

Abstract
Thrombotic microangiopathy (TMA) develops from various etiologies, and it is often difficult to distinguish the etiology of TMA in kidney transplantation. Antiphospholipid syndrome (APS) is one of the differential diagnoses for TMA that may cause acute loss of graft function or fatal thrombotic complications. This report details a 66-year-old male patient with polycythemia after ABO-incompatible kidney transplantation. Antibody screening tests were negative before transplant. Despite administration of an adequate desensitization therapy including plasmapheresis and rituximab, he developed acute graft dysfunction on postoperative day 112 and graft biopsy revealed prominent microvascular inflammation in the glomerular capillaries without immunoglobulin deposits. Flow cytometric panel-reactive antibody screening failed to detect donor-specific antibodies at both pre-transplant and episode biopsies. Anticardiolipin antibody was repeatedly positive, but neither thrombosis nor previous thrombotic episodes were detected. After excluding several differential diagnoses, the graft dysfunction with unexplained TMA was treated with steroid pulse, plasmapheresis and rituximab re-induction. Anticardiolipin antibody disappeared after this intensive treatment and graft function recovered gradually and stabilized for 52 months. This report suggests that asymptomatic anticardiolipin antibody may be associated with acute graft dysfunction. Even if thrombotic episodes are not observed, an exist of anticardiolipin antibody may be one of the risk factors of renal TMA after kidney transplantation.
Keywords: Living-donor, Antiphospholipid syndrome, Myeloproliferative disorders, Rituximab, Plasma exchange, Plasmapheresis, Graft biopsy
Introduction
Thrombotic microangiopathy (TMA) is an important complication of kidney transplantation and could determine graft survival. TMA in kidney transplantation develops from various etiologies such as antibody-mediated rejection, calcineurin inhibitor toxicity, and atypical hemolytic uremic syndrome [1–4]. Antiphospholipid antibody syndrome (APS) is an autoimmune diseases often associated with systematic lupus erythematosus, characterized by development of circulating antiphospholipid antibodies and recurrent thrombotic episodes. APS is a relatively rare cause of TMA in kidney transplantation, but may lead to severe complications that cause graft failure and patient death shortly after transplantation [5–12]. Manifestations of APS are quite variable, some of which do not meet the current criteria and are classified as “non-criteria” clinical manifestations [13, 14]. TMA in renal small vessels is one such “non-criteria” manifestation of APS [14–16]. In this case report, a kidney transplant recipient with polycythemia experienced acute graft dysfunction 4 months after ABO-incompatible kidney transplantation. Graft biopsy revealed TMA in glomerular capillary walls, suggesting APS with “non-criteria” manifestation.
Case report
A 66-year-old male was diagnosed as essential polycythemia at the age of 59 years and treated with antiplatelet agent. He developed anaphylactoid purpura at the age of 62 years, diagnosed by skin biopsy in a previous hospital and treated with temporal oral steroid. He initially visited our institution because of urinary abnormalities and gradual renal dysfunction at 63 years old. Kidney biopsy was not performed because of severe lumbar pain from disk herniation. Hydroxyurea was added to his treatment regimen for essential polycythemia. He developed end-stage kidney disease at 65 years old and started to receive hemodialysis. At 66 years old, the patient with blood type O expressed hope to receive an ABO-incompatible living-donor kidney transplant from his wife with blood type AB, because of frequent vascular access troubles and severe low back pain. Pre-operative screening test revealed positivity for anticardiolipin IgG antibody [20 IU/mL (normal range < 10 IU/mL)] and a mild stenosis of the aortic valve with coronary angiography. However, the patient was not administered specific treatment due to a lack of symptoms. Regarding his immunological background, human leukocyte antigen typing revealed four antigen mismatches in HLA-B and -DR. Complement-dependent cytotoxicity cross match, flow cytometry cross match and flow cytometric panel-reactive antibody screening test were all negative. Anti-A/B IgM showed titers of 64 and 32, respectively, while IgG titers were 512 and 256, respectively. He received antibody induction with basiliximab, and maintenance immunosuppression with tacrolimus, mycophenolate mofetil, and methylprednisolone. Rituximab and four sessions of plasmapheresis were included as desensitization therapy. He received living-donor kidney transplantation from his wife after 16 months of hemodialysis therapy. Serum creatinine decreased to 111 µmol/L and he was discharged on postoperative day 39.
The clinical course following transplantation is shown in Fig. 1. Serum creatinine degraded to 103 µmol/L at 2 months. Although graft function stabilized, a 3-month protocol biopsy revealed glomerulitis in varying degrees; mild in four, moderate in six, and severe in three of 23 glomeruli without peritubular capillaritis (Fig. 2a). On postoperative day 111, he was admitted to our hospital due to a rise in serum creatinine up to 214 µmol/L. Upon physical examination, blood pressure was 120/71 mmHg and consciousness was clear at admission. Neither diarrhea, arthralgia nor purpura were observed. In laboratory tests, no abnormalities were observed in urine or the coagulation and fibrinolytic systems. Although mild anemia and elevation of lactate dehydrogenase was observed (hemoglobin 111 g/L and lactate dehydrogenase 272 IU/L), hemolysis was thought to be absent because haptoglobin was within the normal range. Red-cell fragmentations were not observed. Platelet count was 56.2 × 104 /µL. Trough level of tacrolimus was 4.0 ng/mL. Immunological study found the anti-double-stranded DNA antibody titer to be 16 IU/mL and an anticardiolipin IgG antibody titer of 12 IU/mL (< 10 IU/mL). Antinuclear antibody, anti-neutrophil cytoplasmic antibodies, and cryoglobulin test were all negative and values of complement were within the normal range. Clinical signs of bacterial or viral infection including cytomegalovirus antigenemia and polyomavirus were not observed. An episode biopsy again revealed glomerulitis in varying degrees; mild in eight, moderate in two, and severe in one glomerulus of a total of 23 glomeruli (Fig. 2b–d). Mesangiolysis and double contour of the glomerular capillary was observed in four and five glomeruli, respectively (Fig. 2b, d). Focal and segmental mesangial proliferation was also noted (Fig. 2c). Moderate peritubular capillaritis was observed to be localized in the corticomedullary junction (Fig. 2e, f). Upon immunofluorescent examination, neither significant staining for immunoglobulin nor complement were observed, except for diffuse C4d staining in the peritubular capillary. Under electron microscopy, glomerular endothelial cells were swollen and lost fenestration with inflammatory cell infiltration (Fig. 3a, b). In the peritubular capillary, multilayering of the basement membrane was not observed (Fig. 3c, d). Re-examination with screening and single antigen test of flow cytometric panel-reactive antibody did not suggest the presence of donor-specific antibody. Thromboses were not detected in brain magnetic resonance imaging, body computed tomography and venous echography of the lower extremities. Although the symptoms did not meet the classification criteria for APS, the renal manifestation and episode of aortic stenosis did not conflict for “non-criteria” manifestation of APS in addition to repeatedly positive tests for anticardiolipin antibody. This patient with unexplained TMA was treated with steroid pulse, plasmapheresis and rituximab re-induction. Anticardiolipin antibody disappeared immediately and renal function stabilized slowly. Final follow-up of serum creatinine was 142 µmol/L and anticardiolipin IgG antibody decreased with a titer of 1 U/mL at 52 months after transplantation. During the observation period, platelet count stabilized and remained within the normal range.
Fig. 1.
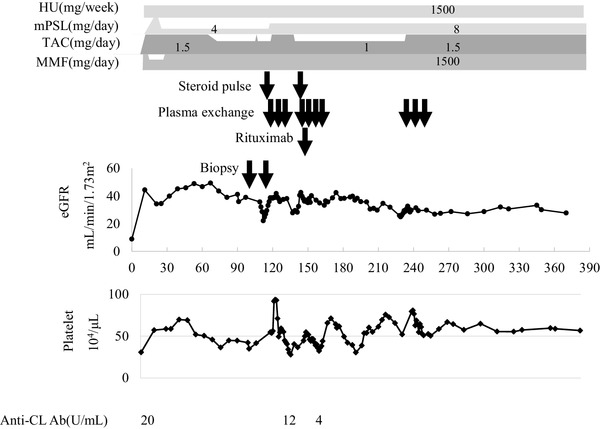
Clinical course. HU hydroxyurea, mPSL methylprednisolone, TAC tacrolimus, MMF mycophenolate mofetil, Anti-CL Ab, anticardiolipin antibody
Fig. 2.
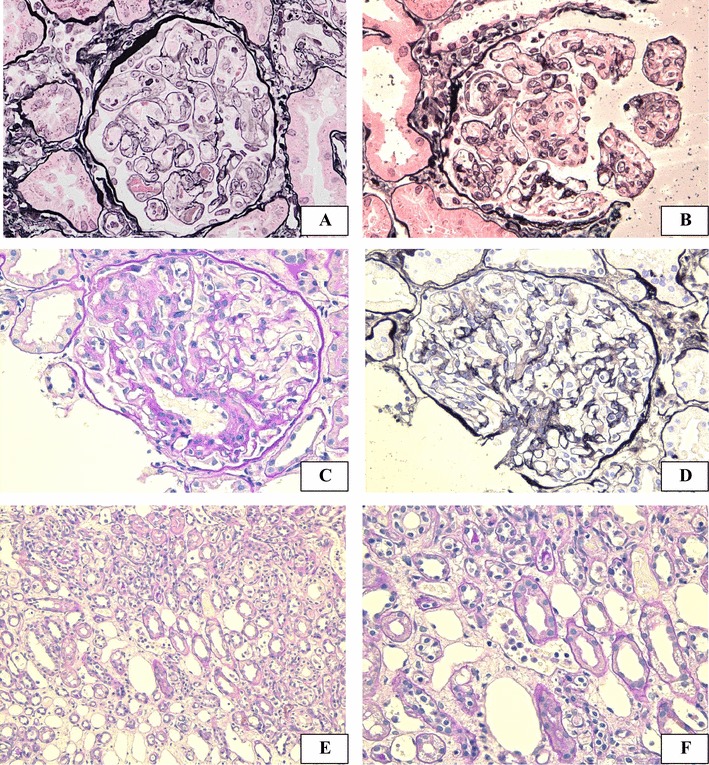
Light microscopy findings. Light microscopy of protocol biopsy at 3 months (a) and of episode biopsy at 4 months (b–f). The routine biopsy showed prominent glomerulitis, and double contour of glomerular basement membrane was seen (a). The episode biopsy showed glomerulitis with mesangiolysis (b), segmental increase in mesangial matrix (c) and double contour of glomerular basement membrane (d) was observed. Focal moderate peritubular capillaritis was observed in a localized area of the corticomedullary junction (e, f). Magnification: PAM × 200 in a, b and d, PAS × 200 in c, f, PAS × 100 in e
Fig. 3.
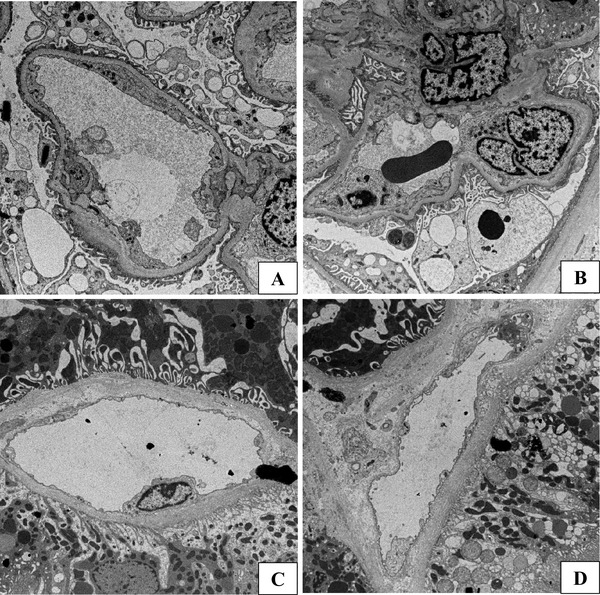
Electron microscopy findings. Electron microscopy of the episode biopsy at 4 months. In the glomerulus, swollen endothelial cells and loss of fenestration were observed (a, b) and an inflammatory cell infiltrated the capillary wall (b). In the peritubular capillary, injury to the endothelial cell was very slight and multilayering of the peritubular capillary wall was not observed (c, d). Magnification: ×1300 in a, ×1200 in b and × 1000 in c, d
Discussion
In this case report, a kidney transplant recipient experienced acute graft dysfunction in the glomerular capillaries after ABO-incompatible kidney transplantation with desensitization therapy consisting of plasmapheresis and rituximab. Episode biopsy at postoperative day 112 revealed TMA lesions mainly in the glomerular capillaries. Donor-specific antibodies were not detected either before transplantation or at the time of episode biopsy. Although no thrombotic episodes, hemolysis, or thrombocytopenia had been observed, anticardiolipin antibody was repeatedly positive. The patient did not fulfill the current criteria for APS, but following scrutiny, other differential diagnoses as causal factors of TMA were not apparent. We concluded that renal TMA lesion and aortic valve stenosis presented as a “non-criteria” manifestation of APS and thus treated with antibody depletion therapy. Graft function was improved and stabilized for 52 months.
TMA after kidney transplantation develops from various etiologies. The major causes are antibody-mediated rejection, calcineurin inhibitor-associated arteriolopathy, and atypical hemolytic uremic syndrome [1–4]. The majority of TMA after kidney transplantation demonstrates systematic manifestation with thrombocytopenia and hemolysis (62%), but some cases can be localized to grafts only without these hematological disorders (38%) [17]. Hence, the unexplained graft dysfunction in this case did not fulfill the classical criteria for APS and some differential diagnoses must be excluded. Antibody-mediated rejection is one of the most frequent causes of TMA after kidney transplantation. Acute glomerulitis and focal peritubular capillaritis in our case might suggest microvascular inflammation, a typical pathological feature of acute antibody-mediated rejection. Diffuse C4d staining in peritubular capillaries in this patient did not support the diagnosis of antibody-mediated rejection, because of ABO-incompatible transplantation. Despite the limitation that non-HLA antibodies were undetectable by flow cytometric panel-reactive antibody test, donor-specific HLA antibodies were not detected either before transplantation or at the time of episode biopsy. In addition, this episode of graft dysfunction occurred only 3 months after transplantation in spite of desensitization therapy for ABO-incompatible transplantation, including plasmapheresis and induction of rituximab. Histopathology showed that microvascular inflammation was localized to the glomeruli only, and peritubular capillaritis was minimal. Thus, we concluded that it was unlikely that antibody-mediated rejection contributed to the TMA lesions. Trough level of tacrolimus was within an appropriate range, and vacuolar degeneration of tubular epithelial cells was absent. Thus, tacrolimus toxicity was also unlikely. Although the patient had received a diagnosis of anaphylactoid purpura previously, a diagnosis of IgA vasculitis was easily excluded as there was no mesangial IgA deposition detectable by immunofluorescence or electron microscopy. Other types of immune-complex glomerulonephritis were also excluded for the same reason, including lupus nephritis and membranoproliferative glomerulonephritis. Focal segmental glomerulosclerosis was also an important differential diagnosis because of early recurrence after kidney transplantation. However, proteinuria usually precedes light microscopic changes in a recurrence of focal segmental glomerulosclerosis and diffuse foot process effacement under electron microscopy should be observable as a specific change [18]. Although the lesions in this case were focal and segmental, proteinuria did not precede graft dysfunction and diffuse foot process effacement was not observed. Thus, this case was unlikely to be recurrence of focal segmental glomerulosclerosis. Polycythemia was complicated in the patient and glomerulopathy associated with polycythemia must be reviewed. A few reports review myeloproliferative neoplasm-related glomerulopathies. A case series study identified 11 patients with myeloproliferative neoplasm-related glomerulopathy, two of which were polycythemia [19]. Proteinuria and mesangial proliferation and/or sclerosis was found in all the patients. Intracapillary hematopoietic cells were observed in four patients and chronic TMA in nine. Compared with this previous report, the degree of proteinuria was extremely low and mesangial proliferation was not observed in our case. Megakaryocytes were not observed under light microscopy and the histological findings were not typical, as seen in the previous report. In addition, recurrence of glomerulopathy after kidney transplantation has not been previously reported. Thus, a direct causal relationship between polycythemia and renal TMA in this case could not be confirmed. However, the development of renal TMA was possibly affected by endothelial injury induced by polycythemia.
APS is a relatively rare cause of TMA in kidney transplantation. The current classification criteria needs to satisfy clinical criteria, including vascular thrombotic episodes and/or pregnancy morbidity, and positive laboratory testing for lupus anticoagulant, anti-β2 glycoprotein I (β2GPI), or anticardiolipin antibody [13]. β2GPI-dependent anticardiolipin antibody was stimulated in a clotting-prone context such as in the presence of an inflammatory stimulus or pregnancy hormones, activated endothelial cells, monocytes and/or platelets and led to thrombotic events in large vessels [20]. However, manifestations of APS were quite variable, some of which did not meet the classification criteria and so were classified as “non-criteria” clinical manifestations [14]. Renal microangiopathy, so-called TMA in renal small vessels, was one such “non-criteria” manifestation of APS [14–16]. TMA associated with APS is often observed in patients without thrombosis or pregnancy loss. In addition, TMA lesions are not usually found in other thrombophilia disorders arising from genetic mutations. Thus, pathogenesis of thrombosis in large vessels and TMA may be result from distinct etiologies [21].
Previous reports have revealed fifteen cases of post-kidney transplant APS [5–12]. In four patients, APS was complicated with systematic erythematosus [11, 12]. Ten patients were diagnosed as APS before kidney transplantation, with positive tests for APS-associated auto-antibody and previous thrombotic episodes, such as deep venous thromboses or spontaneous abortions. However, renal TMA with anticardiolipin antibody appeared in five hepatitis C-positive recipients without previous thrombotic episodes or abortion [6]. Although onset of TMA was within 30 days of transplantation in most patients, the symptoms appeared 6 months after kidney transplantation in two patients. Surprisingly, extra-renal thrombotic complications after transplantation were observed in only three patients. In the report, narrowed capillary lumens in glomerular capillary wall were observed in all patients and marked swelling of endothelium in capillary wall was observed in one patient. Only one patient showed arterial platelet thrombus. In our case, although typical arterial lesions were not observed, the glomerular lesion is resembling with our case. Three patients died because of thrombotic complications and graft failure occurred in two patients. Renal prognosis of patients treated with plasma exchange and induction of eculizumab tends to be good. Recent studies have revealed that TMA lesions in APS are closely associated with complement protein C5 activation and attenuated after plasma exchange and induction of eculizumab [11, 22]. In Japan, use of eculizumab for APS is an expensive treatment which is not covered by public health insurance and so is currently used hesitantly. Our case indicates efficacy of plasmapheresis and rituximab for treatment of recurrent APS.
In conclusion, a kidney transplant recipient experienced acute graft dysfunction with TMA lesions in renal biopsy. The TMA lesions suggested latent APS with “non-criteria” manifestation. Screening tests for anticardiolipin antibody should be recommended for potential kidney transplant recipients with unidentified original disease.
Acknowledgements
We thank Gillian Campbell, PhD, from Edanz Group (http://www.edanzediting.com/ac) for editing a draft of this manuscript.
Abbreviations
- APS
Antiphospholipid syndrome
- β2GPI
β2glycoprotein I
- TMA
Thrombotic microangiopathy
Author contributions
TA participated in planning this research and data collection, MK, MY and UK analyzed the renal pathology, NT and DA participated in data collection, and YO, NM, TK, NT, and KT participated in the management of this research. All authors contributed to the writing of the manuscript.
Funding
Honoraria Takanari Kitazono (Bayer Pharmaceutical Co., Bristol-Myers Squibb Co., Daiichi-Sankyo Co.), Kazuhiko Tsuruya (Chugai Pharmaceutical Co., Kyowa Hakko Kirin Co.). Donations Takanari Kitazono (Astellas Pharma Inc., Daiichi-Sankyo Co., Eisai Co., Kyowa Hakko Kirin Co., Mitsubishi Tanabe Pharma Co., MSD K.K., Ono Pharmaceutical Co., Otsuka Pharmaceutical Co., Sanofi-Aventis Pharmaceutical Co., Takeda Pharmaceutical Co.), Kazuhiko Tsuruya (Chugai Pharmaceutical Co., Kyowa Hakko Kirin Co., Otsuka Pharmaceutical Co., Takeda Pharmaceutical Co.). Endowed department Kazuhiko Tsuruya (Baxter).
Ethics
The clinical and research activities of the study were consistent with the principles of the Declaration of Istanbul, as outlined in the Declaration of Istanbul on Organ Trafficking and Transplant Tourism. Informed consent was acquired from the participant included in the study.
Conflict of interest
The authors have no conflict of interest to declare.
Research involving human participants
Ethical Approval: All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee at which the studies were conducted (IRB approval number #24–54) and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
References
- 1.Kawaguchi K, Kawanishi K, Sato M, Itabashi M, Fujii A, Kanetsuna Y, et al. Atypical hemolytic uremic syndrome diagnosed four years after ABO-incompatible kidney transplantation. Nephrology (Carlton) 2015;20(Suppl 2):61–65. doi: 10.1111/nep.12465. [DOI] [PubMed] [Google Scholar]
- 2.Meehan SM, Kremer J, Ali FN, Curley J, Marino S, Chang A, et al. Thrombotic microangiopathy and peritubular capillary C4d expression in renal allograft biopsies. Clin J Am Soc Nephrol. 2011;6:395–403. doi: 10.2215/CJN.05870710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Takeda A, Ohtsuka Y, Horike K, Inaguma D, Goto N, Watarai Y, et al. A case of tacrolimus-associated thrombotic microangiopathy after ABO-blood-type-incompatible renal transplantation. Clin Transplant. 2011;25(Suppl 23):15–18. doi: 10.1111/j.1399-0012.2011.01453.x. [DOI] [PubMed] [Google Scholar]
- 4.Satoskar AA, Pelletier R, Adams P, Nadasdy GM, Brodsky S, Pesavento T, et al. De novo thrombotic microangiopathy in renal allograft biopsies-role of antibody-mediated rejection. Am J Transpl. 2010;10:1804–1811. doi: 10.1111/j.1600-6143.2010.03178.x. [DOI] [PubMed] [Google Scholar]
- 5.Mondragon-Ramirez G, Bochicchio T, Garcia-Torres R, Amigo MC, Martinez-Lavin M, Reyes P, et al. Recurrent renal thrombotic angiopathy after kidney transplantation in two patients with primary antiphospholipid syndrome (PAPS) Clin Transpl. 1994;8:93–96. [PubMed] [Google Scholar]
- 6.Baid S, Pascual M, Williams WW, Jr, Tolkoff-Rubin N, Johnson SM, Collins B, et al. Renal thrombotic microangiopathy associated with anticardiolipin antibodies in hepatitis C-positive renal allograft recipients. J Am Soc Nephrol. 1999;10:146–153. doi: 10.1681/ASN.V101146. [DOI] [PubMed] [Google Scholar]
- 7.Chew CG, Bannister KM, Mathew TH, Russ G, Seymour A. Thrombotic microangiopathy related to anticardiolipin antibody in a renal allograft. Nephrol Dial Transpl. 1999;14:436–438. doi: 10.1093/ndt/14.2.436. [DOI] [PubMed] [Google Scholar]
- 8.Erkan D, Leibowitz E, Berman J, Lockshin MD. Perioperative medical management of antiphospholipid syndrome: hospital for special surgery experience, review of literature, and recommendations. J Rheumatol. 2002;29:843–849. [PubMed] [Google Scholar]
- 9.Ruffatti A, Marson P, Valente M, Ciprian M, Tonello M, Marchini F, et al. Plasma exchange in a patient with primary antiphospholipid syndrome undergoing kidney transplantation. Transpl Int. 2007;20:475–477. doi: 10.1111/j.1432-2277.2007.00454.x. [DOI] [PubMed] [Google Scholar]
- 10.Hadaya K, Ferrari-Lacraz S, Fumeaux D, Boehlen F, Toso C, Moll S, et al. Eculizumab in acute recurrence of thrombotic microangiopathy after renal transplantation. Am J Transpl. 2011;11:2523–2527. doi: 10.1111/j.1600-6143.2011.03696.x. [DOI] [PubMed] [Google Scholar]
- 11.Canaud G, Kamar N, Anglicheau D, Esposito L, Rabant M, Noel LH, et al. Eculizumab improves posttransplant thrombotic microangiopathy due to antiphospholipid syndrome recurrence but fails to prevent chronic vascular changes. Am J Transpl. 2013;13:2179–2185. doi: 10.1111/ajt.12319. [DOI] [PubMed] [Google Scholar]
- 12.Barbour TD, Crosthwaite A, Chow K, Finlay MJ, Better N, Hughes PD, et al. Antiphospholipid syndrome in renal transplantation. Nephrology (Carlton) 2014;19:177–185. doi: 10.1111/nep.12217. [DOI] [PubMed] [Google Scholar]
- 13.Wilson WA, Gharavi AE, Koike T, Lockshin MD, Branch DW, Piette JC, et al. International consensus statement on preliminary classification criteria for definite antiphospholipid syndrome: report of an international workshop. Arthritis Rheum. 1999;42:1309–1311. doi: 10.1002/1529-0131(199907)42:7<1309::AID-ANR1>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 14.Abreu MM, Danowski A, Wahl DG, Amigo MC, Tektonidou M, Pacheco MS, et al. The relevance of “non-criteria” clinical manifestations of antiphospholipid syndrome: 14th International congress on antiphospholipid antibodies technical task force report on antiphospholipid syndrome clinical features. Autoimmun Rev. 2015;14:401–414. doi: 10.1016/j.autrev.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 15.Hughson MD, Nadasdy T, McCarty GA, Sholer C, Min KW, Silva F. Renal thrombotic microangiopathy in patients with systemic lupus erythematosus and the antiphospholipid syndrome. Am J Kidney Dis. 1992;20:150–158. doi: 10.1016/S0272-6386(12)80543-9. [DOI] [PubMed] [Google Scholar]
- 16.Levine JS, Branch DW, Rauch J. The antiphospholipid syndrome. N Engl J Med. 2002;346:752–763. doi: 10.1056/NEJMra002974. [DOI] [PubMed] [Google Scholar]
- 17.Schwimmer J, Nadasdy TA, Spitalnik PF, Kaplan KL, Zand MS. De novo thrombotic microangiopathy in renal transplant recipients: a comparison of hemolytic uremic syndrome with localized renal thrombotic microangiopathy. Am J Kidney Dis. 2003;41:471–479. doi: 10.1053/ajkd.2003.50058. [DOI] [PubMed] [Google Scholar]
- 18.Okamoto M, Koshino K, Sakai K, Nobori S, Matsuyama M, Ushigome H, et al. A case of recurrent focal segmental glomerulosclerosis (FSGS) involving massive proteinuria (> 50 g/day) immediately after renal transplantation. Clin Transpl. 2011;25(Suppl 23):53–58. doi: 10.1111/j.1399-0012.2011.01455.x. [DOI] [PubMed] [Google Scholar]
- 19.Said SM, Leung N, Sethi S, Cornell LD, Fidler ME, Grande JP, et al. Myeloproliferative neoplasms cause glomerulopathy. Kidney Int. 2011;80:753–759. doi: 10.1038/ki.2011.147. [DOI] [PubMed] [Google Scholar]
- 20.Meroni PL, Chighizola CB, Rovelli F, Gerosa M. Antiphospholipid syndrome in 2014: more clinical manifestations, novel pathogenic players and emerging biomarkers. Arthritis Res Ther. 2014;16:209. doi: 10.1186/ar4549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bienaime F, Legendre C, Terzi F, Canaud G. Antiphospholipid syndrome and kidney disease. Kidney Int. 2017;91:34–44. doi: 10.1016/j.kint.2016.06.026. [DOI] [PubMed] [Google Scholar]
- 22.Seshan SV, Franzke CW, Redecha P, Monestier M, Mackman N, Girardi G. Role of tissue factor in a mouse model of thrombotic microangiopathy induced by antiphospholipid antibodies. Blood. 2009;114:1675–1683. doi: 10.1182/blood-2009-01-199117. [DOI] [PMC free article] [PubMed] [Google Scholar]