS (diagnostic measure), CGI‐S (for reduction of anxiety), HRSD (for reduction of depression), CGI‐I (for treatment efficacy), FQ (for reduction of anxiety), SIAS (for reduction of anxiety), SPS (for reduction of anxiety) and SDS (for reduction of functional disability) Time points: Quote: "The study had 4 phases. The first phase (acute treatment) lasted 12 weeks. Medication visits occurred weekly for 4 weeks, then every 2 weeks during this phase" The second, third and fourth phase was also 12 weeks each |
|
| Notes | Industry funded: no. Quote: "This study was supported in part by grants DA023200 (Dr Blanco), MH44119 (Dr Heimberg), and MH57148 (Dr Liebowitz) from the National Institutes of Health; by the New York State Psychiatric
Institute (Drs Blanco, Schneier, Campeas, and Liebowitz and Ms Vermes); and in part by General Clinical Research Center grant RR00349 from the National Center for Research Resources, National Institutes of Health, to Temple University". Medication provided by industry: no Any of the authors work for industry: yes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomized according to a table of pseudorandom numbers by the New York site data manager (A.B.S.), who had no patient contact". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Medication and placebo was provided in identical capsules. However, it is not clear whether the participants were blinded. Quote: "Pharmacotherapy patients began with phenelzine sulfate, 15 mg/d, or matching placebo for 3 days ... Patient allocation was concealed from all other research personnel at both sites before randomization and from independent evaluators providing the clinician administered ..." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Measures administered by independent evaluators blinded to treatment condition". |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Similar proportion of participants withdrew from the phenelzine sulfate group (13/45; 29%) compared to the placebo group (5/27; 19%). No information was provided regarding the reasons for treatment withdrawal. Nevertheless, participants did not differ by group characteristics at baseline. Quote: "Of the 166 individuals randomized, 12 from the placebo group and 10 from the phenelzine group withdrew from the study before receiving any treatment and were excluded from the analyses ... Groups did not differ significantly in demographic characteristics ... Rates of discontinuation were 37.1% (13 of 35) in the phenelzine group and 18.5% (5 of 27) in the placebo group. Those rates were not significantly different when examining all groups jointly or in pairwise treatment comparisons". Overall 25% of the participants dropped out of the study. Quote: "Using linear mixed‐effects models ... Response and remission rates were compared between groups using 2 tests of independence, using the last observation carried forward for individuals who dropped out before the endpoint". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Low risk | No other sources of bias was identified. Quote: "There were some baseline differences across treatment groups and sites. However, the results remained significant after appropriate statistical adjustments, suggesting the robustness of the findings". |
Blomhoff 2001.
| Methods | Design: multicentre, randomised, placebo‐controlled, parallel, flexible dose, double‐blind, maintenance study Duration of intervention: 24 weeks Post‐treatment: no follow‐up Placebo run‐in: 1 week single blind placebo period |
|
| Participants | Sample size: 196 randomised to sertraline or placebo (387 randomised: 98 sertraline, 98 sertraline and exposure therapy, 93 exposure therapy, 98 placebo) Mean age (SD): 40.4 (10.4) years for all 4 groups, not specified for the sertraline and placebo separately Sex: 153 men and 234 women (for all 4 groups, not specified for the sertraline and placebo separately) Diagnostic measure: DSM‐IV Inclusion criteria: quote: "Subjects aged 18‐65 years with GSP according to DSM‐IV criteria study of at least 1 years duration and rated as moderately ill were included in the study ... Patients with comorbid dysthymia or specific phobias were allowed to enter the study". Exclusion criteria: quote: "Patients with panic disorder with onset before social phobia or any other current anxiety, major depression, substance use or eating disorder were not eligible. In addition, patients with a lifetime history of bipolar disorder or psychosis were excluded". Dropouts: 16/196 (9/98 in the sertraline and 7/98 in the placebo groups) |
|
| Interventions | Pharmacological intervention: all participants received either 1 tablet of sertraline 50 mg or placebo once daily, the dose was increased to 100 mg at 4 weeks and 150 mg at 8 and 12 weeks | |
| Outcomes | Primary outcomes: CGI (for treatment efficacy) and SPS (for reduction of anxiety) Secondary outcomes: BSPS (for reduction of anxiety), MFQ (for reduction of anxiety), FNES (for reduction of anxiety), SDS (for reduction of functional disability) and SF‐36 (quality‐of‐life measure) Time points: Quote: "Investigators made intermediate efficacy ratings after 4, 8, 12, and 16 weeks, and final efficacy assessment after 24 weeks of treatment". |
|
| Notes | Industry funded: yes. Quote: "Funding was obtained from Pfizer Inc". Medication provided by industry: yes Any of the authors work for industry: no |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Three hundred and eighty‐seven patients were randomly assigned by a computer to receive double‐blind sertraline or placebo in blocks of eight subjects so that four patients in each block were randomised to each of the treatments". |
| Allocation concealment (selection bias) | Low risk | Quote: "Sealed envelopes of allocations from this list were kept by the investigators and opened after the inclusion of the patient into the study ... Tablets were packaged and numbered by the sponsor and personally delivered to each investigator". |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Medication and placebo was provided in sealed envelopes. However, it is not clear whether both participants and personnel were blinded. Quote: "Sealed envelopes of allocations from this list were kept by the investigators and opened after the inclusion of the patient into the study". |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Outcome assessors were not blinded to treatment. Quote: "Since many of the general practitioners included as investigators worked in single practices, it was not possible to obtain blinded efficacy assessment". |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | A small proportion of participants withdrew from the sertraline (9/98; 9%) and placebo groups (7/98; 7%). Reasons for withdrawal were not clearly stated by treatment group. Participants did not differ by group characteristics at week 24. Quote: "Two hundred and fifty‐three patients completed 24 weeks of treatment (65%). Three hundred and fifty‐four patients were included in the intent‐to‐treat efficacy population (93%) ... In individual analyses, no interaction was observed between response and each of the variables gender, age, country, recruitment method, medication or exposure therapy". Overall 8% of the participants dropped out of the study. |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Medication and study funded by industry. No other sources of bias was identified. |
Book 2008.
| Methods | Design: single‐centre, double‐blind, placebo‐controlled study (NCT00246441) Duration of intervention: 16 weeks Post‐treatment: no follow‐up Placebo run‐in: no |
|
| Participants | Sample size: 42 randomised to paroxetine or placebo Mean age (SD): 29 (7.4) years Sex: 22 men and 20 women Diagnostic measure: Structured Clinical Interview for DSM‐IV (SCID). Inclusion criteria: quote: "To be included in the study, individuals had to (1) be 18–65 years old; (2) have sufficiently severe social anxiety disorder, as defined by a total score of at least 60 on the Liebowitz Social Anxiety Scale; (3) report using alcohol to cope with social anxiety; and (4) consume at least 15 standard drinks in the previous 30‐day period". Exclusion criteria: quote: "Medical exclusion factors included: (1) history of prior medical detoxification from alcohol; (2) current use of psychotropic medications; (3) seeking treatment for alcohol problems; (4) urine drug screen positive for illicit drugs other than marijuana; and (5) liver enzymes greater than three times normal levels. History of prior medical detoxification or treatment seeking for alcohol problems was exclusionary for ethical reasons since no explicit alcohol intervention was provided ... They were excluded if they had current bipolar disorder, schizophrenia, substance abuse or dependence other than alcohol, nicotine, marijuana, or presence of significant suicidality". Dropouts: 4 (insufficient information to determine dropout rates for the 2 groups separately) |
|
| Interventions | Pharmacological intervention: quote: "All subjects were initiated at a dose of 10 mg per day of paroxetine or matching placebo. Active medication and placebo were over‐encapsulated by the investigational pharmacy with 100 mg of riboflavin, a biomarker used to measure medication compliance. The titration plan in the protocol was to increase the dose weekly over 4 weeks from 10 to 20 to 40 to 60 mg daily, pending tolerability". | |
| Outcomes | Primary outcome: LSAS (for reduction of anxiety) Secondary outcomes: LSAS (for reduction of anxiety), CGI‐I (for treatment efficacy), and SPIN (for reduction of anxiety) Time points: Quote: "At weekly visits throughout the trial the clinician also rates improvement in social anxiety severity as compared to baseline on the same 1–7 point scale (CGI‐I)" |
|
| Notes | Industry funded: no. Quote: "This work was supported by grants R01 AA013379 (CLR), K24 AA013314 (CLR), P50 AA010761, and K23 AA014430 (SWB) from the National Institute on Alcohol Abuse and Alcoholism". Medication provided by GlaxoSmithKline Any of the authors work for industry: no |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were randomised using a computerised urn randomisation programme. Quote: "Following determination of eligibility, subjects were randomized to either paroxetine or matching capsule placebo, using a computerized urn randomization program". |
| Allocation concealment (selection bias) | Low risk | Quote: "Group assignment was maintained by an investigational pharmacist, who also prepared each week’s supply of study medication". |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Medication and placebo was provided in identical capsules. However, it is not clear whether the participants were blinded. Quote: "Subjects were randomized to either paroxetine or matching capsule placebo ... All individuals involved in direct care or evaluation of study subjects, or who were involved in study supervision, were blind to group assignment". |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "All individuals involved in direct care or evaluation of study subjects, or who were involved in study supervision, were blind to group assignment ... Clinical and research ratings were collected independently". |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 38 (90%) of the 42 participants completed the study. No information was provided on the reasons for study withdrawal. Participants did not differ by group characteristics at baseline. Nevertheless, the total proportion of dropouts (10%) is relatively low, suggesting that dropout rates may not have biased the outcomes. Quote: "All but four participants provided week 16 (end of trial) data, for a 90% research data completion rate ... There were no significant differences at baseline between groups, including age, gender, ethnicity, social anxiety severity, and alcohol use severity ... There were no significant differences between groups, all p values >.05 ... The number of subjects who dropped out of the trial because of side effects were 1 and 0 for the paroxetine and placebo group, respectively" Quote: "Using a mixed model analysis ... Data from all subjects who were randomised to treatment were included in the analysis, according to intent to treat (ITT) standards". |
| Selective reporting (reporting bias) | High risk | Pre‐specified secondary outcomes (i.e. for quality of life and depression) were not mentioned or measured in the study. |
| Other bias | Unclear risk | Medication was provided by industry. No other sources of bias was identified. |
Connor 1998.
| Methods | Design: single‐centre, randomised, placebo‐controlled, flexible dose, double‐blind Duration of intervention: 20 weeks or to undergo discontinuation treatment every 2 weeks Post‐treatment: no follow‐up Placebo run‐in: no |
|
| Participants | Sample size: 36 randomised to clonazepam or placebo Mean age (SD): 40.05 (7.6) years Sex: 23 men and 13 women Diagnostic measure: DSM‐III‐R. Inclusion criteria: quote: "Subjects entered the study if they fulfilled DSM‐III‐R criteria for a principal diagnosis of social phobia, granted informed consent, and were between the ages of 18 and 55". Exclusion criteria: quote: "Exclusion criteria were as follows: a history of schizophrenia, bipolar disorder, organic brain syndrome, antisocial personality disorder, mental retardation, major depression within the past 12 months, panic disorder, alcohol or substance abuse; the concomitant need for other psychotropic drugs; or any ongoing psychotherapy". Dropouts: 8/36 (2/17 in the clonazepam and 6/19 in the placebo group) |
|
| Interventions | Pharmacological intervention: quote: "At week 24, all subjects exhibiting good clinical response on the CGI‐Improvement scale were randomly assigned to receive either continuation treatment (CT) at the same clonazepam dose for 5 additional months, or discontinuation treatment (DT), which required a fixed‐dose taper of 0.25 mg every 2 weeks. Therefore, 6 weeks of tapered doses were required for the group receiving 1.0 mg/day to reach 0.0 mg, 10 weeks for the 1.5‐mg group, 14 weeks for the 2‐mg group, and 18 weeks for the 2.5‐mg group". | |
| Outcomes | Primary and secondary outcomes: CGI‐S (for reduction of anxiety), BSPS (for reduction of anxiety), MSPSS (for reduction of anxiety), BWC (side effects measure), and fear was measured on a 0‐10 scale, and avoidance was measured along a 5‐point scale. Time points: Quote: "After patients were randomly assigned at week 24, all scales were administered at 2‐week intervals until study completion" |
|
| Notes | Industry funded: yes. Quote: "This work was supported by a grant from Hoffmann‐LaRoche to Dr. Jonathan Davidson". Medication provided by industry: unclear Any of the authors work for industry: no |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "All subjects exhibiting good clinical response on the CGI‐Improvement scale were randomly assigned to receive either continuation treatment (CT) at the same clonazepam dose for 5 additional months, or discontinuation treatment (DT)". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Participants were blinded to treatment however there was insufficient evidence to determine if personnel were blinded. Quote: "Subjects received the same number of pills at each visit, with the diminishing dose supplemented by means of matching placebo. From weeks 24 to 26, all subjects received their usual dosage in double‐blind packaging to allow for adjustment to the double‐blind form of medication, having received the regular, marketed brand of the drug up to that time". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | A larger proportion of participants discontinued the study in the placebo group (6/19; 32%) compared to the clonazepam (2/17; 12%) group. Similiar withdrawals were reported across groups. The 2 groups did not differ by treatment characteristics at baseline. Quote: "Within the group of 36 subjects providing discontinuation data, no significant differences were observed between subjects assigned to CT vs. DT groups in age, gender, or ethnic status ... Two subjects in the CT and six in the DT group dropped out of the study for reasons either related to relapse or to other circumstances. The two CT dropouts were a result of side effects and loss to follow‐up. The six DT dropouts were a result of relapse, marital problems that became aggravated during the time of taper, and work obligations". Overall 22% of the participants dropped out of the study. Quote: "In the event of occasional missing measurement points, the immediately prior observation was carried forward (LOCF)". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. No other source of bias was identified for this study. |
Davidson 1993.
| Methods | Design: single‐centre, randomised, placebo‐controlled, parallel, flexible dose, double‐blind Duration of intervention: 10 weeks Post‐treatment: no follow‐up Placebo run‐in: 2 week medication washout period |
|
| Participants | Sample size: 75 randomised to clonazepam or placebo Mean age (SD): 37.2 (8.45) years Sex: 43 men and 32 women Diagnostic measure: DSM‐III‐R Inclusion criteria: quote: "To be eligible for the study, subjects were required to fulfil DSM‐III‐R criteria for social phobia, with absence of major depression or panic disorder in the last 6 months. Additionally, at least 12 months absence of alcohol or substance abuse was required". Exclusion criteria: quote: "Histories of bipolar disorder, psychotic illness, or organic brain disease represented exclusion. Dropouts: 19/75 (10/39 in the clonazepam and 9/36 in the placebo group) |
|
| Interventions | Pharmacological intervention: quote: "Subjects were assigned to receive either clonazepam or placebo ... The initial dose was 0.25 mg per day for days 1 to 3, increasing to 0.25 mg twice daily on days 4 to 7.05 mg twice daily from days 8 to 14, 0.5 mg in the morning and 1mg at bedtime on days 15 to 17.1 mg twice daily on days 18 to 21, 1 mg in the morning and 1.5mg at bedtime on days 22 to 25, and 1.5 mg twice daily after day 25". | |
| Outcomes | Primary and secondary outcomes: CGI‐S (for reduction of anxiety), LSAS (for reduction of anxiety), FQ (for reduction of anxiety), FNES (for reduction of anxiety), HAMD (for reduction of depression) and Marks‐Kelly Disability Scale (SDS) (for reduction of functional disability) Time points: Quote: "All scales were administered at baseline and at weeks 2, 4, 6, 8, and 10, except for the FQ, which was administered at baseline and at weeks 6 and 10, and the Hamilton Rating Scale for Depression, which was administered at baseline and at week 10" |
|
| Notes | Industry funded: no Medication provided by industry: no Any of the authors work for industry: no |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Author correspondence: quote: "The randomization was determined by a list of computer‐generated numbers". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Author correspondence: quote: "For the clonazepam trial, I can say that the only person who was not blinded was the statistician, and we never had any talk or contact with him about the matter of blinding during the trial. Neither patients, staff nor raters were unblinded". |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Author correspondence: quote: "For the clonazepam trial, I can say that the only person who was not blinded was the statistician, and we never had any talk or contact with him about the matter of blinding during the trial. Neither patients, staff nor raters were unblinded". |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Similar proportions of participants withdrew from the clonazepam (10/39; 26%) and placebo group (9/36; 25%). Common withdrawals were a result of poor response. No information was provided on sample characteristics and how groups differed at end point. Quote: "The numbers of subjects remaining in treatment with clonazepam and placebo were at week 19, n=29 and n=27. 75% of clonazepam subjects and 75% of placebo subjects competed the full course of treatment ... Dropout rates at week 8 were generally the result of poor response". Overall 25% of the participants dropped out of the study. Both intention‐to‐treat and last observation carried forward (LOCF) was carried out. |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Low risk | No other source of bias was identified for this study. |
Davidson 2004a.
| Methods | Design: multicentre, randomised, placebo‐controlled, double‐blind, parallel group study Duration of intervention: 12 weeks Post‐treatment: no follow‐up Placebo run‐in: no, however, quote: "patients taking psychotropic medications were required to discontinue medication 14 days (fluoxetine 30 days) prior to baseline". |
|
| Participants | Sample size: 279 randomised to fluvoxamine or placebo Mean age (SD): 37.25 (0.95) years Sex: 179 men and 100 women Diagnostic measure: DSM‐IV Inclusion criteria: quote: "Patients had to meet the following inclusion criteria: male or female aged 18 to 70 years, Diagnostic and Statistical Manual, 4th Edition (DSM‐IV) diagnosis of GSAD according to the modified Structured Clinical Interview for the DSM‐IV, minimum score of 60 on the Liebowitz Social Anxiety Scale (LSAS) at the screening visit, a score of less than 18 on the Montgomery‐Asberg Depression Rating Scale at the screening visit, and fluency in English. Women with less than 1 year postmenopausal were required to use an acceptable form of birth control. Pregnant or lactating women were not eligible". Exclusion criteria: quote: "Patients were excluded from study participation if they had any of the following comorbid psychiatric disorders deemed to be primary in clinical significance: major depressive disorder, dysthymic disorder, or panic disorder. Patients with a history or current diagnosis of schizophrenia, psychotic disorder, obsessive compulsive disorder, bipolar affective disorder, or borderline personality disorder were also excluded. Patients with evidence of substance or alcohol abuse within the previous 6 months, patients with positive results on a urine drug screen, and patients requiring cognitive behavioral therapy to treat social anxiety disorder within the previous month were also excluded from participation. Patients taking psychotropic medications were required to discontinue medication 14 days (fluoxetine 30 days) prior to baseline. Patients were also excluded if they had a clinically significant medical condition or required medications that could put them at risk for taking fluvoxamine CR". Dropouts: 119/279 (66/139 in the fluvoxamine and 53/140 in the placebo group) |
|
| Interventions | Pharmacological intervention: quote: "Patients randomized to receive fluvoxamine CR began treatment at 100 mg/d at day 1 (baseline). The dose could be increased, based on efficacy and tolerability, in increments of 50 mg/d at 1‐week intervals up to a maximum dose of 300 mg/d. The dose remained constant during weeks 6 through 12. The minimum dose allowed at any time during the study was 100 mg/d". | |
| Outcomes | Primary outcome: LSAS (for reduction of anxiety) Secondary outcomes: CGI (for treatment efficacy), SDS (for reduction of functional disability), PGI (for treatment efficacy), ASEX (assesses sexual experiences), and MADRS (for reduction of depression) Time points: Quote: "Efficacy measures were assessed at baseline, weeks 2, 4, 6, 8, 10, and 12, or upon early termination" |
|
| Notes | Industry funded: yes. Quote: "This study was supported by a grant from Solvay Pharmaceuticals, Inc". Medication provided by industry: unclear Any of the authors work for industry: yes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Author correspondence: quote: "For fluvoxamine, randomization was determined for each site by the sponsor (Solvay) from a central source". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Author correspondence: quote: "All site study personnel and the patients remained blind throughout the study". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Similar proportions of participants withdrew from the fluvoxamine CR (66/139; 47%) and placebo group (53/140; 39%). Reasons for withdrawal were similar between groups except for lack of efficacy and adverse events. No information was provided on sample characteristics at endpoint, although groups did not differ significantly on demographics and clinical history at baseline. Quote: "Of these 279 patients, 73/139 (53%) in the fluvoxamine CR treatment group and 87/140 (62%) in the placebo treatment group completed the study, a non statistically significant difference. The reasons for withdrawal were similar between treatment groups with the exception of lack of efficacy (8% of the placebo group compared with <1% of the fluvoxamine CR group) and adverse events (26% of the fluvoxamine CR group vs. 1% of the placebo group)". Overall 43% of the participants dropped out of the study. Quote: "All analyses of response refer to the conventional last observation carried forward algorithm for all patients who had at least 1 dose of study medication, evaluable efficacy data at baseline and at least 1 post baseline efficacy assessment (intent‐to‐treat efficacy population, fluvoxamine CR = 121 patients; placebo = 126 patients)". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias was identified. |
Davidson 2004b.
| Methods | Design: multicentre, randomised, double‐blind, placebo‐controlled parallel group trial Duration of intervention: 14 weeks Post‐treatment: no follow‐up Placebo run‐in: no |
|
| Participants | Sample size: 117 randomised to fluoxetine or placebo (295 randomised: 57 fluoxetine, 60 comprehensive cognitive behaviour therapy group, 59 combinations of comprehensive cognitive behaviour therapy and fluoxetine, 59 combinations of comprehensive cognitive behaviour therapy and placebo, 60 placebo group) Mean age (SD): 36.6 (10.65) years Sex: 66 men and 51 women Diagnostic measure: DSM‐IV Inclusion criteria: quote: "Inclusion criteria were: (1) DSM‐IV diagnosis of GSP; (2) age between 18 and 65 years; (3) fluency in English; and (4) provision of written informed consent". Exclusion criteria: quote: "Exclusion criteria were: (1) a primary comorbid anxiety disorder (defined by which disorder was the more debilitating and clinically salient); (2) lifetime history of schizophrenia, bipolar disorder, or organic brain syndrome; (3) major depression within the last 6 months; (4) substance abuse or dependence within the past year; (5) mental retardation or pervasive developmental disability; (6) unstable medical condition; (7) prior failure of response to fluoxetine at 60 mg/d for at least 4 weeks or to 12 weekly sessions of CCBT for GSP; (8) concurrent psychiatric treatment or other psychoactive medications; (9) positive urine drug screen results; (10) inability to maintain 2 weeks’ psychotropic drug‐free washout; and (11) pregnancy or lactation". Dropouts: 33/117 (13/57 in the fluoxetine and 20/60 in the placebo group) |
|
| Interventions | Pharmacological intervention: quote: "Fluoxetine was started at 10 mg/d, increasing on day 8 to 20 mg/d, on day 15 to 30 mg/d, and on day 29 to 40 mg/d". | |
| Outcomes | Primary outcomes: CGI (for treatment efficacy) and BSPS (for reduction of anxiety) Secondary outcome: SPAI (for reduction of anxiety) Time points: Quote: "Independent evaluator ratings were conducted at baseline and at weeks 4, 8, and 14" |
|
| Notes | Industry funded: no. Quote: "This study was supported by grant R10‐ MH49339‐05A1 from the National Institute of Mental Health, Bethesda, Md (Drs Davidson and Foa)". Medication provided by industry: quote: "Medication and matching placebo were provided by Eli Lilly, Indianapolis, Ind". Any of the authors work for industry: no |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Subjects were assigned to treatment by block randomisation, which was generated by computer program". |
| Allocation concealment (selection bias) | Low risk | Author correspondence: quote: "The fluoxetine study ‐ I believe we provided medication in bottles which carried a pre‐numbered label based on the randomization". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Author correspondence: quote: "Patients, raters and medical staff were blind as to whether drug or placebo was given". |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "An independent rater, blinded to treatment assignments, conducted the primary outcome assessments". |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Similar proportions of participants withdrew from the clonazepam (13/57; 23%) and placebo group (20/60; 33%). Reasons for withdrawals were similar across groups (i.e. adverse effects, unclear, depression, not improving, treatment too difficult etc). No information was provided on sample characteristics at end point. Quote: "The overall significance for rate of dropout by treatment type was not statistically significant". Overall 28% of the participants dropped out of the study. Quote: "Linear mixed‐effect model analyses included all randomised subjects and were conducted using pretreatment and posttreatment behavioral measures, with the behavioral measure as the dependent variable". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Medication and study funded by industry. No other sources of bias were identified. |
Fahlen 1995.
| Methods | Design: multicentre, randomised, placebo‐controlled, parallel, fixed dose, double‐ blind Duration of intervention: 12 weeks Post‐treatment: 6, 9 and 12 months follow‐up Placebo run‐in: no |
|
| Participants | Sample size: 77 randomised to brofaromine or placebo Mean age (SD): 37.8 (10.3) years Sex: 45 men and 32 women Diagnostic measure: DSM‐III‐R Inclusion criteria: quote: "Patients with comorbid DSM‐III‐R generalized anxiety disorder, simple phobia or dysthymia were accepted in the study". Exclusion criteria: quote: "Patients with a history of DSM‐III‐R major depressive episode, a total score of 15 or more on the HDRS, those with other Axis I disorders, suicidal ideation, severe sleep disturbances, organic brain diseases, alcohol or drug abuse within the last 5 years, pregnancy or lactation or some other clinically relevant medical condition that might interfere with the study were excluded". Dropouts: 8/77 (5/37 in the brofaromine and 3/40 in the placebo group). |
|
| Interventions | Pharmacological intervention: quote: "Brofaromine or placebo was given twice daily; the first week 2 x 25 mg, the second week 2 x 75 mg to the dose of 150 mg/day". | |
| Outcomes | Primary outcomes: LSAS (for reduction of anxiety) and CGI (for treatment efficacy) Secondary outcomes: HRSD (for reduction of depression), MADRS (for reduction of depression), HARS (for reduction of anxiety), STAI (for reduction of anxiety) and SCL‐90 (for reduction of anxiety) Time points: Quote: "Assessments were made before treatment and at weeks 1, 2, 4, 6, 8 and 12. The HRSD was administered before treatment and at week 12 or when the patient prematurely discontinued the trial (end‐point). The Montgomery‐Asberg Depression Rating Scale (MADRS) was used at every visit" |
|
| Notes | Industry funded: yes. Quote: "Grants for the study were given by Ciba, Pharmaceuticals Division, Sweden". Medication provided by industry: unclear Any of the authors work for industry: yes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "The study was double blind and patients were randomised (1:1) to brofaromine or placebo and treated for 12 weeks". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | A small proportion of participants withdrew from the brofaromine (5/37; 14%) and placebo group (3/40; 8%). More participants in the brofaromine group withdrew due to side effects compared to the placebo group. However, the 2 groups did not differ significantly. Quote: "Five patients in the brofaromine group and 3 in the placebo group withdrew prematurely from the study. One brofaromine patient withdrew because of untolerable side effects, 1 after 3 days (increased anxiety) and 3 between week 2 and 10 (sleep disturbance, nausea and diarrhoea, and irritability and hyperactivity). The 3 placebo patients withdrew for administrative reasons, poor compliance and unsatisfactory therapeutic effect, respectively ... In all 35 different adverse symptoms were reported. The total number of such reports was 192 in the brofaromine group (n=36) and 94 in the placebo group (n=40). Most of the reported symptoms did not differ significantly between groups". Overall 10% of the participants dropped out of the study. ITT population was assessed and LOCF was used for missing data. |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias was identified. |
Feltner 2011.
| Methods | Design: multicentre, randomised, double‐blind, fixed dose study Duration of intervention: 11 weeks, with a 6‐day titration period and 1‐week taper period Post‐treatment: no follow‐up Placebo run‐in: no |
|
| Participants | Sample size: 329 randomised to pregabalin or placebo Mean age (SD): 35.4 (5.68) years Sex: 195 men and 134 women Diagnostic measure: DSM‐IV Inclusion criteria: quote: "Patients were enrolled if they were at least 18 years of age and met the DSM‐IV criteria for SAD, generalized subtype, confirmed using the Mini‐International Neuropsychiatric Interview, with a LSAS score of at least 50 at both screening and baseline. Women were enrolled if they were using a hormonal or barrier method of contraception, or were menopausal or surgically sterilized, and had a negative pregnancy test at the screening visit, and were not lactating". Exclusion criteria: quote: "Patients were excluded from the study for any of the following reasons: a current DSM‐IV diagnosis of panic disorder, with or without agoraphobia, GAD, anorexia, bulimia, delirium, dementia, or any other clinically significant cognitive disorders, major depressive disorder, obsessive‐compulsive disorder, posttraumatic stress disorder, or borderline or antisocial personality disorder; a current or past history of schizophrenic or psychotic disorder, bipolar disorder, or factitious disorder; a diagnosis of substance abuse/dependence unless in full remission for at least 6 months or a positive urine drug screen; a score of at least 3 on item 1 (depressed mood) at screening of the Hamilton Depression Rating Scale (HAM‐D); a creatinine clearance of less than 60 ml/min; any clinically significant or unstable hematological, autoimmune, endocrine, cardiovascular, renal, hepatic, gastrointestinal, or neurological disorder; electrocardiogram (ECG) changes indicating acute ischemia; a recent history of seizure disorder, and any need for treatment with anti‐convulsants; any previous treatment with pregabalin, or use of gabapentin or benzodiazepines within 2 weeks of baseline; or current use of any psychotropic medications". Dropouts: 96/329 (25/78, 25/86, 26/82 in the pregabalin and 20/82 in the placebo groups) |
|
| Interventions | Pharmacological intervention: quote: "Patients who continued to meet eligibility criteria at the end of the screening phase were randomized to double‐blind, parallel‐group treatment with one of three fixed daily doses of pregabalin, 300 mg [administered 100 mg three times daily (TID)], 450 mg (administered 150 mg TID), 600 mg (administered 200 mg TID), or matching placebo". | |
| Outcomes | Primary outcome measure: LSAS (for reduction of anxiety) Secondary outcome measures: HARS (for reduction of anxiety), HAM‐D (for reduction of depression), CGI‐I (for treatment efficacy), MFQ (for reduction of anxiety) and the SF‐36 Health Survey (measure of health status) Time points: Quote: "The LSAS and the MFQ were administered at screening, baseline, and weeks 1, 2, 4, 6, 8, and 10 (the LSAS was also administered at a follow‐up visit). The CGI‐I was administered on the same schedule starting at week 1. Other secondary measures were obtained at screening, baseline, and at week 10 (or the time of early termination)" |
|
| Notes | Industry funded: yes. Quote: "This study was funded by Pfizer Inc". Medication provided by industry: unclear Any of the authors work for industry: no |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "Patients who continued to meet eligibility criteria at the end of the screening phase were randomized to double‐blind, parallel‐group treatment with one of three fixed daily doses of pregabalin...". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Medication and placebo was provided in identical capsules. However, it is not clear whether both participants and personnel were blinded. Quote: "Patients who continued to meet eligibility criteria at the end of the screening phase were randomized to double‐blind, parallel‐group treatment with one of three fixed daily doses of pregabalin, 300 mg [administered 100 mg three times daily (TID)], 450 mg (administered 150 mg TID), 600 mg (administered 200 mg TID), or matching placebo". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | A similar proportion of participants withdrew from the pregabalin groups (25/78 300mg; 25/86 450mg; 26/82 600mg, 32%, 29%, and 31% respectively) and placebo group (20/82; 24%). More participants in the pregabalin groups withdrew due to side effects. Other reasons for withdrawal were similar across groups (i.e. discontinued, adverse events, lack of efficacy, withdrew consent, lost to follow‐up and miscellaneous). No information was provided on sample characteristics at endpoint; however, the groups were comparable on baseline characteristics. Quote: "The proportion of patients completing study treatment was slightly lower for patients in the pregabalin 300 (67.9%), 450 (70.9%), and 600 mg (68.3%) dosage groups compared with the placebo group (75.65) ... Baseline demographic and clinical characteristics were comparable among the four treatment groups ... The majority of patients in all treatment groups experienced adverse events during the double‐blind treatment phase. The proportion of patients experiencing at least one adverse event was higher in the pregabalin treatment groups than in the placebo group, though the rates among the three pregabalin dose groups were similar". Overall 29% of the participants dropped out of the study. Quote: "Efficacy measures were analysed using the intent‐to‐treat (ITT) population ... Endpoint was defined as last observation carried forward (LOCF)". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. Quote: "The relatively high attrition rate in the pregabalin treatment groups (29–32%) may have biased this analysis". |
Furmark 2005.
| Methods | Design: multicentre, randomised, double‐blind, placebo controlled, experimental trial Duration of intervention: 6 weeks Post‐treatment: no follow‐up Placebo run‐in: no |
|
| Participants | Sample size: 36 randomised to NK1 antagonist GR205171, citalopram or placebo Mean age (SD): 31.6 (7.7) years Sex: 17 men and 19 women Diagnostic measure: DSM‐IV Inclusion criteria: DSM‐IV criteria for social phobia with no other serious psychiatric disorders Exclusion criteria: quote: "Main criteria for exclusion were treatment of social anxiety in the past 6 months, current serious or dominant psychiatric disorder other than social phobia (e.g., psychosis, major depressive or bipolar disorder), neurological disorders, somatic disease, chronic use of prescribed medication, abuse of alcohol/narcotics, pregnancy, menopause, left handedness, previous PET examination, and positive family history of cancer". Dropouts: 0 |
|
| Interventions | Pharmacological intervention: quote: "The NK1 group received a daily oral dose of 5 mg GR205171, which started after 14 days of placebo because of limited available safety data on repeated dosing. GR205171 was taken as 4 mL solution made up to 100 mL in orange juice. The SSRI group was treated with 40 mg citalopram (one tablet), starting with 20 mg (half tablet) during the first week". | |
| Outcomes | Primary outcome measures: CGI‐I (for treatment efficacy), STAI‐S (for reduction of anxiety) and LSAS‐SR (for reduction of anxiety) Secondary outcome measures: CGI‐S (for reduction of anxiety), CGI‐I (for treatment efficacy), SPSQ (for reduction of anxiety), SPS (for reduction of anxiety), SIAS (for reduction of anxiety), GAF (for reduction of functional disability), PRCS (for performance anxiety), and SDI (for reduction of functional disability) Time points: Quote: "Response rate was determined by the Clinical Global Impression improvement item (CGI‐I) administered by a psychiatrist (K.W.) at weeks 2, 4, and 6 and at follow‐ups" |
|
| Notes | Industry funded: yes. Quote: "This research was funded by GlaxoSmithKline, with additional support from the Swedish Research Council (MF and TF), the Bank of Sweden Tercentenary Foundation (MF), and the Swedish Brain Foundation (TF)". Medication provided by industry: quote: "GlaxoSmithKline (Verona, Italy) supplied the study drugs for a 6‐week treatment period". Any of the authors work for industry: yes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Author correspondence: quote: "Randomization was performed by the statisticians at their Verona (Italy) research unit. Only the randomization list was provided to us, in my recollection a blocked randomization as the sample sizes were equal across the three arms". |
| Allocation concealment (selection bias) | Low risk | Author correspondence: quote: "The study codes (opaque envelopes) were locked in and kept safe by a Quintiles confederate during the whole study period until the study was unblinded". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Author correspondence: quote: "Personnel involved in the study only had access to a randomization list containing randomization numbers. The randomization list was created in Verona and the allocation was kept secret there in accordance with GSK research standards, also see above regarding the study codes. All participants and personnel involved in the study (planning, treatment, data collection, imaging, analyses, CRO activities etc) were blinded". |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Author correspondence: quote: "All participants and personnel involved in the study (planning, treatment, data collection, imaging, analyses, CRO activities etc) were blinded". |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There were no dropouts reported during this study. |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Medication and study funded by industry. No other sources of bias was identified. |
Heimberg 1998.
| Methods | Design: multicentre, randomised, placebo‐controlled, parallel, flexible dose, double‐blind Duration of intervention: 12 weeks Post‐treatment: 6 months follow‐up Placebo run‐in: no |
|
| Participants | Sample size: 64 randomised to phenelzine sulfate or pill‐placebo (133 randomised: 36 CBGT, 31 phenelzine sulfate, 33 pill‐placebo, 33 educational‐supportive group therapy) Mean age (SD): 34.1 (9.3) years Sex: 36 men and 28 women Diagnostic measure: DSM‐III‐R Inclusion criteria: quote: "For study inclusion, prospective patients had to meet criteria for social phobia and had to be between 18 and 65 years old, fluent in English, willing to provide written informed consent, and able to participate responsibly in treatment". Exclusion criteria: quote: "Exclusions included schizophrenia, major depression, prominent risk of self‐harm, organic mental disorder, history of bipolar I disorder, alcohol or substance abuse (within the past six months), a previous adequate trial of cognitive behavioural therapy (> 6 sessions) or MAOI treatment for social phobia, or any serious medical condition that would increase the patients chances of being harmed by study participation". Dropouts: 11/26 of 64 (5/31 in the phenelzine sulfate and 6/33 in the placebo groups, the additional dropouts were found in the CBGT (n=8) and ES group (n=7)) |
|
| Interventions | Pharmacological intervention: quote: "Patients received 15 mg phenelzine sulfate tablets (n=31) or matching placebo (n=33) in 1 morning dose; dosages of 60 mg/d and greater were split between morning and noontime. Dosage started at 15 mg/d and increased to 30 mg/d on day 4, to 45 mg/d on day 8, and to 60 mg/d on day 15. After 4 weeks dosages could be raised to 75 mg/d depending on symptoms and adverse effects. After 5 weeks dosages could be raised to 90 mg/d". | |
| Outcomes | Primary and secondary outcome measures: CGI‐I (SPDS) (for treatment efficacy), LSAS (for reduction of anxiety), ADIS‐R (for reduction of anxiety), FNES (for reduction of anxiety), FQ (for reduction of anxiety), SIAS (for reduction of anxiety), SPS (for reduction of anxiety) and SCL‐90 (for reduction of anxiety) Time points: Quote: "Assessments were repeated after 6 (interviews and questionnaires only) and 12 weeks of treatment" |
|
| Notes | Industry funded: no. Quote: "Supported by grant MH44 119 and MH40 121 from the National Institute of Mental Health, Bethesda, Md, and grant PO5 MH30906 from the New York State Psychiatric Institute Mental Health Clincal Research Center, New York". Medication provided by industry: quote: "Parke‐Davis Pharmaceuticals, Morris Plains, NJ, supplied Nardil and matching placebo". Any of the authors work for industry: no |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Author correspondence: quote: "The randomization sequence was generated by using a printed random numbers table from a statistics text book and was prepared before the study began, separately for each of the two study sites. The last digit of each number sequence was used to determine treatment allocation for cohorts of approximately 6 patients at a time (this was a study of group psychotherapy versus phenelzine)". |
| Allocation concealment (selection bias) | Low risk | Author correspondence: quote: "Cohorts of approximately 6 patients included both phenelzine and placebo patients randomly intermixed". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Author correspondence: quote: "Blinding was carried out by separating of functions between personnel, by separation of location of offices used for different purposes, and by the mixing of drug and placebo patients in the same cohort. Regarding medication/placebo status, patients, physicians, and assessors were blinded". |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Author correspondence: quote: "Regarding medication/placebo status, patients, physicians, and assessors were blinded. We conducted regular assessments of the integrity of blinding, and on the few occasions when it appeared necessary, we switched patients to different assessors. Regarding psychotherapy status, this was clearly known to patients and therapists". |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Similar proportions of participants withdrew from the phenelzine sulfate (5/31; 16%) and pill‐placebo group (6/33; 18%). Reasons for withdrawal were similar across groups (i.e. noncompliance, lack of efficacy, adverse effects, non treatment effects, unknown reasons, and positive effects) and groups did not differ by sample characteristics at week 12. Quote: "Attrition (n=26) did not differ across conditions. Eight patients discontinued CBGT, 5 discontinued phenelzine sulfate therapy, 6 discontinued placebo use, and 7 discontinued ES. Five patients were noncompliant, 5 patients discontinued therapy because of positive treatment effects, 3 because of lack of efficacy, 5 because of adverse effects, 2 because of non treatment‐related events, and 6 because of unknown reasons. There were no severe adverse effects ... Completers and dropouts did not differ on demographic or pretreatment clinical measures or group cohesion". Overall 17% of the participants dropped out of the study. All analyses were ITT with LOCF for dropouts. |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Medication was provided by industry. No other sources of bias was identified. |
Kasper 2005.
| Methods | Design: multicentre, randomised, placebo‐controlled, parallel, flexible dose, double‐blind Duration of intervention: 12 weeks Post‐treatment: 1 month follow‐up Placebo run‐in: 1‐week, single‐blind, placebo lead‐in period |
|
| Participants | Sample size: 358 randomised to escitalopram or placebo Mean age (SD): 38 (11) years Sex: 195 men and 163 women Diagnostic measure: DSM‐IV and MINI Inclusion criteria: quote: "The patient population comprised female and male out‐patients with a primary diagnosis of generalised social anxiety disorder established by means of a diagnostic interview following DSM–IV criteria, using the MINI to assist in the exclusion of disallowed comorbidity. At the screening visit, patients 18–65 years old were selected if they had a total score of at least 70 on the LSAS; with exhibited fear or avoidance traits in at least four social situations, and were otherwise healthy based on a physical examination". Exclusion criteria: quote: "Patients were excluded if they had another Axis I disorder that was considered the primary diagnosis within the previous 6 months, if the investigator diagnosed a serious risk of suicide or if the MADRS total score was higher than 19. Patients were also excluded if they had a DSM–IV diagnosis of alcohol or drug misuse during the past 6 months, or if they had taken a psychoactive drug (including any type of antidepressant, beta‐blocker, benzodiazepine, narcotic, analgesic, antipsychotic, or herbal remedy) within 2 weeks (5 weeks for fluoxetine and 6 months for depot neuroleptics) before screening, or if the patient had a positive urine drug screen for opiates, methadone, cocaine, amphetamines or benzodiazepines. The only allowed concomitant use of a psychotropic drug during the study was chloral hydrate taken as a hypnotic but not for more than three consecutive nights. Furthermore, patients with a diagnosis of mania or hypomania, body dysmorphic disorder, schizophrenia/other psychotic disorder, eating disorders, mental retardation or any Axis II cluster diagnosis were also excluded. Patients with a known drug (including citalopram) allergy or hypersensitivity or a known lack of therapeutic response to an adequate trial with citalopram were also excluded. Patients participating in a formal psychotherapy programme that went beyond medical counselling were not included". Dropouts: 68/358 (36/181 in the escitalopram and 32/177 in the placebo groups) |
|
| Interventions | Pharmacological intervention: quote: "The initial dosage of escitalopram was 10 mg per day. The dosage could be increased to 20 mg per day after 4, 6 or 8 weeks of treatment in case of an unsatisfactory response, judged as a score above 5 on the CGI–S rating for severity or no decrease in CGI–S score since baseline. The mean daily dose of escitalopram was 17.6mg at week 12". | |
| Outcomes | Primary outcome measure: LSAS (for reduction of anxiety) Secondary outcome measures: LSAS subscales (for reduction of anxiety), CGI‐I (for treatment efficacy), CGI‐S (for reduction of anxiety), SDS (for reduction of functional disability), and MADRS (for reduction of depression) Time points: Quote: "Efficacy and tolerability were assessed at baseline and after 1, 2, 3, 4, 6, 8 and 12 weeks of treatment" |
|
| Notes | Industry funded: yes. Quote: "The study was sponsored by H. Lundbeck A/S." Medication provided by industry: unclear Any of the authors work for industry: yes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "Patients with generalised social anxiety disorder were randomised to receive placebo (n=177) or 10‐20 mg escitalopram (n=181) in a 12‐week, double‐blind trial". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Medication and placebo was provided in identical capsules. However, it is not clear whether both participants and personnel were blinded. Quote: "Patients who met selection criteria entered a 1‐week, single‐blind, placebo lead‐in period before being randomised to 12 weeks of double‐blind treatment with escitalopram or matched placebo capsules". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Similar proportions of participants withdrew from the escitalopram (36/181; 20%) and placebo group (32/177; 18%). Withdrawals differed slightly, although not significantly, across groups (i.e. withdrew treatment, adverse effects, lack of efficacy, consent withdrawn, protocol violation, other administrative forms). More participants in the escitalopram group withdrew due to adverse effects, and more participants withdrew due to lack of efficacy in the placebo. The 2 groups did not differ by sample characteristics at baseline. Quote: "A total of 68 patients (19%) withdrew from the study, with no overall between group difference (18% in the placebo group and 20% in the escitalopram group). However, numerically more patients in the escitalopram group (8.8%) than in the placebo group (4.5%) withdrew because of adverse events and numerically more patients in the placebo group (6.2%) than in the escitalopram group (2.2%) withdrew because of lack of efficacy, with the latter difference approaching statistical significance ... There were slightly more men than women in both treatment groups. Baseline characteristics were similar for the two treatment groups with the exception of age and duration of the disorder, both of which were slightly higher in the escitalopram group". Overall 19% of the participants dropped out of the study. All analyses were ITT with LOCF for missing data. Observed cases analysed. |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias was identified. |
Katschnig 1997.
| Methods | Design: multicentre, randomised, placebo‐controlled, parallel, fixed dose, double‐blind Duration of intervention: 12 weeks Post‐treatment: no follow‐up Placebo run‐in: 1‐week placebo run‐in period |
|
| Participants | Sample size: 578 randomised to moclobemide or placebo Mean age (SD): 36.4 (9.9) years Sex: 329 men and 249 women Diagnostic measure: SCID‐Ro (adapted from the DSM‐IV) Inclusion criteria: quote: "Patients to be included in the study were adult men and non‐pregnant, non‐lactating women who satisfied the DSM IV criteria for social phobia". Exclusion criteria: quote: "Patients with any of the following disorders concurrently or within the prior 6 months were excluded from the study: panic disorder, agoraphobia, obsessive‐compulsive disorder, or major depression. Patients who met SCID‐Ro criteria for probable or definite substance abuse within the prior 6 months, as well as those who met lifetime criteria for bipolar disorder, schizophrenia or any other psychotic disorder, were also excluded. The patients were free of any significant unstabIe or uncontrolled medical disease, physical or psychological condition, medication, or treatment that might put them at risk or obscure or confound the effects of treatment". Dropouts: 133/578 (insufficient information to determine dropout rates for the 2 groups separately) |
|
| Interventions | Pharmacological intervention: quote: "After a 1‐week placebo run‐in period, patients fulfilling the entry criteria were randomly assigned to one of the three treatment groups to receive either 300 mg moclobemide, 600 mg moclobemide, or placebo in two divided daily doses for a 12‐week period. Patients were to take their tablets in the morning and in the evening after a meal. The patients of the 600 mg treatment group started with a reduced daily dose of 300 mg for the first 3 days, increasing to 600 mg on the 4th day". | |
| Outcomes | Primary and secondary outcome measures: LSPS (for reduction of anxiety), CIC‐SP (for treatment efficacy), SDS (for reduction of functional disability), CIS‐SP (for reduction of anxiety), PIC‐SP (for reduction of anxiety), HAM‐A (for reduction of anxiety), and the MADRS (for reduction of depression) Time points: Quote: "Assessments were performed at screen, on baseline and on weeks 1, 2, 3, 4, 6, 8, 10, and 12" |
|
| Notes | Industry funded: no Medication provided by industry: medication was supplied by F Hoffmann‐La Roche Ltd Any of the authors work for industry: no |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "After a 1‐week placebo run‐in period, patients fulfilling the entry criteria were randomly assigned to one of the three treatment groups to receive either 300 mg moclobemide, 600 mg moclobemide, or placebo in two divided daily doses for a 12‐week period". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. Quote: "Moclobemide was supplied by F. Hoffmann‐La Roche Ltd as an oval, cylindrical, biconvex, film‐ coated tablet light yellow in colour and scored on one side, containing 150 mg moclobemide. Placebo tablets were identical both in appearance and composition". |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Medication and placebo was provided in identical capsules. However, it is not clear whether both participants and personnel were blinded. Quote: "Moclobemide was supplied by F. Hoffmann‐La Roche Ltd as an oval, cylindrical, biconvex, film‐ coated tablet light yellow in colour and scored on one side, containing 150 mg moclobemide. Placebo tablets were identical both in appearance and composition". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | There was insufficient information to determine dropout rates for the 2 groups separately, although the study reported that dropouts were similar across groups. Common reasons for withdrawal across group were insufficient therapeutic response, withdrawal of consent, and adverse events. Quote: "The ITT population comprised 578 patients, who had received treatment and had at least one assessment after baseline; 445 patients completed the study through week 12. The most frequently cited reasons for discontinuation were insufficient therapeutic response (63 patients), withdrawal of consent (22 patients), and adverse events (19 patients). Attrition rates were similar among the three treatment groups (< 30%). The groups did not differ with respect to reasons for early termination, except that insufficient therapeutic response was somewhat more frequent in the placebo group (26 patients) than in the moclobemide 300 mg (18 patients) or 600 mg groups (19 patients). The three treatment groups were similar with respect to their demographic data and baseline characteristics of social phobia and concurrent psychiatric illnesses". Quote: "Demographic results presented in this paper are based on the ITT population. Efficacy results of weeks 8 and 12 were analysed with the last observation carried forward in the case of missing observations (LOCF analysis)". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Medication was provided by industry. No other sources of bias were identified. |
Katzelnick 1995.
| Methods | Design: single‐centre, randomised, placebo‐controlled, crossover, flexible dose, double‐blind Duration of intervention: 10 weeks Post‐treatment: 3 and 4 months follow‐up Placebo run‐in: no |
|
| Participants | Sample size: 12 randomised to sertraline or placebo Mean age (SD): 42.62 (7.54) years Sex: 8 men and 4 women Diagnostic measure: DSM‐III‐R Inclusion criteria: men and women with a DSM‐III‐R social phobia diagnosis. Exclusion criteria: not specified Dropouts: 2/12 (2/6 in the sertraline and 0/6 in the placebo groups) |
|
| Interventions | Pharmacological intervention: quote: "The subjects were randomly assigned to receive either sertraline (N=6) (50‐200 mg/day, flexible dosing) or placebo (N=6) for 10 weeks, followed by taper and no treatment for 2 weeks. The subjects were then crossed over to the other treatment for a further 10 weeks. The sertraline dose was begun at 50 mg/day and was increased 50 mg/day every 2 weeks if there was no treatment response, except if the drug was not tolerated". | |
| Outcomes | Primary outcome measure: LSAS (for reduction of anxiety) Secondary outcome measures: BSPS (for reduction of anxiety), FQ (for reduction of anxiety), SDS (for reduction of functional disability), SF‐36 (MOS) (to measure general health), MADRS (for reduction of depression), and the Liebowitz Social Phobic Disorders Rating Form (for reduction of anxiety) change and severity scales Time points: Quote: "The patients were seen for administration of the outcome measures at baseline and at the end of weeks 2, 6, 10, 12, 14, 18, and 22" |
|
| Notes | Industry funded: yes. Quote: "Supported in part by a grant from Pfizer Pharmaceuticals". Medication provided by industry: unclear Any of the authors work for industry: no |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "The subjects were randomly assigned to receive either sertraline (N=6) (50‐200 mg/day, flexible dosing) or placebo (N=6) for 10 weeks ...". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | A larger proportion of participants discontinued the study in the sertraline (2/6; 33%) group compared to the placebo group (0/6; 0%). One participant withdrew because of an adverse effect and the other withdrew because of lack of efficacy. No information was provided on whether groups differed by sample characteristics at week 10. Quote: "Overall, sertraline was well tolerated. Only one patient left the study early because of adverse events (e.g., queasiness, anxiety, and insomnia); that patient did so after treatment with sertraline, before crossover to placebo. The only other patient to discontinue early dropped out 1 week after crossover to placebo, because of a lack of efficacy after a substantial clinical response to sertraline during the first half of the study". Overall 17% of the participants dropped out of the study. All analyses were intention‐to‐treat. |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias were identified. |
Kobak 2002.
| Methods | Design: single‐centre, randomised, placebo‐controlled, parallel, flexible dose, double‐blind Duration of intervention: 14 weeks Post‐treatment: 1 month follow‐up Placebo run‐in: 1‐week, single‐blind, placebo lead‐in |
|
| Participants | Sample size: 60 randomised to fluoxetine or placebo Mean age (SD): 39.47 (12.84) years Sex: 25 men and 35 women. Diagnostic measure: DSM‐IV Inclusion criteria: quote: "The sample consisted of 60 subjects with a primary DSM‐IV diagnosis of generalized social phobia with a duration of at least 6 months". Exclusion criteria: quote: "Patients were excluded from the study for the following reasons: other concurrent Axis I disorders within the past 12 months; intolerance or nonresponse to previous fluoxetine treatment; previous participation in a fluoxetine study; concurrent use of psychotropic or centrally acting drugs, anticonvulsants, corticosteroids, or tryptophan; pregnancy or lactation; serious suicide risk; serious medical illness or abnormal lab or electrocardiogram results; history of severe allergies, multiple adverse drug reactions, or seizure disorder (with seizure during the last 12 months); or treatment with any form of psychotherapy during the trial". Dropouts: 12/60 (5/30 in the fluoxetine and 7/30 in the placebo groups) |
|
| Interventions | Pharmacological intervention: quote: "During the first 8 weeks of the 14‐week initial treatment phase, patients were started on either placebo or a fixed dose of fluoxetine 20 mg/day. During this time, a patient’s dose could be decreased to 10 mg/day at any time if an adverse event occurred that would have caused the patient to withdraw from the study. The patient could be increased back to 20 mg/day at the investigator’s discretion if the patient did not experience adverse events while receiving 10 mg/day and was not improving. During the last 6 weeks of the 14‐week initial phase, the patient’s dose could be increased every 2 weeks in 20 mg/day increments to a maximum of 60 mg/day, except for patients who were on 10 mg/day, who must first have been increased to 20 mg/day. A patient’s dose could also be reduced in decrements of 20 mg/day (or from 20 to 10 mg/day) at any time at the investigator’s discretion if an adverse event occurred that would have caused the patient to discontinue the study. Subsequently, a patient’s dose could be increased in increments of 20 mg/day (at scheduled visits only) back to a maximum of 60 mg/day (or from 10 to 20 mg/day followed by increments of 20/mg day to a maximum of 60 mg/day) if the patient did not experience adverse events at a subsequent visit and was not improving. This dosing schedule was used in order to evaluate whether patients would respond to 20 mg (an effective dose for depression) after 8 weeks, and if not, whether increasing the dose to as much as 60 mg would result in a positive response" | |
| Outcomes | Primary outcome measure: LSAS (for reduction of anxiety) Secondary outcome measures: the Social Phobia Subscale of the FQ (for reduction of anxiety), CGI‐S (for reduction of anxiety), CGI‐I (for treatment efficacy), PGI (for treatment efficacy), HAM‐A (for reduction of anxiety), BSPS (for reduction of anxiety), HAM‐D (for reduction of depression), GAF (for reduction of functional disability) and SF‐36 (for quality of life) Time points: Quote: "The LSAS, CGI‐S, FQ, and BSPS were administered at all visits. The CGI‐I and PGI scales were administered at visits 2 through 17. The HAM‐A, HAM‐D, GAF, and SF‐36 were administered at visits 2, 5, 7, 10, and 17 (or final visit if discontinued early). The HAM‐A and HAM‐D were also administered at visit 1" |
|
| Notes | Industry funded: yes. Quote: "This work was supported by a grant from Eli Lilly & Co". Medication provided by industry: unclear Any of the authors work for industry: no |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "randomly assigned to one of three treatment conditions". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Similar proportions of participants withdrew from the fluoxetine (5/30; 17%) and placebo groups (7/30; 23%). The proportions of women to men was 35:25. Common withdrawals included: participant moved, protocol violation, clinical relapse and adverse events. No information was provided on sample characteristics at end point, however. Quote: "Five (16%) of the 30 fluoxetine patients and 7 (23%) of the 30 placebo patients discontinued before completion of the 14‐week, double‐blind initial therapy phase. Fluoxetine was discontinued because the patient moved (1) or there was a protocol violation (2), clinical relapse (1), or adverse event (1) (palpitations). Placebo was discontinued because the patient moved (1) or there was a lack of efficacy (2), clinical relapse (1), or adverse event (3) (diarrhea, depression, and rash, respectively)". Overall 23% of the participants dropped out of the study. |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. There was a high placebo response in this trial. Quote: "By comparison, the change on placebo in the current trial (23.37) was greater than any other individual trial and much greater than the mean placebo change found in the previous studies (11.13). The reason for the high placebo response in this trial is unknown". |
Lader 2004.
| Methods | Design: multicentre randomised, placebo‐controlled, fixed‐dose, active‐reference study Duration of intervention: 12 and 24 weeks Post‐treatment: 1 month follow‐up Placebo run‐in: 1‐week, single‐blind, placebo lead‐in period |
|
| Participants | Sample size: 839 randomised to escitalopram, paroxetine, or placebo Mean age (SD): 36.98 (11.2) years Sex: 394 men and 445 women Diagnostic measure: DSM‐IV Inclusion criteria: quote: "The selection criteria were chosen to select physically healthy female and male outpatients with a primary diagnosis of generalised SAD according to DSM‐IV criteria. At the screening visit, patients between 18–65 years of age were included if they had a total score > 70 on the Liebowitz Social Anxiety Scale (LSAS), demonstrable fear and avoidance traits in at least four social situations, and a score > 5 on one or more of the Sheehan Disability Scale (SDS) subscales". Exclusion criteria: quote: "Patients were excluded if: a) they had another Axis I disorder designated the primary diagnosis within the previous 6 months; b) they had a MADRS total score > 18; c) they had a DSM‐IV diagnosis of schizophrenia/other psychotic disorder, mania or hypomania or history thereof, or were currently suffering from alcohol or drug abuse, eating disorders, MDD, panic disorders (patients with panic attacks not due to panic disorders could be included), obsessive compulsive disorders (OCD), body dysmorphic disorder; d) they had an Axis II Cluster B diagnosis; e) they had learning difficulties or had other cognitive disorder; f) the investigator detected a serious risk of suicide or the patient had a score > 5 in the MADRS item 10 (suicidal tendencies); g) they had a known lack of therapeutic response to any SSRI; h) they had a known hypersensitivity to citalopram or escitalopram or a history of severe drug allergy or hypersensitivity; i) they had taken a psychoactive drug (including antidepressants, beta‐blockers, benzodiazepines, antipsychotics, and psychoactive herbal remedies), monoamine oxidase inhibitors (MAOI), or prophylactic treatment (lithium, valproate, or carbamazepine) within 2 weeks (5 weeks for fluoxetine) before screening, an investigational drug (within 3 months before), or triptans; or j) they were receiving (or planning to initiate) formal psychotherapy". Dropouts: 242/839 (42/167, 56/167, and 49/170 in the escitalopram groups, 45/169 in the paroxetine group, and 50/166 in the placebo group, 24 weeks). |
|
| Interventions | Pharmacological intervention: quote: "After screening, patients entered a 1‐week, single‐blind, placebo lead‐in period before being randomised equally to 24 weeks of double‐blind treatment with fixed doses of escitalopram (5, 10, or 20 mg/day), paroxetine (20 mg/day), or placebo. Patients who completed double‐blind treatment entered a 2‐week, single‐blind, placebo run‐out period". | |
| Outcomes | Primary outcome measure: LSAS (for reduction of anxiety) Secondary outcome measures: LSAS (fear/anxiety, avoidance) scores (for reduction of anxiety), CGI‐S (for reduction of anxiety), CGI‐I (for treatment efficacy), SDS (for reduction of functional disability) and DESS (assess side effects) Time points: Quote: "Efficacy and tolerability were assessed at baseline and after 1, 2, 4, 6, 8, 10, 12, 16, 20, 24, 25, and 26 weeks of treatment; tolerability was also assessed 30 days after the last double‐blind dose of study product. Adverse events were assessed at all visits and the clinical assessments were made at the screening visit, and at Weeks 12 and 24" |
|
| Notes | Industry funded: yes. Quote: "Contract grant sponsor: H. Lundbeck A/S" Medication provided by industry: unclear Any of the authors work for industry: yes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participantubjects were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "After screening, patients entered a 1‐week, single‐blind, placebo lead‐in period before being randomised equally to 24 weeks of double‐blind treatment with fixed doses of escitalopram". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Similar proportions of participants withdrew from the escitalopram (42/167, 25.1%; 56/167, 33.5%; 49/170, 28.8%) paroxetine (45/169, 26.6%) and placebo groups (50/166, 30.1%). Primary reasons for withdrawals were adverse events, lack of efficacy, withdrawal of consent and lost to follow‐up. The groups did not differ by sample characteristics at baseline. Quote: "There were no clinically relevant differences in patient demographics or baseline values between the five treatment groups. The treatment groups did not differ significantly in age of SAD onset or duration of SAD, baseline height, weight, or BMI ... Two hundred forty‐two patients (29%) withdrew from the study during the 24‐week, double‐blind period, with similar withdrawal rates in all treatment groups; 22% of all patients had withdrawn by Week 12. Withdrawals due to adverse events were lowest in the 5 mg escitalopram group, whereas withdrawals due to lack of effect were highest in the placebo group. Withdrawal of consent and loss to follow‐up each accounted for < 7% of withdrawals in any treatment group. Because most of the withdrawals occurred in the first 12 weeks of the study, the remaining 12 weeks was too long a period to carry observations forward, so most of the efficacy analyses are based on observed case (OC) analysis". Overall 29% of the participants dropped out of the study. |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias were identified. |
Lepola 2004.
| Methods | Design: multicentre, randomised, double‐blind, placebo‐controlled, flexible‐dose (GSK protocol ID: 29060/790) Duration of intervention: 12 weeks Post‐treatment: no follow‐up Placebo run‐in: 1‐week, single‐blind, placebo run‐in period |
|
| Participants | Sample size: 370 randomised to paroxetine or placebo (375 randomised: 5 participants withdrew prior to treatment and were excluded from the study, the ITT sample comprised of 370 participants) Mean age (SD): 38.85 (11) years Sex: 270 men and 100 women Diagnostic measure: MINI (according to DSM‐IV criteria) Inclusion criteria: quote: "The Mini‐International Neuropsychiatric Interview (Version 5.0; MINI) was used to screen for social anxiety disorder according to DSM‐IV criteria. Outpatients (≥ 18 years of age) who met the criteria as their primary diagnosis were enrolled. Patients older than 65 years were included if they did not have renal or hepatic impairment". Exclusion criteria: quote: "Patients with a Clinical Global Impressions (CGI)‐Global Improvement score of 1 or 2 at baseline (following the placebo run‐in period) or a score on the 17‐item Hamilton Rating Scale for Depression (HAM‐D) of ≥ 15 at baseline were excluded. Patients evaluated with the MINI who met DSM‐IV criteria for Axis I disorders such as major depressive disorder, obsessive‐compulsive disorder, or panic disorder as a primary diagnosis currently or within 6 months prior to the screening visit were excluded. Also excluded were patients with substance abuse within 3 months of screening or substance dependence within 6 months of screening and patients considered a current homicidal or suicidal risk. Patients with a history of seizures (except febrile seizures), schizophrenia, or bipolar disorder or a current diagnosis of body dysmorphic disorder or a serious medical illness were excluded. In addition, patients who had been treated with psychotropic medications or antidepressants within 14 days of screening, monoamine oxidase inhibitors or fluoxetine within 4 weeks of screening, depot neuroleptics within 12 weeks of screening, or electroconvulsive therapy within the past 3 months were excluded. Patients requiring concomitant therapy with β‐adrenergic blockers, monoamine oxidase inhibitors, benzodiazepines, or other psychoactive medications were excluded. Women who were pregnant, lactating, or of childbearing potential and not practicing a clinically accepted method of contraception were ineligible". Dropouts: 77/370 (30/186 in the paroxetine group and 47/184 in the placebo groups) |
|
| Interventions | Pharmacological intervention: quote: "All patients randomly assigned to paroxetine CR began therapy at 12.5 mg and remained at this daily dose for the first 2 weeks of treatment. Dose elevation was permitted in 12.5‐mg/day increments no more frequently than every 7 days to a maximum of 37.5 mg/day. One dose reduction was permitted only when made necessary by the development of an adverse event. Patients completing the study (or withdrawing prematurely) at doses of 37.5 mg/day received 1 week of taper phase medication at a daily dose of 25 mg before stopping treatment". | |
| Outcomes | Primary outcome measures: LSAS (for reduction of anxiety) and CGI‐I (for treatment efficacy) Secondary outcome measures: CGI‐S (for reduction of anxiety), SADS (for reduction of anxiety), SDS (for reduction of functional disability) and HAM‐D (for reduction of depression) Time points: Quote: "After the initial screening visit, these efficacy assessments were administered at baseline and weeks 1, 2, 3, 4, 6, 8, and 12 (or at the time of early withdrawal from the study). In addition, the 17‐item HAM‐D was administered by a clinician at baseline and at week 12 (or at the time of early withdrawal)" |
|
| Notes | Industry funded: yes. Quote: "This study was funded by GlaxoSmithKline, Research Triangle Park, N.C". Medication provided by industry: unclear Any of the authors work for industry: yes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "Eligible patients were then randomly assigned at baseline to receive paroxetine CR (paroxetine hydrochloride) (flexible dose range of 12.5–37.5 mg/day) or placebo once daily in a 1:1 ratio for a treatment duration of 12 weeks". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Medication and placebo was provided in identical capsules. However, it is not clear whether both participants and personnel were blinded. Quote: "Patients randomly assigned to placebo medication received placebo throughout the study and were dosed in an identical manner to patients randomly assigned to paroxetine CR". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Similar proportions of participants withdrew from the paroxetine (30/186, 16%) and placebo groups (47/184, 26%). Five participants withdrew prior to treatment and were excluded from the ITT analysis. Common withdrawals include adverse events, protocol deviation, loss to follow‐up and other. More participants in the placebo group discontinued due to lack of efficacy. The 2 groups did not differ by sample characteristics a baseline. Quote: "The treatment groups were generally comparable with respect to age, gender, and race ... A total of 156 patients (83.9%) in the paroxetine CR group and 137 patients (74.5%) in the placebo group completed the 12‐week study. Dropout rates due to adverse events were low and comparable in the 2 treatment groups (2.7% in the paroxetine CR group and 1.6% in the placebo group). A greater proportion of patients in the placebo group withdrew from the study prematurely due to lack of efficacy (2.2% in the paroxetine CR group and 15.8% in the placebo group)". Overall 21% of the participants dropped out of the study. The ITT population was assessed and LOCF and observed cases were used for primary and or secondary missing data. |
| Selective reporting (reporting bias) | Low risk | All outcomes were reported. |
| Other bias | Unclear risk | Funding for study provided by industry. The study had various limitations with regards to comparisons of paroxetine CR and IR, as well as dosage ranges different to prior studies conducted. The high percentage of dropouts in the placebo group due to lack of efficacy, however, raises the question of whether blinding of participants was broken. Quote: "It must be emphasized that this study did not include a comparison of paroxetine CR and paroxetine IR. Moreover, the dose ranges studied in the current study were not identical to the dose ranges employed in prior studies, hence conclusions regarding their relative tolerability and efficacy profiles cannot be drawn from these trials ... A greater proportion of patients in the placebo group withdrew from the study prematurely due to lack of efficacy (2.2% in the paroxetine CR group and 15.8% in the placebo group)". |
Liebowitz 1992.
| Methods | Design: single‐centre, randomised, placebo‐controlled, parallel, flexible dose, double‐blind Duration of intervention: 8 weeks Post‐treatment: no follow‐up Placebo run‐in: 1‐week, single‐blind, placebo run‐in |
|
| Participants | Sample size: 85 randomised to phenelzine, atenolol or placebo (data however is reported for 74 completers) Mean age (SD): 34.3 (8.5) years Sex: 51 men and 23 women Diagnostic measure: DSM‐III Inclusion criteria: quote: "Medically healthy patients aged 18 to 50 with a DSM‐III criteria for social phobia". Exclusion criteria: quote: "Current major depression or substance abuse, a history of schizophrenia, organicity, or bipolar disorder, avoidant personality disorder, and other medical conditions (e.g. benign essential tumours), as well as prior treatment of phenelzine or atenolol". Dropouts: 11/85 (4/29 in the phenelzine, 5/28 in the atenolol and 2/28 in the placebo groups) |
|
| Interventions | Pharmacological intervention: "Treatment with atenolol began at 50 mg/d given in the morning and raised to 100 mg/d, if tolerated, after 2 weeks. Treatment with phenelzine sulfate was begun at 15mg/d and increased to 39 mg/d on day 4, to 45 mg/d on day 8, and to 60 mg/d on day 15. After 4 weeks, depending on clinical state and side effects, the dose of phenelzine sulfate could be optimally raised to 75 mg/d, and to 90 mg/d after 5 weeks. In addition to the 8 week short term treatment phase, the study had maintenance (8 weeks) and discontinuation phases (8 weeks)". | |
| Outcomes | Primary and secondary outcome measures: self‐report versions of the CGI‐S (for reduction of anxiety), LSPS (for reduction of anxiety), CGI‐I (for treatment efficacy), HSC (for reduction of anxiety), SADS (for reduction of anxiety), FNES (for reduction of anxiety), FQ (for reduction of anxiety), WPI (personality measure), HAM‐D (for reduction of depression), HAM‐A (for reduction of anxiety) and SDS (for reduction of functional disability) Time points: Quote: "These occurred before placebo washout, before randomisation, and at 4‐week intervals thereafter. Patients also underwent weekly physician and self‐ratings" |
|
| Notes | Industry funded: no. Quote: "This study was supported in part by grant MH 40121 from the National Institute of Mental Health, Bethesda, Md". Medication provided by industry: yes. Quote: "Parke‐Davis Pharmaceuticals Co, Morris Plains, NJ, kindly supplied phenelzine, and Stuart Pharmaceuticals, Wilmington, Del, kindly supported atenolol for the study". Any of the authors work for industry: no |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "randomised to an 8 week short term comparison" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | A larger proportion of participants discontinued the study in the phenelzine (4/25; 16%) and atenolol groups (5/23; 22%) compared to the placebo group (2/26; 8%). Common withdrawals included rash and other sides effects and change of mind. Groups did not differ by sample characteristics at end point, however. Quote: "Of the 85 patients randomised, 74 completers met prospectively determined criteria for inclusion in end‐phase analyses of at least four weeks of randomised treatment with 2 weeks of phenelzine or atenolol. Eleven other, including two placebo‐, four phenelzine‐, and five atenolol‐blind treatment failed to complete at least four weeks of double‐blind treatment. Reasons for doing so included rash, and cheese rash, change of mind, rediagnosis of schizophrenia,and painful erection, rash and other side effects and non compliance with study procedures ... There were no significant difference in demographic or baseline scores amongst the 74 completers". Overall 15% of the participants dropped out of the study. |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Medication was provided by industry. No other sources of bias were identified. |
Liebowitz 2002.
| Methods | Design: multicentre, randomised, placebo‐controlled, parallel, fixed dose, double‐blind Duration of intervention: 12 weeks Post‐treatment: no follow‐up Placebo run‐in: 1 week, single blind, placebo run‐in |
|
| Participants | Sample size: 384 randomised to paroxetine or placebo Mean age (SD): 36.95 (40.2) years Sex: 225 men and 159 women Diagnostic measure: SCID (modified version of the DSM‐IV) Inclusion criteria: quote: "Adult outpatients (>18 years of age) who met the criteria for the generalised sub‐type of social anxiety disorder were enrolled; patients older than 65 years were permitted if they did not have renal or hepatic impairment and could tolerate paroxetine starting dose of at least 20 mg/day". Exclusion criteria: quote: "Patients who scored 1 or 2 on the CGI‐I scale at baseline or who had a score greater than or equal to 15 at baseline on the HAM‐D scale were excluded. Patients with comorbid psychiatric disorders such as major depression, OCD, generalised anxiety disorder, and panic disorder were excluded using the SCID if comorbid disorder occurred within the past 6 months and was predominant. Also excluded were patients with substance abuse or dependence within 6 months of baseline, body dysmorphic disorder, schizophrenia, bipolar disorder, homicidal/suicidal tendencies, serious medical illness, or a history of seizures, as well as patients who had started psychotherapy within 6 months of baseline or who had been treated with other psychotropic medications or antidepressants within 14 days of baseline, fluoxetine within 5 weeks of baseline, electroconvulsive therapy within the past 3 months. Patients requiring concomitant therapy with beat‐adrenergic medications, warfarin, anticoagulants, digitalis glycosides, phenytoin, cimetidine, or sumatriptan were not included. Women who were pregnant, lactating, or of child‐bearing potential and not practicing a clinically accepted method of contraception were ineligible". Dropouts: 142/384 (31/97, 40/95, 43/97 in the paroxetine and 28/95 in the placebo groups) |
|
| Interventions | Pharmacological intervention: quote: "All patients randomly assigned to paroxetine began therapy at 20 mg/day. Patients were instructed to take 2 capsules each morning irrespective of treatment assignment. Those randomly assigned to paroxetine 20 mg, remained at that dose for the duration of the study. At week 1, patients randomly assigned to the 40 mg paroxetine group were titrated to that daily dose. Doses for the 60 mg paroxetine group were titrated to 40 mg at week 1 and 60 mg at week 2". | |
| Outcomes | Primary outcome measures: LSAS (for reduction of anxiety) and CGI (for treatment efficacy) Secondary outcome measures: LSAS (fear and anxiety subscales) (for reduction of anxiety), CGI‐S (for reduction of anxiety), SADS (for reduction of anxiety), SDS (for reduction of functional disability) and HAM‐D (for reduction of depression) Time points: Quote: "After the initial screening visit, all other tests were administered to patients at baseline and weeks 1, 2, 3, 4, 6, 8, and 12 (at the time of discontinuation). Safety was assessed by monitoring adverse experiences and vital signs at weeks 1, 2, 3, 4, 6, 8, and 12 (or at discontinuation)" |
|
| Notes | Industry funded: yes. Quote: "[s]upported by SmithKline Beecham Pharmaceuticals" Medication provided by industry: unclear Any of the authors work for industry: yes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "After a one week placebo run‐in: "Eligible patients then began a 12 week double blind treatment phase and were randomly assigned at baseline to receive paroxetine, 20, 40, or 60mg or placebo once daily in a 1:1:1:1 ratio". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | The medication and placebo groups did not differ significantly in attrition rate nor by sample characteristics at baseline. The proportion of dropouts increased with increasing dose of paroxetine (20 mg/d: 31/97, 31.9%; 40 mg/d: 40/95, 42.1%; 60 mg/d: 43/97, 44.3%), with the dropout rate in the arm receiving the lowest dose of medication similar to that observed amongst participants receiving placebo (28/95, 29.5%). The proportion of dropouts were higher in the paroxetine group due to adverse experience compared to the placebo group due to lack of efficacy. Withdrawals similar across groups included withdrawn, protocol violation, loss to follow‐up, and other reasons. Quote: "The demographic characteristics of the 4 treatment groups were well matched ... Approximately 63% of the randomly assigned subjects (242/384) completed the 12‐week study. There were no significant differences between the 20‐,40‐, or 60‐mg paroxetine groups and the placebo group in the overall attrition rates ... patients remaining in the study beyond week 2, discontinuation rates were comparable between the paroxetine (8%‐23%) and placebo (20%) groups. The primary reason for early discontinuation in any of the paroxetine groups was adverse experiences (17.5%, 21.1% and 23.7%) , whereas the primary reason for early discontinuation in the placebo group was lack of efficacy (10%) ... However there was no difference in the overall incidence of reported adverse experiences between 20‐mg (92%), 40 ‐mg (91%) and 60‐mg (88%) paroxetine groups ane the placebo group (83%)". Overall 37% of the participants dropped out of the study. Efficacy data are presented at both LOCF and OC for missing points. |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias were identified. |
Liebowitz 2003.
| Methods | Design: multicentre, randomised, placebo‐controlled, parallel, flexible dose, double‐blind Duration of intervention: 12 weeks Post‐treatment: no follow‐up Placebo run‐in: 1 week, single blind, placebo lead‐in period |
|
| Participants | Sample size: 415 randomised to sertraline or placebo Mean age (SD): 35.05 (10.6) years Sex: 247 men and 168 women Diagnostic measure: DSM‐IV Inclusion criteria: quote: "Study entry criteria required patients to be aged 18 years or over with a primary diagnosis of generalised social phobia of at least 2 years duration and a LSAS score > 68 at baseline. Social phobia was diagnosed using the DSM‐IV. In addition to meeting DSM‐IV criteria for social phobia, patients were required to exhibit fear and/or avoidance of at least 4 social situations. Women of childbearing potential were required to have negative results on a serum beta‐human chorionic gonadotropin pregnancy test and to be using a medically accepted form of contraception". Exclusion criteria: quote: "Patients were excluded if they met DSM‐IV criteria in the previous 6 months for substance dependence, body dysmorphic disorder, major depressive disorder, dysthymia, panic disorder, post‐traumatic stress disorder, or an eating disorder; if they reported any current or past diagnosis of schizophrenia, psychotic disorder, bipolar disorder, or obsessive‐compulsive disorder (OCD); or if they met criteria for a primary diagnosis of generalised anxiety disorder. Patients were also excluded for the following reasons: (1) HAM‐D score of 14 or items 1 rating moderate or greater in severity; (2) currently reporting serious suicidal or homicidal risk; (3) currently receiving specific behavioural or supportive therapy for social phobia or another anxiety disorder; (4) any history of seizure disorder; (5) any serious or uncontrolled medical illness or condition that preludes sertraline use; (6) women who were pregnant, nursing, or lactating; (7) receiving any concomitant therapy with any psychotropic drug or with any drug with a psychotropic component, except zolpidem for insomnia". Dropouts: 122/415 (59/211 in the sertraline and 63/204 in the placebo groups) |
|
| Interventions | Pharmacological intervention: quote: "Sertraline treatment was initiated at a daily dose of 25 mg, which was increased at week 1 to 50 mg. After 2 weeks at a dally dose of 50 mg, patients with sufficient clinical response but good tolerability were permitted to increase to 100 mg, and then by 50 mg increments per week to a maximum dose of 200 mg/day. | |
| Outcomes | Primary outcome measures: LSAS (for reduction of anxiety) Secondary outcome measures: CGI‐I (for treatment efficacy), CGI‐S (for reduction of anxiety) and SPI (for reduction of anxiety) Time points: Quote: "Patients were evaluated for medication safety and efficacy at weeks 1, 2, 3, 4, 6, 8, and 12" |
|
| Notes | Industry funded: yes. Quote: "Funded by Pfizer Inc, New York, N.Y". Medication provided by industry: unclear Any of the authors work for industry: yes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "Patients who continued to meet all inclusion and exclusion criteria were then randomly assigned in a double blind fashion to 12 weeks of double blind treatment with flexible doses of sertraline or matching placebo". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Medication and placebo was provided in identical capsules. However, it is not clear whether both participants and personnel were blinded. Quote: "Patients who continued to meet all inclusion and exclusion criteria were then randomly assigned in a double blind fashion to 12 weeks of double blind treatment with flexible doses of sertraline or matching placebo". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Similar proportions of patients withdrew from the sertraline (59/211; 28%) and placebo groups (63/204; 31%). More participants in the sertraline group withdrew due to adverse events compared to others reasons in the placebo groups. Quote: "Patient characteristics were similar in both groups at baseline ... One hundred fifty‐two (72%) of the 211 patients treated with sertraline and 141 (69%) of the patients treated with placebo completed 12 weeks of double‐blind treatment. Reasons for premature discontinuation during treatment with sertraline and placebo, respectively, included the following: withdrawal of consent, 11 (5.2%) versus 17 (8.3%); lost to follow‐up, 17 (8.1%) versus 10 (4.9%); adverse events, 16 (7.6%) versus 6 (2.9%); insufficient clinical response, 5 (2.4%) versus 9 (4.4%); protocol violation, 3 (1.4%) versus 3 (1.5%); and miscellaneous other reasons, 7 (3.3%) versus 18 (8.8%)". Overall 29% of the participants dropped out of the study. All analyses were ITT and LOCF. |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias were identified. |
Liebowitz 2005a.
| Methods | Design: multicentre, randomised, double blind, placebo controlled, parallel group, flexible dose study Duration of intervention: 12 weeks Post‐treatment: no follow‐up Placebo run‐in: 1 week, single blind, placebo lead‐in period |
|
| Participants | Sample size: 279 randomised to venlafaxine ER or placebo Mean age (SD): 35.4 (11.55) years (271 randomised, LOCF scores) Sex: 148 men and 123 women (271 randomised, LOCF scores) Diagnostic measure: DSM‐IV Inclusion criteria: quote: "Outpatients at least 18 years of age who met DSM‐IV criteria for social anxiety disorder for a least 6 months before study initiation were eligible for screening. Inclusion was dependent on a CGI‐S baseline score > 50 with a decrease of < 30% between prestudy and baseline, and a Covi Anxiety Scale score greater than Raskin Depression Scale score". Exclusion criteria: quote: "Individuals with a history of hepatic or medical disease, mental disorder due to a general medical condition, psychotic disorder or organic brain disorder, seizure disorder, or head trauma were excluded from enrolment. Patients with clinically important Axis I or Axis II comorbidities were excluded from study participation if the disorder was current or was predominant with 6 months of the start of the study. Also excluded were patients with a history of alcohol abuse within 1 year of the study, those who regularly used alcohol, and those with a urine drug screen positive for drugs of abuse. Those with multiple drug allergies or a clinically meaningful abnormality in vital signs and findings from physical examination, ECG, laboratory tests, or urine drug screen were not included. Individuals who used the investigational drugs, antipsychotics, sedative hypnotic drugs, antidepressants, anxiolytics, or migraine medication or received electroconvulsive therapy within 6 months before the study or formal psychotherapy within 30 days of the study day 1 were excluded, as were women of childbearing potential who were pregnant or breastfeeding or who did not utilise a medically acceptable form of contraception". Dropouts: 106/279 (51/139 in the venlafaxine ER and 55/140 in the placebo groups) |
|
| Interventions | Pharmacological intervention: quote: "The venlafaxine ER regimen was potentially 3‐step dose‐escalation process with an initial 75 mg/day dose during the first week ± 3 days. During the second week, the dose was increased to 150 mg/day if clinically indicated to enhance response. The dose was increased to 225 mg/day if clinically indicated on study day 15 ± 3 days. At study completion (or early termination), patients who had been taking more than 1 capsule daily for more than 1 week had their dose tapered". | |
| Outcomes | Primary outcome measure: LSAS (for reduction of anxiety) Secondary outcome measures: CGI‐S (for reduction of anxiety), CGI‐I (for treatment efficacy), SPIN (for reduction of anxiety), LSAS subscales (for reduction of anxiety), SDS (for reduction of functional disability) and HAM‐D (for reduction of depression) Time points: Quote: "The primary efficacy variable was the LSAS Total score, which was assessed at weeks 1, 2, 3, 4, 6, 8, 10, and 12. The LSAS, CGI‐S, CGI‐I and SPIN assessments were performed at baseline and on study days 7, 14, 21, 28, 42, 56, 70, and 84; the SDS was administered at baseline and on study days 28 and 84; and the HAM‐D and Covi‐Raskin scales were administered at the prestudy visit and on study days 42 and 84 as ancillary evaluations. Final ratings for efficacy were obtained on the last day of full dose administration before tapering or within 3 days of the last full dose" |
|
| Notes | Industry funded: yes. Quote: "This study was supported by Wyeth research, Collegeville, Pa". Medication provided by industry: unclear Any of the authors work for industry: yes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "eligible patients were randomly assigned to receive either venlafaxine ER or placebo for up to 12 weeks". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Similar proportions of participants withdrew from the venlafaxine ER (51/139; 37%) and placebo groups (55/140; 39%). More participants in the venlafaxine group discontinued due to adverse events compared to unsatisfactory response in the placebo group. No information was provided on the other reasons for withdrawal, however the 2 groups did not differ by sample characteristics at baseline or at week 12. Quote: "A comparable number of patients in each treatment group completed the study, i.e., 88 in the venlafaxine group and 85 in the placebo group. Significantly more patients in the placebo group (15%) than in the venlafaxine group (2%) discontinued treatment because of unsatisfactory response, while significantly more patients in the venlafaxine group discontinued treatment because of adverse events (17%) ... There were no significant differences between treatment groups for any of the demographic or baseline characteristics, nor did the demographic and baseline characteristics of the ITT patient population differ appreciably from those of the safety population". Overall 38% of the participants dropped out of the study. All analyses were ITT and LOCF. |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias were identified. |
Liebowitz 2005b.
| Methods | Design: multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group comparison Duration of intervention: 12 weeks Post‐treatment: no follow‐up Placebo run‐in: 1 week, single blind, placebo lead‐in period |
|
| Participants | Sample size: 440 randomised to venlafaxine ER, paroxetine or placebo Mean age (SD): 36.27 (11.3) years (for 413 participants) Sex: 221 men and 192 women (for 429 participants) Diagnostic measure: DSM‐IV Inclusion criteria: quote: "Outpatients 18 years and older who fulfilled the DSM‐IV criteria for SAD for 6 months or longer at screening were eligible to participate in the study. In addition, patients were required to have a LSAS score of 50 or more at prestudy and baseline evaluations; a score of 4 or more on the CGI‐S scale; a prestudy Covi Anxiety Scale total score greater than the Raskin Depression Scale total score; and a prestudy HAM‐D score of less than 15, with a score of 2 or less on the depressed mood items". Exclusion criteria: quote: "Patients with a clinically important Axis I or Axis II disorder other than SAD or avoidant personality disorder were excluded, as were those with a history or current diagnosis of any psychotic illness, patients who were suicidal, and those with a history of drug or alcohol dependence within 1 year of study start. In addition, patients were ineligible if they had used any psychopharmacologic medications within 7 days before study day 1; used antidepressants, anxiolytics, or herbal products intended to treat anxiety or depression within 14 days of the study; received electroconvulsive therapy within 6 months of the study; or used antipsychotic medication or fluoxetine or received treatment with formal psychotherapy within 30 days of the study. Patients with clinically significant abnormal findings on laboratory tests, electrocardiograms, or physical examinations; those with abnormal vital signs; those with a history or presence of clinically important medical conditions; and women of childbearing potential who were pregnant, breastfeeding, or not using a medically acceptable form of contraception were prohibited from participating". Dropouts: 111/440 (insufficient information to determine dropout rates for the three groups separately) |
|
| Interventions | Pharmacological intervention: quote: "After a 7 day, single‐blind, placebo lead‐in period, eligible patients were randomly assigned to receive flexible doses of venlafaxine ER (75‐225 mg/d), paroxetine (20‐50 mg/d), or placebo for up to 12 weeks, followed by a taper period of up to 2 weeks. Starting doses were 75 mg/d for venlafaxine ER and 20 mg/d for paroxetine if clinically indicated to improve response, daily doses could be titrated upward each week by 75 mg for venlafaxine ER or 20 mg for paroxetine to a maximum 225 mg/d of venlafaxine ER and 50 mg/d of paroxetine. The dosage could be reduced at any time to improve tolerance; however, the minimum allowed dosages after day 7 were 75 mg/d for venlafaxine ER and 20 mg/d for paroxetine". | |
| Outcomes | Primary outcome measure: LSAS (for reduction of anxiety) Secondary outcome measures: CGI‐I (for treatment efficacy), CGI‐S (for reduction of anxiety), and SPI (for reduction of anxiety) Time points: Quote: "The primary efficacy time point was the final on‐therapy (defined as observations that occurred within 3 days of the patient’s last full dose of study medication) LSAS total score. The week 12 last‐observation‐carried‐forward values were the final on‐therapy observations" |
|
| Notes | Industry funded: yes. Quote: "This study was supported by Wyeth research, Collegeville, Pa". Medication provided by industry: unclear Any of the authors work for industry: yes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "...patients were randomly assigned to receive flexible doses of venlafaxine hydrochloride ER (75‐225 mg/d), paroxetine (20‐50 mg/d), or placebo for up to 12 weeks". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | There was insufficient information to determine the proportion of dropouts for the three treatment groups. Nevertheless, the three groups did not differ by sample characteristics at baseline nor did they significantly differ by withdrawals. More participants in the venlafaxine and paroxetine group discontinued due to adverse events compared to unsatisfactory response in the placebo group. Quote: "There were no statistically significant differences between the treatment groups for any of the demographic or baseline characteristics ... Three hundred eighteen patients (74.1%) completed the 12‐week double‐blind treatment period and 111 (25.9%) withdrew from the study. Significantly more patients in the active treatment groups (venlafaxine ER and paroxetine) withdrew because of adverse events than in the placebo group, while significantly more patients in the placebo group withdrew because of lack of efficacy; however, there were no significant differences between the active treatment groups. Statistical analyses were performed using last‐observation carried‐forward values for the intent‐to‐treat population". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias were identified. |
Lott 1997.
| Methods | Design: multicentre, randomised, placebo‐controlled, parallel, flexible dose, double‐blind Duration of intervention: 10 weeks Post‐treatment: no follow‐up Placebo run‐in: 1‐week single‐blind placebo washout |
|
| Participants | Sample size: 106 randomised to brofaromine or placebo Mean age (SD): 36.5 (9.8) years Sex: 62 men and 40 women (for 102 ITT participants) Diagnostic measure: DSM‐III‐R Inclusion criteria: quote: "Participants were 102 outpatients (62 men and 40 women) with a primary DSM‐III‐R diagnosis of social phobia of at least 6 months' duration. Primary social phobia was defined as dominating the clinical picture and temporally preceding any secondary diagnosis ... A minimum score of 8 was required on the Avoidance Subscale of the LSAS ... A minimum score of 4 (moderately ill) was required on the Liebowitz Social Phobia Global Severity Rating Scale. A maximum score of 15 was allowed on the MADRS". Exclusion criteria: quote: "Patients were excluded for the following reasons: alcohol or drug abuse in the past 6 months; a DSM‐III‐R diagnosis of organic mental syndrome, organic anxiety syndrome, or caffeinism; clear and immediate suicide risk; personality pathology that might interfere with trial compliance; previous participation in another brofaromine trial; a previous failure to respond to an adequate trial of an SRI or MAO inhibitor; clinically significant comorbid medical disease; concurrent use of other psychotropic drugs, opiates, or certain dietary or prescription amines (patients were allowed to take chloral hydrate, diphenhydramine, and doxylamine); and patients who in the judgment of the investigator were likely to be noncompliant". Dropouts: 31/106 (14/52 in the brofaromine and 17/54 in the placebo groups). |
|
| Interventions | Pharmacological intervention: quote: "Brofaromine dosage began at 50 mg/day and was titrated up to a maximum of 150 mg/day, depending on response to treatment. Patient visits were scheduled at weekly intervals for the first 2 weeks of double‐blind treatment and biweekly thereafter. After 10 weeks of double‐blind treatment (or at early termination), patients entered a 1‐to 2‐week double‐blind weaning period during which the trial drug dose was tapered and discontinued". | |
| Outcomes | Primary outcome measure: LSAS (for reduction of anxiety) Secondary outcome measures: CGI (for treatment efficacy), GAF (for reduction of functional disability), HAM‐A (for reduction of anxiety), MADRS (for reduction of depression), FONE (for reduction of anxiety), LSAS (for reduction of anxiety), CSPS (for reduction of anxiety), and SDS (for reduction of functional disability) Time points: Quote: "Efficacy measures were obtained at bassline and at end of 2, 6, and 10 week of double‐blind treatment" |
|
| Notes | Industry funded: yes. Quote: "This study was supported by the Ciba‐Geigy Corporation, Summit, NJ". Medication provided by industry: unclear Any of the authors work for industry: yes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "patients who still met inclusion criteria and and did not respond to placebo ... were randomly assigned to 10 weeks of treatment with either brofaromine or place". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Similar proportions of participants withdrew from the brofaromine (14/52; 27%) and placebo groups (17/50; 34%). More participants in the brofaromine group discontinued due to adverse events compared to withdrawal of consent in the placebo group. The 2 groups did not differ by sample characteristics at baseline. Quote: "Fourteen brofaromine patients and 17 placebo patients discontinued the trial prematurely. Eleven of the 14 brofaromine early terminators discontinued because of adverse experiences, as did four of the 17 placebo early terminators. Other reasons for early termination included unsatisfactory therapeutic effect (N = 3, placebo); did not meet protocol criteria (N = 1, placebo); patient noncompliance (N = 2, placebo); patient withdrew consent (N = 1, brofaromine; N = 6 placebo); and lost to follow‐up (N = 2, brofaromine; N = 1, placebo) ... No significant baseline differences were found between treatment groups on race, gender, age, or LSAS total score". Overall 30% of the participants dropped out of the study. Quote: "An intent‐to‐treat analysis was employed, with the last evaluable visit past baseline carried forward as endpoint". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias were identified. |
Moghadam 2015.
| Methods | Design: single‐centre, quasi‐experimental, randomised‐controlled, wait‐list study Duration of intervention: 12 weeks Post‐treatment: no follow‐up Placebo run‐in: no |
|
| Participants | Sample size: 30 randomised to sertraline or placebo (the additional 15 participants were randomised to psychotherapy) Mean age (SD): 24.5 (2.2) years Sex: 30 men Diagnostic measure: DSM‐IV‐TR (based on SCID) Inclusion criteria: quote: "SPIN score ≥ 24, age between 18 to 50 years, and meeting the DSM‐IV‐TR criteria for social phobia based on SCID" Exclusion criteria: quote: "being psychotic, or obsessive‐compulsive; having bipolar, or organic brain disorders; drug and alcohol dependency; having impulse control disorders, cluster A and B personality disorders, active disorder on axis III, a history of suicidal thoughts and actions, a history of violent behavior; being on psychotropic medications or receiving psychotherapy for the treatment of social phobia during the last 6 months, or experiencing the symptoms of social phobia as part of other psychiatric disorders". Dropouts: not specified |
|
| Interventions | Pharmacological intervention: quote: "Members of the MED group received pharmacotherapy (sertraline) for 12 weeks. Patients in the WL group received no intervention. However, after the waiting period, they received preferred treatment services. Each group was evaluated 4 times during the study". | |
| Outcomes | Primary outcome measure: SPIN (for reduction of anxiety) Secondary outcome measures: GAF (for reduction of functional disability), CGI‐I (for treatment efficacy), CGI‐S (for reduction of anxiety) Time points: Quote: "In this randomized‐controlled trial study, 13 male students were treated with short‐term dynamic psychotherapy (McCullough method) lasting 25 sessions, 11 students received sertraline for 12 weeks, and 14 students, as the waiting list, received no intervention for 8 weeks. Participants completed the Social Phobia Inventory (SPIN) as primary efficacy variable 4 times, and were rated with Clinical Global Impression scale (CGI) and Global Assessment of Functioning (GAF) as secondary efficacy variables". Time points were not specified |
|
| Notes | Industry funded: no. Quote: "This research was funded by Tehran University of Medical Sciences and Mental Health Research Network (MHRN)". Medication provided by industry: unclear Any of the authors work for industry: no |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "The participants were randomly assigned into 3 groups: 1‐psychotherapy (STDP), 2‐ medical therapy (MED) and 3‐ waiting list (WL)". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No information was provided to determine if participants and personnel were blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | No information was provided to determine if outcome assessors were blinded. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | No dropout rates were reported. |
| Selective reporting (reporting bias) | Low risk | All outcomes were reported on as specified in the protocol. |
| Other bias | Low risk | No other sources of bias were identified. |
Muehlbacher 2005.
| Methods | Design: single‐centre, randomised, double‐blind, fixed dose, placebo‐controlled study Duration of intervention: 10 weeks Post‐treatment: no follow‐up Placebo run‐in: no |
|
| Participants | Sample size: 66 randomised to mirtazepine or placebo Mean age (SD): 24 (3.45) years Sex: 66 women Diagnostic measure: DSM‐IV Inclusion criteria: quote: "Women aged 18 or older who have social phobia were included in the study". Exclusion criteria: quote: "Exclusion criteria were psychotic symptoms, a severe major depressive episode (according to the DSM‐IV criteria), the current use of mirtazepine or other psychotropic medication, and psychotherapy. Potential subjects were also excluded if they currently were pregnant (or planning to be or not using contraception), severely somatically ill, currently suicidal, or abusing alcohol or drugs". Dropouts: 7/66 (insufficient information to determine dropout rates for the 2 groups separately) |
|
| Interventions | Pharmacological intervention: quote: "Subjects received blinded capsule per day, either 30 mg of mirtazepine or matching placebo. The dosage of mirtazepine stayed constant". | |
| Outcomes | Primary outcome measures: SPI (for reduction of anxiety), LSAS (for reduction of anxiety) and SF‐36 (for quality of life) Secondary outcome measures: none Time points: Weekly examinations. |
|
| Notes | Industry funded: no. Quote: "This study was not funded and was not influenced by outside economic interests". Medication provided by industry: no Any of the authors work for industry: no |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Author correspondence: quote: "We used an excel sheet with random number generator". |
| Allocation concealment (selection bias) | Low risk | Quote: "Subjects received blinded capsule per day, either 30 mg of mirtazepine or matching placebo. The dosage of mirtazepine stayed constant. Tablets were supplied in numbered boxes". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Both subjects and clinicians were blinded to mirtazepine/placebo assignment". |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Both subjects and clinicians were blinded to mirtazepine/placebo assignment". |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There was insufficient information to determine the proportion of dropouts for the three treatment groups, though a relatively small number dropped out overall (N = 7, 11%). No information was provided on the reasons for study withdrawal. There was also no information on group characteristics. Quote: "Seven patients who failed to appear more than twice for the weekly evaluations dropped out of the study". All analyses were ITT. |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Low risk | Self reported measures were used but No other sources of bias were identified. Quote: "The questionnaires were filled out by the patients both independently and anonymously". |
NCT00273039.
| Methods | Design: multicentre, randomised, double‐blind, placebo‐controlled, parallel, forced‐dose titration study (NKF100110) Duration of intervention: 12 weeks Post‐treatment: follow‐up not specified Placebo run‐in: not specified |
|
| Participants | Sample size: 107 randomised to paroxetine or placebo (the additional participants were randomised to NCE, the results will be available once NCE is approved by the FDA). Mean age (SD): 34.4 (10.83) years Sex: 60 men and 44 women (for 104 participants) Diagnostic measure: DSM‐IV Inclusion criteria: quote: "Male or female subjects between 18‐65 years of age; A primary diagnosis of Generalized Social Anxiety Disorder/Social Phobia (DSM‐IV, 300.23)". Exclusion criteria: quote: "CGI‐I item score of 1 or 2 at baseline; Montgomery‐Asberg Depression Rating Scale (MADRS) score of 18 or more at screening; DSM‐IV criteria for any other Axis I disorder as a current primary disorder or within 6 months prior to the screening visit; Subjects with Body Dysmorphic Disorder; History of Schizophrenia, Schizoaffective Disorder, or a Bipolar Disorder; Current, serious suicidal or homicidal risk or suicide attempt within the past 6 months or have ever been homicidal; Unstable medical disorder; or a disorder that would interfere with the action, absorption, distribution, metabolism, or excretion of the NCE or paroxetine; may pose a safety concern; or interfere with the accurate assessment of safety or efficacy". Dropouts: 44/107 (14/36 in the paroxetine and 30/71 in the placebo group) |
|
| Interventions | Pharmacological intervention: quote: "After completion of a screening period, subjects fulfilling the inclusion/exclusion criteria were randomised (2:2:2:1) to two dose ranges of an NCE, placebo, or paroxetine (20‐30 mg/day). A forced‐flexible dose titration scheme was employed". | |
| Outcomes | Primary outcome measures: LSAS (for reduction of anxiety) Secondary outcome measures: CGI‐I (for treatment efficacy), CGI‐S (for reduction of anxiety), LSAS Fear and Avoidance subscales (for reduction of anxiety), SDS (for reduction of functional disability), MOS‐12 (assess changes in sleep quality), and LSEQ (assess changes in sleep quality) Time points: Week 1 and 12. Quote: "Adverse events (AEs) and Serious Adverse Events (SAEs) were reported during treatment weeks 1 through 12, the taper visit, and at the 14‐day follow‐up visit or early withdrawal visit. This summary includes data for the paroxetine and placebo groups. Results for the unmarketed NCE will be added, if and when the NCE is approved and marketed" |
|
| Notes | Industry funded: not specified Medication provided by industry: not specified Any of the authors work for industry: not specified |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "After completion of a screening period, subjects fulfilling the inclusion/exclusion criteria were randomized (2:2:2:1) to two dose ranges of an NCE, placebo, or paroxetine (20‐30 mg/day). A forced‐flexible dose titration scheme was employed". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Similar proportions of participants withdrew from the paroxetine (14/36; 39%) and placebo groups (30/71; 42%). No information was provided on the reasons for study withdrawal and how participants differed by sample characteristics at week 12. Overall 41% of the participants dropped out of the study. ITT population was assessed and LOCF was used for missing data. |
| Selective reporting (reporting bias) | Unclear risk | Not all the required information is presented in the protocol to determine if selective reporting occurred. |
| Other bias | Unclear risk | It is unclear if any other bias occurred and if the study was funded. |
NCT00318669.
| Methods | Design: multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group, comparative study (PIR104776) Duration of intervention: 12 weeks Post‐treatment: follow‐up not specified Placebo run‐in: 2 weeks placebo run in |
|
| Participants | Sample size: 400 randomised to paroxetine or placebo Mean age (SD): 36.96 (9.98) years Sex: 186 men and 209 women (for 395 participants) Diagnostic measure: DSM‐IV‐TR Inclusion criteria: quote: "Diagnosis of Social Anxiety Disorder (SAD) according to DSM‐IV‐TR criteria; Must have given written informed consent. But if the subject was under 20 years of age, both the subject and his/her proxy consenter had to have given written informed consent; Outpatients; Age: ≥18 years, < 65 years (at the time of informed consent); Sex: no restriction; Had to have a LSAS total score of ≥60 at baseline". Exclusion criteria: quote: "Subjects diagnosed with Axis I disorders other than SAD (e.g., major depression, dysthymic disorder, specific phobia (simple phobia), obsessive compulsive disorder, panic disorder) as a primary diagnosis according to DSM‐IV‐TR within 24 weeks prior to the Week ‐2 visit; Subjects with a history of or concurrent schizophrenia or bipolar disorder; Subjects with concurrent body dysmorphic disorder; Subjects who met the DSM‐IV‐TR criteria for substance abuse (alcohol or drugs) or substance dependence within 24 weeks prior to the Week ‐2 visit; Subjects who started psychotherapy, other than supportive psychotherapy, or cognitive‐behavioural therapy within 24 weeks prior to the Week ‐2 visit; Subjects who received electro‐convulsive therapy (ECT) within 12 weeks prior to the Week ‐2 visit; Subjects who were pregnant, lactating, might be pregnant, or were planning a pregnancy during the study period; Subjects who scored 3 or more on HAM‐D Item No.3 or, who, in the investigator's clinical judgement, were at acute risk of suicide attempt; Subjects with a history of or concurrent cancer or malignant tumor; Subjects who received MAO inhibitors (FP®) within 14 days prior to the scheduled Week 0 visit". Dropouts: 44/107 (43/267 in the paroxetine and 19/133 in the placebo group) |
|
| Interventions | Pharmacological intervention: quote: "Subjects received paroxetine or placebo once daily after an evening meal for 12 weeks. Subjects randomized to paroxetine 20mg or 40mg were started on 10mg/day for the first week and the dose was then increased according to a fixed dosing schedule. Subjects in the paroxetine 20mg group remained on 20mg/day for 11 weeks. Subjects in the paroxetine 40mg group received 20mg/day for 1 week, 30mg/day for 1 week, and then 40mg/day for 9 weeks. Subjects randomized to placebo received the same number of placebo tablets in the same manner as for those randomized to paroxetine treatment. Taper phase: participants who completed the treatment phase underwent a 3‐week taper phase, and participants who withdrew prematurely underwent a taper phase of 1 to 3 weeks depending upon their final level of study medication. The dose was decreased by 10mg/day weekly until 10mg/day was reached". ITT population was assessed and LOCF was used for missing data. | |
| Outcomes | Primary outcome measures: LSAS (for reduction of anxiety) Secondary outcome measures: change from baseline in LSAS total score (for reduction of anxiety), CGI‐I (for treatment efficacy), CGI‐S (for reduction of anxiety), and HAM‐D (for reduction of depression) Time points: Quote: "Change from baseline in LSAS total score (at Weeks 2, 3, 4, 6, 8 and 10). Proportion of CGI Global Improvement responders (i.e., subjects rated as either "very much improved" or "much improved") at Week 12. Change from baseline in LSAS Fear/Anxiety and Avoidance subscale scores (at Weeks 2, 3, 4, 6, 8, 10 and 12). Change from baseline in CGI Severity of Illness score (at Weeks 2, 3, 4, 6, 8, 10 and 12). Change from baseline in HAM‐D total score at Weeks 12 (score at Week 12 minus Score at Week 0)" |
|
| Notes | Industry funded: not specified Medication provided by industry: not specified Any of the authors work for industry: not specified |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Similar proportions of participants withdrew from the paroxetine (43/267; 16%) and placebo groups (19/133; 14%). No information was provided on the reasons for study withdrawal and how participants differed by sample characteristics at week 12. Overall 16% of the participants dropped out of the study. |
| Selective reporting (reporting bias) | Low risk | All outcomes were reported as specified in the protocol. |
| Other bias | Unclear risk | It is unclear if any other bias occurred and if the study was funded. |
NCT00397722.
| Methods | Design: multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group, active comparator study (CRH103390) Duration of intervention: 12 weeks Post‐treatment: follow‐up not specified Placebo run‐in: not specified |
|
| Participants | Sample size: 294 randomised to paroxetine, GW876008 or placebo Mean age (SD): 37.35 (11.86) years Sex: 155 men and 139 women Diagnostic measure: DSM‐IV Inclusion criteria: quote: "Male and female subjects 18‐64 years of age inclusive with a primary diagnosis of SocAD/Social Phobia diagnosed using criteria established in the DSM‐V participated in the study". Exclusion criteria: quote: "Subjects who scored 1 or 2 on the Clinical Global Impression‐Global Improvement (CGI‐I) score item at the randomisation visit, or who met the DSM‐V criteria for major depressive disorder, or who scored ≥ 15 on the 17‐item Hamilton Rating Scale for Depression (HAMD‐17), at the screening visit were excluded from the study". Dropouts: 44/107 (13/42 in the paroxetine, 36/164 in the GW876008 and 25/88 in the placebo group) |
|
| Interventions | Pharmacological intervention: quote: "Subjects were randomised to a 2:2:2:1 ratio such that 81 subjects received GW876008 25‐50mg/day, 83 received GW876008 100‐125 mg/day, 88 received placebo and 42 received paroxetine 20‐30 mg/day". | |
| Outcomes | Primary outcome measures: LSAS (for reduction of anxiety), SADS (for reduction of anxiety) Secondary outcome measures: CGI‐I (for treatment efficacy) Time points: Baseline and week 12. |
|
| Notes | Industry funded: not specified Medication provided by industry: not specified Any of the authors work for industry: not specified |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "Subjects were randomised to a 2:2:2:1 ratio such that 81 subjects received GW876008 25‐50mg/day, 83 received GW876008 100‐125 mg/day, 88 received placebo and 42 received paroxetine 20‐30 mg/day". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Similar proportions of participants withdrew from the paroxetine (13/42; 31%), GW876008 (36/164; 22%) and placebo groups (25/88; 28%). No information was provided on the reasons for study withdrawal and how participants differed by sample characteristics at week 12. Overall 25% of the participants dropped out of the study. Quote: "Key efficacy analyses were intention to treat (ITT) using the mixed‐effects model repeated‐measure analysis for comparisons". |
| Selective reporting (reporting bias) | Unclear risk | Not all the required information is presented in the protocol to determine if selective reporting occurred. |
| Other bias | Unclear risk | It is unclear if any other bias occurred and if the study was funded. |
NCT00403962.
| Methods | Design: multicentre, randomised, double‐blind, placebo‐controlled, fixed dose, parallel group study (NKP103401). Duration of intervention: 12 weeks Post‐treatment: follow‐up not specified Placebo run‐in: not specified |
|
| Participants | Sample size: 133 randomised to paroxetine or placebo (the additional participants were randomised to NCE, the results will be available once NCE is approved by the FDA) Mean age (SD): 40.3 (11.21) years Sex: 54 men and 74 women (for 128 participants) Diagnostic measure: DSM‐IV and MINI Inclusion criteria: quote: "Male and female subjects, 18‐65 years of age, with a primary diagnosis of Generalised Social Anxiety Disorder/Social Phobia as defined in the Diagnostic and Statistical Manual of Mental Disorders, 4th Edition (DSM‐IV), diagnosed using psychiatric confirmation of diagnosis in conjunction with the Mini International Neuropsychiatric Interview (MINI), Clinician Rated version 5.0". Exclusion criteria: quote: "Subjects were excluded if they scored 1 or 2 on the CGI‐I item at baseline; subjects with a score 15 or more on the Hamilton Depression (HAMD) Rating Scale 17 (HAMD‐17) item at screening were also excluded. In addition, subjects were excluded if they had a history of myocardial infarction within one year prior to the screening visit, or had body dysmorphic disorder or had a history of schizophrenia, schizoaffective disorder, or a bipolar disorder". Dropouts: 42/133 (23/68 in the paroxetine and 19/65 in the placebo group) |
|
| Interventions | Pharmacological intervention: quote: "NCE/paroxetine combination, paroxetine monotherapy (7.5 mg/day, fixed dose) or placebo for a period of 12 weeks". | |
| Outcomes | Primary outcome measures: LSAS (for reduction of anxiety) Secondary outcome measures: LSAS (for reduction of anxiety), CGI‐I (for treatment efficacy), SADS (for reduction of anxiety), SDS (for reduction of functional disability) Time points: Baseline and week 12. |
|
| Notes | Industry funded: not specified Medication provided by industry: not specified Any of the authors work for industry: not specified |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "A 12‐week randomised, multicentre, double‐blind, placebo‐controlled, fixed dose, parallel group study". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Similar proportions of participants withdrew from the paroxetine (23/68; 34%) and placebo groups (19/65; 29%). No information was provided on the reasons for study withdrawal and how participants differed by sample characteristics at week 12. Overall 32% of the participants dropped out of the study. ITT population was assessed and LOCF was used for missing data. |
| Selective reporting (reporting bias) | Unclear risk | Not all the required information is presented in the protocol to determine if selective reporting occurred. |
| Other bias | Unclear risk | It is unclear if any other bias occurred and if the study was funded. |
NCT00470483.
| Methods | Design: single‐centre, double‐blind, randomised, placebo‐controlled, parallel group (TMT106386) Duration of intervention: 8 weeks Post‐treatment: follow‐up not specified Placebo run‐in: not specified |
|
| Participants | Sample size: 33 randomised to paroxetine or placebo (the additional participants were randomised to no treatment (HVT)) Mean age: 22.9 years Sex: 5 men and 28 women Diagnostic measure: DSM‐IV (using the SCID‐I) Inclusion criteria: quote: "Male or female subjects of 18 to 60 years of age, with a primary diagnosis of SAD (DSM‐IV, 300.23) diagnosed using psychiatric confirmation of diagnosis in conjunction with the structured clinical diagnostic interview (SCID‐I) were included into the study. Healthy participants free from significant cardiac, pulmonary, gastrointestinal, hepatic, renal, haematological, neurological and psychiatric disease as determined by history, physical examination, MRI and clinical laboratory test results (for healthy volunteers only) were also included in to the study". Exclusion criteria: not specified Dropouts: 1/33 (0/17 in the paroxetine and 1/16 in the placebo group) |
|
| Interventions | Pharmacological intervention: quote: "Each eligible subject was assigned to receive following treatments in a 1:1 ratio as per the randomisation schedule: 1. Paroxetine 20 mg, oral capsules once a day repeatedly administered for 8 weeks 2. Placebo to match paroxetine capsules, once a day, for 8 weeks The subjects, after the last treatment dose, entered in a tapering phase during which subjects received following regimen, after which subjects definitively discontinued the study drug. 3. Paroxetine 10 mg, oral capsules (2 x 5 mg capsules) once a day repeatedly administered for a week 4. Placebo to match paroxetine 5 mg capsules (2 capsules), once a day, for a week." | |
| Outcomes | Primary and secondary outcome measures: STAI‐S (for reduction of anxiety), CGI (for treatment efficacy), LSAS (for reduction of anxiety) Time points: Week 2, 4, 6, 8. |
|
| Notes | Industry funded: not specified Medication provided by industry: not specified Any of the authors work for industry: not specified |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "Each eligible subject was assigned to receive following treatments in a 1:1 ratio as per the randomisation schedule". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | More participants withdrew from the placebo group (1/16; 6%) compared to the paroxetine (0/17; 0%) and venlafaxine (7/129; 5%) group. No information was provided on whether participants differed in terms of characteristics by group at week 12, however. Nevertheless, the total proportion of dropouts (6%) is relatively low, suggesting that dropout rates may not have biased the outcomes. Overall 0.6% of the participants dropped out of the study. |
| Selective reporting (reporting bias) | Unclear risk | Not all the required information is presented in the protocol to determine if selective reporting occurred. |
| Other bias | Unclear risk | It is unclear if any other bias occurred and if the study was funded. |
Nordahl 2016.
| Methods | Design: single‐centre, randomised, double‐blind, placebo‐controlled, comparative, flexible dose study Duration of intervention: 12 weeks Post‐treatment: 12‐month follow‐up Placebo run‐in: no |
|
| Participants | Sample size: 52 randomised to paroxetine or placebo (102 randomised: 26 paroxetine, 24 CT, 26 combination, 26 placebo) Mean age (range): 30.85 (18‐65) years Sex: 26 men and 26 women Diagnostic measure: DSM‐IV Inclusion criteria: quote: "Inclusion criteria were as follows: age of 18–65 years, fulfillment of DSM‐IV criteria for SAD, and symptoms present for at least 6 months". Exclusion criteria: quote: "Exclusion criteria were any form of physical disease, psychotic illness, acute suicidality, a primary diagnosis of major depressive disorder, diagnosis of body dysmorphic disorder, drug or alcohol dependence, and cluster A or cluster B personality disorders. Subjects not willing to accept random allocation were also excluded. We excluded patients who had been exposed to CT or to SSRIs previously in order to eliminate any bias of negative expectations to the treatment offered. Participants who were pregnant or were planning to become pregnant during the next 6 months were excluded due to the drug condition". Dropouts: 8/52 (5/26 in the paroxetine and 3/26 in the placebo group; 12 month follow‐up 4 dropouts) |
|
| Interventions | Pharmacological intervention: quote: "Following the clinical guideline by Stein et al. [25] , drug treatment was administered over 26 weeks, and tapering of medications/placebo commenced at week 23, tapering 10 mg per week or alternatively 25% of dosage per week. Medication was administered adhering to best prescribing practices for social phobia as suggested by the manufacturer. The recommended initial dosage was 20 mg per day, and minimum–maximum dosage was 20– 60 mg/day. The target range of paroxetine in the blood serum was set between 80 and 450 μmol/l. After 4 and 12 weeks of medication, blood serum was tested in all patients receiving paroxetine or pill placebo to monitor treatment compliance and ensure the target range of the drug was achieved. If needed, medication could be titrated upwards by 20 mg/day in steps until reaching the defined target level. The laboratory communicated serum levels outside the targeted range to the psychiatrist, and medications were added. Changes of medications were always counter balanced in a 1: 1 format so that changes in dosage were done simultaneously in both the active and the placebo arms in order to maintain the blinding of the treatment. The mean dosage of paroxetine in the overall group was 28 ± 5.5 mg/day". | |
| Outcomes | Primary outcome measure: FNES (for reduction of anxiety) Secondary outcome measures: LSAS (for reduction of anxiety), BAI (for reduction of anxiety), IIP‐64 (for reduction of functional disability) Time points: Quote: "Patients were assessed pretreatment, posttreatment at 12 weeks (post‐acute), and at the 12‐month follow‐up" |
|
| Notes | Industry funded: no. Quote: "The study was financially supported by the Departments of Psychology and Neuroscience at the Norwegian University of Science and Technology (NTNU), Trondheim". Medication provided by industry: yes. Quote: "It was administered as capsules manufactured by the pharmaceutical laboratory at St. Olav’s University Hospital to make them identical to the placebo". Any of the authors work for industry: no |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "The participants were randomly assigned to 1 of 4 conditions. The randomization used gender and diagnosis of APD as stratification variables in blocks of 10 to ensure equal distribution". |
| Allocation concealment (selection bias) | Low risk | Quote: "The medication used was paroxetine (paroxetine hydrochloride). It was administered as capsules manufactured by the pharmaceutical laboratory at St. Olav’s University Hospital to make them identical to the placebo. The placebo capsules contained lactose. The paroxetine and the placebo were identical in size, color, smell, taste, and appearance. The pharmaceutical laboratory at St. Olav’s University Hospital provided the medication to the psychiatrists". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "The participants, independent diagnosticians, psychiatrists, and the principal investigator remained blinded to the paroxetine alone and pill placebo conditions. In addition, specific instructions were given to all participants to avoid disclosing information about their treatment to the evaluators". |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Blinding was conducted for the treatment conditions using medication or placebo and achieved for the primary outcome measures by using independent evaluators who were blinded to the treatment assignment". |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | A similar proportion of participants withdrew from the paroxetine group (5/26, 19%) and placebo group (3/26; 12%). Quote: "All data were analyzed based on an intention‐to‐treat approach, and missing data were treated using last observation carried forward on the primary measure. We used a linear mixed model analysis". |
| Selective reporting (reporting bias) | High risk | Secondary outcomes where not specified in the protocol for LSAS, IIP‐64 and BAI measures as well as for relapse |
| Other bias | Unclear risk | Medication was provided by industry. Quote: "We are not sure whether a different setting of the interviews could bias the data, but this must be considered ... There is a possibility that drug treatment in our study may have been adversely affected by bias produced by the blinding process. Specifically, in completing self‐report measures, those in the drug arm may have doubted that they received the active treatment rather than placebo". |
Noyes 1997.
| Methods | Design: multicentre, randomised, placebo‐controlled, parallel, fixed dose, double‐blind Duration of intervention: 12 weeks Post‐treatment: no follow‐up Placebo run‐in: 1‐week, single‐blind, placebo run‐in |
|
| Participants | Sample size: 523 randomised to moclobemide or placebo Mean age (SD): 38.1 (10.4) years (ITT sample of 506) Sex: 290 men and 216 women (ITT sample of 506) Diagnostic measure: DSM‐III‐R Inclusion criteria: quote: "Subjects were screened using the Structured Clinical Interview for DSM‐III‐R, Roche version and were required to meet DSM‐III‐R criteria for social phobia (primary diagnosis). Subjects were included who had coexisting generalized anxiety disorder or avoidant personality disorder, but those with coexisting panic disorder, agoraphobia, or obsessive‐compulsive disorder were excluded. Subjects were required to achieve a score of 4 (moderate) or more on the Clinical Impression of Severity‐Social Phobia scale". Exclusion criteria: quote: "Any subjects who had mental retardation, organic mental disorders including dementia, psychoses including schizophrenia, bipolar disorder, or borderline personality disorder were excluded. Subjects were also excluded who had major depressive disorder within 6 months of the study, suicidal ideation, substance abuse within 6 months of the study, or positive urine drug screening. Also excluded were subjects with uncontrolled physical disease or significant laboratory abnormalities. Women of childbearing potential were required to take adequate contraceptive precautions". Dropouts: 158/523 (25/84 in the moclobemide and 33/85 in the placebo groups) |
|
| Interventions | Pharmacological intervention: quote: "Subjects were then randomly assigned to placebo or one of five doses of moclobemide (75 mg, 150 mg, 300 mg, 600 mg, or 900 mg). Those assigned to 75 mg and 150 mg received the full dose from the time of randomization, and those assigned to 300 mg, 600 mg, and 900 mg received 150 mg initially followed by increments of 150 mg every 4 days until the full dose was achieved". | |
| Outcomes | Primary outcome measure: CGI‐I (for treatment efficacy) Secondary outcome measures: CGI‐SP (for reduction of anxiety), BSPS (for reduction of anxiety), LSAS (for reduction of anxiety) and SDS (for reduction of functional disability) Time points: Quote: "The primary measure of efficacy was the global rating of improvement made on the Clinical Impression of Change scale at week 12. Efficacy measures also included patient‐rated scales completed at 4, 6, and 12 weeks. Adverse events, as observed or elicited by the clinician, were recorded at baseline and at each subsequent visit" |
|
| Notes | Industry funded: yes. Quote: "This study was sponsored by Hoffmann‐La Roche, Inc., Nutley, NJ ". Medication provided by industry: unclear Any of the authors work for industry: yes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "Subjects were then randomly assigned to placebo or one of five doses of moclobemide". |
| Allocation concealment (selection bias) | Unclear risk | The study was described as "double‐blind" however there was insufficient evidence to determine if study medication was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Similar proportions of participants withdrew from the moclobemide (25/84; 29.7%) and placebo groups (33/85; 38.8%). No information was provided on the reasons for study withdrawal and how participants differed by sample characteristics at week 12. Nevertheless, the study reported that 158 participants dropped out of the study and these participants did not differ significantly. Quote: "One hundred fifty‐eight (31.2%) subjects dropped out before week 12. The proportion of placebo‐treated subjects who dropped out (38.8%) was higher than, but not significantly different from, the percentage of moclobemide‐treated subjects who failed to complete the trial (29.7%)". Overall 34% of the participants dropped out of the study. Quote: "The main analyses involved intent‐to‐treat subjects defined as those who received at least one dose of study medication and had at least one efficacy assessment after baseline. For these analyses, last observations were carried forward to replace missing values". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias were identified. |
Oosterbaan 2001.
| Methods | Design: multicentre, randomised, placebo‐controlled, parallel, flexible dose, double‐blind Duration of intervention: 15 weeks Post‐treatment: 2 and 15 months follow‐up Placebo run‐in: no |
|
| Participants | Sample size: 54 randomised to moclobemide or placebo (82 out of 86 randomised: 27 moclobemide, 28 cognitive therapy, 27 placebo, four participants refused participation after randomisation because they were not allocated to the condition they preferred) Mean age (SD): 36.5 (11) years (for the moclobemide or placebo groups; 82 randomised) Sex: 31 men and 23 women (for the moclobemide or placebo groups; 82 randomised) Diagnostic measure: DSM‐III‐R Inclusion criteria: quote: "After a general diagnostic interview by an experienced clinician, a main diagnosis of social phobia was confirmed by a semi‐structured interview ... Patients were included when aged 18 to 65 years, and had no serious medical problems as revealed by medical history and laboratory screening tests". Exclusion criteria: quote: "Patients with comorbid panic disorder with or without agoraphobia, obsessive‐compulsive disorder, major depressive disorder, psychotic and organic mental disorder were excluded, as were patients suffering from psychoactive substance‐use disorder and borderline personality disorder". Dropouts: 11/54 for the moclobemide or placebo groups (3/27 in the moclobemide and 8/27 in the placebo groups). |
|
| Interventions | Pharmacological intervention: quote: "In the pharmacotherapy condition, patients started either with placebo or moclobemide 450 mg/day, which was also the target dose. After 2 weeks, the dose was increased to 600 mg/day in case of insufficient efficacy and good tolerability or decreased to 300 mg/day in case of severe side effects. Moclobemide and placebo were supplied in matching tablets of 150 mg by Hoffman–La Roche Ltd., Basel, Switzerland". | |
| Outcomes | Primary and secondary outcome measures: LSAS (for reduction of anxiety), ADS (modified) (for reduction of anxiety), a 4‐point scale for the tolerability of moclobemide and placebo, MADRS (for reduction of depression), CIC (for treatment efficacy), IIS (for treatment efficacy), SCI (for reduction of functional disability), FQ (for reduction of anxiety), SDS (for reduction of functional disability), and SCL‐90 (for reduction of anxiety) Time points: Quote: "Measurements took place at pre‐test and post‐test (week 0 and 15), 2‐month follow‐up (week 23) as well as 15 months after completion of the trial" |
|
| Notes | Industry funded: yes. Quote: "This study was sponsored by Hoffmann‐La Roche, Ltd., Basel, Switzerland". Medication provided by industry: yes. Quote: "Moclobemide and placebo were supplied in matching tablets of 150mg by Hoffman – LaRoche Ltd., Basel, Switzerland". Any of the authors work for industry: no |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "patients were randomly assigned to one of the three treatment conditions". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Pharmacotherapy was administered under double‐blind conditions ... Moclobemide and placebo were supplied in matching tablets of 150mg by Hoffman – LaRoche Ltd., Basel, Switzerland ... To check double‐blindness, patients and therapists were asked at post‐test to estimate whether moclobemide or placebo had been administered. Correct classifications did not differ from chance. These results indicated that in our study double‐blindness was maintained throughout". |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Independent raters, blind to therapeutic conditions, assessed social anxiety and avoidance with the LSAS ... and MADRS ..." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | More participants discontinued in the placebo group (8/27; 30%) compared to the moclobemide group (3/27; 11%), largely due to non‐compliance with the protocol and inadequate treatment response. The 2 groups did not differ by sample characteristics at baseline and endpoint nor did the dropout rates differ significantly across groups. Quote: "A total of 67 patients completed the 15 weeks of treatment. Completers and dropouts were divided into the three treatment groups as follows: cognitive therapy, 24 completers and four dropouts; moclobemide, 24 completers and three dropouts; placebo, 19 completers and eight dropouts. The proportion of dropouts did not differ significantly between these conditions. All four patients in the cognitive therapy condition dropped out due to insufficient therapeutic response compared with time‐investment. In the moclobemide group reasons for dropping out were: extreme fatigue (n = 1) and insufficient therapeutic response (n = 2). Reasons for dropping out in the placebo group were: increase of migraine complaints (n = 1); insufficient therapeutic response (n = 3); increase of social phobic complaints (n = 1) and non‐compliance to the treatment protocol (n = 3). No other significant differences between completers and dropouts emerged on other demographic variables or baseline clinical scores. Insomnia was the only side‐effect that was reported significantly more often in the moclobemide group compared to the placebo group (22 vs. 4%)". Overall 20% of the participants dropped out of the study. Quote: "For this analysis, the last observation of patients who dropped out was carried forward to serve as post‐test or follow‐up test". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Medication and study funded by industry. No other sources of bias were identified. |
Pande 1999.
| Methods | Design: multicentre, randomised, placebo controlled, parallel, flexible dose, double‐blind Duration of intervention: 14 weeks Post‐treatment: no follow‐up Placebo run‐in: 1‐week, single‐blind placebo lead‐in |
|
| Participants | Sample size: 69 randomised to gabapentin or placebo Mean age (SD): 35.6 (9.6) years Sex: 40 men and 29 women Diagnostic measure: DSM‐IV Inclusion criteria: quote: "The study sample consisted of outpatients of either gender who were aged 18 years or older and who had received a diagnosis of social phobia according to DSM‐IV criteria ... To exclude those with mild social anxiety limited to few situations that clinicians may or may not consider for drug therapy, study patients were required to have a minimum score of 50 on the LSAS at entry into the study". Exclusion criteria: quote: "Patients were excluded if they suffered from uncontrolled medical illnesses, had prominent depressive symptoms measured by a HAM‐D Depressed Mood subscale score of > 3 at baseline, met criteria for a current diagnosis of alcohol or substance abuse, or were taking other psychotropic agents". Dropouts: 30/69 (13/34 in the gabapentin and 17/35 in the placebo groups) |
|
| Interventions | Pharmacological intervention: quote: "Gabapentin was dispensed as 300 mg capsules. All patients initiated the randomized portion of the study with one capsule twice a day (either placebo or gabapentin 300 mg) and had to reach a dose of 1 capsule three times a day by the end of the first week Thereafter, as long as symptoms of social phobia were present and there were no limiting adverse effects, the dose was required to be escalated in increments of no more than 300 mg each day, up to the maximum of 3,600 mg/day. At the conclusion of the double‐blind phase of the trial, treatment was discontinued by reducing the dose by two capsules daily". | |
| Outcomes | Primary outcome measure: LSAS (for reduction of anxiety) Secondary outcome measures: BSPS (for reduction of anxiety), MMFQ (for reduction of anxiety), SPIN (for reduction of anxiety), CGIC (for treatment efficacy), HAM‐D (for reduction of depression) and HAM‐A (for reduction of anxiety) Time points: Quote: "Patients were evaluated at weekly visits for the first 4 weeks, biweekly for the next 4 weeks, and monthly thereafter. Various efficacy and safety assessments were made during each visit according to a predetermined schedule" |
|
| Notes | Industry funded: yes. Quote: "This study was supported by the Parke‐Davis Division of Warner‐Lambert Company". Medication provided by industry: unclear Any of the authors work for industry: yes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "After a 1‐week, single‐blind placebo lead‐in during which patients had to continue to meet entry criteria, they were randomly assigned to receive double‐blind treatment with either gabapentin or placebo for 14 weeks". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | A smaller proportion of participants withdrew from the gabapentin (13/34; 38%) than the placebo group (17/35; 49%). The primary reason for withdrawal in the gabapentin was adverse events compared to lack of efficacy and adverse events in the placebo group. The 2 groups did not differ by sample characteristics at baseline. Quote: "More patients on gabapentin (62%) completed the study than did those on placebo (51%). Adverse events were the primary reason for early withdrawal from the gabapentin group during the study. Patients on placebo withdrew early for a variety of reasons, including adverse events and lack of efficacy. Overall, the rate of withdrawal was gradual over the 14‐week study (averaging two patients per visit per treatment group) and was similar for both treatment groups ... Patients in each treatment group were comparable with respect to demographics". Overall 43% of the participants dropped out of the study. Quote: "For all analyses, the last observation carried forward (LOCF) was used as the endpoint measurement". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. |
Pande 2004.
| Methods | Design: multicentre, randomised, double‐blind, placebo‐controlled, multiple fixed dose, clinical trial Duration of intervention: 10 weeks Post‐treatment: no follow‐up Placebo run‐in: 1‐week, single‐blind, placebo lead‐in |
|
| Participants | Sample size: 135 randomised to pregabalin or placebo Mean age (SD): 38.4 (11.5) years Sex: 79 men and 56 women Diagnostic measure: DSM‐IV Inclusion criteria: quote: "The study sample consisted of outpatient men and women, 18 years or older, suffering from social anxiety disorder ... Patients were also required to have a total score of 50 or greater on the LSAS to be included in the study". Exclusion criteria: quote: "Patients with any of the following axis I DSM‐IV diagnoses were excluded from the study: delirium, dementia, amnestic, or other cognitive disorders; schizophrenia; bipolar or schizoaffective disorder; substance abuse disorder active in the last 6 months; and panic disorder, agoraphobia, or obsessive‐compulsive disorders. Patients with a secondary diagnosis of major depressive disorder (based on DSM‐IV criteria) were not excluded, however, patients with a HAM‐D Rating Scale Item 1 (depressed mood) score 3 at screening were excluded. Patients with borderline or antisocial personality disorder, or ongoing psychodynamic or behavioral psychotherapy for social anxiety disorder were excluded from the study. In addition, patients were excluded if they suffered from uncontrolled medical illnesses, or if they were taking other psychotropic agents". Dropouts: 41/135 (20/47 and 11/42 in the pregabalin and 10/46 in the placebo groups) |
|
| Interventions | Pharmacological intervention: quote: "The study had 3 phases: screening, double‐blind treatment, and taper. Following a 1‐week, single‐blind, placebo lead‐in, patients who continued to meet entry criteria were randomized to double‐blind treatment with either pregabalin 600 mg/d (200 mg TID), pregabalin 150 mg/d (50 mg TID), or placebo. Study medication was titrated to the full assigned dose over the first 6 days of the double‐blind phase. Following 10 weeks of double‐blind treatment, the final efficacy assessments were made (termination visit). Patients then entered the taper phase, study medication dose was tapered off over 6 days, and a final follow‐up visit was conducted". | |
| Outcomes | Primary outcome measure: LSAS (for reduction of anxiety) Secondary outcome measures: BSPS (for reduction of anxiety), HAM‐A (for reduction of anxiety), SPI (for reduction of anxiety), FQ (for reduction of anxiety), CGI‐I (for treatment efficacy), CGI‐S (for reduction of anxiety) and the SF‐3 (for quality of life) Time points: Quote: "Patients were evaluated every 1 to 2 weeks for the duration of the study" |
|
| Notes | Industry funded: yes. Quote: "This study was supported by Parke‐Davis Pharmaceutical Research Division of Warner‐Lambert". Medication provided by industry: unclear Any of the authors work for industry: yes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "The safety and efficacy of pregabalin for the treatment of social anxiety disorder was evaluated in a double‐blind, multicenter clinical trial in which 135 patients were randomized to 10 weeks of double‐blind treatment with either pregabalin 150 mg/d, pregabalin 600 mg/d, or placebo". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | A higher proportion of participants withdrew from the 150 mg/d and 600 mg/d (16/45; 36%) groups compared to the placebo group (10/46; 22%). Most participants dropped out of the study in the 600 mg/d pregabalin group because of adverse events and lack of compliance compared to the 150 mg/d pregabalin and placebo groups. No information was provided on if the 2 groups differed by sample characteristics at week 10. Quote: "Of the 135 randomized patients, 94 completed the study. One patient receiving pregabalin 600 mg/d had no postrandomisation efficacy assessment and was excluded from the efficacy analysis ... More patients receiving pregabalin 600 mg/d (n = 11, 23.4%) withdrew due to adverse events than patients receiving either pregabalin 150 mg/d (n = 4, 9.5%) or placebo (n = 4, 8.7%)". Overall 29% of the participants dropped out of the study. Quote: "The primary efficacy measure was changed in the LSAS total score from baseline (randomization visit) to end point (termination visit or last observation carried forward during the 10‐week double‐blind phase)". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias were identified. |
Randall 2001.
| Methods | Design: single‐centre, randomised, placebo‐controlled, parallel, flexible dose, double‐blind Duration of intervention: 8 weeks Post‐treatment: no follow‐up Placebo run‐in: no |
|
| Participants | Sample size: 15 randomised to paroxetine or placebo (18 randomised: 3 participants were excluded due to various reasons) Mean age (SD): 35.5 (8.4) years Sex: 13 men and 2 women Diagnostic measure: DSM‐IV Inclusion criteria: quote: "All individuals were required to meet DSM‐IV criteria for both current social anxiety disorder (social phobia) as well as alcohol abuse or dependence criteria. Other inclusion criteria included an age range between 18 and 70 years, willingness to attend 8 weekly outpatient study visits, and consumption of at least 15 standard drinks in the past 30 days". Exclusion criteria: quote: "An individual was excluded if (s)he had a primary Axis I DSM‐IV diagnosis other than alcohol abuse/dependence and social anxiety disorder (including dependence on another drug of abuse excepting nicotine). Other exclusion criteria included the presence of significant medical problems, current use of any prescribed psychotropic medicine on a regular basis, transportation problems, abnormal electrocardiogram, or elevated liver enzymes". Dropouts: 2/15 (1/6 in the paroxetine and 1/9 in the placebo groups) |
|
| Interventions | Pharmacological intervention: quote: "Paroxetine or visually matched placebo was delivered in 20‐mg pills for 8 weeks. Patients were instructed to take 1 pill per day in week 1, 2 pills per day in week 2, and 3 pills per day thereafter, unless there were dose‐limiting side effects. The targeted maintenance dosage was 60 mg/d". | |
| Outcomes | Primary outcome measures: LSAS (for reduction of anxiety), CGI‐I (for treatment efficacy) and SPIN with TLFB drinking measures (for reduction of anxiety) Secondary outcome measures: BDI (for reduction of depression), ASI (for addiction) and ADS (for alcohol dependence) Time points: Quote: "After randomization and the meeting with the physician, the patient met weekly with the research assistant to fill out the assessments noted above, except for the SPIN, ASI, and BDI, which were administered only twice (baseline and at week 8)" |
|
| Notes | Industry funded: yes. Quote: "This work was supported by an investigator‐initiated award from SmithKline Beecham (to J.R.D.)". Medication provided by industry: yes. Quote: "SmithKline Beecham supplied the drug and matched placebo". Any of the authors work for industry: no |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "Patients were randomized according to a predetermined order prepared by the pharmaceutical company". |
| Allocation concealment (selection bias) | Low risk | Quote: "Paroxetine or visually matched placebo was delivered in 20‐mg pills for 8 weeks ... The institutional research pharmacy maintained the blind and dispensed all study medications". |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Medication and placebo was provided in identical capsules. However, it is not clear whether both participants and personnel were blinded. Quote: "Paroxetine or visually matched placebo was delivered in 20‐mg pills for 8 weeks". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Similar proportions of participants withdrew from the paroxetine (1/6; 17%) and placebo groups (1/9; 11%). No information was provided on the reasons for withdraws for the 2 groups and the groups did not differ by sample characteristics at baseline. Quote: "Treatment groups were similar at baseline on alcohol use measures, social anxiety severity, and demographic variables ... Week 8 SPIN data were available only for 12 of the 15 patients". Overall 13% of the participants dropped out of the study. Quote: "Two patients (1 from each treatment group) had missing data for 1 or more of the weekly assessments, and a last‐point‐carried‐forward approach was used to provide values for the missing data". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Medication and study funded by industry. The study sample comprised of more men than women. Quote: "Most patients were male (87%), about 36 years of age, and all were of white ethnicity". |
Ravindran 2009.
| Methods | Design: single‐centre, randomised, double‐blind, flexible dose, placebo‐controlled trial (NCT00260533) Duration of intervention: 10 weeks Post‐treatment: no follow‐up Placebo run‐in: no |
|
| Participants | Sample size: 27 randomised to atomoxetine (ATM) or placebo Mean age (SD): 42.05 (10.15) years Sex: 21 men and 6 women Diagnostic measure: DSM‐IV Inclusion criteria: quote: "Medically healthy outpatients aged 18 to 65 years, with a clinically predominant diagnosis of DSM‐IV GSAD ... The subjects were required to have a score of 60 or higher on the LSAS and a total score of 14 or lower on the HAM‐D to be included in the study". Exclusion criteria: quote: "Other exclusion criteria included the presence of comorbid ADHD or another primary axis‐I disorder (including DSM‐IV diagnosis of another anxiety, eating, or substance use disorder in the previous 6 months), failure to respond to prior adequate trials of 2 or more medications to treat GSAD, and the subjects were on concomitant psychotropic medications in the previous 2 weeks (fluoxetine in the previous 4 weeks) or those receiving concurrent formal psychotherapy targeted at GSAD". Dropouts: 6/27 (3/14 in the atomoxetine and 3/13 in the placebo groups) |
|
| Interventions | Pharmacological intervention: quote: "The study medication was titrated on a fixed schedule over the first 2 weeks starting with 20 mg per day for 1 week and 40 mg for another week and then flexibly titrated based on response and tolerability in 20‐mg increments every 2 weeks up to a maximum dose of 100 mg. For the final 4 weeks of the study, the dose of medication was held stable". | |
| Outcomes | Primary outcome measures: LSAS (for reduction of anxiety) Secondary outcome measures: change in LSAS (for reduction of anxiety), CGI‐C (for treatment efficacy), HAM‐D (for reduction of depression) and SDS (for reduction of functional disability) Time points: Quote: "All measures were completed at each study visit (baseline and weeks 2, 4, 6, 8, and 10)" |
|
| Notes | Industry funded: yes. Quote: "This study was supported by an investigator‐initiated research grant from Eli Lilly and Company". Medication provided by industry: unclear Any of the authors work for industry: yes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "Twenty‐seven outpatients with clinically prevailing diagnoses of GSAD by the DSM‐IV were randomized in a 1:1 ratio to 10 weeks of double‐blind flexible‐dose treatment with either ATM 40‐100 mg per day (n = 14) or placebo (n = 13)". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Medication and placebo was provided in identical capsules. However, it is not clear whether both participants and personnel were blinded. Quote: "Within 1 week of the screening visit, eligible subjects were then randomized to receive a double‐blind treatment with either ATM or a matching placebo in a 1:1 ratio". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Similar proportions of participants withdrew from the atomoxetine (3/14; 21%) and placebo groups (3/13; 23%). Participants withdrew for the following reasons: adverse events, poor compliance, worsening depression, and lost to follow‐up. The 2 groups did not differ by sample characteristics at baseline nor did they significantly differ by attrition rates. Quote: "Overall, 21 of the 27 randomised subjects completed the study (n = 11 [78.6%], ATM; n = 10 [76.9%], placebo), with no significant difference in attrition rates for each group (ATM, 21.4%; placebo, 23.1%). No significant differences were found in any of the other baseline demographic or clinical characteristics between treatment groups. Reasons for early termination included adverse events (n = 1, ATM), poor compliance (n = 1, ATM), worsening depression (n = 1, placebo), and 3 participants lost to follow‐up (n = 1, ATM; n = 2, placebo)". Overall 22% of the participants dropped out of the study. Quote: "Analyses of results were performed for the efficacy on intention‐to‐treat (ITT) population (all randomised subjects who took any study medication and returned for 1 or more post baseline evaluation) using the last observation carried forward (LOCF), and for observed cases, a completer analysis was performed". |
| Selective reporting (reporting bias) | Low risk | All outcomes were reported as specified in the protocol. |
| Other bias | Unclear risk | Funding for study provided by industry. Their were more men than women in the study sample (21:6). |
Rickels 2004.
| Methods | Design: multicentre, double‐blind, randomised, flexible dose, placebo‐controlled trial Duration of intervention: 12 weeks Post‐treatment: no follow‐up Placebo run‐in: 1 week single‐blind, placebo treatment period |
|
| Participants | Sample size: 272 randomised to venlafaxine ER or placebo Mean age (SD): 41.5 (12.25) years (for 261 participants) Sex: 150 men and 111 women (for 261 participants) Diagnostic measure: DSM‐IV Inclusion criteria: quote: "Patients could be included in the study if they were a male or female outpatient at least 18 years of age, had met the DSM‐IV criteria for (generalized) SAD for at least 6 months before the commencement of the study, had anxiety severe enough to warrant anxiolytic therapy, had a CGI‐S score of 4, and had a minimum score of 50 on the LSAS at the prestudy evaluation and on day 1 of the study, with a decrease of 30% between prestudy and baseline tests. In addition, the Covi Anxiety scale score had to be greater than the Raskin Depression total score (with a Raskin total score of not more than 9) at the prestudy evaluation". Exclusion criteria: quote: "Patients were excluded from the study if they had a clinically important medical condition, seizure disorder, mental disorder due to medical condition, myocardial infarction during the previous 6 months, laboratory or electrocardiographic abnormality, positive pregnancy test result at prestudy evaluation, pregnancy or lactation during the study, prestudy HAM‐D score of 15, Axis I or Axis II disorder current or predominant during last 6 months, alcohol or substance abuse within last year, suicidality, regular alcohol use, or drug abuse. Patients who failed to respond to prior treatment were eligible to participate; however, those who had received venlafaxine (ER or IR) within 6 months of study day 1 or had a known hypersensitivity to venlafaxine or related compounds were excluded. In addition, psychotherapy, electroconvulsive therapy, investigational drugs or procedures, sedative hypnotic agents, sumatriptan, naratriptan or zolmitriptan or similar agents, herbal remedies, monoamine oxidase inhibitors, benzodiazepines, anxiolytics, and antidepressants and antipsychotics were not permitted during the study and for a specified period before the start of the study". Dropouts: 100/272 (insufficient information to determine dropout rates for the 2 groups separately) |
|
| Interventions | Pharmacological intervention: quote: "Patients were randomly assigned to receive venlafaxine ER (75 to 225 mg/d) or placebo for a 12‐week period, followed by a 2‐week (±3 days) taper period ...Patients were randomly assigned to take either venlafaxine ER 75 mg (1 capsule) or placebo on study days 1 to 7 (±3 days). On study days 8 (±3 days) to 14, the dose was increased to 2 capsules (placebo or 150 mg venlafaxine ER). If clinically indicated, on day 15 (±3 days), the dose was increased to 225 mg (3 capsules) or placebo (3 capsules). The dose could also be reduced to 75 mg to improve tolerability if needed. Upon completion of or discontinuation from the study, the number of capsules taken daily was reduced by 1 during each week of the taper period. Patients taking only 1 capsule did not need to taper their dose". | |
| Outcomes | Primary outcome measures: LSAS (for reduction of anxiety) Secondary outcome measures: CGI‐S (for reduction of anxiety), CGI‐I (for treatment efficacy), SPIN (for reduction of anxiety), LSAS fear and avoidance subscale (for reduction of anxiety) Time points: Quote: "Safety and efficacy measures as well as vital signs (including supine pulse rate, and supine and standing blood pressure) were recorded at baseline and weeks 1, 2, 3, 4, 6, 8, 10, and 12; adverse events were recorded at a poststudy evaluation. Patients were also evaluated 4 to 10 days after the last dose of medication" |
|
| Notes | Industry funded: no Medication provided by industry: no Any of the authors work for industry: yes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "Two hundred seventy‐two outpatients were randomly assigned to receive either a flexible dose of venlafaxine ER (75 to 225 mg/d) or placebo for 12 weeks". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The proportion of dropouts per treatment group could not be determined due to the lack of information provided by the study. More participants in the venlafaxine group withdrew to adverse events compared to those in the placebo group. The two group differed by sample characteristics at baseline and at week 12. Quote: "No significant differences were observed between treatment groups in any of the characteristics or baseline scores, and the characteristics were similar to those of the safety population ... Thus, 261 patients were included in the intent‐to‐treat efficacy analysis. There were 100 discontinuations. One hundred seventy‐two patients completed the study. Significantly more patients in the venlafaxine ER group (15%) than the placebo group (4%) withdrew because of adverse events". Quote: "Analyses of observed cases and last observation carried forward (LOCF) data were performed". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Low risk | No other sources of bias were identified, although there were more men than women who participated in the study. Quote: "The group was predominantly White, had more men than women, and the mean duration of the current episode of SAD was 25.9 to 28.6 years, underscoring the chronic nature of untreated SAD". |
Schneier 1998.
| Methods | Design: single‐centre, randomised, placebo‐controlled, parallel, flexible dose, double‐blind Duration of intervention: 8 weeks Post‐treatment: 1 month follow‐up Placebo run‐in: one week of single‐blind placebo |
|
| Participants | Sample size: 78 randomised to moclobemide or placebo (77 ITT sample, due to data missing for one participant) Mean age (SD): 34.95 (17.3) years (for 77 ITT sample) Sex: 46 men and 31 women (for 77 ITT sample) Diagnostic measure: DSM‐III‐R Inclusion criteria: quote: "Out‐patients aged 18‐65 years, with a principal diagnosis of social phobia, absence of major depression, psychotic disorders, substance misuse (in the past six months) and other major psychiatric disorders were included in the study". Exclusion criteria: quote: "Past history of major depression or substance misuse, and current dysthymia were permitted". Dropouts: 20/77 (10/40 in the moclobemide and 10/37 in the placebo groups) |
|
| Interventions | Pharmacological intervention: quote: "Moclobemide was initiated at 100 mg twice daily and increased over a period of two weeks to a maximum dose of 400 mg twice daily. Dosage could be adjusted as clinically indicated to manage adverse effects". | |
| Outcomes | Primary outcome measures: LSAS (for reduction of anxiety), LSPDS (for reduction of anxiety), CGI (for treatment efficacy), SADS (for reduction of anxiety) and FNES (for reduction of anxiety) Secondary outcome measures: FQ (for reduction of anxiety), SDS (for reduction of functional disability), BDI (for reduction of depression), HAM‐D (for reduction of depression), HAM‐A (for reduction of anxiety) and SCL‐90 (for reduction of anxiety) Time points: Quote: "Major evaluations were done at randomisation and after 4, 8 and 16 weeks of randomised treatment" |
|
| Notes | Industry funded: no. Quote: "This study was supported by National Institute of Mental Health grant 5 R29 MH 47831‐04 to F.R.S". Medication provided by industry: yes. Quote: "Hofmann‐LaRoche, Nutley, NJ kindly supplied moclobemide and matching placebo for the study". Any of the authors work for industry: no |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Author correspondence: quote: "A random number sequence, generated by a data manager with no patient contact, was used to generate the randomization sequence". |
| Allocation concealment (selection bias) | Low risk | Author correspondence: quote: "Yes, allocation sequence was concealed by keeping codes in sealed opaque envelopes". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Integrity of the blind design was assessed at week 8 by questionnaires which asked patients and independent evaluators to make a forced choice regarding their belief about which treatment each patient had received ... Neither patients nor independent evaluators identified the treatment condition correctly at a rate greater than chance ... Hofmann‐LaRoche, Nutley, NJ kindly supplied moclobemide and matching placebo for the study". |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Assessment instruments included patient self‐ratings and ratings by an independent evaluator who, in addition to being blind to randomisation status, was blind to adverse effects and dosage adjustments". |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Similar proportions of participants withdrew from the moclobemide (10/40; 25%) and placebo groups (10/37; 27%). Reasons for withdrawal were similar across groups. The 2 groups did not differ by sample characteristics at baseline and no statistically significant difference was found between attrition rates. Quote: "The treatment groups did not differ in demographic features, social phobia subtype, comorbidity or rates of prior drug treatment. In the moclobemide group, 33 patients (82.5%) completed at least four weeks of randomised treatment, and 30 (75.0%) completed at least eight weeks. In the placebo group, 32 (86.5%) and 27 (73.0%) completed at least four and at least eight weeks, respectively. Rates of dropout by week 4 and by week 8 did not differ between moclobemide and placebo groups. A comparison of patients who dropped out to week 4 versus patients who completed at least 4 weeks yielded no significant differences between the 2 groups on any baseline variables ... Number of dropouts in each group, by reason for dropout, were: adverse effects (four moclobemide, three placebo), unknown (two moclobemide, three placebo), non‐compliance with appointments (one moclobemide), lack of efficacy (one moclobemide, two placebo), non‐compliance with medication (two moclobemide), personal reasons unrelated to the study (two placebo)". Overall 26% of the participants dropped out of the study. Quote: "Outcome over the first eight weeks of treatment was analysed for the following three samples: (a) intention‐to‐treat, including all subjects randomised and carrying last observations forward for dropouts ..." |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Medication was provided by industry. No other sources of bias were identified. |
Schutters 2010.
| Methods | Design: single‐centre, randomised, double‐blind, parallel, flexible dose, placebo‐controlled study Duration of intervention: 12 weeks Post‐treatment: no follow‐up Placebo run‐in: no |
|
| Participants | Sample size: 60 randomised to mirtazepine or placebo Mean age (SD): 38.6 (10.5) years Sex: 26 men and 34 women Diagnostic measure: DSM‐IV Inclusion criteria: quote: "Men and women aged between 18 and 65 years of age with generalized SAD according to the DSM‐IV classification and MINI". Exclusion criteria: quote: "Patients were excluded from the study if they had a current comorbid axis I diagnosis according to the DSM‐IV classification; in case of an actual risk for suicide according to the investigator, pregnancy or instable chronic physical conditions as assessed by the medical history and physical examination at screening. The HAM‐D was used to screen for comorbid depressive symptoms and patients were excluded if they had a score of 15 or more. ... Psychotropic medication or any psychotherapeutic interventions in the last month preceding or during the trial period were not allowed". Dropouts: 3/60 (2/30 in the mirtazepine and 1/30 in the placebo groups) |
|
| Interventions | Pharmacological intervention: quote: "The patients received an initial dose of 30mg/day of mirtazepine or an identical looking and tasting placebo. From day 15 onwards, the dose was increased to 45mg of mirtazepine or placebo ODT". | |
| Outcomes | Primary outcome measures: LSAS (for reduction of anxiety) and CGI (for treatment efficacy) Secondary outcome measures: FNES (for reduction of anxiety), SDS (for reduction of functional disability), and ASEX (for sexual functioning) Time points: Quote: "The patients were evaluated at screening, baseline, week 2, week 4, week 8 and week 12 with the LSAS, the FNES, the SDS, and the CGI" |
|
| Notes | Industry funded: yes. "Quote: This study was funded by an unrestricted grant by Organon". Medication provided by industry: unclear Any of the authors work for industry: no |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "Patients were randomly assigned to the double‐blind treatment with mirtazepine or placebo for 12 weeks". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Medication and placebo was provided in identical capsules. However, it is not clear whether both participants and personnel were blinded. Quote: "The patients received an initial dose of 30mg/day of mirtazepine or an identical looking and tasting placebo". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Similar, low proportions of participants withdrew from the mirtazepine (2/30; 7%) and placebo groups (1/30; 3%). More participants in the mirtazepine group withdrew due to adverse events compared to lost for continuation in the placebo group. The 2 groups did not differ by sample characteristics at baseline. Quote: "Three patients (0.05%) did not complete the study, two patients from the mirtazepine group dropped out because of side effects (mainly sedation) and one patient from the placebo group was lost for continuation (reason unknown). There were no significant differences between the two groups in age, sex and age of onset". Overall 10% of the participants dropped out of the study. Quote: "Last observation carried forward (LOCF) efficacy analyses were conducted on all the patients who had received any double‐blind medication and from whom at least one valid post baseline efficacy evaluation was obtained". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias were identified. |
Stein 1996.
| Methods | Design: single‐centre, open‐label trial, followed by randomised, parallel, flexible dose, double‐blind placebo‐controlled discontinuation, and a relapse prevention phase Duration of intervention: 12 weeks Post‐treatment: no follow‐up Placebo run‐in: no |
|
| Participants | Sample size: 16 randomised to paroxetine or placebo (36 in the open label phase, 16 of which was randomised in the 12 week phase) Mean age: not specified Sex: 24 men and 6 women (for the 30 completers in the open label phase) Diagnostic measure: DSM‐IV Inclusion criteria: quote: "Patients who met DSM‐IV criteria for social phobia, generalized type, according to a semi‐structured interview derived from the Structured Clinical Interview for DSM‐III‐R ... The presence of current (past 6 months) comorbid major depression was an exclusion criterion, although patients with past histories of major depression were included". Exclusion criteria: quote: "Patients with comorbid panic disorder were excluded, unless the panic disorder was felt to be clearly secondary to the social phobia in terms of severity and current impact on functioning. Patients who were currently abusing substances, including alcohol, were excluded from the study". Dropouts: 6/16 (1/8 in the paroxetine and 5/8 in the placebo groups) |
|
| Interventions | Pharmacological intervention: quote: "Eligible subjects began treatment with 10 mg of paroxetine at bedtime. Treating clinicians (M.B.S., C.D.L.K., and R.A.C.) followed a protocol that mandated 10‐mg weekly increments to a maximum of 50 mg/day. If subjects experienced side effects, a once‐only dosage reduction of 10 mg/day was permitted, with the option of reinstituting the prior dose if clinically indicated 1 week later. Only one such dosage adjustment was allowed per patient. Patients who could not tolerate a minimum of 20 mg/day were withdrawn from the study. Treatment lasted for a total of 11 weeks ... Patients were then randomized to either continue paroxetine with no change in dose or to taper and then discontinue paroxetine with placebo substitution for a total of 12 weeks. Regardless of the starting dose, the rate of taper consisted of 1 week at 20 mg and then discontinuation. The taper and all subsequent dosing were conducted under double‐blind conditions wherein all subjects took the same number of turquoise‐colored tablets for the duration of the study". | |
| Outcomes | Primary and secondary outcome measures: MADRS (for reduction of depression), LSAS (for reduction of anxiety), DSPS (for reduction of anxiety), CGI‐S (for reduction of anxiety), CGI (patient‐rated) (for treatment efficacy), FNES (for reduction of anxiety), SIAS (for reduction of anxiety), SPS (for reduction of anxiety), and SDS (for reduction of functional disability) Time points: Quote: "The following ratings were conducted by an experienced clinician at baseline, week 4, week 8, and week 12 in the double‐blind discontinuation study: Montgomery‐Asberg Depression Rating Scale, Liebowitz Social Anxiety Scale, Duke Social Phobia Scale, and the Clinician‐Rated Clinical Global Impressions Scale (CGI; severity version at baseline and change version subsequently). Self‐ratings conducted pre‐ and posttreatment included a patient‐rated version of the CGI, the Fear of Negative Evaluation Scale, the Social Interactional Anxiety Scale, and the Social Performance Scale; the Sheehan Disability Scale (which has three subscales: work, social, and family disability) was completed at baseline, weeks 4, 8, and 11. In the double‐blind discontinuation study, a subject was considered "relapsed" if the clinician‐rated CGI was rated as "no improvement" or "worse" (compared to pre‐treatment) on two consecutive visits. At each visit, subjects were systematically questioned about possible side effects' |
|
| Notes | Industry funded: no Medication provided by industry: no Any of the authors work for industry: no |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "Sixteen responders were randomized to an additional 12 weeks of either paroxetine (with no dosage change) or placebo (after a taper period) on a double‐blind basis". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Medication and placebo was provided in identical capsules. However, it is not clear whether both participants and personnel were blinded. Quote: "The taper and all subsequent dosing were conducted under double‐blind conditions wherein all subjects took the same number of turquoise‐colored tablets for the duration of the study". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Information was not provided for number of dropouts during the randomised relapse‐prevention phase of the study. |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Low risk | No other sources of bias were identified. |
Stein 1998.
| Methods | Design: multicentre, randomised, placebo‐controlled, parallel, flexible dose, double‐blind (GSK protocol ID: 29060/382). Duration of intervention: 12 weeks. Post‐treatment: no follow‐up. Placebo run‐in: 1 week, single‐blind, placebo, run‐in period. |
|
| Participants | Sample size: 187 randomised to paroxetine or placebo (184 efficacy population, lost to follow‐up before efficacy evaluation). Mean age (range): 36.3 (18‐76) years Sex: 81 men and 106 women. Diagnostic measure: DSM‐IV. Inclusion criteria: quote: "Eligible patients were 18 years of age or older; adults older than 65 years were permitted if they were able to tolerate a starting paroxetine dose of at least 20 mg/d". Exclusion criteria: quote: "Patients who required concurrent psychoactive medication (except chloral hydrate for insomnia), narcotic analgesics, warfarin sodium, digitalis glycosides, phenytoin, cimetidine, or sulfonylurea derivatives were excluded. Patients who had taken any psychotropic agent or beta‐blockers within 14 days prior to the study were ineligible, as were those who had received depot neuroleptics within the previous 12 weeks. Also excluded were patients with any other Axis I diagnosis that was considered to be clinically predominant within the previous 6 months. Patients who met DSM‐IV criteria for substance abuse or dependence within 3 or 6 months prior to this study, respectively, were excluded, as were those judged to be serious suicidal or homicidal risks. Additional reasons for exclusion were body dysmorphic disorder, history of seizure disorder, schizophrenia or bipolar affective disorder, any serious or uncontrolled medical illness or condition that precluded paroxetine use, electroconvulsive therapy within the previous 3 months, investigational drug use or participation in a clinical trial within the previous 12 months, and previous intolerance or lack of response to paroxetine (no subject was excluded on this basis). Women who were pregnant, lactating, or not using a clinically acceptable method of birth control also were ineligible. Finally patients with clinically significant abnormal laboratory or electrocardiographic findings that could not be resolved prior to baseline evaluations were not included". Dropouts: 53/187 (32/94 in the paroxetine and 21/93 in the placebo groups). |
|
| Interventions | Pharmacological intervention: quote: "For the purposes of blinding, dosage was referred to as level 1 (20 mg), level 2 (30 mg), level 3 (40 mg), or level 4 (50 mg). Patients received an initial dose of level 1 medication once daily. After 2 weeks, the dosage could be increased to the next level (i.e. 50 mg/d) based on clinical response as determined by the treating physician. Dosage could be reduced to a minimum of level 1 (i.e. 20 mg/d) at any time if the physician felt that adverse effects warranted this adjustment". | |
| Outcomes | Primary outcome measures: CGI‐I (for treatment efficacy) and LSAS (for reduction of anxiety) Secondary outcome measures: SADS (for reduction of anxiety), SDI (for reduction of functional disability) and LSAS (for reduction of anxiety) Time points: Quote: "Patients were evaluated for safety and efficacy at weeks 1, 2, 3, 4, 6, 8, and 12. Adverse events were also assessed telephone at week 10" |
|
| Notes | Industry funded: yes. Quote: "Financial support for this study was provided by SmithKline Beecham Pharmaceuticals. Collegeville, Pa". Medication provided by industry: unclear Any of the authors work for industry: yes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomly assigned in a balanced fashion to the 2 treatment groups (in blocks of 4) using a computer‐generated randomisation code for up to 300 patients". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Medication and placebo was provided in identical capsules. However, it is not clear whether both participants and personnel were blinded. Quote: "Patients who were rated no more than minimally improved and who continued to meet all inclusion criteria were randomised to receive paroxetine or placebo (identical in appearance) ... For the purposes of blinding, dosage was referred to as level 1 (20 mg), level 2 (30 mg), level 3 (40 mg) or level 4 (50 mg)". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Higher proportions of participants withdrew from the paroxetine (32/94; 34%) than the placebo groups (21/93; 23%). More participants in the paroxetine group withdrew due to adverse events, whereas being lost to follow‐up was cited as the being reasons for study dropout in the placebo group. The 2 groups did not differ by sample characteristics at baseline. Quote: "Of the 187 patients randomised in this study, 4 patients received the drug but were lost to follow‐up prior to the first efficacy evaluation. Therefore, the efficacy data for the remaining 183 patients (i.e., the efficacy population) are reported ... No statistically significant differences between groups were detected with regards to demographic characteristics or mean baseline rating scale scores ... In the paroxetine group, 62 (66%) of 94 patients completed the 12‐week trial. The most common reasons for patient withdrawal were adverse events (15%; 14/94) or lost to follow‐up (13%, 12/94). In the placebo group, 72 (77%) of 93 patients completed the trial. The most common reason for discontinuation was lack of efficacy (11%, 10/93)". Overall 28% of the participants dropped out of the study. Quote: "For patients who did not complete the entire study, the last evaluation during treatment was used as an estimate of the missing data (i.e. last observation carried forward)". |
| Selective reporting (reporting bias) | Low risk | All outcomes were reported as specified in the protocol. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias were identified. |
Stein 1999.
| Methods | Design: multicentre, randomised, placebo‐controlled, parallel, flexible dose, double‐blind Duration of intervention: 12 weeks Post‐treatment: no follow‐up Placebo run‐in: no |
|
| Participants | Sample size: 92 randomised to fluvoxamine or placebo Mean age (SD): 39.4 (10.55) years Sex: 59 men and 32 women Diagnostic measure: DSM‐IV Inclusion criteria: quote: "Eligible participants, in addition to meeting the DSM‐IV criteria for social phobia and having a minimum score of 20 on the Brief Social Phobia Scale, were required to be between 18 and 65 years of age and to have no serious medical conditions (and be taking no medications medications) that might put them at risk were they to receive fluvoxamine". Exclusion criteria: quote: "Patients taking psychotropic medications within the 7 days before the study were precluded from participating, as were patients with other psychiatric disorders that were deemed to be primary in terms of clinical significance (e.g., bipolar disorder, schizophrenia) or patients judged to be at serious suicidal or homicidal risk. Patients receiving a specific form of psychotherapy for social phobia were ineligible. Women who were pregnant, lactating, or not using an acceptable method of birth control were ineligible. Patients with clinically significant abnormal laboratory or ECG findings were also ineligible". Dropouts: 24/92 (14/48 in the fluvoxamine and 10/44 in the placebo groups) |
|
| Interventions | Pharmacological intervention: quote: "After the screening procedures, each patient was randomly assigned to either placebo or fluvoxamine (at an initial dose of 50 mg of fluvoxamine per day). After 1 week the daily dose could be increased by 50 mg each week to a maximum of 300 mg/day and could be reduced at any time if side effects necessitated this (i.e., flexible dosing)". | |
| Outcomes | Primary outcome measure: CGI‐I (for treatment efficacy) Secondary outcome measures: BSPS (for reduction of anxiety), SPI (for reduction of anxiety), LSAS (for reduction of anxiety) and SDS (for reduction of functional disability) Time points: Quote: "The patients were evaluated at weeks 1, 2, 3, 4, 6, 8, 10, and 12" |
|
| Notes | Industry funded: yes. Quote: "Sponsored by Solvay Pharmaceuticals, Inc., Marietta, Ga.; funding provided by The Pharmacia & Upjohn Co., Kalamazoo, Mich". Medication provided by industry: unclear Any of the authors work for industry: yes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "After the screening procedures, each patient was randomly assigned to either placebo or fluvoxamine". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Similar proportions of participants withdrew from the fluvoxamine (14/48; 29%) and placebo groups (10/44; 23%). More participants in the fluvoxamine group withdrew due to adverse events compared to those in the placebo group. No other information was provided the reasons for treatment withdrawal nor was information reported on differences between sample characteristics at week 12. Quote: "Six patients did not return for at least one subsequent assessment, leaving 86 patients (42 taking fluvoxamine and 44 taking placebo) in the evaluable study group ... Fluvoxamine was generally well tolerated by the patients in the study, although more fluvoxamine‐treated patients (12, 25.0%) than placebo treated patients (N=4, 9.1%) discontinued the study early because of adverse events ... Final assessment was conducted at week 12 or earlier if the patient dropped out prematurely. Scores were available for 34 patients taking fluvoxamine and 34 patients taking placebo". Overall 26% of the participants dropped out of the study. Quote: "Analyses of response refer to the last observation carried forward for all subjects who had evaluable efficacy data at baseline and with treatment and who had taken at least one dose of study medication". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. Not all the participants in the sample had generalised disorder. Quote: "Although this was not an a priori criterion for entry into the study, nearly all of the patients (91.3%) suffered from the generalised type of the disorder". |
Stein 2002a.
| Methods | Design: multicentre, randomised, placebo‐controlled, parallel, flexible dose, double‐blind Duration of intervention: 12 weeks Post‐treatment: no follow‐up Placebo run‐in: 1‐week, single‐blind placebo run‐in |
|
| Participants | Sample size: 390 randomised to moclobemide or placebo (390 participants were randomised, of whom data was available for 384; seven participants randomised did not receive medication, so the ITT population for the 12‐week treatment trial comprised 377 participants (188 moclobemide, 189 placebo) Mean age (SD): 34.4 (10.5) years (for 384 participants) Sex: 204 men and 180 women (for 384 participants) Diagnostic measure: DSM‐IV Inclusion criteria: quote: "All subjects met Diagnostic and Statistical Manual (DSM‐IV) diagnostic criteria for social anxiety disorder as the primary diagnosis, had a minimum score of 4 (moderate) on the Clinical Global Impressions scale severity item (CIS‐SP) and were aged 18–65 years. Subjects with comorbid panic disorder, generalized anxiety disorder, agoraphobia or other phobias, were eligible". Exclusion criteria: quote: "Subjects were to be excluded on a number of grounds, including: (i) comorbid psychiatric conditions, i.e. if there were current mood disorders (excluding dysthymia), obsessive–compulsive disorder, or substance use disorder (excluding nicotine); if there was a history of psychosis, schizophrenia, or bipolar disorder (type I or II); an organic mental disorder, dementia or mental retardation; or if there was suicidality; (ii) comorbid medical conditions, i.e. if there was hyperthyroidism, thyrotoxicosis, pheochromocytoma, neuroblastoma, epilepsy, significant liver disease or any significant unstable or uncontrolled medical disease; or if there were any clinically significant physical examination, laboratory, or electrocardiogram findings that would put the patient at risk or obscure the effects of treatment; (iii) treatment requirements and history, i.e. if there was treatment with moclobemide during the past 6 months; treatment with classical MAOIs in the 5 weeks preceding baseline; concomitant use of other psychotropic medications in the 2 weeks preceding baseline; or any investigational drug or experimental procedure in the 4 weeks preceding baseline; and (iv) response during placebo run‐in, i.e. if between the screening and baseline visits, clinical response was rated as 1 (very much improved) or 2 (much improved) on the Clinical Global Impression scale change item (CIC‐SP)". Dropouts: 64/390 (insufficient information to determine dropout rates for the 2 groups separately) |
|
| Interventions | Pharmacological intervention: quote: "The moclobemide was initiated at 600 mg/day (300 mg a.m. +300 mg p.m.) for 1 week. This dose could be titrated downward to 450 mg/day (300 mg a.m. +150 mg p.m.) (minimum dose) if tolerability was less than moderate. Alternatively, dosage could be titrated upwards to 750 mg/day (450 mg a.m. +300 mg p.m.) (maximum dose) if tolerability was moderate or better and CIC‐SP was 2 or more. This dose range is at the higher end of the range suggested in most moclobemide package inserts (which describe initiation of medication at 300 mg daily and indicate that medication may be raised to 600 mg daily where necessary)". | |
| Outcomes | Primary outcome measure: CIC‐SP (for treatment efficacy) Secondary outcome measures: CIS‐SP (for reduction of anxiety), LSAS (for reduction of anxiety), MADRS (for reduction of depression), HAM‐A (for reduction of anxiety), SDS (for reduction of functional disability), PIC‐SP (for treatment efficacy), SAS (for reduction of anxiety), and MMFQ (for reduction of anxiety) Time points: Quote: "Clinic visits then took place at weeks 1, 2, 4, 8 and 12 for assessments of efficacy, tolerability, concurrent medications and compliance with study procedures. After an initial 12 weeks, it was at the discretion of study clinicians to invite patients to complete an additional 6 months of treatment. Monitoring visits to each site were performed in order to ensure investigator adherence to the protocol" |
|
| Notes | Industry funded: no Medication provided by industry: no Any of the authors work for industry: yes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "At the baseline visit, non‐responders (i.e. CIC‐SP of 3 or more) were randomised to moclobemide or placebo". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Medication and placebo was provided in identical capsules. However, it is not clear whether both participants and personnel were blinded. Quote: "Moclobemide and placebo were provided as identical tablets". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The proportion of dropouts per treatment group could not be determined due to the lack of information provided by the study. Reasons for withdrawal were not reported on for 4 participants, nor were the groups divided to determine the proportion of withdrawals for others reasons for discontinuation. It appears that more participants in the moclobemide group withdrew to adverse events compared to those in the placebo group. The 2 groups did not differ by sample characteristics at baseline. Quote: "Three hundred and ninety patients were randomised, of whom data was available for 384. The two treatment groups were well matched with regard to demographic characteristics ... Seven patients randomised did not receive medication, so the ITT population for the 12‐week treatment trial comprised 377 subjects (188 moclobemide, 189 placebo) ... Sixty‐four patients discontinued during the 12‐week treatment trial; the most frequent reasons were insufficient response (11 in the placebo group, eight in the moclobemide group) and adverse events (10 in the moclobemide group, five in the placebo group). Other reasons for withdrawal included failure to return to follow‐up (n=9), not cooperating with the study (n=7), withdrawal of consent (n=6) and protocol violation (n=4)". Overall 18% of the participants dropped out of the study. Quote: "The intent‐to‐treat (ITT) sample, defined as patients randomized and having received post randomization medication, was used for the efficacy analyses, with last observation carried forward for the 12‐week acute treatment study". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Low risk | No other sources of bias were identified. |
Stein 2002b.
| Methods | Design: multicentre, randomised, placebo‐controlled, parallel, flexible dose, double‐blind, maintenance study Duration of intervention: 24 weeks Post‐treatment: no follow‐up Placebo run‐in: 1‐week, single‐blind placebo run‐in |
|
| Participants | Sample size: 323 randomised to paroxetine or placebo Mean age (SD): 38.15 (11.45) years Sex: 128 men and 195 women Diagnostic measure: MINI and DSM‐IV Inclusion criteria: quote: "Eligible patients were aged at least 18 years and had a primary diagnosis of social anxiety disorder, as assessed by psychiatrists and other qualified health care professionals who received training using the MINI for DSM‐IV. No attempt was made to categorise patients as having generalised vs nongeneralised social anxiety disorder. Those older than 65 years had to be able to tolerate a paroxetine starting dosage of at least 20 mg/d and to be without renal or hepatic impairment". Exclusion criteria: quote: "Patients were excluded from the study if they had any Axis I disorder other than generalized anxiety disorder or agoraphobia during the 6 months before screening, a primary diagnosis of panic disorder during the previous 6 months, or a history of schizophrenia or bipolar affective disorder. Subscale or dependence according to DSM‐IV criteria also excluded patients from the study. Concomittant therapy with beta‐adrenergic blockers, monoamine oxidase inhibitors, benzodiazepines, or other psychoactive medication or scleroatrophic or antidepressant therapy within 14 days of baseline also precluded patients from entering the study. Patients who had previously received a therapeutic dosage of an SSRI for social anxiety disorder from an adequate duration to achieve a clinical response or who had received paroxetine for any indication but had not responded were also prevented from participating in the study". Dropouts: 66/323 (26/162 in the paroxetine and 40/161 in the placebo groups) |
|
| Interventions | Pharmacological intervention: quote: "All patients entering the 12‐week single blind acute phase received paroxetine for 12 weeks. The initial dosage was 20 mg/d with food, and patients continued receiving this dosage for at least 2 weeks. Therafter, dose titration (2‐8 weeks, by 10 mg increments) up to a maximum of 50 mg/d was permitted at the investigators discretion ... Responders at week 12 then continued with paroxetine or gradually switched to placebo for a further 24 weeks, depending on their randomization. Patients receiving paroxetine were to remain at the same dosage level as that of week 12. However, a single dosage reduction was permitted in the event of adverse experiences. Patients receiving placebo were dispensed medication to reduce their paroxetine dosage gradually during a 3‐week down titration period, depending on their level of medication at week 12, and then received placebo for the remainder of the study. The daily dosage was reduced to 10 mg each week to 20 mg/d and was maintained at this dosage until week 15, after which all patients randomised to placebo received medication for the remainder of the study". | |
| Outcomes | Primary outcome measure: CGI‐S (for reduction of anxiety) Secondary outcome measures: CGI‐I (for treatment efficacy), LSAS (for reduction of anxiety), SCL‐90 (for reduction of anxiety), EuroQol (for quality of life) and HAM‐D (for reduction of depression), Time points: Quote: "Assessments using the CGI improvement scale, LSAS, and Social phobia Inventory were made at baseline; at weeks 1, 2, 3, 4, 8, and 12 during the acute phase of the study; and at weeks 16, 20, 24, 28, 32, and 36 during the 24‐week maintenance treat phase. In addition, an evaluation using the Sheehan Disability Scale was made at each visit. Symptom Checlist‐90 (SCL‐90) and EuroQol (EQ‐5D) questionnaires were completed at baseline, at week 12 (end of acute phase), and during the maintenance phase at weeks 24 and 36 (study end point). Hamilton Depression Rating Scale assessments were performed at baseline, at the end of the acute phase (week 12), and at the end of the study (week 36) to evaluate the presence of depressive symptoms. To assess tolerability, adverse events were monitored throughout the study by asking patients non leading questions such as “Have you felt different in any way since your last visit?” |
|
| Notes | Industry funded: yes. Quote: "This study was supported by SmithKline Beecham Pharmaceuticals". Medication provided by industry: unclear Any of the authors work for industry: yes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "A computer‐generated randomisation list was used to randomise patients in a 1:1 ratio to receive either paroxetine or placebo". |
| Allocation concealment (selection bias) | Low risk | Quote: "Each investigator and centre was allocated a block of consecutively numbered treatment packs, which were dispensed in strict sequential order. The paroxetine and placebo capsules were identical in appearance and packaged to maintain blinding". |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Medication and placebo was provided in identical capsules. However, it is not clear whether both participants and personnel were blinded. Quote: "The paroxetine and placebo capsules were identical in appearance and packaged to maintain blinding". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Similar proportions of participants withdrew from the paroxetine (26/162; 16%) and placebo groups (40/161; 25%). Withdrawals were similar across groups (i.e. adverse events, lack of efficacy, deviation from protocol, lost to follow‐up and other. No information was provided on differences between sample characteristics at week 12. Quote: "Of the 323 patients continuing into the double‐blind maintenance phase of the study, 257 (136 paroxetine‐treated [84%] and 121 placebo‐treated [75%] patients) completed the study. Sixty‐six patients withdrew (26 paroxetine‐treated [16%] and 40 placebo‐treated [25%] patients). There were 26 withdrawals (8 in the paroxetine group and 18 in the placebo group) because of lack of efficacy. Withdrawals because of adverse events were more common in the placebo group (8 [5%]) than in the paroxetine (3 [2%]). Further reasons for withdrawal included deviation from protocol (4 paroxetine‐treated [3%] and 7 placebo‐treated [4%] patients, lost to follow‐up (6 paroxetine treated [4%] and 3 placebo‐treated [2%] patients), and other (5 paroxetine‐treated [3%] and 4 placebo‐treated [3%] patients). This last group comprised patients who moved away, those who withdrew consent, and those suspected to being pregnant". Overall 20% of the participants dropped out of the study. Quote: "Primary inferences were based on intention‐to‐treat last observation carried forward". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias were identified. |
Stein 2003.
| Methods | Design: multicentre, randomised, placebo‐controlled, parallel, flexible dose, double‐blind, maintenance study Duration of intervention: 12 weeks Post‐treatment: 4, 5 and 6 months follow‐up Placebo run‐in: no |
|
| Participants | Sample size: 112 randomised to fluvoxamine CR or placebo Mean age (SD): 37.15 (1.5) years (109 ITT sample) Sex: 58 men and 51 women (109 ITT sample) Diagnostic measure: DSM‐IV Inclusion criteria: quote: "Subjects were outpatients, aged 18–70 yr, had a predominant DSM‐IV diagnosis of GSAD according to the modified SCID, and a minimum score of 60 on the LSAS at screening. Women of childbearing potential or less than 1 yr post‐menopausal were required to use a medically acceptable method of birth control, while pregnant and lactating women were not eligible". Exclusion criteria: quote: "Subjects were excluded if they met any of the following criteria: Subjects with psychiatric disorders deemed to be predominant in the last 6 months including major depressive disorder, dysthymic disorder, or panic disorder; subjects with history or current diagnosis of schizophrenia, other psychotic disorders, bipolar affective disorder, borderline personality, or obsessive–compulsive disorder; subjects who had a score of 18 on the MADRS at screening, and subjects at serious suicidal risk; subjects with evidence of substance abuse disorder or dependence within the past 6 months, and subjects with positive results on a urine drug screen; subjects with unstable or serious medical conditions; subjects who required formal CBT to treat social anxiety symptoms within the previous month; and subjects taking psychotropic medications". Dropouts: 22/112 (10/57 in the fluvoxamine CR and 12/55 in the placebo groups) |
|
| Interventions | Pharmacological intervention: quote: "In the acute phase, dosage was titrated weekly during the first 5 wk of the study, from 100 mg up to 300 mg at bedtime, in 50‐mg increments, as tolerated". | |
| Outcomes | Primary outcome measure: LSAS (for reduction of anxiety) Secondary outcome measures: CGI‐I (for treatment efficacy), CGI‐S (for reduction of anxiety) and SDS (for reduction of functional disability) Time points: Quote: "In the extension phase, these measures were administered every 4 wk (i.e. weeks 12, 16, 20 and 24), or on early termination. Safety assessments comprised adverse‐event monitoring, concomitant medication monitoring, and vitalsign measurement (at weeks 12, 16, 20, 24 or early termination) as well as physical examination, 12‐lead electrocardiogram, and clinical laboratory evaluation (haematology, serum chemistry, urinalysis, and urine drug screening, serum b‐HCG in females of childbearing potential) (at weeks 12 and endpoint)" |
|
| Notes | Industry funded: yes. Quote: "This study was supported by a grant from Solvay Pharmaceuticals, Inc". Medication provided by industry: unclear Any of the authors work for industry: yes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "Subjects were randomized to fluvoxamine CR or placebo according to a centrally generated random allocation sequence, with concealment of the sequence at participating sites, which were provided with numbered packs of fluvoxamine CR or placebo that were identical in appearance". |
| Allocation concealment (selection bias) | Low risk | Quote: "Subjects were randomized to fluvoxamine CR or placebo according to a centrally generated random allocation sequence, with concealment of the sequence at participating sites, which were provided with numbered packs of fluvoxamine CR or placebo that were identical in appearance". |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Medication and placebo was provided in identical capsules. However, it is not clear whether both participants and personnel were blinded. Quote: "Subjects were randomized to fluvoxamine CR or placebo according to a centrally generated random allocation sequence, with concealment of the sequence at participating sites, which were provided with numbered packs of fluvoxamine CR or placebo that were identical in appearance". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Similar proportions of participants withdrew from fluvoxamine CR (10/57; 18%) and placebo groups (12/55; 22%). Reasons for withdrawal were not provided by the study. The 2 groups did not differ by sample characteristics at baseline. Quote: "Of the 112 subjects who enrolled, 47 (82 %) in the fluvoxamine CR‐treatment group and 43 (78%) in the placebo‐treatment group completed the extension phase ... At baseline (day 1 of the acute phase), most demographic (age, gender, ethnicity, marital status, years in school, occupational status) and clinical (GSAD duration, LSAS score, presence of Axis II disorders, family history of psychiatric disorder) variables did not significantly differ in subjects in the fluvoxamine CR (n=56) and placebo (n=53) groups". Overall 20% of the participants dropped out of the study. Quote: "Statistical analyses were nevertheless performed on the primary and secondary efficacy parameters for the intent‐to‐treat (ITT) population, using both last observation carried forward (LOCF) and observed cases". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias were identified. |
Stein 2005.
| Methods | Design: multicentre, randomised, placebo‐controlled, parallel, fixed and flexible dose, double‐blind study Duration of intervention: 28 weeks Placebo run‐in: yes |
|
| Participants | Sample size: 395, with 261 receiving venlafaxine ER (131 fixed dose, 130 flexible dose), and 134 randomised to placebo Mean age (SD): 36.9 (11.6) years (364 ITT sample) Sex: 212 men and 152 women (364 ITT sample) Diagnostic measure: DSM‐IV, as assessed using the MINI Inclusion criteria: quote: "Eligible outpatients were those aged 18 years and older who fulfilled DSM‐IV criteria for GSAD for ≥6 months before the study. Additional inclusion criteria included a Liebowitz Social Anxiety Scale (LSAS) (Heimberg et al. 1999) score ≥50, with a decrease of ≤30% between pre‐study and baseline evaluations; a Clinical Global Impressions (CGI) Scale (Guy 1976) (severity of illness item 1) score ≥4; and pre‐study 17‐item Hamilton Depression (HAM‐D‐17) (Hamilton 1960) score <15, with a score ≤2 on the depressed mood item". Exclusion criteria: quote: "Patients were excluded if they had comorbid major depression, panic disorder with or without agoraphobia, or generalized anxiety disorder, had a history or current diagnosis of any psychotic illness, were acutely suicidal, used alcohol regularly (>24 oz of beer/day or the equivalent), or had a history of drug or alcohol dependence within 1 year of the study. In addition, patients who used psychopharmacologic medications within the 7 days before the study, used antidepressants or herbal products intended to treat anxiety or depression within 14 days of the study, received venlafaxine or ECT within 6 months of the study, or received cognitive behavioral therapy within 30 days of the study were ineligible. Also prohibited from participating were patients with clinically significant abnormal findings on laboratory tests, ECG, vital signs, or physical examination; those with a history or presence of clinically important medical conditions (including head trauma and seizure disorders); and women of childbearing potential who were pregnant, lactating, or not using a medically acceptable form of contraception". Dropouts: 218/368 (133/239 in the venlafaxine ER and 85/129 in the placebo groups) |
|
| Interventions | Pharmacological intervention: a fixed dose (75 mg/d) and flexible dose (150 mg/d ‐ 225 mg/d) intervention compared to placebo x 28 weeks | |
| Outcomes | Primary outcome measure: LSAS (for reduction of anxiety) Secondary outcome measures: CGI‐I (for treatment efficacy), SPIN (for reduction of anxiety), LSAS fear and avoidance subscales (for reduction of anxiety), SDS (for reduction of functional disability), WPAI (for reduction of functional disability), clinical response (CGI‐I < 3) (for treatment efficacy), remission (LSAS total score < 30) (for treatment efficacy) Time points: Quote: "Patients were evaluated at baseline (study day −1), and on days 7, 14, 21, 28, 42, 56, 84, 112, 140, 168, and 196. Safety assessments were based on reports of adverse events and measurements of vital signs that were recorded at each visit; laboratory determinations and ECGs, which were assessed at the prestudy (or baseline), week 12, and final visits; and routine physical examinations performed at the prestudy (or baseline) and final visits" |
|
| Notes | Industry funded: yes. This study was supported by a grant from Wyeth. Medication provided by industry: yes Any of the authors work for industry: yes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information was provided on how the random sequence was generated. |
| Allocation concealment (selection bias) | Low risk | It appears as if a random number, and the individual package of medication to which it corresponded, were both supplied by the study sponsor. Quote: "Each participating center was pre‐assigned a specified block of patient numbers. A number was given to a patient at the screening visit after the informed consent was signed. At the baseline visit, once eligibility was confirmed, the patient was given a randomization number and the accompanying treatment supplies. The study sponsor supplied study medication as identical appearing capsules, packaged individually and label‐coded for each subject." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | The medication and placebo was provided in identical capsules. Quote: "The study sponsor supplied study medication as identical appearing capsules, packaged individually and label‐coded for each subject." |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | The clinician rated both outcome and side effects, with the high proportion of drug‐related adverse events in the combined venlfaxine groups compared to the placebo group (e.g. approximately 36% versus 10% for nausea) increasing the risk that the clinician could guess treatment allocation. Quote: "A second limitation of the present study is the possibility that raters may have been unblinded by the occurrence of particular medication‐related side effects (e.g. nausea or somnolence)". |
| Incomplete outcome data (attrition bias) All outcomes | High risk | A greater proportion of participants withdraw from placebo, primarily due to lack of effect (possibly resulting from the placebo run‐in component of the study design). Though the demographic and symptom severity characteristics of participants in the treatment arms were comparable at baseline, no information on the composition of the participants at endpoint was provided. Quote: "The first 12 weeks of the protocol were completed by 234 (60.6% of) patients; the entire 28 weeks treatment protocol was completed by 164 (43%). The proportion of patients who withdrew from the protocol for any reason was significantly greater in the placebo group (66%) than in either active treatment group (48% and 56%, respectively; both P<0.05); this difference was primarily attributable to the greater number of placebo patients withdrawing due to lack of effect (26% of placebo patients versus 9% and 10% of the two venlafaxine treated groups). At week 28, the mean daily dose (taking into account missed doses) of venlafaxine ER was 72.2 mg. Withdrawals due to adverse events were significantly more likely to occur in the venlafaxine ER 75 mg group (15%) and the venlafaxine 150–225 mg group (21%) than in the placebo group (6%; P=0.026 and P=0.001, respectively)". Quote: "Statistical analyses were performed using last‐observation carried‐forward (LOCF) values". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias were identified. |
Stein 2010.
| Methods | Design: multicentre, randomised, double‐blind, placebo‐controlled, 2 arm, parallel group study (NCT00612859) Duration of intervention: 12 weeks Post‐treatment: 4, 5 and 6 months follow‐up Placebo run‐in: 1 week, single blind, placebo‐run‐in period |
|
| Participants | Sample size: 216 randomised to levetiracetam or placebo (217 randomised, one participant dropped out of the levetiracetam group prior to medication intake and is not included in the analyses) Mean age (SD): 38.5 (11.75) years Sex: 133 men and 83 women Diagnostic measure: DSM‐IV Inclusion criteria: quote: "Following the consent process, subjects were screened for study eligibility. The screening visit involved confirmation of GSAD diagnosis and evaluation of other psychiatric diagnoses with the MINI. In addition, medical history, physical exam including vital signs, and routine blood and urine tests were also conducted for safety monitoring and to ensure participants did not suffer from clinically significant medical conditions. Subjects were also required to have a score of > 60 on the LSAS and a total score of < 17 to be included in the study". Exclusion criteria: quote: "Female patients of childbearing potential were required to have a negative serum pregnancy test at screening and negative urine pregnancy tests administered periodically throughout the study. Other exclusion criteria included the presence of another primary Axis I disorder, failure to respond to adequate trials of > 2 medications to great GSAD, and concomitant psychotropic medications in the previous week". Dropouts: 70/216 (34/111 in the levetiracetam and 36/106 in the placebo groups) |
|
| Interventions | Pharmacological intervention: quote: "Study medication was titrated on a fixed schedule over the first 2 weeks from 250 mg/d up to 500 mg bid and then flexibly titrated over the next 4 weeks up to a maximum of 3,000 mg daily (1,500 mg bid). The dosage was then held stable for the remaining 6 weeks. Follow‐up was weekly for 2 weeks and then 2‐week intervals until the end of the study (week 12)". | |
| Outcomes | Primary outcome measure: LSAS (for reduction of anxiety) Secondary outcome measures: CGI‐C (for treatment efficacy), HDRS (for reduction of depression), and SDS (for reduction of functional disability) Time points: Quote: "Follow‐ups was weekly for 2 weeks and then at 2‐week intervals until the end of the study (week 12)" |
|
| Notes | Industry funded: yes. Quote: "This study was funded in its entirety by UCB Pharma, USA". Medication provided by industry: unclear Any of the authors work for industry: no |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "Those who maintained an LSAS score > 60 and a CGI‐C >2 (score range: 0‐7) on their return visit (baseline‐week 0) were then randomly assigned to double‐blind treatment with either levetiracetam or matching placebo in a 1:1 ratio. Randomization was stratified according to LSAS scores at baseline (≤ 80, > 80) and age (≤ 40 years, > 40 years)". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Medication and placebo was provided in identical capsules. However, it is not clear whether both participants and personnel were blinded. Quote: "Those who maintained an LSAS score > 60 and a CGI‐C >2 (score range: 0‐7) on their return visit (baseline‐week 0) were then randomly assigned to double‐blind treatment with either levetiracetam or matching placebo in a 1:1 ratio". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Similar proportions of participants withdrew from levetiracetam (34/111; 31%) and placebo groups (36/106; 34%). With exception of adverse events, and lack of efficiency the reasons for dropouts were similar between groups. The 2 groups did not differ by sample characteristics at baseline, nor did they differ significantly in attrition rates. Quote: "Overall, 148 of the 217 randomly assigned patients (N = 77 [70%], levetiracetam; N=71 [67%], placebo) completed the treatment period, with no statistically significant difference in attrition rates for each group. There was no differences in demographic or clinical characteristics of subjects who terminated the study prematurely. Reasons for early termination included adverse events (n=11, levetiracetam; n‐6, placebo), lack/loss of efficacy (n‐5, levetiracetam; n=4, placebo), withdrawal of consent not related to adverse events/lack of efficacy (n=5, levetiracetam; n=1, placebo), and other reasons (n=7, levetiracetam; n=5, placebo)". Overall 32% of the participants dropped out of the study. The ITT sample was assessed using the LOCF and OCs. |
| Selective reporting (reporting bias) | Low risk | All outcomes were reported as specified in the protocol. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias were identified. |
Tauscher 2010.
| Methods | Design: multicentre, randomised, double‐blind, placebo‐controlled, fixed dose, parallel group trial (NCT00191022) Duration of intervention: 12 weeks Post‐treatment: 2 week follow‐up Placebo run‐in: 1 week, single blind, placebo‐run‐in period |
|
| Participants | Sample size: 189 randomised to LY686017, paroxetine or placebo Mean age: not specified Sex: not specified Diagnostic measure: DSM IV‐TR Inclusion criteria: quote: "Patients meeting DSM‐IV‐TR diagnostic criteria for Generalized SAD as confirmed by the MINI were included in the study if they were outpatients between 18 and 65 years of age and presented with a CGI‐Severity score of ≥4". Exclusion criteria: quote: "Patients were excluded from the study if they exhibited any comorbid Axis I disorders within the last 6 months; or suffered from any Axis II disorder, except avoidant personality disorder. Further exclusion criteria were a history of substance or alcohol abuse or dependence within the past year and prior non‐responders to either selective serotonin reuptake inhibitors (SSRI) or serotonin norepinephrine reuptake inhibitors (SNRI)". Dropouts: 69/189 (insufficient information to determine dropout rates for the 2 groups separately) |
|
| Interventions | Pharmacological intervention: quote: "At Visit 3, all patients were randomized to treatment with LY686017, 50 mg QD, placebo, or paroxetine, 10 mg QD for up to 12 weeks ... One week after the beginning of the acute therapy phase (at Visit 4), patients randomized to paroxetine received a dose escalation from 10 mg QD [4 times daily] to 20 mg QD. At the end of the acute therapy phase (Visit 9), patients in the LY686017 and placebo arms received a 2‐week supply of placebo. Patients in the paroxetine arm received paroxetine 10 mg QD for one week, followed by placebo for 1 week. All patients were discontinued at Visit 10". | |
| Outcomes | Primary outcome measure: LSAS (for reduction of anxiety) Secondary outcome measures: LSAS (for reduction of anxiety), CGI‐I (for treatment efficacy), CGI‐S (for reduction of anxiety), HAM‐A (for reduction of anxiety), and SDS (for reduction of functional disability) Time points: Time points were not specified |
|
| Notes | Industry funded: yes. Quote: "Funding for this study was provided by Eli Lilly and Company (“Lilly”), Indianapolis, IN". Medication provided by industry: unclear Any of the authors work for industry: yes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "189 outpatients suffering from SAD were randomly assigned to 12‐weeks treatment with 50 mg/d LY686017 (N=77), placebo (N=74), or 20 mg/d paroxetine (N=38)". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The proportion of dropouts per treatment group could not be determined due to the lack of information provided by the study. The most common reasons for early discontinuation were adverse events and personal conflict or other participant decisions. This, however, was not specified by group. The 2 groups did not differ by sample characteristics at baseline. Quote: "189 patients were randomized to treatment and received at least one dose of the study drug, with 120 patients completing the study. The most common reasons for early discontinuation were adverse events (21 patients) and personal conflict or other patient decisions (21 patients). There were no statistically significant differences between treatment groups in patients who discontinued early ... The last‐observation‐carried‐forward (LOCF) method was used to impute missing data for the ANCOVA model". |
| Selective reporting (reporting bias) | High risk | Information was only provided for the primary outcome (i.e. LSAS) |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias were identified. |
Turner 1994.
| Methods | Design: multicentre, randomised, placebo‐controlled, parallel, flexible dose, double‐blind (NCT00191022) Duration of intervention: 12 weeks Post‐treatment: 6‐month follow‐up Placebo run‐in: no |
|
| Participants | Sample size: 46 randomised to atenolol or placebo (72 randomised: 25 atenolol, 26 flooding and 21 placebo) Mean age (range): 35.4 (18‐56) years Sex: 28 men and 34 women (for all three groups). Diagnostic measure: Initial Evaluation Form and Anxiety Disorders Interview Schedule‐Revised Inclusion criteria: quote: "Patients had to have a primary diagnosis of social phobia and could not have a secondary Axis I diagnosis other than generalized anxiety disorder, simple phobia, or dysthymia. In these cases, the additional diagnoses clearly had to be secondary to the social phobia with respect to chronology of onset and degree of impairment of daily functioning". Exclusion criteria: quote: "Patients who had an Axis II diagnosis of schizotypal, schizoid, borderline, paranoid, or antisocial personality disorder were excluded. Fifty‐five patients were excluded because of the presence of exclusionary Axis I or Axis II diagnoses, or medical conditions contraindicating the use of atenolol". Dropouts: 5/46 (4/25 in the atenolol and 1/21 in the placebo groups) |
|
| Interventions | Pharmacological intervention: quote: "Patients took 25 mg/day (in the morning) during the first week, 50 mg during the second and third weeks and 100 mg during the fourth and subsequent weeks". | |
| Outcomes | Primary and secondary outcome measures: SPAI (for reduction of anxiety), SAD (for reduction of anxiety), FNE (for reduction of anxiety), FQ (for reduction of anxiety), STAI (for reduction of anxiety), ISPI (for reduction of anxiety), and SPEFI (for reduction of functional disability) Time points: Quote: "Self‐report instruments were administered at pre‐ and posttreatment and at each follow‐up" |
|
| Notes | Industry funded: no. Quote: "This study was supported in part by Grant MH 41852 from the National Institute of Mental Health and was conducted in the Anxiety Disorders Clinic, Western Psychiatric Institute and Clinic, Pittsburgh, Pennsylvania". Medication provided by industry: yes. Quote: "Also, we extend our appreciation to Stuart Pharmaceutical Company for supplying the atenolol and placebo medication that was used". Any of the authors work for industry: no |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "Patients were assigned randomly to one of three groups: flooding, atenolol, or placebo". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Medication and placebo was provided in identical capsules. However, it is not clear whether both participants and personnel were blinded. Quote: "Medications were given in double‐blinded fashion ... Atenolol tablets of different doses and placebo tablets had an identical appearance ... The monitoring physician received all assessment data independent of the treating nurse clinician and made all medication decisions independently". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. Quote: "Multiple measures of outcome were used, including self‐report, clinician ratings (including assessment by independent evaluators), behavioral assessment, and performance on composite indexes". |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | A larger proportion of participants discontinued the study in the atenolol (4/25; 16%) group compared to the placebo group (1/21; 5%), though a Chi2 test reported that the difference in dropout proportions across all 3 treatment arms was not significant (Chi2(2, N = 71) = 2.19, P > 0.05). No information was provided regarding the reasons for treatment withdrawal nor was there information reported on if the 2 groups differed by sample characteristics at week 12. Quote: "Nine patients (12.1%) dropped out during the course of the 12‐week treatment; 5 from flooding, 3 from atenolol, and 1 from placebo. In addition, 1 patient was removed from atenolol treatment because of orthostatic hypotension. Thus, there were 62 patients who completed the study". Overall 11% of the participants dropped out of the study. |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Medication was provided by industry. No other sources of bias were identified. |
Vaishnavi 2007.
| Methods | Design: single‐centre, randomised, double‐blind, flexible dose, placebo‐controlled treatment trial (NCT00215254) Duration of intervention: 8 weeks Post‐treatment: no follow‐up Placebo run‐in: no |
|
| Participants | Sample size: 15 randomised to quetiapine or placebo Mean age (SD): 32.93 (8.64) years Sex: 7 men and 8 women Diagnostic measure: DSM IV Inclusion criteria: quote: "The inclusion criteria were as follows: (1) adult outpatients 18–65 years of age, (2) a primary diagnosis of SAD using DSM‐IV criteria, (3) a minimum Clinical Global Impression Severity score (CGI‐S) of 4 and minimum Brief Social Phobia Scale (BSPS) score of 20 at baseline, (4) written informed consent, and (5) a negative serum pregnancy test for women of childbearing potential". Exclusion criteria: quote: "The exclusion criteria were as follows: (1) current DSM IV diagnosis of bipolar disorder, schizophrenia or other psychotic disorder, mental retardation or other pervasive developmental disorder, or cognitive disorder due to a general medical condition, (2) any current primary anxiety disorder other than SAD, (3) current primary diagnosis of major depressive disorder, (4) history of substance abuse or dependence within the last 6 months, (5) suicidal risk or serious suicide attempt within the last year, (6) clinically significant medical condition or laboratory abnormality, (7) women of childbearing potential who are unwilling to practice an acceptable method of contraception, (8) concomitant use of medication with psychotropic effects, and (9) history of hypersensitivity to quetiapine". Dropouts: not specified |
|
| Interventions | Pharmacological intervention: quote: "The titration schedule of quetiapine or matching placebo was a flexibly‐dosed regimen as follows: 25 mg twice a day for the first 3 days, 50 mg twice a day after that until the end of the first week, 100 mg twice a day for the second week, 150 mg twice a day for the third week, and 200 mg twice a day for the fourth week, with the actual doses prescribed dependent on the tolerability for each patient. Upon completion of the study, patients were tapered off the medication over 3 days. Patients who had a significant worsening of symptoms (i.e., increase in CGI‐S of 2 or more compared to baseline at 2 consecutive visits) were removed from the study and referred for treatment as clinically indicated. Patients who missed 5 consecutive days of treatment were discontinued from the trial". | |
| Outcomes | Primary outcome measures: BSPS (for reduction of anxiety) and CGI‐I (for treatment efficacy) Secondary outcome measures: SPIN (for reduction of anxiety) and SDI (for reduction of functional disability), as well as the CGI‐S (for reduction of anxiety), SOSS (for reduction of anxiety), BAS (for reduction of anxiety), and SAS (for reduction of anxiety) Time points: Quote: "At baseline and weeks 1, 3, and 5, efficacy assessments (BSPS, SPIN, CGI) and safety measures (vital signs, BAS, SAS, and SOSS ) were performed. At week 3, SDI was repeated. The full battery of assessments were performed at week 8 (the final visit) and, for women of childbearing potential, a serum pregnancy test was repeated" |
|
| Notes | Industry funded: yes. Quote: "Funding was provided to the last author by AstraZeneca to conduct the study". Medication provided by industry: unclear Any of the authors work for industry: no |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were assessed for eligibility at a screening visit, with eligible patients returning for a baseline assessment in approximately 1 week, at which time they were randomized 2:1 to either quetiapine (10 patients) or placebo (5 patients); this was done by utilizing a computer code generated by a study statistician who did not have contact with subjects". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Medication and placebo was provided in identical capsules. However, it is not clear whether both participants and personnel were blinded. Quote: "This was an eight week, randomized, double‐blind, placebo controlled treatment trial of SAD with quetiapine (50–400 mg/day) or matching placebo." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The study did not report attrition. Quote: "Data analysis was performed on the intent‐to‐treat (ITT) population using the last‐observation‐carried‐forward (LOCF) method for missing data". |
| Selective reporting (reporting bias) | Low risk | All outcomes were reported as specified in the protocol. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias were identified. |
Van Ameringen 2001a.
| Methods | Design: multicentre, randomised, placebo‐controlled, parallel, flexible dose, double‐blind Duration of intervention: 20 weeks Post‐treatment: no follow‐up Placebo run‐in: 1‐week, single‐blind, placebo run‐in |
|
| Participants | Sample size: 204 randomised to sertraline or placebo Mean age (range): 35.65 (19‐56) years Sex: 114 men and 90 women Diagnostic measure: DSM IV Inclusion criteria: quote: "Inclusion criteria for the study required patients to meet DSM‐IV criteria for primary generalized social phobia of at least 1‐year duration at screening. Patients had to have a CGI severity rating of 4 or less (i.e., moderately ill or worse) and to be between 18 and 60 years of age without any serious or uncontrolled medical illness or condition that precluded sertraline use. Patients with an additional diagnosis of avoidant personality were allowed to participate. Patients with comorbid major depression were permitted to enter the study provided their diagnosis was secondary to social phobia, their baseline Montgomery Åsberg Depression Rating Scale score was 19 or less, the onset of social phobia predated onset of the current episode of depression by 5 years or more, and the patient did not represent a substantial suicide risk". Exclusion criteria: quote: "Patients were excluded if they had another primary axis I disorder or fulfilled criteria in the previous 6 months for panic disorder, agoraphobia, obsessive‐compulsive disorder, eating disorders, body dysmorphic disorder, or substance abuse. Other exclusion criteria included concomitant use before study screening of psychotropic medication within a period of 5 half lives, neuroleptics within 7 months, serotonergic antidepressants or an antianxiety medication for 3 or more weeks within 3 months, and cognitive behavior therapy within 4 weeks. Patients receiving benzodiazepines were permitted to enter the study after completing a minimum 2–4‐week tapered discontinuation. Additional reasons for exclusion included a urinary screen positive for benzodiazepines at baseline, treatment with β‐blockers or clonidine, and participation in a clinical trial within the previous 12 months. Women who were pregnant, lactating, or not using an acceptable method of contraception were excluded, as were patients who had had a major life event in the last 3 months that, in the investigators' opinion, was influencing their current condition". Dropouts: 46/204 (31/135 in the sertraline and 15/69 in the placebo groups) |
|
| Interventions | Pharmacological intervention: quote: "Patients received an initial dose of 50 mg/day of sertraline or matching placebo. After 4 weeks, the dose could be increased by 50 mg/day every 3 weeks in the absence of satisfactory response (CGI improvement score indicating much or very much improved) up to a maximum allowable dose of 200 mg/day by week 10. The dose could be reduced to a minimum of 50 mg/day if required by the presence of intolerable side effects". | |
| Outcomes | Primary outcome measures: CGI‐I (for treatment efficacy), FQ (for reduction of anxiety), and BSPS (for reduction of anxiety) Secondary outcome measures: CGI‐I (for treatment efficacy), CGI‐S (for reduction of anxiety), Liebowitz Panic and Social Phobic Disorders Rating Form (for reduction of anxiety), SPAI (for reduction of anxiety), SADS (for reduction of anxiety), FNE (for reduction of anxiety), MADRS (for reduction of depression), CAS (for reduction of anxiety), and SDS (for reduction of functional disability) Time points: Quote: "Subjects were evaluated at weeks 1, 2, 4, 7, 10, 13, 16, and 20" |
|
| Notes | Industry funded: yes. Quote: "Financial support for this study was provided by Pfizer Inc". Medication provided by industry: unclear Any of the authors work for industry: yes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "Patients who continued to meet all inclusion criteria and did not have a CGI severity score decline of 2 points or more were randomly assigned to receive sertraline or placebo in a ratio of 2:1". |
| Allocation concealment (selection bias) | Unclear risk | Study medication was identical; however, the method of how this concealment took place was not discussed. Quote: "Patients received an initial dose of 50 mg/day of sertraline or matching placebo". |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Medication and placebo was provided in identical capsules. However, it is not clear whether both participants and personnel were blinded. Quote: "Patients received an initial dose of 50 mg/day of sertraline or matching placebo". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Similar proportions of participants withdrew from sertraline (31/135; 23%) and placebo groups (15/69; 22%). Similar reasons for withdrawal were found across the 2 groups, although more participants withdrew in the sertraline group because of adverse events compare to those participants in the placebo groups. The 2 groups did not differ by sample characteristics at baseline. Quote: "There were no statistically significant differences between groups in demographic characteristics or mean baseline rating scale scores ... In the sertraline group, 104 (77%) of 135 patients completed the 20‐week trial. In the placebo group, 54 (78%) of 69 patients completed the trial. The reasons for patient discontinuation in the sertraline and placebo groups, respectively, were adverse events (N=16 versus N=1), lack of efficacy (N=4 versus N=4), withdrew consent (N=4 versus N=7), lost to follow‐up (N=3 versus N=1), protocol violation (N=1 versus N=0) and administrative reasons (N=3 versus N=2)". Overall 22% of the participants dropped out of the study. Quote: "Random regression was used to compare improvement slopes because it makes fewer assumptions about missing data while optimizing the use of available data". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias were identified. |
Van Ameringen 2007.
| Methods | Design: multicentre, randomised, flexible dose, double‐blind, placebo‐controlled, parallel‐group trial Duration of intervention: 14 weeks Post‐treatment: no follow‐up Placebo run‐in: 1‐week, single‐blind, placebo run‐in |
|
| Participants | Sample size: 105 randomised to nefazodone or placebo Mean age (SD): 35.8 (10.65) years Sex: 50 men and 55 women Diagnostic measure: DSM IV Inclusion criteria: quote: "Inclusion criteria for the study required subjects to be psychiatric outpatients between the ages of 18 and 65 years, to fulfil DSM‐IV criteria for GSP for more than 1 year, and to be of at least moderate illness severity on the basis of the CGI‐S rating. Patients with comorbid secondary major depression were permitted to participate in the study provided that their baseline score on the MADRS was 19 or less, there was no risk to suicidality on the basis of mental status examination, and the onset of their social phobia predated the major depressive disorder by at least 5 years". Exclusion criteria: quote: "Current comorbid Axis I disorders such a panic disorder with agoraphobia, obsessive‐compulsive disorder, body dysmorphic disorder, or alcohol/substance abuse were excluded from this study. Those with a lifetime history of bipolar affective disorder, schizophrenia, psychoses, delirium, dementia, or other cognitive disorders were also excluded, as were individual reporting 2 previous treatment failures for GSP". Dropouts: 22/102 (15/51 in the nefazodone and 7/51 in the placebo groups; three participants were randomly assigned to treatment but did not take at least 1 dose of study medication). |
|
| Interventions | Pharmacological intervention: quote: "Nefazodone or placebo was started at an initial dose of 100 mg/day in divided doses. Doses were increased to 200 mg/day week 2, and up to 300 mg/day by week 4. Further increments of 100 mg were added every 2 weeks, until a maximum dose of 600 mg/ day was reached". | |
| Outcomes | Primary outcome measures: CGI‐I (for treatment efficacy) and LSAS (for reduction of anxiety) Secondary outcome measures: CGI‐S (for reduction of anxiety), SPI (for reduction of anxiety), SPS (for reduction of anxiety), SIAS (for reduction of anxiety), BDI (for reduction of depression), BAS (for reduction of anxiety), SDS (for reduction of functional disability), and the RAND 36‐Item Health Survey (for general health) Time points: Quote: "Patients were evaluated at weeks 1, 2, 3, 5, 7, 9, 12, and 16" |
|
| Notes | Industry funded: yes. Quote: "Partial funding for this study was provided by an investigator‐initiated research grant from Bristol‐Myers Squibb, Montreal, Quebec, Canada". Medication provided by industry: unclear Any of the authors work for industry: no |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "Those subjects who continued to meet inclusion criteria were randomly assigned on a 1:1 basis to receive either nefazodone or placebo for 14 weeks". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: "Because of the distinct side effect profiles of placebo and any active medication, it is quite possible that the raters were not blind to experimental condition". |
| Incomplete outcome data (attrition bias) All outcomes | High risk | A larger proportion of participants discontinued the study in the nefazodone (15/52; 29%) group compared to the placebo group (7/53; 13%). No information was provided regarding the reasons for treatment withdrawal except for adverse events. More participants in the nefazodone compared to the placebo group withdrew because of reported adverse events. No information was reported on if the 2 groups differed by sample characteristics at week 14. Quote: "Thirty‐six (70.6%) of 51 patients in the nefazodone group completed the trial compared with 44 (86.3%) of 51 in the placebo group". Overall 21% of the participants dropped out of the study. Quote: "All efficacy analyses were carried out on the intention‐to‐treat sample using the last‐observation carried forward method" . |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias were identified. |
Van Vliet 1992.
| Methods | Design: single‐centre, randomised, placebo‐controlled, parallel, fixed dose, double‐blind Duration of intervention: 12 weeks Post‐treatment: 3 months follow‐up Placebo run‐in: no |
|
| Participants | Sample size: 30 randomised to brofaromine or placebo Mean age (SE): 32.8 (2.0) years Sex: 9 men and 21 women. Diagnostic measure: DSM‐III‐R Inclusion criteria: quote: "Included in the study were patients suffering from social phobia according to DSM‐III‐R criteria". Exclusion criteria: quote: "Excluded were patients with another anxiety disorder, major affective disorder or psychotic disorder, alcohol abuse and those patients suffering from medical problems on the basis of a complete medical evaluation". Dropouts: 1/30 (0/15 in the brofaromine and 1/15 in the placebo groups) |
|
| Interventions | Pharmacological intervention: quote: "The dose of brofaromine was gradually increased from 50 to 150 mg daily (75 mg b.i.d.) in 3 weeks". | |
| Outcomes | Primary and secondary outcome measures: SCL‐90 (for reduction of anxiety), HAM‐D (for reduction of depression), SPS (for reduction of anxiety), STAI (for reduction of anxiety), and HAM‐A (for reduction of anxiety) Time points: Quote: "At the onset and the end of the study period. Adverse events were assessed by open questioning at week 1, 2, 4, 8 and 12" |
|
| Notes | Industry funded: no Medication provided by industry: no Any of the authors work for industry: no |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "Patients were randomly allocated to one of the two treatment groups". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | A single participant of the 30 discontinued the study, in the placebo condition (1/15; 7%). |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Low risk | No other sources of bias were identified. |
Van Vliet 1994.
| Methods | Design: single‐centre, randomised, placebo‐controlled, parallel, fixed dose, double‐blind Duration of intervention: 12 weeks Post‐treatment: no follow‐up Placebo run‐in: no |
|
| Participants | Sample size: 30 randomised to fluvoxamine or placebo Mean age (SD): 35.2 (9.5) years Sex: 13 men and 17 women Diagnostic measure: DSM‐III‐R Inclusion criteria: quote: "Included in the study were patients suffering from social phobia according to DSM‐III‐R criteria". Exclusion criteria: quote: "Excluded were patients with another anxiety disorder, major affective disorder or psychotic disorder, alcohol or drug abuse, those patients suffering from medical problems on the basis of a complete medical evaluation and patients who were pregnant or lactating. A score of 15 or higher on the HAM‐D Scale was an exclusion criterion. Patients with personality disorders according to DSM‐III‐R criteria were also excluded". Dropouts: 2/30 (1/15 in the fluvoxamine and 1/15 in the placebo groups) |
|
| Interventions | Pharmacological intervention: quote: "The dose of fluvoxamine was gradually increased from 50 mg to 150 mg daily (50 mg t.i.d. [3 times daily]) in 3 weeks" | |
| Outcomes | Primary and secondary outcome measures: SAS (for reduction of anxiety and performance), HAS (for reduction of anxiety), SCL‐90 (for reduction of anxiety), and HDS (for reduction of depression) Time points: Quote: "Efficacy of the treatment was assessed using the Social Anxiety Scale (SAS) and the Hamilton Anxiety Scale (HAS) on baseline and at weeks 1, 2, 4, 8, and 12. At the outset and the end of the study period, patients completed the 90‐Item Symptom Checklist (SCL‐90). The Hamilton Depression Scale (HDS) was completed on baseline and at the end of treatment. Averse events were assessed by open questioning at weeks 1, 2, 4, 8 and 12" |
|
| Notes | Industry funded: no Medication provided by industry: no Any of the authors work for industry: no |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "Patients were randomly allocated to one of the two treatment groups". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Similar, low proportions of participants withdrew from fluvoxamine (1/16; 6%) and placebo groups (1/14; 7%). One participant withdrew due to a side effect in the fluvoxamine group compared to lack of efficacy in the placebo group. The 2 groups did not differ by sample characteristics at baseline. Quote: "From the patients who were recruited, one dropped out in the second week due to severe side effects (treated with fluvoxamine); another patient dropped out in week 8 due to lack of efficacy (treated with placebo) ... The two treatment groups did not differ in mean age, mean age of onset and sex". Overall 7% of the participants dropped out of the study. |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Low risk | No other sources of bias were identified. |
Van Vliet 1997.
| Methods | Design: single‐centre, randomised, placebo‐controlled, parallel, fixed dose, double‐blind Duration of intervention: 12 weeks Post‐treatment: no follow‐up Placebo run‐in: no |
|
| Participants | Sample size: 30 randomised to buspirone or placebo Mean age (SD): 37.25 (8.85) years Sex: 19 men and 11 women Diagnostic measure: DSM‐IV Inclusion criteria: quote: "Included in the study were patients suffering from social phobia, specific or generalised subtype, according to DSM‐IV criteria". Exclusion criteria: quote: "Excluded were patients with another anxiety disorder, major affective disorder or psychotic disorder, alcohol or drug abuse; and pregnancy or lactation and those patients suffering from medical evaluation. Patients with a personality disorder according to the DSM‐IV were also excluded. A score of 15 or higher on the HAM‐D was an exclusion criterion". Dropouts: 3/30 (0/15 in the buspirone and 3/15 in the placebo groups) |
|
| Interventions | Pharmacological intervention: quote: "The dose of buspirone was gradually increased from 15 mg in the first week to 30 mg from the third week on (10 mg t.i.d. [3 times daily])". | |
| Outcomes | Primary and secondary outcome measures: SPS (for reduction of anxiety), HAM‐A (for reduction of anxiety), SCL‐90 (for reduction of anxiety) and HAM‐D (for reduction of depression) Time points: Quote: "Efficacy of the treatment was assessed using the Social Phobia Scale (SPS) and the Hamilton Rating Scale for Anxiety (HAM‐A) at baseline and at Weeks 1, 2, 4, 8, and 12. At the outset and the end of the study period, patients completed the 90‐item Symptom Checklist (SCL‐90). The HAM‐D was completed at baseline and at the end of treatment. Averse events were assessed by open questioning at weeks 1, 2, 4, 8, and 12" |
|
| Notes | Industry funded: no Medication provided by industry: yes. Quote: "The authors thank Bristol‐Myers Squibb for their technical support and providing the trial medication". Any of the authors work for industry: no |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "Patients were randomly allocated to one of the two treatment groups". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | More participants withdrew from the placebo group (3/15; 20%) compared to the buspirone (0/15; 0%) group. Participants dropped out for reasons of inefficacy or distance to the hospital. No information was provided on whether participants differed in terms of characteristics by group at week 12, however. Nevertheless, the total proportion of dropouts (10%) is relatively low, suggesting that dropout rates may not have biased the outcomes. Quote: "Of the 15 patients randomly assigned to receive placebo, 3 patients dropped out for reasons of inefficacy or distance to the hospital ... There were no dropouts among the 15 patients randomly assigned to receive buspirone". Overall 10% of the participants dropped out of the study. |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Medication provided by industry. No other sources of bias were identified. Quote: "One third of the total patient sample used alcoholic beverages to reduce social phobic anxiety and symptoms in social situations". |
Versiani 1992.
| Methods | Design: single‐centre, randomised, placebo‐controlled, parallel, flexible dose, double‐blind, cross‐over trial Duration of intervention: 8 weeks Post‐treatment: no follow‐up Placebo run‐in: no |
|
| Participants | Sample size: 78 randomised to phenelzine, moclobemide and placebo Mean age: not specified Sex: not specified Diagnostic measure: DSM‐III‐R (SCID) Inclusion criteria: quote: "The patients were of either sex, and aged 19‐60 years. The disorder had to meet the following criteria: by CGI severity score of > 4; (ii) global score on the SDSof 3; and clinical judgement that a drug treatment was indicated. All patients met the DSM‐III‐R criteria for social phobia, as diagnosed, by the Structured Clinical Interview for DSM‐III‐R. They had to have been free from any psychotropic medication for at least one month". Exclusion criteria: quote: "Patients were excluded if they had, or had a history of, any other DSM‐III‐R diagnoses to which social phobia could have been secondary. These included organic mental disorders, abuse of psychoactive substances, other anxiety disorders except generalised anxiety disorder, panic disorder (with a more stringent criterion than those of DSM‐III‐R, i.e. history of a single unexpected panic attack), and psychosis. Patients with significant medical illness e.g. essential tremor or Parkinson's disease that could mimic certain social phobic symptoms were also excluded. Inability to fill in self‐rating scales or to adhere to the study requirements, as well as concomitant psychotherapy or lack of protection against pregnancy, were other exclusion criteria". Dropouts: 4/78 (1/26 in the phenelzine, 0/26 in the moclobemide and 3/26 in the placebo groups; these rates are for phase I) |
|
| Interventions | Pharmacological intervention: quote: "Medication was provided in capsules of identical appearance containing either moclobemide (100mg), phenelzine (15mg), or placebo. The initial dose was one capsule twice daily, morning, and afternoon; if tolerated, this dose was increased on day 4 to four capsules a day ‐ two in the morning, one in the afternoon, and one at bedtime. This dose was maintained until the end of week 4. At week5, if the dose was tolerated, it was increased again to five capsules per day ‐ two in the morning, two in the afternoon, and one at bedtime. At week 6, there was a further option to increase the dose to two capsules thrice daily; attempts were made to reach this maximum dose (600 mg/day moclobemide, 90 mg/day phenelzine) in all cases, irrespective of the degree of improvement". | |
| Outcomes | Primary outcome measures: CGI (for treatment efficacy), CGI‐S (for reduction of anxiety), WPI (for reduction of anxiety and measure of personality), SADS (for reduction of anxiety), and FNE (for reduction of anxiety) Secondary outcome measures: HRSD (for reduction of depression), HAM‐A (for reduction of anxiety), SCL‐90 (for reduction of anxiety) and SDS (for reduction of functional disability) Time points: Quote: "The CGI, the Social Phobia Scale, and a form to assess and record side‐effects were administered each week during phase I, and thereafter every four weeks until week 24. The other rating scales were administered at baseline and then every four weeks throughout the study. A battery of laboratory tests and an electrocardiogram were performed in all patients, immediately before inclusion and at weeks 8 and 16" |
|
| Notes | Industry funded: no Medication provided by industry: no Any of the authors work for industry: yes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "Blindness was maintained throughout by using capsules of identical appearance; these were administered according to a randomisation list". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Medication and placebo was provided in identical capsules. However, it is not clear whether both participants and personnel were blinded. Quote: "Medication was provided in capsules of identical appearance containing either moclobemide (100mg), phenelzine (15mg), or placebo". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | A larger proportion of participants discontinued the study in the placebo group (3/26; 12%) compared to the phenelzine (1/26; 4%) and moclobemide group (0/26; 0%). Participants in the placebo group dropped out due to lack of efficacy compared to adverse events in the phenelzine group. Participants did not differ by group characteristics at baseline. Quote: "Seventy‐eight patients, 26 in each treatment group, entered phase I of the trial, while 45 patients (7 in the placebo group; 17 in the moclobemide group; 21 in the phenelzine group) entered phase II. Seventeen patients in the moclobemide group and 20 patients in the phenelzine group entered phase III ... There were no significant differences between the three treatment groups regarding demographic characteristics or diagnostic features. In phase I, during the first week, two patients refused to continue the study and were replaced. During weeks 4 to 8, four patients left the study three from the placebo group (for lack of efficacy) and one from the phenelzine group (for side‐effects ‐ dizziness, loss of libido, and headache). At the end of phase I, 21 non‐responders (16 from the placebo group, and 5 from the moclobemide group) were withdrawn from the study". Overall 8% of the participants dropped out of the study. |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Low risk | No other sources of bias were identified. |
Versiani 1997.
| Methods | Design: single‐centre, placebo‐controlled, parallel, flexible dose, double‐blind Duration of intervention: 12 weeks Post‐treatment: no follow‐up Placebo run‐in: no |
|
| Participants | Sample size: 60 randomised to bromazepam and placebo Mean age (SD): 36.7 (9.9) years Sex: 39 men and 21 women Diagnostic measure: DSM‐III‐R Inclusion criteria: quote: "The patients were of either sex and aged 19‐60 years. The disorder had to meet the following criteria: a CGI Severity score equal to or greater than 4 and a Sheehan Govla Disabilities score of at least 3. All patients met criteria for social phobia as diagnosed by the structured clinical interview for DSM‐III‐R. They had to have been free from any psychotropic medication for at least one month". Exclusion criteria: quote: "Patients were excluded if they had a history of any other DSM‐III‐R diagnoses to which social phobia could have been secondary. These excluded organic mental disorders, abuse of psychoactive substances, other anxiety disorders, except generalised anxiety disorder, panic disorder and psychotic disorders. Relative to mood disorders only past major depression or secondary dysthymia were allowed. Personailty disorders of cluster A or cluster B were excluded. Significant medical illnesses were also excluded. Inability to fill in self‐rating scales or to adhere to the study requirements were also reasons for exclusion". Dropouts: 3/60 (1/30 in the bromazepam and 2/30 in the placebo groups). |
|
| Interventions | Pharmacological intervention: 3‐9 mg bromazepam or placebo | |
| Outcomes | Primary and secondary outcome measures: CGI (for treatment efficacy), LSAS (for reduction of anxiety), WPI (for reduction of anxiety and measure of personality), SADS (for reduction of anxiety), FNE (for reduction of anxiety), HAM‐D (for reduction of depression), HAM‐A (for reduction of anxiety), SCL‐90 (for reduction of anxiety) and SDS (for reduction of functional disability) Time points: Quote: "Medical visits were performed every week for dosage adjustments, assessments, recording of viral signs and evaluation and recording of treatment emergent adverse events" |
|
| Notes | Industry funded: no Medication provided by industry: no Any of the authors work for industry: no |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The study did not report on if the participants were randomly assigned to treatment and comparison nor was the procedure specified. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Medication and placebo was provided in identical capsules. However, it is not clear whether both participants and personnel were blinded. Quote: "Blindnesss was maintained throughout by using tablets of identical appearance". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Similar, low proportions of participants withdrew from bromazepam (1/30; 3%) and placebo groups (2/30; 7%). Pateients in the bromazepam group withdrew due to lack of efficacy and sedation compared to lack of efficacy and nausea in the placebo group. No information was provided on if groups differed by sample characteristics at week 12. Quote: "There were three dropouts. One in the placebo group at week 6, was due to lack of efficacy. Another in the placebo group was due to lack of efficacy and nausea, at week 5. One dropout occurred in the bromazepam group, at week 5, due to lack of efficacy and sedation". Overall 10% of the participants dropped out of the study. |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Low risk | No other sources of bias were identified. |
Walker 2000.
| Methods | Design: multicentre, double‐blind, placebo‐controlled, parallel, flexible dose, relapse prevention study Duration of intervention: 24 weeks Post‐treatment: no follow‐up Placebo run‐in: no |
|
| Participants | Sample size: 50 randomised to sertraline continuation and placebo switch (65 eligible) Mean age (range): 36.6 (21‐57) years Sex: 32 men and 18 women Diagnostic measure: DSM‐IV Inclusion criteria: quote: "Inclusion criteria for the study required patients to meet DSM‐IV criteria for primary generalized social phobia of at least 1 year's duration at screening with a CGI‐S score of 4 or greater and to be between 18 and 60 years of age without any serious or uncontrolled medical illness or condition that precluded sertraline use. Patients with an additional diagnosis of avoidant personality were allowed to participate. Patients with comorbid major depression were permitted to enter the study provided that their diagnosis was the result of social phobia; their baseline MADRS score had to be 19 or less; the onset of social phobia had to predate the onset of the current episode of depression by 5 years or more; and they did not represent a significant suicide risk". Exclusion criteria: quote: "Patients were excluded if they had another primary axis I disorder or if they fulfilled criteria in the previous 6 months for panic disorder, agoraphobia, obsessive‐compulsive disorder, eating disorders, body dysmorphic disorder, or substance abuse. Other exclusion criteria included concomitant use prior to study screening of psychotropic medication within a period of five half‐lives, neuroleptics within 7 months, serotonergic antidepressants or an antianxiety medication for 3 or more weeks within 3 months, and cognitive‐behavior therapy within 4 weeks. Patients receiving benzodiazepines were permitted to enter the study after completing a minimum of 2 to 4 weeks of tapered discontinuation. Additional reasons for exclusion included a urinary screen positive for benzodiazepines at baseline; treatment with [beta]‐blockers, methyldopa, guanethidine, or clonidine; and participation in a clinical trial within the previous 12 months. Women who were pregnant, lactating, or not using an acceptable method of contraception were excluded, as were patients who had had a major life event in the last 3 months that, in the investigator's opinion, was influencing their current condition". Dropouts: 18/50 (3/25 in the sertraline continuation and 15/25 in the placebo switch groups) |
|
| Interventions | Pharmacological intervention: quote: "In the initial short‐term treatment study, patients who met all inclusion criteria were randomly assigned to receive flexible‐dose sertraline (50‐200 mg/day) or placebo in a ratio of 2:1. After completion of the 20‐week double‐blind study, patients who had responded (Clinical Global Impression Scale of Improvement [CGI‐I] score of much or very much improved) were eligible to enter the 24‐week study. Patients who had been receiving sertraline were randomly assigned again in a double‐blind fashion in a ratio of 1:1 to either continue sertraline or switch to placebo for another 24 weeks. Patients who had been receiving placebo continued to receive double‐blind placebo. The only sleep medications permitted during the study were chloral hydrate (500‐1,000 mg per night) or zopiclone (3.75‐7.5 mg per night). Patients who developed persistent side effects could have their study medication dosage reduced to the next lower level". | |
| Outcomes | Primary outcome measures: CGI‐S (for reduction of anxiety), MFQ (for reduction of anxiety) and BSPS (for reduction of anxiety) Secondary outcome measures: SPAI (for reduction of anxiety), SADS (for reduction of anxiety), FNE (for reduction of anxiety), MADRS (for reduction of depression), CAS (for reduction of anxiety) and SDI (for reduction of functional disability) Time points: Quote: "The final visit of the initial 20‐week study served as the baseline visit of the continuation study. Subjects were also evaluated at weeks 24, 28, 32, 36, 40, and 44. Safety assessments included the evaluation, at each visit, of vital signs (weight, blood pressure, and heart rate) and the recording of spontaneously reported or observed adverse events. In addition, the use of concomitant medication was recorded and compliance was monitored by pill counts of returned medication" |
|
| Notes | Industry funded: yes. Quote: "Financial support for this study was provided by Pfizer Inc". Medication provided by industry: unclear Any of the authors work for industry: yes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Author response: quote: "We know that there was random assignment but we did not take care of that. As patients were recruited for the study they were assigned the next study number in the sequence and received the medication over the course of the study assigned to that sequence number". |
| Allocation concealment (selection bias) | Low risk | Author response: quote: "The pharmaceutical firm provided the placebo and medication to us and these were taken to the hospital pharmacy for storage. The pharmacy dispensed the packaged medication from the group of medicine bottles for the participants ‐ week 1, week 2, and so on. The dose was monitored by the treating psychiatrist and patients returned unused medications". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Author response: quote: "All of these persons were blinded". |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Author response: quote: "All of these persons were blinded". |
| Incomplete outcome data (attrition bias) All outcomes | High risk | A larger proportion of participants discontinued the study in the placebo switch (15/25; 60%) group compared to the sertraline continuation group (3/25; 12%). The most common reason for treatment discontinuation in the placebo group was due to lack of efficacy and adverse events. The 2 groups did not differ by sample characteristics at baseline. Quote: "3 patients in the sertraline continuation group, 15 in the placebo switch group, and 9 in the placebo responder groups failed to complete the study (see table 3 page 641) ... There were no statistically significant differences between groups in demographic characteristics or mean baseline rating scale scores". Overall 36% of the participants dropped out of the study. ITT was assessed for efficacy outcomes. |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias were identified. |
Westenberg 2004.
| Methods | Design: multicentre, randomised, double‐blind, flexible dose, placebo‐controlled study Duration of intervention: 12 weeks Post‐treatment: no follow‐up Placebo run‐in: no |
|
| Participants | Sample size: 300 randomised to fluvoxamine CR and placebo Mean age (SE): 32.95 (0.9) years Sex: 143 men and 157 women Diagnostic measure: DSM‐IV and DSM‐IV Axis I Disorders (SCID‐I) Inclusion criteria: quote: "To be eligible to participate in this study, outpatients had to meet the following inclusion criteria: male or female aged 18 to 70 years, a predominant DSM‐IV diagnosis of GSAD according to the modified SCID‐I, and a minimum score of 60 on the LSAS at screening. Women of childbearing potential or less than 1 year postmenopausal were required to use a medically acceptable method of birth control. Pregnant or lactating women were not eligible". Exclusion criteria: quote: "Subjects were excluded from the study if they met any of the following criteria: subjects with psychiatric disorders other than GSAD deemed to be predominant in the last 6 months including major depressive disorder, dysthymic disorder, or panic disorder were excluded. Subjects with history or current diagnosis of schizophrenia, other psychotic disorders, bipolar affective disorder, borderline personality, or obsessive compulsive disorder were excluded. Subjects who had a score 18 on the MADR Scale at screening were excluded. Subjects with evidence of substance abuse disorder or dependence with the past 6 months, subjects with positive results on a urine drug screen, subjects at serious suicidal risk, subjects with unstable or serious medical conditions, and subjects who required formal cognitive‐behavioral therapy to treat social anxiety symptoms within the previous month were also excluded". Dropouts: 101/300 (57/149 in the fluvoxamine CR and 44/151 in the placebo groups). |
|
| Interventions | Pharmacological intervention: quote: "Subjects randomized to receive, fluvoxamine CR began at a bedtime dose of 100 mg at day 1. The dose was titrated weekly in 50 mg increments based on clinical judgment of response and tolerance up to a maximum of 300 mg/d, once daily, over the first 5 weeks of treatment. Thereafter, the dose was to remain constant for the duration of the double‐blind period. One decrease of 50 mg/d was permitted after week 1 and through the end of week 5. The minimum dose allowed at any time during the study was 100 mg/d". | |
| Outcomes | Primary outcome measure: LSAS (for reduction of anxiety) Secondary outcome measures: CGI‐I (for treatment efficacy), CGI‐S (for reduction of anxiety), SDS (for reduction of functional disability), PGI (for treatment efficacy) and ASEX (to measure sexual experience) Time points: Quote: "The LSAS was administered at screening, baseline, and weeks 2, 4, 6, 8, 10, and 12; the CGI‐S, SDS, and ASEX were administered at baseline, weeks 2, 4, 6, 8, 10, and 12; the CGI‐I and PGI were administered at weeks 2, 4, 6, 8, 10, and 12. Safety measures obtained at every visit included vital signs, weight, adverse events, and concomitant medications. A 12‐lead ECG and physical examination were performed at the screening visit and week 12; laboratory testing was performed at screening, baseline (if the screening period was more than 10 days in length), and week 12. All week 12 assessments were performed upon early discontinuation" |
|
| Notes | Industry funded: yes. Quote: "This study was supported by a grant from Solvay Pharmaceuticals, Inc". Medication provided by industry: unclear Any of the authors work for industry: yes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "A total of 300 subjects with GSAD were randomly assigned to receive either fluvoxamine CR (N = 149) or placebo (N = 151) for 12 weeks". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Similar proportions of participants withdrew from fluvoxamine CR (57/149; 38%) and placebo groups (44/151; 29%). The reasons for withdrawal were similar across treatment groups. More participants in the fluvoxamine CR group withdrew due to adverse events compared to lack of efficacy in the placebo group. The 2 groups did not differ by sample characteristics at baseline. Quote: "Of the subjects who were randomized, 92 subjects (62%) in the fluvoxamine CR treatment group and 107 subjects (71%) in the placebo treatment group completed the study. The reasons for withdrawal were similar across treatment groups except for withdrawal due to lack of efficacy and withdrawal due to adverse experience. Fourteen subjects (9%) in the placebo treatment group but no subjects in the fluvoxamine CR treatment group withdrew due to lack of efficacy. A higher percentage of subjects in the fluvoxamine CR treatment group (38 subjects, 26%) than in the placebo treatment group (8 subjects, 5%) discontinued due to adverse events. Most subjects in the fluvoxamine CR group (20 subjects) discontinued within the first 3 weeks of treatment. There were no statistically significant differences between the 2 groups in demographics or disorder characteristics at baseline". Overall 34% of the subjects dropped out of the study. Quote: "Statistical analyses were performed using the last observation carried forward (LOCF) and observed case (OC) algorithms". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias were identified. |
Zhang 2005.
| Methods | Design: single‐centre, randomised, double blind, flexible dose placebo controlled pilot study Duration of intervention: 7 weeks Post‐treatment: no follow‐up Placebo run‐in: no |
|
| Participants | Sample size: 19 randomised to levetiracetam and placebo Mean age (SD): 37.5 (12.7) years Sex: 9 men and 10 women Diagnostic measure: DSM‐IV and MINI Inclusion criteria: quote: "Inclusion criteria were as follows: (1) age 18–65; (2) social anxiety disorder according to DSM‐IV criteria; (3) minimum baseline score of 20 on the BSPS; (4) medically stable; and (5) provision of written, informed consent". Exclusion criteria: quote: "Subjects who met the following criteria were excluded from the study: (1) history of psychosis or bipolar disorder; (2) substance use disorder or other primary anxiety disorder in the past six months; (3) primary diagnosis of major depression in the last year; (4) the use of other psychotropic medication in the previous week (14 days for MAOI and 28 days for fluoxetine); (5) need for ongoing use of psychotropic medications; or (6) pregnancy or lactation". Dropouts: 4/18 (4/11 in the levetiracetam and 0/7 in the placebo groups; one participant dropped out immediately after baseline for unrelated medical reasons, presenting a protocol violation, and was therefore replaced to provide 18 appropriately enrolled participants as per the study design). |
|
| Interventions | Pharmacological intervention: quote: "Study medication was started at 500 mg at bedtime for 4 days, and increased as tolerated at the rate of 500mg every 3–4 days, to 2000 mg/day by day 14, and to a maximum daily dose of 3000 mg (1500 mg BID)". | |
| Outcomes | Primary outcome measure: BSPS (for reduction of anxiety) and CGI‐I (for treatment efficacy) Secondary outcome measures: LSAS (for reduction of anxiety) and SPIN (for reduction of anxiety) Time points: Quote: "Following the screening assessment, eligible subjects returned for a baseline visit at which they were randomly assigned to double‐blind treatment with either LEV or matching PBO. Subjects returned for followup at weeks 1, 2, 3, 5 and 7" |
|
| Notes | Industry funded: yes. Quote: "Acknowledgement is due to a grant from UCB Pharma, Inc to Dr Davidson". Medication provided by industry: unclear Any of the authors work for industry: no |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "The study design called for 18 subjects to randomly receive double‐blind treatment with either LEV or matching PBO, in a 2:1 ratio, for 7 weeks". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Medication and placebo was provided in identical capsules. However, it is not clear whether both participants and personnel were blinded. Quote: "The study design called for 18 subjects to randomly receive double‐blind treatment with either LEV or matching PBO, in a 2:1 ratio, for 7 weeks". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | More participants withdrew from the levetiracetam group (4/11; 36%) compared to the placebo (0/7; 0%) group. The 2 groups did not differ by sample characteristics at baseline. Quote: "Twenty‐four subjects were screened and 19 subjects were randomized to double‐blind treatment. Reasons for screen failure included voluntary withdrawal after reading the informed consent (n=3), a primary diagnosis of post‐traumatic stress disorder (n=1), and not meeting criteria for social anxiety disorder (n=1). Among those who were enrolled, one subject dropped out immediately after baseline for unrelated medical reasons, presenting a protocol violation, and was therefore replaced to provide 18 appropriately enrolled subjects as per the study design. Two subjects dropped out following the baseline visit due to early side effects from LEV (muscle spasms and pain, n=1; severe headache, n=1) and failed to return for efficacy ratings, thus leaving 16 subjects (n=9 LEV, n=7 PBO) available for the ITT LOCF analysis. Fourteen subjects (n=7 each LEV and PBO) completed the 7‐week treatment period ... No between treatment differences were observed in the baseline demographic characteristics". Overall 22% of the participants dropped out of the study. Quote: "Analyses were performed on the intention to treat (ITT) sample, which included all subjects who returned for at least one post‐baseline assessment, using the last observation carried forward (LOCF)". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias were identified. |
ADIS‐R: Anxiety Disorder Interview Schedule ‐ Revised; AIMS: Abnormal Involuntary Movements Scale; APD: avoidant personality disorder; ASEX: Arizona Sexual Experience Scale; ATM: atomoxetine; BAI: Beck Anxiety Inventory; BAS: Brief Assessment Scale; BSPS: Brief Social Phobia Scale; CAS: Clinical Anxiety Scale; CGI‐I: Clinical Global Impressions Scale ‐ Improvement item; CGI‐S: Clinical Global Impressions Scale ‐ Severity item; CIC‐SP: Clinical Impression of Change ‐ Social Phobia Scale; CIS‐SP: The Clinical Impression of Severity ‐ Social Phobia; CR: controlled release; CSPS: Cornell Social Phobia Scale; CT: Cognitive Therapy; DESS: Discontinuation Emergent Signs and Symptoms; DSM: Diagnostic and Statistical Manual of Mental Disorders; DSPS: Duke Social Phobia Scale; ECG: electrocardiogram; ECT: electroconvulsive therapy; ER: extended release; FONE/FNES: Fear of Negative Evaluation Scale; FQ: Fear Questionnaire; GAF: Global Assessment of Functioning; GSAD: generalised social anxiety disorder; GSK: Glaxo‐Smith Kline; GSP: generalised social phobia; HAM‐A/HARS: Hamiton Anxiety Rating Scale; HAM‐D/HDS/HDRS: Hamilton Depression Rating Scale; HSC: Hopkins Symptoms Checklist; IEs: Independent evaluator’s; IIP‐64: Inventory of Interpersonal Problems; IIS: Inventory of Interpersonal Situations; ISPI: Index of Social Phobia Improvement; ITT: intention‐to‐treat; LOCF: last observation carried forward; LSAS: Liebowitz Social Anxiety Scale; LSEQ: Leeds Sleep Evaluation Questionnaire; LSPDS: Liebowitz Social Phobic Disorders Scale; LSPS: The Liebowitz Social Phobia Symptom Scale; MADRS: Montgomery‐Asberg Depression Rating Scale; MFQ: Marks Fear Questionnaire; MINI: Mini‐International Neuropsychiatric Interview; MMFQ: Marks‐Mathews' Fear Questionnaire; MOS‐12: Medical Outcomes Study, 12‐item sleep module; NCE: New Chemical Entity; OC: observed case; PGI: Patient Global Impression of Improvement scale; PIC‐SP: The Patient's Impression of Change ‐Social Phobia; PRCS: Personal Report of Confidence as a Speaker Questionnaire; SADS: Social Avoidance and Distress Scale; SAS: symptom assessment scale; SCI: Social Cognitions Inventory; SCID: Structured Clinical Interview for DSM; SCID‐P: Structured Clinical Interview for DSM, Patient edition; SCL‐90: symptom checklist; SDI: social difficulties inventory; SDS: Sheehan Disability Scale; SF‐36: 36 Short‐Form Health Survey; SIAS: Social Interaction Anxiety Scale; SOSS: Severity of Symptoms Scale; SPAI: Social Phobia and Anxiety Inventory; SPEFI: Social Phobia Endstate Functioning Index; SPIN: Social Phobia Inventory; SPS: Social Phobia Scale; SPW: scale of psychological well‐being; SSRI: selective serotonin reuptake inhibitor; SSS: Severity of Symptoms Scale; STAI: State‐Trait Anxiety Inventory; TLFB: timeline follow‐back; VAS: Visual Analogue Scale; WPAI: Work Productivity and Impairment Questionnaire; WPI: WiIloughby Personality Inventory.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| ACTRN12608000363381 | No placebo group |
| ACTRN12609000091202 | No placebo group |
| Allsopp 1984 | No placebo control |
| Angelini 1989 | Open‐label pilot study, social phobia participants not separately analysed |
| Atmaca 2002 | No placebo control |
| Blank 2006 | No placebo control |
| Brantigan 1982 | No diagnosis of social phobia |
| Bystritsky 2005 | No placebo control |
| Clark 2003 | Concomitant behavioural therapy |
| Clark‐Elford 2015 | Measures performance anxiety |
| Coupland 2000 | Main focus is on panic attacks |
| Dempsey 2009 | Secondary analysis of a previous study |
| Dodhia 2014 | Focus on measuring regions of the brain through imaging |
| Donahue 2009 | Concomitant exposure therapy |
| Dunlop 2007 | Open‐label pilot study not controlled from beginning |
| EUCTR2004‐001894‐24‐DE | No placebo group |
| Falloon 1981 | Concomitant behavioral therapy |
| Fang 2014 | Measures performance anxiety |
| Faria 2014 | Assessment of brain function using fMRI |
| Feifel 2011 | Augmentation study |
| Gale 2007 | Commentary |
| Gates 1985 | No diagnosis of social phobia |
| Gelernter 1991 | Concomitant behavioral therapy |
| Gorka 2015 | Measures performance anxiety with brain imaging |
| Greenhill 1999 | Variety of anxiety disorders measured |
| Grosser 2012 | Measures GAD not GSAD |
| Guastella 2009 | Concomitant exposure therapy |
| Hartley 1983 | No diagnosis of social phobia |
| Haug 2003 | Concomitant exposure therapy. |
| Heun 2013 | Not specific to social phobia diagnosis |
| Hofmann 2006 | Augmentation design (of exposure therapy) |
| Ionescu 2013 | Adolescent population |
| James 1977 | No diagnosis of social phobia |
| James 1983 | Measures performance anxiety |
| James 1984 | No diagnosis of social phobia |
| Krishman 1976 | Examination anxiety and no placebo control |
| Liappas 2003 | Combination psychotherapy and pharmacotherapy compared to psychotherapy, no placebo control |
| Liden 1974 | No diagnosis of social phobia |
| Liebowitz 1999 | No placebo control (continuation study of Heimberg 1998) |
| Liebowitz 2014 | Measures performance anxiety |
| Malcolm 1992 | Variety of social anxiety disorders |
| Mangano 2003 | Review |
| Mortberg 2007 | Concomitant individual and group cognitive therapy |
| Mountjoy 1977 | Social phobia participants not separately analysed |
| NCT00118833 | Herbal medication |
| NCT00248612 | Participants with social phobia, GAD and or panic disorder |
| NCT00308724 | CBT for the treatment of GAD |
| NCT00332046 | Assessment of brain function using fMRI |
| NCT00343707 | Measures performance anxiety |
| Neftel 1982 | No diagnosis of social phobia |
| Oosterbaan 1997 | No placebo control |
| Otto 2000 | No placebo control |
| Pecknold 1982 | Augmentation design (clomipramine + tryptophan vs clomipramine + placebo) |
| Phan 2015 | Brain imaging study |
| Pine 2001 | Social phobia participants not separately analysed |
| Prasko 2004 | Moclobemide + supportive guidance versus CBT + placebo pills versus combination of CBT + moclobemide |
| Ravindran 2014 | Augmentation study, medication added to psychotherapy |
| Rickels 1978 | No placebo control |
| Rynn 2008 | Measured treatment‐emergent effects |
| Schuurmans 2004 | Social phobia participants not separately analysed |
| Seedat 2003 | Augmentation trial of pindolol as adjunctive treatment to the SSRI paroxetine and incomplete results (cross‐over data not reported separately for first leg of treatment) |
| Shlik 2002 | Measures neuroendocrine and behavioural responses |
| Siitonen 1976 | No diagnosis of social phobia |
| Silverstone 1973 | Concomitant behavioural therapy |
| Simon 2010 | No placebo control |
| Solyom 1973 | Combined population and combined intervention (psychotherapy and medication) |
| Solyom 1981 | Combined population |
| Tubaki 2012 | No placebo control |
| Tyrer 1973 | Social phobia participants not separately analysed |
| Wardle 2012 | Measures performance anxiety and is an augmentation study – medication added to psychotherapy |
CBT: cognitive behavioural therapy; GAD: generalised anxiety disorder; GSAD: generalised social anxiety disorder; fMRI: functional magnetic resonance imaging.
Characteristics of studies awaiting assessment [ordered by study ID]
Asakura 2016.
| Methods | Randomized, double‐blind placebo‐controlled study, 12 weeks |
| Participants | Quote: "Patients aged 18–64 years with a primary diagnosis of DSM‐IV‐TR defined SAD, a Liebowitz Social Anxiety Scale Japanese version (LSAS‐J) total score ≥60 and a Clinical Global Impression–Severity (CGI‐S) score ≥4 at baseline were randomly assigned (1:1:1) to placebo, escitalopram 10 mg or escitalopram 20 mg" |
| Interventions | Placebo Escitalopram 10 mg or 20 mg |
| Outcomes | Quote: "The primary endpoint was change from baseline to Week 12 in the LSAS‐J total score for both escitalopram 10 mg and 20 mg versus placebo (ANCOVA, FAS, LOCF), using a hierarchical testing procedure. Pre‐specified secondary endpoints included LSAS‐J sensitivity analyses' |
| Notes | Clinical trial identifier: JapicCTI‐121842 |
Careri 2015.
| Methods | Quote: "The study was a 12‐week double‐blind, placebo‐controlled, flexible‐dose trial; daily doses of vilazodone 20 mg/d to 40 mg/d or matching placebo were administered in a 1:1 ratio. Data were collected between November 2012 and April 2014" |
| Participants | Quote: "Enrollment was planned for 30 subjects who achieved the prospectively determined minimum adequate treatment of at least 6 consecutive weeks on ≥ 20 mg/d of vilazodone or the placebo equivalent. Subjects included men and women, aged 18–75 years, who met DSM‐IV‐TR criteria for generalized social anxiety disorder and had a minimum total Liebowitz Social Anxiety Scale (LSAS) score at screening and baseline of 70 and a minimum Clinical Global Impressions–Severity scale (CGI‐S) score of 4 (moderately ill). Subjects also had to agree to practice effective contraception methods. Exclusion criteria included lifetime bipolar disorder, schizophrenia, and body dysmorphic disorder, as well as posttraumatic stress disorder, obsessive‐compulsive disorder, panic disorder, and substance dependence within the past 24 weeks. Comorbid major depression, dysthymia, generalized anxiety disorder, and specific phobias were allowed if generalized social anxiety disorder was the primary disorder (the major clinical problem for which the subjects sought treatment). Subjects who were suicidal, who were medically unstable, who had a history of cancer or treatment‐refractory generalized social anxiety disorder (failure to respond to adequate trials of 2 effective agents), or who were in active cognitive‐behavioral therapy or were currently pregnant or lactating were excluded. Zolpidem as needed was allowed for insomnia if not taken more than 3 times per week. Other psychotropic drugs had to be discontinued at least 2 weeks before the baseline visit" |
| Interventions | Quote: "Subjects started at baseline on vilazodone 10 mg/d or placebo, taken in the morning with food, and increased to 20 mg/d or placebo after 1 week and to 40 mg/d or placebo after the second week. Dose increases could be delayed or reversed for problems of tolerability; however, attempts were made to raise all subjects to 40 mg/d. Noncompliance was defined as < 80% or > 120% of prescribed drug taken during any evaluation period. Subjects who were noncompliant at more than 2 consecutive study visits could be terminated" |
| Outcomes | Quote: "The MINI was used for diagnostic assessment of DSM‐IV disorders. The CGI‐S, CGI‐I, PGIC, and LSAS were administered to assess social anxiety disorder severity and change and global improvement. The LSAS is a 24‐item instrument developed by Liebowitz that assesses anxiety and avoidance in a variety of commonly encountered performance and social situations and was found by Heimberg et al to be reliable and valid. The HDRS‐17 and HARS were used to quantify depressive and anxiety symptoms at baseline and endpoint. Safety measures included routine laboratory tests, ECGs, and physical examinations. Subjects were asked about adverse events and concomitant medications at each study visit" |
| Notes | ClinicalTrials.gov identifier: NCT01712321 Funding/support: Quote: "This study was supported by an investigator‐initiated grant from Forest Research Institute, Inc, Jersey City, New Jersey, to The Medical Research Network, LLC" |
De la Barquera 2008.
| Methods | Double‐blind controlled study |
| Participants | Patients with social phobia |
| Interventions | Clonazepam and placebo |
| Outcomes | — |
| Notes | — |
Frick 2015.
| Methods | Randomised, double‐blind treatment for 6 weeks |
| Participants | 18 SAD patients. Serotonin synthesis rate capacity was assessed before and after treatment in the patients and 17 age and sex‐matched healthy controls (HC; only scanned once) using positron emission tomography imaging with the radiotracer [11C]5‐HTP. |
| Interventions | Experimental: SSRI Citalopram or NK1R antagonist GR205171 Control: placebo |
| Outcomes | Primary outcome: Liebowitz Social Anxiety Scale (LSAS) was used to index symptom severity |
| Notes | Funding/Support: Quote: "This study was supported by the Swedish Research Council, the Swedish Brain Foundation, Riksbankens Jubileumsfond–the Swedish Foundation for Humanities and Social Sciences, and the Swedish Research Council for Health,Working Life, andWelfare. Ligand production of 5‐hydroxytryptophan for the patients was supported by GlaxoSmithKline" |
Krylov 1996.
| Methods | Double‐blind, parallel group trial |
| Participants | 66 participants with social phobia |
| Interventions | Alprazolam, buspirone, or placebo |
| Outcomes | — |
| Notes | — |
NCT00114127.
| Methods | Study type: intervention Study design: randomised allocation Intervention model: parallel assignment Masking: double‐blind (participant, caregiver) Primary purpose: treatment |
| Participants | Enrollment: 28 Ages eligible for study: > 18 years (adult, senior). Genders eligible for study: all Accepts healthy volunteers: no Inclusion criteria: quote: "Male or female outpatients > 18 years of age with a primary psychiatric diagnosis of generalized social anxiety disorder as defined by DSM‐IV criteria and an LSAS score > 50; physical examination, electrocardiogram, and laboratory findings without clinically significant abnormalities; willingness and ability to comply with the requirements of the study protocol". Exclusion criteria: quote: "Patient has a history of intolerance or lack of response to a treatment trial of duloxetine at highest tolerated dose (<120mg/day); patients with acute narrow angle glaucoma; pregnant women, lactating women, and women of childbearing potential who are not using medically accepted forms of contraception (e.g., IUD, oral contraceptives, barrier devices, condoms and foam, or implanted progesterone rods stabilized for at least 3 months); concurrent use of other psychotropic medications. Patients must discontinue regular benzodiazepine or antidepressant therapy at least one week (5 weeks for fluoxetine) prior to baseline. Concomitant beta‐blockers are proscribed unless prescribed for a medical indication (e.g., hypertension, at a stable daily dose for > 1 month); patients with a history of failure to satisfactorily respond to >2 prior adequate treatment trials; significant personality dysfunction likely to interfere with study participation; serious medical illness or instability for which hospitalization may be likely within the next year; Seizure disorders with the exception of a history of febrile seizures if they occurred during childhood, were isolated, and did not recur in adulthood; Concurrent psychotherapy initiated within 2 months of baseline is prohibited. Ongoing psychotherapy of any duration directed specifically toward treatment of the social anxiety disorder is excluded. Prohibited psychotherapy includes cognitive behavioural therapy or psychodynamic therapy that focuses on exploring specific, dynamic causes of the phobic symptomatology and provides skills for their management. General supportive individual, couples, or family therapy greater than 2 months duration is acceptable; diagnosis of any of the following mental disorders as defined by the DSM‐IV: a lifetime history of schizophrenia or any other psychosis, mental retardation, organic medical disorders or bipolar disorder; eating disorders in the past 6 months; alcohol or substance abuse in the past 3 months or dependence within the past 6 months; entry of patients with major depression, dysthymia, panic disorder, generalized anxiety disorder, post‐traumatic stress disorder or obsessive‐compulsive disorder will be permitted if the social anxiety disorder is judged to be the predominant disorder, in order to increase accrual of a clinically relevant sample; patients with significant suicidal ideation (MADRS item 10 score > 3) or who have enacted suicidal behaviours within 6 months prior to intake will be excluded from study participation and referred for appropriate clinical intervention. |
| Interventions | Active comparator: duloxetine 60 mg/day for 6 weeks; in phase 1 all participants entered an open trial Active comparator: duloxetine 120 mg for 18 weeks; in phase 2 participants were randomised to 60 mg duloxetine + placebo or 120 mg duloxetine Placebo comparator: duloxetine 60 mg + placebo for 18 weeks; in phase 2 participants were randomised to 60 mg duloxetine + placebo or 120 mg duloxetine |
| Outcomes | Primary outcome measures: anxiety symptoms as assessed by Liebowitz Social Anxiety Scale (time frame: 6 months)
Secondary outcome measures: CGI‐S (time frame: 6 months) Baseline collected for phase 1 at week 0 and for phase 2 at week 6 |
| Notes | Responsible party: Naomi M Simon, Director, Center for Anxiety and Traumatic Stress Disorders, Massachusetts General Hospital ClinicalTrials.gov Identifier: NCT00114127 Other study ID numbers: 2004‐P‐001384 Study first received: 13 June 2005 Last updated: 5 June 2014 Locations: Massachusetts General Hospital, Boston, Massachusetts 02114, USA Sponsors and collaborators: Massachusetts General Hospital. Investigators: principal investigator: Naomi M Simon, MD, Massachusetts General Hospital. |
NCT00208741.
| Methods | Study type: intervention Study design: randomised allocation Intervention model: single group assignment Masking: double‐blind Primary purpose: treatment |
| Participants | Estimated enrollment: 50 Ages eligible for study: 18‐65 years (adult) Genders eligible for study: all Accepts healthy volunteers: no Inclusion criteria: quote: "primary diagnosis of SAD; CGI (S) ≥ 4 at screen; LSAS ≥ 50 at baseline; Covi Anxiety Scale score greater than the Raskin depression Scale total score at screen". Exclusion criteria: quote: "non‐responsive to adequate trials of two or more treatment medications, if previously treated for SAD; HAM‐D ≥15 or a score of >2 on Item 1 at baseline; serious or unstable medical condition; alcohol or substance use disorder within 6 months prior to study". |
| Interventions | This study consists of two parts. The first part consists of 12 weeks of open‐label treatment with Gabitril. If the study doctor determines that the patients condition has improved and they have completed the initial 12 weeks of treatment they may be eligible for the second part of the study. This part is a 24‐week double‐blind treatment period with either Gabitril or placebo (inactive medication). There will also be a follow‐up visit about 1 to 3 weeks after they have completed taking the study medication. Altogether study participation is expected to last approximately 37 weeks. |
| Outcomes | Primary outcome measures: Liebowitz Social Anxiety Scale (LSAS); Clinical Global Impression‐Change (CGI‐C) Secondary outcome measures: Hamilton Anxiety Scale (HAM‐A); Social Phobia Inventory (SPIN); Pittsburgh Sleep Quality Index (PSQI); 36‐Item Short‐Form Health Survey (SF‐36); Clinical Global Impression‐S (CGI‐S) |
| Notes | Responsible Party: Emory University ClinicalTrials.gov Identifier: NCT00208741 Other study ID numbers: 0337‐2002 Study first received: 13 September 2005 Last updated: 8 November 2013 Locations: Emory University School of Medicine, Atlanta, GA 30329, USA Hillside Hospital of the North Shore‐Long Island Jewish Health System, Long Island, NY 10032, USA Columbia/New York State Psychiatric Institute, New York, NY 10032, USA Sponsors and collaborators: Emory University, Cephalon |
NCT00215254.
| Methods | Study type: intervention Study design: randomised allocation Intervention model: parallel assignment Masking: double‐blind Primary purpose: treatment |
| Participants | Ages eligible for study: 18‐65 years (adult) Genders eligible for study: all Accepts healthy volunteers: no Inclusion criteria: quote: "adult outpatients 18‐65 years of age, primary diagnosis of social anxiety disorder, using DSM‐IV criteria; minimum CGI severity score of 4 at baseline; minimum BSPS score of 20 at baseline; written informed consent; negative serum pregnancy test for women of childbearing potential". Exclusion criteria: quote: "current DSM‐IV diagnosis of bipolar disorder, schizophrenia or other psychotic disorder, or cognitive disorder due to a general medical condition, any current primary anxiety disorder other than SAD or current primary depression; history of substance abuse or dependence with the last 6 months; suicide risk or serious suicide attempt within the last year; clinically significant medical condition or laboratory abnormality; women of childbearing potential who are unwilling to practice an acceptable method of contraception; concomitant medication use for psychotropic purposes, history of hypersensitivity to quetiapine". |
| Interventions | Quote: "This is an eight week, randomized, double‐blind, placebo‐controlled trial of quetiapine (100‐400 mg/day) in social anxiety disorder". |
| Outcomes | Primary outcome measures: Brief Social Phobia Scale (BSPS) Secondary outcome measures: Clinical Global Impressions of Severity (CGI‐S); Clinical Global Impressions of Improvement (CGI‐I); Social Phobia Inventory (SPIN); Hospital Anxiety and Depression Scale (HADS); Connor Davidson Resilience Scale (CD‐RISC); Sheehan Disability Inventory (SDI); Barnes Akathisis Scale (BAS); Simpson‐Angus Scale (SAS) |
| Notes | ClinicalTrials.gov Identifier: NCT00215254 Other study ID numbers: 5639‐04‐3R0 Study first received: 20 September 2005 Last updated: 18 December 2006 Locations: Duke University Medical Center, Durham, North Carolina, 27705, USA Sponsors and collaborators: Duke University, AstraZeneca Investigators: principal investigator: Jonathan Davidson, MD, Duke University |
NCT00246441.
| Methods | Study type: intervention Study design: randomised allocation Intervention model: parallel assignment Masking: double‐blind (participant, caregiver, investigator) Primary purpose: treatment |
| Participants | Enrollment: 42 Ages eligible for study: 18‐65 years (adult) Genders eligible for study: all Accepts healthy volunteers: no Inclusion criteria: quote: "Meets DSM‐IV criteria for current social anxiety disorder; reports social anxiety in most situations (generalized type); treatment seeking for relief of social anxiety; meets DSM‐IV criteria for current alcohol use disorder; reads at the 6th grade level or above; endorses using alcohol to cope with social anxiety either "very often" or "always"; reports no prior medical alcohol detoxification; willingness to be randomized to the placebo group; willingness to attend 16 weekly medication management visits and one alcohol‐related therapy session; Liebowitz Social Anxiety Scale Total score (modified version) of at least 60; endorses drinking at least 15 standard drinks in a typical 30 day period or reports drinking heavily (defined as greater‐than‐or‐equal‐to 4 standard drinks on one occasion for women; greater‐than‐or‐equal‐to 5 standard drinks on one occasion for men, respectively) on at least 2 days in a typical 30 day period". Exclusion criteria: quote: "Abuse or dependence on drugs other than nicotine or marijuana in last 90 days; current or past diagnosis of bipolar disorder or schizophrenia; significant suicide risk as assessed by the SCID; current use of psychotropic medications; treatment seeking for alcohol problems; any unstable medical condition that might interfere with safe participation in the trial; elevated liver enzymes (3 x greater than normal levels); history of adverse reaction to paroxetine; history of failure to respond to adequate trial or dose of paroxetine for social phobia (60 mg/day for at least 6 weeks); history of heart problems or abnormal ECG recording; pregnancy, nursing, or refusal to use effective birth control if sexually active and premenopausal; history of one or more alcohol detoxifications". |
| Interventions | Drug: paroxetine 16 weeks treatment; dosing will start at 20 mg/day paroxetine and will increase gradually to a maximum dose of 60 mg/day Drug: placebo treatment phase will last 16 weeks; dosing will start at 20 mg/day (placebo) and will increase gradually to a maximum dose of 60 mg/day |
| Outcomes | Primary outcome measures: social anxiety severity; alcohol use, quantity and frequency; drinking to cope, quantity and frequency Secondary outcome measures: quality of life, depressive symptoms Time frame: 16 weeks treatment; 6 month and 12 month follow‐up interviews |
| Notes | Responsible Party: Medical University of South Carolina ClinicalTrials.gov Identifier: NCT00246441 Other study ID numbers: NIAAARAN013379; R01AA013379; NIH Grant R01 AA013379 Study first received: 28 October 2005 Last updated: 1 December 2016 Locations: Medical University of South Carolina, Institute of Psychiatry; Charleston, South Carolina, 29425, USA Sponsors and collaborators: Medical University of South Carolina, National Institute on Alcohol Abuse and Alcoholism (NIAAA) Investigators: principal investigator: Carrie L Randall, PhD, Medical University of South Carolina |
NCT00294346.
| Methods | Study type: intervention Study design: randomised allocation Intervention model: parallel assignment Masking: double‐blind Primary purpose: treatment |
| Participants | Estimated enrollment: 180 Ages eligible for study: 18‐65 years (adult) Genders eligible for study: all Accepts healthy volunteers: no Inclusion criteria: quote: "The subject is male or female, 18 ‐ 65 years of age (inclusive); The subject meets current DSM‐IV‐TR (American Psychiatric Association, 2000) criteria for social phobia (300.23), generalized subtype, as confirmed by the Mini‐International Neuropsychiatric Interview at Screening (visit 1); The subject has had symptoms of SAD (social phobia) present for at least 6 months prior to screening (visit 1); The subject has a total score ≥ 60 on the LSAS at both screening (visit 1) and baseline (visit 2); The subject has a score ≥ 4 on the Clinical Global Impression ‐ Severity (CGI‐S) scale at both screening (visit 1) and baseline (visit 2); The subject has a score ≤ 15 on the 17‐item Hamilton Rating Scale for Depression (HAM‐D) at screening; The subject, if female and of child‐bearing potential (not 2 years post‐menopausal or surgically sterilized), must have a negative serum pregnancy test at screening (Visit 1) and be willing to avoid pregnancy and practice adequate birth control from the time of study enrollment until 30 days after the last dose of study medication. Adequate methods of birth control are: oral contraception, intrauterine device, implantable contraceptive device, depot contraceptive, or a barrier method plus spermicide. Additional serum pregnancy tests will be administered at visit 6, visit 8, and visit 9; The subject, if engaged in ongoing psychotherapy for SAD or any other mental health condition, must have been attending therapy regularly for at least 3 months prior to screening (Visit 1) and must agree to continue the same type and frequency of psychotherapy throughout the course of the study; The subject agrees to refrain from blood donation during the course of the study; The subject has written and oral fluency in English or Spanish; The subject is willing to participate in the study, as evidenced by a signed and dated written Informed Consent Form (ICF)". Exclusion criteria: quote: "The subject has a decrease >15 points on the LSAS total score between screening (visit 1) and baseline (visit 2); The subject has a clinically significant abnormality or clinically significant unstable medical condition as indicated by medical history, physical examination, ECG results, clinical laboratory testing, or the investigator's judgment at screening (visit 1) or baseline (visit 2); The subject has a QTc interval of 450 msec or greater at screening (visit 1) if male or a QTc interval of 470 msec or greater at screening (visit 1) if female; The subject has current hypothyroidism or hyperthyroidism or laboratory findings consistent with thyroid dysfunction. Subjects who are being treated for thyroid disorder are eligible if they have been on stable doses of thyroid hormone for at least 6 months and are currently euthyroid; The subject has any history of schizophrenia or other psychotic disorder, bipolar disorder, post‐traumatic stress disorder, borderline personality disorder, or antisocial personality disorder; The subject has a history within the previous 5 years of obsessive‐compulsive disorder or an eating disorder; The subject exhibits evidence of a clinically predominant DSM‐IV‐TR Axis I or II disorder other than social phobia or avoidant personality disorder within the 6 months prior to screening (visit 1); The subject, in the opinion of the investigator, presents a significant risk of doing harm to himself, herself, or others; The subject has met DSM‐IV‐TR criteria for alcohol or substance dependence (other than nicotine or caffeine dependence) within 6 months of screening (visit 1); The subject has met DSM‐IV‐TR criteria substance abuse (other than alcohol, nicotine or caffeine abuse) within 3 months of screening (visit 1); The subject tests positive on the urine drug screen conducted at screening (visit 1) for illicit drugs, including opiates, barbiturates, amphetamines, cocaine, and phencyclidine; The subject is a pregnant or lactating female; The subject has previously participated in a clinical trial for AV608 (previously identified as NKP608 and CGP608); The subject has used any prohibited medications, or has any anticipated need or intended use of these medications during the study; The subject has used any investigational drugs, products, or devices in the 3 months prior to screening (visit 1); The subject is a member of the investigative site staff or an immediate family member; The subject has any other condition that the investigator believes would jeopardize the safety or rights of the subject or would render the subject unable to comply with the trial protocol". |
| Interventions | Quote: "The purpose of this study is to look at the safety and effectiveness of an investigational drug (AV608) when used in subjects who have Social Anxiety Disorder. AV608 is an NK‐1 receptor antagonist that exhibits central nervous system activity after oral administration. The study will compare AV608 to placebo (a medically inactive substance) to see if AV608 helps the symptoms of Social Anxiety Disorder. Eligible subjects will be assigned by chance to take either AV608 or placebo for 12 weeks. During the study, subjects will be asked about their overall health and mood and their Social Anxiety Disorder". |
| Outcomes | Primary outcome measures: Liebowitz Social Anxiety Scale (LSAS) |
| Notes | Study start date: February 2006 Study completion date: December 2006 Locations: Alabama, Arizona, California, Florida, Maryland, New York, Ohio, Oklahoma, Oregon, Texas (USA) Sponsors and collaborators: Avera Pharmaceuticals ClinicalTrials.gov Identifier: NCT00294346 Other study ID numbers: AV608‐105 Study first received: 17 February 2006 Last updated: 15 February 2008 |
NCT00485888.
| Methods | Allocation: randomised Endpoint classification: efficacy study Intervention model: parallel assignment Masking: double‐blind (participant, caregiver, investigator) Primary purpose: treatment |
| Participants | Enrollment: 71 Age: 18‐75 years (both sexes) Inclusion criteria: quote: "The patient has provided signed informed consent. Outpatients aged 18‐65 (extremes included). Patients with a primary diagnosis of Social Phobia according to DSM IV (300.23) criteria (diagnosis to be made using the Mini International Neuropsychiatric Interview (MINI)). On the basis of a physical examination, medical history and basic laboratory screening, the patient is, in the investigators opinion, in a suitable condition. Willing and able to attend study appointments in the correct time windows". Exclusion criteria: quote: "Any other axis I diagnosis that was a primary disorder in the previous six months. Continuation or commencement of formal psychotherapy. Alcohol or drug abuse as defined in the DSM IV within the last six months. Mania or hypomania as defined in the DSM IV. Current use of or commencement of antidepressant and anxiolytic medications. Patients, who have been on an antidepressant or other anxiolytic prior to the study, will have discontinued it more than two weeks prior to entry into the study. Those who have been on fluoxetine, will have been off of it for at least 5 weeks. Patients who have been on an herbal or alternative treatment judged to be potentially anxiolytic or with psychobiological activity, will have terminated usage of the agent more than two weeks prior to entering the study. Previous reaction to niacin administration, use of a non‐steroidal anti‐inflammatory, any psychotic disorder. Eating disorders as defined in the DSM IV. Mental retardation or other cognitive disorder. Clinical interpretation of apparent suicide risk. Laboratory values at screening or in medical history that may be considered through clinical interpretation to be significant. Diseases which could, through clinical interpretation, interfere with the assessments of safety, tolerability and efficacy. Serious illness: liver or renal insufficiency, cardiac, vascular, pulmonary, gastrointestinal, endocrine, neurological, infectious, neoplastic or metabolic disturbance. The patient is, in the opinion of the investigator, unlikely to be able to comply with the clinical trial protocol, or is unsuitable for any other reasons". |
| Interventions | Drug: cipralex Drug: placebo |
| Outcomes | Primary outcome measures: changes in intensity of the vasodilatory response to 10 mM topical m‐N over 16 weeks. (time frame: 20 weeks) (designated as safety issue: no). Secondary outcome measures: mean change from baseline on the LSAS, HAM‐A, SPIN, BAI, SPS, SIAS, BTS‐Q, BPS, Sheehan Disability Scale, Euroquol SF‐36, PSWQ (time frame: 20 weeks) (designated as safety issue: no) |
| Notes | Sponsors and collaborators: START Clinic for Mood and Anxiety Disorders, H Lundbeck A/S. Principal investigator: Martin A Katzman, MD; START Clinic for the Mood and Anxiety Disorders |
NCT00612859.
| Methods | Study type: intervention Study design: randomised allocation Intervention model: parallel assignment Masking: double‐blind Primary purpose: treatment |
| Participants | Enrollment: 217 Ages eligible for study: 18‐70 years (adult, senior) Genders eligible for study: all Accepts healthy volunteers: no Inclusion criteria: quote: "Male or female outpatients between 18 and 70 years old inclusive; symptoms of social anxiety disorders (generalized type) present for at least 1 year prior to Selection Visit; had a score of ≥60 on the LSAS at the Selection Visit and at the Randomization Visit. Additionally, the clinician's global impression of change score must have been ≥ 2 at the randomization visit; had a telephone where they could be directly contacted". Exclusion criteria: quote: "History of autism or Asperger's disease; had another primary axis I disorder or fulfilled diagnostic and statistical manual of mental disorders‐4th edition (DSM‐IV) criteria in the 6 months prior to Screening; major depression as measured by a Hamilton Depression Rating Scale (HAM‐D‐17 items) total score of > 17 and/or a suicide subscale score on the HAM‐D‐17 items of > 2 at the selection or randomization visit; history of electroconvulsive therapy within the prior 3 months; history of psychotherapy which was not stable and ongoing for at least 6 months prior to visit 1; clinical history of significantly impaired renal function with an estimated creatinine clearance below 80 mL/min; clinically significant medical condition; history of any clinically significant allergic condition or allergy to LEV or pyrrolidone derivatives; neutrophil count of less than 1800/µL" |
| Interventions | Quote: "A multicenter, randomized, double‐blind, PBO‐controlled, parallel group study to assess the efficacy and safety of levetiracetam versus PBO for the treatment of social anxiety disorder (generalized type)". |
| Outcomes | Primary outcome measures: change in Liebowitz Social Anxiety Scale (LSAS) score from Visit 2 to the last evaluation period visit attended using last observation carried forward (LOCF) methods; safety: monitoring of AEs, clinical laboratory tests, physical examination and vital signs. |
| Notes | Study start date: September 2003 Study completion date: June 2004 Sponsors and collaborators: UCB Pharma Investigators: study director: UCB Clinical Trial Call Center +1 877 822 9493 (UCB) ClinicalTrials.gov Identifier: NCT00612859 Other study ID numbers: N01086 Study first received: 14 January 2008 Last updated: 25 November 2013 |
NCT01316302.
| Methods | Study type: intervention Study design: randomised Intervention model: parallel assignment Masking: double‐blind (participant, investigator, outcomes assessor). Primary purpose: treatment |
| Participants | Enrollment: 63 Ages eligible for study: 18‐75 years (adult, senior) Genders eligible for study: all Accepts healthy volunteers: no Inclusion criteria: quote: "Subjects must give written informed consent prior to any study procedures. Diagnosis of Social Anxiety Disorder (SAD) (300.23 Social Phobia/Social Anxiety Disorder, Generalized Subtype) according to DSM‐IV‐TR criteria, as determined by psychiatric evaluation with the Principal Investigator. A minimum score of 60 on the LSAS total score at both screening and baseline visits. A total HAM‐D score of less than 15 at the screening visit. CGI Severity score of 4 or greater at both screening and baseline visits. Female participants of childbearing potential must commit to an effective form of contraception for the duration of the trial. Effective forms of contraception include: condoms with spermicide, diaphragm with spermicide, hormonal contraceptive agents (oral, transdermal, or injectable), and implantable contraceptive devices". Exclusion criteria: quote: "An Axis I disorder other than SAD (e.g., post‐traumatic stress disorder, obsessive compulsive disorder, panic disorder) within 24 weeks of the baseline visit. Subjects with co‐morbid MDD, GAD, dysthymia, or specific phobias will be allowed if GSAD is the primary disorder in terms of clinical severity, as determined by the investigator; Any history or complication of schizophrenia or bipolar disorder; any complication of body dysmorphic disorder; substance dependence, as defined by DSM‐IV‐TR criteria, within 24 weeks of the baseline visit; subjects who are currently pregnant, lactating, or of childbearing potential and not practicing an effective method of contraception; Subjects scoring >2 on item #3 of the HAM‐D, or who, in the opinion of the PI, are at a clinically significant risk for suicide; systolic blood pressure ≥165 and/or diastolic blood pressure ≥95; positive urine drug screen at the screening visit; any current unstable and/or clinically significant medical condition, based on history or as evidenced in screening laboratory and ECG assessments; Any history or complication of cancer or malignant tumor; fluoxetine within 28 days of baseline; MAO inhibitors within 14 days of baseline ‐ any other psychotropics (including SSRIs, SNRIs, and benzodiazepines) within 14 days of baseline. Zolpidem (Ambien®) PRN is allowed for insomnia if not taken more than 3 times per week; subjects who started psychotherapy or cognitive‐behavioural therapy within 24 weeks of the baseline visit, except for supportive psychotherapy; electro‐convulsive therapy (ECT) within 12 weeks of the baseline visit; treatment refractory GSAD." |
| Interventions | Drug: desvenlafaxine (Pristiq); flexible dose, 50‐100 mg 4 times daily for 12 weeks Drug: matching placebo, taken 4 times daily for 12 weeks |
| Outcomes | Primary outcome measures: Change in the LSAS total score (time frame: baseline to study endpoint (week 12)) LSAS measuring social anxiety symptoms, possible total scores ranging from 0‐144, with higher scores indicating greater severity of symptoms Secondary outcome measures: Clinical Global Impression of Improvement Scale (CGI‐I) (time frame: baseline to week 12); CGI‐I: one item, measuring overall improvement of illness; possible scores range from 1‐7, with lower scores representing greater improvement CGI‐I responders: defined as having a CGI‐I scores of 1 or 2 at week 12/study endpoint; Patient Global Impression of Change (time frame: baseline to week 12) Participant‐rated global outcome scale, people who rated themselves as 1 (very much improved) or 2 (much improved) on the PGIC were considered self‐rated responders |
| Notes | Locations: the Medical Research Network, LLC New York, New York, USA, 10128 Sponsors and collaborators: the Medical Research Network, Pfizer Investigators: principal investigator Michael R Liebowitz, MD, the Medical Research Network |
AEs: Adverse events; ANCOVA: Analysis of covariance; BAI: Beck Anxiety Inventory; BPS: Propensity Scale; BSPS: Brief Social Phobia; BTS‐Q: Blushing, Trembling, and Sweating Questionnaire; CGI: Clinical Global Impression; CGI‐S: Clinical Global Impression – Severity Scale; DSM: Diagnostic and Statistical Manual for Mental Health Disorders; ECG: electrocardiogram; Euroquol SF‐36: Euroquol (quality of life) and the Medical Outcomes Study Short‐Form‐36 (SF‐36); FAS: full analysis set; GAD: Generalised Anxiety Disorder; GSAD: Generalised Social Anxiety Disorder; HDRS‐17: Hamilton Depression Rating Scale, 17 items; HAM‐A/HARS: Hamilton Anxiety Rating Scale; HAM‐D: Hamilton Rating Scale for Depression; Inc.: Incorporation; LOCF: Last Observation Carried Forward; LSAS: Liebowitz Social Anxiety Scale;LSAS‐J: Liebowitz Social Anxiety Scale ‐ Japanese Version; MADRS: Montgomery–Åsberg Depression Rating Scale; MAO: Monoamine oxidase; MDD: Major Depressive Disorder; mg/d: Milligram per day;MINI: Mini‐International Neuropsychiatric Interview; PGIC: Patient Global Impression of Change; PSWQ: Penn State Worry Questionnaire; SAD: Social Anxiety Disorder; SF‐36: 36‐item Short‐Form Health Survey; SIAS: Social Interaction Anxiety Scale; SNRIs: Serotonin–norepinephrine reuptake inhibitors; SPIN: Social Phobia Inventory; SPS: Social Phobia Scale; SSRIs: Selective serotonin re‐uptake inhibitors.
Characteristics of ongoing studies [ordered by study ID]
NCT00182533.
| Trial name or title | Sertraline in the treatment of generalized social phobia with comorbidity |
| Methods | Randomised placebo control trial |
| Participants | Expected enrolment: 170 Inclusion criteria: ‐ outpatient with primary Diagnostic and Statistical Manual of Mental Disorders, 4th Edition (DSM‐IV) GSP plus at least one of the following comorbid DSM‐IV anxiety disorders: panic disorder with agoraphobia, obsessive compulsive disorder, major depressive disorder, generalised anxiety disorder ‐ score on LSAS > 50 ‐ score on MADRS < 25. Locations: Canada |
| Interventions | Sertraline and placebo |
| Outcomes | Primary outcome: Clinical Global Impression ‐ Improvement = 2 Liebowitz Social Anxiety Scale (LSAS) (mean change from baseline) Secondary outcome: mean change from baseline on the following scales: Quality of Life and Employment Satisfaction Questionnaire, Sheehan Disability Scale, Social Phobia Scale, Brief Social Phobia Scale, Penn State Worry Questionnaire, Panic and Agoraphobia Scale, Davidson Trauma Scale, Social Anxiety Spectrum Self‐Report (SHY‐SR), Yale‐Brown Obsessive Compulsive Scale, Montgomery‐Asberg Depression Rating Scale (MADRS) |
| Starting date | Expected completion August 2006 |
| Contact information | Van Ameringen 2006 |
| Notes | The recruitment status of this study is unknown because the information has not been verified recently. |
NCT01712321.
| Trial name or title | Vilazodone in the treatment of social anxiety disorder: a double blind study |
| Methods | 12 week double‐blind, placebo‐controlled trial |
| Participants | 30 outpatients aged 18‐75 years with SAD, generalised subtype who return for at least one postrandomisation visit where efficacy evaluations are conducted Inclusion criteria: diagnosis of social anxiety disorder, generalised subtype; LSAS total score of 70 at visits 1 and 2. Exclusion criteria: lifetime history of bipolar disorder or schizophrenia; current suicidal risk; current unstable medical condition |
| Interventions | Daily doses of vilazodone 20 mg/d to 40 mg/day or matching placebo |
| Outcomes | All participants randomised to drug or placebo and returning for at least one subsequent visit will be included in the primary efficacy analyses. Primary outcome measures: Change in Liebowitz Social Anxiety Scale (LSAS) ‐ total score (time frame: change from baseline to final study visit: minimum 1 week ‐ maximum 12 weeks). Randomised participants taking minimum target dose (20 mg or matching placebo daily) for at least six consecutive weeks will be considered a minimum adequate trial for the purposes of secondary analyses. Secondary outcome measures (time frame: study endpoint: minimum 6 weeks ‐ maximum 12 weeks): Responder rate, as defined by Clinical Global Impression of Improvement score of 1 or 2 Change in the Clinical Global Impression of Severity of Illness score Change on the LSAS anxiety and avoidance subscales Change in Hamilton Depression scale total Change in Hamilton Anxiety scale total Participant‐assessed responder rate, as defined by a Patient Global Impression of Change score of 1 (very much improved) or 2 (much improved) at study endpoint |
| Starting date | Date of registration: 18 October 2012 |
| Contact information | The Medical Research Network and Forest Laboratories |
| Notes | This study is ongoing, but not recruiting participants. |
NCT02083926.
| Trial name or title | Ketamine infusion for social anxiety disorder |
| Methods | Randomised, placebo‐controlled crossover study |
| Participants | Ages eligible for study: 18‐65 years Genders eligible for study: both Accepts healthy volunteers: no Inclusion criteria:
Exclusion criteria:
|
| Interventions | Pharmacological intervention: Experimental: ketamine (ketamine will be given at a dose of 0.5 mg/kg over 40 minutes. This dose is identical to that used in previous anti‐depressant studies of ketamine) Placebo comparator: saline (saline will be given at a dose of 0.5 mg/kg over a 40 minute period) |
| Outcomes | Primary outcome measures (time frame: first 2 weeks following infusion), at screening, 1 hour prior to infusion, 1, 2 and 3 hours after infusion, 1, 2, 3, 5, 7, 10, and 14 days following a single ketamine/saline infusion: Visual analogue scale (VAS) of anxiety states Secondary outcome measures (time frame: first 2 weeks following infusion): Anxiety severity, according to Beck Anxiety Inventory (BAI) at screening, 1 hour prior to infusion, 3 hours after infusion, 1, 2, 3, 5, 7, 10, and 14 days following a single ketamine/saline infusion Depression severity, according to Hamilton Depression Rating scale (HAM‐D) at screening, 1 hour prior to infusion, 3 hours after infusion, 1, 2, 3, 5, 7, 10, and 14 days following a single ketamine/saline infusion Clinical Global Impressions, according to Clinical Global Impressions (CGI) ratings of overall severity of SAD symptoms at screening, 1 hour prior to infusion, 3 hours after infusion, 1, 7 and 14 days following a single ketamine/saline infusion Brief Psychiatric Rating Scale, Positive Symptom Subscale (BPRS‐PS), with regard to thought content, conceptual disorganisation, hallucinatory behavior, and grandiosity at screening, 1 hour prior to infusion,1‐3 hours after infusion, 1 day following a single ketamine/saline infusion Clinician‐Administered Dissociative States Scale, according to Clinician‐Administered Dissociative States Scales (CADSS) ratings of dissociative symptoms at screening, 1 hour prior to infusion, 3 hours after infusion, 1 day following a single ketamine/saline infusion Self‐Statement During Public Speaking Scale (SPSS) (time frame: first week following infusion), according to Self‐Statement During Public Speaking Scale (SPSS) ratings of cognitions that occurred during a speech 1 hour prior to infusion, 3 hours after infusion, 1, 7, and days following a single ketamine/saline infusion Impromptu Speech Behavioral Assessment Test, according to the Impromptu Speech Behavioral Assessment Test (BAT) of social anxiety symptoms during public speaking at 1 hour prior to infusion, 1 and 7 days following a single ketamine/saline infusion Attention bias, according to the dot‐probe paradigm 1 hour before infusion, 3 hours after infusion, 1, 2, 3, 5, 7, 10 and 14 days following a single ketamine/saline infusion SAD severity, according to the Liebowitz Social Anxiety Scale (LSAS) ratings of SAD severity at screening, 1 hour before infusion, 3 hours after infusion, 1, 2, 3, 5, 7, 10 and 14 days following a single ketamine/saline infusion Positive and negative affect symptoms, according to positive and negative affect schedule (PANAS) ratings of positive and negative symptoms at screening, 1 hour before infusion, 1 and 7 days following a single ketamine/saline infusion State‐Trait Anxiety Inventory, at screening, 1 hour before infusion, 3 hours after infusion, 1, 2, 3, 5, 7, 10 and 14 days following a single ketamine/saline infusion |
| Starting date | Study start date: March 2014 Estimated study completion date: March 2018 |
| Contact information | Contact: Angeli Landeros, MD 203‐737‐4809. angeli.landeros@yale.edu |
| Notes | This study is currently recruiting participants. |
NCT02294305.
| Trial name or title | Vortioxetine versus placebo in major depressive disorder comorbid with social anxiety disorder |
| Methods | 12 week randomised, double‐blind, parallel, placebo‐controlled study |
| Participants | Estimated enrollment: 40 Ages eligible for study: 18‐70 years Genders eligible for study: both Accepts healthy volunteers: no Inclusion criteria: quote: "Male and female adults between 18 and 70 years of age (inclusive); subjects must give written informed consent prior to any study procedures; Diagnosis of Major Depressive Disorder (MDD), single episode (296.2) or recurrent (296.3), according to Diagnostic and Statistical Manual of Mental Disorders, version 5 (DSM‐5) criteria, as determined by psychiatric evaluation with the investigator and confirmed by the Mini‐International Neuropsychiatric Interview (MINI); duration of current major depressive episode must be at least 4 weeks; diagnosis of social anxiety disorder (SAD) (300.23 social phobia) according to DSM‐5 criteria, as determined by psychiatric evaluation with the investigator and confirmed by the MINI; duration of current SAD must be at least 6 months, and SAD should be observable in subjects' lives when they are not suffering from MDD, if such periods have occurred. Subjects must have a minimum total score of 60 on the Liebowitz Social Anxiety Scale (LSAS) at both screening and baseline visits; subjects must have a minimum total Montgomery Asberg Depression Rating Scale (MADRS) score of 20 at both screening and baseline visits; subjects must have a Clinical Global Inventory (CGI) Severity score of 4 or greater at both screening and baseline visits, where the CGI is based on a composite of MDD and SAD; male and female subjects of childbearing potential must commit to an effective form of contraception for the duration of the trial (screening/visit 1 through follow‐up/visit 10); effective forms of contraception include: condoms with spermicide, diaphragm with spermicide, hormonal contraceptive agents (oral, transdermal, or injectable), or implantable contraceptive devices; true abstinence will also be considered an effective form of contraception". Exclusion criteria: quote: "Subjects with any lifetime history of bipolar disorder, schizophrenia, schizoaffective disorder, obsessive compulsive disorder, eating disorders, or body dysmorphic disorder. Subjects with comorbid generalized anxiety disorder, dysthymia, or specific phobias can be included in the study provided that MDD and SAD are considered to be the primary clinical conditions in terms of need for treatment; subjects with substance abuse, panic disorder, or post‐traumatic stress disorder, in the past 6 months before screening; subjects who started psychotherapy for SAD or MDD or had electroconvulsive therapy (ECT) in the past 6 months before screening. Subjects who have been receiving psychotherapy or cognitive behavioral therapy for more than 24 weeks prior to the baseline visit are eligible provided that the therapy continues at the same frequency for the duration of the trial; subjects who are currently pregnant or lactating, or who are of childbearing potential and not able and willing to practice an effective method of contraception (as outlined in Inclusion criterion #10) for the duration of the trial (screening/visit 1 through follow‐up/visit 10; subjects who, in the opinion of the investigator, are at a clinically significant risk for suicide. This would include prominent suicidal ideation or suicidal behavior in the past 6 months before screening; systolic blood pressure ≥165 and/or diastolic blood pressure ≥95, as measured at screening and baseline visits; positive urine drug screen at the screening visit, unless due to prescribed medication; any current unstable and/or clinically significant medical condition, based on history or as evidenced in screening laboratory or electrocardiogram (ECG) assessments; subjects with a history or complication of cancer or malignant tumor not in remission for at least 5 years. Basal cell skin cancers are not exclusionary; subjects receiving fluoxetine within 28 days of the baseline visit; subjects receiving monoamine oxidase inhibitors (MAOIs) within 14 days of the baseline visit; subjects receiving any other psychotropic medication (including selective serotonin re‐uptake inhibitors (SSRIs), serotonin‐norepinephrine reuptake inhibitors (SNRIs), and benzodiazepines) within 14 days of the baseline visit. Zolpidem (Ambien) PRN is allowed for insomnia if not taken more than 3 times per week for the duration of the trial; treatment refractory SAD: subjects who have a history of two or more failed treatment trials with an FDA‐approved SAD treatment, each given for at least 6 weeks, during which the subject received an adequate dosage; treatment refractory MDD: subjects who have a history of two or more failed treatment trials with an FDA‐approved MDD treatment in the current episode" |
| Interventions | Experimental: vortioxetine Vortioxetine 10 to 20 mg orally 4 times daily for 12 weeks Placebo comparator: placebo Placeboorally 4 times daily for 12 weeks |
| Outcomes | Primary outcome measures: CGI‐I responder rate (time frame: 12 weeks) Secondary outcome measures: Change in total MADRS score (time frame: baseline and 12 weeks); change in total LSAS score (time frame: baseline and 12 weeks) |
| Starting date | Study start date: December 2014 Estimated primary completion date: September 2015 |
| Contact information | Contact: Ann E Draine 212 595‐5012 ext 222 ADraine@MedicalResearchNetwork.com Contact: Michael R. Liebowitz, MD 212 595‐5012 MLiebowitz@MedicalResearchNetwork.com |
| Notes | This study is not yet open for participant recruitment. |
NCT02432703.
| Trial name or title | A safety and efficacy study of JNJ‐42165279 in participants with social anxiety disorder |
| Methods | 12 week randomised, double‐blind, placebo‐controlled, parallel‐group, multicentre study |
| Participants | Estimated enrollment: 122 Ages eligible for study: 18‐64 years Genders eligible for study: both. Accepts healthy volunteers: no Inclusion criteria: quote: "must have a primary DSM‐5 diagnosis of Social anxiety disorder (SAD) except those with performance only as a specifier; participants with a diagnosis of comorbid generalized anxiety disorder (GAD) or major depressive disorder (MDD) may be included if the investigator considers SAD to be the predominant diagnosis; participants with current or lifetime history of attention deficit hyperactivity disorder (ADHD) and specific phobia may be included as well. Must have a Liebowitz Social Anxiety Scale score greater than or equal (≥) 70 at screening and baseline; participants with a current episode of MDD must have a HDRS17 total score less than or equal to (≤) 18. Must have a body mass index (BMI) between 18 and 35 kilogram per meter square (kg/m2), inclusive, at screening; female participants must be either postmenopausal or surgically sterile". Exclusion criteria: quote: "Participants who have performance only SAD are excluded. Participants with other current significant psychiatric condition(s) (Axis 1 under DSM‐IV), including, but not limited to, MDD with psychotic features (lifetime), bipolar disorder (including lifetime diagnosis), obsessive‐compulsive disorder, borderline personality disorder, eating disorder (e.g., bulimia, anorexia nervosa), autism spectrum disorders, post‐traumatic stress disorder (PTSD) or schizophrenia are excluded. Participants with a diagnosis of comorbid GAD or MDD may be included; participants currently receiving specific psychotherapy for SAD; has a history of more than two unsuccessful adequate pharmacological treatment trials for SAD, defined as lack of response to at least 10 weeks of treatment at adequate doses (e.g., paroxetine ≥ 40 milligram per day (mg/day) or its equivalent; or clonazepam ≥ 2.5 mg/day or its equivalent); concurrent use of psychotropic medications; has a history of or current thyroid disease, thyroid dysfunction and is currently untreated for it". |
| Interventions | Experimental: JNJ‐42165279. Participants will receive 25 milligram (mg) JNJ‐42165279 orally once daily from day 1 up to 12 weeks Placebo comparator: placebo. Participants will receive a matching placebo orally once daily from day 1 up to 12 weeks |
| Outcomes | Primary outcome measures: Change from baseline in the Liebowitz Social Anxiety Scale (LSAS) total score at week 12 Secondary outcome measures: Change from baseline in Liebowitz Social Anxiety Scale (LSAS) fear/anxiety and avoidance subscales at week 12 Number of participants who are responders and remitters on Liebowitz Social Anxiety Scale (LSAS) total score at week 12 Percentage of participants who are responders and remitters on Liebowitz Social Anxiety Scale (LSAS) total score at week 12 (time frame: baseline and week 12) (designated as safety issue: no) Change from baseline in structured interview guide for the Hamilton Anxiety Rating Scale (SIGH‐A) total score at week 12 Change from baseline in Hamilton Anxiety Rating scale (HAM‐A6) score at week 12 Change from baseline in Hamilton Depression Rating Scale (HDRS17) total score at week 12 Change from baseline in HDRS17 anxiety/somatisation factor total score at week 12 Change from baseline in 6‐Item Hamilton Depression Scale (HAM‐D6) score at week 12 Clinical Global Impression ‐ Improvement (CGI‐I) score from baseline at week 12 Number of participants who are responders on SIGH‐A total score at Week 12 Percentage of participants who are responders on SIGH‐A total score at week 12 |
| Starting date | Study start date: June 2015 Estimated study completion date: February 2017 |
| Contact information | Janssen Research & Development, LLC |
| Notes | This study has suspended participant recruitment. |
ADHD: attention deficit hyperactivity disorder; BAI: Beck Anxiety Inventory; BAT: Behavioural Assessment Test; BMI: body mass index; BPRS‐PS: Brief Psychiatric Rating Scale, Positive Symptom Subscale; CADSS: Clinician‐Administered Dissociative States Scales; CGIC/CGI‐I: Clinical Global Impressions Improvement Scale; CGI‐S: Clinical Global Impression of Severity of Illness; DSM: Diagnostic and Statistical Manual of Mental Disorders; DSM 5: Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition; DSM‐IV: Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition; DSM‐IV‐TR: Diagnostic and Statistical Manual of Mental Disorders; FDA: Food and Drug Administration (USA); GAD: Generalised Anxiety Disorder; GSP: Generalised Social Phobia; HAM‐A6: 6‐item Hamilton Anxiety Rating scale; HDRS17: Hamilton Rating Scale for Depression (17 items); LSAS: Liebowitz Social Anxiety Scale; MADRS: Montgomery Asberg Depression Rating Scale; MAOI: monoamine oxidase inhibitor; MDD: Major Depressive Disorder; MINI: Mini‐International Neuropsychiatric Interview; PANAS: positive and negative affect schedule; PO: Placebo; PRN: as needed; PTSD: post‐traumatic stress disorder; SAnD: Social Anxiety Disorder; SCID: structured clinical interview for DSM; SIGH‐A: Structured Interview Guide for the Hamilton Anxiety Rating Scale; SNRI: serotonin‐norepinephrine reuptake inhibitor; SPSS: Self‐Statement During Public Speaking Scale; SSRI: selective serotonin re‐uptake inhibitor; STAI: State‐Trait Anxiety Inventory.
Differences between protocol and review
We did not conduct a survival analysis to obtain time‐to‐event data or hazard ratios.
We included post hoc additional comparisons involving 5HT partial agonists, anticonvulsants with and without GABAs, antipsychotics, NARIs, NaSSAs, and SARIs.
We conducted a post hoc analysis for all medications for the treatment of SAnD, with the removal of three studies. We also conducted a post hoc analysis for the RIMA moclobemide given that brofaromine is no longer available.
We included mean change scores, where provided, for the LSAS outcome for symptom reduction, rather than the approach taken in the previous version of this review of conducting separate analyses of endpoint and change scores.
This review incorporates the GRADE approach with 'Summary of findings' tables.
We have moved treatment tolerability (i.e. dropouts due to side effects) to a primary outcome for this review. This is in keeping with recommendations from section 4.5 of the Cochrane Handbook for Systematic Reviews of Interventions that primary outcomes of a review should include negative as well as positive outcomes (Higgins 2011).
We added subgroup analyses (i.e. multicentre compared to single centre trials; generalised SAnD compared to inclusive SAnD; industry funding compared to no industry funding; and whether or not the sample included or excluded patients diagnosed with major depressive disorder (MDD)) and sensitivity analyses (worst case versus best case: support for robustness of evidence).
Contributions of authors
Dan Stein, Taryn Williams and Jonathan Ipser co‐ordinated the work on the update of this review. Jonathan Ipser and Taryn Williams compiled the updated version of the review, including rechecking all studies for eligibility and risk of bias, completing all GRADE tables, analysing the data, and updating the Abstract, Results, Discussion and Authors' conclusion sections of the review. Catherine Kariuki, Sean Tromp, and Coenraad Hattingh reviewed the final draft and made comments where relevant.
Sources of support
Internal sources
No sources of support supplied
External sources
MRC Research Unit on Anxiety and Stress Disorders, Cape Town, South Africa.
Declarations of interest
Dan Stein has received research grants and/or consultancy honoraria from AMBRF, Biocodex, Cipla, Lundbeck, National Responsible Gambling Foundation, Novartis, Servier, and Sun.
Taryn Williams: none known.
Jonathan Ipser: none known.
Coenraad J Hattingh: none known.
Catherine M Kariuki: none known.
Sean A Tromp: none known.
Anton J van Balkom: none known.
Edited (no change to conclusions), comment added to review
References
References to studies included in this review
Allgulander 1999 {published data only}
- Allgulander C. Efficacy of paroxetine in social phobia ‐ a single‐center double‐blind study of 96 symptomatic volunteers randomized to treatment with paroxetine 20‐50 mg or placebo for 3 months. 11th European College of Neuropsychopharmacology Congress; 1998 Oct 31‐Nov 4. Paris, France, 1998:P.3.005.
- Allgulander C. Paroxetine in social anxiety disorder: a randomised placebo‐controlled study. Acta Psychiatrica Scandinavica 1999;100(3):193‐8. [DOI] [PubMed] [Google Scholar]
Allgulander 2004 {published data only}
- Allgulander C, Mangano R, Zhang J, Dahl AA, Lepola U, Sjödin I, Emilien G. Efficacy of venlafaxine ER in patients with social anxiety disorder: a double‐blind, placebo‐controlled, parallel‐group comparison with paroxetine. Human Psychopharmacology 2004;19(6):387‐96. [DOI] [PubMed] [Google Scholar]
- Mangano R, Liebowitz MR, Allgulander C. Comparison of venlafaxine extended release and paroxetine in short‐term treatment of SAD. New Research Abstracts. 156th Annual Meeting of the American Psychiatric Association; 2003 May 17‐22. San Francisco (CA), 2003.
Asakura 2007 {published data only}
- Asakura S, Tajima O, Koyama T. Fluvoxamine treatment of generalized social anxiety disorder in Japan: a randomized, double‐blind, placebo‐controlled study. International Journal of Neuropsychopharmacology 2007;10(2):263‐74. [DOI] [PubMed] [Google Scholar]
Baldwin 1999 {published data only}
- 29060/502. Randomised, double‐blind study of paroxetine and placebo in the treatment of social phobia. www.gsk‐clinicalstudyregister.com/study/29060/502#rs (first received 30 September 2005).
- Baldwin D, Bobes J, Stein DJ, Scharwaechter I, Faure M. Paroxetine in social phobia/social anxiety disorder. Randomised, double‐blind, placebo‐controlled study. British Journal of Psychiatry 1999;175(2):120‐6. [DOI] [PubMed] [Google Scholar]
- Stein DJ, Berk M, Els C, Emsley RA, Gittelson L, Wilson D, et al. A double‐blind placebo‐controlled trial of paroxetine in the management of social phobia (social anxiety disorder) in South Africa. South African Medical Journal 1999;89(4):402‐6. [PubMed] [Google Scholar]
Barnett 2002 {published data only}
- Barnett SD, Kramer ML, Casat CD, Connor KM, Davidson JR. Efficacy of olanzapine in social anxiety disorder: a pilot study. Journal of Psychopharmacology 2002;16(4):365‐8. [DOI] [PubMed] [Google Scholar]
Blanco 2010 {published data only}
- Blanco C, Heimberg RG, Schneier FR, Fresco DM, Chen H, Turk CL, et al. A placebo‐controlled trial of phenelzine, cognitive behavioral group therapy, and their combination for social anxiety disorder. Archives of General Psychiatry 2010;67(3):286‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
Blomhoff 2001 {published data only}
- Blomhoff S, Haug TT, Hellstrom K, Holme I, Humble M, Madsbu HP, et al. Randomised controlled general practice trial of sertraline, exposure therapy and combined treatment in generalised social phobia. British Journal of Psychiatry 2001;179(1):23‐30. [DOI] [PubMed] [Google Scholar]
- Blomhoff S, Haug TT, Humble M, Hallstrom K, Madsbu HP, Wold JE, et al. Treatment of generalized social phobia. New Research Abstracts. 152nd Annual Meeting of the American Psychiatric Association; May 15‐20. Washington DC, 1999:NR650.
- Haug TT, Blomhoff S, Hellstrom K, Holme I, Humble M, Madsbu HP, et al. Exposure therapy and sertraline in social phobia: 1‐year follow‐up of a randomised controlled trial. British Journal of Psychiatry 2003;182(4):312‐8. [DOI] [PubMed] [Google Scholar]
- Walker JR. The benefits of exposure therapy alone may last longer than sertraline alone or sertraline plus exposure therapy in social phobia. Evidence‐based Mental Health 2003;6(3):90. [DOI] [PubMed] [Google Scholar]
Book 2008 {published data only}
- Book SW, Thomas SE, Randall PK, Randall CL. Paroxetine reduces social anxiety in individuals with a co‐occurring alcohol use disorder. Journal of Anxiety Disorders 2008;22(2):310‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas SE, Randall PK, Book SW, Randall CL. A complex relationship between co‐occurring social anxiety and alcohol use disorders: what effect does treating social anxiety have on drinking?. Alcoholism, Clinical and Experimental Research 2008;32(1):77‐84. [DOI] [PubMed] [Google Scholar]
Connor 1998 {published data only}
- Connor KM, Davidson JR, Potts NL, Tupler LA, Miner CM, Malik ML, et al. Discontinuation of clonazepam in the treatment of social phobia. Journal of Clinical Psychopharmacology 1998;18(5):373‐8. [DOI] [PubMed] [Google Scholar]
Davidson 1993 {published data only}
- Davidson JR, Potts N, Richichi E, Krishnan R, Ford SM, Smith R, et al. Treatment of social phobia with clonazepam and placebo. Journal of Clinical Psychopharmacology 1993;13(6):423‐8. [PubMed] [Google Scholar]
- Sutherland SM, Tupler LA, Colket JT, Davidson JR. A 2‐year follow‐up of social phobia. Status after a brief medication trial. Journal of Nervous and Mental Disease 1996;184(12):731‐8. [DOI] [PubMed] [Google Scholar]
Davidson 2004a {published data only}
- Davidson J, Yaryura‐Tobias J, DuPont R, Stallings L, Barbato LM, Hoop RG, et al. Fluvoxamine‐controlled release formulation for the treatment of generalized social anxiety disorder. Journal of Clinical Psychopharmacology 2004;24(2):118‐25. [DOI] [PubMed] [Google Scholar]
- Davidson JR, Hemby LW, Barbato L, Hoop RG. Fluvoxamine controlled release for the treatment of generalized social anxiety disorder. 39th Annual Meeting of the American College of Neuropsychopharmacology; Dec 10‐14;. San Juan, Puerto Rico, 2000:161.
Davidson 2004b {published data only}
- Aderka IM, McLean CP, Huppert JD, Davidson JRT, Foa EB. Fear, avoidance and physiological symptoms during cognitive‐behavioral therapy for social anxiety disorder. Behaviour Research and Therapy 2013;51(7):352‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson JRT, Foa EB, Huppert JD, Keefe FJ, Franklin ME, Compton JS, et al. Fluoxetine, comprehensive cognitive behavioral therapy, and placebo in generalized social phobia. Archives of General Psychiatry 2004;61(10):1005‐13. [DOI] [PubMed] [Google Scholar]
- Huppert JD, Schultz LT, Foa EB, Barlow DH, Davidson JR, Gorman JM, et al. Differential response to placebo among patients with social phobia, panic disorder, and obsessive‐compulsive disorder. American Journal of Psychiatry 2004;161(8):1485‐7. [DOI] [PubMed] [Google Scholar]
- Ledley DR, Huppert JD, Foa EB, Davidson JRT, Keefe FJ, Potts NLS. Impact of depressive symptoms on the treatment of generalized social anxiety disorder. Depression and Anxiety 2005;22(4):161‐7. [DOI] [PubMed] [Google Scholar]
Fahlen 1995 {published data only}
- Fahlen T, Humble M, Koczkas C, Nilsson H. Social phobia and its treatment with brofaromine, efficacy regarding social phobia symptoms and personality traits. Clinical Neuropharmacology 1992;15(1 Pt B):64. [Google Scholar]
- Fahlen T, Nilsson Hl, Borg K, Humble M, Pauli U. Social phobia: the clinical efficacy and tolerability of the monoamine oxidase‐A and serotonin uptake inhibitor brofaromine. A double‐blind placebo‐controlled study. Acta Psychiatrica Scandinavica 1995;92(5):351‐8. [DOI] [PubMed] [Google Scholar]
Feltner 2011 {published data only}
- Feltner D, Bielski R, Liu‐Dumaw M. Efficacy of pregabalin in generalised social anxiety disorder: results of a placebo‐controlled, fixed‐dose study. European Neuropsychopharmacology 2010;20(Suppl 3):S525–6. [DOI] [PubMed] [Google Scholar]
- Feltner DE, Liu‐Dumaw M, Schweizer E, Bielski R. Efficacy of pregabalin in generalized social anxiety disorder: results of a double‐blind, placebo‐controlled, fixed‐dose study. International Clinical Psychopharmacology 2011;26(4):213–20. [DOI] [PubMed] [Google Scholar]
Furmark 2005 {published data only}
- Furmark T, Appel L, Michelgard A, Wahlstedt K, Ahs F, Zancan S, et al. Cerebral blood flow changes after treatment of social phobia with the neurokinin‐1 antagonist GR205171, citalopram or placebo. Biological Psychiatry 2005;58(2):132‐42. [DOI] [PubMed] [Google Scholar]
Heimberg 1998 {published data only}
- Heimberg RG, Liebowitz MR, Hope DA, Schneier FR, Holt CS, Welkowitz LA, et al. Cognitive behavioural group therapy vs phenelzine therapy for social phobia: 12 week outcome. Archives of General Psychiatry 1998;55(12):1133‐41. [DOI] [PubMed] [Google Scholar]
Kasper 2005 {published data only}
- Kasper S, Loft H, Nil R. Escitalopram in the treatment of social anxiety disorder. XII World Congress of Psychiatry; Aug 24‐9. Yokohama, Japan, 2002.
- Kasper S, Loft H, Smith JR. Escitalopram is efficacious and well tolerated in the treatment of social anxiety disorder. New Research Abstracts. 155th Annual Meeting of the American Psychiatric Association; May 18‐23;. Philadelphia (PA), 2002.
- Kasper S, Stein DJ, Loft H, Nil, R. Escitalopram in the treatment of social anxiety disorder: randomised, placebo‐controlled, flexible‐dosage study. British Journal of Psychiatry 2005;186(3):222‐6. [DOI] [PubMed] [Google Scholar]
Katschnig 1997 {published data only}
- Katschnig K, Stein MB, Buller R. Moclobemide in social phobia. A double‐blind, placebo‐controlled clinical study. European Archives of Psychiatry and Clinical Neuroscience 1997;247(2):71‐80. [PubMed] [Google Scholar]
Katzelnick 1995 {published data only}
- Katzelnick DJ, Kobak KA, Greist JH, Jefferson JW, Mantle JM, Serlin RC. Sertraline for social phobia: a double‐blind, placebo‐controlled crossover study. American Journal of Psychiatry 1995;152(9):1368‐71. [DOI] [PubMed] [Google Scholar]
Kobak 2002 {published data only}
- Kobak KA, Greist JH, Jefferson JW, Katzelnick DJ. Fluoxetine in social phobia: a double‐blind, placebo‐controlled pilot study. Journal of Clinical Psychopharmacology 2002;22(3):257‐62. [DOI] [PubMed] [Google Scholar]
Lader 2004 {published data only}
- Lader M, Stender K, Burger V, Nil R. Efficacy and tolerability of escitalopram in 12‐ and 24‐week treatment of social anxiety disorder: randomised, double‐blind, placebo‐controlled, fixed dose study. Depression and Anxiety 2004;19:241‐8. [DOI] [PubMed] [Google Scholar]
Lepola 2004 {published data only}
- Lepola U, Bergtholdt B, Lambert JS, Davy KL, Ruggiero L. Controlled‐release paroxetine in the treatment of patients with social anxiety disorder. Journal of Clinical Psychiatry 2004;65:222‐9. [DOI] [PubMed] [Google Scholar]
Liebowitz 1992 {published data only}
- Liebowitz MR, Gorman JM, Fyer AJ, Campeas R, Levin AP, Sandberg D, et al. Pharmacotherapy of social phobia: an interim report of a placebo‐controlled comparison of phenelzine and atenolol. Journal of Clinical Psychiatry 1988;49(7):252‐7. [PubMed] [Google Scholar]
- Liebowitz MR, Schneier F, Campeas R, Gorman J, Fyer A, Hollander E, et al. Phenelzine and atenolol in social phobia. Psychopharmacology Bulletin 1990;26(1):123‐5. [PubMed] [Google Scholar]
- Liebowitz MR, Schneier F, Campeas R, Hollander E, Hatterer J, Fyer A, et al. Phenelzine vs atenolol in social phobia: a placebo‐controlled comparison. Archives of General Psychiatry 1992;49(4):290‐300. [DOI] [PubMed] [Google Scholar]
Liebowitz 2002 {published data only}
- Liebowitz MR, Stein MB, Tancer M, Carpenter D, Oakes R, Pitts CD. A randomized, double‐blind, fixed‐dose comparison of paroxetine and placebo in the treatment of generalized social anxiety disorder. Journal of Clinical Psychiatry 2002;63(1):66‐74. [DOI] [PubMed] [Google Scholar]
Liebowitz 2003 {published data only}
- Liebowitz MR, DeMartinis NA, Weihs KL, Londborg PD, Smith WT, Chung H, et al. Efficacy of sertraline in severe generalized social anxiety disorder: results of a double‐blind, placebo‐controlled study. Journal of Clinical Psychiatry 2003;64(7):785‐92. [DOI] [PubMed] [Google Scholar]
Liebowitz 2005a {published data only}
- Liebowitz MR, Mangano RM, Bradwejn J, Asnis G. A randomized controlled trial of venlafaxine extended release in generalized social anxiety disorder. Journal of Clinical Psychiatry 2005;66(2):238‐47. [DOI] [PubMed] [Google Scholar]
Liebowitz 2005b {published data only}
- Liebow3149018itz MR, Gelenberg AJ, Munjack D. Venlafaxine extended release vs placebo and paroxetine in social anxiety disorder. Archives of General Psychiatry 2005;62(2):190‐8. [DOI] [PubMed] [Google Scholar]
Lott 1997 {published data only}
- Lott M, Greist JH, Jefferson JW, Kobak KA, Katzelnick DJ, Katz RJ, Schaettle SC. Brofaromine for social phobia: a multicenter, placebo‐controlled, double‐blind study. Journal of Clinical Psychopharmacology 1997;17(4):255‐60. [DOI] [PubMed] [Google Scholar]
Moghadam 2015 {published data only}
- Moghadam MNM, Atef‐Vahid MK, Asgharnejad‐Farid AA, Shabani A, Lavasni F. Effectiveness of short‐term dynamic psychotherapy versus sertraline in treatment of social phobia. Iranian Journal of Psychiatry and Behavioral Sciences 2015;9(2):e228. [DOI] [PMC free article] [PubMed] [Google Scholar]
Muehlbacher 2005 {published data only}
- Muehlbacher M, Nickel MK, Nickel C, Kettler C, Lahmann C, Gil FP, et al. Mirtazapine treatment of social phobia in women. A randomized, double‐blind, placebo‐controlled study. Journal of Clinical Psychopharmacology 2005;25(6):580‐3. [DOI] [PubMed] [Google Scholar]
NCT00273039 {unpublished data only}
- NCT00273039. A Study Of A New Medicine (GW679769) For The Treatment Of Social Anxiety Disorder [A Randomized, Double‐Blind, Parallel Group, Placebo‐Controlled, Forced Dose Titration Study Evaluating the Efficacy and Safety of GW679769 and Paroxetine in Subjects With Social Anxiety Disorder]. clinicaltrials.gov/ct2/show/record/NCT00273039 (first received 5 January 2006).
NCT00318669 {unpublished data only}
- NCT00318669. Clinical Evaluation of BRL29060A (Paroxetine Hydrochloride Hydrate) in Social Phobia/Social Anxiety Disorder (SAD) ‐A Double‐blind, Placebo‐controlled Study‐ <Phase III Study> [Social Anxiety Disorder Study Of Paroxetine]. clinicaltrials.gov/ct2/show/NCT00318669 (first received 24 April 2006).
NCT00397722 {unpublished data only}
- NCT00397722. Study CRH103390: A 12 Week Flexible Dose Study of GW876008, Placebo and Active Control (Paroxetine) in the Treatment of Social Anxiety Disorder (SocAD) [Treatment of patients with social anxiety disorder]. clinicaltrials.gov/ct2/show/results/NCT00397722 (first received 8 November 2006).
NCT00403962 {unpublished data only}
- NCT00403962. A Randomised, Double‐Blind, Parallel‐Group, Placebo‐Controlled Fixed Dose Study Comparing the Efficacy and Safety of GW597599/Paroxetine Combination or Paroxetine Monotherapy to Placebo in Patients With Social Anxiety Disorder (SAD) [A Combination Therapy In Patients With Social Anxiety Disorder]. clinicaltrials.gov/ct2/show/NCT00403962 (first received 29 August 2005).
NCT00470483 {unpublished data only}
- NCT00470483. A Double‐Blind, Randomized, Placebo‐Controlled, Parallel Group, fMRI and PET Study Comparing Emotional Challenge‐induced Regional Cerebral Blood Flow Changes Before and After 8 Weeks of Treatment With Placebo and Paroxetine in Subjects With Social Anxiety Disorder [A Study To Compare Emotional Changes In Subjects With Social Anxiety Disorder]. clinicaltrials.gov/ct2/show/results/NCT00470483 (first received 3 May 2007).
Nordahl 2016 {published data only}
- Nordahl HM, Vogel PA, Morken G, Stiles TC, Sandvik P, et al. Paroxetine, cognitive therapy or their combination in the treatment of social anxiety disorder with and without avoidant personality disorder: a randomized clinical trial. Psychotherapy and Psychosomatics 2016;85(6):346–56. [DOI] [PubMed] [Google Scholar]
Noyes 1997 {published data only}
- Noyes R Jr, Moroz G, Davidson JR, Liebowitz MR, Davidson A, Siegel J, et al. Moclobemide in social phobia: a controlled dose response trial. Journal of Clinical Psychopharmacology 1997;17(4):247‐54. [DOI] [PubMed] [Google Scholar]
Oosterbaan 2001 {published data only}
- Oosterbaan DB, Balkom AJLM, Spinhoven P, Oppen P, Dyck R. Cognitive therapy versus moclobemide in social phobia: a controlled study. Clinical Pscyhology and Psychotherapy 2001;8(4):263‐73. [Google Scholar]
Pande 1999 {published data only}
- Pande AC, Davidson JR, Jefferson JW, Janney CA, Katzelnick DJ, Weisler RH, Greist JH, Sutherland SM. Treatment of social phobia with gabapentin: a placebo‐controlled study. Journal of Clinical Psychopharmacology 1999;19(4):341‐8. [DOI] [PubMed] [Google Scholar]
Pande 2004 {published data only}
- Feltner DE, Davidson JR, Pollack MH, Stein MB, Futterer R, Jefferson JW, et al. A placebo‐controlled, double‐blind study of pregabalin treatment of social anxiety disorder: outcome and predictors of response. 39th Annual Meeting of the American College of Neuropsychopharmacology; Dec 10‐14. San Juan, Puerto Rico, 2000.
- Feltner DE, Pollack MH, Davidson JR, Stein MB, Futterer R, Jefferson JW, et al. A placebo‐controlled study of pregabalin in social phobia (social anxiety disorder) [abstract]. International Journal of Neuropsychopharmacology 2000;3(Suppl 1):284. [Google Scholar]
- Feltner DE, Pollack MH, Davidson JR, Stein MB, Futterer RA, Jefferson JW, et al. Pregabalin treatment of social phobia. New Research Abstracts. 153rd Annual Meeting of the American Psychiatric Association; May 13‐18. Chicago (IL), 2000.
- Feltner DE, Pollack MH, Davidson JRT, Stein MB, Futterer R, Jefferson JW, et al. A placebo‐controlled, double‐blind study of pregabalin treatment of social phobia: outcome and predictors of response [abstract]. European Neuropsychopharmacology 2000;10(Suppl 3):S345. [Google Scholar]
- Pande AC, Feltner DE, Jefferson JW, Davidson JRT, Pollack M, Stein MB, et al. Efficacy of the novel anxiolytic pregabalin in social anxiety disorder: a placebo‐controlled, multicenter study. Journal of Clinical Psychopharmacology 2004;24(2):141‐9. [DOI] [PubMed] [Google Scholar]
Randall 2001 {published data only}
- Randall CL, Johnson MR, Thevos AK, Sonne SC, Thomas SE, Willard SL, et al. Paroxetine for social anxiety and alcohol use in dual‐diagnosed patients. Depression & Anxiety 2001;14(4):255‐62. [DOI] [PubMed] [Google Scholar]
- Randall CL, Johnson MR, Thevos AK, Sonne SC, Willard SL, Thomas SE, et al. Paroxetine improves both social anxiety and alcohol use in dual‐diagnosed patients. 39th Annual Meeting of the American College of Neuropsychopharmacology; 2000 Dec 10‐14; San Juan, Puerto Rico. 2000.
Ravindran 2009 {published data only}
- Ravindran LN, Kim DS, Letamendi AM, Stein MB. A randomized controlled trial of atomoxetine in generalized social anxiety disorder. Journal of Clinical Psychopharmacology 2009;29(6):561‐4. [DOI] [PubMed] [Google Scholar]
Rickels 2004 {published data only}
- Rickels K, Mangano R, Khan A. A double‐blind, placebo‐controlled study of a flexible dose of venlafaxine ER in adult outpatients with generalized social anxiety disorder. Journal of Clinical Psychopharmacology 2004;24(5):488‐96. [DOI] [PubMed] [Google Scholar]
Schneier 1998 {published data only}
- Schneier FR, Goetz D, Campeas R, Fallon B, Marshall R, Liebowitz MR. Placebo‐controlled trial of moclobemide in social phobia. British Journal of Psychiatry 1998;172:70‐7. [DOI] [PubMed] [Google Scholar]
Schutters 2010 {published data only}
- Schutters SIJ, Megen HJGM, Veen JF, Denys DAJP, Westenberg HGM. Mirtazapine in generalized social anxiety disorder: a randomized, double‐blind, placebo‐controlled study. International Clinical Psychopharmacology 2010;25(5):302–4. [DOI] [PubMed] [Google Scholar]
Stein 1996 {published data only}
- Stein MB, Chartier MJ, Hazen AL, Kroft CD, Chale RA, Coté D, et al. Paroxetine in the treatment of generalized social phobia: open‐label treatment and double‐blind placebo‐controlled discontinuation. Journal of Clinical Psychopharmacology 1996;16(3):218‐22. [DOI] [PubMed] [Google Scholar]
Stein 1998 {published data only}
- Stein MB, Liebowitz MR, Lydiard RB, Pitts CD, Bushnell W, Gergel I. Paroxetine treatment of generalized social phobia (social anxiety disorder): a randomized controlled trial. JAMA 1998;280(8):708‐13. [DOI] [PubMed] [Google Scholar]
Stein 1999 {published data only}
- Stein MB, Fyer AJ, Davidson JR, Pollack MH, Wiita B. Fluvoxamine treatment of social phobia (social anxiety disorder): a double‐blind, placebo‐controlled study. American Journal of Psychiatry 1999;156(5):756‐60. [DOI] [PubMed] [Google Scholar]
Stein 2002a {published data only}
- Stein DJ, Cameron A, Amrein R, Montgomery SA. Moclobemide is effective and well tolerated in the long‐term pharmacotherapy of social anxiety disorder with or without comorbid anxiety disorder. International Clinical Psychopharmacology 2002;17(4):161‐70. [DOI] [PubMed] [Google Scholar]
Stein 2002b {published data only}
- 29060/595. A study of the maintained efficacy and safety of paroxetine versus placebo in the long‐term treatment of social phobia. www.gsk‐clinicalstudyregister.com/study/29060/595#rs (first received 28 September 2008).
- Stein DJ, Versiani M, Hair T, Kumar R. Efficacy of paroxetine for relapse prevention in social anxiety disorder: a 24‐week study. Archives of General Psychiatry 2002;59(12):1111‐8. [DOI] [PubMed] [Google Scholar]
Stein 2003 {published data only}
- Stein DJ, Westenberg HGM, Yang H, Li D, Barbato LM. Fluvoxamine CR in the long‐term treatment of social anxiety disorder: the 12‐ to 24‐week extension phase of a multicentre, randomized, placebo‐controlled trial. International Journal of Neuropsychopharmacology 2003;6(4):317‐23. [DOI] [PubMed] [Google Scholar]
Stein 2005 {published data only}
- Stein MB, Pollack MH, Bystritsky A, Kelsey JE, Mangano RM. Efficacy of low and higher dose extended‐release venlafaxine in generalized social anxiety disorder: a 6‐month randomized controlled trial. Psychopharmacology 2005;177(3):280‐8. [DOI] [PubMed] [Google Scholar]
- Stein MB, Pollack MH, Mangano R. Long‐term treatment of generalized SAD with venlafaxine extended release. 156th Annual Meeting of the American Psychiatric Association; May 17‐22. San Francisco (CA), 2003:NR251.
Stein 2010 {published data only}
- Stein MB, Ravindran LN, Simon NM, Liebowitz MR, Khan A, Brawman‐Mintzer O, et al. Levetiracetam in generalized social anxiety disorder: a double‐blind, randomized controlled trial. Journal of Clinical Psychiatry 2010;71(5):627‐31. [DOI] [PubMed] [Google Scholar]
Tauscher 2010 {published data only}
- Tauscher J, Kielbasa W, Iyengar S, Vandenhende F, Peng X, Mozley D, et al. Development of the 2nd generation neurokinin‐1 receptor antagonist LY686017 for social anxiety disorder. European Neuropsychopharmacology 2010;20(2):80‐7. [DOI] [PubMed] [Google Scholar]
Turner 1994 {published data only}
- Turner S, Beidel DC, Jacob RG. Social phobia: a comparison of behaviour therapy and atenolol. Journal of Consulting and Clinical Psychology 1994;62(2):350‐8. [DOI] [PubMed] [Google Scholar]
Vaishnavi 2007 {published data only}
- Vaishnavi S, Alamy S, Zhang W, Connor KM, Davidson JRT. Quetiapine as monotherapy for social anxiety disorder: a placebo‐controlled study. Progress in Neuro‐Psychopharmacology and Biological Psychiatry 2007;31(7):1464‐9. [DOI] [PubMed] [Google Scholar]
Van Ameringen 2001a {published data only}
- Lane R, Walker J, Swinson R, Ameringen M. Sertraline in generalised social phobia. 11th World Congress of Psychiatry; Aug 6‐11. Hamburg, Germany, 1999.
- Pieters G. Sertraline was effective and well tolerated for generalised social phobia. Evidence‐based Mental Health 2001;4(3):91. [Google Scholar]
- Ameringen M, Oakman JM, Farvolden PG, Mancini C, Walker JR, Lane RM. Predictors of sertraline response in social phobia. 39th Annual Meeting of the American College of Neuropsychopharmacology; Dec 10‐14. San Juan, Puerto Rico, 2000:163.
- Ameringen MA, Lane RM, Walker JR, Bowen RC, Chokka PR, Goldner EM, et al. Sertraline treatment of generalised social phobia: a 20‐week, double‐blind, placebo‐controlled study. American Journal of Psychiatry 2001;158(2):275‐81. [DOI] [PubMed] [Google Scholar]
- Ameringen MA, Swinson R, Walker JR, Lane RM. A placebo‐controlled study of sertraline in generalized social phobia [abstract]. European Neuropsychopharmacology 2000;10(Suppl 3):S335. [Google Scholar]
Van Ameringen 2007 {published data only}
- Ameringen M, Mancini C, Oakman J, Walker J, Kjernisted K, Chokka P, et al. Nefazodone in the treatment of generalized social phobia: a randomized, placebo‐controlled trial. Journal of Clinical Psychiatry 2007;68(2):288‐95. [DOI] [PubMed] [Google Scholar]
- Ameringen MA, Mancini CL, Pipe B, Oakman J, Walker JR, Kjernisted KD, et al. Not all serotonergic agents are created equal in social phobia: nefazodone, a placebo‐controlled trial. 156th Annual Meeting of the American Psychiatric Association; May 17‐22. San Francisco (CA), 2003:NR789.
Van Vliet 1992 {published data only}
- Vliet IM, Boer JA, Westenberg HG. Psychopharmacological treatment of social phobia: clinical and biochemical effects of brofaromine, a selective MAO‐A inhibitor. European Neuropsychopharmacology 1992;2(1):21‐9. [DOI] [PubMed] [Google Scholar]
Van Vliet 1994 {published data only}
- Vliet IM, Boer JA, Westenberg HG. Psychopharmacological treatment of social phobia; a double blind placebo controlled study with fluvoxamine. Psychopharmacology 1994;115(1‐2):128‐34. [DOI] [PubMed] [Google Scholar]
Van Vliet 1997 {published data only}
- Vliet I, Boer J, Westenberg H, Ho Pian K. Clinical effects of buspirone in social phobia, a double‐blind placebo controlled study. 9th European College of Neuropsychopharmacology Congress; Sept 21‐25. Amsterdam, Netherlands, 1996.
- Vliet IM, Boer JA, Westenberg HG, Pian KL. Clinical effects of buspirone in social phobia: a double‐blind placebo‐controlled study. Journal of Clinical Psychiatry 1997;58(4):164‐8. [DOI] [PubMed] [Google Scholar]
Versiani 1992 {published data only}
- Versiani M, Nardi AE, Mundim FD, Alves AB, Liebowitz MR, Amrein R. Pharmacotherapy of social phobia. A controlled study with moclobemide and phenelzine. British Journal of Psychiatry 1992;161:353‐60. [DOI] [PubMed] [Google Scholar]
Versiani 1997 {published data only}
- Versiani M, Nardi AE, Figueira I, Mendlowicz M, Marques C. Double‐blind placebo controlled trial with bromazepam in social phobia. Jornal Brasileiro de Psiquiatria 1997;46(3):167‐71. [Google Scholar]
Walker 2000 {published data only}
- Walker JR, Ameringen MA, Swinson R, Bowen RC, Chokka PR, Goldner E, et al. Prevention of relapse in generalized social phobia: results of a 24‐week study in responders to 20 weeks of sertraline treatment. Journal of Clinical Psychopharmacology 2000;20(6):636‐44. [DOI] [PubMed] [Google Scholar]
- Walker JR, Ameringen MA, Swinson RP, Lane RM. A 24‐week prevention of relapse of generalized social phobia study in responders to 20 weeks of sertraline treatment. 153rd Annual Meeting of the American Psychiatric Association; May 13‐18. Chicago (IL), 2000:NR259.
Westenberg 2004 {published data only}
- Westenberg HGM, Stein DJ, Yang H, Li D, Barbato LM. A double‐blind placebo‐controlled study of controlled release fluvoxamine for the treatment of generalized social anxiety disorder. Journal of Clinical Psychopharmacology 2004;24(1):49‐55. [DOI] [PubMed] [Google Scholar]
Zhang 2005 {published data only}
- Zhang W, Connor KM, Davidson JRT. Levetiracetam in social phobia: a placebo controlled pilot study. Journal of Psychopharmacology 2005;19(5):551‐3. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
ACTRN12608000363381 {published data only}
- ACTRN12608000363381. Dopaminergic challenges in social anxiety disorder: evidence for dopamine D3 desensitisation following successful treatment with serotonergic antidepressants [Dopaminergic challenges in social anxiety disorder: evidence for dopamine D3 desensitisation following successful treatment with serotonergic antidepressants]. apps.who.int/trialsearch/Trial2.aspx?TrialID=ACTRN12608000363381 (first received 9 July 2008).
ACTRN12609000091202 {published data only}
- ACTRN12609000091202. The use of tiagabine and gabapentin in the treatment of social anxiety disorder in 8 patients [Gabapentin and tiagabine for treatment of social anxiety disorder in 8 patients in a random double blind crossover study with outcome measured as changes in the Leibowitz Social Anxiety Scale]. apps.who.int/trialsearch/Trial2.aspx?TrialID=ACTRN12609000091202 (first received 18 December 2008).
Allsopp 1984 {published data only}
- Allsopp LF, Cooper GL, Poole PH. Clomipramine and diazepam in the treatment of agoraphobia and social phobia in general practice. Current Medical Research and Opinion 1984;9(1):64‐70. [DOI] [PubMed] [Google Scholar]
Angelini 1989 {published data only}
- Angelini G, Bichisao E, Catapano F, Cerreta A, Ferini Strambi L, Maj M, et al. Ketazolam, a new long‐acting benzodiazepine, in the treatment of anxious patients. A multicenter study of 2,056 patients. Current Therapeutic Research, Clinical and Experimental 1989;45(2):294‐304. [Google Scholar]
Atmaca 2002 {published data only}
- Atmaca M, Kuloglu M, Tezcan E, Unal A. Efficacy of citalopram and moclobemide in patients with social phobia: some preliminary findings. Human Psychopharmacology 2002;2002(8):401‐5. [DOI] [PubMed] [Google Scholar]
Blank 2006 {published data only}
- Blank S, Lenze EJ, Mulsant BH, Dew MA, Karp JF, Shear MK, et al. Outcomes of late‐life anxiety disorders during 32 weeks of citalopram treatment. Journal of Clinical Psychiatry 2006;67(3):468‐72. [DOI] [PubMed] [Google Scholar]
Brantigan 1982 {published data only}
- Brantigan CO, Brantigan TA, Joseph N. Effects of beta‐blockade and beta‐stimulation on stage fright. American Journal of Medicine 1982;72(1):88‐94. [DOI] [PubMed] [Google Scholar]
Bystritsky 2005 {published data only}
- Bystritsky A, Kerwin L, Eiduson S, Vapnik T. A pilot controlled trial of bupropion vs. escitalopram in generalized anxiety disorder (GAD). Neuropsychopharmacology 2005;30 Suppl(1):S101. [PubMed] [Google Scholar]
Clark 2003 {published data only}
- Clark DM, Ehlers A, McManus F, Hackmann A, Fennell M, Campbell H, et al. Cognitive therapy versus fluoxetine in generalized social phobia: a randomized placebo‐controlled trial. Journal of Consulting and Clinical Psychology 2003;71(6):1058‐67. [DOI] [PubMed] [Google Scholar]
Clark‐Elford 2015 {published data only}
- Clark‐Elford R, Nathan PJ, Auyeung B, Mogg K, Bradley, BP, Sule A, et al. Effects of oxytocin on attention to emotional faces in healthy volunteers and highly socially anxious males. International Journal of Neuropsychopharmacology 2014;18(2):1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Coupland 2000 {published data only}
- Coupland NJ, Bell C, Potokar JP, Dorkins E, Nutt DJ. Flumazenil challenge in social phobia. Depression and Anxiety 2000;11(1):27‐30. [DOI] [PubMed] [Google Scholar]
Dempsey 2009 {published data only}
- Dempsey JP, Randall PK, Thomas SE, Book SW, Carrigan MH. Treatment of social anxiety with paroxetine: mediation of changes in anxiety and depression symptoms. Comprehensive Psychiatry 2009;50(2):135‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
Dodhia 2014 {published data only}
- Dodhia S, Hosanagar A, Fitzgerald DA, Labuschagne I, Wood AG, Nathan PJ, et al. Modulation of resting‐state amygdala‐frontal functional connectivity by oxytocin in generalized social anxiety disorder. Neuropsychopharmacology 2014;39(9):2061‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Donahue 2009 {published data only}
- Donahue CB, Kushner MG, Thuras PD, Murphy TG, Demark JB, Adson DE. Effect of quetiapine vs. placebo on response to two virtual public speaking exposures in individuals with social phobia. Journal of Anxiety Disorders 2009;23(3):362‐8. [DOI] [PubMed] [Google Scholar]
Dunlop 2007 {published data only}
- Dunlop BW, Papp L, Garlow SJ, Weiss PS, Knight BT, Ninan PT. Tiagabine for social anxiety disorder. Human Psychopharmacology: Clinical and Experimental 2007;22(4):241‐4. [DOI] [PubMed] [Google Scholar]
EUCTR2004‐001894‐24‐DE {unpublished data only}
- EUCTR2004‐001894‐24‐DE. A randomised, double‐blind, parallel‐group, placebo‐controlled fixed dose study comparing the efficacy and safety of GW597599/paroxetine combination or paroxetine monotherapy to placebo in patients with social anxiety disorder (SAD). apps.who.int/trialsearch/Trial2.aspx?TrialID=EUCTR2004‐001894‐24‐DE 2012.
Falloon 1981 {published data only}
- Falloon IR, Llody GG, Harpin RE. The treatment of social phobia: real‐life rehearsal with nonprofessional therapists. Journal of Nervous and Mental Disease 1981;169(3):180‐4. [DOI] [PubMed] [Google Scholar]
Fang 2014 {published data only}
- Fang A, Hoge EA, Heinrichs M, Hofmann SG. Attachment style moderates the effects of oxytocin on social behaviors and cognitions during social rejection: applying a research domain criteria framework to social anxiety. Clinical Psychological Science 2014;2(6):740‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Faria 2014 {published data only}
- Faria V, Ahs F, Appel L, Linnman C, Bani M, Bettica P, et al. Amygdala‐frontal couplings characterizing SSRI and placebo response in social anxiety disorder. International Journal of Neuropsychopharmacology 2014;17(8):1149‐57. [DOI] [PubMed] [Google Scholar]
Feifel 2011 {published data only}
- Feifel D, Macdonald K, McKinney R, Heissere N, Serrano V. A randomized, placebo controlled investigation of intranasal oxytocin in patients with anxiety. Neuropsychopharmacology 2011;36:S421. [Google Scholar]
Gale 2007 {published data only}
- Gale C. Escitalopram 10 mg daily is more effective than paroxetine and placebo for generalised anxiety disorder. Evidence Based Mental Health 2007;10(2):45. [DOI] [PubMed] [Google Scholar]
Gates 1985 {published data only}
- Gates GA, Saegert J, Wilson N, Johnson L, Shepherd A, Hearne EM 3rd. Effect of beta‐blockage on singing performance. Annals of Otology, Rhinology and Laryngology 1985;94(6 Pt 1):570‐4. [DOI] [PubMed] [Google Scholar]
Gelernter 1991 {published data only}
- Gelernter CS, Uhde TW, Cimbolic P, Arnkoff DB, Vittone BJ, Tancer ME, et al. Cognitive‐behavioural and pharmacological treatments of social phobia: a controlled study. Archives of General Psychiatry 1991;48(10):938‐45. [DOI] [PubMed] [Google Scholar]
Gorka 2015 {published data only}
- Gorka SM, Fitzgerald DA, Labuschagne I, Hosanagar A, Wood AG, Nathan PJ, et al. Oxytocin modulation of amygdala functional connectivity to fearful faces in generalized social anxiety disorder. Neuropsychopharmacology 2015;40(2):278‐86. [] [DOI] [PMC free article] [PubMed] [Google Scholar]
Greenhill 1999 {published data only}
- Greenhill LL. A multisite treatment of anxiety disorders. 152nd Annual Meeting of the American Psychiatric Association; May 15‐20;. Washington DC, 1999.
Grosser 2012 {published data only}
- Grosser B, Monti‐Bloch L, Jennings‐White C. Rapid anti‐anxiety effects in women of picogram quantities of a 19‐carbon steroid [abstract]. International Journal of Neuropsychopharmacology 2012:230‐1. [Google Scholar]
Guastella 2009 {published data only}
- Guastella AJ, Howard AL, Dadds MR, Mitchell P, Carson DS. A randomized controlled trial of intranasal oxytocin as an adjunct to exposure therapy for social anxiety disorder. Psychoneuroendocrinology 2009;34(6):917‐23. [DOI] [PubMed] [Google Scholar]
Hartley 1983 {published data only}
- Hartley LR, Ungapen S, Davie I, Spencer DJ. The effect of beta adrenergic blockade on speaker's performance and memory. British Journal of Psychiatry 1983;142(5):512‐7. [DOI] [PubMed] [Google Scholar]
Haug 2003 {published data only}
- Haug TT, Blomhoff S, Hellstrom K, Holme I, Humble M, Madsbu HP, et al. Exposure therapy and sertraline in social phobia: 1‐year follow up of a randomised controlled trial. British Journal of Psychiatry 2003;182:312‐8. [DOI] [PubMed] [Google Scholar]
Heun 2013 {published data only}
- Heun R, Corral RM, Ahokas A, Nicolini H, Teixeira JM, Dehelean P. Efficacy of agomelatine in more anxious elderly depressed patients. A randomized, double‐blind study vs placebo. European Psychiatry 2013;28(Suppl 1):1. [Google Scholar]
Hofmann 2006 {published data only}
- Hofmann SG, Meuret AE, Smits JAJ, Simon NM, Pollack MH, Eisenmenger K, et al. Augmentation of exposure therapy with D‐cycloserine for social anxiety disorder. Archives of General Psychiatry 2006;63(3):298‐304. [DOI] [PubMed] [Google Scholar]
Ionescu 2013 {published data only}
- Ionescu D. Clinical study on the efficacy and tolerability of escitalopram in patients diagnosed with social anxiety [abstract]. European Psychiatry 2013;28(1):1. [Google Scholar]
James 1977 {published data only}
- James IM, Griffith DN, Pearson RM, Newbury P. Effect of oxprenolol on stage‐fright in musicians. Lancet 1977;2(8045):952‐4. [DOI] [PubMed] [Google Scholar]
James 1983 {published data only}
- James IM, Burgoyne W, Savage IT. Effect on pindolol on stress‐related disturbances of musical performance: preliminary communication. Journal of the Royal Society of Medicine 1983;76(3):194‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
James 1984 {published data only}
- James I, Savage I. Beneficial effect of nadolol on anxiety‐induced disturbances of performance in musicians: a comparison with diazepam and placebo. American Heart Journal 1984;108(4 Pt 2):1150‐5. [DOI] [PubMed] [Google Scholar]
Krishman 1976 {published data only}
- Krishman G. Oxprenolol in the treatment of examination stress. Current Medical Research and Opinion 1976;4(3):241‐3. [DOI] [PubMed] [Google Scholar]
Liappas 2003 {published data only}
- Liappas J, Paparrigopoulos T, Tzavellas E, Christodoulou G. Alcohol detoxification and social anxiety symptoms: a preliminary study of the impact of mirtazepine administration. Journal of Affective Disorders 2003;76(1‐3):279‐84. [DOI] [PubMed] [Google Scholar]
Liden 1974 {published data only}
- Liden S, Gottfries CG. Beta‐blocking agents in the treatment of catecholamine‐induced symptoms in musicians. Lancet 1974;2(7879):529. [DOI] [PubMed] [Google Scholar]
Liebowitz 1999 {published data only}
- Liebowitz MR, Heimberg RG, Schneier FR, Hope DA, Davies S, Holt CS, et al. Cognitive‐behavioral group therapy versus phenelzine in social phobia: long‐term outcome. Depression and Anxiety 1999;10(3):89‐98. [PubMed] [Google Scholar]
Liebowitz 2014 {published data only}
- Liebowitz MR, Salman E, Nicolini H, Rosenthal N, Hanover R, Monti L. Effect of an acute intranasal aerosol dose of PH94B on social and performance anxiety in women with social anxiety disorder. American Journal of Psychiatry 2014;171(6):675‐82. [DOI] [PubMed] [Google Scholar]
Malcolm 1992 {published data only}
- Malcolm R, Anton RF, Randall CL, Johnston A, Brady K, Thevos A. A placebo‐controlled trial of buspirone in anxious inpatient alcoholics. Alcoholism Clinical and Experimental Research 1992;16(6):1007‐13. [DOI] [PubMed] [Google Scholar]
Mangano 2003 {published data only}
- Mangano R, Liebowitz MR, Allgulander C. Comparison of venlafaxine extended release and paroxetine in short‐term treatment of SAD. 156th Annual Meeting of the American Psychiatric Association, May 17‐22. San Francisco (CA), 2003.
Mortberg 2007 {published data only}
- Mortberg E, Clark DM, Sundin O, Aberg Wistedt A. Intensive group cognitive treatment and individual cognitive therapy vs treatment as usual in social phobia: a randomized controlled trial. Acta Psychiatrica Scandinavica 2007;115(2):142‐54. [DOI] [PubMed] [Google Scholar]
Mountjoy 1977 {published data only}
- Mountjoy CQ, Roth M, Garside RF, Leitch IM. A clinical trial of phenelzine in anxiety depressive and phobic neuroses. British Journal of Psychiatry 1977;131:486‐92. [DOI] [PubMed] [Google Scholar]
NCT00118833 {published data only}
- NCT00118833. St. John's Wort for the Treatment of Generalized Social Anxiety Disorder (GSAD) [Placebo‐Controlled Trial of Hypericum Perforatum in the Treatment of Generalized Social Anxiety Disorder (GSAD)]. clinicaltrials.gov/ct2/show/NCT00118833 (first received 7 July 2005).
NCT00248612 {published data only}
- NCT00248612. Psychosocial and Medication Treatment for Anxiety in Alcoholism. clinicaltrials.gov/ct2/show/results/NCT00248612 (first received 2 November 2005).
NCT00308724 {published data only}
- NCT00308724. Treating Late‐Life Generalized Anxiety Disorder (GAD) in Primary Care. clinicaltrials.gov/ct2/show/NCT00308724 (first received 28 March 2006).
NCT00332046 {published data only}
- NCT00332046. fMRI Study Comparing BOLD Activation Patterns Using GW679769 In Subjects With Social Anxiety Disorder [A Double‐Blind, Randomized, Placebo‐Controlled, Double‐Dummy, Parallel Group, fMRI Study Comparing BOLD Activation Patterns Before and After 12 Weeks of Treatment With Placebo, Comparator and GW679769 in Subjects With Social Anxiety Disorder (SAD)]. clinicaltrials.gov/ct2/show/NCT00332046 (first received 30 May 2006).
NCT00343707 {published data only}
- NCT00343707. PET (Positron Emission Tomography)/Public Speaking Study With A Combination Of 2 Medications In Social Anxiety Patients [A Double‐blind, Triple Dummy, Placebo‐controlled, Randomised, Parallel Group Positron Emission Tomography Study to Investigate the Effects of a 8 Week Administration of GW597599 and Paroxetine Either Alone or in Combination on Regional Cerebral Blood Flow During a Public Speaking Test in Subjects Affected by Social Phobia]. clinicaltrials.gov/ct2/show/NCT00343707 (first received 21 June 2006).
Neftel 1982 {published data only}
- Neftel KA, Adler RH, Kappeli L, Rossi M, Dolder M, Kaser HE, et al. Stage fright in musicians: a model illustrating the effect of beta blockers. Psychosomatic Medicine 1982;44(5):461‐9. [DOI] [PubMed] [Google Scholar]
Oosterbaan 1997 {published data only}
- Oosterbaan D, Balkom A, Spinhoven, Dyk R. Cognitive behavior therapy versus moclobemide in social phobia. The World Psychiatric Association's (WPA) Thematic Congress; Nov. Jerusalem, 1997.
Otto 2000 {published data only}
- Otto MW, Pollack MH, Gould RA, Worthington JJ, McArdle ET, Rosenbaum JF. A comparison of the efficacy of clonazepam and cognitive‐behavioural group therapy for the treatment of social phobia. Journal of Anxiety Disorders 2000;14(4):345‐8. [DOI] [PubMed] [Google Scholar]
Pecknold 1982 {published data only}
- Pecknold JC, McClure DJ, Appeltauer L, Allan T, Wrzesinski L. Does tryptophan potentiate clomipramine in the treatment of agoraphobic and agoraphobic and social phobic patients. British Journal of Psychiatry 1982;140:484‐90. [DOI] [PubMed] [Google Scholar]
Phan 2015 {published data only}
- Phan KL. Oxytocin modulation of amygdala‐frontal reactivity and connectivity to threat and at rest in social anxiety disorder [abstract]. Biological Psychiatry 2015;75(9 Suppl 1):27S. [Google Scholar]
Pine 2001 {published data only}
- Pine DS, Walkup JT, Labellarte MJ, Riddle MA, Greenhill L, Klein R, et al. Research Unit on Pediatric Psychopharmacology Anxiety Study Group. Fluvoxamine for the treatment of anxiety disorders in children and adolescents. New England Journal of Medicine 2001;344(17):1279‐85. [DOI] [PubMed] [Google Scholar]
Prasko 2004 {published data only}
- Prasko J. Moclobemide and/or cognitive‐behavior therapy in the treatment of social phobia. 157th Annual Meeting of the American Psychiatric Association; May 1‐6. New York (NY), 2004.
Ravindran 2014 {published data only}
- Ravindran AV, Abraham G, Johnson S, Chandrasena R. Randomized, placebo‐controlled effectiveness study of quetiapine XR in co‐morbid depressive and anxiety disorders [abstract]. International Journal of Neuropsychopharmacology 2014;46(1):91. [Google Scholar]
Rickels 1978 {published data only}
- Rickels K, Case WG, Chung H. Diazepam and halazepam in anxiety: some prognostic indicators. International Pharmacopsychiatry 1978;13(2):118‐25. [DOI] [PubMed] [Google Scholar]
Rynn 2008 {published data only}
- Rynn M, Russell J, Erickson J, Detke MJ, Ball S, Dinkel J, et al. Efficacy and safety of duloxetine in the treatment of generalized anxiety disorder: a flexible‐dose, progressive‐titration, placebo‐controlled trial. Depression and Anxiety 2008;25(3):182‐9. [DOI] [PubMed] [Google Scholar]
Schuurmans 2004 {published data only}
- Schuurmans J, Comijs HC, Emmelkamp PM, Dyck R. A randomized controlled trial of the effectiveness of cognitive‐behavioral therapy and sertraline versus a waiting list control group for anxiety disorders in older adults. 32nd Congress of the British Association for Behavioural and Cognitive Psychotherapies (jointly with the European Association of Behavioural and Cognitive Therapies); Sept 7‐11. Manchester, UK, 2004.
Seedat 2003 {published data only}
- Seedat S, Stein M. Pindolol potentiation of paroxetine for generalized social phobia: a double‐blind, placebo‐controlled, crossover study. American Journal of Psychiatry 2001;158(10):1725‐7. [DOI] [PubMed] [Google Scholar]
Shlik 2002 {published data only}
- Shlik J, Maron E, Aluoja A, Vasar V, Toru I. Citalopram challenge in social anxiety disorder. European Neuropsychopharmacology 2002;12(Suppl 3):S339. [DOI] [PubMed] [Google Scholar]
Siitonen 1976 {published data only}
- Siitonen L, Janne J. Effect of beta‐blockade during bowling competition. Annals of Clinical Research 1976;8(6):393‐8. [PubMed] [Google Scholar]
Silverstone 1973 {published data only}
- Silverstone JT, Salkind MR. Controlled evaluation of intravenous drugs in the specific desensitization of phobias. Canadian Psychiatric Association Journal 1973;18(1):47‐53. [DOI] [PubMed] [Google Scholar]
Simon 2010 {published data only}
- Simon NM, Worthington JJ, Moshier SJ, Marks EH, Hoge EA, Brandes M, et al. Duloxetine for the treatment of generalized social anxiety disorder: a preliminary randomized trial of increased dose to optimize response. CNS Spectrums 2010;15(7):436‐43. [PubMed] [Google Scholar]
Solyom 1973 {published data only}
- Solyom L, Heseltine GF, McClure DJ, Solyom C, Ledwidge B, Steinberg G. Behaviour therapy versus drug therapy in the treatment of phobic neurosis. Canadian Psychiatric Association Journal 1973;18(1):25‐32. [DOI] [PubMed] [Google Scholar]
Solyom 1981 {published data only}
- Solyom C, Solyom L, LaPierre Y, Pecknold J, Morton L. Phenelzine and exposure in the treatment of phobias. Biological Psychiatry 1981;16(3):239‐47. [PubMed] [Google Scholar]
Tubaki 2012 {published data only}
- Tubaki BR, Chandrashekar CR, Sudhakar D, Prabha TN, Lavekar GS, Kutty BM. Clinical efficacy of manasamitra vataka (an Ayurveda Medication) on generalized anxiety disorder with comorbid generalized social phobia: a randomized controlled study. Journal of Alternative & Complementary Medicine 2012;18(6):612‐21. [DOI] [PubMed] [Google Scholar]
Tyrer 1973 {published data only}
- Tyrer P, Candy J, Kelly D. A study of the clinical effects of phenelzine and placebo in the treatment of phobic anxiety. Psychopharmacologica 1973;32(3):237‐54. [DOI] [PubMed] [Google Scholar]
Wardle 2012 {published data only}
- Wardle MC, Frye CG, Wit H. MDMA buffers against cues of social rejection [abstract]. Neuropsychopharmacology 2012:S414‐5. [Google Scholar]
References to studies awaiting assessment
Asakura 2016 {published data only}
- Asakura S, Hayano T, Hagino A, Koyama T. A randomized, double‐blind, placebo‐controlled study of escitalopram in patients with social anxiety disorder in Japan. Current Medical Research and Opinion 2016;32(4):749‐57. [DOI: 10.1185/03007995.2016.1146663] [DOI] [PubMed] [Google Scholar]
Careri 2015 {published data only}
- Careri JM, Draine AE, Hanover R, Liebowitz MR. A 12‐Week Double‐Blind, Placebo‐Controlled, Flexible‐Dose Trial of Vilazodone in Generalized Social Anxiety Disorder. The Primary Care Companion for CNS Disorders 2015;17(6):e1‐e6. [DOI: 10.4088/PCC.15m01831] [DOI] [PMC free article] [PubMed] [Google Scholar]
De la Barquera 2008 {published data only}
- Barquera JAOS. Double‐blind controlled study with clonazepam and placebo in social anxiety disorder. Salud Mental 2008;31(4):299‐306. [Google Scholar]
Frick 2015 {published data only}
- Frick A, Ahs F, Appel L, Jonasson M, Linnman C, Faria V, et al. Reduced serotonin synthesis after pharmacological treatment of social anxiety disorder [abstract]. Biological Psychiatry 2015;77(9):236. [Google Scholar]
Krylov 1996 {published data only}
- Krylov V. Pharmacological treatment of social phobia: a controlled study with alprazolam and buspirone. 9th European College of Neuropsychopharmacology (ECNP) Congress. Amsterdam, Netherlands, 1996.
NCT00114127 {published data only}
- NCT00114127. Duloxetine for Social Anxiety Disorder: Prediction of Long Term Outcome [Duloxetine for Social Anxiety Disorder: Prediction of Long Term Outcome]. clinicaltrials.gov/ct2/show/NCT00114127 (first received 13 June 2005).
NCT00208741 {published data only}
- NCT00208741. Study To Evaluate The Effects Of Gabitril™ In Patients With Social Anxiety Disorder [A 12‐Week Open‐Label Study Followed By A 24‐Week Double‐Blind Discontinuation Exploratory Study To Evaluate The Effects Of Gabitril™ (Tiagabine Hydrochloride) In Patients With Social Anxiety Disorder (SAD)]. clinicaltrials.gov/ct2/show/NCT00208741 (first received 13 September 2005).
NCT00215254 {published data only}
- NCT00215254. Quetiapine in Social Anxiety Disorder [A Randomized, Double‐Blind, Placebo‐Controlled Pilot Study of Quetiapine in Social Anxiety Disorder]. clinicaltrials.gov/ct2/show/NCT00215254 (first received 20 September 2005).
NCT00246441 {published data only}
- NCT00246441. Paroxetine for Comorbid Social Anxiety Disorder and Alcoholism [Paroxetine for Comorbid Social Anxiety Disorder and Alcoholism]. clinicaltrials.gov/ct2/show/NCT00246441 (first received 28 October 2005).
NCT00294346 {published data only}
- NCT00294346. Safety and Efficacy Study of AV608 in Subjects With Social Anxiety Disorder [A Phase 2, Double‐Blind, Placebo‐Controlled Trial to Investigate the Safety and Efficacy of AV608 in Subjects With Social Anxiety Disorder]. clinicaltrials.gov/ct2/show/NCT00294346 17 February 2006).
NCT00485888 {published data only}
- NCT00485888. Flushing in Social Anxiety Disorder on Cipralex [Changes in the Vasodilatory Response to Methyl‐nicotinate in Response to S‐citalopram Treatment in Social Phobia Patients]. clinicaltrials.gov/ct2/show/NCT00485888 (first received 12 June 2007).
NCT00612859 {published data only}
- NCT00612859. Study to Assess the Efficacy and Safety of Levetiracetam for the Treatment of Social Anxiety Disorder (Generalized Type) [A Multicenter, Randomized, Double‐Blind, PBO‐Controlled, Parallel Group Study to Assess the Efficacy and Safety of Levetiracetam Versus PBO for the Treatment of Social Anxiety Disorder (Generalized Type)]. clinicaltrials.gov/ct2/show/NCT00612859 (first received 14 January 2008).
NCT01316302 {published data only}
- NCT01316302. 12‐Week Study of Pristiq (Desvenlafaxine) Social Anxiety Disorder [A 12‐Week Double‐Blind, Placebo‐Controlled, Flexible‐Dose Trial of Pristiq® (Desvenlafaxine) Extended‐Release Tablets in Generalized Social Anxiety Disorder]. clinicaltrials.gov/ct2/show/NCT01316302 (first received 14 March 2011).
References to ongoing studies
NCT00182533 {published data only}
- NCT00182533. Sertraline in Generalized Social Phobia With Co‐Occurring Anxiety and Mood Disorders [Sertraline in the Treatment of Generalized Social Phobia With Comorbidity]. clinicaltrials.gov/ct2/show/NCT00182533 (first received 14 September 2005).
NCT01712321 {published data only}
- NCT01712321. Study of Vilazodone to Treat Social Anxiety Disorder [Vilazodone in the Treatment of Social Anxiety Disorder: A Double Blind Study]. clinicaltrials.gov/ct2/show/NCT01712321 (first received 18 October 2012).
NCT02083926 {published data only}
- NCT02083926. Ketamine Infusion for Social Anxiety Disorder [Ketamine Infusion for Social Anxiety Disorder]. clinicaltrials.gov/show/NCT02083926 (first received 5 March 2014).
NCT02294305 {published data only}
- NCT02294305. ortioxetine Versus Placebo in Major Depressive Disorder Comorbid With Social Anxiety Disorder. clinicaltrials.gov/show/NCT02294305 (first received 11 November 2014).
NCT02432703 {published data only}
- NCT02432703. A Safety and Efficacy Study of JNJ‐42165279 in Participants With Social Anxiety Disorder [A Phase 2a Randomized, Double‐blind, Placebo‐Controlled, Parallel‐Group, Multi‐center Study Investigating the Efficacy, Safety, and Tolerability of JNJ‐42165279 in Subjects With Social Anxiety Disorder]. clinicaltrials.gov/show/NCT02432703 (first received 29 April 2015).
Additional references
Aarre 2003
- Aarre TF. Phenelzine efficacy in refractory social anxiety disorder: a case series. Nordic Journal of Psychiatry 2003;57(4):313‐5. [DOI] [PubMed] [Google Scholar]
Als‐Nielsen 2003
- Als‐Nielsen B, Chen W, Gluud C, Kjaergard LL. Association of funding and conclusions in randomized drug trials ‐ a reflection of treatment effect or adverse events?. JAMA 2003;290(7):921‐8. [DOI] [PubMed] [Google Scholar]
Bailar 1999
- Bailar JC 3rd. The promise and problems of meta‐analysis. New England Journal of Medicine 1999;337(8):559‐61. [DOI] [PubMed] [Google Scholar]
Baker 2003
- Baker CB, Johnsrud MT, Crismon ML, Rosenheck RA, Woods SW. Quantitative analysis of sponsorship bias in economic studies of antidepressants. British Journal of Psychiatry 2003;183(6):498‐506. [DOI] [PubMed] [Google Scholar]
Baldwin 2014
- Baldwin DS, Anderson IM, Nutt DJ, Allgulander C, Bandelow B, Boer JA, et al. Evidence‐based pharmacological treatment of anxiety disorders, post‐traumatic stress disorder and obsessive‐compulsive disorder: a revision of the 2005 guidelines from the British Association for Psychopharmacology. Journal of Psychopharmacology 2014;28(5):403‐39. [DOI: 10.1177/0269881114525674] [DOI] [PubMed] [Google Scholar]
Ballenger 1998
- Ballenger JC, Davidson JR, Lecrubier Y, Nutt DJ, BobesJ, Beidel DC, et al. Consensus statement on social anxiety disorder from the international consensus group on depression and anxiety. Journal of Clinical Psychiatry 1998;59(Suppl 17):54–60. [PubMed] [Google Scholar]
Bandelow 2002
- Bandelow B, Zohar J, Hollander E, Kasper S, Moller HJ. WFSBP Task Force on Treatment Guidelines for Anxiety O‐CaPSD. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for the pharmacological treatment of anxiety, obsessive‐compulsive and posttraumatic stress disorders. World Journal of Biological Psychiatry 2002;3(4):171‐99. [DOI] [PubMed] [Google Scholar]
Bandelow 2012
- Bandelow B, Sher L, Bunevicius R, Hollander E, Kasper S, Zohar J, et al. WFSBP Task Force on Mental Disorders in Primary Care, WFSBP Task Force on Anxiety Disorders, OCD, PTSD. Guidelines for the pharmacological treatment of anxiety disorders, obsessive – compulsive disorder and posttraumatic stress disorder in primary care. International Journal of Psychiatry in Clinical Practice 2012;16:77–84. [DOI: 10.3109/13651501.2012.667114] [DOI] [PubMed] [Google Scholar]
Bandelow 2015
- Bandelow B, Michaelis S. Epidemiology of anxiety disorders in the 21st century. Dialogues in Clinical Neuroscience 2015;17(3):327‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
Beck 1961
- Beck AT, Ward CH, Mendelson M, Mock J, Erbaugh J. An inventory for measuring depression. Archives of General Psychiartry 1961;4(6):561‐71. [DOI] [PubMed] [Google Scholar]
Blanco 2002
- Blanco C, Antia SX, Liebowitz MR. Pharmacotherapy of social anxiety disorder. Biological Psychiatry 2002;51(1):109‐20. [DOI] [PubMed] [Google Scholar]
Blanco 2003
- Blanco C, Schneier FR, Schmidt A, Blanco‐Jerez C, Marshall RD, Sanchez‐Lacay A, et al. Pharmalogical treatment of social anxiety disorder: a meta‐analysis. Depression and Anxiety 2003;18(1):29‐40. [DOI] [PubMed] [Google Scholar]
Blanco 2013
- Blanco C, Bragdon LB, Schneier FR, Liebowitz MR. The evidence‐based pharmacotherapy of social anxiety disorder. International Journal of Neuropsychopharmacology 2013;16(1):235‐49. [DOI] [PubMed] [Google Scholar]
Chamberlain 2007
- Chamberlain SR, Campo N, Dowson J, Müller U, Clark L, Robbins TW, et al. Atomoxetine improved response inhibition in adults with attention deficit/hyperactivity disorder. Biological Psychiatry 2007;62(9):977–84. [DOI] [PubMed] [Google Scholar]
Cowen 1997
- Cowen PJ. Pharmacotherapy for anxiety disorders: drugs available. Advances in Psychiatric Treatment 1997;3:66‐71. [Google Scholar]
Curtiss 2017
- Curtiss J, Andrews L, Davis M, Smits J, Hofmann SG. A meta‐analysis of pharmacotherapy for social anxiety disorder: anexamination of efficacy, moderators, and mediators. Expert Opinion on Pharmacotherapy 2017;18(3):243‐51. [hdl.handle.net/2144/21703] [DOI] [PubMed] [Google Scholar]
Davidson 1991
- Davidson JR, Ford SM, Smith RD, Potts NL. Long‐term treatment of social phobia with clonazepam. Journal of Clinical Psychiatry 1991;52(Suppl 1):16‐20. [PubMed] [Google Scholar]
Davidson 1998
- Davidson JR. Pharmacotherapy of social anxiety disorder. Journal of Clinical Psychiatry 1998;59(Suppl 17):47‐51. [PubMed] [Google Scholar]
Davidson 2006
- Davidson JR. Pharmacotherapy of social anxiety disorder: what does the evidence tell us?. Journal of Clinical Psychiatry 2006;67(Suppl 12):20–6. [PubMed] [Google Scholar]
Davies 2015
- Davies SJ, Champion C, Dawson S, Sharp J, Churchill R. The Subway Map ‐ a new way to visualize antidepressant classification (a Cochrane CCDAN initiative). 14th ICGP and 19th JSNP Joint Congress; 3 October. Tsukuba, Japan, 2014:78. [Abstract P14]
Davis 2014
- Davis ML, Smits JAJ, Hofmann SG. Update on the efficacy of pharmacotherapy for social anxiety disorder: a meta‐analysis. Expert Opinion on Pharmacotherapy 2014;15(16):2281‐91. [DOI] [PubMed] [Google Scholar]
De Menezes 2011
- Menezes GB, Coutinho ESF, Fontenelle LF, Vigne P, Figueira I, Versiani Má. Second‐generation antidepressants in social anxiety disorder: meta‐analysis of controlled clinical trials. Psychopharmacology 2011;215(1):1‐11. [DOI] [PubMed] [Google Scholar]
Dechartres 2011
- Dechartres A, Boutron I, Trinquart L, Charles P, Ravaud P. Single‐center trials show larger treatment effects than multicenter trials: evidence from a meta‐epidemiologic study. Annals of Internal Medicine 2011;155(1):39–51. [DOI] [PubMed] [Google Scholar]
Deeks 200
- Deeks JJ, Altman DG, Bradburn MJ. Statistical methods for examining heterogeneity and combining results from several studies in meta‐analysis. In: Egger M, Davey SG, Altman DG editor(s). Systematic Reviews in Health Care: Meta‐Analysis in Context. 2nd Edition. BMJ Publication Group, 2008:285‐312. [Google Scholar]
Deeks 2002
- Deeks JJ. Issues in the selection of a summary statistic for meta‐analysis of clinical trials with binary outcomes. Statistics in Medicine 2002;21(11):1575‐600. [DOI] [PubMed] [Google Scholar]
Deeks 2011
- Deeks JJ, Altman DG, Bradburn MJ. Statistical methods for examining heterogeneity and combining results from several studies in meta‐analysis. In: Egger M, Davey SG, Altman DG editor(s). Systematic Reviews in Health Care: Meta‐Analysis in Context. 2nd Edition. BMJ Publication Group, 2008:285‐312. [Google Scholar]
Dickersin 1992
- Dickersin K, Min YI, Meinert CL. Factors influencing the publication of research results: follow‐up of applications submitted to two substantial review boards. JAMA 1992;267(3):374‐8. [PubMed] [Google Scholar]
Dorrepaal 2014
- Dorrepaal E, Thomaes K, Hoogendoorn AW, Veltman DJ, Draijer N, Balkom AJLM. Evidence‐based treatment for adult women with child abuse‐related Complex PTSD: a quantitative review. European Journal of Psychotraumatology 2014;14(5):236‐413. [DOI] [PMC free article] [PubMed] [Google Scholar]
DSM‐III 1980
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 3rd Edition. Washington, DC: American Psychiatric Press, 1980. [Google Scholar]
DSM‐IV 2004
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4th Edition. Washington, DC: American Psychiatric Association, 1994. [Google Scholar]
Easterbrook 1991
- Easterbrook PJ, Berline JA, Gopalan R, Mathews DR. Publication bias in clinical research. Lancet 1991;337(8746):867‐72. [DOI] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Smith GD, Schneider M, Minder C. Bias in meta‐analysis detected by a simple, graphical test. BMJ 1997;13(315(7109)):629–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne DR, Altman DG, Higgins JPT, Curtin F, Worthington HV, Vail A. Meta‐analyses involving cross‐over trials: methodological issues. International Journal of Epidemiology 2002;31(1):140‐9. [DOI] [PubMed] [Google Scholar]
Fedoroff 2001
- Fedoroff IC, Taylor S. Psychological and pharmacological treatments of social phobia. Journal of Clinical Psychopharmacology 2001;21(3):311‐24. [DOI] [PubMed] [Google Scholar]
Ford 2014
- Ford C, Law F. Guidance for the use and reduction of misuse of benzodiazepines and other hypnotics and anxiolytics in general practice. www.emcdda.europa.eu/attachements.cfm/att_248926_EN_UK59_benzos (accessed prior to 26 July 2017).
Furmark 2002
- Furmark T, Tillfors M, Marteinsdottir I, Fischer H, Pissiota A, Långström B, et al. Common changes in cerebral blood flow in patients with social phobia treated with citalopram or cognitive‐behavioral therapy. Archives of General Psychiatry 2002;59(5):425‐33. [DOI] [PubMed] [Google Scholar]
Gorman 1987
- Gorman JM, Gorman LK. Drug treatment of social phobia. Journal of Affective Disorders 1987;13(2):183‐92. [DOI] [PubMed] [Google Scholar]
Gorman 2003
- Gorman JM. Treating generalized anxiety disorder. Journal of Clinical Psychiatry 2003;64(Suppl 2):24‐9. [PubMed] [Google Scholar]
Gould 1997
- Gould RA, Buckminister S, Pollack MH, Otto MW, Yap L. Cognitive‐behavioral and pharmacological treatments of social phobia. Clinical Psychology ‐ Science & Practice 1997;4(4):291‐306. [Google Scholar]
Gury 1999
- Gury C, Cousin F. Pharmacokinetics of SSRI antidepressants: half‐life and clinical applicability. Encephale 1999;25(5):470‐6. [PubMed] [Google Scholar]
Guy 1976
- Guy W. ECDEU Assessment Manual for Psychopharmacology. Washington, DC: US National Institute of Health, 1976. [Google Scholar]
Hamilton 1959
- Hamilton M. The assessment of anxiety states by rating. British Journal of Medical Psychology 1959;32(1):50‐5. [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JT, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327(6):557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Greens S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org. The Cochrane Collaboration. Available from www.cochrane‐handbook.org, 2011.
Hubbard 2011
- Hubbard CS, Labus JS, Bueller J, Stains J, Suyenobu B, Dukes GE, et al. Corticotropin‐releasing factor receptor 1 antagonist alters regional activation and effective connectivity in an emotional arousal circuit during expectation of abdominal pain. Journal of Neuroscience 2011;31(35):12491–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ipser 2009a
- Ipser JC, Stein DJ, Hawkridge S, Hoppe L. Pharmacotherapy for anxiety disorders in children and adolescents. Cochrane Database of Systematic Reviews 2009, Issue 3. [DOI: 10.1002/14651858.CD005170.pub2] [DOI] [PubMed] [Google Scholar]
Ipser 2009b
- Ipser JC, Sander C, Seifan A, Stein DJ. Augmentation of psychotherapy with d‐cycloserine for anxiety disorders. Cochrane Database of Systematic Reviews 2009, Issue 2. [DOI: 10.1002/14651858.CD007803] [DOI] [Google Scholar]
Irwig 1998
- Irwig L, Macaskill P, Berry G, Glasziou P. Bias in meta‐analysis detected by a simple, graphical test. Graphical test is itself biased. BMJ 1998;316(7109):470‐1. [PMC free article] [PubMed] [Google Scholar]
Kelsey 1995
- Kelsey JE. Venalafaxine in social phobia. Psychopharmacology Bulletin 1995;31(4):767‐71. [PubMed] [Google Scholar]
Kessler 1994
- Kessler RC, McGonagle KA, Zhao S, Nelson CB, Hughes M, Ehsleman S, et al. Lifetime and 12‐month prevalence of DSM III‐R psychiatric disorders in the United States. Results from the National Comorbidity Survey. Archives of General Psychiatry 1994;51(1):8‐19. [DOI] [PubMed] [Google Scholar]
Kessler 2005
- Kessler RC, Berglund P, Demler O, Jin R, Merikangas KR, Walters EE. Lifetime prevalence and age‐of‐onset distributions of DSM‐IV disorders in the National Comorbidity Survey Replication. Archives of General Psychiatry 2005;62(6):593‐602. [DOI] [PubMed] [Google Scholar]
Kobak 2002
- Kobak KA, Greist JH, Jefferson JW, Katzelnick DJ. Fluoxetine in social phobia: a double‐blind, placebo‐controlled pilot study. Journal of Clinical Psychopharmacology 2002;22(3):257‐62. [DOI] [PubMed] [Google Scholar]
Licata 2008
- Licata SC, Rowlett JK. Abuse and dependence liability of benzodiazepine‐type drugs: GABAA receptor modulation and beyond. Pharmacology Biochemistry & Behavior 2008;90(1):74–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
Liebowitz 1987
- Liebowitz MR. Social Phobia. Modern Problems in Pharmacopsychiatry 1987;22:141‐73. [DOI] [PubMed] [Google Scholar]
Lumley 2002
- Lumley, T. Network meta‐analysis for indirect treatment comparisons. Statistics in Medicine 2002;21(16):2313‐24. [DOI] [PubMed] [Google Scholar]
Marks 1966
- Marks IM, Gelder MG. Different ages of onset in varieties of phobias. American Journal of Psychiatry 1966;123(2):218‐21. [DOI] [PubMed] [Google Scholar]
Michelson 2001
- Michelson D, Faries D, Wernicke J, Kelsey D, Kendrick K, Sallee FR, et al. the Atomoxetine ADHD Study Group. Atomoxetine in the treatment of children and adolescents with attention‐deficit/hyperactivity disorder: a randomized, placebo‐controlled, dose‐response study. Pediatrics 2001;108(5):1‐9. [DOI] [PubMed] [Google Scholar]
Moher 1999
- Moher D, Cook DJ, Eastwood S, Olkin I, Rennie D, Stroup DF. Improving the quality of reports of meta‐analyses of randomized controlled trials: the QUOROM statement. Lancet 1999;354(9193):1896‐900. [DOI] [PubMed] [Google Scholar]
Mulrow 1997
- Mulrow CD, Oxman AD. Cochrane Collaboration Handbook. Oxford: Update Software, 1997. [Google Scholar]
Nemeroff 2008
- Nemeroff CB, Entsuah R, Benattia I. Comprehensive Analysis of Remission (COMPARE) with venlafaxine versus SSRIs. Biological Psychiatry 2008;63(4):424‐34. [DOI] [PubMed] [Google Scholar]
NICE 2011
- National Institute for Health and Care Excellence. Common mental health problems: identification and pathways to care. NICE Clinical Guideline; 2011. Available from www.nice.org.uk.
Pecknold 1997
- Pecknold JC. A risk‐benefit assessment of buspirone in the treatment of anxiety disorders. Drug Safety 1997;16(2):118–32. [DOI] [PubMed] [Google Scholar]
Peters 2008
- Peters JL, Sutton AJ, Jones DR, Abrams KR, Rushton L. Contour‐enhanced meta‐analysis funnel plots help distinguish publication bias from other causes of asymmetry. Journal of Clinical Epidemiology 2008;61(10):991‐6. [DOI] [PubMed] [Google Scholar]
RevMan 2014 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Roest 2015
- Roest AM, Jonge P, Williams CD, Vries YA, Schoevers RA, Turner EH. Reporting bias in clinical trials investigating the efficacy of second‐generation antidepressants in the treatment of anxiety disorders: a report of 2 meta‐analyses. JAMA Psychiatry 2015;72(5):500‐10. [DOI: ] [DOI] [PubMed] [Google Scholar]
Scherer 1994
- Scherer RW, Dickersin K, Langenberg P. Full publication of results initially presented in abstracts. A meta‐analysis. JAMA 1994;272(2):158‐62. [PubMed] [Google Scholar]
Sheehan 1996
- Sheehan DV, Harnett‐Sheehan K, Raj BA. The measurement of disability. International Clinical Psychopharmacology 1996;11(Suppl 3):89‐95. [DOI] [PubMed] [Google Scholar]
Song 2003
- Song F, Altman DG, Glenny A‐M, Deeks JJ. Validity of indirect comparison for estimating efficacy of competing interventions: empirical evidence from published meta‐analyses. BMJ 2003;326(7387):472. [DOI] [PMC free article] [PubMed] [Google Scholar]
Stahl 2008
- Stahl SM. Stahl's Essential Psychopharmacology: Neuroscientific Basis and Practical Applications. 3rd Edition. Cambridge University Press, 2008. [Google Scholar]
Stein 2001
- Stein MB, Sareen J, Hami S, Chao J. Pindolol potentiation of paroxetine for generalized social phobia: a double‐blind, placebo‐controlled, crossover study. American Journal of Psychiatry 2001;158(10):1725‐7. [DOI] [PubMed] [Google Scholar]
Stein 2002c
- Stein DJ, Stein MB, Pitts CD, Kumar R, Hunter B. Predictors of response to pharmacotherapy in social anxiety disorder: an analysis of 3 placebo‐controlled paroxetine trials. Journal of Clinical Psychiatry 2002;63(2):152‐5. [DOI] [PubMed] [Google Scholar]
Stein 2002d
- Stein DJ, Hunter B, Rolfe T, Oakes, R. Paroxetine in social anxiety disorder: a remission analysis. European College of Neuropsychopharmacology (ECNP) Congress; Oct. Barcelona, Spain, 2002.
Stein 2004
- Stein DJ, Ipser JC, Balkom AJ. Pharmacotherapy for social phobia. Cochrane Database of Systematic Reviews 2004, Issue 4. [DOI: 10.1002/14651858.CD001206.pub2] [DOI] [PubMed] [Google Scholar]
Stein 2008
- Stein MB, Stein DJ. Social anxiety disorder. Lancet 2008;271(9618):1115‐25. [DOI] [PubMed] [Google Scholar]
Stephen 2013
- Stahl M, Moore BA. Anxiety Disorders: A Guide for Integrating Psychopharmacology and Psychotherapy (Clinical Topics in Psychology and Psychiatry). 1st edition. Routledge, 2013. [Google Scholar]
Sterne 2011
- Sterne JAC, Egger M, Moher D. Chapter 10: Addressing reporting biases. In: Higgins JP, Green S, editor(s). Cochrane Handbook of Systematic Reviews of Interventions. Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Thompson 1994
- Thompson SG. Why sources of heterogeneity in meta‐analysis should be investigated. BMJ 1994;309(6965):1351‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Van Ameringen 1996
- Ameringen M, Mancini C, Wilson C. Busiprone augmentation of selective serotonin reuptake inhibitors (SSRIs) in social phobia. Journal of Affective Disorders 1996;39(2):115‐21. [DOI] [PubMed] [Google Scholar]
Van der Linden 2000
- Linden GJ, Stein DJ, Balkom AJ. The efficacy of the selective serotonin reuptake inhibitors for social anxiety disorder (social phobia): a meta‐analysis of randomized controlled trials. International Clinical Psychopharmacology 2000;15(Suppl 2):15‐24. [DOI] [PubMed] [Google Scholar]
Verbeke 2000
- Verbeke M, Molenberghs G. Linear Mixed Models for Longitudinal Data. New York: Springer, 2000. [Google Scholar]
Viechtbauer 2010
- Viechtbauer W. Conducting meta‐analyses in R with the metafor package. Journal of Statistical Software 2010;36(3):1‐48. [Google Scholar]
Wancata 2009
- Wancata J, Fridl M, Friedrich F. Social phobia: epidemiology and health care. Psychiatrica Danubina 2009;21(4):520‐4. [PubMed] [Google Scholar]
References to other published versions of this review
Stein 2001b
- Stein DJ, Ipser JC, Balkom AJ. Pharmacotherapy for social anxiety disorder. Cochrane Database of Systematic Reviews 2000, Issue 4. Art. No.: CD001206. [DOI: 10.1002/14651858.CD001206.pub2] [DOI] [PubMed] [Google Scholar]