

Abstract
DB2277, a heterocyclic diamidine, is a successful design for mixed base pair (bp) DNA sequence recognition. The compound has a central aza-benzimidazole group that forms two H-bonds with a GC bp that has flanking AT bps. The NMR structure of the DB2277-DNA complex with an AAGATA recognition site sequence was solved and here we report extended molecular dynamics (MD) simulations of the structure. DB2277 has two terminal phenyl-amidine groups, one of which is directly linked to the DB2277 heterocyclic core while the other is linked through a flexible -OCH2- group. The flexibly-linked phenyl is too far from the minor groove floor to make direct H-bonds but it is linked to an AT bp through water-mediated H-bonds. The flexibly linked phenyl-amidine with water mediated H-bonds to the bases at the floor of the minor groove suggested that it might rotate in time-spans accessible in MD. To test this idea, we have conducted multi-microsecond MD simulations to determine if these phenyl rotations could be observed for a compound bound to DNA. In a 3 μs simulation, highly dynamic torsional motions were observed for the –OCH2– linked phenyl but not for the other phenyl. The dynamics periodically reached a level to allow 180° rotation of the phenyl while still bound in the minor groove. This is the first observation of rotation of a phenyl bound to DNA and the results provide mechanistic details of how a rotation can occur as well as how mixed bp recognition can occur for monomer compounds bound to the minor groove.
Graphical Abstract
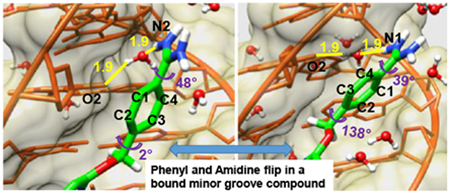
Introduction:
We are systematically designing entirely new heterocyclic amidine derivatives with the goal of recognizing a much broader range of biologically important DNA sequences than has been previously possible with these and related agents.1–8 Additional goals, which are addressed in the research described in this paper, are to better understand the dynamics of bound minor groove complexes, and the roles that waters of interaction play in the structure, binding selectivity and energetics of complex formation. Heterocyclic amidine cations, such as pentamidine, furamidine and berenil, whose DNA complexes have been extensively investigated, are uniformly specific for binding to AT DNA sequences. To also recognize G•C base pairs, it is necessary to incorporate H-bond acceptor units, as noted many years ago by Dickerson and Lown.9–12 To broaden the applications of heterocyclic amidines, particularly in therapeutic applications, it is essential to expand their sequence recognition abilities.1–8 It should be noted that pentamidine and berenil are already used in therapy of humans and animals and furamidine has recently reached Phase III clinical trials against human parasitic diseases. These therapeutic successes suggest that the heterocyclic amidines are excellent prospects for expanded uses and this has recently been confirmed in leukemia.13
The compounds listed above have H-bond donors for recognition of A•T base pair AN3 and T2 C=O groups but no available acceptors. DB2277 (Figure 1) is a member of a new class of designed heterocyclic diamidines that specifically recognize a single G•C bp with a broad range of flanking AT sequences.1,2,5,7 There are quite a large number of NMR and crystal structures of heterocyclic amidines binding to pure AT DNA sequences but no previous structures with a centrally bound GC base pair.14–20 To begin to understand GC recognition by the new agents, a number of mixed sequence DNAs were used in NMR studies to understand DNA sequence-dependent effects of GC recognition by DB2277 in a minor groove complex.5 The formation of a unique 1:1 complex for NMR studies was achieved with a DNA sequence that has a central –AAGATA– binding site.7
Figure 1.
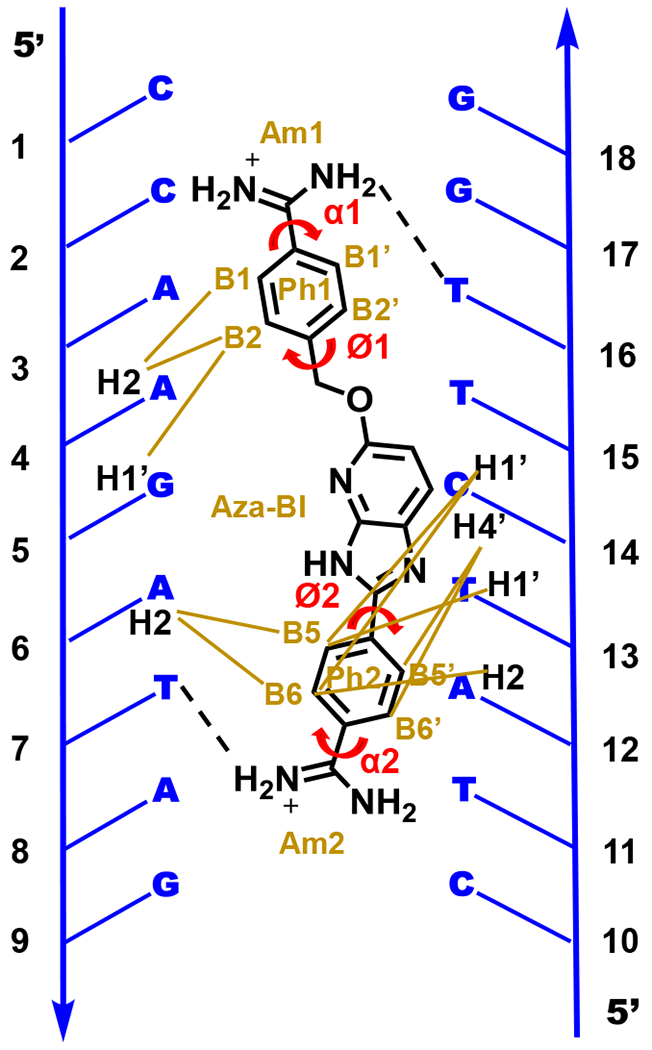
Graphic of the molecular structure of DB2277 with the target DNA sequence. Abbreviations for aromatic rings are listed along with their torsion angles. Brown lines represent the NOE interactions of DB2277 protons with specific DNA protons in the minor groove.7 Broken lines illustrates the H-bond interaction of amidines with T-O2 in the minor groove of DNA.
The AAGATA–DB2277 complex structure was solved by 2D NMR and was found to be quite unusual. The aza-benzimidazole-phenyl-amidine end of the complex fits tightly into the minor groove with numerous van-der-Waals contacts, H-bonds and solvent interactions. The experimental structure models (Figure 2) show that the G5-NH in the minor groove of AAGATA forms an excellent H-bond with the aza-N of the aza-BI group, as expected from the compound design.7 The DB2277-G•C interaction is further locked down by the unexpected formation of an H-bond between the aza-BI–NH group and the C14-O2 (cytidine at position 14) in the minor groove.7 The flanking AT sequences also strongly interact with DB2277. The amidine on the phenyl-amidine attached to the aza-BI group forms a direct H-bond in the minor groove to an AT base pair (T=O●●●H-N-amidine). The amidine-DNA interaction is additionally stabilized by an ensemble of dynamic water H-bonds in the minor groove.7 The complex is also stabilized by phenyl C-H protons that are near the floor of the minor groove. These aromatic protons carry a small positive charge and can form stabilizing interactions with A-N3 and T=O groups. The DB2277-DNA interactions coupled to the dynamic stabilizing hydration network and van-der-Waals interactions provide a very favorable complex and specific DNA recognition for the aza-BI-phenyl-amidine module.
Figure 2.
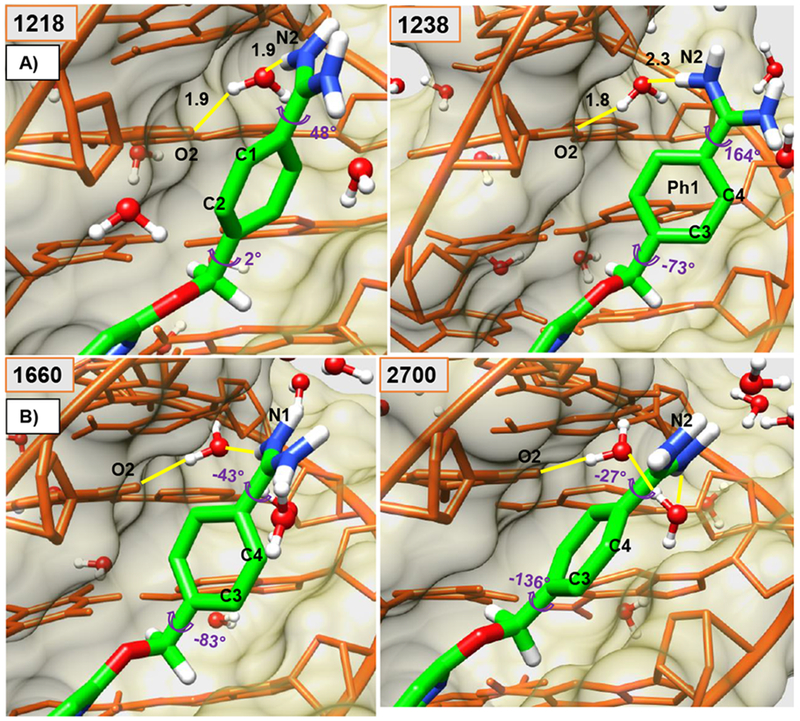
Minor groove views of the DB2277-DNA complex are shown in panel A and B. Ø1 and α1 torsion angles are shown at several key times (frame-number on top-left) along with water-mediated H-bonds (yellow) (Å). Panel A shows larger twists for α1 and Ø1, compare 1218-frame with the orthogonal position of Ph1 relative to other aromatic groups of bound DB2277 in frame 1238. Starting at frame 1218 with C1 and C2 pointed out in panel A and end at the frame 2700 with C3 and C4 pointed out in Panel B. The DNA backbone is represented in orange-colored-stick with khaki-colored-space fill. The DB2277 molecule is shown in stick (green-C). Water molecules in ball and stick.
The other phenyl-amidine of the complex is connected by a flexible -OCH2- to the aza-benzimidazole (Figure 1). As a result of the overall complex structure, this terminal phenyl-amidine rises away from the floor of the minor groove such that it cannot form direct interactions with the A•T bps at the floor of the groove. Instead, there is an interfacial water molecule that links the inner facing amidine-NH to T16-O2 (Figure 1) at the floor of the groove (-NH●●●O-H●●●O=T). This water molecule, unlike the other waters that are well known to externally stabilize minor groove complexes21,22, is an integral part of a trimer DB2277-DNA-water complex. Both amidines are also stabilized by a dynamic, external, extended water-network in the groove (Figure 2). Similar waters of stabilization are seen in essentially all minor groove complexes that have been solved at sufficient resolution and are clearly a common feature of DNA minor groove interactions.23,24 The unique feature of the DB2277-water interactions is the interfacial water interaction which is a very rare feature of small molecule-DNA complexes. It should be emphasized that the complexes that we are designing for GC bp recognition bind to the DNA minor groove as monomers and are quite different from the extensively studied polyamides that form stacked dimer or hairpin binding units.25–28
To better understand the DB2277 binding to a mixed base pair DNA sequence, we have conducted long time scale MD investigations of with several goals: (i) investigate the local dynamics of both AT and GC bp interactions of DB2277 to better characterize the critical design features that are essential for strong, specific complex formation with mixed base pair DNA sequences; (ii) determine whether the water-linked phenyl could rotate while DB2277 was bound tightly in the minor groove and if so, determine what changes in the bound molecule, interfacial water and DNA are required to allow the flip; (iii) in long MD simulations could rotation of the other, directly H-bonded phenyl in DB2277 be observed; (iv) investigate the influence on complex structure and stability of water molecules that are external to bound DB2277. By conducting multi-microsecond MD calculations starting with the NMR-restrained DB2277 structure, we were able to observe the rotation of the flexible-linked phenyl as well as how this rotation affects the interfacial water component of the complex. To our knowledge, this is the first observation of an aromatic group rotation when bound to the DNA minor groove and the mechanistic characteristics of the dynamic transitions are described here.
Materials and Methods:
Molecular Dynamics (MD) Simulations for 3.0 microseconds were performed without any restraints on the DB2277-AAGATA complex. All energy minimizations and molecular dynamics (MD) simulations were conducted on the PMEMD CUDA module of the Assisted Model Building with an Energy Refinement (AMBER14) program.29 The initial structure of the DB2277-DNA complex used in MD simulations was based on the restrained MD NMR structure (PDB ID, 6AST).7
To conduct the molecular dynamics simulations of the DB2277 complex in the AMBER package, all the parameters of the molecules are required. Amber contains the parameters for DNA but parameters of DB2277 used for MD simulation must be determined and the values are reported in detail in the Supplementary Information. The procedure used is similar to that described by the Cheatham group30 and our group for minor groove binders.31 The AMBER14 package was used to equilibrate the DB2277-DNA complex system using “ff14SB” and ff14SB/chi/ez (i.e., parmbsc0_chiOL4_ezOL1) force field modifications for DNA.32 Significant improvement in the development of empirical force fields and MD simulation methods has helped to describe the structure, energetics and dynamics of nucleic acids more accurately.33–35 The parm94-99 force field was parametrized for simulation on the 1-10 ns time scale but severe distortions of DNA structure was found in 50 ns simulations due to irreversible transitions in α and γ torsion angles. Thus parmbsc036 contains the reparametrized α and γ dihedral terms using the high-level quantum mechanical (QM) calculations on models of sugars and phosphates. Similarly, the χ distribution using relevant model systems was subsequently improved using various levels of theory in QM methods. The most tested χ modifications are the “OL” modifications used in ff14SB. Therefore, the combination of ff14 + ε/ζOL1 + χOL4, which includes ε/ζOL1 [Ref] and χOL4 [Ref] modifications to ff14 force field, is suitable to use for DNA.37
MD simulation of the complex was performed in explicit solvation conditions using a truncated octahedron periodic box filled with TIP3P water. The box wall was extended to a distance of 10.0 Å from any solute atom in each dimension. The system was neutralized by sodium cations using the LEAP module of AMBER. The Particle Mesh Ewald method with a 10.0 Å cutoff was used for the correct treatment of electrostatic interactions. During the equilibration, the SHAKE procedure was applied to constrain all bonds involving hydrogen atoms to their correct values. The system was relaxed with 1000 steps of energy minimization in the initial phase of equilibration. Then the system was heated from 0 K to 300 K over 10 ps under constant-volume conditions. A production run on the system was subsequently performed for 3.0 μs for the DB2277-DNA complex with NPT conditions (isothermal, isobaric ensemble). For the detailed investigation of the dynamic motions of the phenyl rings of DB2277, the time interval for each frame was decreased to 0.25 ps for a nanosecond between 1020 to 1021 ns of MD simulation. Helical analysis of the DNA was done using the CURVES+ module38 and analysis of trajectories was performed using various scripts in the cpptraj module.39 All of the molecular graphics were generated using VMD 1.9 and UCSF Chimera molecular visualization software.40–42
Results:
NOESY spectra obtained for the DB2277-AAGATA complex in D2O buffer show degeneracy in the chemical shift (δ) observed for all pairs of phenyl protons B1/B1’, B2/B2’, B5/B5’ and B6/B6’ phenyl proton signals of DB2277 as labeled in Figure 1.7 Observed averaging of chemical shifts of phenyl protons signals could be caused by rapid phenyl rotation or by an unlikely complete chemical shift overlap of all four sets of phenyl protons on the NMR chemical shift timescale. NMR data reported by Embrey and Searle et al. for Hoechst 33258 indicates that the similar overlap observed for phenyl protons of that molecule are caused by rotational averaging of the signals.43,44 Intramolecular exchange rate calculations from EXSY NMR between the two opposite binding orientations of DB2277 in the DB2277-AAGTTT complex (5’-3’ and 3’-5’) at 1:1 binding ratio are 6.8 s−1 at 303 K and 2.8 s−1 at 285 K.5 The half-life observed for the exchange between major and minor binding species is around 100 ms by NMR whereas the half-life for dissociation of the DB2277-AAGTTT complex is in the range of 30-60 s by SPR. Therefore, the long dissociation rate of DB2277 from the complex allows microscopic exchange between major and minor binding species of DB2277 while bound to DNA.5 It is also clearly evident from all of these observations that internal phenyl dynamics of DB2277 in the bound state are much faster than the exchange of DB2277 between binding orientations, end to end rotation, in the more symmetric minor groove complexes. With the –AAGATA- sequence only one favored bound form of DB2277 is observed and this allowed determination of the DB2277-DNA complex structure.7
The initial conformation for the DB2277-AAGATA complex for MD simulations was based on the restrained NMR structure.7 The equilibrated structure obtained after 2 ns of MD with restraints obtained from NMR was used as an initial conformation for MD (Figure 1).7 The three aromatic groups of DB2277 were initially very close to one plane while the amidine groups are twisted about 30° out of the phenyl plane due to a steric clash of protons on the phenyls and amidines.7
The DNA sequence used in the restrained NMR structure was retained in the MD calculations and the DB2277 complex is shown in a schematic graphic in Figure 1. By labeling the DB2277 atoms in the 3.0-μs MD simulations, it was obvious that the phenyl at the top of Figure 1 (Ph1), attached to the flexible -CH2O- had rotated 180°, several times while no rotation of Ph2 was observed in this timespan. The two phenyl-amidine groups of DB2277 are both H-bonded to T-O2 groups at the floor of the minor groove, but the Ph1 phenyl-amidine (Figure 1) is coupled through an interfacial water that is an integral part of the complex and the MD results show that it is very dynamic (Figure 2). The observation that the phenyl amidine rotates 180° during the MD simulation is the first observation of this type for a small molecule bound to DNA and allows us to evaluate the mechanism of flipping.
The lower phenyl (Ph2), linked to BI, is constrained in the complex through an optimum H-bond of the inner facing –NH2 group of the amidine (Am2) with T7-O2, as well as strong van-der-Waals interactions with the walls and floor groups of the minor groove (Figure 3). Due to these constraints, no 180° rotational motion was observed for phenyl Ph2 throughout the MD simulations. Beginning and ending views of the flipped structures of Ph1 (C1 and C2 point out in frame 1218 while C3 and C4 point out in frame 3763) and the constant orientation of Ph2 (C17 and C19 are pointed out) are seen in Figure 3A. Clearly the dynamics of these two phenyls are very different. The average values calculated for torsional angles Ø2 and α2 (Figure 3B and Table 1) show no major fluctuations from the standard geometry of the conformation in the DB2277-DNA complex with Ø2 close to 17° and α2 at 43° (average). Figure 4 shows major fluctuations in torsional angles for Ph1 and these are discussed below.
Figure 3.
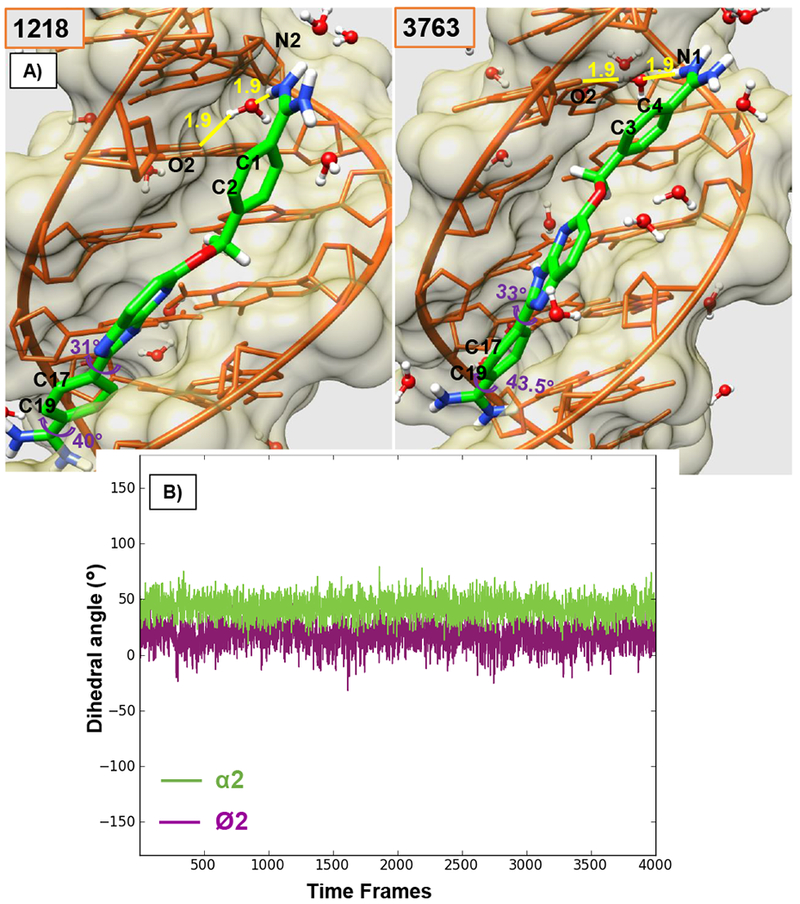
A) Minor groove views of the DB2277-DNA complex with reported Ø2 and α2 torsion angles of Phenyl Ph2 and amidine Am2 respectively, for the initial frame and the end frame for 1 ns trajectory of Ph1 flip. Colors as in Figure 2. B) Torsion angle plot of Ø2 and α2 angles of Ph2 in the bound DB2277 molecule for 1 ns trajectory. The time interval for each frame in the plot is 0.25 ps of 1 ns trajectory (1020-1021 ns of 3 μs MD simulation).
Table 1.
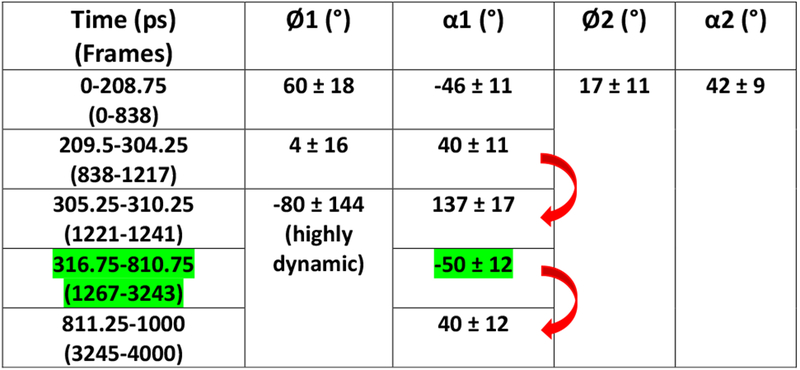
Phenyl and amidine torsion angles Ø1, Ø2, α1 and α2 of the bound DB2277 molecule during the 1-ns trajectory of Ph1 flip. Frame numbers are shown in parenthesis below the time-steps (in ps) where each frame is 0.25 ps. Zero picosecond of the flip period points at the 1020 ns of the 3 μs MD simulations. Significant shifts in the α1 value are marked in red arrow and the time range that shows stable α1 value is highlighted in green.
|
Figure 4.
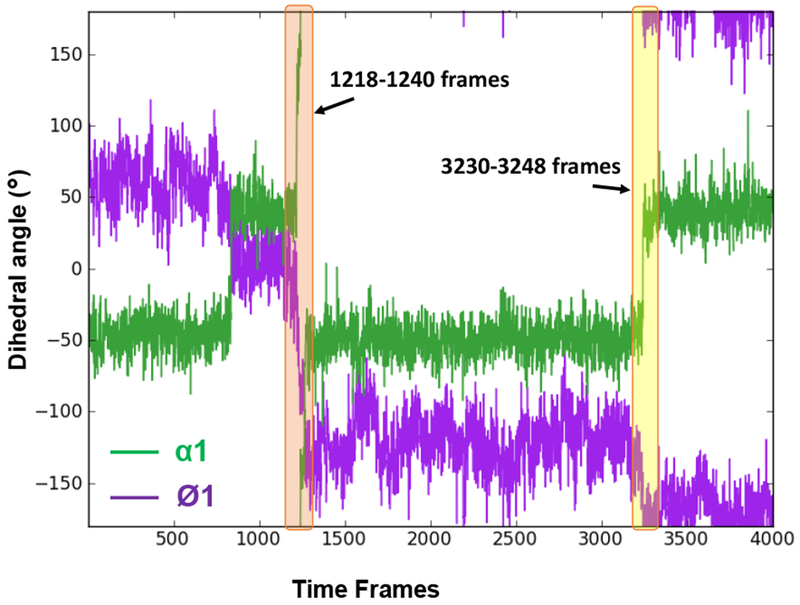
Torsion angle plot of Ø1 and α1 angles of Phenyl Ph1 and amidine Am1 respectively, of the bound DB2277 molecule for 1 ns trajectory of Ph1 flip. Major transitions of Ø1 and α1 torsions are highlighted and illustrated in time frames in Figures 2 and 5. The time interval for each frame in the plot is 0.25 ps of 1 ns trajectory (1020-1021 ns of 3 μs MD simulation).
Dynamic internal motions of Ph1 phenyl:
Ph1 was investigated in detail to understand the interplay of water, DB2277 and DNA in the flipping motion of this phenyl. The 180° rotation of Ph1 attached to –OCH2- is observed 16 times through the MD simulation. To analyze the flipping mechanism of the Ph1 phenyl, the MD simulation frames related to a typical flip, 180° rotation, at around 1 μs in the simulations, of Ph1 were observed in detail. Similar mechanisms of phenyl rotation in DB2277, however, were observed for all phenyl rotations in the simulation. Interestingly, the rotations observed for Ph1 phenyl were observed to happen in less than 1 ns and the results shown here are captured for the rotation of Ph1 in a nanosecond time period between 1020 and 1021 ns of the MD simulation.
For a high resolution investigation of the rotational motion of Ph1, the time-interval for each frame was decreased from our standard 2 ps to 0.25 ps. Up to 1218 frame of the observed nanosecond, the DB2277-AAGATA complex (Figure 2), stays close to the initial low energy conformation which is similar to the published NMR structure of the complex (PDB ID 6AST).7 In this conformation, N2 of the planar amidine, Am1, points above the plane of the aromatic groups of DB2277 (Figure 2) to facilitate the water-mediated H-bond with O2 of T16 (N2-H--O-H--O-T16). C1 and C2 Ph1 phenyl atoms (Figure 2) project out of the minor groove in this structure.
As a build up to full phenyl rotations, substantial dynamic changes in Ø1 and α1 torsion angles of Ph1 phenyl are observed within a few picoseconds i.e. around 304.5 – 310 ps range which relate to frames starting from 1218 and ending up at ~1240 (highlighted in orange block in Figure 4 and shown in Figure 2) while maintaining H-bonds of the Am1 group in the optimum range with DNA (Table 1). A large change in α1 torsional angle is observed at the 305 ps time step in the one ns flip region, i.e. from ~40° to ~137° (averaged) is highlighted by red arrow in Table 1 and Figure 4. Concerted flexible twists of the highly dynamic Ø1 and α1 torsions, in this time-range, play a key-role in maintaining the water-mediated H-bond contact with DNA that must be released before a 180° rotation can occur. The perpendicular position of Ph1 with respect to other aromatic rings of DB2277 caused by dynamic thermal motions of the atoms at 309.75 ps/1238 frame is shown in Figure 2 with the large phenyl Ø1 twist of −73° and Am close to ~150°. Highly dynamic states of Ø1 and α1 torsion angles of Ph1 between 305.25 ps and ~800 ps are evident in the Table 1 and Figure 4. This dynamic range is well illustrated by frames 1238, 1660 and 2700 in Figure 2 where a water-mediated H-bond contact of Am1 with T16-O2 (T16-O2--H-O--H-N-Am1) helps to stabilize the complex energetics in frame 1660 and 1238. The configuration of DB2277-DNA complex in frame 2700 is stabilized by a dynamic H-bond water network of the amidine and T-O2 in the minor groove of DNA (Figure 2B). These large motions of Ph1 clearly set it up for a complete rotation while no such motions are observed for Ph2 (Figure 3 and 4).
Role of Am1 twist in Ph1 flip:
Dynamic internal motions of the Am1 group play a vital role in stabilizing Ph1 in DB2277. The α1 torsion angle of amidine Am1 remains stable around ~ −50° for almost 500 ps time-span in the 1 ns trajectory as reported in Table 1 (highlighted in green) and Figure 4 before a transition in amidine group rotation. As a result of the large torsional fluctuations, the H-bond to the interfacial water is lost (Figure 5A) and Ph1 rapidly flips so that C3, C4 and N2 are pointed out of the minor groove in frame 3244 as compared to the orthogonal position of Ph1 relative to other aromatic moieties in the molecule in frame 1238 as shown in Figure 2. The total time span for 1238 to 3244 frame is around 500 ps. In the conformation shown in frame 3238 in Figure 5, N1 points inside the minor groove but is placed below the plane of Ph1 with no H-bond and water-mediated contact possible with DNA. Although the three aromatic rings of DB2277 are roughly planar in this structure, there is no H-bond stabilization of Am1 group and this structure has a higher than optimum energy. The Am1 amidine rotates to a position in the plane of Ph1 in frame 3244 and a water molecule forms an H-bond with T16-O2, but there is still no H-bond contact of the amidine Am1 with DNA (Figure 5). Significant rotation of α1 torsion angle of Ph1 of DB2277 (3244-3248 frames, Figure 5) helps to place Am1 in an optimal position to form a water-mediated H-bond contact of Am1 with T16-O2. Between time-frame 3244 and 3248 (Figure 5), the structure moves to lower energy by forming the water-mediated H-bond contact of amidine group with T16-O2 (T16-O2--2.2 Å--H-O--1.9 Å--H-N-Am1). The three aromatics are close to planar and water-mediated H-bond contacts help to stabilize this conformation. The dramatic transition of α1 torsion from ~ −50° to ~ +40° within 0.5 ps (3244 and 3245 frames) is evident in the reported 810.75 to 811.25 ps time-span marked with red arrow in Table 1. This large twist observed in α1 of the Am1 group (Table 1 and Figure 4) facilitates the full 180° rotation in Ph1 as seen in 3763 frame at 940.75 ps (Figure 3A) which is quite similar to 3248 frame (Figure 5).
Figure 5.
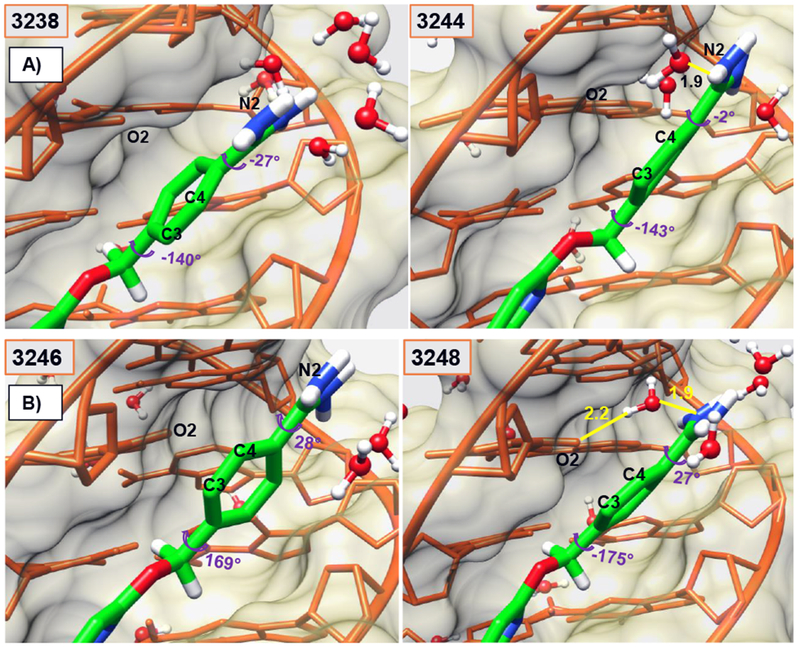
Minor groove views of the DB2277-DNA complex capturing the concerted motions of water molecules and Am1 twist to facilitate the Ph1-flip within few ps (3238-3248-frames). Flipped Ph1 in complex with C3 and C4 atoms pointed out of the minor groove. Water-mediated H-bond network of Am1 group with the minor groove of DNA helps to stabilize the structure in 3248-frame as it approaches the minimum energy compared to other frames. Water-mediated H-bonds (yellow) between T16-O2 and –NH of Am1 at the floor of the minor groove are reported in Å in different frames. Ø1 and α1 torsion angles are also reported. Colors as in Figure 2.
C3 and C4 atoms of the flipped phenyl point out of the minor groove after rotation in Figure 5. This low energy conformation of DB2277 is similar to the initial structure and remains stable for several hundred nanoseconds when Ph1 undergoes a reverse 180° rotation back to the initial state. Two key H-bonds responsible for the specificity and stability of DB2277-DNA complex (Figure 3A) remain in an optimal range in the flipped phenyl conformations of DB2277 during rotation of Ph1: G5-NH2 with N of aza-BI (3.2 Å) and C14-O2 (cytidine at position 14) with NH-aza-BI (3.2 Å). The lower phenyl Ph2 remains tightly bound in the minor groove of DNA with H-bond contact of Am2-NH with T7-O2 (2.9 Å).
The variations in the relative motions of the base pairs in DB2277-DNA complex over the 1 ns trajectory of the Ph1 flip are represented in terms of their helical parameters (minor groove width, roll, and twist-angles) displayed in Figure S1. Dynamic motions of the minor groove as the bound DB2277 phenyl-amidine undergoes large torsional fluctuations cause increases in the minor groove width of DNA (Figure S1). Thus, transient breathing motions of the complex assist in the dynamic flipping of Ph1. Helical twist calculations for DNA are consistent with the published helical analysis in the NMR structure of DB2277-DNA complex.
In summary, we have shown how the flexible -OCH2- linker in DB2277 provides freedom for the Ph1 phenyl to project away from the floor of the minor groove of DNA. The flexibility of -OCH2- Ph1 helps DB2277 to track along the minor groove curvature through forming indirect and dynamic water-mediated H-bond contacts of Am1 with the bases at the floor of the minor groove. The flexibility and water-mediated contact allow Ph1 to rotate several times in a microsecond so that we are able to observe the rotation in a bound compound for the first time. During these flips of Ph1, the aza-benzimidazole H-bonds to G-NH2 and C-O2 (cytidine) of the central GC bp, that account for the mixed sequence recognition by DB2277 and H-bonds of the amidine of Ph2 remain quite constant. Neither the aza-BI nor Ph2 show the large dynamic motions as seen with Ph1 or a 180° rotation.
Supplementary Material
ACKNOWLEDGMENT
We thank Professor David W. Boykin for DB2277 and helpful comments, and Professor Markus Germann for NMR advice and assistance.
Funding Sources
This work was supported by the National Institutes of Health [NIH-R01-GM111749 to W.D.W] and to the Molecular Basis of Disease for a Fellowship (N.K.H).
Footnotes
Supporting Information
Force field parameters of the DB2277 molecule, helical analysis of DNA bound to the DB2277 molecule.
The Supporting Information is available free of charge on the ACS Publications website.
The authors declare no competing financial interests.
REFERENCES
- 1).Chai Y, Paul A, Rettig M, Wilson WD, and Boykin DW (2014) Design and Synthesis of Heterocyclic Cations for Specific DNA Recognition: From AT-Rich to Mixed-Base-Pair DNA Sequences, J. Org. Chem 79, 852–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2).Paul A, Chai Y, Boykin DW, and Wilson WD (2015) Understanding Mixed Sequence DNA Recognition by Novel Designed Compounds: The Kinetic and Thermodynamic Behavior of Azabenzimidazole Diamidines, Biochemistry 54, 577–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3).Paul A, Nanjunda R, Kumar A, Laughlin S, Nhili R, Depauw S, Deuser SS, Chai Y, Chaudhary AS, David-Cordonnier MH, Boykin DW, and Wilson WD (2015) Mixed up minor groove binders: Convincing A.T specific compounds to recognize a G.C base pair, Bioorg. Med. Chem. Lett 25, 4927–4932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4).Guo P, Paul A, Kumar A, Farahat AA, Kumar D, Wang S, Boykin DW, and Wilson WD (2016) The Thiophene "Sigma-Hole" as a Concept for Preorganized, Specific Recognition of GC Base Pairs in the DNA Minor Groove, Chem. Eur. J 22, 15404–15412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5).Harika NK, Paul A, Stroeva E, Chai Y, Boykin DW, Germann MW, and Wilson WD (2016) Imino proton NMR guides the reprogramming of A*T specific minor groove binders for mixed base pair recognition, Nucleic Acids Res 44, 4519–4527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6).Guo P, Paul A, Kumar A, Harika NK, Wang S, Farahat AA, Boykin DW, and Wilson WD (2017) A modular design for minor groove binding and recognition of mixed base pair sequences of DNA, Chem. Commun 53, 10406–10409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7).Harika NK, Germann MW, and Wilson WD (2017) First Structure of a Designed Minor Groove Binding Heterocyclic Cation that Specifically Recognizes Mixed DNA Base Pair Sequences, Chem. Eur. J 23, 17612–17620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8).Paul A, Kumar A, Nanjunda R, Farahat AA, Boykin DW, and Wilson WD (2017) Systematic synthetic and biophysical development of mixed sequence DNA binding agents, Org. Biomol. Chem 15, 827–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9).Goodsell D, and Dickerson RE (1986) Isohelical analysis of DNA groove-binding drugs, J. Med. Chem 29, 727–733. [DOI] [PubMed] [Google Scholar]
- 10).Kopka ML, Goodsell DS, Han GW, Chiu TK, Lown JW, and Dickerson RE (1997) Defining GC-specificity in the minor groove: side-by-side binding of the di-imidazole lexitropsin to C-A-T-G-G-C-C-A-T-G, Structure 5, 1033–1046. [DOI] [PubMed] [Google Scholar]
- 11).Kissinger K, Krowicki K, Dabrowiak JC, and Lown JW (1987) Molecular recognition between oligopeptides and nucleic acids. Monocationic imidazole lexitropsins that display enhanced GC sequence dependent DNA binding, Biochemistry 26, 5590–5595. [DOI] [PubMed] [Google Scholar]
- 12).S. Goodsell D, Ng HL., Kopka M, William Lown J, and E. Dickerson R (1996) Structure of a dicationic monoimidazole lexitropsin bound to DNA, Vol. 34, p. 16654–16661. [DOI] [PubMed] [Google Scholar]
- 13).Antony-Debré I, Paul A, Leite J, Mitchell K, Kim HM, Carvajal LA, Todorova TI, Huang K, Kumar A, Farahat AA, Bartholdy B, Narayanagari S-R, Chen J, Ambesi-Impiombato A, Ferrando AA, Mantzaris I, Gavathiotis E, Verma A, Will B, Boykin DW, Wilson WD, Poon GMK, and Steidl U (2017) Pharmacological inhibition of the transcription factor PU.1 in leukemia, Eur J Clin Invest 127, 4297–4313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14).Campbell NH, Evans DA, Lee MP, Parkinson GN, and Neidle S (2006) Targeting the DNA minor groove with fused ring dicationic compounds: comparison of in silico screening and a high-resolution crystal structure, Bioorg. Med. Chem. Lett 16, 15–19. [DOI] [PubMed] [Google Scholar]
- 15).Joubert A, Sun XW, Johansson E, Bailly C, Mann J, and Neidle S (2003) Sequence-selective targeting of long stretches of the DNA minor groove by a novel dimeric bis-benzimidazole, Biochemistry 42, 5984–5992. [DOI] [PubMed] [Google Scholar]
- 16).Neidle S (2001) DNA minor-groove recognition by small molecules, Nat. Prod. Rep 18, 291–309. [DOI] [PubMed] [Google Scholar]
- 17).Rettig M, Germann MW, Ismail MA, Batista-Parra A, Munde M, Boykin DW, and Wilson WD (2012) Microscopic Rearrangement of Bound Minor Groove Binders Detected by NMR, J. Phys. Chem. B 116, 5620–5627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18).Glass LS, Nguyen B, Goodwin KD, Dardonville C, Wilson WD, Long EC, and Georgiadis MM (2009) Crystal Structure of a Trypanocidal 4,4′-Bis(imidazolinylamino)diphenylamine Bound to DNA, Biochemistry 48, 5943–5952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19).Lane AN, Jenkins TC, and Frenkiel TA (1997) Hydration and solution structure of d(CGCAAATTTGCG)2 and its complex with propamidine from NMR and molecular modelling, Biochim. Biophys. Acta 1350, 205–220. [DOI] [PubMed] [Google Scholar]
- 20).Lane AN, Jenkins TC, Brown T, and Neidle S (1991) Interaction of berenil with the EcoRI dodecamer d(CGCGAATTCGCG)2 in solution studied by NMR, Biochemistry 30, 1372–1385. [DOI] [PubMed] [Google Scholar]
- 21).Degtyareva NN, Wallace BD, Bryant AR, Loo KM, and Petty JT (2007) Hydration changes accompanying the binding of minor groove ligands with DNA, Biophys. J 92, 959–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22).Spitzer GM, Fuchs JE, Markt P, Kirchmair J, Wellenzohn B, Langer T, and Liedl KR (2008) Sequence-specific positions of water molecules at the interface between DNA and minor groove binders, ChemPhysChem 9, 2766–2771. [DOI] [PubMed] [Google Scholar]
- 23).Nguyen B, Neidle S, and Wilson WD (2009) A Role for Water Molecules in DNA−Ligand Minor Groove Recognition, Acc. Chem. Res 42, 11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24).Savelyev A, and MacKerell AD Jr. (2015) Differential Deformability of the DNA Minor Groove and Altered BI/BII Backbone Conformational Equilibrium by the Monovalent Ions Li(+), Na(+), K(+), and Rb(+) via Water-Mediated Hydrogen Bonding, J. Chem. Theory Comput 11, 4473–4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25).Lee M, Krowicki K, Shea RG, Lown JW, and Pon RT (1989) Molecular recognition between oligopeptides and nucleic acids. Specificity of binding of a monocationic bis-furan lexitropsin to DNA deduced from footprinting and 1H NMR studies, J. Mol. Recognit 2, 84–93. [DOI] [PubMed] [Google Scholar]
- 26).Kawamoto Y, Bando T, and Sugiyama H (2018) Sequence-specific DNA binding Pyrrole-imidazole polyamides and their applications, Bioorg. Med. Chem 26, 1393–1411. [DOI] [PubMed] [Google Scholar]
- 27).Dervan PB, Doss RM, and Marques MA (2005) Programmable DNA Binding Oligomers for Control of Transcription, Curr. Med. Chem 5, 373–387. [DOI] [PubMed] [Google Scholar]
- 28).Wemmer DE, and Dervan PB (1997) Targeting the minor groove of DNA, Curr Opin Struct Biol 7, 355–361. [DOI] [PubMed] [Google Scholar]
- 29).Amber 14 (2014) by Case DA, Babin V, Berryman JT, et al.
- 30).Špačková N.a., Cheatham TE, Ryjáček F, Lankaš F, van Meervelt L, Hobza P and Šponer J (2003) Molecular Dynamics Simulations and Thermodynamics Analysis of DNA−Drug Complexes. Minor Groove Binding between 4‘, 6-Diamidino-2-phenylindole and DNA Duplexes in Solution. J. Am. Chem. Soc 125, 1759–1769. [DOI] [PubMed] [Google Scholar]
- 31).Athri P and Wilson WD (2009). “Molecular Dynamics of Water-Mediated Interactions of a Linear Benzimidazole–Biphenyl Diamidine with the DNA Minor Groove.” J. Am. Chem. Soc 131(22): 7618–7625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32).Galindo-Murillo R, Robertson JC, Zgarbová M, Šponer J, Otyepka M, Jurečka P, and Cheatham TE (2016) Assessing the Current State of Amber Force Field Modifications for DNA, J. Chem. Theory Comput 12, 4114–4127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33).Auffinger P, and Westhof E (1998) Simulations of the molecular dynamics of nucleic acids, Curr Opin Struct Biol 8, 227–236. [DOI] [PubMed] [Google Scholar]
- 34).Beveridge DL, and McConnell KJ (2000) Nucleic acids: theory and computer simulation, Y2K, Curr Opin Struct Biol 10, 182–196. [DOI] [PubMed] [Google Scholar]
- 35).Cheatham TE 3rd, and Kollman PA (2000) Molecular dynamics simulation of nucleic acids, Annu. Rev. Phys. Chem 51, 435–471. [DOI] [PubMed] [Google Scholar]
- 36).Perez A, Marchan I, Svozil D, Sponer J, Cheatham TE 3rd, Laughton C and Orozco M (2007) Refinement of the AMBER force field for nucleic acids: Improving the description of alpha/gamma conformers. Biophys. J, 92, 3817–3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37).Cheatham TE 3rd, and Case DA (2013) Twenty-five years of nucleic acid simulations, Biopolymers 99, 969–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38).Lavery R, Moakher M, Maddocks JH, Petkeviciute D, Zakrzewska K; Conformational analysis of nucleic acids revisited: Curves+. Nucleic Acids Res 2009; 37 (17): 5917–5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39).Roe DR and Cheatham TE (2013) PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput 9, 3084–3095. [DOI] [PubMed] [Google Scholar]
- 40).Humphrey W, Dalke A and Schulten K (1996) VMD: Visual molecular dynamics. J. Mol. Graph 14, 33–38. [DOI] [PubMed] [Google Scholar]
- 41).Couch GS, Hendrix DK and Ferrin TE (2006) Nucleic acid visualization with UCSF Chimera. Nucleic Acids Res 34, e29–e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42).Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC and Ferrin TE (2004) UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem, 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- 43).Embrey KJ, Searle MS, and Craik DJ (1993) Interaction of Hoechst 33258 with the minor groove of the A+T-rich DNA duplex d(GGTAATTACC)2 studied in solution by NMR spectroscopy, Eur. J. Biochem 211, 437–447. [DOI] [PubMed] [Google Scholar]
- 44).Searle MS, and Embrey KJ (1990) Sequence-specific interaction of Hoechst 33258 with the minor groove of an adenine-tract DNA duplex studied in solution by 1H NMR spectroscopy, Nucleic Acids Res 18, 3753–3762. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.