

Abstract
Severe malaria is caused by the apicomplexan parasite Plasmodium falciparum. Despite decades of research the unique biology of these parasites has made it challenging to establish high throughput genetic approaches for identification of therapeutic targets. Using transposon mutagenesis of P. falciparum in an approach that exploited its AT-rich genome we generated >38,000 mutants, saturating the genome and defining fitness costs for 95% of genes. Of 5,399 genes we found ~3,000 genes are essential for optimal growth of asexual blood-stages in vitro. Our study defines ∼1000 essential genes, including genes associated with drug resistance, vaccine candidates, and conserved proteins of unknown function. We validated this approach by testing proteasome pathways for individual mutants associated with artemisinin sensitivity.
Original
Transposon mutagenesis of Plasmodium falciparum was used to generate >38,000 mutants, saturating the genome and defining fitness costs for 95% of genes. We functionally define the relative fitness cost of disruption for 5,399 genes, and find that ~3,000 genes, ~62% of the genome, are essential for optimal asexual blood-stage in vitro growth. Our study defines ∼1000 essential genes, including genes associated with drug resistance, leading vaccine candidates, and hundreds of Plasmodium-conserved proteins of unknown function that are now potential therapeutic intervention targets. We experimentally validated the essentiality of proteasome pathways with drug studies of individual mutants associated with artemisinin sensitivity. This study defines high-priority targets and pathways and points the way for the future of P. falciparum high throughput genetics.
One Sentence Summary:
Saturation-scale mutagenesis of Plasmodium falciparum reveals a core set of genes essential for asexual blood-stage growth in vitro.
Graphical Abstract
INTRODUCTION:
Malaria remains a devastating global parasitic disease, with the majority of malaria deaths caused by the highly virulent Plasmodium falciparum. The extreme AT-bias of the P. falciparum genome has hampered genetic studies through targeted approaches such as homologous recombination or CRISPR-Cas9, and only a few hundred P. falciparum mutants have been experimentally generated in the past decades. In this study, we have used high throughput piggyBac transposon insertional mutagenesis and Quantitative Insertion Site Sequencing (QIseq) to reach saturation-level mutagenesis of this parasite.
RATIONALE:
Our study exploits the AT-richness of P. falciparum genome, which provides numerous piggyBac transposon insertion targets within both gene coding and non-coding flanking sequences, to generate over 38,000 P. falciparum mutants. At this level of mutagenesis, we could distinguish essential genes as non-mutable and dispensable genes as mutable. Subsequently, we identified 3,357 genes essential for in vitro asexual blood-stage growth.
RESULTS:
We calculated Mutagenesis Index Scores (MIS) and Mutagenesis Fitness Scores (MFS) to functionally define the relative fitness cost of disruption for 5,399 genes. A competitive growth phenotype screen confirmed that MIS and MFS were predictive of the fitness cost for in vitro asexual growth. Genes predicted to be essential included genes implicated in drug resistance, such as the “K13” Kelch propeller, mdr and dhfr-ts, as well as targets considered to be high-value for drugs development such as pkg, and cdpk5. The screen revealed essential genes that are specific to human Plasmodium parasites but absent from rodent-infective species, such as lipid metabolic genes that may be crucial to transmission commitment in human infections. MIS and MFS profiling provides a clear ranking of the relative essentiality of gene ontology (GO) functions in P. falciparum. GO pathways associated with translation, RNA metabolism, and cell cycle control are more essential, whereas genes associated with protein phosphorylation, virulence factors, and transcription are more likely to be dispensable. Finally, we confirm that the proteasome-degradation pathway is a high-value druggable target based on its high ratio of essential:dispensable genes, and by functionally confirming its link to the mode of action of artemisinin, the current front-line antimalarial.
CONCLUSION:
Saturation-scale mutagenesis allows prioritization of intervention targets in the genome of the most important cause of malaria. The identification of the essential genome, consisting of over 3000 genes, will be valuable for antimalarial therapeutic research.
Saturation-scale mutagenesis of Plasmodium falciparum reveals genes essential and dispensable for asexual blood-stage development.
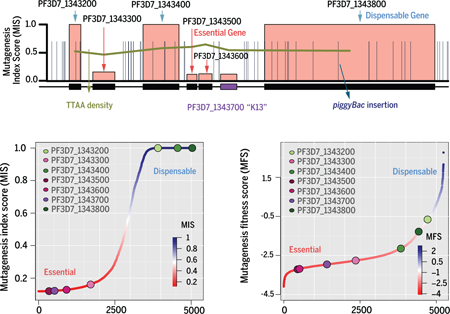
(A) The Mutagenesis Index Score (MIS) rates the potential mutability of P. falciparum genes based on the number of recovered CDS insertions relative to the potential number that could be recovered by large-scale mutagenesis. Genes highlighted are those previously validated to be dispensable (var, eba175, orc-1) or essential (cdpk5, kelch K13, dhfr-ts, pkg). (B) A high-resolution map of a ~50 KB region of chromosome 13 depicts an essential gene cluster, including K13, that lack insertions in the CDS but is flanked by dispensable genes with multiple CDS-disrupting insertions. (C) Conserved essential genes of known (PF3D7_1032900, RNA polymerase II-associated protein 1, putative, PF3D7_1033100, S-adenosylmethionine decarboxylase/ornithine decarboxylase) and unknown function (PF3D7_1033000) were concentrated in syntenic blocks whereas dispensable genes were significantly more common in the syntenic breaks, revealing an evolutionarily-conserved genomic architecture.
Malaria caused by Plasmodium falciparum remains an insidious global health problem, with hundreds of thousands of deaths each year. Recently there have been significant reductions in disease intensity, in part through concerted recent use of artemisinin combination therapies (ACTs), but these gains are now threatened by emerging ACT treatment failures spreading across South East Asia (1, 2). If ACT resistance reaches Africa, a devastating rebound of disease is expected, as occurred with chloroquine resistance in the 1970s. The development of new antimalarial therapies, and identification and prioritization of new targets, is a priority. More than a decade after the completion of the P. falciparum genome, a significant fraction of its genome still lacks functional annotation (3). Although CRISPR-Cas9 and other targeted endonucleases will accelerate functional genomics studies (4–6), their usefulness for genome-scale applications is restricted by the absence of non-homologous end joining in Plasmodium parasites, and by the extreme AT-richness of the P. falciparum genome which reduces gRNA target site abundance. Therefore, a critical need remains for large-scale genetic analysis to systematically identify essential genes and prioritize parasite metabolic pathways for drug discovery (7).
Large-scale genetic screening methods in model organisms rely on efficient scalable methods for genome engineering. Transposon mutagenesis using the piggyBac transposon, which preferentially inserts at the tetranucleotide target sequence TTAA, has been used to carry out whole-genome loss-of-function screens in many organisms (8–11). The highly skewed nucleotide composition of the P. falciparum genome, with >81% AT content, is an advantage for the application of piggyBac mutagenesis. The skewed composition results in a high density of TTAA sites, averaging one site per 70 bp over both coding and non-coding regions, in theory allowing systematic and saturation-level mutagenesis of the whole genome. While piggyBac mutagenesis has been developed and optimized for P. falciparum, and previously used for small-scale phenotypic screens and functional characterization of loss-of-function mutants, it has not been used for large-scale screening (12–16). To scale up piggyBac mutagenesis in P. falciparum, we developed high-throughput transfection mutagenesis methods that mostly create a single insertion per genome, and combined them with an Illumina-based sequencing method for identifying transposon insertion sites (17, 18). This approach, known as Quantitative Insertion-site Sequencing (QIseq), thus allowed whole-genome experimental mutagenesis analysis of P. falciparum (fig. S1, A and B).
Achieving saturation-level mutagenesis
In a preliminary study we carried out large-scale transfections followed by short-term in vitro growth of mixed pools of drug-selected P. falciparum parasites and identified insertion sites by QIseq. This pilot identified a total of 3,651 insertions across the P. falciparum genome (table S1)(17, 19). Based on the density and distribution of those insertions modeled by a Negative Binomial Distribution, we predicted that recovery of ≥33,000 insertions would be sufficient to achieve saturation-level mutagenesis in the compact genome of the malaria parasite (fig. S1C-F); this number is sufficient for there to be a high probability that multiple transposon insertions would occur within the CDS of every protein-coding gene larger than 500 bp. We therefore scaled our transfection methods to achieve this number of insertions. Transfected parasite populations were drug-selected briefly to isolate only mutant parasites with integrated piggyBac elements carrying the drug resistance gene hdhfr, before insertion-site locations were identified by QIseq. Computational analyses were used to verify the sequence reads of the QIseq libraries and the consistency of the raw data (fig. S2A-F); nearly all QIseq-defined insertions occurred at the characteristic TTAA target sequence. All insertions sites that were not flanked by the consensus TTAA (2.49%) insertion site were removed from subsequent analysis (fig. S2A). Previously validated individual piggyBac mutant clones were included in each QIseq run to act as a control for the accuracy and sensitivity of each insertion-site identification run (fig. S3A-C and table S2).
The saturation mutagenesis approach identified ~38,000 independent piggyBac insertions at distinct TTAA target sites, covering 5,399 nuclear protein-coding genes across all 14 chromosomes of the P. falciparum genome (Fig. 1A and table S3). These randomly distributed insertions exceeded those predicted to be required to achieve saturation-level mutagenesis (Fig. 1A), but there were numerous genes and regions encompassing several genes that had significantly fewer insertions than would be expected based purely on the distribution of TTAA-sites across the genome (Fig. 1B, C and D). As well as discrepancies in non-random spatial distribution, coding regions overall lacked insertions compared with intergenic regions (p > 2.2e-16, Fisher test) (Fig. 1D). A more detailed analysis of piggyBac insertion density within transcriptional units revealed that insertions in CDS were threefold less common than in flanking intergenic regions, and even within intergenic regions, insertion-site density decreased with proximity to CDS (Fig. 1E). This significant bias towards recovering intergenic insertions in surviving blood-stage parasites, with many CDS having no insertions, are indications that genes lacking insertions are lethal when disrupted by piggyBac insertion.
Fig. 1. A genome-wide saturation mutagenesis screen for Plasmodium falciparum.
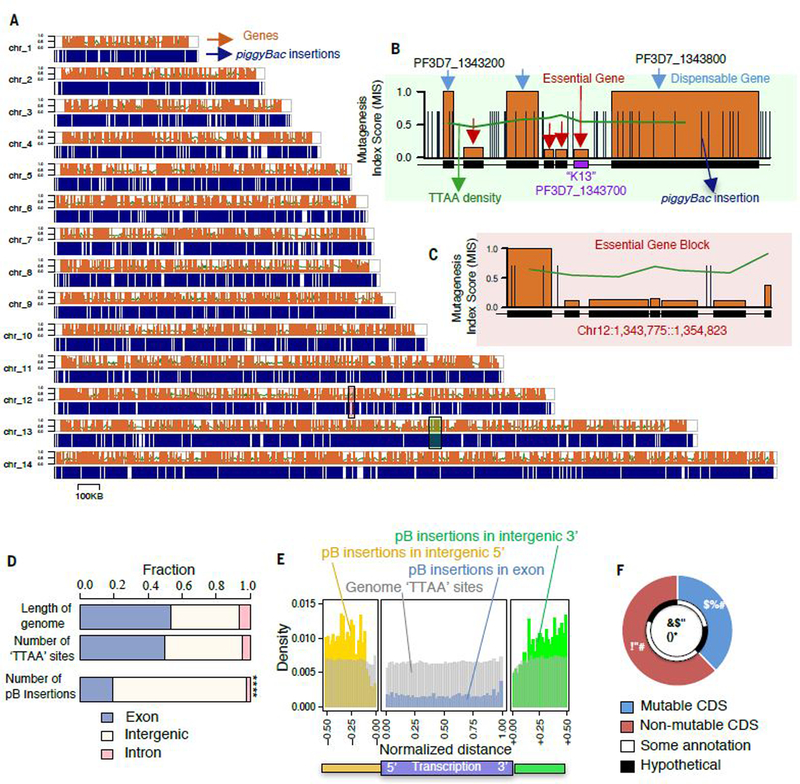
(A) Chromosomal map displays 38,173 piggyBac insertion sites from all mutants evenly distributed throughout the genome. (B) High-resolution map of a ~50 KB region of chromosome 13 depicts an essential gene cluster, including K13, flanked by dispensable genes with multiple CDS-disrupting insertions. (C) High-resolution map of a ~20 KB region without insertions includes three conserved genes of unknown function (PF3D7_1232700, PF3D7_1232800, PF3D7_1232900) and a putative nucleotidyltransferase (PF3D7_1232600) (see also fig. S5). (D) A plot of all piggyBac insertions revealed significantly fewer insertions were recovered from exon-intron regions compared to the proportion of available TTAA sites (see also fig. S1D) (p < 2.2e-16, Fisher’s Exact test). (E) Density of piggyBac insertion-site distribution revealed threefold fewer insertions recovered in transcriptional regions (blue) than intergenic 5ʹ (yellow) and 3ʹ (green) regions, depicted as relative distance upstream and downstream to a gene, respectively. (F) This study determined that under ideal culture conditions for asexual blood-stage growth, 38% of genes in the P. falciparum genome have mutable CDSs, while 62% of genes have non-mutable CDSs.
Defining gene dispensability using mutagenesis index scores
While there was a bias against insertions in CDSs, 2,042 genes were disrupted by at least one piggyBac insertion (~38% of genes in the P. falciparum genome) (Fig. 1F). The remainder, 3,357 genes (~62% of the genome) had no insertions in their CDSs, and some of these were also completely devoid of insertions in the surrounding intergenic regions (2.9% of genes) (Fig. 1C, fig. S4A and table S4). In the 2,042 mutable genes, insertion sites were distributed uniformly along the gene body CDSs, indicating all disruptions have equivalent consequences for the disrupted gene (Fig. 1E). The uniform distribution of transposon insertions throughout the genome is independent from chromatin structure and gene locations (fig. S4B). We therefore reasoned that the recovery of one or more insertions within the CDS of disrupted genes was an indicator of gene dispensability for in vitro asexual blood-stage growth. Therefore, mutable genes are subsequently referred to as dispensable, while the absence of any insertions in the CDS could be considered an indicator that disruptions are lethal, and subsequently these genes are referred to as essential. To quantify the evidence for dispensability or essentiality of each gene we developed a mutagenesis index score (MIS) based on the number of identified piggyBac insertions relative to the number of available TTAA target sites within that gene (Fig. 2A and table S5). Genes with higher MIS (on a scale of 0 to 1) were considered to have a higher possibility of dispensability, while those with lower MIS were considered to have a higher possibility of essentiality. The 2000 genes with highest and lowest MIS represent mutability characterization with the strongest confidence.
Fig. 2. Identification of dispensable and essential genes through Mutagenesis Index Score (MIS) and Fitness Score (MFS).
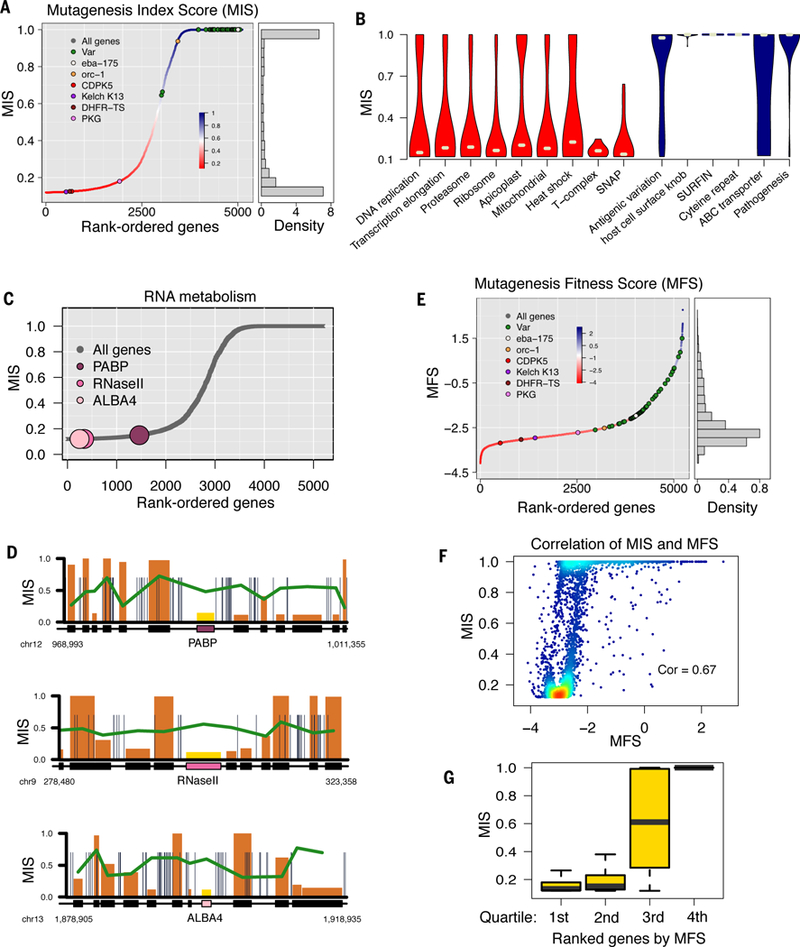
(A) The Mutagenesis Index Score (MIS) rates the potential mutability of P. falciparum genes based on the number of recovered CDS insertions relative to the potential number that could be recovered. Genes known as dispensable or essential are highlighted. (B) MIS violin plots of GO processes grouped from lowest to highest dispensability, according to gene functional annotations. (C) MIS plots and (D) high-resolution chromosome maps highlighting important genes of interest for RNA metabolism [−20kb, +20kb] (see also fig. S4 for MIS plots of other genes of interest. (E) The Mutagenesis Fitness Score (MFS) estimates the relative growth fitness cost for mutating a gene based on its normalized QIseq sequencing reads distribution. (F) MIS has significant correlation to MFS (Pearson’s R = 0.67, p < 2.2e-16 compared with permutation). (G) The first and second MFS quartiles were comprised primarily of non-mutable genes, the 4th quartile was comprised mostly of mutable genes, and 3rd quartile had nearly equal numbers of both.
Biological processes identified as dispensable and essential
Mutants in the lowest MIS quartile were enriched in core metabolic processes shared across eukaryotes, which would be predicted to be essential. Mutants in the highest MIS quartile were enriched in parasite-specific multi-gene families that interact directly with the host, and which are known to be partially functionally overlapping and contain many redundant genes (Fig. 2B). It is important to note that this screen was carried out using in vitro cultured parasites, and many of these in vitro dispensable genes, such as those for antigenic variation and cytoadherence, have greater importance in vivo in human infections (fig. S5). The dispensability of parasite processes involved in host-parasite interactions contrasted with the essentiality of internal parasite processes, such as those associated with RNA metabolism (Fig. 2C-D, fig. S6 and table S6). RNase II, a gene implicated in a post-transcriptional regulatory mechanism relevant to severe malaria, is an example with a low MIS score, consistent with previous reports of the locus being refractory to disruption (20). Other genes involved in general RNA metabolism that have low MIS include both widely-conserved RNA-binding proteins (such as PABP) and apicomplexan-specific RNA-binding proteins likely to have more specific, mostly unidentified, regulatory targets. The P. falciparum genome has an abundance of RNA-binding proteins that are largely functionally uncharacterized and our analysis suggests that many of these genes are likely to be essential.
MIS therefore correlates with what is known broadly about the importance or redundancy of metabolic pathways. To further validate that MIS is a good predictor of essentiality, we analyzed genes that have been the focus of drug or vaccine development. Genes strongly predicted to be essential based on MIS included the “K13” Kelch propeller implicated in artemisinin resistance, as well as other genes implicated in drug resistance, such as DHFR-TS, MDR and AAC2, involved in pyrimethamine, mefloquine and atovaquone reistance, respectively. Other genes classified as essential based on MIS included high priority blood-stage drug targets such as PKG, and CDPK5. By contrast, most blood-stage vaccine candidates were dispensable, with the notable exception of RH5. Not unexpectedly, sporozoite vaccine candidates CSP and TRAP, which are essential for sporozoite development in mosquitoes but are not required in blood-stage development (21), had high MIS. In contrast, the gene for pore-forming protein Cell Traversal of Ookinetes and Sporozoites (CelTOS) had low MIS. CelTOS is important for these parasite migratory phases and is emerging as a pre-erythrocytic-transmission blocking vaccine candidate; its low MIS indicates it may also have an essential function in blood-stage infections, making it a potential multi-stage vaccine target. MIS analysis also revealed likely essential genes that are specific to human Plasmodium parasites but absent from rodent-infective species, such as the lipid metabolic genes (PCD, PMT) that are crucial in the development of mosquito-transmissible sexual-stages in P. falciparum (22, 23).
Mutant growth fitness as a measure of gene dispensability
We previously developed a phenotyping method in which a pool of piggyBac mutants were grown as mixed-mutant pools over multiple generations, and the number of reads for each piggyBac insertion site quantified using next-gen sequencing to measure parasite growth rates (17). We adapted this method to generate a second quantitative measure of gene dispensability independent from MIS, by comparing the normalized number of reads from each insertion site to the total pool of reads across all P. falciparum mutants in the saturation mutagenesis screen (Fig. 2E and table S5). This mutagenesis fitness score (MFS) serves as a proxy for mutant growth fitness. MFS scores strongly correlated with MIS scores, despite the fact that they are independent of each other (Fig. 2F). The MFS of mutable genes was high, in keeping with the MIS prediction of dispensability, while predicted essential non-mutable genes had a low MFS (Fig. 2G). Intermediate MIS and MFS scores were weakly correlated, in keeping with the lower confidence that we can place on the essentiality or dispensability of genes with these intermediate scores.
A competitive in vitro growth screen (17) was used to phenotype four separate pools of mixed-mutant populations to provide additional validation of the correlation between MIS and a gene’s importance for growth fitness. Mutable genes with high MIS had low or minimal fitness cost were competitive growth ‘winners’, whereas growth fitness ‘losers’ had relatively low MIS (Fig. 3A and table S7). Relatively few mutations had little or no fitness cost, as measured by MFS, resulting in a disproportionate number of mutant ‘losers’ (Fig. 3B). Overall both the MIS and MFS were predictive of the fitness cost for in vitro asexual growth (Fig. 3C), and between these metrics we were able to predict separate 5,399 genes in the P. falciparum genome into non-mutable and mutable categories (Fig. 3D and E).
Fig. 3. Validation of mutagenesis score through phenotype screen.
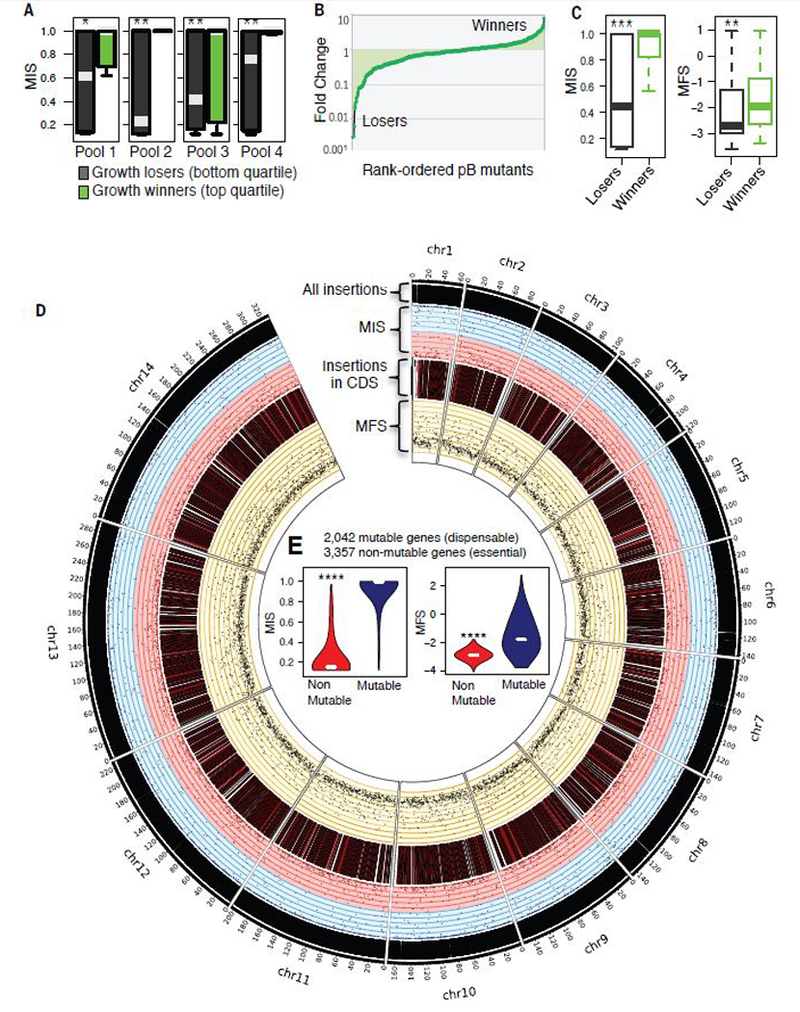
(A) Competitive growth assays of asexual blood-stage growth under ideal in vitro culture conditions: phenotypes of four independent mixed-population pools grown for three cycles confirmed losers (left, bottom quantile) and winners (right, top quartile) had significantly different MIS. (B) Overall rank-ordered plot of competitive growth phenotypes shows losers and winners. (C) Competitive growth ‘losers’ had significantly lower MIS and MFS, respectively, validating MIS and MFS as predictor of gene essentiality and dispensability. (D) Circos plot from outer to inner shows the distribution of all piggyBac insertions, MIS (pink indicates MIS < 0.5, while blue is >0.5), CDS insertions, and MFS along each chromosome of P. falciparum genome. (E) Violin plots indicate non-mutable genes had significantly lower MIS and MFS (‘****’ represents Wilcoxon p < 2.2e-16).
Association of essentiality with genome structure and transcription patterns
Dispensable and essential genes rarely occurred as single isolated genes, but rather occurred as multi-gene clusters reminiscent of conserved syntenic blocks (Fig. 1A, C and fig. S4A)(24). We therefore assessed the relationship between evolutionary conservation of genome structure and essentiality/dispensability by using previously defined syntenic relationships between Plasmodium spp. (25). Syntenic genes indeed had lower MIS, suggesting conserved essential functions across Plasmodium spp.; conversely, non-syntenic genes were more likely to be dispensable (Fig. 4A). Mapping chromosomal regions with synteny breaks, which typically harbor gene duplications and paralogs, showed patterns of clustering of essential and dispensable genes in syntenic and non-syntenic chromosomal regions, as seen in examples for chromosomes 13 and 10 (Fig. 4B-E).
Fig. 4. Chromosomal syntenic breakpoints are enriched in dispensable genes.
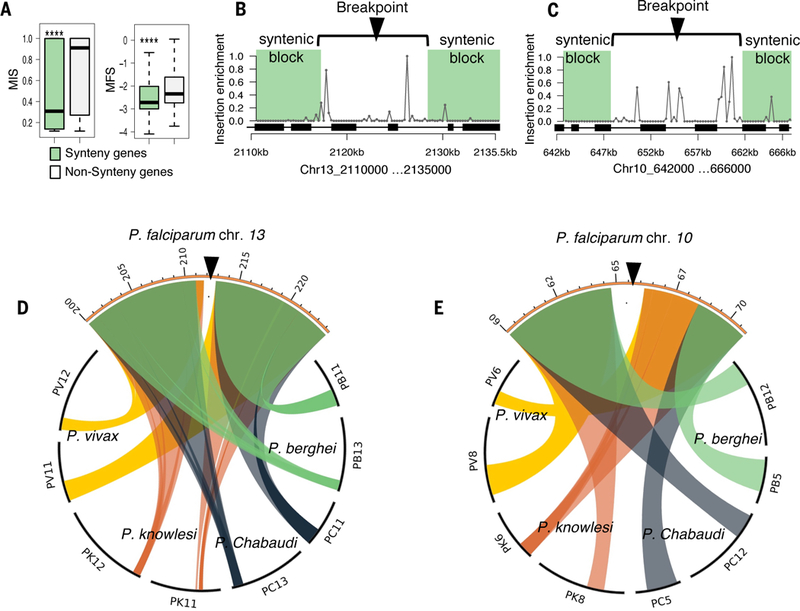
(A) Genes within conserved syntenic blocks have significantly lower MIS and MFS (Wilcoxon p < 2.2e-16). Syntenic genes or “syntenic block” is defined as at least three genes in the same order on the same chromosome as their orthologs in another species within a 25-kb search window. (B and C) Scatter plots show the insertion site enrichment along two syntenic breakpoints (Ch13:2,110,000 −2,135,000, Chr10: 642000–666000). Each gap in synteny (white area) is enriched for piggyBac insertions while flanked by essential regions (green shading); black boxes represent the location of CDS. (D and E) Circos plots indicate the syntenic blocks of P. falciparum in relation to other Plasmodium spp. (P. berghei, P. chabaudi, P. knowlesi, P. vivax).
Transcription metrics for each gene based on maximum FPKM values (26) were examined for correlations with essentiality and dispensability (Fig. 5A). This analysis showed that essential genes are expressed at significantly higher levels throughout both the asexual and sexual life cycle phases, indicating they may have critical functional roles throughout both the mosquito and human stages and therefore represent potential multi-stage drug targets (Figs. 5B and C). Within the intraerythrocytic cycle, genes with peak expression during the trophozoite stage were more likely to be essential than those expressed during other stages (Fig. 5C). During the trophozoite stage, P. falciparum must acquire nutrients from the host and remodel the infected erythrocyte to avoid host defenses, such as clearance by the spleen. In contrast, dispensable processes were enriched at either end of intraerythrocytic development, reflecting parasite stages with a greater need for functional redundancy, such as those involved in host interaction (e.g., erythrocyte invasion, antigenic variation), and are hence more likely to undergo duplication/paralog evolution. Not unexpectedly, about 41% of the dispensable genes are predominantly expressed in sexual stages (Fig. 5C). These genes may be essential for transmission to or from the mosquito, but do not appear to have an essential function for asexual blood-stage development. The high coverage of sexual-stage genes in this screen emphasizes the potential for piggyBac-based screens for identifying sexual phenotypes.
Fig. 5. Distinct biological process and evolutionarily conservation segregate the tendency of dispensable and essential genes.
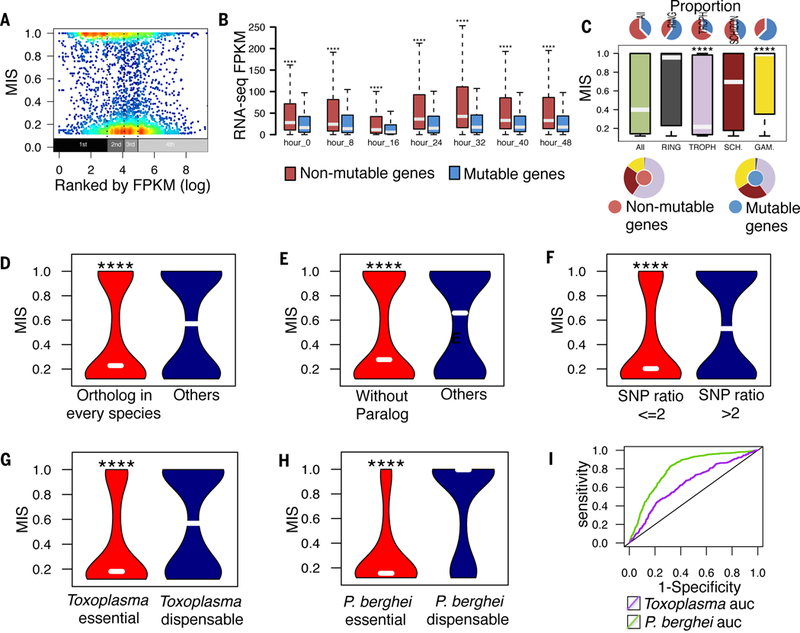
(A) The genes with lowest FPKM expression value (first quantile) among different stages were enriched for dispensable genes (Wilcoxon P < 2.2e-16 compared with other quantiles)(26). The expression level cut off is set at 20 FPKM. (B) Non-mutable essential genes had significantly higher expression value for blood-stage development. (C) The group of trophozoite-stage genes had the highest proportion of essential genes (red) whereas gametocyte genes had the highest proportion of dispensable genes (blue) (Wilcoxon p < 1e-12). (D to F) Characteristics of essential genes significantly different from dispensable genes include: (D) 1:1 ortholog conserved among Plasmodium spp; (E) absence of paralogs; and (F) reduced rate of non-synonymous to synonymous SNPs. Bars indicate the group median (‘****’ indicates Wilcoxon p < 2.2e-16). (G and H) Essential genes reported in (G) Toxoplasma and (H) P. berghei showed significantly lower MIS in this mutagenesis screen of P. falciparum (‘****’ indicates Wilcoxon p < 2.2e-16). (I). Plot of Receiver Operating Characteristics (ROC) indicate the level of retention of essential genes across species. The MIS of P. falciparum more strongly correlates with the essentiality phenotype of P. berghei than Toxoplasma.
Evolutionary conservation of apicomplexan gene essentiality
We compared the data from our genome-wide saturation screen with data from large-scale genome sequencing studies of P. falciparum, as well as data from recent large-scale mutagenesis studies of apicomplexan genomes (6, 18, 27–29) to evaluate the conservation of gene essentiality across the organisms’ lineages. Essential P. falciparum genes were more highly conserved across Plasmodium spp. (Fig. 5D and fig. S8A), are more likely to have a paralog (Fig. 5E and fig. S8B), and encode genes with less genetic variation among P. falciparum clinical isolates (Fig. 5F and fig. S8C). Of the P. falciparum genes studied here, 2,083 and 1,998 have orthologs in the more distantly-related parasites Toxoplasma gondii (6) and P. berghei (29), respectively. Overall, there was a strong correlation in gene function between species, particularly with genes predicted as essential (Fig. 5G and H and fig. S8D and E). Genes that are dispensable in P. falciparum were also more likely to be dispensable in P. berghei. By contrast, the correlation between the dispensable genes of P. falciparum and T. gondii was weaker, indicating that genus-specific gene functions are enriched for dispensability. Overall, these comparisons show that while phenotype classifications from the large-scale mutagenesis screens in apicomplexan species are also broadly predictive of the P. falciparum phenotype classifications, the closer evolutionary relationship of P. berghei provides a higher level of sensitivity and specificity for the orthologs that could be disrupted (Fig. 5I and fig. S8F).
Consistent with this assessment that non-mutable genes represented core essential functions in P. falciparum, GO enrichment analysis indicated genes with functions associated with translation, RNA metabolism, and cell cycle control were more likely to be essential, whereas genes associated with protein phosphorylation, virulence factors, and transcription were more likely to be dispensable (Fig. 6A). Comparing the relative number of essential, versus dispensable, genes within specific GO biological processes, molecular functions, and cellular components provided a ranking of the essentiality of potential druggable targets and pathways (Figs. 6A-C, figs. S9A-C and table S8). For example, nearly all genes annotated as being involved in ubiquitin-dependent degradation processes were experimentally defined as essential, while individual genes linked to microtubule motility and antigenic variation are experimentally defined as dispensable based on MIS. RNA metabolism and translation-related processes are also highly essential, supporting emerging evidence for the importance of post-transcriptional and translational control (30).
Fig. 6. Differentiating dispensable and essential genes and discovering high-priority druggable targets and pathways.
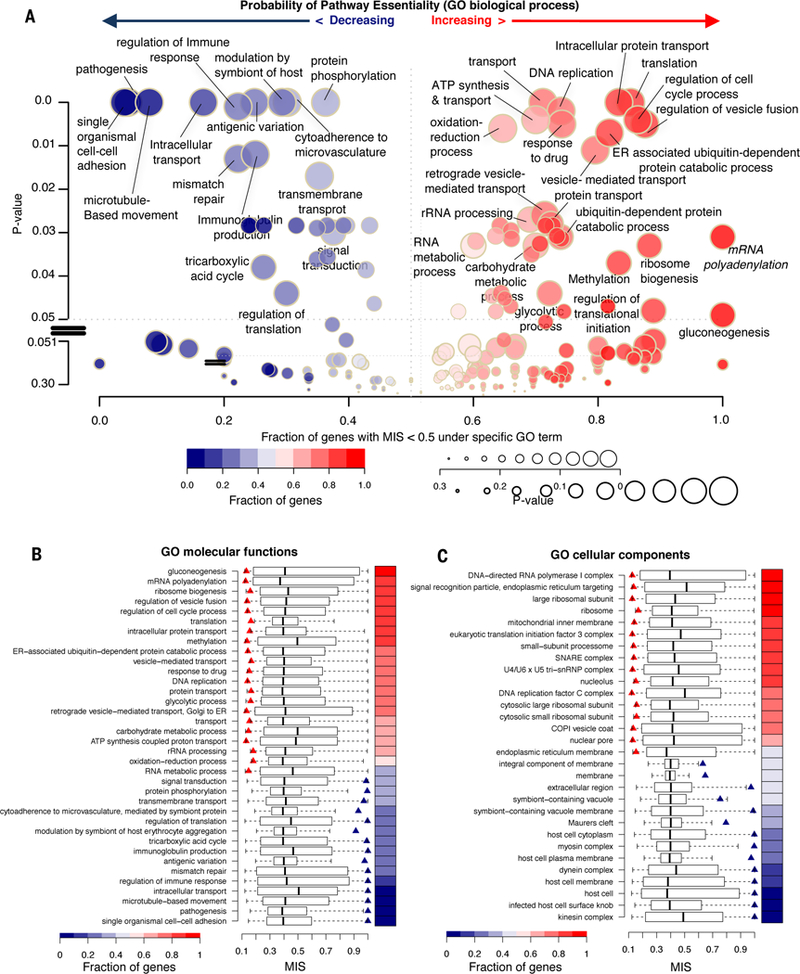
(A) Functional annotations of biological processes are represented by the p-value and the X-axis shows the fraction of the genes with MIS > 0.5. Each GO term is assigned a p-value on the Y-axis to represent the tendency to be essential or dispensable. Essentiality is indicated on a spectrum of red (essential) to blue (dispensable) and circle sizes indicate the GO term enrichment. (B and C) Boxplot of (B) molecular processes and (C) cellular components shows the MIS distribution generated by 1000× sampling of the number of genes in the query GO-term category. Left (red) and right (blue) triangles indicate GO terms with significantly lower or higher MIS (p-value < 0.05 compared to background), respectively; the heatmap represents the essentiality defined as the fraction of genes per GO term with MIS > 0.5.
Essentiality of the proteasome-degradation pathway
Recent genetic analysis of artemisinin-combination therapy (ACT) resistance has linked drug resistance to cellular stress-response mechanisms involving the P. falciparum ubiquitin/proteasome system (31, 32). Genes of the proteasome-degradation pathway were well represented in the piggyBac insertion set, and 54 of the 72 genes could be classified as essential genes based on our data (fig. S10A), further strengthening the priority of the proteasome degradation pathway as a high-value druggable target (33). We validated this link between ART sensitivity and proteasome inhibition sensitivity using a set of single-insertion piggyBac mutants previously defined by chemogenomic profiling to be part of an ART-sensitivity cluster, including a mutant of the K13 Kelch propeller gene (16, 34). We found ten-fold increased sensitivity to the proteasome inhibitor Bortezomib in mutants of the ART-sensitivity cluster; significantly different relative to wild-type NF54 or mutants with distinct chemogenomic profiles (fig. S10B-E), providing additional support for the association between the ART mechanism of action and proteasome function.
Summary
In conclusion, our genome-wide saturation mutagenesis screen in the major human pathogen, P. falciparum has defined genes essential for parasite survival during the blood stage and provided critical functional data for prioritizing high-value drug targets and pathways. The complete characterization of the essential genome with high-throughput saturation mutagenesis will help open new frontiers for antimalarial therapeutic research. The methodology also opens the way for new systematic functional screens for other phenotypes, such as transmission and cytoadherence.
Materials and Methods
Parasite culture
All P. falciparum parasite NF54 and piggyBac mutants were cultured in complete RPMI 1640 with 5% hematocrit (medium containing 0.5% Albumax II, 0.25% sodium bicarbonate and 0.01 mg/ml gentamicin). All parasite cultures were maintained by standard methods (35).
piggyBac Transfection (36)
High-efficiency transfection (96-well plate method) was carried out on NF54 schizonts purified by magnetic column (MiltyenyiBiotec, CS column) using a transposon plasmid (pLBacII-HDH, containing selection marker human dhfr) and a transposase-expressing helper plasmid (pDCTH)(19). RBCs were first loaded with plasmid DNA by electroporation using Gene PulserXCell+CE Module (BioRad). A total of 10 million schizonts, 1200 μg plasmid DNA and 600 μg helper plasmid DNA were used per 96-well plate to achieve maximal transfection efficiency. The drug WR99210 (final concentration 2.5 nM) was added to each plate for selecting transfected parasites, using a robot (Integra VIAFLO 96) to change media with drug every day for 5 days. Transfections were performed in batches over the course of 4 to 6 weeks. Each 96-well plate was cultured in triplicate and cryopreserved in duplicate. One hundred 96-well plates were used to generate the large pools for sequencing (fig. S1A).
For maximum recovery of unique mutants, the transfected culture was distributed into 96-well plates by robot (200μl each well) and grown under two rounds of drug selection. Wells were screened for parasites by PCR and Giemsa-stained thin blood films. >50% of wells became parasite-positive within three weeks. Each positive well with ≥9 unique mutants (one well = one “Mixed Population”, or MP) was cryopreserved in duplicate (two 96 well plates). Additional 96-well plates were used to generate the pools that were then cultured in a T75 flask and harvested after two cycles growth by standard methods for genomic DNA isolation for QIseq (37). Although mutants grow at different rates, the pools can be expanded for at least 12 asexual life cycle generations (“cycles”) without significant loss in detectable diversity. Importantly, each mutant pool can be regenerated and cloned from their original MPs.
Datasets of reference genomes, transcriptome data and epigenetics data
The NF54 reference genome can be downloaded from (ftp://ftp.sanger.ac.uk/pub/project/pathogens/Plasmodium/falciparum/NF54/Assembly/V1_morphed/). The QIseq sequencing mapping followed previously published methods (37). Ortholog counts, paralog counts, and non-synonymous/synonymous (NonSyn/Syn) ratios can be downloaded from PlasmoDB (http://plasmodb.org/plasmo/) (38). Previously published RNA-seq expression data (26, 39) are used in computational analysis. Formaldehyde-Assisted Isolation of Regulatory Elements (FAIRE-seq) data of seven time points (0, 6, 12, 18, 24, 30 and 36 hour) were used to measure chromatin accessibility (40). The motif information of transcription factor ApiAP2 binding sites is available in previously published work (41). The genome synteny across five Plasmodium species is available in published work (25).
Identification of > 38,000 mutants carrying piggyBac insertion
The QIseq sequencing generated a total of 3,301,112 raw reads. First, all the raw reads were filtered by the criteria of matching expected transposon target site ‘TTAA’. This filtering step yielded 2,042,147 reads, which is >6-fold genome coverage. The insertion site QIseq signal was calculated as the reads count of the filtered QIseq reads. To compare the insertion signals between different QIseq runs, we normalized for each insertion i as
| (1) |
, where Sj represents the signal of the insertions i with reads counts ci in run R. All the piggyBac insertions with reads in both 5ʹ and 3ʹ ends were treated as true insertions by using an extremely high level of accuracy in target site (target motif TTAA ~ 99%). To identify true insertions with reads only in single 5ʹ or 3ʹ ends, we used a method that selected the most accurate signal cutoff based on the ratio of the number of input reads/ the number of output disrupted genes. We computed the number of gene CDSs targeted by the insertions upper cutoff (i.e. more accurate). We noticed that there was a single large and sharp increase in the number of CDSs targeted by the insertions with reads number increasing from cutoff 2.0 to cutoff 2.3 (Fig. S2E, p > 0.05 compared with changing another cutoff). The result indicated a large amount of false positive may be included when setting cutoff ≥ 2.3. Therefore, the normalized reads count = 2.3 was set as the lower bound for identifying true insertions.
Mutagenesis saturation computational validation
To evaluate the levels of whole-genome mutagenesis and eventual saturation, we used a computational method based on sampling. First, a specific number of insertions were randomly extracted and the fraction of genome elements targeted were counted. This procedure was repeated 1000 times for each query insertion number in fig. S1C. A log-based curve was observed by plotting the mutational events and the median number of genome elements targeted by insertions. The plateauing of the curve at about 20,000 mutational events indicated that a very small number of new genome elements could be targeted even with very large amount of insertions. Importantly, the real data-based sampling curve was very similar to our independent mathematical predictions of Negative Binomial distributions prior to the start of the saturation mutagenesis experiments (fig. S1D). Together, both our mathematical model and real-data computational sampling show that > 33,000 mutants represent saturation-level mutagenesis of the entire set of protein-encoding genes in the P. falciparum genome.
Sampling methods for accounting for fragment length differences and nucleoside composition bias
For genes in the P. falciparum genome, 5ʹ upstream and 3ʹ downstream intergenic regions displayed similar TTAA densities (median number of TTAA sites per 100bp were 1.82 and 1.89, respectively). However, they have significantly different region length distributions (median values are 1681bp and 926bp in 5ʹ and 3ʹ direction, respectively; p< 0.001 Wilcoxon test). Therefore, there are different numbers of genomic TTAA sites in 5ʹ and 3ʹ intergenic regions, as an intrinsic feature of the P. falciparum genome. To account for this region-length and total-TTAA bias in our QIseq calculations, the TTAA sites in 3ʹ intergenic regions were sampled based on the TTAA number in 5ʹ intergenic regions, so that our comparison was based on two identical background target-site distributions. Distribution of insertions and TTAA sites (Fig. 1E) were plotted based on these sampling data.
Mutagenesis index score (MIS) calculation
To quantify the mutability of every protein-coding gene in the P. falciparum genome, we first checked the number of piggyBac insertions located inside the gene CDS regions. To keep the criteria stringent, the insertions located on the very end (distance from insertion to TSS > 99% of the CDS length) of the CDS were not considered, due to a significantly higher number of insertions at the last 1% of the CDS compared with other regions of the CDS (fig. S2F). We calculated the initial score MIS of gene MSg based on the equation
| (2) |
where Ng represents the number of insertions on gene is the TTAA density of the geneg, which could be represented as the number of TTAA per kb of the CDS. We found that the MS could be decomposed into two mixed Gaussian distributions (Fig. S11). We reasoned that dispensable and essential genes exhibit different MS distribution. Therefore, a binary variable was used to model whether the gene g is dispensable or not: corresponds to dispensable and vice versa. The probability of observing a MSg of gene g is mixture of two Gaussian distributions:
| (3) |
Subsequently, the EM algorithm was implemented to search for the optimized value of parameters. The posterior distribution after the EM optimization procedures could be calculated as:
| (4) |
,where Θ is the parameter space. The MIS was defined as the posterior distribution to be dispensable.
Mutagenesis fitness score (MFS) calculation
For the mutants that were subjected to competitive growth essay, we calculated MFS to represent the comparative growth fitness of a mutant. The QIseq sequencing reads distribution reflects the fitness of unique piggyBac mutants in the competitive-growth assays. The start and the end of the growth assay could be represented as t0 and t. At the end of the assay, indicateed the relative abundance of each mutant m, targeted into the CDS of gene g in sample v. Therefore, the abundance of the mutant of the gene g at the end of the assay could be represented as . The was directly proportional to the QIseq reads number . Here, represents the normalized reads number of mutant m targeting gene g on CDS, which is . The QIseq reads starting with ‘TTAA’ were used to measure . To normalize different in different sample v, the average value of was calculated as
| (5) |
Here, I(·) is the indicator function. was the number of mutants targeting gene g in all samples. At the start of the assay t0, the relative abundance of each mutant equal to the background TTAA density Dg. Finally, the MFS could be calculated as:
| (6) |
Therefore, the MFS calculation based on individual mutant abundance is a proxy for competitive growth fitness in in vitro blood stage.
GO analysis
All the Gene Ontology (GO) terms were downloaded from (http://plasmodb.org/plasmo/)27. For each specific GO term g, the MIS distribution in the term g could be represented as MISg. The number of genes in term g is NUMg. We asked whether the genes in the term g are more prone to be dispensable or essential. To answer this question, we compared the median value of MISg with sampled data distribution as background. The genes with the same number of NUMg were sampled out and represented asSg. This procedure was repeated 1000 times and the median MIS of each was calculated as . The p-value was calculated as
| (7) |
Here, I(·) is the indicator function.
Half-maximal inhibitory concentration (IC50) estimation
To calculate IC50, the dose-response data (e.g., drug concentrations c1, c2, …cn and growth inhibition q1, q2, … qn) were used to fit a Hill Equation.
| (8) |
where c was drug concentrations in logarithmic form. The parameter C was the estimate of IC50. The goodness of fit was calculated from dose-response data and the Hill Equation.
Supplementary Material
Acknowledgements
Funding: This work was supported by the Wellcome Trust grant 098051 (J.C.R.), the National Institutes of Health grants R01 AI094973, R01 AI117017 (J.H.A.), and F32 AI112271 (J.O.).
Footnotes
Competing interests: U.S. patent 7932088 (26 April 2011), “High efficiency transformation of Plasmodium falciparum by the Lepidopteran transposon, piggyBac,” is held by inventors J.H.A., M. J. Fraser Jr., B. Balu, and D. A. Shoue. The invention relates to use of piggyBac as a tool for genetic manipulation of the Plasmodium genome.
Data and materials availability: All data and code to understand and assess the conclusions of this research are available in the main text and supplementary materials and are deposited into the European Nucleotide Archive. Accession numbers for individual experiments and libraries are listed in table S9. piggyBac transfection plasmids (MRA911/912) and mutants are deposited with the Malaria Research Reagent and Reference Repository (BEI Resources). piggyBac transfection plasmids and mutant parasites may be obtained directly from the authors at the University of South Florida, using a standard academic materials transfer agreement based on the Uniform Biological Material Transfer Agreement.
Supplementary Materials
References and Notes:
- 1.Ashley EA et al. , Spread of artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med 371, 411–423 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Woodrow CJ, White NJ, The clinical impact of artemisinin resistance in Southeast Asia and the potential for future spread. FEMS Microbiol Rev, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gardner MJ et al. , Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 419, 498–511 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ghorbal M et al. , Genome editing in the human malaria parasite Plasmodium falciparum using the CRISPR-Cas9 system. Nature biotechnology, (2014). [DOI] [PubMed] [Google Scholar]
- 5.Wagner JC, Platt RJ, Goldfless SJ, Zhang F, Niles JC, Efficient CRISPR-Cas9-mediated genome editing in Plasmodium falciparum. Nature methods 11, 915–918 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sidik SM et al. , A Genome-wide CRISPR Screen in Toxoplasma Identifies Essential Apicomplexan Genes. Cell 166, 1423–1435 e1412 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Koning-Ward TF, Gilson PR, Crabb BS, Advances in molecular genetic systems in malaria. Nat Rev Microbiol 13, 373–387 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Thibault ST et al. , A complementary transposon tool kit for Drosophila melanogaster using P and piggyBac. Nat Genet 36, 283–287 (2004). [DOI] [PubMed] [Google Scholar]
- 9.Fraser MJ, Smith GE, Summers MD, Acquisition of Host Cell DNA Sequences by Baculoviruses: Relationship Between Host DNA Insertions and FP Mutants of Autographa californica and Galleria mellonella Nuclear Polyhedrosis Viruses. J Virology 47, 287–300 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fraser MJ, Brusca JS, Smith GE, Summers MD, Transposon-mediated mutagenesis of a baculovirus. Virology 145, 356–361 (1985). [DOI] [PubMed] [Google Scholar]
- 11.Cary LC et al. , Transposon mutagenesis of baculoviruses: analysis of Trichoplusia ni transposon IFP2 insertions within the FP-locus of nuclear polyhedrosis viruses. Virology 172, 156–169 (1989). [DOI] [PubMed] [Google Scholar]
- 12.Fu J et al. , Targeted disruption of maebl in Plasmodium falciparum. Molecular and biochemical parasitology 141, 113–117 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ikadai H et al. , Transposon mutagenesis identifies genes essential for Plasmodium falciparum gametocytogenesis. Proceedings of the National Academy of Sciences of the United States of America 110, E1676–1684 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Balu B et al. , Atypical mitogen-activated protein kinase phosphatase implicated in regulating transition from pre-S-Phase asexual intraerythrocytic development of Plasmodium falciparum. Eukaryot Cell 12, 1171–1178 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Balu B et al. , CCR4-associated factor 1 coordinates the expression of Plasmodium falciparum egress and invasion proteins. Eukaryot Cell 10, 1257–1263 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pradhan A et al. , Chemogenomic profiling of Plasmodium falciparum as a tool to aid antimalarial drug discovery. Sci Rep 5, 15930 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bronner IF et al. , Quantitative insertion-site sequencing (QIseq) for high throughput phenotyping of transposon mutants. Genome Res 26, 980–989 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hart T et al. , High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell 163, 1515–1526 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Balu B et al. , piggyBac is an effective tool for functional analysis of the Plasmodium falciparum genome. BMC microbiology 9, 83 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Q et al. , Exonuclease-mediated degradation of nascent RNA silences genes linked to severe malaria. Nature 513, 431–435 (2014). [DOI] [PubMed] [Google Scholar]
- 21.Menard R et al. , Circumsporozoite protein is required for development of malaria sporozoites in mosquitoes. Nature 385, 336–340 (1997). [DOI] [PubMed] [Google Scholar]
- 22.Bobenchik AM et al. , Plasmodium falciparum phosphoethanolamine methyltransferase is essential for malaria transmission. Proceedings of the National Academy of Sciences of the United States of America 110, 18262–18267 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brancucci NMB et al. , Lysophosphatidylcholine Regulates Sexual Stage Differentiation in the Human Malaria Parasite Plasmodium falciparum. Cell 171, 1–13 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kooij TW et al. , A Plasmodium whole-genome synteny map: indels and synteny breakpoints as foci for species-specific genes. PLoS Pathog 1, e44 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DeBarry JD, Kissinger JC, Jumbled genomes: missing Apicomplexan synteny. Mol Biol Evol 28, 2855–2871 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Otto TD et al. , New insights into the blood-stage transcriptome of Plasmodium falciparum using RNA-Seq. Mol Microbiol 76, 12–24 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blomen VA et al. , Gene essentiality and synthetic lethality in haploid human cells. Science 350, 1092–1096 (2015). [DOI] [PubMed] [Google Scholar]
- 28.Wang T et al. , Identification and characterization of essential genes in the human genome. Science 350, 1096–1101 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bushell E et al. , Functional Profiling of a Plasmodium Genome Reveals an Abundance of Essential Genes. Cell 170, 260–272 e268 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vembar SS, Droll D, Scherf A, Translational regulation in blood stages of the malaria parasite Plasmodium spp.: systems-wide studies pave the way. WIREs RNA 7, 772–792 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dogovski C et al. , Targeting the cell stress response of Plasmodium falciparum to overcome artemisinin resistance. PLoS Biol 13, e1002132 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mbengue A et al. , A molecular mechanism of artemisinin resistance in Plasmodium falciparum malaria. Nature 520, 683–687 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li H et al. , Structure- and function-based design of Plasmodium-selective proteasome inhibitors. Nature 530, 233–236 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Van Voorhis WC et al. , Open Source Drug Discovery with the Malaria Box Compound Collection for Neglected Diseases and Beyond. PLoS Pathog 12, e1005763 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moll K, Kaneko A, Scherf A, Wahlgren M (EVIMalaR, MR4/BEI Resources, Glasgow, UK; Manassas, Virginia, 2013). [Google Scholar]
- 36.Maher SP, Zhang M, Balu B, Adams JH, Moll K, Kaneko A, Scherf A, Wahlgren M, Eds. (EVIMalR; MR4/ATCC, Glasgow, UK; Manassas, Virginia, 2013), pp. 391–396. [Google Scholar]
- 37.Bronner IF et al. , Quantitative Insertion-site Sequencing (QIseq) for high throughput phenotyping of transposon mutants. Genome research, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aurrecoechea C et al. , EuPathDB: the eukaryotic pathogen database. Nucleic acids research 41, D684–691 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.López-Barragán MJ et al. , Directional gene expression and antisense transcripts in sexual and asexual stages of Plasmodium falciparum. BMC Genomics 12, 587–587 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ponts N et al. , Nucleosome landscape and control of transcription in the human malaria parasite. Genome Res 20, 228–238 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Campbell TL, De Silva EK, Olszewski KL, Elemento O, Llinás M, Identification and Genome-Wide Prediction of DNA Binding Specificities for the ApiAP2 Family of Regulators from the Malaria Parasite. PLoS Pathog 6, e1001165 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.